 Sabarna Bhattacharyya1
Sabarna Bhattacharyya1 Maya Giridhar1,2
Maya Giridhar1,2 Bastian Meier3
Bastian Meier3 Edgar Peiter3
Edgar Peiter3 Ute C. Vothknecht1†
Ute C. Vothknecht1† Fatima Chigri1*†
Fatima Chigri1*†- 1Institute for Cellular and Molecular Botany, University of Bonn, Bonn, Germany
- 2Leibniz Institute for Food Systems Biology at the Technical University of Munich, Freising, Germany
- 3Institute of Agricultural and Nutritional Sciences, Faculty of Natural Sciences III, Martin Luther University Halle-Wittenberg, Halle, Germany
In cereal crops, such as barley (Hordeum vulgare L.), the ability to appropriately respond to environmental cues is an important factor for yield stability and thus for agricultural production. Reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), are key components of signal transduction cascades involved in plant adaptation to changing environmental conditions. H2O2-mediated stress responses include the modulation of expression of stress-responsive genes required to cope with different abiotic and biotic stresses. Despite its importance, knowledge of the effects of H2O2 on the barley transcriptome is still scarce. In this study, we identified global transcriptomic changes induced after application of 10 mM H2O2 to five-day-old barley plants. In total, 1883 and 1001 differentially expressed genes (DEGs) were identified in roots and leaves, respectively. Most of these DEGs were organ-specific, with only 209 DEGs commonly regulated and 37 counter-regulated between both plant parts. A GO term analysis further confirmed that different processes were affected in roots and leaves. It revealed that DEGs in leaves mostly comprised genes associated with hormone signaling, response to H2O2 and abiotic stresses. This includes many transcriptions factors and small heat shock proteins. DEGs in roots mostly comprised genes linked to crucial aspects of H2O2 catabolism and oxidant detoxification, glutathione metabolism, as well as cell wall modulation. These categories include many peroxidases and glutathione transferases. As with leaves, the H2O2 response category in roots contains small heat shock proteins, however, mostly different members of this family were affected and they were all regulated in the opposite direction in the two plant parts. Validation of the expression of the selected commonly regulated DEGs by qRT-PCR was consistent with the RNA-seq data. The data obtained in this study provide an insight into the molecular mechanisms of oxidative stress responses in barley, which might also play a role upon other stresses that induce oxidative bursts.
1 Introduction
In aerobic organisms, reactive oxygen species (ROS) are generated as by-products of certain metabolic pathways in plant organelles such as chloroplasts, mitochondria, and peroxisomes (Huang et al., 2019; Smirnoff and Arnaud, 2019). Because of their high reactivity with cellular components, aerobic organisms have developed systems for enzymatic ROS removal based on the activity of ascorbate peroxidase (APX), superoxide dismutase (SOD), and catalase (CAT) as well as non-enzymatic antioxidative systems such as ascorbic acid, proline, and glutathione (GSH) (Foyer and Noctor, 2003; Ahmad et al., 2010). Plants also actively produce ROS as part of signaling cascades that coordinate the appropriate responses to environmental stimuli and contribute to stress tolerance (Pei et al., 2000; Zhu, 2016; Mohanta et al., 2018). It is proposed that systemic communication via redox systems is very fundamental to all photosynthetic organisms.
The ROS species hydrogen peroxide (H2O2) has been shown to play a role in various processes such as cell differentiation, senescence, and cell wall formation (Kärkönen and Kuchitsu, 2015; Ribeiro et al., 2017; Zeng et al., 2017). It is generated from superoxide in various cellular compartments as well as the apoplast as a result of a highly conserved superoxide dismutation reaction (Smirnoff and Arnaud, 2019). H2O2 is also known to be transported across the cell membrane by specific aquaporins (Bienert et al., 2007) and to participate in long distance cell signaling (Mittler et al., 2011). Exogenous treatment with H2O2 has been shown to increase the tolerance of plants to abiotic stress by regulating multiple stress-responsive pathways and expression of genes including heat shock proteins and genes involved in abscisic acid (ABA) biosynthesis (Wahid et al., 2007; Terzi et al., 2014). An activation of ROS-dependent signaling by H2O2 causes the accumulation of defense proteins such as ROS-scavenging enzymes, transcription factors (TFs), and other response factors (Hossain et al., 2015), and it thus increases the tolerance of plants to abiotic stress. For example, certain HEAT SHOCK TRANSCRIPTION FACTORS (HSFs) have been suggested to serve as sensors that perceive H2O2 and regulate the expression of oxidative stress response genes (Miller and Mittler, 2006).
An early transcriptomic approach pursued to elucidate the effect of H2O2 was performed in Arabidopsis thaliana cell suspension cultures and showed that various TFs, hormone-associated pathways, and genes associated with other vital metabolic pathways like photosynthesis and fatty acid biosynthesis were affected (Desikan et al., 2001). Other studies revealed the role of H2O2 as a signaling molecule in a variety of plant species and under various conditions. For instance, H2O2 is involved in the response of plants to a variety of environmental cues, such as salt stress in tomato (Li et al., 2019), heat stress in rice (Wang et al., 2014), chilling stress in mung beans and manila grass (Yu et al., 2003; Wang et al., 2010), copper stress in maize and mung bean (Guzel and Terzi, 2013; Fariduddin et al., 2014), and many more (Khan et al., 2018).
Barley is one of the oldest cultivated cereal crops and has a high tolerance to stresses like salt, drought, and heat (Munns et al., 2006; Rollins et al., 2013; Gürel et al., 2016). Whereas changes in the barley transcriptome upon those stresses have been analyzed (Janiak et al., 2018; Osthoff et al., 2019; Nefissi Ouertani et al., 2021), a global transcriptome analysis in response to H2O2 has not been performed yet.
In the present study, we used RNA sequencing (RNA-Seq) to analyze changes in the transcriptome of barley roots and leaves upon application of H2O2. This analysis identified a total of 1001 and 1883 differentially expressed genes (DEGs) in response to H2O2 in leaves and roots, respectively. Comparative and quantitative analyses of gene expression patterns revealed commonly regulated key genes related to H2O2 stress between both tissues, nine of which were further confirmed by qRT-PCR analysis. The data obtained in this study contribute to the understanding of molecular mechanisms of oxidative stress response in barley, which might also play a role upon other stresses that induce oxidative bursts.
2 Materials and methods
2.1 Plant material and growth conditions
Barley plants (Hordeum vulgare cultivar Golden Promise) were grown in pots filled with water-soaked vermiculite in a climate-controlled growth chamber under long-day conditions with 16 h light at 20°C and a light intensity of 120 µmol photons m-2 s-1 (Philips TLD 18W of alternating 830/840 light color temperature) and 8 h darkness at 18°C for five days.
2.2 H2O2 application and RNA isolation
Five-day-old seedlings were harvested and washed carefully to remove any remaining vermiculite prior to submersion in 10 mM H2O2 (Carl Roth, Germany) or ddH2O (control) for three hours. The duration of H2O2 treatment was selected based on previous studies, which showed that at this time point H2O2 induced the strongest changes in the expression of most of the H2O2-responsive genes (Desikan et al., 2001; Stanley Kim et al., 2005; Hieno et al., 2019). Subsequently, seedlings were carefully rinsed with ddH2O and dissected into roots and leaves. Samples were shock-frozen in liquid nitrogen and homogenized using a sterile, ice-cold mortar and pestle. Total RNA was extracted using the Quick-RNA miniprep Kit (Zymo Research, USA) according to the manufacturer’s instructions. The yield and purity of extracted RNA was determined with a NABI Nanodrop UV/Vis Spectrophotometer (MicroDigital, South Korea). The integrity of the extracted RNA was verified by separation of the 28S and 18S rRNA bands on a 1% agarose gel.
2.3 RNA-sequencing and data analyses
RNA sequencing was performed on three biological replicates for each treatment. Each replicate furthermore consisted of pooled material from three plants. Library preparation and transcriptome sequencing (3’ mRNA sequencing) were carried out at the NGS Core Facility (Medical Faculty at the University of Bonn, Germany) using a NOVASEQ 6000 (Illumina, USA) with a read length of 1x100 bases and an average sequencing depth of >10 million raw reads per sample (Table 1). 3’ end sequencing libraries were prepared using the QuantSeq protocol (Moll et al., 2014). Briefly, oligo dT priming were followed by synthesis of the complementary first strand without any prior removal of ribosomal RNA. After successful introduction of Illumina specific adapter sequences, the resulting cDNA was further purified with magnetic beads. The unpaired reads were processed for quality control using fastQC and cutAdapt (Martin, 2011) in order to trim any remaining adapter sequences. They were then aligned using Tophat2 software (Trapnell et al., 2012) against a H. vulgare IBSC v2 reference genome obtained from Ensembl (http://plants.ensembl.org/info/data/ftp/index.html) using a Bowtie index (Langmead and Salzberg, 2012) created with the help of the reference genome (in FASTA format; the individual FASTA files of the chromosomes were concatenated using the “cat” command in UNIX shell). The alignment with Tophat2 was performed on an Ubuntu 18.04 LTS operating system, in a UNIX shell environment. Every step after alignment was performed using R 4.0.0 (R Core Team, 2020). Gene counts from the aligned BAM files were generated using featureCounts function in RStudio (Liao et al., 2014). Differential gene expression analyses was carried out using DESeq2 (Love et al., 2014). The p-values were corrected using the False Discovery Rate (FDR) method (Benjamini and Hochberg, 1995) and subsequently the FDR and the log2FC cutoffs were set to 0.01 and 1, respectively. Principal Component Analyses (PCA) plots were prepared with the raw gene counts for all samples and replicates using the tidyverse and ggplot2 packages. The volcano plots and heatmaps were generated using the EnhancedVolcano and Pheatmap packages, respectively. In addition, transcript per million (TPM) values of each gene were calculated using a separate function designed in the R environment (Supplementary Table S1). With common regulated DEGs, a clustering was performed with four predefined clusters based on FDR and log2FC cutoffs of 0.01 and 0.5, respectively. The first and second cluster consisted of commonly down- and up-regulated genes, respectively, while the third and fourth cluster contained counter-regulated genes between leaves and roots of barley. The clusters were then represented as heatmaps using the pheatmap package and line plots using the ggpubr package.
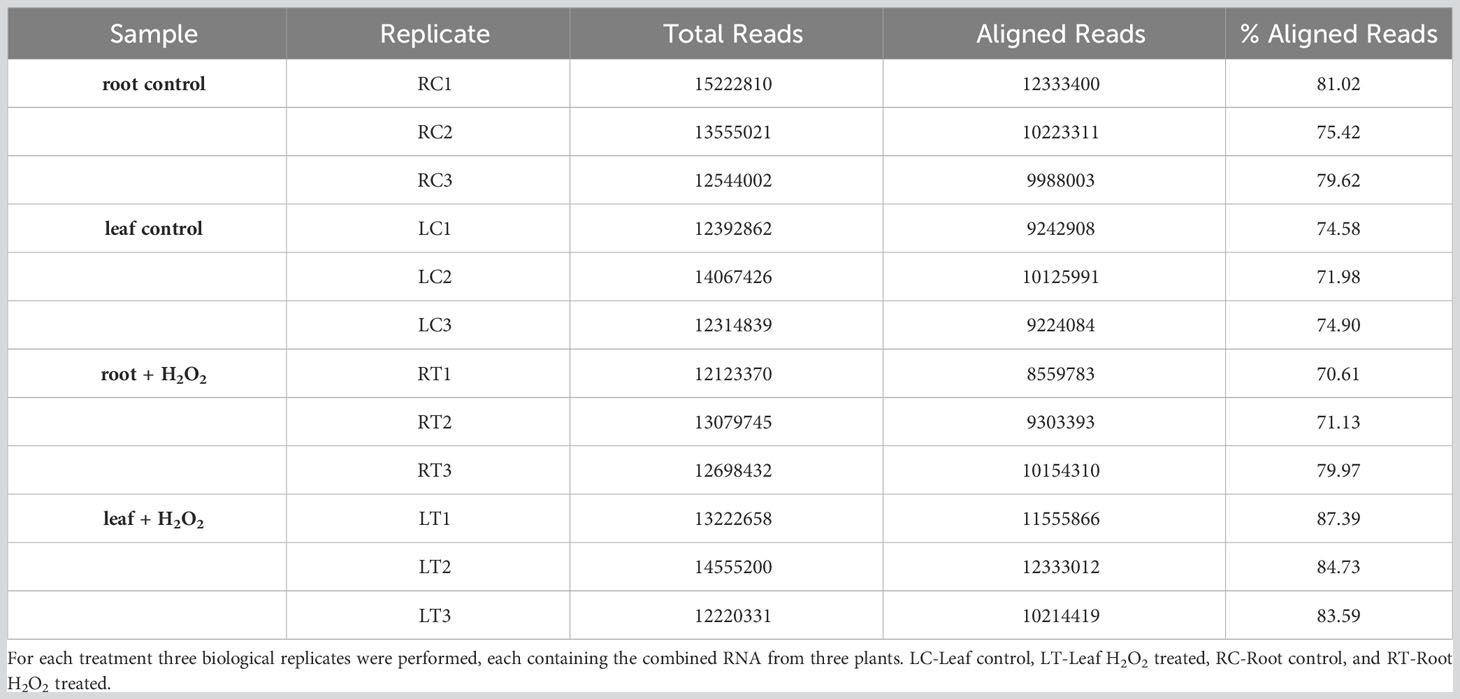
Table 1 Summary of total reads and aligned reads in the RNA-seq samples from barley roots and leaves obtained under H2O2 treatment and control conditions.
