
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 27 July 2023
Sec. Plant Systematics and Evolution
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1224308
 Changying Xia1
Changying Xia1 Youwei Zuo1
Youwei Zuo1 Tiantian Xue2
Tiantian Xue2 Ming Kang3
Ming Kang3 Huan Zhang1
Huan Zhang1 Xiaoxia Zhang2
Xiaoxia Zhang2 Binru Wang1
Binru Wang1 Jiabin Zhang1
Jiabin Zhang1 Hongping Deng1,4*
Hongping Deng1,4*Introduction: Whole-genome resequencing technology covers almost all nucleotide variations in the genome, which makes it possible to carry out conservation genomics research on endangered species at the whole-genome level.
Methods: In this study, based on the whole-genome resequencing data of 101 critically endangered Artocarpus nanchuanensis individuals, we evaluated the genetic diversity and population structure, inferred the demographic history and genetic load, predicted the potential distributions in the past, present and future, and classified conservation units to propose targeted suggestions for the conservation of this critically endangered species.
Results: Whole-genome resequencing for A. nanchuanensis generated approximately 2 Tb of data. Based on abundant mutation sites (25,312,571 single nucleotide polymorphisms sites), we revealed that the average genetic diversity (nucleotide diversity, π) of different populations of A. nanchuanensis was relatively low compared with other trees that have been studied. And we also revealed that the NHZ and QJT populations harboured unique genetic backgrounds and were significantly separated from the other five populations. In addition, positive genetic selective signals, significantly enriched in biological processes related to terpene synthesis, were identified in the NHZ population. The analysis of demographic history of A. nanchuanensis revealed the existence of three genetic bottleneck events. Moreover, abundant genetic loads (48.56% protein-coding genes) were identified in Artocarpus nanchuanensis, especially in genes related to early development and immune function of plants. The predication analysis of suitable habitat areas indicated that the past suitable habitat areas shifted from the north to the south due to global temperature decline. However, in the future, the actual distribution area of A. nanchuanensis will still maintain high suitability.
Discussion: Based on total analyses, we divided the populations of A. nanchuanensis into four conservation units and proposed a number of practical management suggestions for each conservation unit. Overall, our study provides meaningful guidance for the protection of A. nanchuanensis and important insight into conservation genomics research.
The Dalou Mountains, with the famous Jinfo Mountains as the highest peak, are the boundary between the Guizhou Plateau and the Sichuan Basin (López-Pujol et al., 2011; Deng, 2019; Zu et al., 2022; Zu et al., 2023; Supplementary Figure 1). Local orogeny led to the formation of its present complex topography, which is mainly composed of low mountain canyons and middle mountain platforms. The humid and warm climate and high habitat heterogeneity together contributed to the abundant biodiversity of the Dalou Mountains. A recent study indicated that there were more than 5,000 species of vascular plants in the Dalou Mountains, including more than 200 plants for which type specimens were collected from the Dalou Mountains (Ding et al., 2014; Deng, 2019). Moreover, due to the Qinling Mountains-Daba Mountains in the north, the Dalou Mountains were less affected by the Quaternary ice age and retained many ancient relict species, such as Ginkgo biloba, Cathaya argyrophylla and Taxus chinensis (Wang and Ge, 2006; Tang et al., 2012; Qian et al., 2016; Supplementary Figure 1). Although the Dalou Mountains harbour abundant biodiversity, it has been given less attention than Daba Mountains and Wuling Mountain, which are also located in Chongqing City and designated as priority areas for biodiversity conservation. This has greatly hindered the further protection of endangered plants in this region, especially species with small populations.
Currently, many endangered and narrowly distributed plant species are found in the Dalou Mountains (Deng, 2019). For the rescue of endangered plants, ex - situ protection is generally considered to be an important and reliable means to prevent extinction (Huang and Zhang, 2012; Xu and Zang, 2022). However, recent studies have shown that ex - situ conservation of endangered plants with a lack of comprehensive population genetic information often leads to failure to cover the genetic diversity of wild populations and achieve protection of germplasm resources (Wu et al., 2013; Xu and Zang, 2022). Therefore, it is very important to identify conservation units for the protection and management of endangered species, which could provide a clear understanding of the distribution of genetic diversity and the boundary of population units for protection managers (Funk et al., 2012; Li et al., 2020; Xu et al., 2022). With recent developments in sequencing technology and reductions in sequencing costs, whole-genome resequencing has become increasingly popular because it covers almost all nucleotide variations in the genome (Hohenlohe et al., 2021; Ma et al., 2021; Ma et al., 2022). This has greatly helped accurately and clearly reveal the demographic history and genetic structure of endangered species, which are further helpful for the division of conservation units and the development of effective conservation strategies (Ma et al., 2021; Ma et al., 2022; Xu et al., 2022).
Climate change is considered to be the major threat to biodiversity in the 21st century because it may lead to loss of biodiversity, termination of evolutionary potential and disruption of ecological services (Thomas et al., 2004; Pimm, 2008; Dawson et al., 2011). For endangered species that are vulnerable to climate fluctuations, it is difficult to formulate appropriate conservation plans without considering the impact of climate change (Huang, 2011). Recent studies have indicated that climate change greatly threatens the population maintenance of endangered species and also drive the distribution pattern of endangered species to change, affecting the effectiveness of species conservation (Ackerly et al., 2010; Barber et al., 2016; Xue et al., 2022). Species distribution modelling combines species distribution records with environmental variables to predict the potential distribution range of species and has been widely used in the assessment of biodiversity conservation and priority management of many endangered species (Qin et al., 2017a; Tang et al., 2018; Gao et al., 2022; Xue et al., 2022).
Artocarpus nanchuanensis S.S. Chang, S.C. Tan, Z.Y. Liu, which is mainly distributed in the northern section of the Dalou Mountains, is the northernmost species in the natural distribution of the genus Artocarpus (Wu and Zhang, 1989; Figure 1; Supplementary Figure 1). This species is distinguished from other species of Artocarpus by two rows of alternate leaves, elliptic to nearly round leaves, irregular spherical polythalamic fruit with papillary protrusion on the surface, etc (Figure 1). Recent phylogenetic studies have indicated that this species belongs to the basal clade of the subg. Pseudojaca (Ser. Clavati), which contains mostly Chinese species (Gardner et al., 2021). The fruit of A. nanchuanensis is rich in nutrients and can be used for brewing, processing jam and beverages (He, 2014; He et al., 2022a). Straight trunks are often used for shipbuilding and making furniture (He, 2014). Although artificial breeding technologies for A. nanchuanensis have gradually been established in recent years owing to its high potential application value, only breeding bases and several parks currently have cultivation (Tan et al., 2015). In addition, due to its fragmented distribution and small wild population, this species was rated as critically endangered (CR) and as a Wild Plants with Extremely Small Populations (WPESP) in Chongqing City (Qin et al., 2017b). A recent field investigation indicated that A. nanchuanensis was distributed only in 4 counties of Chongqing City, and no more than 100 maternal trees remained (He, 2014). It was also found that adult trees were rare in most populations, and there was a pattern of more seedlings and fewer young trees (field survey data). Therefore, research on conservation biology and molecular genetics for this critically endangered species is urgently needed to support the formulation of further conservation plans.
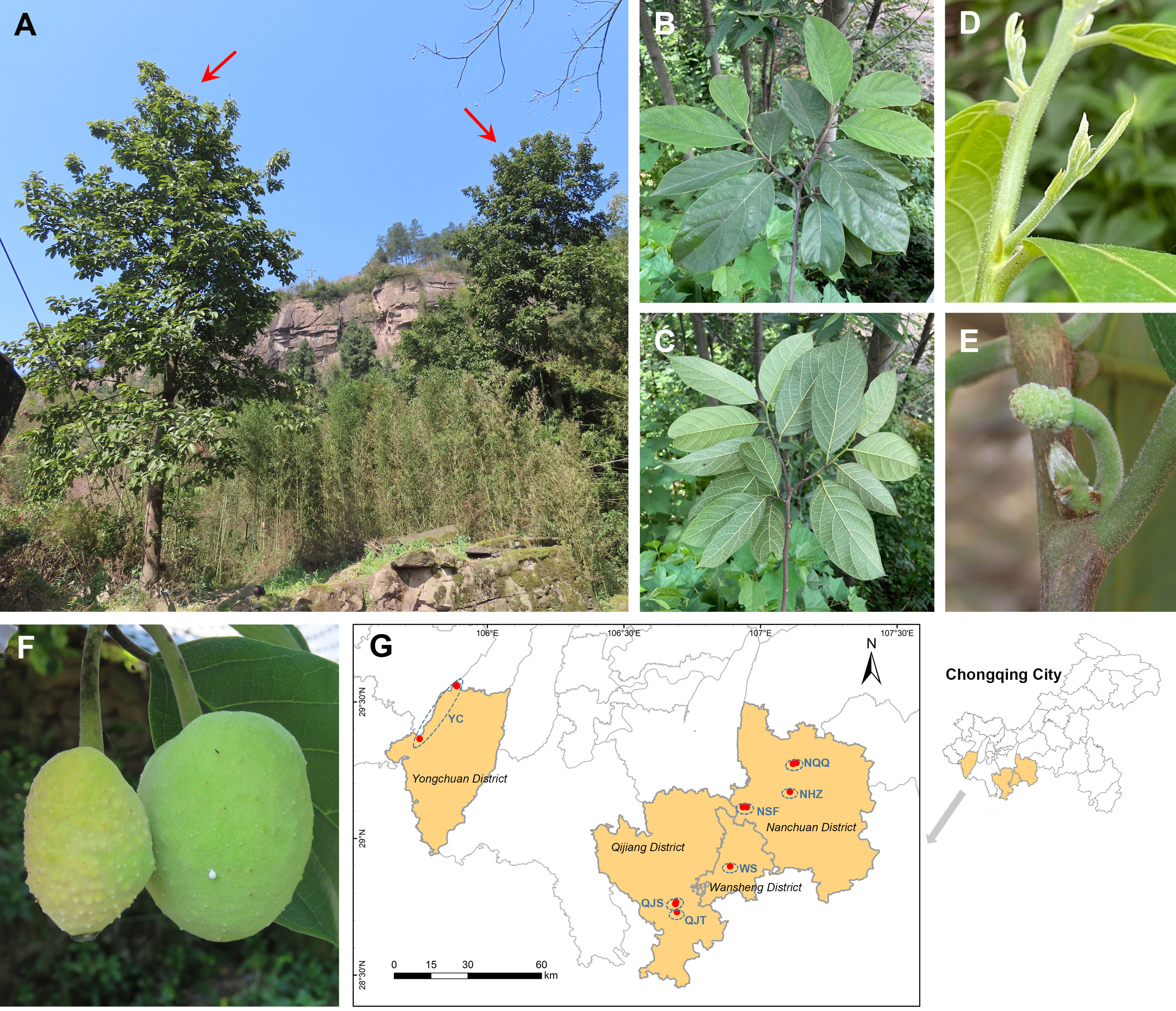
Figure 1 Wild habitats, morphological characters, and geographical distribution of Artocarpus nanchuanensis. (A) Representative photographs of A. nanchuanensis. (B, C) Leaves in front view and back view. (D) Leaf buds and stipules. (E) Inflorescences. (F) Fruits. (G) Locations of the sampled 7 populations.
