 Fei Shen
Fei Shen Chenyang Hu
Chenyang Hu Xin Huang1
Xin Huang1 Deng Yang
Deng Yang Jirong Zhao
Jirong Zhao Xiaozeng Yang
Xiaozeng Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Plant Sci. , 18 September 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1232466
This article is part of the Research Topic Crop Improvement by Omics and Bioinformatics View all 17 articles
In plants, alternative splicing is a crucial mechanism for regulating gene expression at the post-transcriptional level, which leads to diverse proteins by generating multiple mature mRNA isoforms and diversify the gene regulation. Due to the complexity and variability of this process, accurate identification of splicing events is a vital step in studying alternative splicing. This article presents the application of alternative splicing algorithms with or without reference genomes in plants, as well as the integration of advanced deep learning techniques for improved detection accuracy. In addition, we also discuss alternative splicing studies in the pan-genomic background and the usefulness of integrated strategies for fully profiling alternative splicing.
Alternative splicing (AS) is a crucial mechanism for gene expression regulation, which entails the selection of different splice sites, removal of introns, and subsequent combine various exons to generate multiple mature mRNA isoforms in plants (Barbazuk et al., 2008). Plants generate extensive AS to increase the diversity of their transcriptomes, especially faced with complex environmental changes (Nilsen and Graveley, 2010; Szakonyi and Duque, 2018; Jia et al., 2022; Lam et al., 2022). There are several types of AS events in plants, including exon skipping (ES), intron retention (IR), alternative 5′ splice site (AE5′), alternative 3′ splice site (AE3′), mutually exclusive alternate exon splicing (MEE), alternative first exon (AFE), and alternative last exon (ALE) (Filichkin et al., 2010; E et al., 2013; Chen et al., 2020b). Among them, IR is the predominant type (Syed et al., 2012; Zhu et al., 2017).
The spliceosome is a large ribonucleoprotein complex that interacts with various trans-acting factors and is involved in controlling AS in plants (Will and Luhrmann, 2010; Ule and Blencowe, 2019; Liu et al., 2021; Jia et al., 2022). The U2 and U12 spliceosomal RNA are the focus RNAof most studies on the spliceosome (Hartmann, 2007; Reddy et al., 2012; Zhang et al., 2020). The spliceosome splices intron-exon junction sites, which are characterized by the conserved 5′-GT sequence and AG-3′ sequence. Non-snRNA (small nuclear RNA) splicing factors, such as serine/arginine-rich proteins and heterogeneous ribonucleoproteins, are known to facilitate the localization of splicing enhancers and inhibitors, thereby regulating the selection of splice sites (Geuens et al., 2016; Jeong, 2017; Chen et al., 2020a). Pre-mRNA undergoes two consecutive reactions to complete the splicing process: (i) introns form a unique chain-like structure; (ii) intron are rapidly degraded as a chain-like structure, and exons at the left and right ends are joined by phosphodiester bonds, achieving intron excision and exon joining (Black, 2003; Wan et al., 2019).
AS plays a crucial role in regulating plant growth, development and responses to abiotic stresses. AS generally occurs during seed germination, plant growth, and flowering stages. For example, AS of the NAC transcription factor 109 (NACTF109) during maize embryo development regulates seed dormancy by controlling ABA content in seeds (Thatcher et al., 2016). FLOWERING LOCUS C (FLC) is an important repressor of flowering in Arabidopsis (Andersson et al., 2008; Sharma et al., 2020), and AtU2AF65b is a splicing factor involved in ABA-mediated regulation of flowering time in Arabidopsis by splicing FLC pre-mRNA (Xiong et al., 2019; Lee et al., 2023). JASMONATE ZIM-DOMAIN (JAZ) is a key regulators of jasmonate (JA) signaling in plants (Yan et al., 2009). In Arabidopsis, the JAZ protein binds to the transcription factor MYC2 and inhibits JA signaling during quiescence. Binding to the hormone receptor CORONATINE INSENSITIVE 1 (COI1) upon hormone induction leads to degradation of JAZ. This degradation allows AtMED25 to activate MYC2 and promote JA signaling. AtMED25 regulates JAZ gene replacement splicing by recruiting splicing factors PRP39a and PRP40a, preventing excessive desensitization of JA signaling mediated by JAZ splice variants (Pauwels and Goossens, 2011; Wu et al., 2020). In rice (Oryza Sativa), OsDREB2 activates the expression of downstream genes involved in heat shock stress response and tolerance. The direct homolog of OsDREB2B enhances the ability of plants to cope with drought stress through AS by directly producing OsDREB2B2 by splicing I1, E2, and I2 at once under drought stress (Matsukura et al., 2010).
Different gene variants affecting alternative splicing (AS) have been observed in numerous functional gene studies. These variants play a crucial role in phenotypic changes. For instance, in poplar (Populus tomentosa), age-dependent AS triggers an aberrant splicing event in the pre-mRNA encoding PtRD26. This event leads to the production of a truncated protein, PtRD26IR, which acts as a dominant negative regulator of senescence by interacting with multiple senescence-associated NAC family transcription factors, inhibiting their DNA-binding activity (Wang et al., 2021). In Arabidopsis, the RNA-binding splicing factor SUPPRESSOR-OF-WHITE-APRICOT/SURP RNA-BINDING DOMAIN-CONTAINING PROTEIN1 (SWAP1) interacts with the splicing factor complexes SPLICING FACTOR FOR PHYTOCHROME SIGNALING (SFPS) and REDUCED RED LIGHT RESPONSES IN CRY1CRY2 BACKGROUND 1 (RRC1). These complexes regulate pre-mRNA splicing and induce alterations in photo morphology (Kathare et al., 2022). In bread wheat (Triticum aestivum), two variable splicers, Pm4b_V1 and Pm4b_V2, of the powdery mildew resistance gene Pm4b interact. In brief, Pm4b_V2 enhances wheat disease resistance by recruiting Pm4b_V1 from the cytoplasm to the endoplasmic reticulum (ER) by forming an ER-related complex (Sanchez-Martin et al., 2021).