Gene ontology (GO) and enrichment analyses were carried out using shinyGO (Ge et al., 2020). Categories were chosen as significant if the FDR was less than 0.05 (Benjamini and Hochberg, 1995). Homology searches against the A. thaliana genome were carried out using the BaRT (Barley Reference Transcript) tool available on www.ics.hutton.ac.uk (Mascher et al., 2017) based on a E-value cutoff of 1e-30.
2.4 Quantification of transcript levels by qRT-PCR
qRT-PCR was performed with three replicates for each sample. Each replicate consisted of the pooled RNA material from three different plants. Synthesis of first strand cDNA for qRT-PCR was carried out from at least 1 µg of total RNA using the RevertAid first strand cDNA synthesis kit (Thermo Fisher Scientific, USA) with oligo-dT18 primers following the manufacturer’s instructions. The quality of cDNA was assessed using a NABI UV/Vis Nanodrop Spectrophotometer. Gene expression was quantified in 48-well plates using a BioRad CFX 96 real-time PCR detection system (BioRad, Germany) and a SYBR Green PCR master mix (Thermo Fisher Scientific, USA). All forward and reverse primers used for qRT-PCR are listed in Supplementary Table S2. Data were quantified using the BioRad CFX Maestro software, and the expression was estimated using the 2–δδCt method (Livak and Schmittgen, 2001) after normalization against the two reference genes HvACTIN and HvGAPDH, as the Cq values of both genes were unchanged upon H2O2 treatment. Data were analyzed statistically with one-way analysis of variance (ANOVA) and Tukey’ Post-Hoc HSD test using the agricolae and tidyverse packages, respectively. Graphs were prepared using the ggpubr package.
2.5 H2O2 staining and microscopic analyses
Staining of hydrogen peroxide in barley leaves and roots was performed with 2’,7’-dichlorodihydrofluorescein diacetate (H2-DCFDA; Thermo Fisher Scientific, USA) based on a modified protocol (Kaur et al., 2016). Briefly, five-day-old barley seedlings were treated with either 10 mM H2O2 or ddH2O (control) for 3 hours. Afterwards, the seedlings were briefly rinsed and treated with 10 µM H2-DCFDA prepared from a 4 mM stock dissolved in DMSO for 1 hour in the dark. After staining, seedlings were washed, and roots and leaves were mounted separately on a microscopy slide. 2’,7’-Dichlorfluorescein (DCF) fluorescence was analyzed using a Leica SP8 Lightning confocal laser scanning microscope (Leica Microsystems, Germany). For excitation, an argon laser with a wavelength of 488 nm was used, and emission of 517-527 nm was detected using a HyD Detector. Fluorescence intensity was quantified in regions of interest (ROI) using the integrated LASX software.
3 Results
3.1 Differential gene expression in leaves and roots of barley in response to application of H2O2
To investigate the transcriptomic modulation in barley (Hordeum vulgare cv. Golden Promise) in response to oxidative stress, five-day-old plants were exposed for three hours to 10 mM H2O2 or to ddH2O as control (Figure 1A). H2-DCFDA staining confirmed that H2O2 penetrated both roots and leaves (Figures 1B, C and Supplementary Figure 1). RNA was then extracted separately from roots and leaves, and RNA-seq analysis was carried out on three biological replicates per tissue and treatment, each comprising the pooled RNA from three different plants (Supplementary Table S1). On average approximately 13 million total reads were obtained per sample. About 75-85% of these reads could be aligned to the barley reference genome (Table 1). To assess the main variances within the dataset, a principal component analysis (PCA) was performed. The result showed that PC1 (X-axis), which separates the samples by tissue, represents the largest variation in our dataset compared to PC2 (Y-axis), which separates the samples by treatment (Figure 2A). Consequently, the differential gene expression analysis was separately performed for the leaf and root samples.
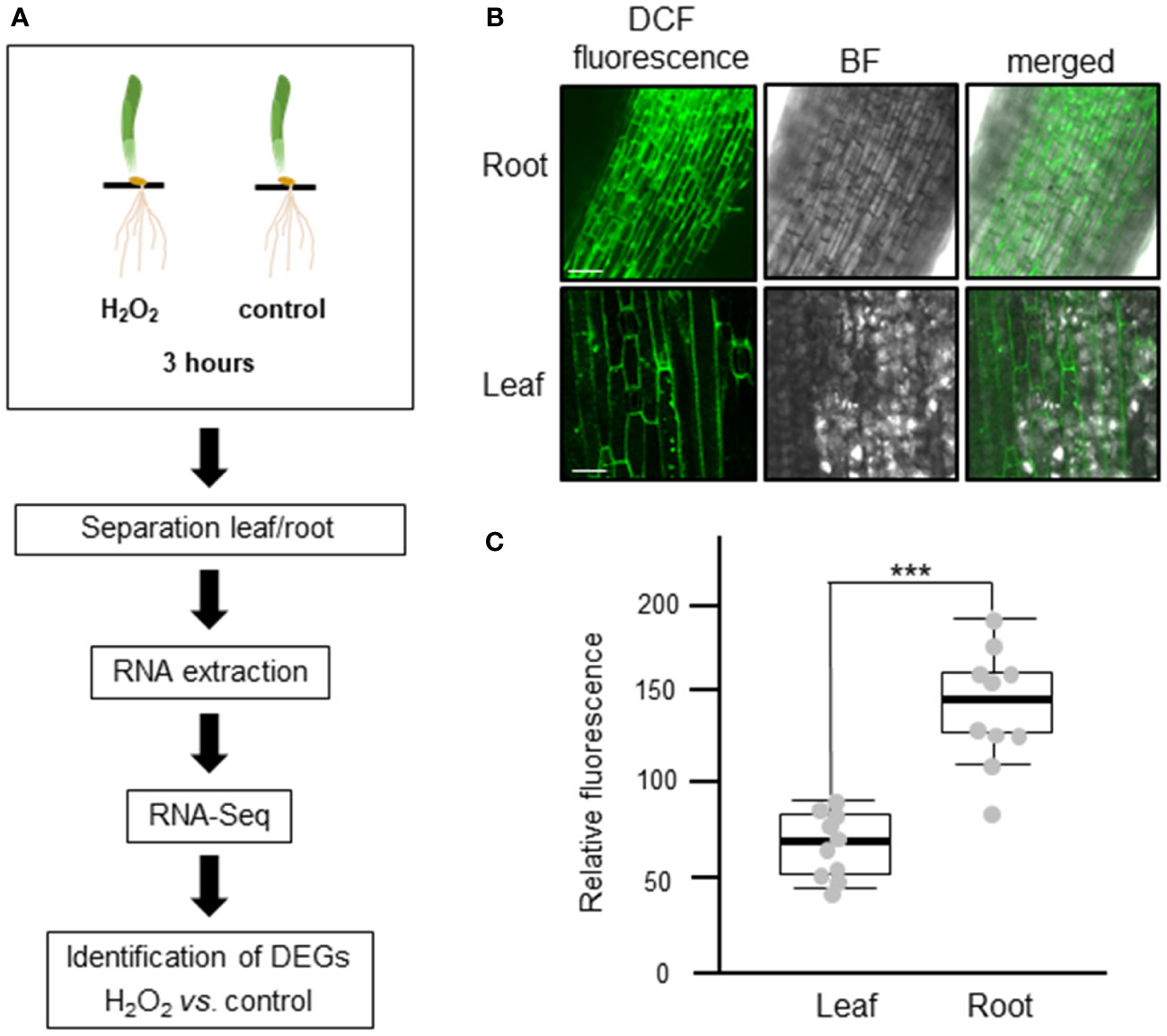
Figure 1 Experimental design to analyze the transcriptional changes of barley plants to oxidative stress. (A) Schematic representation of the study design. Five-day-old barley plants were treated with either 10 mM H2O2 or water (control) for three hours. After the treatment, leaves and roots were separated, RNA was extracted, and three independent biological replicates, each containing the pooled RNA from three plants, were submitted to RNA-Seq analyses. The raw reads obtained were subjected to quality control and aligned against the barley reference genome. Based on raw gene counts, a differential expression analysis was carried out using DESeq2. (B) Uptake of H2O2 in roots (upper panel) and leaves (lower panel) visualized by H2-DCFDA. Green fluorescence of the 2’,7’-Dichlorfluorescein (DCF) was observed using a Leica SP8 lightning confocal laser scanning microscope. BF: bright field; bar: 100 µm. (C) Quantification of fluorescence intensity of H2-DCFDA relative to untreated control tissues. Each dot represents the average of five regions of interests (ROIs). ROIs were taken from two independent images from three biological replicates (n=6). Statistical analysis was carried out using the two-tailed t-test (*** = P<0.001).
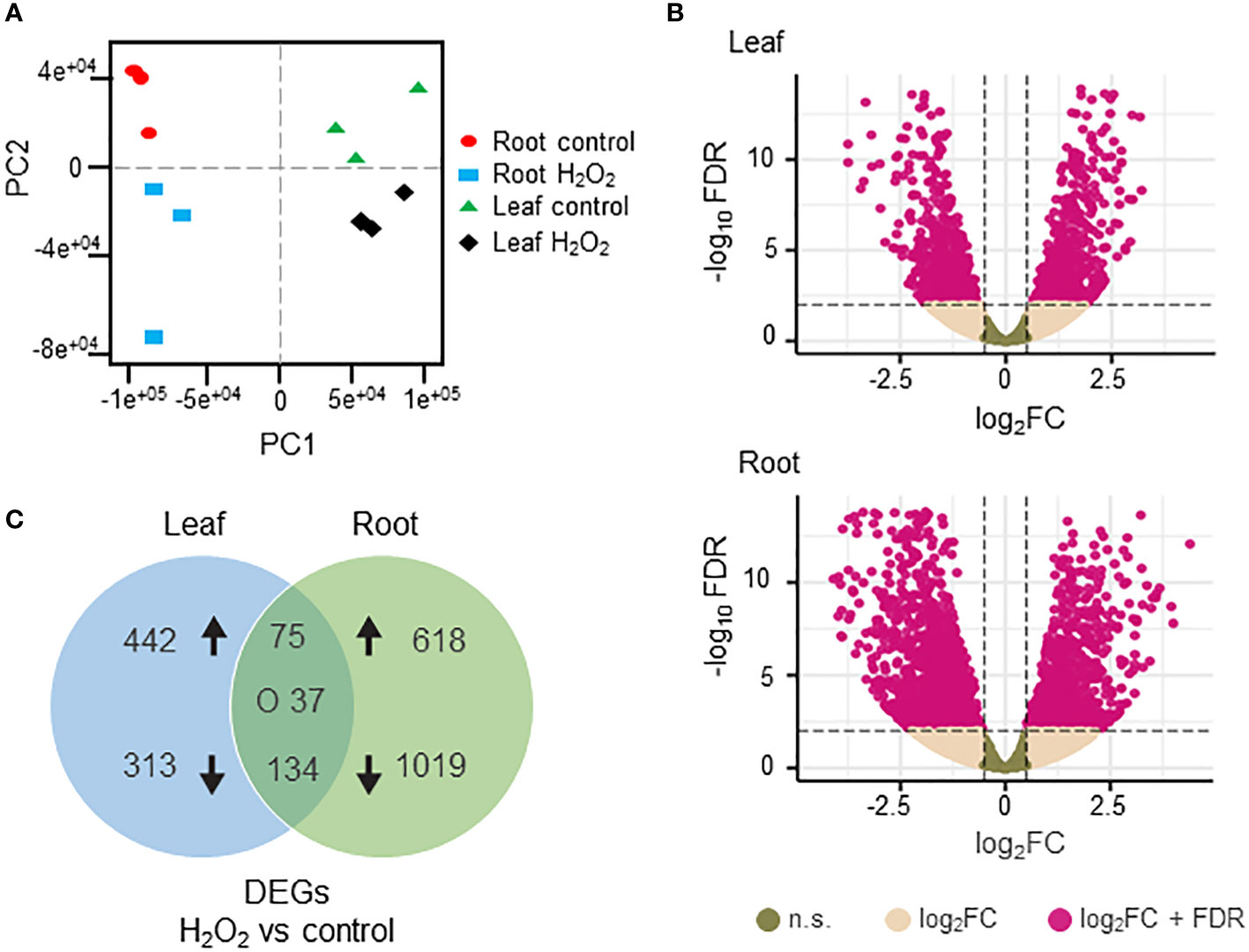
Figure 2 Differentially expressed genes (DEGs) in H2O2-treated and untreated barley plants. (A) Principal component analysis of the RNAseq data showing the homogeneity of the different samples. PC1 (X axis) separates the samples by tissue while PC2 (Y axis) separates the samples by treatment. (B) Volcano plots of the DEGs in leaves (upper panel) and roots (lower panel). The X axis represents the fold change (Log2FC) of the DEGs (H2O2 vs. control), whereas the Y axis represents the statistical significance (log10FDR). Pink dots indicate genes that fit the DESeq criteria of FDRand │Log2FCin│, while beige dots represent DEGs that fit only Log2FC. N.S.: not significant (C) Venn diagram representing DEGs (DESeq, adjusted to FDR<0.01 and │Log2FC│≥1) between H2O2-treated and untreated samples in leaves and roots. Arrows indicate up- and down-regulation. ‘O’ indicates counter-regulated genes.