The recently reported genome assembly at the chromosome level of A. nanchuanensis provided an opportunity to resequence at the whole genome level (He et al., 2022a). To reveal the genetic structure and demographic history of A. nanchuaensis fully and accurately and provide meaningful guidance for its conservation, 101 individuals from 7 populations of A. nanchuanensis were resequenced in this study. We aimed to: (1) evaluate the genetic diversity and population structure of A. nanchuanensis; (2) infer the demographic history of this species and evaluate its genetic load; (3) predict the potential distributions of this species in the past, present and future; and (4) use our analysis results to classify conservation units and propose targeted suggestions for the conservation of this critically endangered species.
We collected 101 individuals from 7 populations of A. nanchuanensis from Yongchuan District, Qijiang District, Wansheng District and Nanchuan District in southwestern Chongqing City, China (Figure 1; Supplementary Table 1). Due to the scarcity of adult and young trees of A. nanchuanensis, we first sampled all adult and young tree individuals from each population, and then sampled seedlings at intervals of three meters. The number of individuals sampled from each population ranged from 11 to 27. Two populations from Yongchuan District were merged due to following reasons. (1) These two populations are located in the same branch range of Dalou Mountains (Supplementary Figure 1). (2) Poorly differentiated and similar genetic backgrounds were detected between them. (3) These two are closest, but far away from populations in other regions.
A tender leaf of each selected individual was collected and taken back to the laboratory for storage at -80 degrees Celsius. Genomic DNA was extracted from samples using the standard cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987). A Qubit4 Fluorometer nucleic acid protein fluorescence quantitative instrument and agarose gel electrophoresis were used to detect the concentration and quality of total genomic DNA, respectively. To construct paired-end libraries, we performed following treatments on the total genomic DNA (1 μg) of each sample: fragmentation, end repairing, adaptor ligation, PCR amplification and PCR product cyclization (Huang et al., 2018). The Agilent 2100 Bioanalyzer (Agilent DNA 1000 Reagents) was used to detect the fragment size and concentration of the constructed library. All libraries were sequenced on the BGISEQ-500 sequencing platform, which uses optimized Combinatorial Probe-Anchor Synthesis (cPAS) and DNA nanospheres (DNB) core sequencing technology, producing 150 bp pair end reads (Huang et al., 2018).
Raw data with adapter sequences or low-quality sequences were filtered using SOAPnuke with the following parameters: -n 0.01 -l 20 -q 0.3 –adaMR 0.25 –ada_trim –polyX 50 –minReadLen 150 (Chen et al., 2018). BWA-MEM (Li, 2013) and default parameters were used to map paired-end clean reads to the reference genome of A. nanchuanensis (He et al., 2022b). SAMtools (Danecek et al., 2021) and Picard (http://picard.sourceforge.net/) were used to sort and delete duplicate reads, respectively. Then, we employed the mpileup + call (–multiallelic-caller –variants-only) and filter modules (-g 3 -G 10 -e “%QUAL<10”) in BCFtools (Danecek et al., 2021) to call and filter variable sites in each sample, and the view module (–types snps) in BCFtools was used to extract single nucleotide polymorphisms sites (SNPs) to obtain Dataset 1, including 25,312,571 SNPs. To ensure high-quality SNPs, VCFtools v0.1.17 (Danecek et al., 2011) was utilized to further filter Dataset 1, with the following settings: –maf 0.01 –max-missing 0.9 –min-alleles 2 –max-alleles 2. After filtering, a total of 18,906,649 SNPs (Dataset 2) remained for downstream analysis. SnpEff 5.0e (Cingolani et al., 2012) was used for SNP annotation. We first used SnpEff 5.0e to construct a new database based on the assembly and annotation file of the A. nanchuanensis genome, and then annotated Dataset 1 by using the constructed database and default parameters.
Principal component analysis (PCA) was conducted by using Plink (Purcell et al., 2007), and the R package (ggplot2; Wickham, 2016) was used for visualization of the analysis results. Population structure was analyzed by Admixture (Alexander et al., 2009), which could infer the stratification relationships of populations. First, PLINK (Purcell et al., 2007) was used to filter the linkage sites and obtain a temporary file containing the sites that needed to be preserved from Dataset 2 with the following parameters: indep pairwise 50 10 0.2. Independent SNPs (Dataset 3) were extracted based on the temporary file and Dataset 2, yielding a total of 5,146,515 SNPs. Then, the bed file converted from the vcf file was used as the input file of Admixture for population structure analysis. According to the total number of natural populations, the number of clusters K was set from 2 to 7. The optimal K value was determined based on the actual distribution information and PCA results. Finally, the results were visualized by Poppelper (Francis, 2017) based on the Q matrix obtained from the Admixture analysis.
Based on the latest research of Artocarpus, we downloaded the resequencing data of five other species of Artocarpus (A. hypargyreus, A. styracifolius, A. tonkinensis, A. petelottii, and A. gomezianus) to explore the phylogenetic status of A. nanchuensis (Gardner et al., 2021; Lin et al., 2022; Supplementary Table 2). The first four species are thought to be in the same clade as A. nanchuensis (ser. Clavati), and the last one belongs to the sisters clade (ser. Peltati) of ser. Clavati (Gardner et al., 2021). These data were processed with the method described in section 2.2 to obtain SNP data. This dataset, along with Dataset 3 of A. nanchuanensis, was used to construct the phylogenetic tree by IQ-TREE (Nguyen et al., 2015). We first converted the vcf file of the combined dataset to phylip format and then used the following settings for tree building: automatic model selection, unpartitioned, and 1000 fast bootstrap analyses. The obtained phylogenetic tree was rooted with A. gomezianus (Gardner et al., 2021). In addition, based on the annotation results from SnpEff 5.0e (Cingolani et al., 2012), we calculated the genetic diversity of synonymous (πS) and nonsynonymous sites (πN) of each population using VCFtools with a 100-kb sliding window and a step size of 10 kb. Genetic diversity was defined as the average number of pairwise nucleotide differences (nucleotide diversity, π) in this study (Nei and Li, 1979; Tajima, 1983; Corbett-Detig et al., 2015). VCFtools v0.1.17 (Danecek et al., 2011) was also used to calculate other population genetic parameters with a 100-kb sliding window and a step size of 10 kb based on Dataset 2, including fixation statistics (FST) and π ratio values between each population pair.
To identify the potential selective sweeps in the two special populations of A. nanchuanensis (NHZ and QJT), we selected the windows with high genetic differentiation (top 1% of FST and top 1% of nucleotide diversity ratio values (πQJT/πNHZ)) as the candidate divergent regions (CDRs) by using Microsoft Excel. Candidate genes were obtained from CDRs and GO enrichment analysis was carried out by using TBtools (Chen et al., 2020). Then we extracted the main genes from the significantly enriched GO items and analyzed the clustering of these genes in the corresponding gene family. The complete members of the gene family in A. nanchuanensis were searched and confirmed by HMMER 3.0 (Finn et al., 2015) and the NCBI Conserved Domain database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). In addition, the genes of the corresponding gene family in Arabidopsis thaliana were downloaded from the Phytozome database (Goodstein et al., 2012) and used together with the genes of A. nanchuanensis to construct a phylogenetic tree.
To reconstruct the detailed demographic history of A. nanchuanensis, BCFtools (mpileup + call module) (Danecek et al., 2021) was used to obtain the consensus sequence between the sample and reference genome, and the fq2psmcfa module embedded in paired sequential Markov condensation (PSMC) (Li and Durbin, 2011) was used to convert its output file into fasta-like format. Then, the demographic history of each population was inferred by a PSMC model (Li and Durbin, 2011) with the following parameter settings: - N25 - t15 - r5 - p “4 + 25 * 2 + 4 + 6. The generation time was set to 11 years on the basis of years of field observation of A. nanchuanensis, and the mutation rate per generation was set to 2.86e-8 (11* 2.6e-9) (Gaut et al., 1996).
It is predicted that deleterious mutations (genetic load) will destroy gene function, which would significantly reduce the fitness of individuals in the population (Mattila et al., 2012). Moreover, loss of function (LOF) was the major type of deleterious mutations. To estimate the genetic load of A. nanchuanensis, we first defined the dominant alleles in all individuals (more than 50% of individuals had homozygous alleles) as the ancestral allele genotype (Hu et al., 2021). Then, the ancestral allele loci of each individual were extracted and annotated by using VCFtools v0.1.17 (Danecek et al., 2011) and SnpEff 5.0e (Cingolani et al., 2012), respectively. For each individual, we calculated the number of homozygous LOF sites and the number of genes containing one or more homozygous LOF sites divided by the total number of protein-coding genes (PCGs). Finally, gene ontology (GO) analysis was applied to characterize the detected genes containing homozygous LOF sites based on the genome annotation results of recent studies by using TBtools (Chen et al., 2020; He et al., 2022a).
