 Naixing Shi
Naixing Shi Zefen Yang1
Zefen Yang1 Ke Miao
Ke Miao Lilei Tang
Lilei Tang Guosong Wen
Guosong Wen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 23 August 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1202634
This article is part of the Research Topic Challenges and Progressions in Herbal Genomics View all 8 articles
Background: Polygonatum kingianum has been widely used as a traditional Chinese medicine as well as a healthy food. Because of its highly variable morphology, this medicinal plant is often difficult to distinguish from other related verticillate leaf types of the Polygonatum species. The contaminants in P. kingianum products not only decrease the products’ quality but also threaten consumer safety, seriously inhibiting the industrial application of P. kingianum.
Methods: Nine complete chloroplast (cp) genomes of six verticillate leaf types of the Polygonatum species were de novo assembled and systematically analyzed.
Results: The total lengths of newly sequenced cp genomes ranged from 155,437 to 155,977 bp, including 86/87 protein-coding, 38 tRNA, and 8 rRNA genes, which all exhibited well-conserved genomic structures and gene orders. The differences in the IR/SC (inverted repeats/single-copy) boundary regions and simple sequence repeats were detected among the verticillate leaf types of the Polygonatum cp genomes. Comparative cp genomes analyses revealed that a higher similarity was conserved in the IR regions than in the SC regions. In addition, 11 divergent hotspot regions were selected, providing potential molecular markers for the identification of the Polygonatum species with verticillate leaf types. Phylogenetic analysis indicated that, as a super barcode, plastids realized a fast and efficient identification that clearly characterized the relationships within the verticillate leaf types of the Polygonatum species. In brief, our results not only enrich the data on the cp genomes of the genus Polygonatum but also provide references for the P. kingianum germplasm resource protection, herbal cultivation, and drug production.
Conclusion: This study not only accurately identifies P. kingianum species, but also provides valuable information for the development of molecular markers and phylogenetic analyses of the Polygonatum species with verticillate leaf types.
Polygonatum kingianum belongs to the perennial herb of Polygonatum Mill in Asparagaceae. Its wild resource distributes widely in southwest China and southeast Asia (Myanmar, Vietnam) (Rix and Rushforth, 2016; Duan et al., 2017). Because of its great medicinal and edible value, P. kingianum, a top medicinal herb, is listed in the Chinese ancient medical book (Ming Yi Bie Lu, Han Dynasty, 220-450 AD) with the Chinese name “Huangjing”. Modern pharmacological studies have shown that its components possess anti-aging, anti-tumor, immune enhancement, sterilization, and anti-inflammatory effects (Zhao and Li, 2015; Zhao et al., 2018b; Zhang et al., 2019). Based on its important value, hundreds of commercial drugs and health products whose raw materials include Polygonati rhizome have been developed in China (Su et al., 2018).
The genus Polygonatum is roughly divided into verticillate leaf types of the Polygonatum species (P. sect. Sibirica + P. sect. Verticillata) and alternate phyllotaxis Polygonatum species (P. sect. Polygonatum) (Liu, 2018; Peng, 2018). According to the description of the genus Polygonum in the Flora of China (FOC), the verticillate leaf type of the Polygonatum species holds a more variable morphology than the alternate phyllotaxis Polygonatum species, and even some morphological intermediates of the species have been found. P. kingianum is a typical representative of the verticillate leaf type of the Polygonatum species. However, the highly variable morphology of P. kingianum makes it difficult to distinguish it from the other verticillate leaf types of the Polygonatum species. Previous surveys have revealed that P. kingianum is generally contaminated with common adulterants, such as P. zanlanscianense, P. cirrhifolium, P. verticillatum, and other verticillate leaf types of the Polygonatum species (Liu, 2018; Zhang et al., 2023). These adulterants are usually of poor quality and some may even impair the clinical safety and efficacy of P. kingianum (Wang et al., 2019). Although the phylogenetic relationships of the Polygonatum genus have been reported recently, some research results were inconsistent, especially regarding the verticillate leaf type of the Polygonatum species (Zhao et al., 2018a; Wang et al., 2022; Xia et al., 2022).
Chloroplast (cp) is an important subcellular organelle for photosynthesis and energy transformation in plant cells (Verma and Daniell, 2008). In addition, cp holds a relatively independent genome. In most land plants, the cp genome belongs to the maternal inheritance, which has the characteristics of stable structure, conserved coding region sequence, and rich information (Wolfe et al., 1987; Wicke et al., 2011). Compared with standard DNA barcodes and ultra-barcodes, plastids can provide more abundant genetic variation information and higher species discrimination capabilities (Hollingsworth et al., 2016). With high throughput sequencing methods, the cp genome has been widely used in plant system evolution, related species identification, genetic diversity analysis, cp genetic engineering, etc. (Zhao et al., 2018c; Li L. et al., 2021; Fang et al., 2023). Therefore, it is of great significance to accurately identify P. kingianum and clarify the complex genetic background of the verticillate leaf type of the Polygonatum species. The use of the cp genome can improve the safety and effectiveness of clinical drug use and the development and utilization of Polygonatum medicinal plants.
In the present study, we reported on the whole cp genomes of six verticillate leaf types of the Polygonatum species. These sequences together with earlier published Polygonatum cp genome data helped us better understand its genome structure, codon usage preference, repeat sequences, mutation hotspots, and phylogenetic relationships. The data acquired in this study not only increased the genomic resources available for the Polygonatum genus but also provided valuable information for the phylogenetic analysis and identification of the verticillate leaf type of the Polygonatum species, as well as for the safe medical applications of P. kingianum.
P. kingianum with different morphological characteristics and other verticillate leaf-type Polygonatum species were obtained from Yunnan Province, China (Supplementary Table 1). Fresh and healthy leaves from these plants were collected and stored in silica gel for DNA extraction. The voucher specimens were identified by Professor Yunheng Ji and deposited in the herbarium of Kunming Institute of Botany (Chinese Academy of Sciences). In addition, 12 Polygonatum plastomes were downloaded from the NCBI database for subsequent analysis (Supplementary Table 2).
Total DNA was extracted from approximately 20 mg silica-gel dried leaf tissues using the modified CTAB method (Porebski et al., 1997). Sequence libraries were constructed using approximately 5 µg of purified genomic DNA with a TruSeq DNA Sample Prep Kit (Illumina, Inc., San Diego, CA, United States) according to the manufacturer’s recommendation. The NGS QC tool kit tool was used to trim the raw reads to obtain clean data (Patel and Jain, 2012).
We used NOVOPlasty v4.1.0 to perform cp genome assembly with a k-mer of 39 (Nicolas et al., 2017). Gene matK sequence from P. kingianum (GenBank: JX185477) as the seed for iterative extension of contigs to recover the whole plastome of each accession. The positions of start and stop codons were adjusted manually with a BLAST search against the NCBI protein database. All tRNA genes were identified by the tRNAscan-SE 2.0 with default parameters (Schattner et al., 2005). Thereafter, the boundaries of IR (inverted-repeat) and SC (single-copy) regions were defined by Geneious v. 9.0.2 (Kearse et al., 2012). Organellar Genome DRAW (Lohse et al., 2007) was used to draw the cp genome maps (http://ogdraw.mpimp-golm.mpg.de/). Additionally, all annotated cp genome sequences were submitted to GenBank on the NCBI website (https://www.ncbi.nlm.nih.gov/).
To verify the expansion or contraction of cp gene regions within verticillate leaf type of the Polygonatum species, the alignment was visualized using online mVISTA (https://genome.lbl.gov/vista/index.shtml) (Mayor et al., 2000) in Shuffle LAGAN mode, with the annotated cp genome of P. kingianum (Genbank number: MW566455) as a reference.
The protein-coding genes and intergenic spacers of the 20 plastomes (16 Polygonatum species) were extracted in Geneious v9.0.2 (Kearse et al., 2012) and aligned using the MAFFT v7.221 software (Katoh and Standley, 2013). Then, nucleotide diversity values (Pi) were calculated to estimate the divergence level of shared genes and intergenic spacers using DnaSP v5 (Librado and Rozas, 2009). The IRscope online program (https://irscope.shinyapps.io/irapp/) was used to evaluate IR expansion and contraction of plastome genetic architecture (Amiryousefi et al., 2018).