Differentially expressed genes (DEGs) between H2O2-treated and control samples were identified based on fold change (FC) │Log2FC ≥ 1│ and FDR < 0.01 (Supplementary Table S3). A total number of 2884 DEGs were detected across both tissues. H2O2 application clearly resulted in stronger transcriptional changes in roots compared to leaves (Figure 2B). Of the 1883 DEGs detected in roots, 701 were up- and 1182 were down-regulated, while in leaves 1001 DEGs were identified with 546 up- and 455 down-regulated (Figure 2C). Among all DEGs only 75 and 134 were commonly up- and down-regulated, respectively, in both tissues, while 37 were counter-regulated.
3.2 Gene ontology analyses
GO classification was used to identify the 20 most significant biological process categories within the DEGs. The results show that not only the number of genes, but also the biological processes affected by H2O2 were clearly different between leaves and roots (Figure 3). In leaves, GO terms associated with genes that showed the highest fold change were related to protein complex oligomerization, response to H2O2 and jasmonate. Further categories with lower fold change but often higher number of genes comprised quite global stress effects associated with different, mostly abiotic stimuli, but also wounding (Figure 3A). In roots, many of the enriched GOs were associated with response to oxygenic stress including H2O2 catabolism, glutathione and ROS metabolism, or cellular oxidant detoxification as well as with cell wall modulation (Figure 3B).
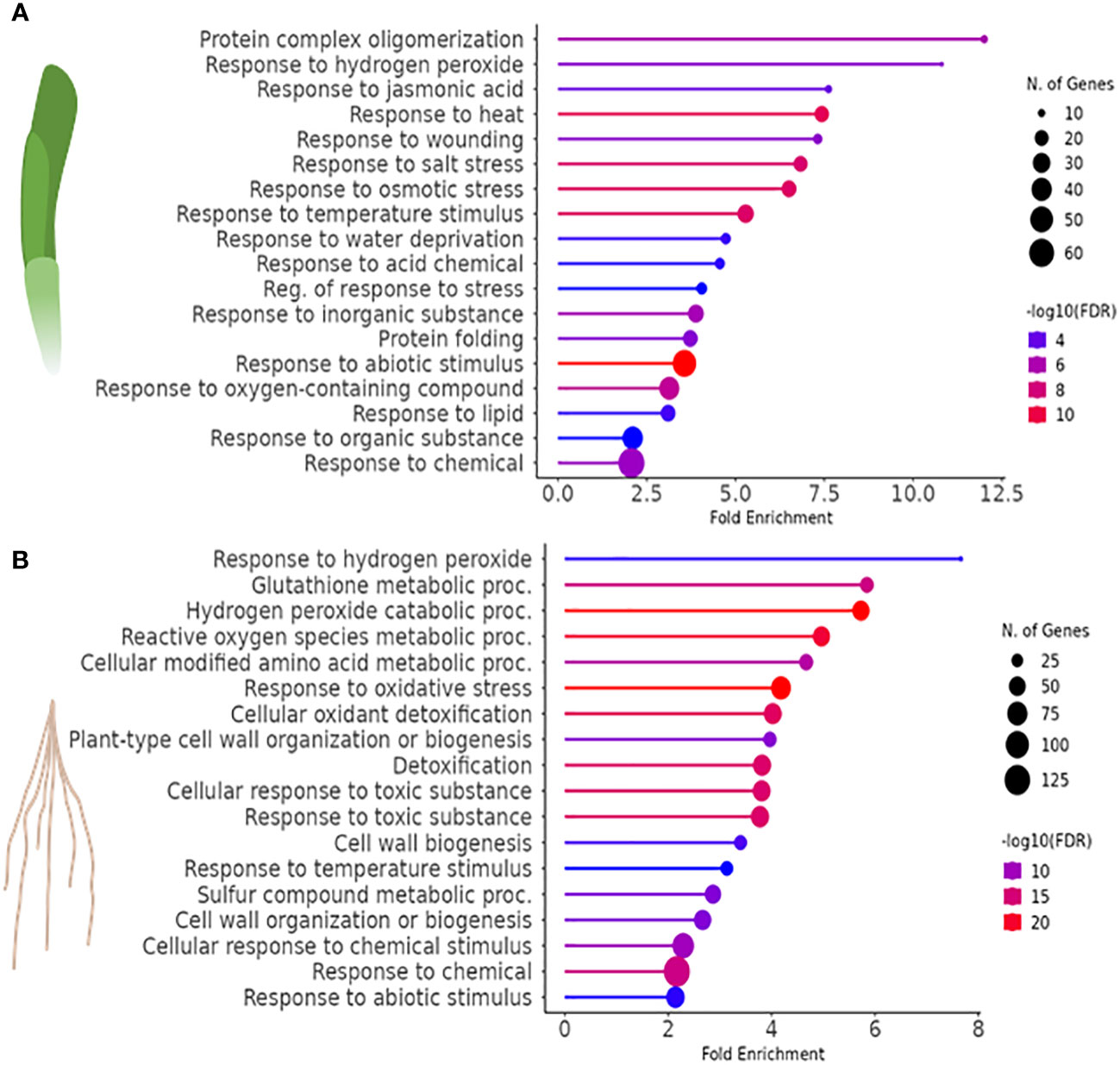
Figure 3 Gene ontology (GO) enrichment analysis to identify biological processes associated with the DEGs (FDR<0.01) of H2O2-treated vs. control samples in (A) leaves and (B) roots of barley.
3.2.1 Differentially expressed genes in barley leaves in response to H2O2
In barley leaves, the most highly enriched GO term category upon exposure to H2O2 was the response to H2O2 and protein complex oligomerization (Figure 3A). Both categories consist of the same SMALL HEAT SHOCK PROTEINS (SHSP domain-containing proteins) (Table 2). SHSPs are ubiquitous in prokaryotic and eukaryotic organisms and function as chaperone proteins involved in the response to many abiotic stresses (Basha et al., 2012; Waters, 2013). Their expression levels were shown in different plant species to increase upon stress and to enhance stress tolerance. Here, barley leaves exposed to H2O2 showed an increased expression of SHSPs, except for the 18.8 kDa class V heat shock protein (HORVU2Hr1G046370), which was down-regulated. All of the differentially regulated SHSPs have close orthologs in Arabidopsis (Li and Liu, 2019) with the majority being orthologous to AtHSP17.6II (At5g12020).
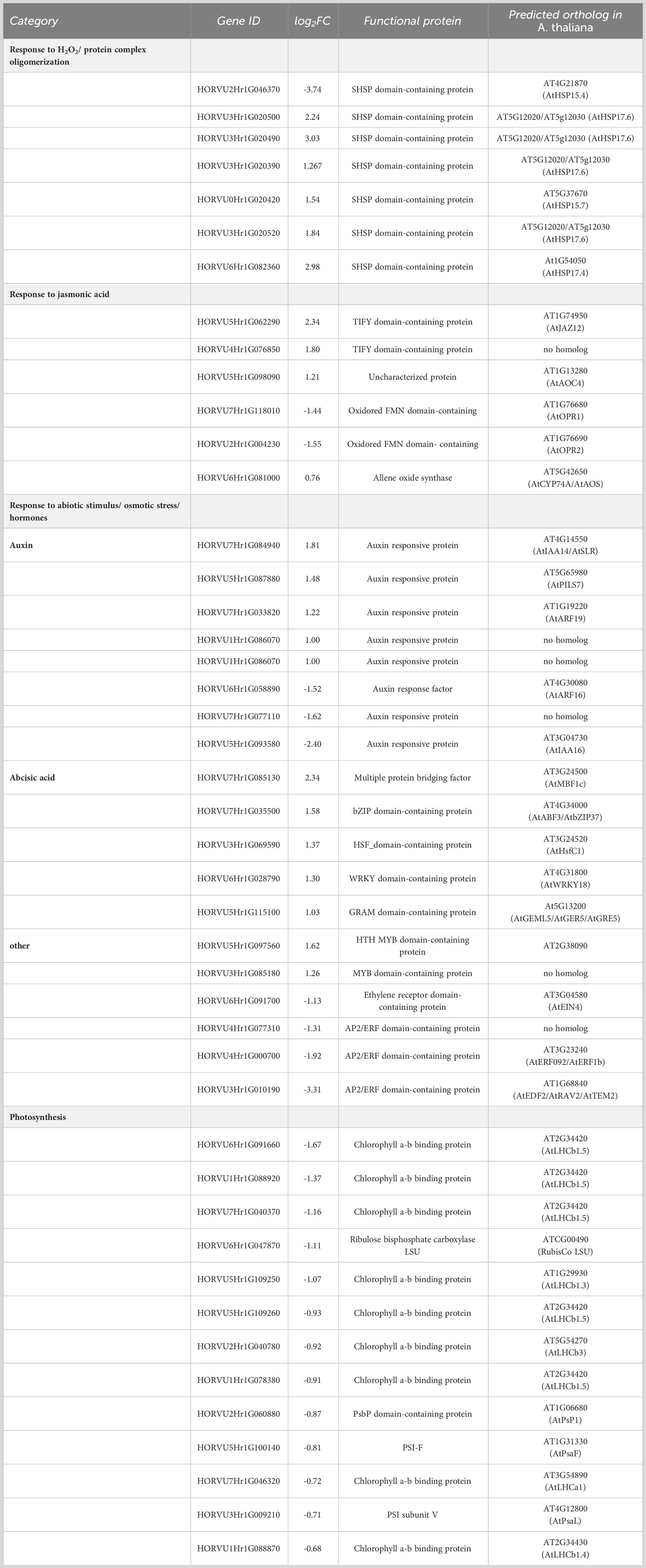
Table 2 Selected DEGs associated with top GO terms in leaves of barley in response to H2O2.
An enrichment was also found for genes involved in hormone biosynthesis and signaling, especially jasmonate, auxin, and abscisic acid (ABA). Jasmonate-related DEGs were represented by the specific GO-term category ‘response to jasmonic acid’. This category comprised two up-regulated TIFY domain-containing proteins with no direct homologs in Arabidopsis (Table 2). The TIFY domain is also known as ZIM domain which is present in members of the transcriptional repressor JASMONATE ZIM-domain (JAZ) family, key elements in the jasmonate signaling pathway (Chung and Howe, 2009; Pauwels and Goossens, 2011). This category also includes genes that encode for enzymes involved in jasmonate biosynthesis (Schaller and Stintzi, 2009; Bittner et al., 2022) such as ALLENE OXIDE CYCLASE (AOC), and OXOPHYTODIENOATE-REDUCTASE (OPR) as well as ALLENE OXIDE SYNTHASE (AOS) but with a FC less than 2 (FC 1.69, Log2FC=0.76). By contrast, genes related to other hormone signaling pathways were found redundantly interspersed in the two GO terms ‘response to abiotic stimulus’ and ‘response to salt stress’ (Figure 3A). With regard to auxin, a number of orthologs to auxin-responsive genes from Arabidopsis, especially IAA-type TFs, were found. Similar to the jasmonate signaling pathway, H2O2 seems to affect the auxin pathway differentially since both, up- and down-regulated DEGs, were identified. All components related to the phytohormone ABA were up-regulated and those related to APETALA2/ETHYLENE RESPONSIVE FACTOR (AP2/ERF) domain-containing proteins, known to be involved in abiotic stress responses and associated with various hormones, were down-regulated. Similar to the GO term categories related to auxin, both sets comprise mostly orthologs to TFs or co-regulators known in Arabidopsis (Table 2).
In leaves, genes associated with photosynthesis light harvesting in photosystem I, were also affected, however, the category did not appear in the top GOs since for several of the genes the FC was less than 2 but mostly higher than 1.5 (Table 2; Log2FC between 0.5 and 1). This category contained mostly down-regulated DEGs, including several orthologs of Arabidopsis LHCII trimer components, i.e., genes encoding for LHCb1 and LHCb3, and the LHCa1 protein. It furthermore comprised orthologs to the photosystem I subunits PSAF and PSAL but also the oxygen evolving complex subunit PSBP-1 and the large subunit of RIBULOSE-1,4-BISPHOSPHATE-CARBOXYLASE/OXYGENASE (Rubisco) (Table 2).
3.2.2 Differentially expressed genes in barley roots in response to H2O2
In barley roots, the most enriched GO terms are associated with response to oxidative stress and detoxification (Figure 3B). This is also evident by the fact that many DEGs within those GO terms are class-III peroxidases, catalases, or genes related to glutathione metabolism, which were grouped together as a category named ‘Detoxification of H2O2’ (Table 3). In plants, class-III peroxidases have been described in association with a wide variety of biotic and abiotic stresses along with plant defense mechanisms (Almagro et al., 2009; Shigeto and Tsutsumi, 2016). While most peroxidases were up-regulated, some were down-regulated along with a number of glutathione transferases, an ascorbate peroxidase (APX), and CATALASE 1. We also found strong up-regulation of the genes for two putative detoxification efflux carriers/multidrug and toxic compound extrusion (DTX/MATE) transporters. These metabolite transporters have been described to be associated with plant stress responses and overexpression of a gene encoding a cotton DXT protein in Arabidopsis reduced stress-induced levels of H2O2 (Lu et al., 2019).