To further reveal the change in the distribution pattern of A. nanchuanensis, MaxEnt (version 3.3) (Phillips et al., 2006) was used to predict the past (mid Pliocene warm period - ca. 3.205 Ma, and MIS19-ca. 787 ka), current (1979-2013) and future (2070 years, RCP4.5) potential suitable habitat areas. Since five bioclimatic variables were missing in the two past periods, only 14 bioclimatic variables were downloaded for the prediction (Supplementary Table 3). The 14 bioclimatic variables of current climate data and paleoclimate data were downloaded from the Paleoclim database (Brown et al., 2018) with 2.5 min resolution. The future climate data (2070, BCC-CSM1-1 model, RCP 45) were downloaded from the WorldClim database (https://worldclim.org/data/v1.4/cmip5_2.5m.html). To minimize the impact of multicollinearity and overfitting on the stability and quality of the model, the 14 bioclimatic variables were analyzed with the Pearson correlation test. When the correlation coefficient was > |0.80|, one of the two variables was removed. Ultimately, six bioclimatic variables were selected to predict the suitable habitat areas for four time periods, including BIO1 (annual mean temperature), BIO4 (temperature seasonality), BIO8 (mean temperature of wettest quarter), BIO12 (annual precipitation), BIO14 (precipitation of driest month), and BIO15 (precipitation seasonality; Supplementary Table 3). We obtained 23 distribution records from the field investigations, which covered most of the known living areas of A. nanchuanensis (Supplementary Table 4). We also removed duplicate records in the same grid cell at a spatial resolution of 2.5 arc minutes to reduce spatial autocorrelation (Boria et al., 2014). Finally, a total of 18 distribution records were used in the analysis. In addition, we extracted the six bioclimate variables from these 18 records for principal component analysis (R package). The first two principal components (PC1 and PC2) were extracted from the analysis to estimate the main changes in bioclimatic variables represented by climate instability.
The whole-genome resequencing of 101 individuals generated approximately 2 Tb of data. The average coverage, average depth and primary mapping rate of all samples were 98.00%, 39.23 and 97.57%, respectively (Supplementary Table 5). The annotation results of all SNPs indicated that 60.80% of the SNPs were located in intergenic regions, 13.12% were located in exon regions, and 8.18% were located in intron regions (Supplementary Table 6).
The PCA indicated that the first three principal components (PCs) explained 16.2%, 12.9% and 10.7% of the total genetic variance, respectively. PC1, PC2, and PC3 significantly separated the NHZ and QJT populations from all other populations, but did not separate the remaining five populations (Figure 2A; Supplementary Figures 2, 3). The analysis of population structure showed that for the optimal K value (K=4), all samples were divided into four groups, including QJT, NHZ, QJS and four other populations (Figure 2C). It is worth noting that the genetic backgrounds of the NHZ and QJT populations were quite simple, with only a few genetic mixings compared to other populations (Figure 2C). The QJS population, which was geographically close to the QJT population (only separated by a 30-meter-wide river), had a distinct genetic mixture of QJT and the other four populations (NQQ, NSF, WS and YC). However, there was no significant difference in genetic background among the four populations from Nanchuan District, Wansheng and Yongchuan District (NQQ, NSF, WS and YC) (Figure 2C). The FST analysis indicated that the degrees of differentiation between various populations of A. nanchuanensis were relatively low, all less than 0.01 (Supplementary Table 7).
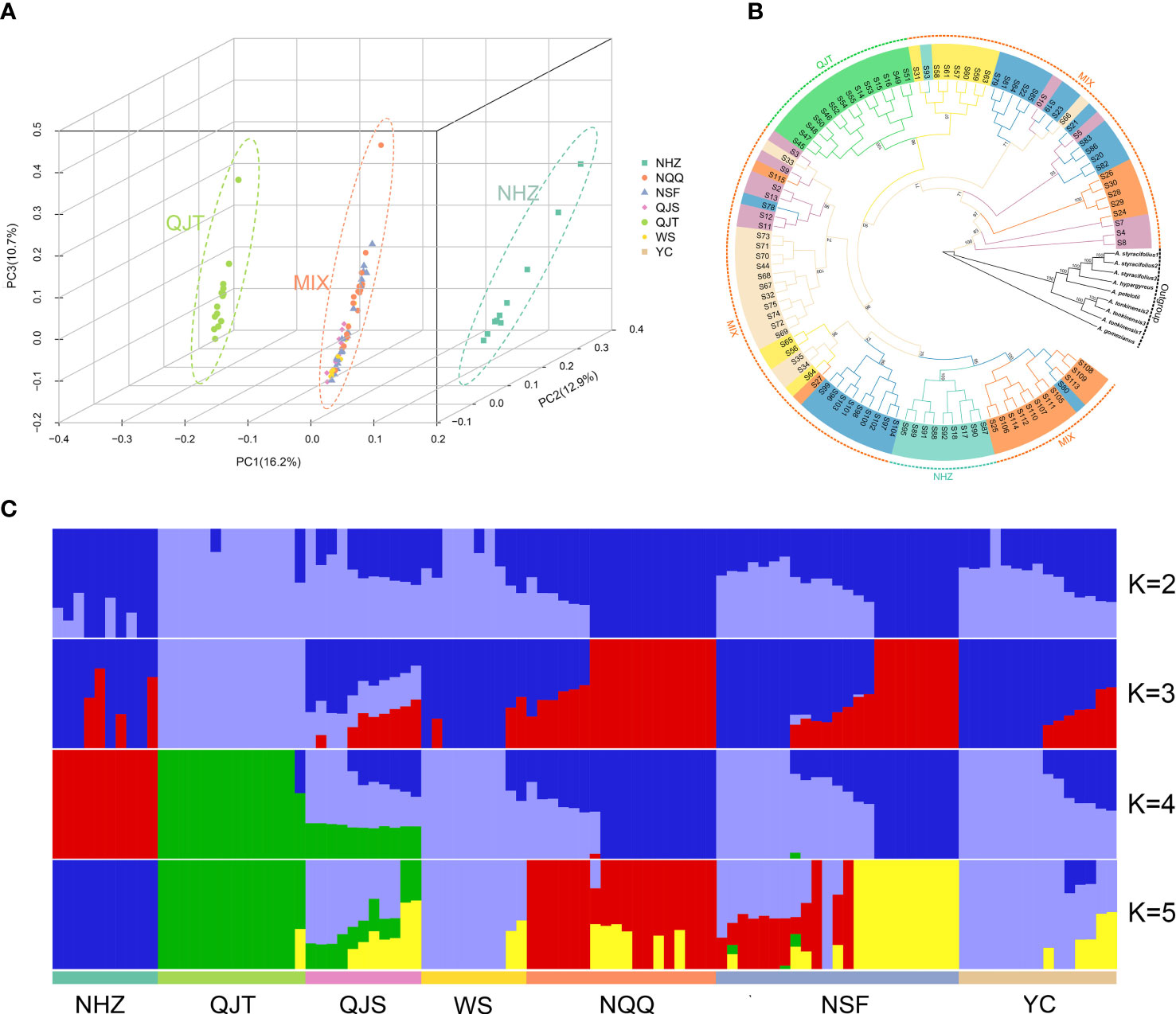
Figure 2 Genetic structure and phylogeny of Artocarpus nanchuanensis. (A) Principal component analysis (PCA) plot for the 101 A. nanchuanensis individuals based on PC1, PC2, and PC3. (B) Phylogenetic tree based on 101 A. nanchuanensis individuals and outgroups, with numbers on branches representing bootstrap percentages. Different colours of the branches represent individuals of A nanchuanensis from different populations. (C) Population structure of 101 A. nanchuanensis individuals based on K=2-5.
Phylogenetic analysis showed that A. nanchuanensis and four species of ser. Clavati (A. tonkinensis, A. petelotii, A. hypargyreus, and A. styracifolius) gathered together and formed a sister branch (Figure 2B). Consistent with the PCA results, the NHZ and QJT populations were significantly separated from the other populations in the phylogenetic tree, while the remaining five populations (MIX) were mixed to varying degrees (Figure 2B). In addition, the average genetic diversity of different populations of A. nanchuanensis was relatively low, with πS values ranging from 1.3×10-3 to 1.33×10-3 and πN values ranging from 2.26×10-3 to 2.32×10-3, and there were no significant differences between populations (Supplementary Table 8).
In the selective sweeps analysis, we detected 376 genes of 375 CDRs that were under clear positive selection in the NHZ population (Supplementary Table 9; Supplementary Table 10). These genes were significantly enriched in biological processes related to the synthesis of terpenes (Supplementary Figure 5), mainly involving five terpene synthase (TPS) genes (TPS38, TPS39, TPS40, TPS41, and TPS42) (Figure 3; Supplementary Table 11). The phylogenetic tree constructed based on 57 detected TPS genes of A. nanchuanensis and 33 TPS genes of Arabidopsis thaliana indicated that these five TPS genes belong to the TPS-b subfamily, and they were notably clustered into a small clade with high bootstrap support (bootstrap percentage 100%; Figure 3C).