The continuous advancement of RNA sequencing (next generation sequencing) and long-read isoform sequencing (Iso-seq) has significantly enhanced our ability to study alternative splicing comprehensively. Two primary computational approaches have been employed to investigate splicing diversity using RNA-seq data.
Transcript reconstruction methods: These approaches focus on inferring isoform usage frequency by utilizing probabilistic models to reconstruct each isoform based on the read distribution mapped to a specific gene. Typical software packages include Cufflinks (Trapnell et al., 2010), StringTie (Pertea et al., 2015), MISO (Yarden et al., 2010), SpliceGrapher (Mark et al., 2012). Indeed, transcriptome reconstruction is an exceptionally challenging problem in the field of bioinformatics and computational biology (Estefania et al., 2021). Single-molecule long-read sequencing technology has emerged as a valuable tool in transcriptome sequencing due to its ability to generate long reads with high throughput. The utilization of Iso-seq has become a preferred approach for sequencing more comprehensive and full-length transcriptomes, enabling the prediction and validation of gene models with greater accuracy and completeness. By producing long reads that can span entire transcript isoforms, Iso-seq overcomes some of the challenges associated with transcriptome reconstruction, such as accurately detecting complex splicing events and resolving alternative isoforms that may be missed by short-read sequencing. However, they are not suitable to pinpoint splicing events but whole sequences of transcripts. For instance, degraded and immature RNA as well as DNA fragments in the RNA samples can be erroneously identified as novel genes and transcripts in the Iso-seq data. In practice, tools such as TAMA software (Sim et al., 2020) could determine splice junctions and transcription start and end sites accurately. Unfortunately, the current cost of third-generation sequencing is high, and the detection of all transcripts may be limited by the depth of sequencing and the number of samples. Therefore, the development of tools combining RNA-seq and Iso-seq could effectively solve these problems. Regrettably, no mature tools have been released so far.
The second computational approach involves utilizing junction and/or exon information to infer, annotate, and identify novel splicing events (Table 1). Several methods, such as rMATS (Shen et al., 2014), MAJIQ (Vaquero-Garcia et al., 2016), and LeafCutter (Li et al., 2018), utilize junction information to identify these splicing events. On the other hand, DEXSeq (Anders and Huber, 2010) specifically focuses on analyzing the differential usage of exons between different experimental conditions. Two main methodologies are commonly used to quantify alternative splicing (AS) events: the percent spliced-in (PSI) and the splicing index (SI). PSI provides an estimate of the relative usage of each alternative pathway of an AS event. In contrast, the splicing index (SI) measures the relative signal or coverage of an exon or a junction compared to the entire gene.
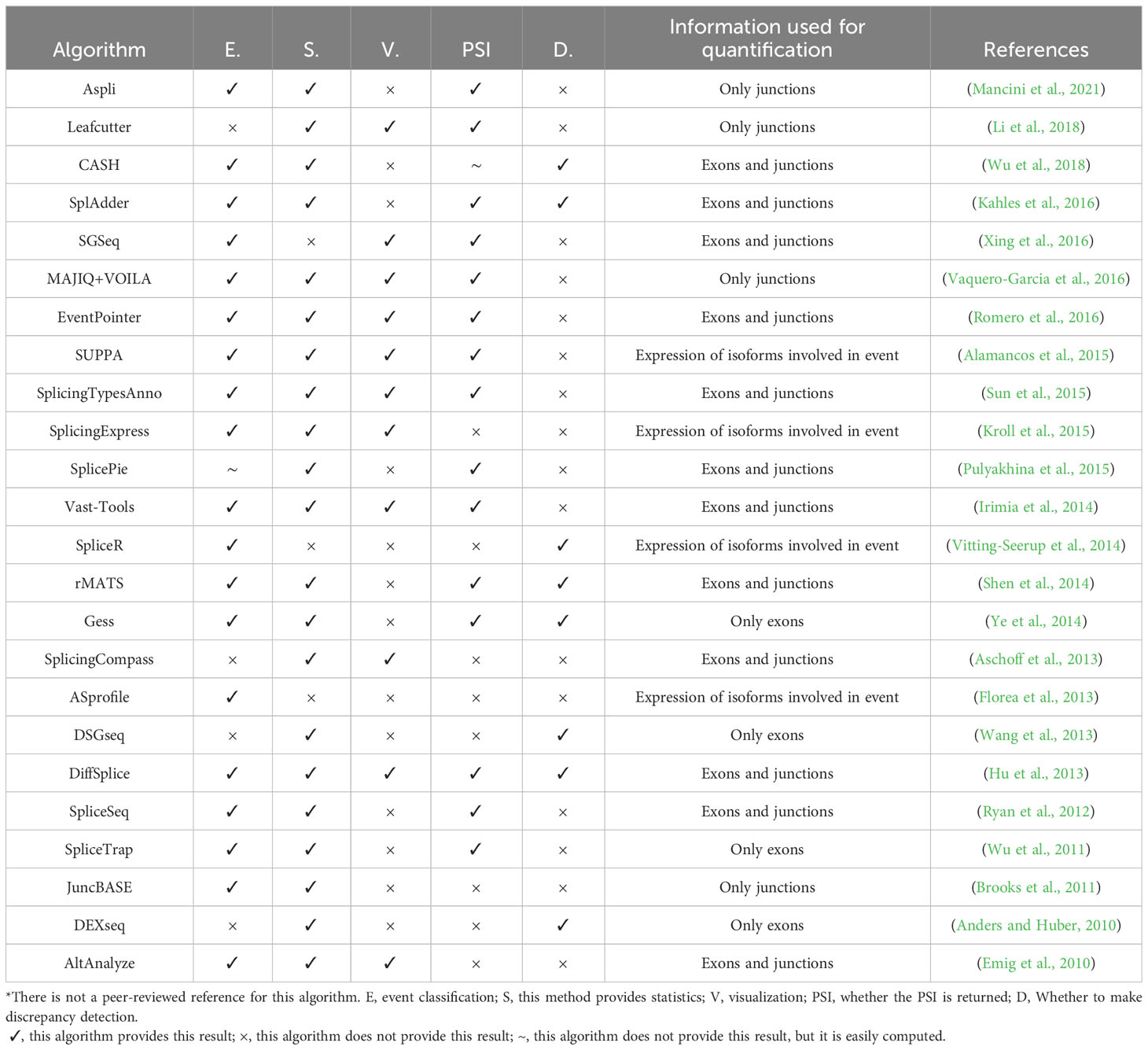
Table 1 Algorithms for the identification of Alternative Splicing events.