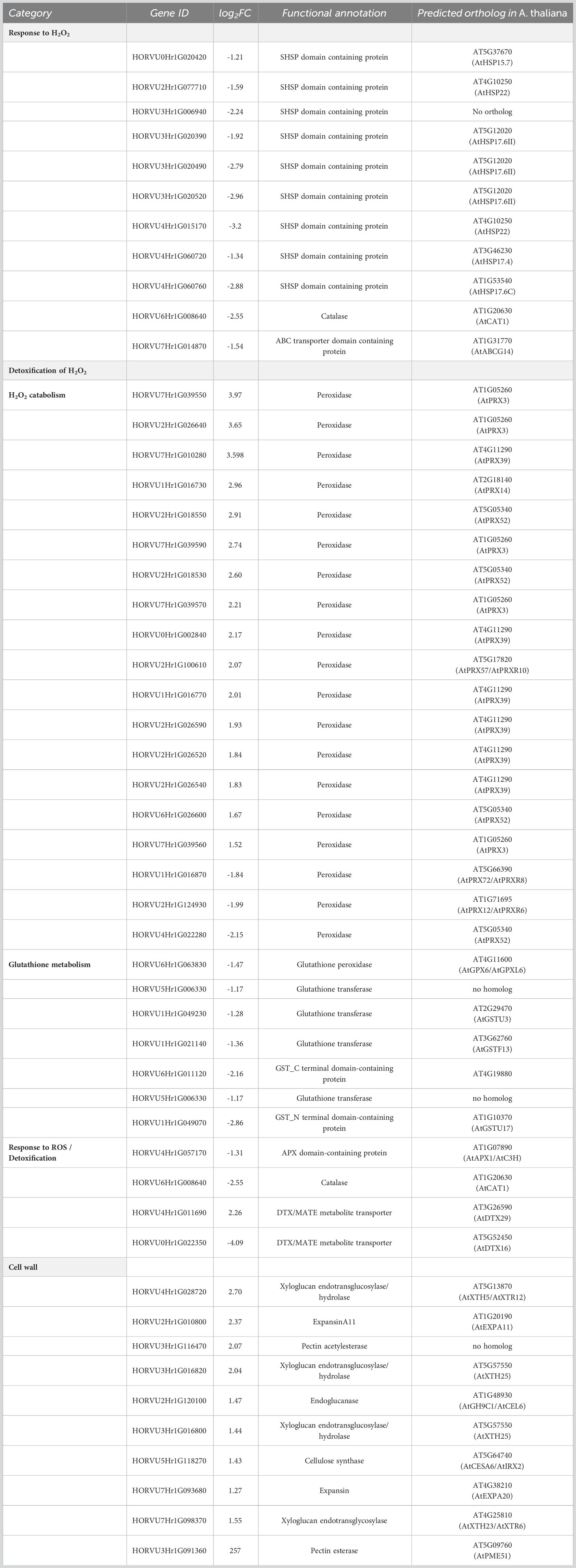
Table 3 Selected DEGs associated with top GO terms in roots of barley in response to H2O2.
As in leaves, the most highly enriched GO term category in roots upon exposure to H2O2 was the response to H2O2, albeit with very few genes (Figure 3B). Similar to leaves, this category includes several SHSP domain-containing proteins, but in contrast to leaves, they were down-regulated (Table 3). All of the differentially regulated SHSPs have close orthologs in Arabidopsis, with several of them being orthologous to AtHSP17.6. This category contains also down-regulated catalase and ABC transporter containing domain proteins.
H2O2 treatment also induced up-regulation of components of cell wall biogenesis and modulation, such as xyloglucan endotransglucosylase/hydrolase, expansin, endo-1,4-beta glucanase, pectin acetyl esterase, and cellulose synthase (Table 3) that were found interspersed in several GO term categories. Indeed, H2O2 and peroxidases were shown to be involved in cell wall remodeling upon environmental stress (Tenhaken, 2015).
3.3 Common DEGs of leaves and roots in response to H2O2
As described above, we identified a total of 246 common DEGs between leaves and roots of barley when using a │log2FC ≥ 1│cutoff (Supplementary Table S3, Figure 2C). For several genes, we noticed that they were differentially regulated in both tissues, however, in one tissue they showed an expression with a FC>2 (│log2FC ≥ 1│) while in the other tissue a FC less than 2 but higher as 1.5 Thus, for (│log2FC between 1 and 0.5│) was detected. determination of commonly regulated genes in leaves and roots we used a cutoff of Log2FC≥0.5 and listed these genes separately in Supplementary Table S3. Using this cut-off, a total 349 common DEGs were identified between roots and leaves of barley (Supplementary Figure S2; Supplementary Table S3). Of these, 116 and 176 genes were up- and down-regulated, respectively, while 58 genes showed counter-regulation. These common DEGs were organized in four clearly distinguishable clusters (Figure 4A), with either commonly down- (cluster 1) and up-regulated (cluster 2) genes or genes up-regulated in leaves but down-regulated in roots (cluster 3) and vice versa (cluster 4). Heat maps and line plots were constructed to visualize the changes in gene expression pattern for each cluster (Figures 4A, B).
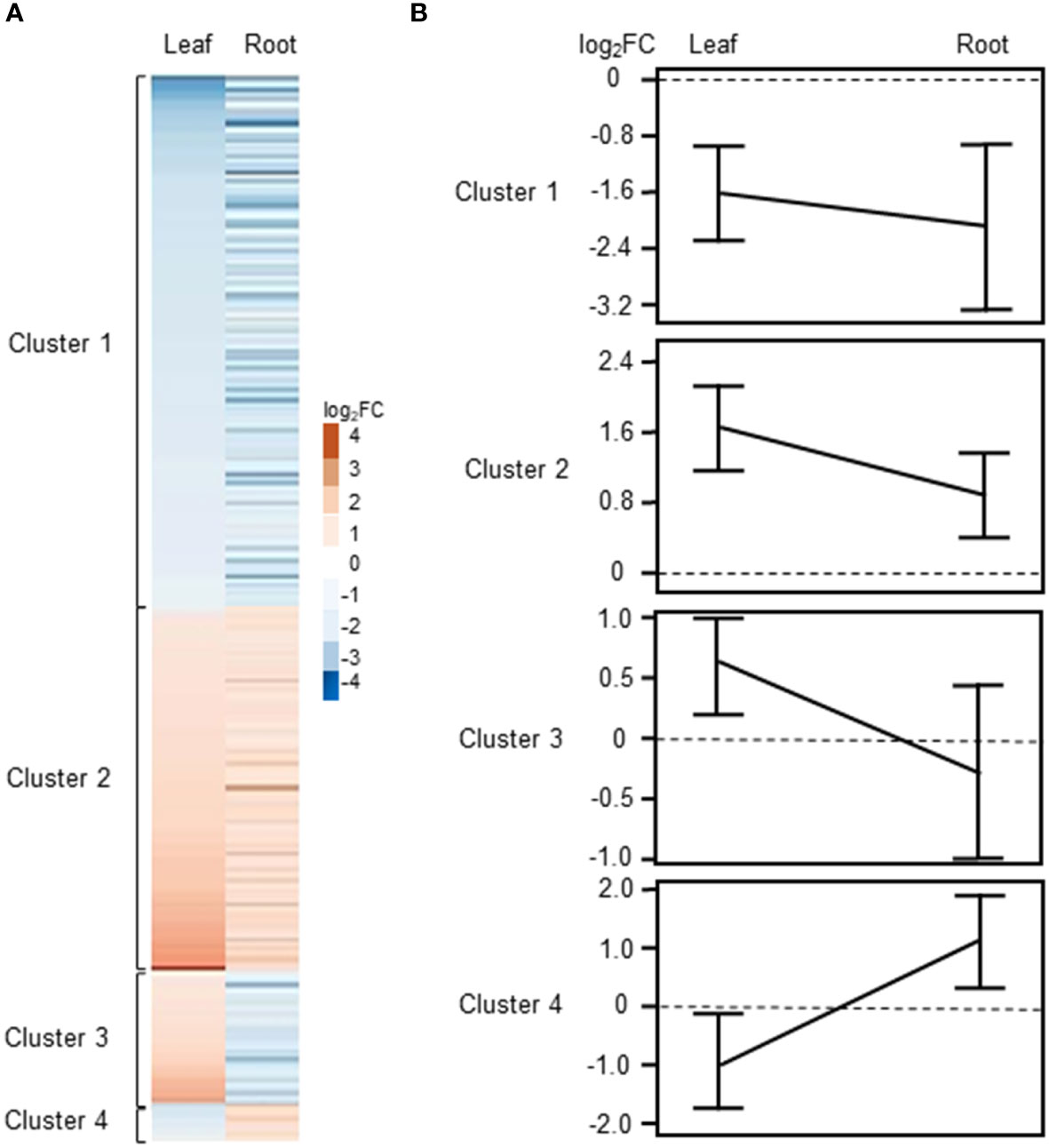
Figure 4 Clustering of DEGs commonly regulated or counter-regulated in leaves and roots of barley upon H2O2 treatment (|Log2FC|≥0.5 and FDR<0.01). (A) Heat map showing the Log2FC associated with each gene in leaves and roots. (B) Line plot showing the mean ± SE of the |Log2FC| associated with each cluster in leaves and roots.
3.3.1 Commonly up- and down-regulated genes
Cluster 1 contains DEGs commonly down-regulated in leaves and roots upon H2O2 treatment (Supplementary Table S3), among them members of important transcription factors such as AP2/ERF, WRKY, CBF1, NAC, and HD-ZIP HOMEOBOX (Supplementary Table S4, Figure 5A). Cluster 1 also comprises orthologs to the Arabidopsis sugar transporters SWEET10 and SWEET5. Other transporters were orthologs to the phosphate transporter PHT1;7 and the aquaporin TIP4;1. TIP aquaporins in plants had been shown to not only transport water molecules but also other molecules like H2O2 (Kurowska et al., 2020). In addition to components of oxidative stress, detoxification or cell wall biogenesis and modification that were already discussed in chapter 3.2.2, cluster 1 also contained several kinases including orthologs to the CYSTEINE-RICH RECEPTOR-LIKE PROTEIN KINASES (CRKs), CRK29 and CRK25. CRKs are presented in Arabidopsis by a large gene family with over 40 members and have been associated with various abiotic and biotic stresses (Bourdais et al., 2015).
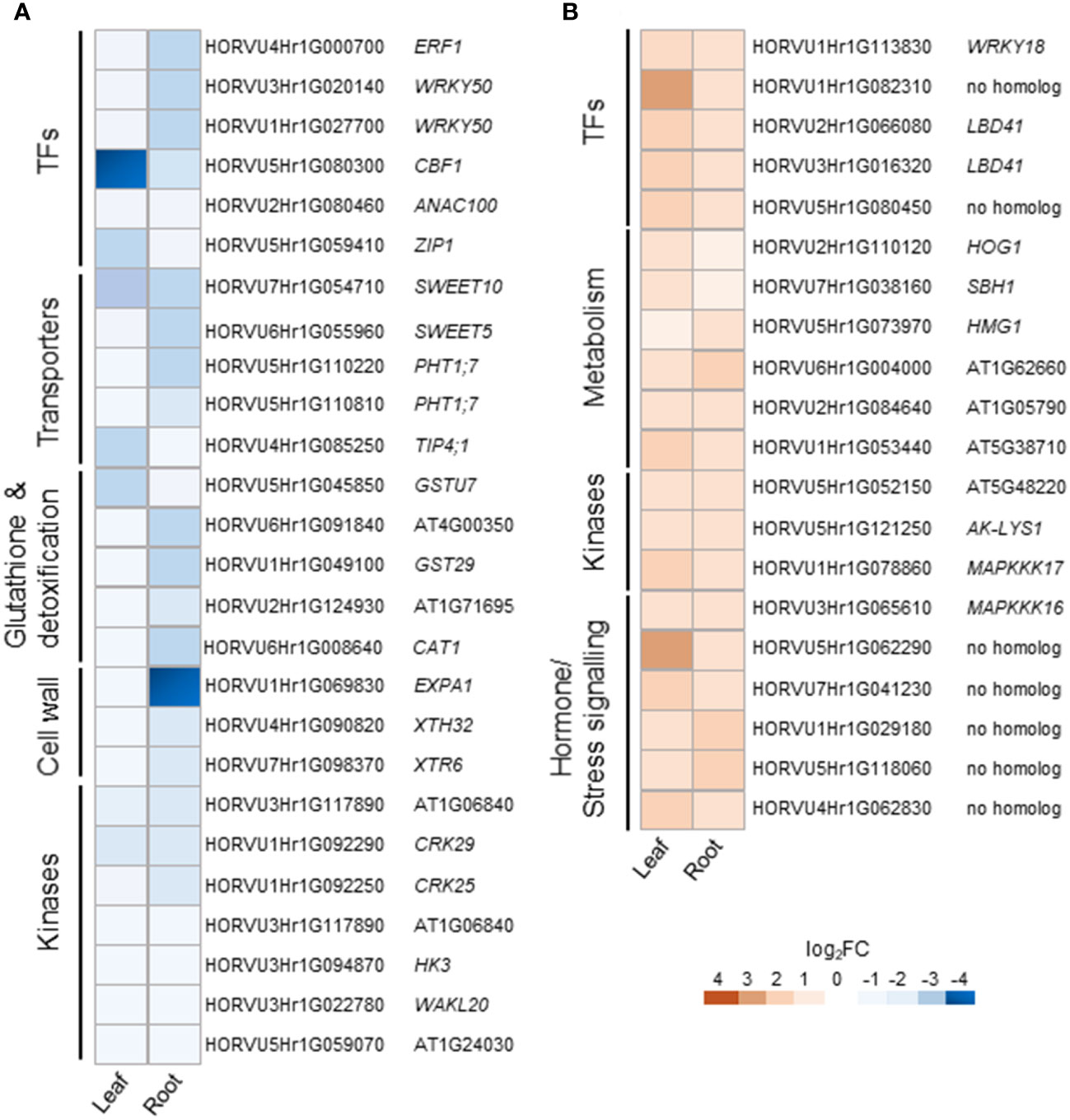
Figure 5 Selected DEGs commonly regulated in leaves and roots of barley upon H2O2 treatment. Down-regulated (A) and up-regulated (B) genes are grouped by functional category and presented with their Arabidopsis orthologs. TFs, transcription factors.