Online program (https://webblast.ipk-gatersleben.de/misa/): MISA software was used to detect simple sequence repeats (SSRs) in the cp genomes (Beier et al., 2017). Parameter setting thresholds of 10, 5, 4, 3, 3, and 3 were set for mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs, respectively.
The amount of codon and relative synonymous codon usage (RSCU) were calculated by CodonW v.1.4.2 with default parameters. Moreover, the heatmap of the RSCU values was shown by using TBtools software (Chen et al., 2020). Following the previous research methods, protein-coding genes (PCGs) of less than 300 nucleotides in length and the repetitive gene sequences were removed to reduce the deviation of the results (Fang et al., 2023).
All 21 available plastome sequences, including 12 published sequences downloaded from NCBI and 9 sequences obtained in this study, were included in the phylogenetic analysis. We constructed the phylogenetic trees by Maximum likelihood (ML) and Bayesian inference (BI) using the entire cp genomes. P. cyrtonema, the relative of P. kingianum, was used as the outgroup to root the phylogenetic trees. The ML tree was built using the CIPRES Science Gateway website (https://www.phylo.org/) under a General Time Reversible + Gamma (GTR + G) model and estimated with 1,000 bootstrap replicates (Stamatakis, 2006). The BI analysis was performed with Mrbayes 3.2.7 under the best substitution models and parameters (Posada and Crandall, 1998; Ronquist and Huelsenbeck, 2003; Posada and Buckley, 2004). The resulting phylogenetic trees were visualized by FigTree v1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/).
The size of newly sequenced plastid genomes within this study varied from 155,437 to 155,977 bp (Supplementary Table 1). All of them exhibited typical quadripartite structures that were similar to the pattern observed in other typical cp genomes of angiosperms, consisting of a pair of inverted repeat regions from 26,278 bp to 26,394 bp, a long single copy region 84,406 bp to 84,742 bp, and a short single copy region 18,415 bp to 18,554 bp (Supplementary Table 1; Figure 1). The GC contents of the nine plastomes were very similar and ranged from 37.6% to 37.7% (Supplementary Table 1). In this study, 133 genes were annotated in these cp genomes, including 86–87 protein-coding genes, 38 genes encoding transfer RNA, and 8 genes encoding ribosomal RNA. All encoded genes were divided into four categories according to their functions, including self-replication genes, genes related to photosynthesis, other genes, and unknown function genes (Table 1). Among these genes, 18 genes were intron-containing genes, 15 genes containing one intron (rpl2, rpl16, rpoC1, rps16, trnA-UGC, trnG-UCC, trnI-GAU, trnK-UUU, trnL-UAA, trnV-UAC, atpF, ndhA, ndhB, petB, and petD), and others containing two introns (rps12, clpP, and ycf3) (Table 1). All infA genes were found as pseudogenes in verticillate leaf types of the Polygonatum species except the P. sibiricum. This is because a G base was inserted into the infA sequence, which caused the infA gene to contain in-frame stop codons. All sample cp genome sequences were deposited in the GenBank database of the NCBI (Supplementary Table 1).
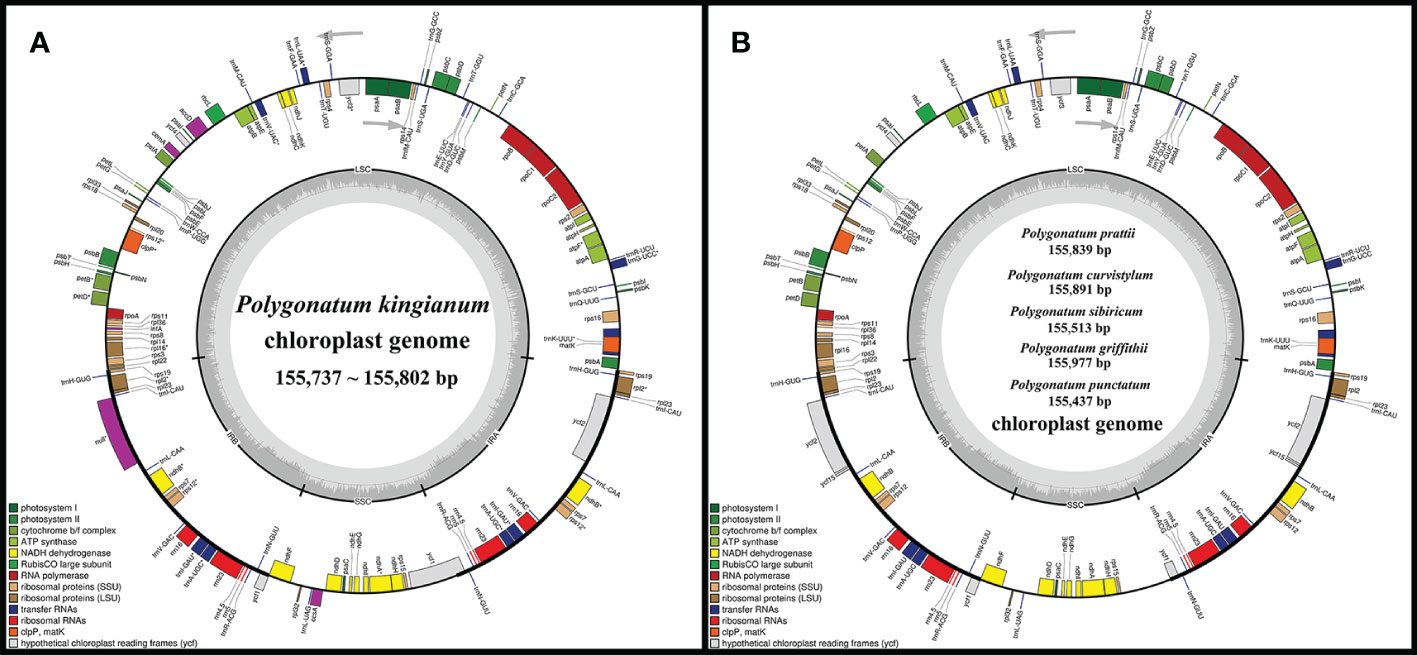
Figure 1 (A) Physical map of the four P. kingianum chloroplast genomes. (B) Chloroplast genome maps of P. prattii, P. curvistylum, P. sibiricum, P. griffithii and P. punctatum. Genes shown outside the circle are transcribed clockwise, while those inside are transcribed counterclockwise. Genes belonging to different functional groups are distinguished by color coding.