In addition to detecting different AS events, it is important to directly compare direct AS differences across samples. The Cuffdiff (Cufflinks) (Trapnell et al., 2010) package can test for differential splicing between isoforms in different samples. In addition, CASH (Wu et al., 2018), DEXseq (Anders and Huber, 2010), DiffSplice (Hu et al., 2013), Gess (Ye et al., 2014), rMATS (Shen et al., 2014), SplAdder (Kahles et al., 2016) and other software can use different algorithms to detect different AS events between different samples. But unfortunately, none of these AS analysis software takes into account the existence of variants. Direct analysis at the allele-aware level cannot be achieved. Allele-aware AS analysis software is of great significance in analyzing the causes of variable AS, such as comparing the differences in AS between different genomic haplotypes.
Several models have been developed for predicting and identifying alternative splicing events combining deep learning approaches (Table 2). For example, DeepASmRNA is a convolutional neural network (CNN) model capable of identifying alternative splicing events with over 90% accuracy (Cao et al., 2022). The Deep Splicing Code model uses raw RNA sequences to classify exons based on their alternative splicing behavior and performs well in identifying splice sites and motifs (Louadi et al., 2019). The deep-learning model AbSplice predicts anomalous splicing, increasing the accuracy of traditional DNA-based anomalous splicing prediction to 48% at a 20% call rate. Furthermore, integrating RNA-Seq raises the accuracy to 60% (Wagner et al., 2023). Additionally, the deep learning based computational framework called DARTS (deep-learning augmented RNA-seq analysis of transcript splicing) utilizes deep neural networks and Bayesian hypothesis testing for identifying exons based on their sequence characteristics, attaining a more than 95% accuracy rate in recognizing alternative splicing (Zhang et al., 2019). Finally the hybrid model combining CNN, recurrent neural network, and Long Short-Term Memory (LSTM) network has a splice locus identification accuracy of 96% (Nazari et al., 2019). In summary, deep learning models for alternative splicing detection have high detection accuracy, event classification, and splice site identification.
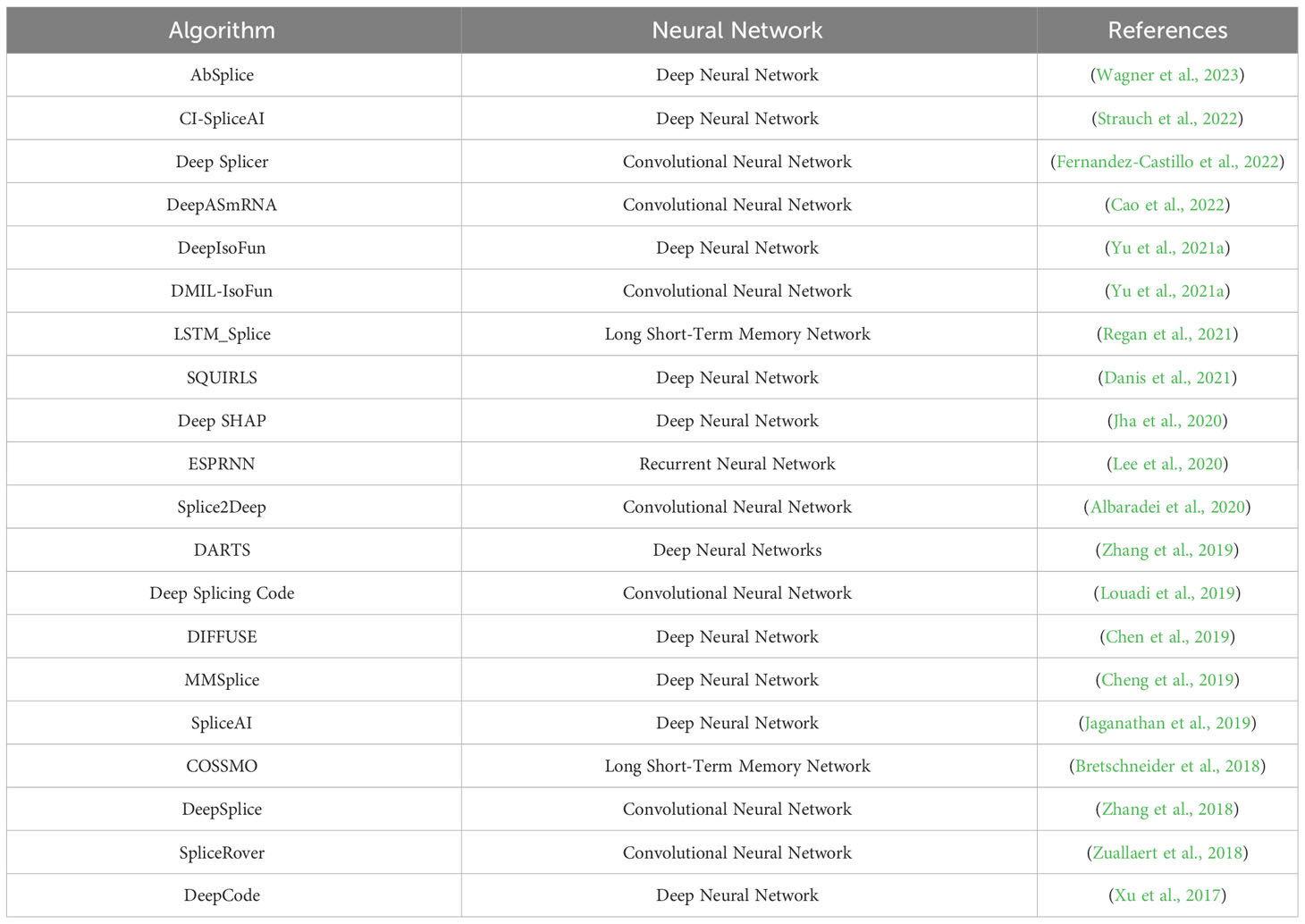
Table 2 Deep learning algorithms for predicting and recognizing Alternative Splicing events.
During the lengthy process of evolution, each plant develops unique genetic influenced by geographical and environmental factors. Consequently, the genome of a single plant can no longer fully represent all the genetic information of a species, and pan-genome of a species encompasses all the genetic information of a species and captures most of its genetic diversity and can help to explore plant genome evolution (Alonge et al., 2020; Liu et al., 2020; Long et al., 2021; Qin et al., 2021), crop molecular breeding (Tao et al., 2019; Yu et al., 2021b), and construction of genotype databases (Gui et al., 2020; Peng et al., 2020; Song et al., 2021). Similarly, the pan-transcriptome is a recalling concept of the pan-genome, which reflects the set of all transcripts of a species or an organism. The aggregation group integrating AS events from different genomes in a species can better represent the whole transcriptomes of the species and can better promote the study of AS biological processes. A tool RPVG (Sibbesen et al., 2023) was released to construct spliced pangenome graphs, to map RNA sequencing data to these graphs, and to perform haplotype-aware expression quantification of transcripts in a pantranscriptome.