Cluster 2 contains DEGs commonly up-regulated in leaves and roots (Supplementary Table S3). Interestingly, it contains TFs of similar families as cluster 1, like WRKY and AP2/ERF but also orthologs of the LOB DOMAIN CONTAINING PROTEIN 41 (LBD41) from Arabidopsis (Supplementary Table S4; Figure 5B). DEGs associated with primary metabolism like amino acid and nucleic acid metabolism were also found in cluster 2. Genes associated with primary metabolism were also shown to be up-regulated in other transcriptome studies associated with abiotic stress (Hirai et al., 2004; Wang et al., 2014) and DEGs found in cluster 2 do not seem to be related to any specific metabolic pathway. Two MITOGEN-ACTIVATED PROTEIN KINASEs (MAPKs) identified in cluster 2 are orthologs to AtMAPKKK16 and AtMAPKKK17, both of which were shown to be regulated by ABA (Wang et al., 2011).
3.3.2 Counter-regulated genes
Cluster 3 consists of 42 DEGs up-regulated in leaves and down-regulated in roots of barley upon H2O2 treatment (Supplementary Table S3). Nine of these DEGs are orthologs to different small heat shock proteins from Arabidopsis (Supplementary Table S4; Figure 6). The cluster furthermore comprises an assorted set of genes whose orthologs in Arabidopsis are connected with various metabolic pathways and hormone signaling.

Figure 6 Selected counter-regulated DEGs in leaves and roots upon H2O2 treatment. Genes up-regulated in leaves and down-regulated in roots are grouped by functional category and presented with their Arabidopsis orthologs. Metabo., metabolism; sig., signaling.
Cluster 4 consists of only 15 genes and no common functional categories were found (Supplementary Table S4). However, they include genes, whose Arabidopsis orthologs have been associated with hormones, or cell wall modification, i.e. the COPPER-CONTAINING AMINE OXIDASE 3 (CUAO3) that was suggested to be involved in stress response since it was up-regulated upon treatment with several hormones or flagellin (Planas-Portell et al., 2013).
Overall, clusters 3 and 4 show very few genes previously described to be associated with oxidative stress.
3.4 qRT-PCR confirmation of selected DEGs
In order to confirm the results obtained from RNA-seq analyses, we performed quantitative RT-PCRs (qRT-PCR) on some of the identified DEGs. For these, we selected several DEGs that showed common regulation in leaves and roots in our dataset and which, based on their functional annotation, could be related to oxidative stress (Supplementary Table S5). Orthologs to some of them had already been shown to play an important role in H2O2 and ROS-related signaling not only in Arabidopsis but also in important crops like wheat, maize, and rice (Polidoros et al., 2005; Mylona et al., 2007; Steffens, 2014; Dudziak et al., 2019). They also represent different levels of regulation, some being among the most highly up- or down-regulated genes and other showing a much more subtle response. These genes represent different gene ontologies, and encode for a catalase, a peroxidase, a glutathione S-transferase, several TFs, a MAPKKK, and a xyloglucan endotransglucosyalase, a protein involved in cell wall modification. As shown in Figure 7 and in Supplementary Table S5, the log2FC changes observed with the different techniques were often quite close and, in all cases, the results of the qRT-PCR matched the trend observed in the RNA-seq data.
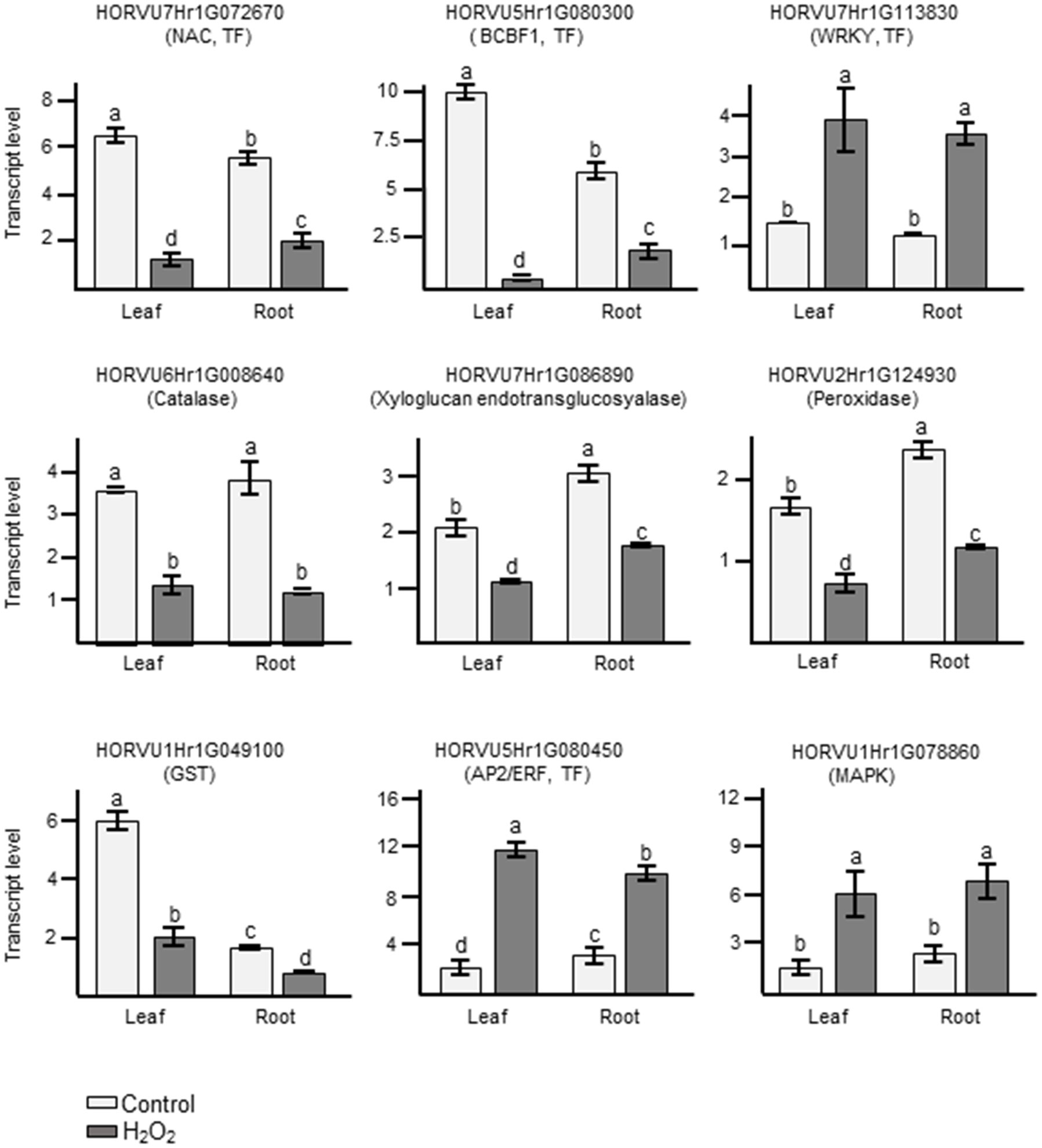
Figure 7 Analyses of transcript levels for selected candidate genes by qRT-PCR. Data represent means ± SE of three biological replicates (n=3), each having two technical repeats. Transcript levels were normalized to HvACTIN and HvGAPDH. Letters represent significant differences estimated using one-way ANOVA and Tukey’s Post-Hoc HSD test (P<0.05). Potential functions of the genes inferred from orthologous genes in Arabidopsis are indicated in brackets.
4 Discussion
In plants, H2O2 is a crucial ROS which plays a dual role as a harmful by-product of cell metabolism and as a secondary messenger that affects development and growth. Complex cross-talk between H2O2 and other signaling molecules, such as Ca2+ ions and hormones, plays a key role in regulating different biological processes that contribute to the response to various biotic and abiotic stresses (Peiter, 2016; Saxena et al., 2016). Despite its importance, very little is known about H2O2-induced changes of the transcriptome in barley. In this study, an analysis of the barley transcriptome in response to H2O2 was performed using next generation sequencing. First, a suitable concentration of H2O2 that was shown to initiate a stress response in barley was selected on basis of previously performed experiments (Dodd et al., 2010; Giridhar et al., 2022). An increase in cytosolic Ca2+ ([Ca2+]cyt) is one of the first responses of plants to most biotic and abiotic stresses (Dodd et al., 2010) that in turn leads to downstream stimulus-specific cellular responses. H2O2 was shown to induce such transient changes of [Ca2+]cyt with 10 mM eliciting the highest response in barley roots and leaves (Giridhar et al., 2022). Staining of intact plants with the ROS indicator H2-DCFDA confirmed that the exogenously applied H2O2 penetrated into both organs (Figures 1B, C, Supplementary Figure 1). To exclude natural degradation of RNA and changes of the transcriptome driven by processes such as senescence or tillering, five-day-old barley plants were used. Growth of monocotyledonous leaves is initiated from the base and the leaf blade shows developmental gradients, i.e., disappearance of poly (A+) RNA levels along the developing blade (Hellmann et al., 1995). Moreover, plant senescence is a natural process known to be initiated by ROS that in turn activates transcription factors interacting with senescence associated genes (Bieker et al., 2012; Shimakawa et al., 2020). Thus, the growth conditions and plant age used in the analysis ensure as much as possible a solely treatment-dependent change of the transcriptome.
Overall, the RNA-seq analysis showed that under the chosen conditions H2O2 caused more transcriptional changes in roots compared to leaves (Figure 2). Most of the identified DEGs were found exclusively in one of the two plant parts, further confirming organ-specific responses. While this difference may be in part due to a difference in H2O2 penetration into roots and leaves, it is more likely caused by differential response of the two tissues to H2O2 signals and/or oxidative stress. Only about 10% of the DEGs were found to be up- and down-regulated in leaves as well as in roots, some of which showed counter-regulation. This difference in response is also mirrored by the GO terms associated with the identified DEGs that only showed a minor overlap (Figure 3).
4.1 Leaf-specific transcriptomic changes in response to H2O2
Our data showed that several genes encoding for small heat shock proteins (SHSPs) were up-regulated by H2O2 in barley leaves (Table 2). In barley, the roles of several HSPs in response to a diverse range of abiotic stimuli have been characterized (Hlaváčková et al., 2013; Chaudhary et al., 2019; Landi et al., 2019). HSPs have also been shown to play crucial roles during abiotic stresses such as cold and heat in other important crop genera, like rice, maize, and wheat (ul Haq et al., 2019). SHSPs are a subgroup of HSPs defined by their size and a conserved α-crystalline C-terminal domain. They are known to form oligomeric complexes and prevent denatured proteins from aggregation until they can be refolded by other HSPs. They have been speculated to interact with transcription factors of the HEAT SHOCK FACTOR (HSF) family to create the HSP-HSF complex, alteration of which can drive essential reactions in response to ROS (Driedonks et al., 2015). The SHSPs in our data set belong to subfamilies with close orthologs in Arabidopsis, i.e. HSP17.6, 15.4, 15.7, and 17.4 (Li and Liu, 2019). HSP17.6 and HSP15.7 have been shown to be localized in the peroxisomes in Arabidopsis (Ma et al., 2006; Li et al., 2017). Peroxisomes are one of the main subcellular compartments in which ROS are produced by processes such as ß-oxidation and photorespiration, and which are crucial for antioxidant defense (Sandalio et al., 2013; del Río and López-Huertas, 2016). Additionally, HSP17.4 and 17.6 have been shown to exhibit increased transcript levels during periods of abiotic stress in Arabidopsis (Swindell et al., 2007). Thus, the induction of these HSPs points to a potential role of these proteins in increasing the tolerance to oxidative stress also in barley leaves. The single down-regulated SHSP is an ortholog to AtHSP15.4, for which this contrary behavior upon stress was already described (Siddique et al., 2008).