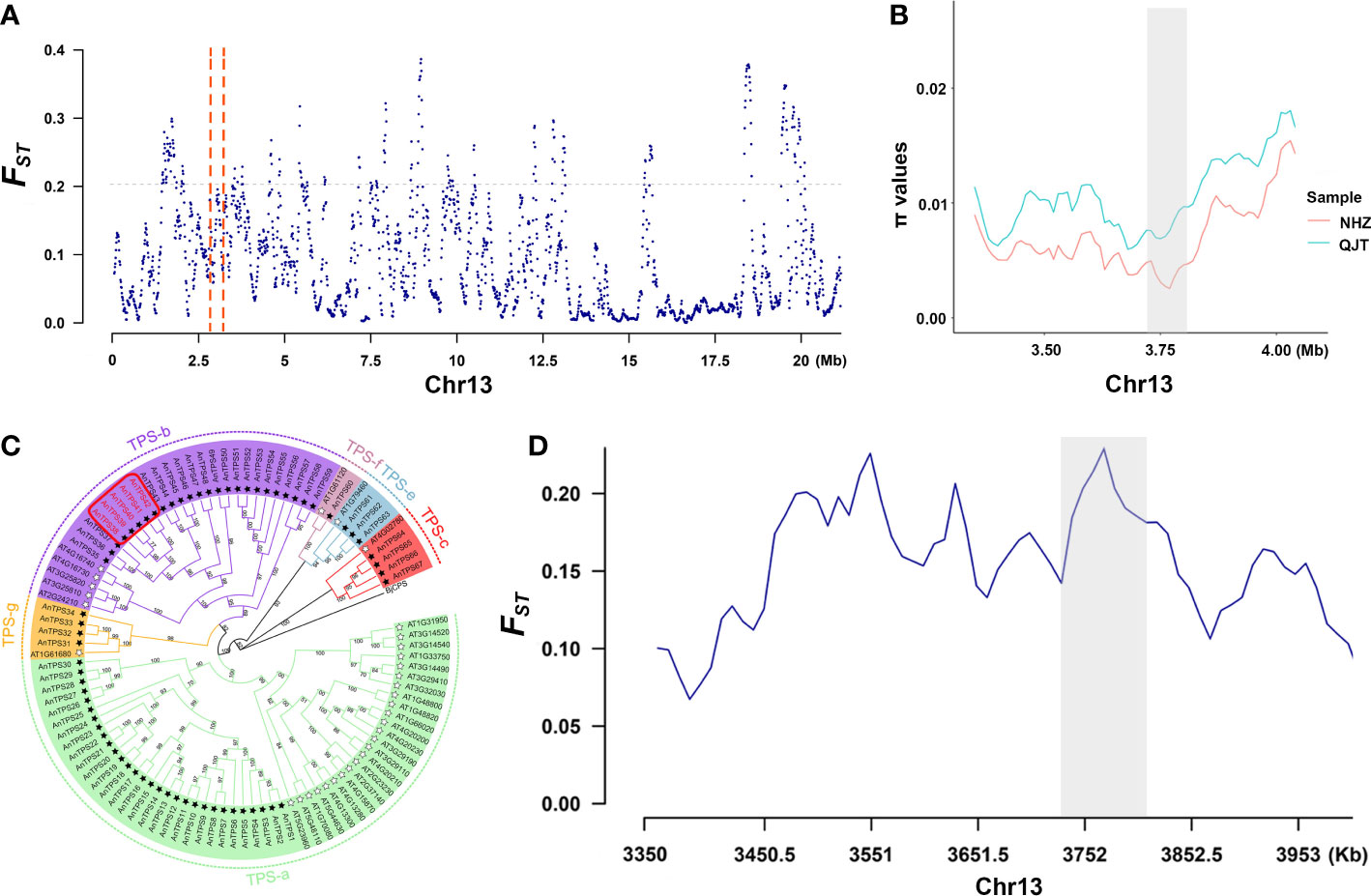
Figure 3 Signals of selective sweep between QJT and NHZ. (A) Genomic landscape of selection signals on chromosome 13 between QJT and NHZ. Vertical red dashed lines represent the 500-kb region around these 5 TPS genes. (B) π values of 5 TPS genes linking region between QJT and NHZ. Red line indicates the nucleotide diversity of NHZ population. Green line indicates the nucleotide diversity of QJT population. The gray shadow represents the location of 5 TPS genes. (C) Phylogenetic analysis of TPS gene families in Artocarpus nanchuanensis and Arabidopsis thaliana. The names of subfamilies are shown outside of the circle. Black star refers to AnTPSs and white star refers to the AtTPSs. The red rectangular box indicates the locations of five significantly enriched TPS genes. (D) FST of 5 TPS genes linking region between QJT and NHZ. The gray shadow represents the location of 5 TPS genes.
The results of PSMC analysis indicated that A. nanchuanensis experienced three genetic bottleneck events in its evolutionary history (Figure 4A). The effective population size (Ne) of A. nanchuanensis began to decline slowly from 10 Ma and encountered the first genetic bottleneck at 2.82 Ma. From 2.2-0.2 Ma, the Ne gradually expanded from 5.4x104 to 1.12x105. However, the Ne has been declining since 0.19 Ma, except for a small increase at 0.09 Ma. Except for the earlier first genetic bottleneck, this species experienced the second and third genetic bottlenecks at 0.14 Ma and 0.06 Ma, respectively. Finally, the Ne of A. nanchuanensis was stable in the range of approximately 6.5 x104 - 8.3x104.
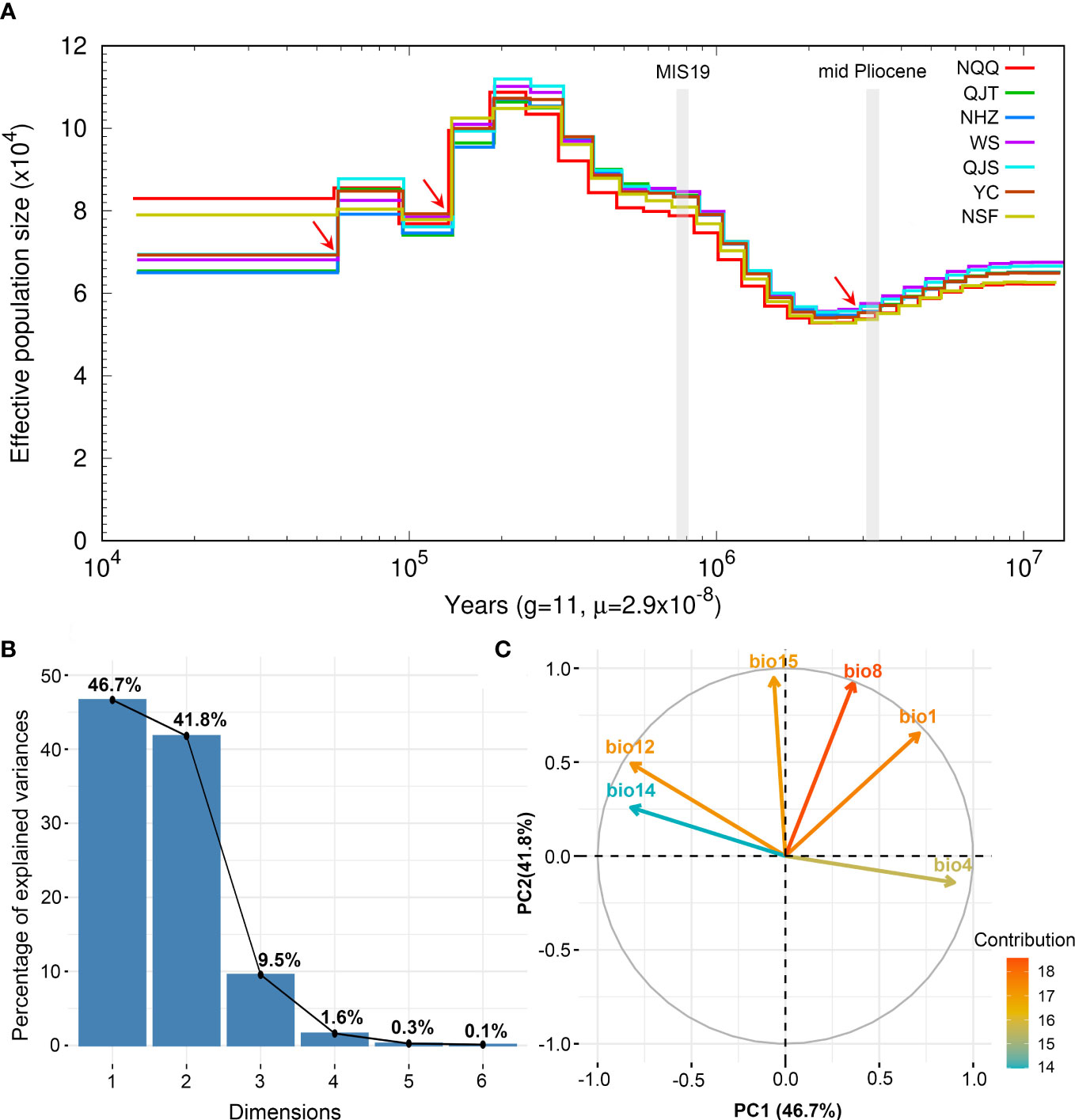
Figure 4 Demographic history of Artocarpus nanchuanensis and PCA results of six bioclimatic variables representing climate lability of A. nanchuanensis. (A) The historical changes in effective population size (Ne) for A. nanchuanensis revealed by PSMC analysis, with a generation time (g) of 11 years, and a mutation rate (μ) of 2.9e-8 per site per generation. The red arrows indicate genetic bottleneck events. (B) Scree plot of the percentage of explained variance. (C) PCA biplot of contribution of six bioclimatic variables to first two PCs.
Loss of function in a homozygous state could be used as an indicator of species fitness. The statistical analysis of the annotation results for each individual revealed no significant differences in genes containing homozygous LOF sites/PCGs between populations (Supplementary Table 12). The mean value for all individuals was 48.56%. A total of 285 GO terms were assigned in the enrichment analysis of genes containing homozygous LOF sites, including 220, 34 and 31 GO terms belonging to the biological process, molecular function and cell component categories, respectively (Supplementary Table 13). In the biological process category, multiple GO terms related to plant development in early stages, as well as potassium ion homeostasis (GO: 0055075) and tyrosine catalytic process (GO: 006572), were significantly enriched (corrected p value < 0.05, Supplementary Figure 6).