The recent the developments of third-generation sequencing technologies and detection algorithms have led to significant advances in the study of alternative splicing. While much has been identified regarding the mechanism of alternative splicing generation and some of its functions, challenges remain in the detection of alternative splicing events without reference genomes. Using the third-generation reconstruction technology can reconstruct the AS version very well, but cannot directly determine the coordinates of the AS sites. Therefore, the algorithm combined with the second generation and the third generation sequencing technologies can solve most of such problems well. Compared with state-of-the-art methods, deep learning-based models have been used to improve the detection accuracy and the number of splicing events. Allele-aware AS analysis software is of great significance in analyzing the causes of variable AS, such as comparing the differences in AS between different genomic haplotypes. In the pan-genome context, it is of great significance to integrate different transcript information from different samples. Exploring the relationship between different alternative splicing events and mutations detected by different algorithms is of great significance for mining the influence of mutations on AS events.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
XY, JZ designed the study and methodology. FS, CH, XH, HH and JZ wrote the manuscript draft. XY performed writing-review, editing and supervision. All authors contributed to the article and approved the submitted version.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (32102339), Beijing Academy of Agriculture and Forestry Sciences (YXQN202203, QNJJ202106).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alamancos, G. P., Pages, A., Trincado, J. L., Bellora, N., Eyras, E. (2015). Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 21 (9), 1521–1531. doi: 10.1261/rna.051557.115
Albaradei, S., Magana-Mora, A., Thafar, M., Uludag, M., Bajic, V. B., Gojobori, T., et al. (2020). Splice2Deep: An ensemble of deep convolutional neural networks for improved splice site prediction in genomic DNA. Gene 763, 100035. doi: 10.1016/j.gene.2020.100035
Alonge, M., Wang, X., Benoit, M., Soyk, S., Pereira, L., Zhang, L., et al. (2020). Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182 (1), 145–161. doi: 10.1016/j.cell.2020.05.021
Anders, S., Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11 (10), R106. doi: 10.1186/gb-2010-11-10-r106
Andersson, C. R., Helliwell, C. A., Bagnall, D. J., Hughes, T. P., Finnegan, E. J., Peacock, W. J., et al. (2008). The FLX gene of Arabidopsis is required for FRI-dependent activation of FLC expression. Plant Cell Physiol. 49 (2), 191–200. doi: 10.1093/pcp/pcm176
Aschoff, M., Hotz-Wagenblatt, A., Glatting, K. H., Fischer, M., Eils, R., Konig, R. (2013). SplicingCompass: differential splicing detection using RNA-seq data. Bioinformatics 29 (9), 1141–1148. doi: 10.1093/bioinformatics/btt101
Barbazuk, W. B., Fu, Y., McGinnis, K. M. (2008). Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res. 18 (9), 1381–1392. doi: 10.1101/gr.053678.106
Black, D. L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336. doi: 10.1146/annurev.biochem.72.121801.161720
Bretschneider, H., Gandhi, S., Deshwar, A. G., Zuberi, K., Frey, B. J. (2018). COSSMO: predicting competitive alternative splice site selection using deep learning. Bioinformatics 34 (13), i429–i437. doi: 10.1093/bioinformatics/bty244
Brooks, A. N., Yang, L., Duff, M. O., Hansen, K. D., Park, J. W., Dudoit, S., et al. (2011). Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res. 21 (2), 193–202. doi: 10.1101/gr.108662.110
Cao, L., Zhang, Q., Song, H., Lin, K., Pang, E. (2022). DeepASmRNA: Reference-free prediction of alternative splicing events with a scalable and interpretable deep learning model. iScience 25 (11), 105345. doi: 10.1016/j.isci.2022.105345
Chen, H., Shaw, D., Zeng, J., Bu, D., Jiang, T. (2019). DIFFUSE: predicting isoform functions from sequences and expression profiles via deep learning. Bioinformatics 35 (14), i284–i294. doi: 10.1093/bioinformatics/btz367
Chen, M. X., Zhang, K. L., Gao, B., Yang, J. F., Tian, Y., Das, D., et al. (2020a). Phylogenetic comparison of 5' splice site determination in central spliceosomal proteins of the U1-70K gene family, in response to developmental cues and stress conditions. Plant J. 103 (1), 357–378. doi: 10.1111/tpj.14735
Chen, M. X., Zhang, K. L., Zhang, M., Das, D., Fang, Y. M., Dai, L., et al. (2020b). Alternative splicing and its regulatory role in woody plants. Tree Physiol. 40 (11), 1475–1486. doi: 10.1093/treephys/tpaa076