Not surprising, considering the well-established juxtaposition between ROS production and photosynthesis, the application of H2O2 negatively affected several photosynthetic components (Table 2). The most affected group represents chlorophyll a/b binding proteins orthologous to various light-harvesting complex proteins of the LHCb-type and to a component of the light-harvesting complex I, LHCa1, of Arabidopsis. Down-regulation of LHCb-type proteins upon oxidative stress has been previously described (Staneloni et al., 2008). It is likely part of an established photoprotection mechanism to alleviate increased ROS levels generated when the photosynthesis reaction becomes unbalanced, e.g., under high light conditions.
The role of phytohormones like ABA and jasmonate in response to several biotic and abiotic stimuli has been extensively studied in plants (Verma et al., 2016). In our data, several genes related to jasmonate signaling were found to be down-regulated (Table 2), including an ortholog of Arabidopsis 12-OXOPHYTODIENOATE REDUCTASE (OPR). The OPR3 protein of Arabidopsis has been denoted as one of the most crucial enzymes in jasmonate synthesis, which converts 12-oxophytodieonic acid (cis-OPDA) to OPC8:0 in peroxisomes (Bittner et al., 2022). However, recent studies highlighted the role of an OPR3-independent pathway for jasmonic acid (JA) biosynthesis, involving an OPR2-mediated alternative bypass via dinor-OPDA (dnOPDA) and 4,5-didehydro-JA, which is then converted to JA (Chini et al., 2018). Interestingly, we found a down-regulation of the barley ortholog of OPR2 in leaves, the consequence of which remains speculative due to the unclear role of the OPR3-independent bypass pathway. By contrast, genes coding for ALLENE OXIDE CYCLASE (AOC) and ALLENE OXIDE SYNTHASE (AOS) were up-regulated in leaves. These enzymes catalyze the generation of both cis-OPDA and dnOPDA, which in turn would increase OPDA production for both pathways. This is interesting, because OPDA is believed to have an independent regulatory function both on transcription (similar to JA-Ile), but also on protein activity by OPDadylation. Moreover, OPDA-mediated signaling seems closely associated with thiol metabolism and redox-mediated processes (Böttcher and Weiler, 2007; Ohkama-Ohtsu et al., 2011; Bittner et al., 2022). Also related to jasmonate signaling are two TIFY domain-containing proteins that were induced in response to H2O2 (Table 2). The TIFY domain is found in members of the JASMONATE ZIM DOMAIN (JAZ)-type transcriptional repressors involved in jasmonate signaling (Chung and Howe, 2009; Pauwels and Goossens, 2011). However, no regulation of TFs associated with jasmonate signaling was detected in our data set.
By contrast, many of the genes associated with other phytohormones, e.g. auxins and ABA, encode TFs or other proteins involved in transcription regulation (Table 2). Several of these genes belong to the large family of AP2/ERF-type TFs, members of which have been associated with environmental stresses including hypoxia and oxidative stress. While mostly associated with ethylene, AP2/ERF function is also connected to ABA, gibberellic acid, cytokinin, and brassinosteroids (Xie et al., 2019). The largest group of genes associated with hormones relates to auxin (Table 2), the role of which is mostly associated with development and growth. However, experimental evidence linked auxin also to oxidative stress, especially auxin-mediated stress-dependent cell proliferation including the RSL-type TF ROOT HAIR DEFECTIVE SIX-LIKE4 (RSL4) that targets NADPH oxidases also known as respiratory burst oxidase homologs (RBOHs) and secreted plant-specific type III peroxidases that impact apoplastic ROS homeostasis and in turn stimulate root hair cell elongation (Pasternak et al., 2005; Iglesias et al., 2010; Mangano et al., 2017).
4.2 Root-specific transcriptomic changes in response to H2O2
In roots, many DEGs were found to be associated with the detoxification of H2O2 (Table 3), especially peroxidases and genes related to glutathione metabolism. GLUTATHIONE TRANSFERASES (GSTs) and GLUTATHIONE PEROXIDASES (GTPs) have both been shown to be involved in plant stress responses (Bela et al., 2015; Nianiou-Obeidat et al., 2017). However, somewhat surprisingly, our data showed clear down-regulation of several GSTs and GTPs along with other key players associated with H2O2 detoxification such as orthologs of Arabidopsis ASCORBATE PEROXIDASE 1 (APX1) and CATALASE 1(CAT1). Moreover, two putative DETOXIFICATION EFFLUX CARRIERS/MULTIDRUG AND TOXIC COMPOUND EXTRUSION (DXT/MATE) proteins were strongly up-regulated in roots. The MATE family proteins facilitate the efflux of various compounds including substances, such as hormones or flavonoids, that improve adaptation to stress (Ku et al., 2022).
The largest set of genes whose expression was affected in response to H2O2 belongs to class III plant type peroxidases (Table 3), whose role in plant defense mechanisms in response to a wide variety of biotic and abiotic stresses is well established. They play an important role in the cellular redox homeostasis upon stress. In addition, they also catalyze the oxidation of a variety of substrates and have been linked to processes involved in cell wall stability, including lignin and suberin polymerization in response to stress (Kidwai et al., 2020). Thus, the up-regulation of these peroxidases in roots upon H2O2 treatment is in line with the up-regulation of genes involved in cell wall metabolism observed in this study. Some components of the cell wall architecture, particularly the xyloglucans, have been shown to play an important role in imparting abiotic stress tolerance by coordinating with hormonal and other signaling cascades. For example, a xyloglucan galactosyl transferase from Arabidopsis, SHORT ROOT IN SALT MEDIUM 3 (RSA3), was shown to play a crucial role under salt stress by assembling actin microfilaments and thus preventing ROS accumulation induced by disruption of actin microfilaments (Cho et al., 2006; Li et al., 2013). Also the role of xyloglucan modifying enzymes along with expansins in loosening and expanding the cell wall network upon abiotic stresses has already been described (Tenhaken, 2015).
4.3 Commonly and counter-regulated DEGs in responses to H2O2
Overall, leaves and roots showed very unique transcriptional responses upon H2O2 treatment. Not only the number of DEGs was much higher in roots compared to leaves, the change in transcription also affected a quite different set of genes (Figures 2, 3). Nevertheless, there are DEGs that were found in both plant parts (Figure 4). These 349 DEGs were further divided into four clusters, depending on their expression pattern. Looking at the two larger clusters, the commonly up- or down-regulated DEGs (Figure 5, Supplementary Table S3 and S4), certain patterns in the functional categories can be observed. Both clusters include TFs from different families. This is not unexpected and highlights their versatility in differentially regulating genes as an important part of all stress responses (Javed et al., 2020). However, of the TFs identified in this study, only few have previously been associated with oxidative stress, such as an Arabidopsis ortholog to HORVU2Hr1G066080 and HORVU3Hr1G016320, the LOB DOMAIN CONTAINING PROTEIN 41 (LBD41), that was previously identified in relation with low-oxygen endurance or high-light-induced increase in H2O2 (Mustroph et al., 2009; Vanderauwera et al., 2011). However, some were found associated with stresses, such as herbivory, that include ROS-mediated signaling or mutations that cause increased levels of ROS (Paudel et al., 2013; Garcia et al., 2016).
Several transporters were found commonly down-regulated (Supplementary Table S4 and Figure 5A). The aquaporin encoded by HORVU4Hr1G085250 is orthologous to the TONOPLAST INTRINSIC PROTEIN 4;1 (TIP4;1) of Arabidopsis and rice. Aquaporins not only transport water but also other molecules including H2O2. TIP4;1 from barley was shown to be up-regulated by ABA in roots and gibberellic acid in shoots (Ligaba et al., 2011). Moreover, its expression was also up-regulated upon drought (Kurowska et al., 2019). Also sugar transporters of the SWEET-type and PHT1.7 phosphate transporters have been demonstrated to play a role in abiotic stress tolerance and showed variable expression patterns under stress conditions (Cao et al., 2020; Gautam et al., 2022).
We also found common down-regulation of orthologs to RECEPTOR-LIKE PROTEIN KINASES(RLKs) from different subfamilies, i.e., WAK, LLR, CRK and RLCK (Supplementary Table S4 and Figure 5A). Experimental evidence suggests that RKLs are a vital part of the growth-defense trade-off, i.e. by facilitating the cross-talk between different phytohormones (Zhu et al., 2023). However, of the specific RLKs found commonly down-regulated in barley leaves and roots, only the pepper ortholog of WAKL20 was described in relation to stress (Zhu et al., 2023). DEGs connected to various facets of primary metabolism were found commonly up-regulated (Supplementary Table S4 and Figure 5B). While several of them are involved in pathways that play a role in stress responses, an obvious connection between these specific DEGs is lacking. Overall, even if no clear connection to oxidative stress exists, many of the commonly regulated DEGs have been described or postulated previously to be involved in stress tolerance mechanisms.
A very small number of DEGs was found counter-regulated upon treatment with H2O2 (Supplementary Table S4 and Figure 6), the majority of which showing up-regulation in leaves and down-regulation in roots. Several of those genes are connected to aspects of metabolism and hormone signaling, and some orthologous genes of other plant species, such as SERAT1, OSM34, and UGT74D1 of tomato, grapevine and Arabidopsis have been previously connected to stress, ABA signaling, or auxin (Tavares et al., 2015; Jin et al., 2021; Park and Kim, 2021; Liu et al., 2022). Remarkably, this cluster also includes a group of nine HSPs, and this different expression in leaves and roots raises questions about their specific role in stress response in the different tissues.
5 Conclusions
Plant adaptation to changing environmental cues requires acclimation, enabling them to fulfil their lifecycle. This adaptation is based to a large extent on substantial changes on transcriptional level. Our data reveal that H2O2 modulates the expression of a wide range of genes within the barley genome. The results provide first insights into the significant role of H2O2 in altering cellular activities in this important crop species. However, in which manner all these genes are coordinated within the cell to provide an appropriate response during stress-induced H2O2 increase is an important question that needs to be addressed in further research. Many of them have previously been associated to stress responses in barley or more often via their orthologs in Arabidopsis or other crops. This reveals a high degree of similarity in the responses of these plants to situations where cellular H2O2 levels increase either as a toxic by-product of stress or as a dedicated signaling molecule. Other genes identified in this screen have so far not been associated with stress. As important redox molecules participating in plant cell signaling, developmental processes stress responses, as well as causing oxidative damage, uncovering the effect of ROS generally and H2O2 specifically on gene expression provides good insights into the molecular mechanisms of oxidative stress responses in barley. Such understanding might increase our ability to improve stress resistance in barley and other crops to optimize crop performance and productivity in present and future environmental climate challenges. Particularly, the highest up- or down-regulated genes in our dataset in both tissues were mostly uncharacterized and information on the exact nature of the genes is missing. These data can be used to guide future studies aimed to functionally characterize novel stress-related genes using state-of-the-art experimental designs including generation of mutants and ectopic expression lines. This will enable us to better understand H2O2 mediated regulation of adaptive processes not only in barley but also in other crops and might thus support targeted breeding of more resilient crops.
Data availability statement
The datasets presented in this study can be found in online repositories (https://www.ncbi.nlm.nih.gov/sra/PRJNA973626).
Author contributions
SB contributed to conceptualization, investigation (responsible for most experimental work), formal analysis (responsible for all bioinformatic analysis), validation, visualization, and writing - original draft as well as review & editing. MG contributed to investigation. BM contributed to validation (qRT-PCR) and writing - review & editing. EP contributed to supervision and writing - review and editing. UV contributed to conceptualization, validation, funding acquisition, project administration, supervision, and writing - review & editing. FC contributed to conceptualization, formal analysis, validation, visualization, supervision, and writing - original draft as well as review & editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, INST 217/939-1 FUGG to UV and GRK 2064 to MG and UV).