The PCA of bioclimate variables showed that the first two PCs (PC1 and PC2) explained 88.5% of the total climate change variation experienced by A. nanchuanensis (Figure 4B; Supplementary Figures 7, 8). Among the six bioclimatic variables, mean temperature of the wettest quarter (BIO8) and annual average temperature (BIO1) contributed most significantly to the first two PCs, followed by annual precipitation (BIO12) and precipitation seasonality (BIO15) (Figure 4B; Supplementary Figure 9). The prediction results showed that the area under the operating characteristic curve (AUC) of the model used in the analysis was 0.998, indicating that the prediction model had reached an excellent threshold (Swets, 1988). This suggested that the model employed in this study could accurately describe the relationship between species and climate. The results of the prediction analysis showed that in the mid-Pliocene warm period (3.205 Ma), the suitable habitat areas of A. nanchuanensis were mainly distributed in central and south-eastern China (Figure 5A). However, the suitable habitat areas were predicted to shift from northern to southern China and were mainly distributed in the south parts of China in MIS19 (Pleistocene, ca. 787 ka; Figure 5B). The prediction results also indicated that the current suitable habitat areas, which were widely distributed in central and south-eastern China, were more extensive than the actual distribution (Figure 5C). Although the future suitable habitat areas were predicted to shrink slightly compared with the current suitable habitat areas, the actual distribution area of A. nanchuanensis (Chongqing City) will still maintain high suitability (Figures 5C, D).
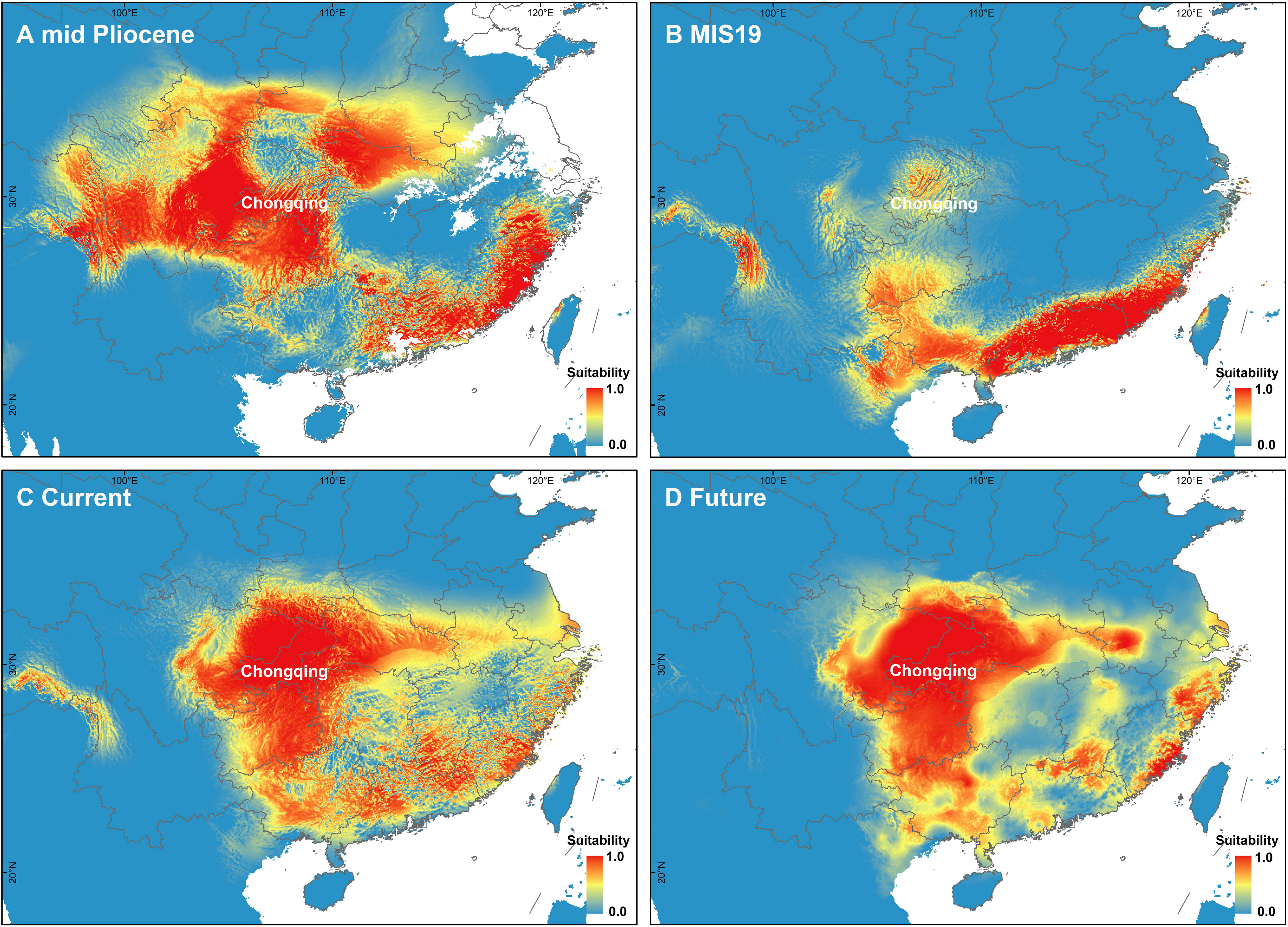
Figure 5 Distribution patterns of potential suitable habitat areas. (A) Potential suitable habitat areas under the paleoclimate scenario of mid-Pliocene warm period (3.205 Ma). (B) Potential suitable habitat areas under the paleoclimate scenario of MIS19 (ca. 787 ka). (C) Potential suitable habitat areas under the current climate scenario (1979-2013). (D) Potential suitable habitat areas under the future climate scenario (2070).
In this study, we sampled 101 individuals of A. nanchuanensis, covering most of the wild distribution areas, and revealed its complex population structure, demographic history, and genetic threats it currently faces by using whole-genome resequencing data. In addition, we identified several key factors that led to the formation of the distribution pattern of A. nanchuanensis by combining the analysis of niche models and demographic history. Our findings provide important guidance for meaningful conservation actions for this critically endangered WPESP.
Compared to that of other trees that have been studied, the genetic diversity of A. nanchuanensis was lower (Corbett-Detig et al., 2015; Hazzouri et al., 2015; Wang et al., 2016; Lin et al., 2022), which may be due to the following factors: (1) Previous studies have shown that genetic bottlenecks may lead to a large loss of genetic diversity due to enhanced genetic drift and a reduction in natural selection efficiency (Jangjoo et al., 2016; Ma et al., 2021; Ma et al., 2022). This seemed to be reasonable in the case of A. nanchuanensis because the demographic history showed three potential bottleneck events, which may have led to a significant reduction in its genetic diversity. (2) The actual Ne found in the field investigation (<200) was significantly smaller than the Ne (>65,000) inferred from demographic history, indicating that the population of A. nanchuanensis has recently encountered strong pressure. During our field investigation in 2019, three small populations of A. nanchuanensis were found in Qianqiu village (Nanchuan district). When we visited the site again in 2022, we found that only a few young trees remained in one of the populations. The main reason was that its straight trunks of A. nanchuanensis are often used by local people for shipbuilding and furniture. Based on the inferred large Ne value and the evidence that multiple existing populations in the four districts shared very similar genetic backgrounds, we speculated that A. nanchuanensis could be widely distributed in the mountains around the four districts. However, this may not be the case, possibly because human activities such as deforestation, road construction, and the expansion of farmland have drastically reduced the Ne of A. nanchuanensis. Human interference and habitat fragmentation will further accelerate the impact of genetic drift, thereby reducing genetic diversity (Ma et al., 2021). (3) Studies on other Artocarpus species indicated that birds and large mammals may be potential seed disseminators (Dickinson et al., 2020). However, considering that most of the existing populations of A. nanchuanensis are located near settlement, the likelihood of the occurrence of large mammals is extremely low. Moreover, field investigations showed that mature individuals of A. nanchuanensis are rare (numbers between 1 and 5) in most populations, and the geographical distances between populations usually exceed 10 kilometers. These factors may together hinder the production of outbred seeds, which may be an important way to increase the genetic diversity of the population.
Artocarpus is a genus mainly distributed in tropical regions, and its diversity center is located in Borneo (Gardner et al., 2021). However, as the northernmost species of this genus, the reason for the formation of the distribution pattern of A. nanchuanensis has always been a mystery. Our study revealed that Quaternary climate change played an important role in forming the current distribution pattern of this species, especially temperature. A recent study indicated that the global temperature has been falling since 10 Ma (Westerhold et al., 2020). Combining the prediction analysis and demographic history, we speculated that due to the decrease of temperature in the early stage (10-2.82 Ma), the suitable habitat areas of A. nanchuanensis shrank, accompanied by a slow decline in Ne (Figures 4A, 5A, B). With the advent of the Quaternary Ice Age, most areas of China were covered by glaciers, except for the mountainous areas in central and southern China, which harboured diverse terrain and a suitable climate, providing long-term stable habitats for plants (Axelrod et al., 1998; López-Pujol et al., 2011; Tang et al., 2012). Thus, A. nanchuanensis might have undergone migration and expansion in this region (south of the Qinling-Daba Mountains) during most of the Pleistocene (2.2-0.2 Ma) (Figures 4A, 5B). However, Ne began to decline rapidly at 0.19 Ma, which coincided with the penultimate glaciation (0.19-0.14 Ma) (Colleoni et al., 2016). In general, during the Pleistocene glaciation (2000-12ka), the significant increase and decrease of Ne detected in A. nanchuanensis was consistent with the published research on other Spermatophytes and Ginkgo biloba, which is located in the same refuge - Dalou Mountains (Qiu et al., 2011; Zhao et al., 2019; Ma et al., 2021; Ma et al., 2022). In addition, the recent comparative analysis of demographic history of Artocarpus species showed that bioclimatic change and habitat were important factors in shaping the population history (Patil et al., 2022). Therefore, we inferred that similar habitat - acid soil of A. nanchuanensis and A. altilis may partly explain their highly consistent demographic histories (Patil et al., 2022).