Cheng, J., Nguyen, T. Y. D., Cygan, K. J., Celik, M. H., Fairbrother, W. G., Avsec, Z., et al. (2019). MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 20 (1), 48. doi: 10.1186/s13059-019-1653-z
Danis, D., Jacobsen, J. O. B., Carmody, L. C., Gargano, M. A., McMurry, J. A., Hegde, A., et al. (2021). Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am. J. Hum. Genet. 108 (9), 1564–1577. doi: 10.1016/j.ajhg.2021.06.014
E, Z., Wang, L., Zhou, J. (2013). Splicing and alternative splicing in rice and humans. BMB Rep. 46 (9), 439–447. doi: 10.5483/BMBRep.2013.46.9.161
Emig, D., Salomonis, N., Baumbach, J., Lengauer, T., Conklin, B. R., Albrecht, M. (2010). AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res. 38 (Web Server issue), W755–W762. doi: 10.1093/nar/gkq405
Estefania, M., Andres, R., Javier, I., Marcelo, Y., Ariel, C. (2021). ASpli: Integrative analysis of splicing landscapes through RNA-Seq assays. Bioinformatics 37 (17), 2609–2616. doi: 10.1093/bioinformatics/btab141
Fernandez-Castillo, E., Barbosa-Santillan, L. I., Falcon-Morales, L., Sanchez-Escobar, J. J. (2022). Deep splicer: A CNN model for splice site prediction in genetic sequences. Genes (Basel) 13 (5), 907. doi: 10.3390/genes13050907
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R., Bryant, D. W., Fox, S. E., et al. (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 20 (1), 45–58. doi: 10.1101/gr.093302.109
Florea, L., Song, L., Salzberg, S. L. (2013). Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res 2, 188. doi: 10.12688/f1000research.2-188.v2
Geuens, T., Bouhy, D., Timmerman, V. (2016). The hnRNP family: insights into their role in health and disease. Hum. Genet. 135 (8), 851–867. doi: 10.1007/s00439-016-1683-5
Gui, S., Yang, L., Li, J., Luo, J., Xu, X., Yuan, J., et al. (2020). ZEAMAP, a comprehensive database adapted to the maize multi-omics era. iScience 23 (6), 101241. doi: 10.1016/j.isci.2020.101241
Hartmann, T. (2007). From waste products to ecochemicals: fifty years research of plant secondary metabolism. Phytochemistry 68 (22-24), 2831–2846. doi: 10.1016/j.phytochem.2007.09.017
Hu, Y., Huang, Y., Du, Y., Orellana, C. F., Singh, D., Johnson, A. R., et al. (2013). DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Res. 41 (2), e39. doi: 10.1093/nar/gks1026
Irimia, M., Weatheritt, R. J., Ellis, J. D., Parikshak, N. N., Gonatopoulos-Pournatzis, T., Babor, M., et al. (2014). A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159 (7), 1511–1523. doi: 10.1016/j.cell.2014.11.035
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell 176 (3), 535–548 e524. doi: 10.1016/j.cell.2018.12.015
Jeong, S. (2017). SR proteins: binders, regulators, and connectors of RNA. Mol. Cells 40 (1), 1–9. doi: 10.14348/molcells.2017.2319
Jha, A., Aicher, J. ,. K., Gazzara, M. ,. R., Singh, D., Barash, Y. (2020). Enhanced Integrated Gradients: improving interpretability of deep learning models using splicing codes as a case study. Genome Biol. 21 (1), 149. doi: 10.1186/s13059-020-02055-7
Jia, Z. C., Yang, X., Hou, X. X., Nie, Y. X., Wu, J. (2022). The importance of a genome-wide association analysis in the study of alternative splicing mutations in plants with a special focus on maize. Int. J. Mol. Sci. 23 (8), 4201. doi: 10.3390/ijms23084201
Kahles, A., Ong, C. S., Zhong, Y., Rätsch, G. (2016). SplAdder: identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 32 (12), 1840–1847. doi: 10.1093/bioinformatics/btw076
Kathare, P. K., Xin, R., Ganesan, A. S., June, V. M., Reddy, A. S. N., Huq, E. (2022). SWAP1-SFPS-RRC1 splicing factor complex modulates pre-mRNA splicing to promote photomorphogenesis in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 119 (44), e2214565119. doi: 10.1073/pnas.2214565119
Kroll, J. E., Kim, J., Ohno-MaChado, L., de Souza, S. J. (2015). Splicing Express: a software suite for alternative splicing analysis using next-generation sequencing data. PeerJ 3, e1419. doi: 10.7717/peerj.1419
Lam, P. Y., Wang, L., Lo, C., Zhu, F. Y. (2022). Alternative splicing and its roles in plant metabolism. Int. J. Mol. Sci. 23 (13), 7355. doi: 10.3390/ijms23137355