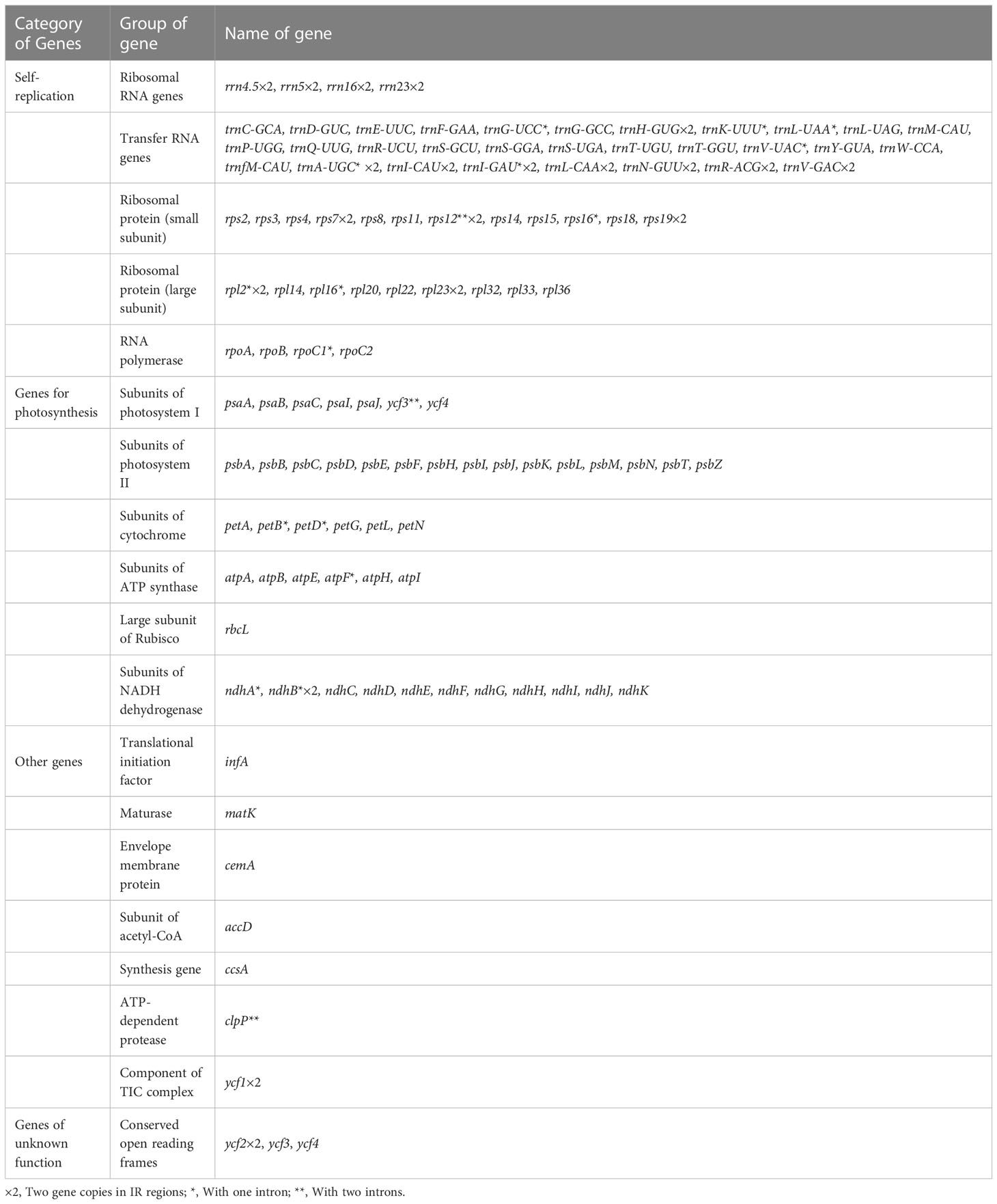
Table 1 List of annotated genes in the newly sequenced chloroplast genomes of the Polygonatum species.
The expansions and contractions of IR borders were considered to be evolutionary events and the main cause of cp genome length changes (Wang et al., 2008). We analyzed the border regions of 16 verticillate leaf types of Polygonatum species plastomes to find similarities and differences (Figure 2). In our study, the rps19 and rpl22 genes were located in the IRb/LSC borders. In the SSC/IRa border, rps19 and psbA genes were found. The ndhF crossed the IRa/SSC region. The ycf1 gene was a cp-genome-encoded giant open reading frame that crossed the IRb/LSC region. Both the pseudogene fragment ψycf1 and the ndhF gene were located at the IRb/SSC border and partially overlapped in the verticillate leaf type of the Polygonatum species. In P. sibiricum, the two rps19 genes extended into the LSC region by 27 and 60 bp, respectively. However, the rps19 genes of other species were entirely located in the IR regions.
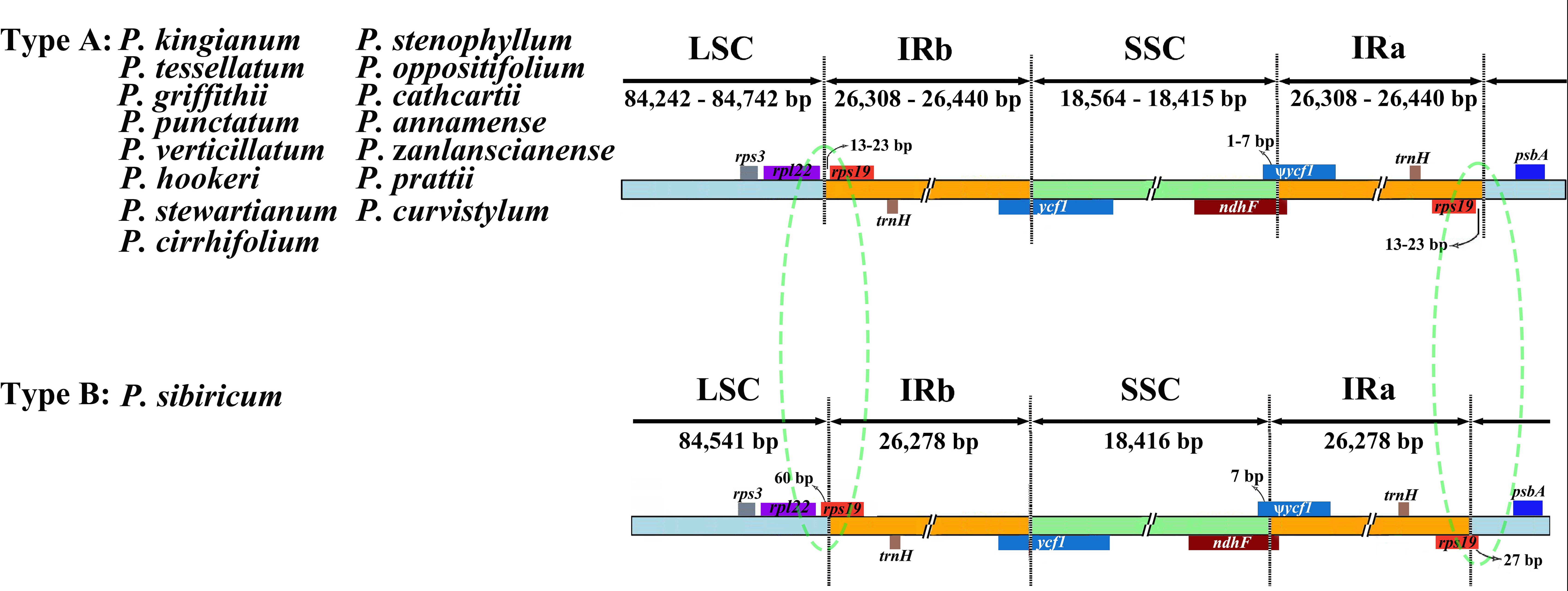
Figure 2 Comparison of LSC, SSC, and IR region boundaries in 16 verticillate leaf types of the Polygonatum species chloroplast genomes. LSC, large single-copy; SSC, small single-copy; IR, inverted repeat. Colored boxes denote genes. The distance between the genes and the boundaries is indicated by the base lengths (bp).
In order to further analyze the results, the 16 verticillate leaf types of the Polygonatum species (20 plastid sequences) were aligned and visualized to determine the overall variations by the program mVISTA. In agreement with other studies (Huang et al., 2021; Wu et al., 2021; Yang et al., 2021), the result of the alignment revealed that the coding regions are more highly conserved than non-coding regions. Most of the variations detected were found in non-coding sequence areas (Figure 3).