The prediction results showed that the current suitable habitat areas of A. nanchuanensis were far larger than its actual distribution area (Figure 5C), which may be due to the rapid climate fluctuations in the late Quaternary (approximately 0.8 - 0.1 Ma, Hofreiter and Stewart, 2009), leading to the disappearance of populations in other areas, while only the populations in the refuge could survive. Previous studies have shown that the Dalou Mountains are an important refuge for plants, harbouring many living fossil plants, such as Ginkgo biloba, Cathaya argyrophylla, and Davidia involucrata. (Wang and Ge, 2006; Tang et al., 2012). Therefore, we speculated that only the remaining population of A. nanchuanensis located in Dalou Mountain survived in the face of the considerable challenges of climate change. In addition, little is known about the pollination and diffusion mechanisms of A. nanchuanensis, and further research is urgently needed in the future to comprehensively infer the underlying reasons for the differences between the current suitable habitat areas and actual distribution area. It is also worth noting that our prediction analysis did not include environmental variables, such as topographical variables and elevation, which should be integrated to yield more accurate prediction results in the future.
The GO annotation of positive selection signal regions of A. nanchuanensis indicated that the NHZ population was significantly enriched in 5 TPS genes, which were detected to have a close genetic relationship (Figure 3). TPS is the key enzyme responsible for terpene biosynthesis (Yu et al., 2020; Jia et al., 2022), which plays an important role in the defense mechanism of plants against herbivores and pathogenic microorganisms (Kessler, 2001; Gershenzon and Dudareva, 2007; Richter et al., 2015) and in the process of attracting pollinators (Yu et al., 2020; Zhao et al., 2021). There are only two adult trees and dozens of annual seedlings in the NHZ population, one of which is a listed ancient tree (DBH 63 cm), and the other adult tree (DBH 25 cm) is inferred as its offspring. This population is relatively isolated and now surrounded by cities (located within the crematorium area in Nanchuan District). Combined with the unique genetic background of the NHZ population revealed by the population structure analysis, it is inferred that the population has undergone a certain degree of differentiation during the long-term isolated growth process, especially in defense-related genes.
Our study also showed that there is a large amount of genetic load in A. nanchuanensis (Hu et al., 2020; Yi et al., 2022; Supplementary Table 12). Moreover, the annotation of genes containing homozygous LOF sites of all populations revealed that they were significantly enriched in multiple GO terms related to plant development in the early stage and plant immunity, such as potassium ion homeostasis (GO: 0055075) and tyrosine catabolic process (GO: 006572; Supplementary Figure S6). Previous studies have shown that potassium (K+) plays an important role in many basic processes in plants, including cell homeostasis, membrane transport, osmotic regulation and the immune response (Amtmann et al., 2008; Wang and Wu, 2015; Shi et al., 2018). Tyrosine phosphorylation has also been shown to be crucial for regulating immune signals in plants (Lin et al., 2014; Macho et al., 2014; Macho et al., 2015; Liu et al., 2018). All of these GO terms were related to deleterious mutations, which may reduce the ability of A. nanchuanensis to adapt to unsuitable environments. Field observations indicated that most populations included numerous newly sprouted seedlings, but perennial seedlings or saplings were rare. Therefore, it could be inferred that the seed germination rate of A. nanchuanensis is high, but the seedling survival rate is low, which may be related to the accumulation of deleterious mutations in its genes about development or immunity. However, further analysis of these deleterious mutations is needed to reach a definite conclusion.
Most of the populations of A. nanchuanensis are located outside nature reserves. In addition, the low genetic diversity and high genetic load, as well as serious human destruction and habitat disturbance, mean that the future survival of A. nanchuanensis may face great challenges. Based on our study results and actual conservation status, we propose to divide all populations into four conservation units, NHZ, QJT, QJS and four other populations (WS, NSF, NQQ and YC), and implement the following protection actions as soon as possible: (1) PCA, genetic structure and phylogenetic analysis revealed the unique genetic backgrounds of QJT and NHZ, which were different from those of other populations (Figures 2A, C). To protect the genetic diversity of A. nanchuanensis, priority should be given to the protection of these two small populations. The geographical distance between QJT and QJS was only 1 km, with the populations separated by a river approximately 30 meters wide. Surprisingly, our analysis showed that QJT and QJS had significant similarities and differences in genetic composition at the same time, which seemed to be an excellent example of geographical isolation and differentiation (Figures 2A, C). Therefore, we suggest establishing plant microreserves for these two small populations in Qijiang District, which harbour many apparent advantages, including lower costs, fast construction, and strong pertinence, and could also improve the public awareness of biodiversity conservation of A. nanchuanensis among local residents and even society (Fos et al., 2014; Laguna et al., 2016; Zhang, 2017). The NHZ population is restricted to flower beds of the crematorium in Nanchuan District, and one of the adult trees is listed as an ancient tree. Therefore, consulting with the management personnel of the crematorium is recommended to increase fencing for the flower beds, allowing this population to grow and breed naturally without human interference.
(2) Our field investigation revealed that the individual with the largest DBH (65 cm vs. 63 cm of the listed ancient tree from NHZ) was located in Honglu Town, Yongchuan District, and there is a sand factory under construction around the ancient individual. It is recommended to contact the local forestry administration to confirm the age of the ancient tree as soon as possible and apply in situ conservation for this small population with only three mature trees. (3) Considering the rarity of adult trees, isolated populations and small FST values between various populations of A. nanchuanensis, artificial cross-pollination among four populations with similar genetic backgrounds should be carried out to maintain the genetic diversity and increase the survival potential of this species. (4) Based on the current status of the extremely small population of A. nanchuanensis, it is strongly recommended to pursue ex situ protection of the populations of the four conservation units to comprehensively preserve the germplasm resources and genetic diversity of this species. Considering the current and future habitat suitability of A. nanchuanensis, large-scale botanical gardens in central China are the preferred choices, such as the Nanshan Botanical Garden in Chongqing City and the Wuhan Botanical Garden of the Chinese Academy of Sciences in Hubei Province.
Based on the whole-genome resequencing data of 101 critically endangered A. nanchuanensis individuals, we revealed that repeated bottleneck events, human disturbance and habitat fragmentation, as well as excessive self-pollination within populations, together contributed to low genetic diversity. Quaternary climate change, especially temperature change, plays an important role in forming the current distribution pattern and endangered status of the species. The Dalou Mountains provided an important refuge for A. nanchuanensis during Quaternary climate fluctuations. Genetic analysis revealed that the NHZ population not only had a special genetic background but also had specific differentiation in defense-related genes. The analysis also indicated that this species has an abundant genetic load, especially in the genes related to the early development and immune function of plants. Based on the study results, we divided the populations of A. nanchuanensis into four conservation units and proposed a number of practical management suggestions for each conservation unit of this critically endangered species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI, PRJNA948357 (SRA).
HD designed the research. CX, BW and JZ collected the plant materials. CX, YZ and TX performed the research and analysed the data. CX and HZ wrote the paper. MK, XZ and HD revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by the Central forestry reform and development fund - Representative National Key Protected Wild Plants Rescue and Protection in the Upper Reaches of the Yangtze River (zlg2021-cq20211210), and the Central forestry reform and development fund - Chengdu-Chongqing economic circle Joint Protection National Key Protected Wild Plants Rescue and Protection (zlg2022-cq20220907).
We are grateful for the help in the sampling and analysis process from Sirong Yi, Jun Zhang, Baocai Han, Xinggu Lin, and Ying Wang.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1224308/full#supplementary-material
Ackerly, D. D., Loarie, S. R., Cornwell, W. K., Weiss, S. B., Hamilton, H., Branciforte, R., et al. (2010). The geography of climate change: Implications for conservation biogeography. Divers. Distrib. 16, 476–487. doi: 10.1111/j.1472-4642.2010.00654.x
Alexander, D. H., Novembre, J., Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19 (9), 1655–1664. doi: 10.1101/gr.094052.109
Amtmann, A., Troufflard, S., Armengaud, P. (2008). The effect of potassium nutrition on pest and disease resistance in plants. Physiol. Plant 133 (4), 682–691. doi: 10.1111/j.1399-3054.2008.01075.x
Axelrod, D. I., Al-Shehbaz, I., Raven, P. H. (1998). “History of the modern flora of China,” in Floristic characteristics and diversity of East Asia plants. Eds. Zhang, A. L., Wu, S. G. (Beijing: China Higher Education Press), 43–55.
Barber, Q. E., Nielsen, S. E., Hamann, A. (2016). Assessing the vulnerability of rare plants using climate change velocity, habitat connectivity, and dispersal ability: A case study in Alberta, Canada. Reg. Environ. Change 16, 1433–1441. doi: 10.1007/s10113-015-0870-6
Boria, R. A., Olson, L. E., Goodman, S. M., Anderson, R. P. (2014). Spatial filtering to reduce sampling bias can improve the performance of ecological niche models. Ecol. Modell. 275, 73–77. doi: 10.1016/j.ecolmodel.2013.12.012
Brown, J. L., Hill, D. J., Dolan, A. M., Carnaval, A. C., Haywood, A. M. (2018). PaleoClim, high spatial resolution paleoclimate surfaces for global land areas. Sci. Data. 5, 180254. doi: 10.1038/sdata.2018.254
Chen, Y. X., Chen, Y. S., Shi, C. M., Huang, Z. B., Zhang, Y., Li, S. K., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7 (1), 1–6. doi: 10.1093/gigascience/gix120
Chen, C. J., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y. H., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cingolani, P., Platts, A., Wang, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6 (2), 80–92. doi: 10.4161/fly.19695
Colleoni, F., Wekerle, C., Naslund, J. O., Brandefelt, J., Masina, S. (2016). Constraint on the penultimate glacial maximum Northern Hemisphere ice topography (140 kyrs BP). Quat. Sci. Rev. 137, 97–112. doi: 10.1016/j.quascirev.2016.01.024
Corbett-Detig, R. B., Hartl, D. L., Sackton, T. B. (2015). Natural selection constrains neutral diversity across a wide range of species. PloS Biol. 13 (4), e1002112. doi: 10.1371/journal.pbio.1002112
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi: 10.1093/bioinformatics/btr330
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10 (2), giab008. doi: 10.1093/gigascience/giab008
Dawson, T. P., Jackson, S. T., House, J. I., Prentice, I. C., Mace, G. M. (2011). Beyond predictions: biodiversity conservation in a changing climate. Science 332 (6025), 53–58. doi: 10.1126/science.1200303
Deng, H. P. (2019). Biodiversity of Chongqing Jinfoshan National Nature Reserve (Beijing: Science Press).