Lee, H. T., Park, H. Y., Lee, K. C., Lee, J. H., Kim, J. K. (2023). Two arabidopsis splicing factors, U2AF65a and U2AF65b, differentially control flowering time by modulating the expression or alternative splicing of a subset of FLC upstream regulators. Plants (Basel) 12 (8), 1655. doi: 10.3390/plants12081655
Lee, D., Zhang, J., Liu, J., Gerstein, M. (2020). Epigenome-based splicing prediction using a recurrent neural network. PloS Comput. Biol. 16 (6), e1008006. doi: 10.1371/journal.pcbi.1008006
Li, Y. I., Knowles, D. A., Humphrey, J., Barbeira, A. N., Dickinson, S. P., Im, H. K., et al. (2018). Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 50 (1), 151–158. doi: 10.1038/s41588-017-0004-9
Liu, Y., Du, H., Li, P., Shen, Y., Peng, H., Liu, S., et al. (2020). Pan-genome of wild and cultivated soybeans. Cell 182 (1), 162–176. doi: 10.1016/j.cell.2020.05.023
Liu, L., Tang, Z., Liu, F., Mao, F., Yujuan, G., Wang, Z., et al. (2021). Normal, novel or none: versatile regulation from alternative splicing. Plant Signaling Behav. 16 (7), e1917170. doi: 10.1080/15592324.2021.1917170
Long, Y., Liu, Z., Wang, P., Yang, H., Wang, Y., Zhang, S., et al. (2021). Disruption of topologically associating domains by structural variations in tetraploid cottons. Genomics 113 (5), 3405–3414. doi: 10.1016/j.ygeno.2021.07.023
Louadi, Z., Oubounyt, M., Tayara, H., Chong, K. T. (2019). Deep splicing code: classifying alternative splicing events using deep learning. Genes (Basel) 10 (8), 587. doi: 10.3390/genes10080587
Mancini, E., Rabinovich, A., Iserte, J., Yanovsky, M., Chernomoretz, A. (2021). ASpli: integrative analysis of splicing landscapes through RNA-Seq assays. Bioinformatics 37 (17), 2609–2616. doi: 10.1093/bioinformatics/btab141
Mark, F. R., Julie, T., Anireddy, S. R., Asa, B. (2012). SpliceGrapher: detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data. Genome Biol. 13 (1), R4. doi: 10.1186/gb-2012-13-1-r4
Matsukura, S., Mizoi, J., Yoshida, T., Todaka, D., Ito, Y., Maruyama, K., et al. (2010). Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol. Genet. Genomics 283 (2), 185–196. doi: 10.1007/s00438-009-0506-y
Nazari, I., Tayara, H., Chong, K. T. (2019). Branch point selection in RNA splicing using deep learning. IEEE Access 7, 1800–1807. doi: 10.1109/access.2018.2886569
Nilsen, T. W., Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463 (7280), 457–463. doi: 10.1038/nature08909
Pauwels, L., Goossens, A. (2011). The JAZ proteins: a crucial interface in the jasmonate signaling cascade. Plant Cell 23 (9), 3089–3100. doi: 10.1105/tpc.111.089300
Peng, H., Wang, K., Chen, Z., Cao, Y., Gao, Q., Li, Y., et al. (2020). MBKbase for rice: an integrated omics knowledgebase for molecular breeding in rice. Nucleic Acids Res. 48 (D1), D1085–D1092. doi: 10.1093/nar/gkz921
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295. doi: 10.1038/nbt.3122
Pulyakhina, I., Gazzoli, I., ‘t Hoen, P.-B., Verwey, N., den Dunnen, J., Aartsma-Rus, A., et al. (2015). SplicePie: a novel analytical approach for the detection of alternative, non-sequential and recursive splicing. Nucleic Acids Res. 43 (12), e80–e80. doi: 10.1093/nar/gkv242
Qin, P., Lu, H., Du, H., Wang, H., Chen, W., Chen, Z., et al. (2021). Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184 (13), 3542–3558 e3516. doi: 10.1016/j.cell.2021.04.046
Reddy, A. S., Rogers, M. F., Richardson, D. N., Hamilton, M., Ben-Hur, A. (2012). Deciphering the plant splicing code: experimental and computational approaches for predicting alternative splicing and splicing regulatory elements. Front. Plant Sci. 3. doi: 10.3389/fpls.2012.00018
Regan, K., Saghafi, A., Li, Z. (2021). Splice junction identification using long short-term memory neural networks. Curr. Genomics 22 (5), 384–390. doi: 10.2174/1389202922666211011143008
Romero, J. P., Muniategui, A., De Miguel, F. J., Aramburu, A., Montuenga, L., Pio, R., et al. (2016). EventPointer: an effective identification of alternative splicing events using junction arrays. BMC Genomics 17, 467. doi: 10.1186/s12864-016-2816-x