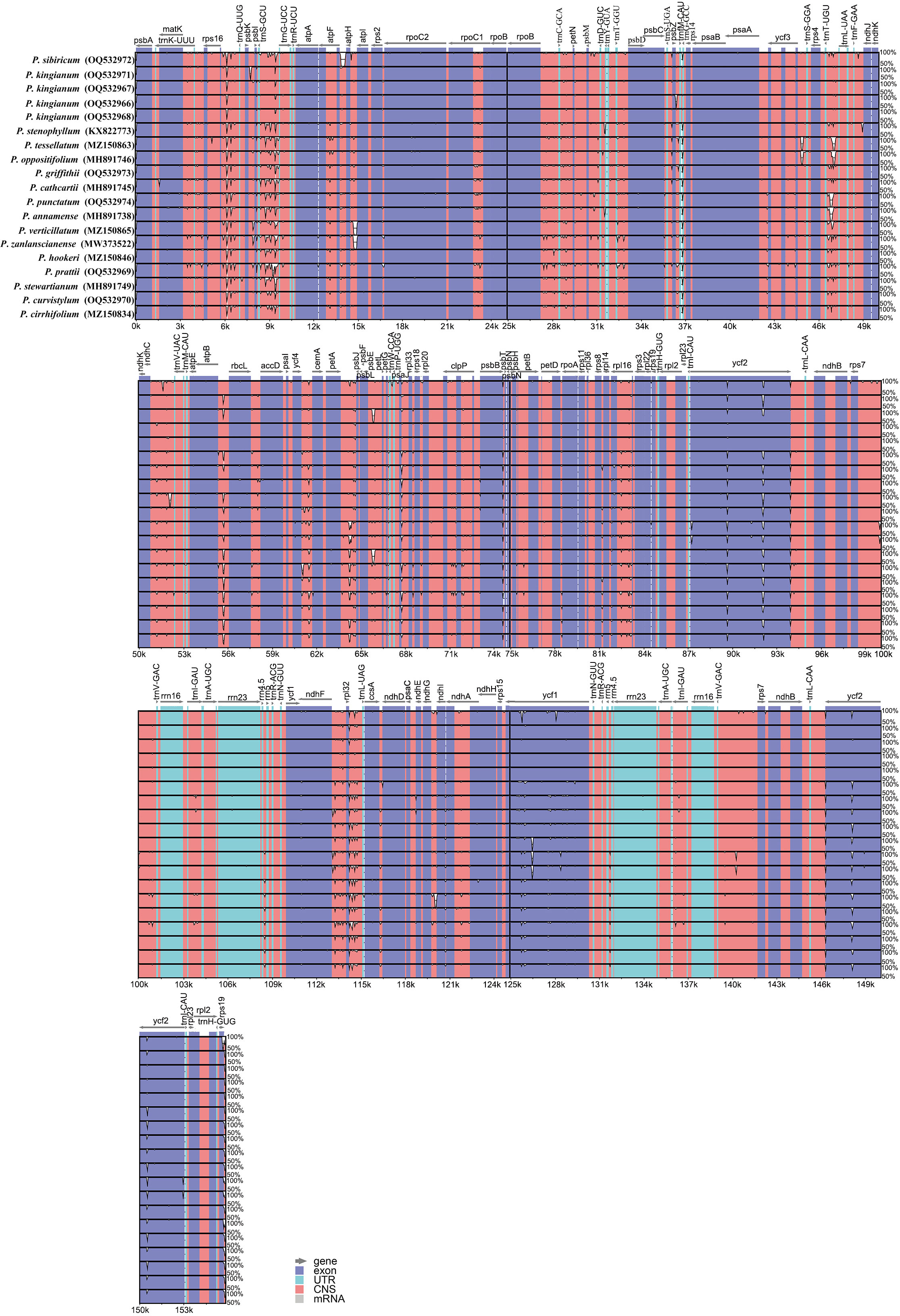
Figure 3 Alignment of the verticillate leaf type of the Polygonatum species chloroplast genome sequences with mVISTA using P. kingianum (Genbank number: MW566455) annotation as a reference. Exon, untranslated region (UTR), conserved noncoding sequences (CNS), and mRNA are color-marked. The direction of the gene transcription is indicated by gray arrows. The vertical scale represents the percentage of identity, ranging from 50 to 100%.
The nucleotide polymorphism (Pi) values of the 20 cp genomes in this study were computed to detect hyper-variable regions using the DnaSP v5 software (Librado and Rozas, 2009). Figure 4 shows that the IR regions were more conserved than the LSC and SSC regions. The average Pi value of shared PCGs ranged from 0 to 0.01113, and the average Pi value of intergenic spacers ranged from 0 to 0.03551 (Supplementary Table 3; Figure 4). These hotspots (PCGs, Pi > 0.0036; Intergenic spacers, Pi > 0.02) were selected including ccsA, psbF, ycf1, psbM, rps19, rpl16, rrn4.5–rrn5, trnG-GCC–trnfM-CAU, ccsA–ndhD, atpA–atpF, and rps19–psbA, which were potentially useful for species identification in P. kingianum with related verticillate leaf type of the Polygonatum species.
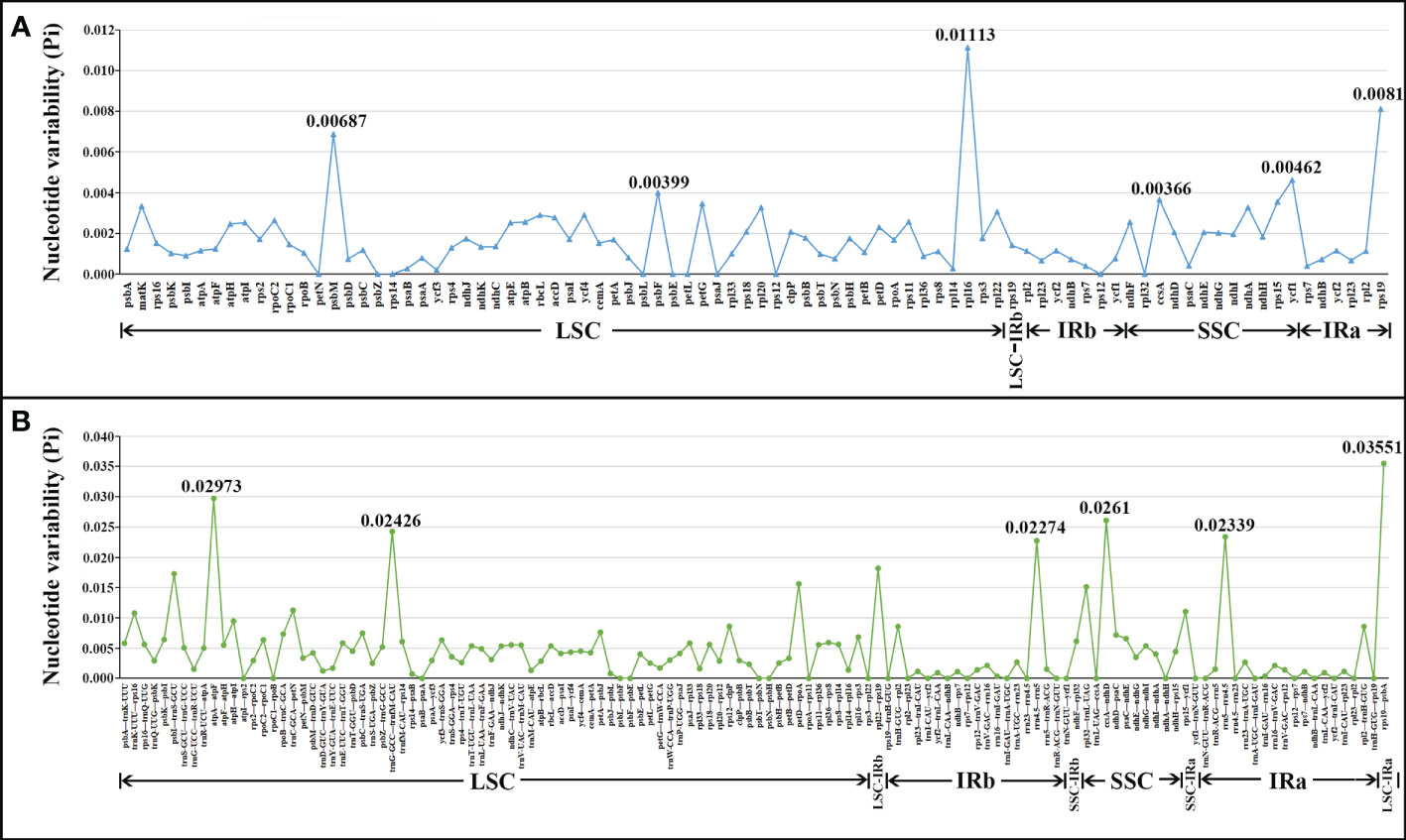
Figure 4 Comparative analysis of the nucleotide diversity (Pi) values among the 20 plastomes (16 Polygonatum species): (A) protein-coding genes; (B) non-coding and intron regions.