Dickinson, L., Noble, H., Gardner, E., Puad, A. S. A., Zakaria, W. N. F. W., Zerega, N. J. C. (2020). Genetic diversity and structure of the critically endangered Artocarpus annulatus, a crop wild relative of jackfruit (A. heterophyllus). PeerJ 8, e9897. doi: 10.7717/peerj.9897
Ding, B., Hua, B., Wen, H. J., Deng, H. P., Ding, X. (2014). On floristic study and scientific name revision of seed type specimens distributed in Jinfo Mountains Nature Reserve. J. Southwest Norm. Univ. (Nat. Sci. Edit.) 39 (12), 47–52. doi: 10.13718/j.cnki.xsxb.2014.12.010
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19 (1), 11–15. doi: 10.1016/0031-9422(80)85004-7
Finn, R. D., Clements, J., Arndt, W., Miller, B. L., Wheeler, T. J., Schreiber, F., et al. (2015). HMMER web server: 2015 update. Nucleic Acids Res. 43 (W1), W30–W38. doi: 10.1093/nar/gkv397
Fos, S., Laguna, E., Jiménez, J. (2014). Plant Micro-Reserves in the Valencian Region (E of Spain): are we achieving the expected results? Passive conservation of relevant vascular plant species. Flora Mediterr. 24, 153–162. doi: 10.7320/FlMedit24.153
Francis, R. M. (2017). Pophelper: an R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 17, 27–32. doi: 10.1111/1755-0998.12509
Funk, W. C., McKay, J. K., Hohenlohe, P. A., Allendorf, F. W. (2012). Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 27 (9), 489–496. doi: 10.1016/j.tree.2012.05.012
Gao, X. X., Liu, J., Huang, Z. H. (2022). The impact of climate change on the distribution of rare and endangered tree Firmiana kwangsiensis using the Maxent modeling. Ecol. Evol. 12 (8), e9165. doi: 10.1002/ece3.9165
Gardner, E. M., Johnson, M. G., Pereira, J. T., Puad, A. S. A., Arifiani, D., Sahromi, et al. (2021). Paralogs and off-target sequences improve phylogenetic resolution in a densely sampled study of the breadfruit genus (Artocarpus, moraceae). Syst. Biol. 70 (3), 558–575. doi: 10.1093/sysbio/syaa073
Gaut, B. S., Morton, B. R., McCaig, B. C., Clegg, M. T. (1996). Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. U. S. A. 93 (19), 10274–10279. doi: 10.1073/pnas.93.19.10274
Gershenzon, J., Dudareva, N. (2007). The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414. doi: 10.1038/nchembio.2007.5
Goodstein, D. M., Shu, S. Q., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic. Acids Res. 40 (D1), D1178–D1186. doi: 10.1093/nar/gkr944
Hazzouri, K. M., Flowers, J. M., Visser, H. J., Khierallah, H. S. M., Rosas, U., Pham, G. M., et al. (2015). Whole genome re-sequencing of date palms yields insights into diversification of a fruit tree crop. Nat. Commun. 6, 8824. doi: 10.1038/ncomms9824
He, L. (2014). Study on the morphological characteristics, modular biomass allocation and photosynthetic characteristics of Artocarpus nanchuanensis seedlings. (Chongqing: Southwest University).
He, J. Y., Bao, S. F., Deng, J. H., Li, Q. F., Ma, S. Y., Liu, Y. R., et al. (2022a). A chromosome-level genome assembly of Artocarpus nanchuanensis (Moraceae), an extremely endangered fruit tree. GigaScience 11, 1–13. doi: 10.1093/gigascience/giac042
He, J. Y., Bao, S. F., Deng, J. H., Li, Q. F., Ma, S. Y., Liu, Y. R., et al. (2022b). Supporting data for “A chromosome-level genome assembly of Artocarpus nanchuanensis (Moraceae), an extremely endangered fruit tree”. GigaSci. Database. doi: 10.5524/102200
Hofreiter, M., Stewart, J. (2009). Ecological change, range fluctuations and population dynamics during the Pleistocene. Curr. Biol. 19, 584–594. doi: 10.1016/j.cub.2009.06.030
Hohenlohe, P. A., Funk, W. C., Rajora, O. P. (2021). Population genomics for wildlife conservation and management. Mol. Ecol. 30 (1), 62–82. doi: 10.1111/mec.15720
Hu, J. Y., Hao, Z. Q., Frantz, L., Wu, S. F., Chen, W., Jiang, Y. F., et al. (2020). Genomic consequences of population decline in critically endangered pangolins and their demographic histories. Natl. Sci. Rev. 7 (4), 798–814. doi: 10.1093/nsr/nwaa031
Hu, J. Y., Kuang, W. M., Wu, H., Yu, L. (2021). Genomic genetic load analysis. Bio101, e1010600. doi: 10.21769/BioProtoc.1010600
Huang, H. W. (2011). Plant diversity and conservation in China: Planning a strategic bioresource for a sustainable future. Bot. J. Linn. Soc 166, 282–300. doi: 10.1111/j.1095-8339.2011.01157.x
Huang, J., Liang, X. M., Xuan, Y. K., Geng, C. Y., Li, Y. X., Lu, H. R., et al. (2018). BGISEQ-500 WGS library construction. (protocols.io). doi: 10.17504/protocols.io.ps5dng6
Huang, H. W., Zhang, Z. (2012). Current status and prospects of ex situ cultivation and conservation of plants in China. Biodivers. Sci. 20 (5), 559–571. doi: 10.3724/SP.J.1003.2012.13124
Jangjoo, M., Matter, S. F., Roland, J., Keyghobadi, N. (2016). Connectivity rescues genetic diversity after a demographic bottleneck in a butterfly population network. Proc. Natl. Acad. Sci. U. S. A. 113, 10914–10919. doi: 10.1073/pnas.1600865113
Jia, Q., Brown, R., Köllner, T. G., Fu, J., Chen, X., Wong, G. K., et al. (2022). Origin and early evolution of the plant terpene synthase family. Proc. Natl. Acad. Sci. U. S. A. 119 (15), e2100361119. doi: 10.1073/pnas.2100361119
Kessler, A. (2001). Defensive function of herbivore-induced plant volatile emissions in nature. Sci. 291 (5511), 2141–2144. doi: 10.1126/science.291.5511.2141
Laguna, E., Fos, S., Jimenez, J., Volis, S. (2016). Role of micro-reserves in conservation of endemic, rare and endangered plants of the Valencian region (Eastern Spain). Isr. J. Plant Sci. 63 (4), 320–332. doi: 10.1080/07929978.2016.1256131
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303, 3997. doi: 10.48550/arXiv.1303.3997
Li, H., Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature 475 (7357), 493–496. doi: 10.1038/nature10231
Li, J. L., Milne, R. I., Ru, D. F., Miao, J. B., Tao, W. J., Zhang, L., et al. (2020). Allopatric divergence and hybridization within Cupressus chengiana (Cupressaceae), a threatened conifer in the northern Hengduan Mountains of western China. Mol. Ecol. 29 (7), 1250–1266. doi: 10.1111/mec.15407
Lin, X. G., Feng, C., Lin, T., Harris, A. J., Li, Y. Z., Kang, M. (2022). Jackfruit genome and population genomics provide insights into fruit evolution and domestication history in China. Hortic. Res. 9, uhac173. doi: 10.1093/hr/uhac173
Lin, W. W., Li, B., Lu, D. P., Chen, S. X., Zhu, N., He, P., et al. (2014). Tyrosine phosphorylation of protein kinase complex BAK1/BIK1 mediates Arabidopsis innate immunity. Proc. Natl. Acad. .Sci. U. S. A. 111, 3632–3637. doi: 10.1073/pnas.1318817111
Liu, J., Liu, B., Chen, S. F., Gong, B. Q., Chen, L. J., Zhou, Q., et al. (2018). A tyrosine phosphorylation cycle regulates fungal activation of a plant receptor ser/thr kinase. Cell Host Microbe 23, 241–253. doi: 10.1016/j.chom.2017.12.005
López-Pujol, J., Zhang, F. M., Sun, H. Q., Ying, T. S., Ge, S. (2011). Centres of plant endemism in China: Places for survival or for speciation? J. Biogeogr. 38, 1267–1280. doi: 10.1111/j.1365-2699.2011.02504.x
Ma, H., Liu, Y. B., Liu, D. T., Sun, W. B., Liu, X. F., Wan, Y. M., et al. (2021). Chromosome-level genome assembly and population genetic analysis of a critically endangered rhododendron provide insights into its conservation. Plant J. 107, 1533–1545. doi: 10.1111/tpj.15399
Ma, Y. P., Liu, D. T., Wariss, H. M., Zhang, R. G., Tao, L. D., Milne, R. I., et al. (2022). Demographic history and identification of threats revealed by population genomic analysis provide insights into conservation for an endangered maple. Mol. Ecol. 31, 767–779. doi: 10.1111/mec.16289
Macho, A. P., Lozano-Durán, R., Zipfel, C. (2015). Importance of tyrosine phosphorylation in receptor kinase complexes. Trends Plant Sci. 20 (5), 269–272. doi: 10.1016/j.tplants.2015.02.005
Macho, A. P., Schwessinger, B., Ntoukakis, V., Brutus, A., Segonzac, C., Roy, S., et al. (2014). A bacterial tyrosine phosphatase inhibits plant pattern recognition receptor activation. Science 343, 1509–1512. doi: 10.1126/science.1248849