Ryan, M. C., Cleland, J., Kim, R., Wong, W. C., Weinstein, J. N. (2012). SpliceSeq: a resource for analysis and visualization of RNA-Seq data on alternative splicing and its functional impacts. Bioinformatics 28 (18), 2385–2387. doi: 10.1093/bioinformatics/bts452
Sanchez-Martin, J., Widrig, V., Herren, G., Wicker, T., Zbinden, H., Gronnier, J., et al. (2021). Wheat Pm4 resistance to powdery mildew is controlled by alternative splice variants encoding chimeric proteins. Nat. Plants 7 (3), 327–341. doi: 10.1038/s41477-021-00869-2
Sharma, N., Geuten, K., Giri, B. S., Varma, A. (2020). The molecular mechanism of vernalization in Arabidopsis and cereals: role of Flowering Locus C and its homologs. Physiol. Plant 170 (3), 373–383. doi: 10.1111/ppl.13163
Shen, S., Park, J. W., Lu, Z. X., Lin, L., Henry, M. D., Wu, Y. N., et al. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U.S.A. 111 (51), E5593–E5601. doi: 10.1073/pnas.1419161111
Sibbesen, J. A., Eizenga, J. M., Novak, A. M., Siren, J., Chang, X., Garrison, E., et al. (2023). Haplotype-aware pantranscriptome analyses using spliced pangenome graphs. Nat. Methods 20 (2), 239–247. doi: 10.1038/s41592-022-01731-9
Sim, M., Lee, J., Lee, D., Kwon, D., Kim, J. (2020). TAMA: improved metagenomic sequence classification through meta-analysis. BMC Bioinf. 21 (1), 185. doi: 10.1186/s12859-020-3533-7
Song, J. M., Liu, D. X., Xie, W. Z., Yang, Z., Guo, L., Liu, K., et al. (2021). BnPIR: Brassica napus pan-genome information resource for 1689 accessions. Plant Biotechnol. J. 19 (3), 412–414. doi: 10.1111/pbi.13491
Strauch, Y., Lord, J., NIranjan, M., Baralle, D. (2022). CI-SpliceAI-Improving machine learning predictions of disease causing splicing variants using curated alternative splice sites. PloS One 17 (6), e0269159. doi: 10.1371/journal.pone.0269159
Sun, X., Zuo, F., Ru, Y., Guo, J., Yan, X., Sablok, G. (2015). SplicingTypesAnno: annotating and quantifying alternative splicing events for RNA-Seq data. Comput. Methods Programs BioMed. 119 (1), 53–62. doi: 10.1016/j.cmpb.2015.02.004
Syed, N. H., Kalyna, M., Marquez, Y., Barta, A., Brown, J. W. (2012). Alternative splicing in plants–coming of age. Trends Plant Sci. 17 (10), 616–623. doi: 10.1016/j.tplants.2012.06.001
Szakonyi, D., Duque, P. (2018). Alternative splicing as a regulator of early plant development. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.01174
Tao, Y., Zhao, X., Mace, E., Henry, R., Jordan, D. (2019). Exploring and exploiting pan-genomics for crop improvement. Mol. Plant 12 (2), 156–169. doi: 10.1016/j.molp.2018.12.016
Thatcher, S. R., Danilevskaya, O. N., Meng, X., Beatty, M., Zastrow-Hayes, G., Harris, C., et al. (2016). Genome-wide analysis of alternative splicing during development and drought stress in maize. Plant Physiol. 170 (1), 586–599. doi: 10.1104/pp.15.01267
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28 (5), 511–515. doi: 10.1038/nbt.1621
Ule, J., Blencowe, B. J. (2019). Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol. Cell 76 (2), 329–345. doi: 10.1016/j.molcel.2019.09.017
Vaquero-Garcia, J., Barrera, A., Gazzara, M. R., Gonzalez-Vallinas, J., Lahens, N. F., Hogenesch, J. B., et al. (2016). A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 5, e11752. doi: 10.7554/eLife.11752
Vitting-Seerup, K., Porse, B. T., Sandelin, A., Waage, a.J. (2014). spliceR: an R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinf. 15, 81. doi: 10.1186/1471-2105-15-81
Wagner, N., Celik, M. H., Holzlwimmer, F. R., Mertes, C., Prokisch, H., Yepez, V. A., et al. (2023). Aberrant splicing prediction across human tissues. Nat. Genet. 55 (5), 861–870. doi: 10.1038/s41588-023-01373-3
Wan, R., Bai, R., Shi, Y. (2019). Molecular choreography of pre-mRNA splicing by the spliceosome. Curr. Opin. Struct. Biol. 59, 124–133. doi: 10.1016/j.sbi.2019.07.010
Wang, W., Qin, Z., Feng, Z., Wang, X., Zhang, X. (2013). Identifying differentially spliced genes from two groups of RNA-seq samples. Gene 518 (1), 164–170. doi: 10.1016/j.gene.2012.11.045
Wang, H. L., Zhang, Y., Wang, T., Yang, Q., Yang, Y., Li, Z., et al. (2021). An alternative splicing variant of PtRD26 delays leaf senescence by regulating multiple NAC transcription factors in Populus. Plant Cell 33 (5), 1594–1614. doi: 10.1093/plcell/koab046