SSRs are useful for detecting genetic diversity and polymorphisms at the population, intraspecific, and cultivar levels, as well as for species-distinguishing (Zaloglu et al., 2015; Guo et al., 2018; Guang et al., 2019). The total number of simple sequence repeats were identified in cp genomes by Perlscript MicroSAtellite (MISA) (Beier et al., 2017) and ranged from 64 to 81, with the most abundant type of SSRs being the mono-nucleotide repeats (Figure 5B; Supplementary Table 4). In each case, the 37–53 mononucleotide SSRs, 13–17 dinucleotide SSRs, 2–5 trinucleotide SSRs, 7–10 tetranucleotide SSRs, and 2 pentanucleotides SSRs were identified. There were no hexanucleotide repeats detected in the verticillate leaf type of the Polygonatum species. Most repeats of all six microsatellite types were of the A/T motif rather than the G/C motif (Figure 5A; Supplementary Table 4). SSRs composed of G/C were not found in the plastomes of P. griffithii, P. tessellatum, P. hookeri, P. zanlanscianense, P. cathcartii, P. sibiricum, P. curvistylum, P. kingianum (OQ532971), P. kingianum (MW566455), and P. kingianum (OQ532967), while trinucleotide SSR AGC/CTG was only present in P. curvistylum, P. stewartianum, P. hookeri, and P. cirrhifolium. The tetranucleotide SSR AAAG/CTTT was only detected in the cp genome of the P. stenophyllum.
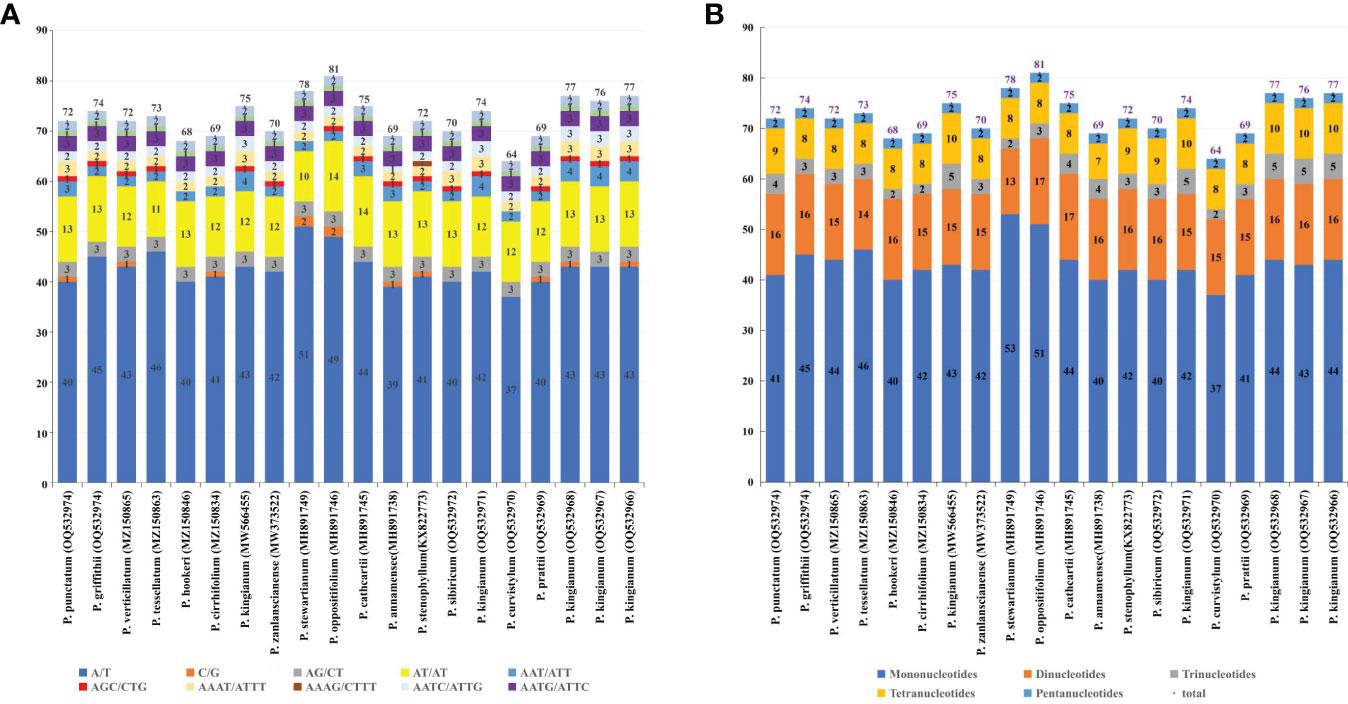
Figure 5 Simple sequence repeats (SSRs) in the 20 cp genomes. (A) Distribution of SSR repeat units in the 20 cp genomes; (B) Distribution of six different types of SSRs in the 20 cp genomes.
A total of 56 protein-coding genes were selected for codon usage analysis. A total of 64 codons were observed, of which 61 codons encoded 20 amino acids and 3 stop codons (Figure 6; Supplementary Table 5). These sequences were 72,513–72,885 bp in length and encoded 24,171–24,295 codons. Of these amino acids, leucine had the highest number of codons, whereas cysteine had the least (Supplementary Table 5). Figure 6 presents the relative synonymous codon usage (RSCU) of each plastome. RSCU values differ slightly in these plastid sequences. Additionally, the results showed that the RSCU values of 31 codons were higher than 1 and that 29 codons presented A/U- endings. Tryptophan and methionine were the amino acids without codon bias (RSCU = 1).
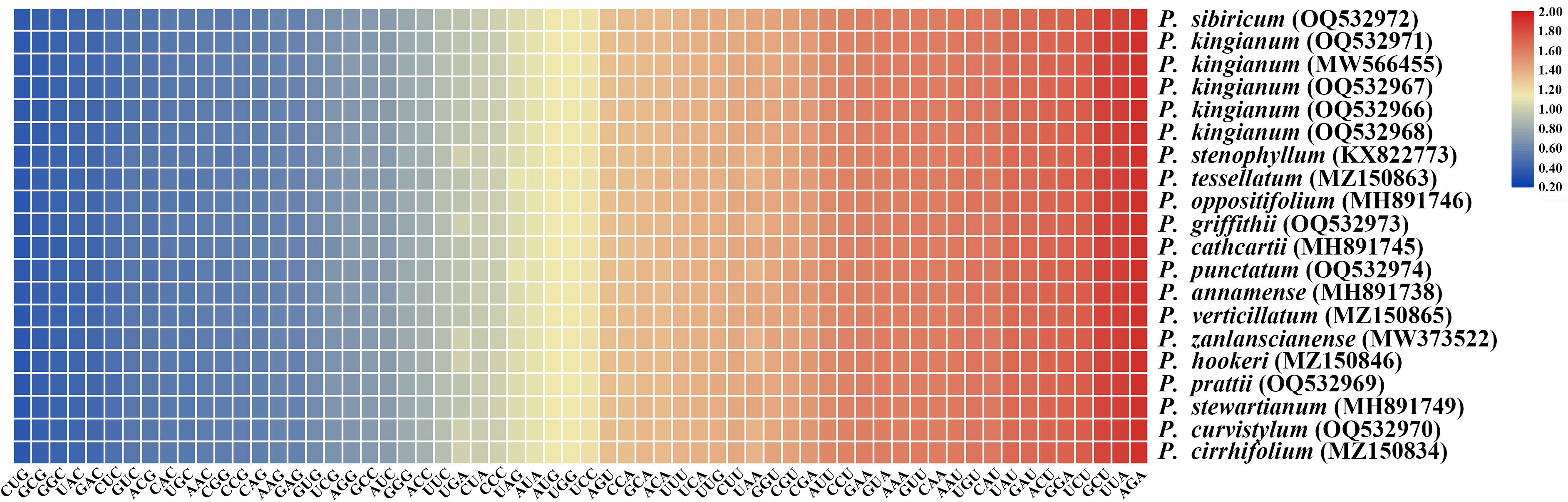
Figure 6 Heat map of the RSCU values of the 20 plastomes in the 16 verticillate leaf types of the Polygonatum species. Color key: Red and blue indicate higher and lower RSCU values, respectively.
We employed the whole cp genome sequences to build the phylogenetic tree to determine the phylogenetic relationship. Maximum likelihood (ML) and Bayesian inference (BI) were used to perform phylogenetic analyses with P. cyrtonema as an outgroup. The topologies of the ML and BI trees were identical (Figure 7). P. kingianum with different morphological characteristics clustered together with high support. P. kingianum was closely related to P. stenophyllum and P. sibiricum (BS = 100, PP = 1.00). P. griffithii and P. cathcartii clustered into a subclade and formed a sister relationship with the subclade of P. oppositifolium and P. tessellatum. P. punctatum and P. annamense clustered into a subclade; P. verticillatum and P. zanlanscianense clustered into a subclade; and P. hookeri, P. prattii, P. stewartianum, P. curvistylum, and P. cirrhifolium clustered into a subclade.