Mattila, A. L., Duplouy, A., Kirjokangas, M., Lehtonen, R., Rastas, P., Hanski, I. (2012). High genetic load in an old isolated butterfly population. Proc. Natl. Acad. .Sci. U. S. A. 109, E2496–E2505. doi: 10.1073/pnas.1205789109
Nei, M., Li, W. H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. .Sci. U. S. A. 76 (10), 5269–5273. doi: 10.1073/pnas.76.10.5269
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Patil, A. B., Vajja, S. S., Raghavendra, S., Satish, B. N., Kushalappa, C. G., Vijay, N. (2022). Jack of all trades: Genome assembly of Wild Jack and comparative genomics of Artocarpus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1029540
Phillips, S. J., Anderson, R. P., Schapire, R. E. (2006). Maximum entropy modelling of species’ geographic distributions. Ecol. Model. 12, 231–259. doi: 10.1016/j.ecolmodel.2005.03.026
Pimm, S. L. (2008). Biodiversity: climate change or habitat loss—which will kill more species? Curr. Biol. 18 (3), R117–R119. doi: 10.1016/j.cub.2007.11.055
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi: 10.1086/519795
Qian, S. H., Yang, Y., Tang, C. Q., Momohara, A., Yi, S. R., Ohsawa, M. (2016). Effective conservation measures are needed for wild Cathaya argyrophylla populations in China: Insights from the population structure and regeneration characteristics. For. Ecol. Manage. 361, 358–367. doi: 10.1016/j.foreco.2015.11.041
Qin, A. L., Liu, B., Guo, Q. S., Bussmann, R. W., Ma, F. Q., Jian, Z. L., et al. (2017a). Maxent modeling for predicting impacts of climate change on the potential distribution of Thuja sutchuenensis Franch., an extremely endangered conifer from southwestern China. Glob. Ecol. Conserv. 10, 139–146. doi: 10.1016/j.gecco.2017.02.004
Qin, H. N., Yang, Y., Dong, S. Y., He, Q., Jia, Y., Zhao, L. N., et al. (2017b). Threatened species list of China’s higher plants. Biodivers. Sci. 25 (7), 696–744. doi: 10.17520/biods.2017144
Qiu, Y. X., Fu, C. X., Comes, H. P. (2011). Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 59, 225–244. doi: 10.1016/j.ympev.2011.01.012
Richter, A., Seidl-Adams, I., Köllner, T. G., Schaff, C., Tumlinson, J. H., Degenhardt, J. (2015). A small, diferentially regulated family of farnesyl diphosphate synthases in maize (Zea mays) provides farnesyl diphosphate for the biosynthesis of herbivore-induced sesquiterpenes. Planta 241, 1351–1361. doi: 10.1007/s00425-015-2254-z
Shi, X. T., Long, Y., He, F., Zhang, C. Y., Wang, R. Y., Zhang, T., et al. (2018). The fungal pathogen Magnaporthe oryzae suppresses innate immunity by modulating a host potassium channel. PloS Pathog. 14 (1), e1006878. doi: 10.1371/journal.ppat.1006878
Swets, J. A. (1988). Measuring the accuracy of diagnostic systems. Science 240 (4857), 1285–1293. doi: 10.1126/science.3287615
Tajima, F. (1983). Evolutionary relationship of DNA sequences in finite populations. Genetics 105, 437–460. doi: 10.1093/genetics/105.2.437
Tan, X. M., Zhou, Y. Q., Deng, A. G., Liu, Y. T., Peng, Y., Zhou, X. Z. (2015). Study on seedling techniques of Artocarpus nanchuanensis. Agr. Sci. Tech. 16 (12), 2754–2757. doi: 10.16175/j.cnki.1009-4229.2015.12.037
Tang, C. Q., Matsui, T., Ohashi, H., Dong, Y. F., Momohara, A., Herrando-Moraira, S., et al. (2018). Identifying long-term stable refugia for relict plant species in East Asia. Nat. Commun. 9, 4488. doi: 10.1038/s41467-018-06837-3
Tang, C. Q., Yang, Y., Ohsawa, M., Yi, S. R., Momohara, A., Su, W. H., et al. (2012). Evidence for the persistence of wild Ginkgo biloba (Ginkgoaceae) populations in the Dalou Mountains, southwestern China. Am. J. Bot. 99 (8), 1408–1414. doi: 10.3732/ajb.1200168
Thomas, C. D., Cameron, A., Green, R. E., Bakkenes, M., Beaumont, L. J., Collingham, Y. C., et al. (2004). Extinction risk from climate change. Nature 427, 145–148. doi: 10.1038/nature02121
Wang, H. W., Ge, S. (2006). Phylogeography of the endangered Cathaya argyrophylla (Pinaceae) inferred from sequence variation of mitochondrial and nuclear DNA. Mol. Ecol. 15, 4109–4122. doi: 10.1111/j.1365-294X.2006.03086.x
Wang, J., Street, N. R., Scofield, D. G., Ingvarsson, P. K. (2016). Variation in linked selection and recombination drive genomic divergence during allopatric speciation of European and American aspens. Mol. Biol. Evol. 33 (7), 1754–1767. doi: 10.1093/molbev/msw051
Wang, Y., Wu, W. H. (2015). Genetic approaches for improvement of the crop potassium acquisition and utilization efficiency. Curr. Opin. Plant Biol. 25, 46–52. doi: 10.1016/j.pbi.2015.04.007
Westerhold, T., Marwan, N., Drury, A. J., Liebrand, D., Agnini, C., Anagnostou, E., et al. (2020). An astronomically dated record of Earth’s climate and its predictability over the last 66 million years. Science 369 (6509), 1383–1388. doi: 10.1126/science.aba685
Wu, X. Q., Xu, G. B., Liang, Y., Shen, X. B. (2013). Genetic diversity of natural and planted populations of Tsoongiodendron odorum from the Nanling Mountains. Biodivers. Sci. 21 (1), 71–79. doi: 10.3724/SP.J.1003.2013.09138
Wu, Z. Y., Zhang, X. S. (1989). Some new taxa of Moraceae in China. Acta Botanica Yunnanica 11, 24–34.
Xu, B., Liao, M., Deng, H. N., Yan, C. C., Lv, Y. Y., Gao, Y. D., et al. (2022). Chromosome-level de novo genome assembly and whole-genome resequencing of the threatened species Acanthochlamys bracteata (Velloziaceae) provide insights into alpine plant divergence in a biodiversity hotspot. Mol. Ecol. Resour. 22, 1582–1595. doi: 10.1111/1755-0998.13562
Xu, Y., Zang, R. G. (2022). Theoretical and practical research on conservation of Wild Plants with Extremely Small Populations in China. Biodivers. Sci. 30, 22505. doi: 10.17520/biods.2022505
Xue, T. T., Yang, X. D., Liu, Q., Qin, F., Zhang, W. D., Janssens, S. B., et al. (2022). Integration of hotspot identification, gap analysis, and niche modeling supports the conservation of Chinese threatened higher plants. J. Syst. Evol. 1–16. doi: 10.1111/jse.12901
Yi, H. Q., Wang, J. Y., Wang, J., Rausher, M., Kang, M. (2022). Genomic insights into inter- and intraspecific mating system shifts in Primulina. Mol. Ecol. 31 (22), 5699–5713. doi: 10.1111/mec.16706
Yu, Z. M., Zhao, C. H., Zhang, G. H., Teixeira da Silva, J. A., Duan, J. (2020). Genome-Wide identification and expression profile of TPS gene family in dendrobium officinale and the role of doTPS10 in linalool biosynthesis. Int. J. Mol. Sci. 21, 5419. doi: 10.3390/ijms21155419
Zhang, F. C. (2017). Nature Small-reserves helps biodiversity conservation. China Environ. 000 (010), 30–33.
Zhao, Y. P., Fan, G., Yin, P. P., Sun, S., Li, N., Hong, X. N., et al. (2019). Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil. Nat. Commun. 10, 4201. doi: 10.1038/s41467-019-12133-5
Zhao, L., Zhao, X. J., Francis, F., Liu, Y. (2021). Genome−wide identifcation and characterization of the TPS gene family in wheat (Triticum aestivum L.) and expression analysis in response to aphid damage. Acta Physiol. Plant 43, 64. doi: 10.1007/s11738-021-03236-y
Zu, K. L., Wang, Z. H., Lenoir, J., Shen, Z. H., Chen, F. S., Shrestha, N. (2022). Different range shifts and determinations of elevational redistributions of native and non-native plant species in Jinfo Mountain of subtropical China. Ecol. Indic. 145, 109678. doi: 10.1016/j.ecolind.2022.109678
Keywords: Artocarpus nanchuanensis, whole-genome resequencing, demographic history, selective sweep, genetic load, conservation unit
Citation: Xia C, Zuo Y, Xue T, Kang M, Zhang H, Zhang X, Wang B, Zhang J and Deng H (2023) The genetic structure and demographic history revealed by whole-genome resequencing provide insights into conservation of critically endangered Artocarpus nanchuanensis. Front. Plant Sci. 14:1224308. doi: 10.3389/fpls.2023.1224308
Received: 17 May 2023; Accepted: 12 July 2023;
Published: 27 July 2023.
Edited by:
Jeremie Benjamin Fant, Chicago Botanic Garden, United StatesReviewed by:
Nagarjun Vijay, Indian Institute of Science Education and Research, Bhopal, IndiaCopyright © 2023 Xia, Zuo, Xue, Kang, Zhang, Zhang, Wang, Zhang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongping Deng, ZGVuZ2hwQHN3dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.