Will, C. L., Luhrmann, R. (2010). Spliceosome structure and function. Cold Spring Harbor Perspect. Biol. 3 (7), a003707–a003707. doi: 10.1101/cshperspect.a003707
Wu, J., Akerman, M., Sun, S., McCombie, W. R., Krainer, A. R., Zhang, M. Q. (2011). SpliceTrap: a method to quantify alternative splicing under single cellular conditions. Bioinformatics 27 (21), 3010–3016. doi: 10.1093/bioinformatics/btr508
Wu, F., Deng, L., Zhai, Q., Zhao, J., Chen, Q., Li, C. (2020). Mediator subunit MED25 couples alternative splicing of JAZ genes with fine-tuning of jasmonate signaling. Plant Cell 32 (2), 429–448. doi: 10.1105/tpc.19.00583
Wu, W., Zong, J., Wei, N., Cheng, J., Zhou, X., Cheng, Y., et al. (2018). CASH: a constructing comprehensive splice site method for detecting alternative splicing events. Brief Bioinform. 19 (5), 905–917. doi: 10.1093/bib/bbx034
Xing, Y., Goldstein, L. D., Cao, Y., Pau, G., Lawrence, M., Wu, T. D., et al. (2016). Prediction and quantification of splice events from RNA-seq data. PloS One 11 (5), e0156132. doi: 10.1371/journal.pone.0156132
Xiong, F., Ren, J. J., Yu, Q., Wang, Y. Y., Lu, C. C., Kong, L. J., et al. (2019). AtU2AF65b functions in abscisic acid mediated flowering via regulating the precursor messenger RNA splicing of ABI5 and FLC in Arabidopsis. New Phytol. 223 (1), 277–292. doi: 10.1111/nph.15756
Xu, Y., Wang, Y., Luo, J., Zhao, W., Zhou, X. (2017). Deep learning of the splicing (epi)genetic code reveals a novel candidate mechanism linking histone modifications to ESC fate decision. Nucleic Acids Res. 45 (21), 12100–12112. doi: 10.1093/nar/gkx870
Yan, J., Zhang, C., Gu, M., Bai, Z., Zhang, W., Qi, T., et al. (2009). The Arabidopsis CORONATINE INSENSITIVE1 protein is a jasmonate receptor. Plant Cell 21 (8), 2220–2236. doi: 10.1105/tpc.109.065730
Yarden, K., Eric, T. W., Edoardo, M. A., Christopher, B. B. (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7 (12), 1009–1015. doi: 10.1038/nmeth.1528
Ye, Z., Chen, Z., Lan, X., Hara, S., Sunkel, B., Huang, T. H., et al. (2014). Computational analysis reveals a correlation of exon-skipping events with splicing, transcription and epigenetic factors. Nucleic Acids Res. 42 (5), 2856–2869. doi: 10.1093/nar/gkt1338
Yu, H., Lin, T., Meng, X., Du, H., Zhang, J., Liu, G., et al. (2021b). A route to de novo domestication of wild allotetraploid rice. Cell 184 (5), 1156–1170. doi: 10.1016/j.cell.2021.01.013
Yu, G., Zhou, G., Zhang, X., Domeniconi, C., Guo, M. (2021a). DMIL-IsoFun: predicting isoform function using deep multi-instance learning. Bioinformatics 37 (24), 4818–4825. doi: 10.1093/bioinformatics/btab532
Zhang, D., Chen, M.-X., Zhu, F.-Y., Zhang, J., Liu, Y.-G. (2020). Emerging functions of plant serine/arginine-rich (SR) proteins: lessons from animals. Crit. Rev. Plant Sci. 39 (2), 173–194. doi: 10.1080/07352689.2020.1770942
Zhang, Y., Liu, X., MacLeod, J., Liu, J. (2018). Discerning novel splice junctions derived from RNA-seq alignment: a deep learning approach. BMC Genomics 19 (1), 971. doi: 10.1186/s12864-018-5350-1
Zhang, Z., Pan, Z., Ying, Y., Xie, Z., Adhikari, S., Phillips, J., et al. (2019). Deep-learning augmented RNA-seq analysis of transcript splicing. Nat. Methods 16 (4), 307–310. doi: 10.1038/s41592-019-0351-9
Zhu, F.-Y., Chen, M.-X., Ye, N.-H., Shi, L., Ma, K.-L., Yang, J.-F., et al. (2017). Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings. Plant J. 91 (3), 518–533. doi: 10.1111/tpj.13571
Keywords: alternative splicing, RNA-seq, Iso-seq, detection algorithm, deep learning, pantranscriptome
Citation: Shen F, Hu C, Huang X, He H, Yang D, Zhao J and Yang X (2023) Advances in alternative splicing identification: deep learning and pantranscriptome. Front. Plant Sci. 14:1232466. doi: 10.3389/fpls.2023.1232466
Received: 31 May 2023; Accepted: 28 August 2023;
Published: 18 September 2023.
Edited by:
Xueqiang Wang, Zhejiang University, ChinaReviewed by:
Sen Yang, Henan Agricultural University, ChinaCopyright © 2023 Shen, Hu, Huang, He, Yang, Zhao and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jirong Zhao, empyNTIwOTk5QDEyNi5jb20=; Xiaozeng Yang, eWFuZ3h6QHNSTkF3b3JsZC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.