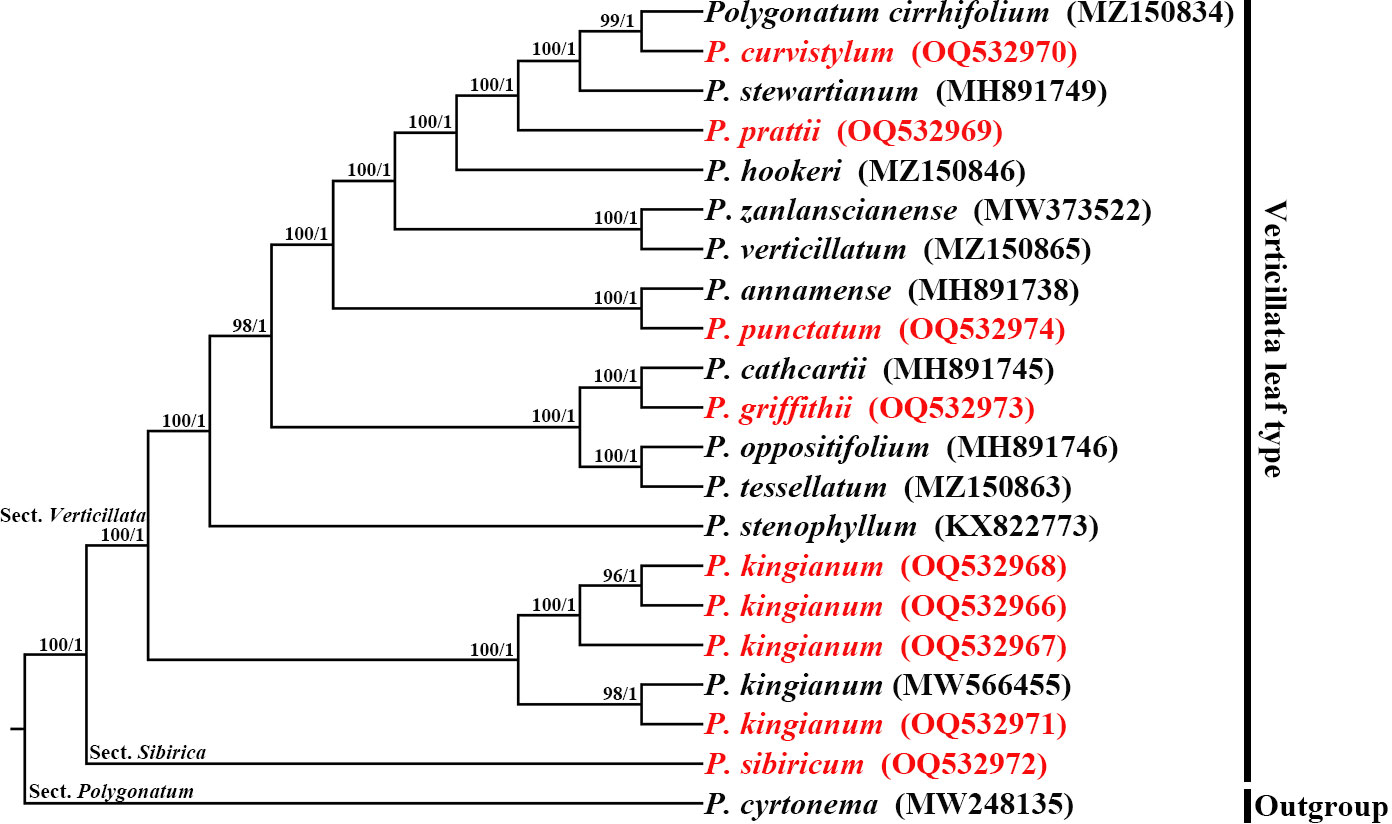
Figure 7 Phylogenetic reconstruction using Maximum likelihood (ML) and Bayesian inference (BI) methods, based on the chloroplast genomes sequences. The numbers above the branches indicate ML bootstrap values (BS)/BI posterior probabilities (PP).
In this study, the cp genomes of nine samples were sequenced by next-generation sequencing (NGS) methods. Similar to previous studies (Huang X. et al., 2022; Zhang et al., 2022; Yang et al., 2023), the newly sequenced cp genomes of the samples were characterized, which displayed the typical quadripartite structure of flowering plants, and cp genomes contained more AT content than GC content. The nine cp genomes were similar both in size and composition. In the current study, we found that most of the infA genes in these cp genomes were pseudogenes except P. sibiricum. Pseudogenization is common in cp genomes ( Abdullah et al., 2021;Li X. et al., 2021; Scobeyeva et al., 2021). In the present study, base insertion was responsible for the pseudogenization of the infA genes. All in all, the cp genomes were highly conservative similar to the majority of plants.
These cp genomes underwent a comparative analysis with sequences from Genebank. We compared the boundary between the SC and two IR regions and their neighboring genes. There was no significant difference at the boundary of most species except in the LSC and IR junctional regions of P. sibiricum. Previous research demonstrated that many rps19 genes of the genus Polygonum were located entirely in the IR region (Wang et al., 2022; Xia et al., 2022). In contrast, the two rps19 genes of P. sibiricum were located partly in the LSC Region, possibly due to IR contraction. Sequence divergence among plastid genomes comparisons based on mVISTA and nucleotide diversity showed that the low-identity region was mainly distributed in the non-coding region of the SC region compared with the IR region through multiple sequence alignment. Mutational hotspots of the cp genome are often developed to distinguish closely related species or genera, which can serve as specific DNA barcodes (Dong et al., 2012). Our study identified eleven regions with a higher evolution rate, namely, ccsA, psbF, ycf1, psbM, rps19, rpl16, rrn4.5–rrn5, trnG-GCC–trnfM-CAU, ccsA–ndhD, atpA–atpF, and rps19–psbA, all of which might be potentially used as candidate markers for identifying P. kingianum and its related verticillate leaf type of the Polygonatum species.
Cp microsatellites (cpSSRs) exist widely in cp genomes and have the advantages of codominant inheritance, high polymorphism, and abundance (Huang et al., 2018). Therefore, cpSSR markers were widely used for species authentication, evolutionary studies, population genetics, and so on (Dzialuk et al., 2009; Jayaswall et al., 2015; Singha et al., 2017; Mahajan et al., 2022).
In our study, we found that cpSSRs abundances in different species were varied. Even within the same species (P. kingianum), the number and type of cpSSRs were different. A or T mononucleotide repetition was the most common primary repetitive type. C or G mononucleotide repetition existed in some species and was rare in number. Previous studies have suggested that this might be due to the convenience of A-T conversion over G-C in the cp genome (Huang Y. et al., 2022). The number of cpSSRs of five kinds of SSRs were discovered: mononucleotide > dinucleotides > tetranucleotide > trinucleotide > pentanucleotide. The cpSSRs detected in this study would be helpful for the identification of the verticillate leaf type of the Polygonatum species, for germplasm resource evaluation, and for the conservation of P. kingianum.
Codon usage biases are found in all eukaryotic and prokaryotic genomes and refer to the unequal use phenomenon of synonymous codons in an organism (Zhou et al., 2016). Codon usage biases are associated with the gene expression level and accuracy of the translation, gene length, gene translation initiation signals, protein amino acid composition, protein structure, tRNA abundance, mutation frequency and patterns, and GC composition (Romero et al., 2000; Xu et al., 2011). These results suggested that all individuals share similar codon usage patterns, with only slight differences in the number of codons and the RSCU value. The AT-rich bias was strongest in the third codon position, and the results of our study are similar to those of other species with cp genome codon usage biases (Huang et al., 2020; Dong et al., 2021; Tang et al., 2021). The research on codon preferences could help us provide a theoretical basis for codon modification of exogenous genes, accuracy of prediction about new members of the cp gene family, and identification of unknown genes.