Acknowledgments
We would like to thank the NSG Core Facility of the Medical Faculty at the University of Bonn for providing support. We would also like to thank Elena Ulland Rodriguez for technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1223778/full#supplementary-material
References
Ahmad, P., Jaleel, C. A., Salem, M. A., Nabi, G., Sharma, S. (2010). Roles of enzymatic and nonenzymatic antioxidants in plants during abiotic stress. Crit. Rev. Biotechnol. 30, 161–175. doi: 10.3109/07388550903524243
Almagro, L., Gómez Ros, L. V., Belchi-Navarro, S., Bru, R., Ros Barceló, A., Pedreño, M. A. (2009). Class III peroxidases in plant defence reactions. J. Exp. Bot. 60, 377–390. doi: 10.1093/jxb/ern277
Basha, E., O’Neill, H., Vierling, E. (2012). Small heat shock proteins and α-crystallins: dynamic proteins with flexible functions. Trends Biochem. Sci. 37, 106–117. doi: 10.1016/j.tibs.2011.11.005
Bela, K., Horváth, E., Gallé, Á., Szabados, L., Tari, I., Csiszár, J. (2015). Plant glutathione peroxidases: Emerging role of the antioxidant enzymes in plant development and stress responses. J. Plant Physiol. 176, 192–201. doi: 10.1016/j.jplph.2014.12.014
Benjamini, Y., Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc Ser. B Methodol. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bieker, S., Riester, L., Stahl, M., Franzaring, J., Zentgraf, U. (2012). Senescence-specific Alteration of Hydrogen Peroxide Levels in Arabidopsis thaliana and Oilseed Rape Spring Variety Brassica napus L. cv. MozartF. J. Integr. Plant Biol. 54, 540–554. doi: 10.1111/j.1744-7909.2012.01147.x
Bienert, G. P., Møller, A. L. B., Kristiansen, K. A., Schulz, A., Møller, I. M., Schjoerring, J. K., et al. (2007). Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes*. J. Biol. Chem. 282, 1183–1192. doi: 10.1074/jbc.M603761200
Bittner, A., Cieśla, A., Gruden, K., Lukan, T., Mahmud, S., Teige, M., et al. (2022). Organelles and phytohormones: a network of interactions in plant stress responses. J. Exp. Bot. 73, 7165–7181. doi: 10.1093/jxb/erac384
Böttcher, C., Weiler, E. W. (2007). cyclo-Oxylipin-galactolipids in plants: occurrence and dynamics. Planta 226, 629–637. doi: 10.1007/s00425-007-0511-5
Bourdais, G., Burdiak, P., Gauthier, A., Nitsch, L., Salojärvi, J., Rayapuram, C., et al. (2015). Large-scale phenomics identifies primary and fine-tuning roles for CRKs in responses related to oxidative stress. PloS Genet. 11, e1005373. doi: 10.1371/journal.pgen.1005373
Cao, M., Liu, H., Zhang, C., Wang, D., Liu, X., Chen, Q. (2020). Functional analysis of stPHT1;7, a solanum tuberosum L. Phosphate transporter gene, in growth and drought tolerance. Plants 9, 1384. doi: 10.3390/plants9101384
Chaudhary, R., Baranwal, V. K., Kumar, R., Sircar, D., Chauhan, H. (2019). Genome-wide identification and expression analysis of Hsp70, Hsp90, and Hsp100 heat shock protein genes in barley under stress conditions and reproductive development. Funct. Integr. Genomics 19, 1007–1022. doi: 10.1007/s10142-019-00695-y
Chini, A., Monte, I., Zamarreño, A. M., Hamberg, M., Lassueur, S., Reymond, P., et al. (2018). An OPR3-independent pathway uses 4,5-didehydrojasmonate for jasmonate synthesis. Nat. Chem. Biol. 14, 171–178. doi: 10.1038/nchembio.2540
Cho, S. K., Kim, J. E., Park, J.-A., Eom, T. J., Kim, W. T. (2006). Constitutive expression of abiotic stress-inducible hot pepper CaXTH3, which encodes a xyloglucan endotransglucosylase/hydrolase homolog, improves drought and salt tolerance in transgenic Arabidopsis plants. FEBS Lett. 580, 3136–3144. doi: 10.1016/j.febslet.2006.04.062
Chung, H. S., Howe, G. A. (2009). A critical role for the TIFY motif in repression of jasmonate signaling by a stabilized splice variant of the JASMONATE ZIM-domain protein JAZ10 in arabidopsis. Plant Cell 21, 131–145. doi: 10.1105/tpc.108.064097
del Río, L. A., López-Huertas, E. (2016). ROS generation in peroxisomes and its role in cell signaling. Plant Cell Physiol. 57, 1364–1376. doi: 10.1093/pcp/pcw076
Desikan, R., A.-H.-Mackerness, S., Hancock, J. T., Neill, S. J. (2001). Regulation of the arabidopsis transcriptome by oxidative stress. Plant Physiol. 127, 159–172. doi: 10.1104/pp.127.1.159
Dodd, A. N., Kudla, J., Sanders, D. (2010). The language of calcium signaling. Annu. Rev. Plant Biol. 61, 593–620. doi: 10.1146/annurev-arplant-070109-104628
Driedonks, N., Xu, J., Peters, J. L., Park, S., Rieu, I. (2015). Multi-level interactions between heat shock factors, heat shock proteins, and the redox system regulate acclimation to heat. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00999
Dudziak, K., Zapalska, M., Börner, A., Szczerba, H., Kowalczyk, K., Nowak, M. (2019). Analysis of wheat gene expression related to the oxidative stress response and signal transduction under short-term osmotic stress. Sci. Rep. 92743. doi: 10.1038/s41598-019-39154-w
Fariduddin, Q., Khan, T. A., Yusuf, M. (2014). Hydrogen peroxide mediated tolerance to copper stress in the presence of 28-homobrassinolide in Vigna radiata. Acta Physiol. Plant 36, 2767–2778. doi: 10.1007/s11738-014-1647-0
Foyer, C. H., Noctor, G. (2003). Redox sensing and signalling associated with reactive oxygen in chloroplasts, peroxisomes and mitochondria. Physiol. Plant 119, 355–364. doi: 10.1034/j.1399-3054.2003.00223.x
Garcia, L., Welchen, E., Gey, U., Arce, A. L., Steinebrunner, I., Gonzalez, D. H. (2016). The cytochrome c oxidase biogenesis factor AtCOX17 modulates stress responses in Arabidopsis. Plant Cell Environ. 39, 628–644. doi: 10.1111/pce.12647
Gautam, T., Dutta, M., Jaiswal, V., Zinta, G., Gahlaut, V., Kumar, S. (2022). Emerging roles of SWEET sugar transporters in plant development and abiotic stress responses. Cells 11, 1303. doi: 10.3390/cells11081303
Ge, S. X., Jung, D., Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Giridhar, M., Meier, B., Imani, J., Kogel, K.-H., Peiter, E., Vothknecht, U. C., et al. (2022). Comparative analysis of stress-induced calcium signals in the crop species barley and the model plant Arabidopsis thaliana. BMC Plant Biol. 22, 447. doi: 10.1186/s12870-022-03820-5
Gürel, F., Öztürk, Z. N., Uçarlı, C., Rosellini, D. (2016). Barley genes as tools to confer abiotic stress tolerance in crops. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01137
Guzel, S., Terzi, R. (2013). Exogenous hydrogen peroxide increases dry matter production, mineral content and level of osmotic solutes in young maize leaves and alleviates deleterious effects of copper stress. Bot. Stud. 54, 26. doi: 10.1186/1999-3110-54-26
Hellmann, A., Meyer, C. U., Wernicke, W. (1995). Tubulin gene expression during growth and maturation of leaves with different developmental patterns. Cell Motil. 30, 67–72. doi: 10.1002/cm.970300108
Hieno, A., Naznin, H. A., Inaba-Hasegawa, K., Yokogawa, T., Hayami, N., Nomoto, M., et al. (2019). Transcriptome analysis and identification of a transcriptional regulatory network in the response to H2O2. Plant Physiol. 180, 1629–1646. doi: 10.1104/pp.18.01426
Hirai, M. Y., Yano, M., Goodenowe, D. B., Kanaya, S., Kimura, T., Awazuhara, M., et al. (2004). Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. 101, 10205–10210. doi: 10.1073/pnas.0403218101
Hlaváčková, I., Vítámvás, P., Šantrůček, J., Kosová, K., Zelenková, S., Prášil, I. T., et al. (2013). Proteins Involved in Distinct Phases of Cold Hardening Process in Frost Resistant Winter Barley (Hordeum vulgare L.) cv Luxor. Int. J. Mol. Sci. 14, 8000–8024. doi: 10.3390/ijms14048000
Hossain, M. A., Bhattacharjee, S., Armin, S.-M., Qian, P., Xin, W., Li, H.-Y., et al. (2015). Hydrogen peroxide priming modulates abiotic oxidative stress tolerance: insights from ROS detoxification and scavenging. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00420
Huang, H., Ullah, F., Zhou, D.-X., Yi, M., Zhao, Y. (2019). Mechanisms of ROS regulation of plant development and stress responses. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00800
Iglesias, M. J., Terrile, M. C., Bartoli, C. G., D’Ippólito, S., Casalongué, C. A. (2010). Auxin signaling participates in the adaptative response against oxidative stress and salinity by interacting with redox metabolism in Arabidopsis. Plant Mol. Biol. 74, 215–222. doi: 10.1007/s11103-010-9667-7
Janiak, A., Kwasniewski, M., Sowa, M., Gajek, K., Żmuda, K., Kościelniak, J., et al. (2018). No time to waste: transcriptome study reveals that drought tolerance in barley may be attributed to stressed-like expression patterns that exist before the occurrence of stress. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.02212
Javed, T., Shabbir, R., Ali, A., Afzal, I., Zaheer, U., Gao, S.-J. (2020). Transcription factors in plant stress responses: challenges and potential for sugarcane improvement. Plants 9, 491. doi: 10.3390/plants9040491
Jin, S., Hou, B., Zhang, G. (2021). The ectopic expression of Arabidopsis glucosyltransferase UGT74D1 affects leaf positioning through modulating indole-3-acetic acid homeostasis. Sci. Rep. 11, 1154. doi: 10.1038/s41598-021-81016-x
Kärkönen, A., Kuchitsu, K. (2015). Reactive oxygen species in cell wall metabolism and development in plants. Mem. G Paul Bolwell Plant Cell Wall Dyn. 112, 22–32. doi: 10.1016/j.phytochem.2014.09.016
Kaur, N., Dhawan, M., Sharma, I., Pati, P. K. (2016). Interdependency of Reactive Oxygen Species generating and scavenging system in salt sensitive and salt tolerant cultivars of rice. BMC Plant Biol. 16, 131. doi: 10.1186/s12870-016-0824-2
Khan, T. A., Yusuf, M., Fariduddin, Q. (2018). Hydrogen peroxide in regulation of plant metabolism: Signalling and its effect under abiotic stress. Photosynthetica 56, 1237–1248. doi: 10.1007/s11099-018-0830-8
Kidwai, M., Ahmad, I. Z., Chakrabarty, D. (2020). Class III peroxidase: an indispensable enzyme for biotic/abiotic stress tolerance and a potent candidate for crop improvement. Plant Cell Rep. 39, 1381–1393. doi: 10.1007/s00299-020-02588-y
Ku, Y.-S., Cheng, S.-S., Cheung, M.-Y., Lam, H.-M. (2022). The roles of multidrug and toxic compound extrusion (MATE) transporters in regulating agronomic traits. Agronomy 12, 878. doi: 10.3390/agronomy12040878
Kurowska, M. Małgorzata, Fahad, S., Saud, S., Chen, Y., Wu, C., Wang, D. (2020). “TIP aquaporins in plants: role in abiotic stress tolerance,” in Abiotic stress in plants(RijekaIntechOpen). doi: 10.5772/intechopen.94165
Kurowska, M. M., Wiecha, K., Gajek, K., Szarejko, I. (2019). Drought stress and re-watering affect the abundance of TIP aquaporin transcripts in barley. PloS One 14, e0226423. doi: 10.1371/journal.pone.0226423
Landi, S., Capasso, G., Ben Azaiez, F. E., Jallouli, S., Ayadi, S., Trifa, Y., et al. (2019). Different Roles of Heat Shock Proteins (70 kDa) During Abiotic Stresses in Barley (Hordeum vulgare) Genotypes. Plants 8, 248. doi: 10.3390/plants8080248
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, W., Guan, Q., Wang, Z.-Y., Wang, Y., Zhu, J. (2013). A bi-functional xyloglucan galactosyltransferase is an indispensable salt stress tolerance determinant in arabidopsis. Mol. Plant 6, 1344–1354. doi: 10.1093/mp/sst062
Li, X., Li, Y., Ahammed, G. J., Zhang, X.-N., Ying, L., Zhang, L., et al. (2019). RBOH1-dependent apoplastic H2O2 mediates epigallocatechin-3-gallate-induced abiotic stress tolerance in Solanum lycopersicum L. Revisiting Role ROS RNS Plants Change Environ. 161, 357–366. doi: 10.1016/j.envexpbot.2018.11.013
Li, G., Li, J., Hao, R., Guo, Y. (2017). Activation of catalase activity by a peroxisome-localized small heat shock protein Hsp17.6CII. J. Genet. Genomics 44, 395–404. doi: 10.1016/j.jgg.2017.03.009
Li, J., Liu, X. (2019). Genome-wide identification and expression profile analysis of the Hsp20 gene family in Barley (Hordeum vulgare L.). PeerJ 7, e6832. doi: 10.7717/peerj.6832
Liao, Y., Smyth, G. K., Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Ligaba, A., Katsuhara, M., Shibasaka, M., Djira, G. (2011). Abiotic stresses modulate expression of major intrinsic proteins in barley (Hordeum vulgare). C. R. Biol. 334, 127–139. doi: 10.1016/j.crvi.2010.11.005
Liu, D., Li, M., Guo, T., Lu, J., Xie, Y., Hao, Y., et al. (2022). Functional characterization of the Serine acetyltransferase family genes uncovers the diversification and conservation of cysteine biosynthesis in tomato. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.913856