Phylogenetic analysis is of great significance for clarifying genetic relationships and species identification (Lan et al., 2022). Despite their morphological diversity, P. kingianum could be clustered into one clade by the phylogenetic tree based on cp genomes. We inferred that the limited geographical distribution, short-distance pollination by insects, and long-distance seed dispersal by birds might contribute to the frequent intraspecies gene flow of P. kingianum. These factors could decrease genetic differences among different morphological P. kingianum, improving the diversity and adaptability of the species. The cp genome of P. griffithii was reported for the first time in this study. The plastome phylogeny found that P. griffithii and P. cathcartii were closely related, which confirmed the speculation of FOC (Flora of China). All verticillate leaf types of the Polygonatum species in this research paper, including P. kingianum, have different degrees of overlap in geographical distribution, and many of them also have morphological intermediates types (e.g., P. sibiricum, P. verticillatum, P. cirrhifolium, and P. zanlanscianense). These species were morphologically indistinguishable. Our high-resolution phylogenetic trees not only distinguished these species but also provided some insight into the verticillate leaf type of the Polygonatum species relationship.
In the past few years, phylogenetic data has accumulated rapidly and has been widely used to analyze phylogenetic relationships, species identification, and so on. Because of their great commercial value of”medicine food homology”, P. kingianum is deemed to be one of the most popular species in the Polygonatum genus. However, the molecular and phylogenetic aspects of P. kingianum and related verticillate leaf types of the Polygonatum species have not been well-researched. Hence, in this study, we compared and analyzed 20 cp genomes from 16 verticillate leaf types of the Polygonatum species, including 9 newly sequenced genomes from P. kingianum, P. curvistylum, P. griffithii, P. prattii, P. punctatum, and P. sibiricum. All the newly sequenced cp genomes have similar genome structures and features, which were shown to be conservative. We also identified 11 highly variable cp regions and 64–81 SSR molecular markers, which may serve as potential molecular markers for phylogenetic relationships as well as for the identification of the Polygonatum species. Phylogenetic analyses showed that cp genomes could provide sufficient information for distinguishing the medicinal plant P. kingianum from morphologically similar congeneric relatives, which also revealed accurate phylogenetic relationships. The findings have great potential for the enhancement of exploitation and conservation strategies of the germplasm resources of P. kingianum.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
GW conceived the study; NS, KM, and PX collected and analyzed the data; NS, NZ, and LT wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1202634/full#supplementary-material
Abdullah, Mehmood, F., Heidari, P., Rahim, A., Ahmed, I., Poczai, P. (2021). Pseudogenization of the chloroplast threonine (trnT-GGU) gene in the sunflower family (Asteraceae). Sci. Rep. 11, 21122. doi: 10.1038/s41598-021-00510-4
Amiryousefi, A., Hyvonen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Beier, S., Thiel, T., Munch, T., Scholz, U., Mascher, M. (2017). MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Dong, F., Lin, Z., Lin, J., Ming, R., Zhang, W. (2021). Chloroplast genome of Rambutan and comparative analyses in Sapindaceae. Plants 10 (2), 283. doi: 10.3390/plants10020283
Dong, W. P., Liu, J., Yu, J., Wang, L., Zhou, S. L. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7 (4), e35071. doi: 10.1371/journal.pone.0035071
Duan, B. Z., Zhang, X. C., Du, Z. F., Tao, A. E., Li, Z. (2017). An exploration of the continual utilization of Polygonatum kingianum resources. J. Dali. Univ. 2 (10), 10–14. doi: 10.3969/j.issn.2096-2266.2017.10.003
Dzialuk, A., Muchewicz, E., Boratyński, A., Montserrat, J. M., Boratyńska, K., Burczyk, J. (2009). Genetic variation of Pinus uncinata (Pinaceae) in the Pyrenees determined with cpSSR markers. Plant Syst. Evol. 277, 197–205. doi: 10.1007/s00606-008-0123-y
Fang, H., Dai, G., Liao, B., Zhou, P., Liu, Y. (2023). Application of chloroplast genome in the identification of Phyllanthus urinaria and its common adulterants. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1099856
Guang, X. M., Xia, J. Q., Lin, J. Q., Yu, J., Wan, Q. H., Fang, S. G. (2019). IDSSR: an efficient pipeline for identifying polymorphic microsatellites from a single genome sequence. Int. J. Mol. Sci. 20 (14), 3497. doi: 10.3390/ijms20143497
Guo, Q., Li, X., Yang, S., Yang, Z., Sun, Y., Zhang, J., et al. (2018). Evaluation of the genetic diversity and differentiation of black locust (Robinia pseudoacacia L.) based on genomic and expressed sequence tagSimple sequence repeats. Int. J. Mol. Sci. 19 (9), 2492. doi: 10.3390/ijms19092492
Hollingsworth, P. M., Li, D. Z., van der Bank, M., Twyford, A. D. (2016). Telling plant species apart with DNA: from barcodes to genomes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 371 (1702), 20150338. doi: 10.1098/rstb.2015.0338
Huang, X., Coulibaly, D., Tan, W., Ni, Z., Shi, T., Li, H., et al. (2022). The analysis of genetic structure and characteristics of the chloroplast genome in different Japanese apricot germplasm populations. BMC Plant Biol. 22 (1), 354. doi: 10.1186/s12870-022-03731-5
Huang, S., Ge, X., Cano, A., Salazar, B. G. M., Deng, Y. (2020). Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): molecular structure, phylogenetic relationships, and adaptive evolution. Peer. J. 8, e8450. doi: 10.7717/peerj.8450
Huang, Y., Li, J., Yang, Z., An, W., Xie, C., Liu, S., et al. (2022). Comprehensive analysis of complete chloroplast genome and phylogenetic aspects of ten Ficus species. BMC Plant Biol. 22, 253. doi: 10.1186/s12870-022-03643-4
Huang, L. S., Sun, Y. Q., Jin, Y. Q., Gao, Q., Hu, X. G., Gao, F. L., et al. (2018). Development of high transferability cpSSR markers for individual identification and genetic investigation in Cupressaceae species. Ecol. Evol. 8 (10), 4967–4977. doi: 10.1002/ece3.4053
Huang, R., Xie, X., Li, F., Tian, E., Chao, Z. (2021). Chloroplast genomes of two Mediterranean Bupleurum species and the phylogenetic relationship inferred from combined analysis with East Asian species. Planta 253, 81. doi: 10.1007/s00425-021-03602-7
Jayaswall, K., Sharma, H., Bhandawat, A., Sagar, R., Jayaswal, D., Kumar, A., et al. (2015). The engineered chloroplast genome just got smarter. Trends Plant Sci. 20 (10), 622–640. doi: 10.1016/j.tplants.2015.07.004
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Lan, Z., Shi, Y., Yin, Q., Gao, R., Liu, C., Wang, W., et al. (2022). Comparative and phylogenetic analysis of complete chloroplast genomes from five Artemisia species. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1049209
Li, L., Hu, Y., He, M., Zhang, B., Wu, W., Cai, P. M., et al. (2021). Comparative chloroplast genomes: insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia. BMC Genom. 22, 138. doi: 10.1186/s12864-021-07427-2
Li, X., Yang, J. B., Wang, H., Song, Y., Corlett, R. T., Yao, X., et al. (2021). Plastid NDH pseudogenization and gene loss in a recently derived lineage from the largest hemiparasitic plant genus Pedicularis (Orobanchaceae). Plant Cell Physiol. 62 (6), 971–984. doi: 10.1093/pcp/pcab074
Librado, P., Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 (11), 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, X. (2018). Study on the biological characteristics in the verticillate leaf type of Polygonatum (CHN: Anhui University of Chinese Medicine). Available at: https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C475KOm_zrgu4lQARvep2SAkWfZcByc-RON98J6vxPv10YrwHReTAcPkdEUX7XYBC_f9R5PG-Hn9cYHJ7aeNWq1m&uniplatform=NZKPT.
Lohse, M., Drechsel, O., Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Mahajan, V., Kumar, S., Singh, M. (2022). Chloroplast derived SSRs reveals genetic relationships in domesticated Alliums and wild relatives. Genet. Resour. Crop Evol. 69, 363–372. doi: 10.1007/s10722-021-01235-z
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046. doi: 10.1093/bioinformatics/16.11.1046
Nicolas, D., Patrick, M., Guillaume, S. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45 (4), e18. doi: 10.1093/nar/gkw955
Patel, R. K., Jain, M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PloS One 7, e30619. doi: 10.1371/journal.pone.0030619
Peng, X. X. (2018). Study on the relationship between biological charactreistics and medicinal morphology of the alternate leaves of rihzoma Polygonatum (CHN: Anhui University of Chinese Medicine). Available at: https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C475KOm_zrgu4lQARvep2SAkWfZcByc-RON98J6vxPv10RlAMd-9PCVKmmx_fN8cS8Vr179v5IYF_Gluhb6EzJWd&uniplatform=NZKPT.
Porebski, S., Bailey, L. G., Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15. doi: 10.1007/BF02772108
Posada, D., Buckley, T. R. (2004). Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 53 (5), 793–808. doi: 10.1080/10635150490522304
Posada, D., Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14 (9), 817–818. doi: 10.1093/bioinformatics/14.9.817
Rix, M., Rushforth, K. (2016). 833. Polygonatum kingianum: asparagaceae-convallariaceae. Curtis’s. Bot. Mag. 33 (2), 142–150. doi: 10.1111/curt.12142
Romero, H., Zavala, A., Musto, H. (2000). Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 28 (10), 2084–2090. doi: 10.1093/nar/28.10.2084
Ronquist, F., Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19 (12), 1572–1574. doi: 10.1093/bioinformatics/btg180
Schattner, P., Brooks, A. N., Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Scobeyeva, V. A., Artyushin, I. V., Krinitsina, A. A., Nikitin, P. A., Antipin, M. I., Kuptsov, S. V., et al. (2021). Gene loss, pseudogenization in plastomes of genus Allium (Amaryllidaceae), and putative selection for adaptation to environmental conditions. Front. Genet. 12. doi: 10.3389/fgene.2021.674783
Singha, N., Pala, A. K., Roya, R. K., Tamtab, S., Ranaa, T. S. (2017). Development of cpSSR markers for analysis of genetic diversity in Gladiolus cultivars. Plant Gene. 10, 31–36. doi: 10.1016/j.plgene.2017.05.003
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analysis with thousands of taxa and mixed models. Bioinformatics 22 (21), 2688–2690. doi: 10.1093/bioinformatics/btl446
Su, W. T., Liu, Y. J., Jiang, Y. F., Xie, J. Q., Pan, X. H., Liu, J. J., et al. (2018). Status of Polygonati Rhizome industry and suggestion for its sustainable development. China J. Chin. Mater. Med. 43 (13), 2831–2835. doi: 10.19540/j.cnki.cjcmm.20180329.002
Tang, D., Wei, F., Cai, Z., Wei, Y., Khan, A., Miao, J., et al. (2021). Analysis of codon usage bias and evolution in the chloroplast genome of Mesona chinensis Benth. Dev. Genes Evol. 231, 1–9. doi: 10.1007/s00427-020-00670-9
Verma, D., Daniell, H. (2008). Chloroplast vector systems for biotechnology applications. Plant Physiol. 145 (4), 1129–1143. doi: 10.1104/pp.107.106690
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8, 36. doi: 10.1186/1471-2148-8-36
Wang, Z. W., Du, F. Q., Zhang, L., Wang, S. M., Wei, S. H. (2019). Advances in classification and identification of the Plants of Polygonati Rhizoma and its adulterants. North. Hortic. 24), 130–136. doi: 10.11937/bfyy.20191881
Wang, J., Qian, J., Jiang, Y., Chen, X., Zheng, B., Chen, S., et al. (2022). Comparative analysis of chloroplast genome and new insights into Phylogenetic relationships of Polygonatum and Tribe Polygonateae. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.882189
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants:gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wolfe, K. H., Li, W. H., Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. P. Natl. Acad. Sci. U.S.A. 84 (24), 9054–9058. doi: 10.1073/pnas.84.24.9054
Wu, L. W., Cui, Y. X., Wang, Q., Xu, Z. C., Wang, Y., Lin, Y. L., et al. (2021). Identification and phylogenetic analysis of five Crataegus species (Rosaceae) based on complete chloroplast genomes. Planta 254, 14. doi: 10.1007/s00425-021-03667-4
Xia, M., Liu, Y., Liu, J., Chen, D., Shi, Y., Chen, Z., et al. (2022). Out of the Himalaya-Hengduan Mountains: Phylogenomics, biogeography and diversification of Polygonatum Mill. (Asparagaceae) in the Northern Hemisphere. Mol. Phylogenet. Evol. 169, 107431. doi: 10.1016/j.ympev.2022.107431
Xu, C., Cai, X., Chen, Q. (2011). Factors affecting synonymous codon usage bias in chloroplast genome of Oncidium gower ramsey. Evol. Bioinforma. 7, 271–278. doi: 10.4137/EBO.S8092
Yang, H., Wang, L., Chen, H., Jiang, M., Wu, W., Liu, S., et al. (2021). Phylogenetic analysis and development of molecular markers for five medicinal Alpinia species based on complete plastome sequences. BMC Plant Biol. 21, 431. doi: 10.1186/s12870-021-03204-1
Yang, C., Wang, K., Zhang, H., Guan, Q., Shen, J. (2023). Analysis of the chloroplast genome and phylogenetic evolution of three species of Syringa. Mol. Biol. Rep. 50, 665–677. doi: 10.1007/s11033-022-08004-w
Zaloglu, S., Kafkas, S., Dogan, Y., Guney, M. (2015). Development and characterization of SSR markers from pistachio (Pistacia vera L.) and their transferability to eight pistacia species. Sci. Hortic. 189, 94–103. doi: 10.1016/j.scienta.2015.04.006
Zhang, J., Huang, H., Qu, C., Meng, X., Meng, F., Yao, X., et al. (2022). Comprehensive analysis of chloroplast genome of Albizia julibrissin Durazz. (Leguminosae sp.). Planta 255, 26. doi: 10.1007/s00425-021-03812-z
Zhang, M. Y., Li, Y. M., Cheng, W. P., Gao, J., Yan, Y. G., Yang, L., et al. (2023). Molecular authentication of medicinal Polygonatum species utilizing the universal DNA barcode sequences. Chin. Tradit. Herb. Drugs 54 (01), 235–244. doi: 10.7501/j.issn.0253-2670.2023.01.025
Zhang, J., Wang, Y. Z., Yang, W. Z., Yang, M. Q., Zhang, J. Y. (2019). Research progress in chemical constituents in plants of Polygonatum and their pharmacological effects. China J. Chin. Mater. Med. 44 (10), 1989–2008. doi: 10.19540/j.cnki.cjcmm.20190222.006
Zhao, X., Li, J. (2015). Chemical constituents of the genus Polygonatum and their role in medicinal treatment. Nat. Prod. Commun. 10 (4). doi: 10.1177/1934578X1501000439
Zhao, P., Zhao, C. C., Li, X., Gao, Q. Z., Huang, L. Q., Xiao, P. G., et al. (2018b). The genus Polygonatum: A review of ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol. 214, 274–291. doi: 10.1016/j.jep.2017.12.006
Zhao, L. H., Zhou, S. D., He, X. J. (2018a). A phylogenetic study of Chinese Polygonatum (Polygonateae, Asparagaceae). Nord. J. Bot. 37, e02019. doi: 10.1111/njb.02019
Zhao, P., Zhou, H. J., Potter, D., Hu, Y. H., Feng, X. J., Dang, M., et al. (2018c). Population genetics, phylogenomics and hybrid speciation of Juglans in China determined from whole chloroplast genomes, transcriptomes, and genotyping-by-sequencing (GBS). Mol. Phylogenet. Evol. 126, 250–265. doi: 10.1016/j.ympev.2018.04.014
Keywords: Polygonatum, verticillate leaf, Polygonatum kingianum, chloroplast genome, comparative analysis, phylogenetic analysis
Citation: Shi N, Yang Z, Miao K, Tang L, Zhou N, Xie P and Wen G (2023) Comparative analysis of the medicinal plant Polygonatum kingianum (Asparagaceae) with related verticillate leaf types of the Polygonatum species based on chloroplast genomes. Front. Plant Sci. 14:1202634. doi: 10.3389/fpls.2023.1202634
Received: 09 April 2023; Accepted: 06 July 2023;
Published: 23 August 2023.
Edited by:
Yinghao Cao, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Mei Yang, Chinese Academy of Sciences (CAS), ChinaCopyright © 2023 Shi, Yang, Miao, Tang, Zhou, Xie and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guosong Wen, d2VuZ3NAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.