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Lu, P., Magwanga, R. O., Kirungu, J. N., Hu, Y., Dong, Q., Cai, X., et al. (2019). Overexpression of cotton a DTX/MATE gene enhances drought, salt, and cold stress tolerance in transgenic arabidopsis. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00299
Ma, C., Haslbeck, M., Babujee, L., Jahn, O., Reumann, S. (2006). Identification and characterization of a stress-inducible and a constitutive small heat-shock protein targeted to the matrix of plant peroxisomes. Plant Physiol. 141, 47–60. doi: 10.1104/pp.105.073841
Mangano, S., Denita-Juarez, S. P., Choi, H.-S., Marzol, E., Hwang, Y., Ranocha, P., et al. (2017). Molecular link between auxin and ROS-mediated polar growth. Proc. Natl. Acad. Sci. 114, 5289–5294. doi: 10.1073/pnas.1701536114
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17 (1), 10–12. doi: 10.14806/ej.17.1.200
Mascher, M., Gundlach, H., Himmelbach, A., Beier, S., Twardziok, S. O., Wicker, T., et al. (2017). A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433. doi: 10.1038/nature22043
Miller, G., Mittler, R. (2006). Could heat shock transcription factors function as hydrogen peroxide sensors in plants? Ann. Bot. 98, 279–288. doi: 10.1093/aob/mcl107
Mittler, R., Vanderauwera, S., Suzuki, N., Miller, G., Tognetti, V. B., Vandepoele, K., et al. (2011). ROS signaling: the new wave? Trends Plant Sci. 16, 300–309. doi: 10.1016/j.tplants.2011.03.007
Mohanta, T. K., Bashir, T., Hashem, A., Abd_Allah, E. F., Khan, A. L., Al-Harrasi, A. S. (2018). Early events in plant abiotic stress signaling: interplay between calcium, reactive oxygen species and phytohormones. J. Plant Growth Regul. 37, 1033–1049. doi: 10.1007/s00344-018-9833-8
Moll, P., Ante, M., Seitz, A., Reda, T. (2014). QuantSeq 3′ mRNA sequencing for RNA quantification. Nat. Methods 11, i–iii. doi: 10.1038/nmeth.f.376
Munns, R., James, R. A., Läuchli, A. (2006). Approaches to increasing the salt tolerance of wheat and other cereals. J. Exp. Bot. 57, 1025–1043. doi: 10.1093/jxb/erj100
Mustroph, A., Zanetti, M. E., Jang, C. J. H., Holtan, H. E., Repetti, P. P., Galbraith, D. W., et al. (2009). Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc. Natl. Acad. Sci. 106, 18843–18848. doi: 10.1073/pnas.0906131106
Mylona, P. V., Polidoros, A. N., Scandalios, J. G. (2007). Antioxidant gene responses to ROS-generating xenobiotics in developing and germinated scutella of maize. J. Exp. Bot. 58, 1301–1312. doi: 10.1093/jxb/erl292
Nefissi Ouertani, R., Arasappan, D., Abid, G., Ben Chikha, M., Jardak, R., Mahmoudi, H., et al. (2021). Transcriptomic analysis of salt-stress-responsive genes in barley roots and leaves. Int. J. Mol. Sci. 22, 8155. doi: 10.3390/ijms22158155
Nianiou-Obeidat, I., Madesis, P., Kissoudis, C., Voulgari, G., Chronopoulou, E., Tsaftaris, A., et al. (2017). Plant glutathione transferase-mediated stress tolerance: functions and biotechnological applications. Plant Cell Rep. 36, 791–805. doi: 10.1007/s00299-017-2139-7
Ohkama-Ohtsu, N., Sasaki-Sekimoto, Y., Oikawa, A., Jikumaru, Y., Shinoda, S., Inoue, E., et al. (2011). 12-oxo-phytodienoic acid–glutathione conjugate is transported into the vacuole in arabidopsis. Plant Cell Physiol. 52, 205–209. doi: 10.1093/pcp/pcq181
Osthoff, A., Donà dalle Rose, P., Baldauf, J. A., Piepho, H.-P., Hochholdinger, F. (2019). Transcriptomic reprogramming of barley seminal roots by combined water deficit and salt stress. BMC Genomics 20, 325. doi: 10.1186/s12864-019-5634-0
Park, E.-J., Kim, T.-H. (2021). Arabidopsis OSMOTIN 34 functions in the ABA signaling pathway and is regulated by proteolysis. Int. J. Mol. Sci. 22, 7915. doi: 10.3390/ijms22157915
Pasternak, T., Potters, G., Caubergs, R., Jansen, M. A. K. (2005). Complementary interactions between oxidative stress and auxins control plant growth responses at plant, organ, and cellular level. J. Exp. Bot. 56, 1991–2001. doi: 10.1093/jxb/eri196
Paudel, J., Copley, T., Amirizian, A., Prado, A., Bede, J. (2013). Arabidopsis redox status in response to caterpillar herbivory. Front. Plant Sci. 4. doi: 10.3389/fpls.2013.00113
Pauwels, L., Goossens, A. (2011). The JAZ proteins: A crucial interface in the jasmonate signaling cascade. Plant Cell 23, 3089–3100. doi: 10.1105/tpc.111.089300
Pei, Z.-M., Murata, Y., Benning, G., Thomine, S., Klüsener, B., Allen, G. J., et al. (2000). Calcium channels activated by hydrogen peroxide mediate abscisic acid signalling in guard cells. Nature 406, 731–734. doi: 10.1038/35021067
Peiter, E. (2016). The ever-closer union of signals: propagating waves of calcium and ROS are inextricably linked. Plant Physiol. 172, 3–4. doi: 10.1104/pp.16.01037
Planas-Portell, J., Gallart, M., Tiburcio, A. F., Altabella, T. (2013). Copper-containing amine oxidases contribute to terminal polyamine oxidation in peroxisomes and apoplast of Arabidopsis thaliana. BMC Plant Biol. 13, 109. doi: 10.1186/1471-2229-13-109
Polidoros, A. N., Mylona, P. V., Pasentsis, K., Scandalios, J. G., Tsaftaris, A. S. (2005). The maize alternative oxidase 1a (Aox1a) gene is regulated by signals related to oxidative stress. Redox Rep. 10, 71–78. doi: 10.1179/135100005X21688
R Core Team. (2020). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for statistical computing. Available at: https://www.R-project.org.
Ribeiro, C. W., Korbes, A. P., Garighan, J. A., Jardim-Messeder, D., Carvalho, F. E. L., Sousa, R. H. V., et al. (2017). Rice peroxisomal ascorbate peroxidase knockdown affects ROS signaling and triggers early leaf senescence. Plant Sci. 263, 55–65. doi: 10.1016/j.plantsci.2017.07.009
Rollins, J. A., Habte, E., Templer, S. E., Colby, T., Schmidt, J., von Korff, M. (2013). Leaf proteome alterations in the context of physiological and morphological responses to drought and heat stress in barley (Hordeum vulgare L.). J. Exp. Bot. 64, 3201–3212. doi: 10.1093/jxb/ert158
Sandalio, L. M., Rodríguez-Serrano, M., Romero-Puertas, M. C., del Río, L. A. (2013). “Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules,” in Peroxisomes and their key role in cellular signaling and metabolism. Ed. Río, L.A.d. (Dordrecht: Springer Netherlands), 231–255. doi: 10.1007/978-94-007-6889-5_13
Saxena, I., Srikanth, S., Chen, Z. (2016). Cross Talk between H2O2 and Interacting Signal Molecules under Plant Stress Response. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00570
Schaller, A., Stintzi, A. (2009). Enzymes in jasmonate biosynthesis – Structure, function, regulation. Jasmonates Stress Responses Dev. 70, 1532–1538. doi: 10.1016/j.phytochem.2009.07.032
Shigeto, J., Tsutsumi, Y. (2016). Diverse functions and reactions of class III peroxidases. New Phytol. 209, 1395–1402. doi: 10.1111/nph.13738
Shimakawa, G., Roach, T., Krieger-Liszkay, A. (2020). Changes in photosynthetic electron transport during leaf senescence in two barley varieties grown in contrasting growth regimes. Plant Cell Physiol. 61, 1986–1994. doi: 10.1093/pcp/pcaa114
Siddique, M., Gernhard, S., von Koskull-Döring, P., Vierling, E., Scharf, K.-D. (2008). The plant sHSP superfamily: five new members in Arabidopsis thaliana with unexpected properties. Cell Stress Chaperones 13, 183–197. doi: 10.1007/s12192-008-0032-6
Smirnoff, N., Arnaud, D. (2019). Hydrogen peroxide metabolism and functions in plants. New Phytol. 221, 1197–1214. doi: 10.1111/nph.15488
Staneloni, R. J., Rodriguez-Batiller, M. J., Casal, J. J. (2008). Abscisic acid, high-light, and oxidative stress down-regulate a photosynthetic gene via a promoter motif not involved in phytochrome-mediated transcriptional regulation. Mol. Plant 1, 75–83. doi: 10.1093/mp/ssm007
Stanley Kim, H., Yu, Y., Snesrud, E. C., Moy, L. P., Linford, L. D., Haas, B. J., et al. (2005). Transcriptional divergence of the duplicated oxidative stress-responsive genes in the Arabidopsis genome. Plant J. 41, 212–220. doi: 10.1111/j.1365-313X.2004.02295.x
Steffens, B. (2014). The role of ethylene and ROS in salinity, heavy metal, and flooding responses in rice. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00685
Swindell, W. R., Huebner, M., Weber, A. P. (2007). Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics 8, 125. doi: 10.1186/1471-2164-8-125
Tavares, S., Wirtz, M., Beier, M. P., Bogs, J., Hell, R., Amâncio, S. (2015). Characterization of the serine acetyltransferase gene family of Vitis vinifera uncovers differences in regulation of OAS synthesis in woody plants. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00074
Tenhaken, R. (2015). Cell wall remodeling under abiotic stress. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00771
Terzi, R., Kadioglu, A., Kalaycioglu, E., Saglam, A. (2014). Hydrogen peroxide pretreatment induces osmotic stress tolerance by influencing osmolyte and abscisic acid levels in maize leaves. J. Plant Interact. 9, 559–565. doi: 10.1080/17429145.2013.871077
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
ul Haq, S., Khan, A., Ali, M., Khattak, A. M., Gai, W.-X., Zhang, H.-X., et al. (2019). Heat shock proteins: dynamic biomolecules to counter plant biotic and abiotic stresses. Int. J. Mol. Sci. 20, 5321. doi: 10.3390/ijms20215321
Vanderauwera, S., Suzuki, N., Miller, G., van de Cotte, B., Morsa, S., Ravanat, J.-L., et al. (2011). Extranuclear protection of chromosomal DNA from oxidative stress. Proc. Natl. Acad. Sci. 108, 1711–1716. doi: 10.1073/pnas.1018359108
Verma, V., Ravindran, P., Kumar, P. P. (2016). Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 16, 86. doi: 10.1186/s12870-016-0771-y
Wahid, A., Gelani, S., Ashraf, M., Foolad, M. R. (2007). Heat tolerance in plants: An overview. Environ. Exp. Bot. 61, 199–223. doi: 10.1016/j.envexpbot.2007.05.011
Wang, Y., Li, J., Wang, J., Li, Z. (2010). Exogenous H2O2 improves the chilling tolerance of manilagrass and mascarenegrass by activating the antioxidative system. Plant Growth Regul. 61, 195–204. doi: 10.1007/s10725-010-9470-0
Wang, R., Liu, S., Zhou, F., Ding, C. (2014). Exogenous ascorbic acid and glutathione alleviate oxidative stress induced by salt stress in the chloroplasts of oryza sativa L. Z. Naturforsch C J Biosci 69, 226–236. doi: 10.5560/znc.2013-0117
Wang, R.-S., Pandey, S., Li, S., Gookin, T. E., Zhao, Z., Albert, R., et al. (2011). Common and unique elements of the ABA-regulated transcriptome of Arabidopsis guard cells. BMC Genomics 12, 216. doi: 10.1186/1471-2164-12-216
Waters, E. R. (2013). The evolution, function, structure, and expression of the plant sHSPs. J. Exp. Bot. 64, 391–403. doi: 10.1093/jxb/ers355
Xie, Z., Nolan, T. M., Jiang, H., Yin, Y. (2019). AP2/ERF transcription factor regulatory networks in hormone and abiotic stress responses in arabidopsis. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00228
Yu, C.-W., Murphy, T. M., Lin, C.-H. (2003). Hydrogen peroxide-induced chilling tolerance in mung beans mediated through ABA-independent glutathione accumulation. Funct. Plant Biol. 30, 955–963. doi: 10.1071/FP03091
Zeng, J., Dong, Z., Wu, H., Tian, Z., Zhao, Z. (2017). Redox regulation of plant stem cell fate. EMBO J. 36, 2844–2855. doi: 10.15252/embj.201695955
Zhu, J.-K. (2016). Abiotic stress signaling and responses in plants. Cell 167, 313–324. doi: 10.1016/j.cell.2016.08.029
Keywords: barley, H2O2, oxidative stress, RNA-sequencing, reactive oxygen species (ROS), transcriptome profiling, stress response
Citation: Bhattacharyya S, Giridhar M, Meier B, Peiter E, Vothknecht UC and Chigri F (2023) Global transcriptome profiling reveals root- and leaf-specific responses of barley (Hordeum vulgare L.) to H2O2. Front. Plant Sci. 14:1223778. doi: 10.3389/fpls.2023.1223778
Received: 16 May 2023; Accepted: 23 August 2023;
Published: 12 September 2023.
Edited by:
Umesh K. Reddy, West Virginia State University, United StatesReviewed by:
Sareena Sahab, Department of Economic Development Jobs Transport and Resources, AustraliaManohar Chakrabarti, The University of Texas Rio Grande Valley, United States
Copyright © 2023 Bhattacharyya, Giridhar, Meier, Peiter, Vothknecht and Chigri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fatima Chigri, ZmNoaWdyaUB1bmktYm9ubi5kZQ==
†These authors share senior authorship