
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 22 June 2023
Sec. Plant Metabolism and Chemodiversity
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1200253
 Charlotte De Bruyn1,2,3*‡
Charlotte De Bruyn1,2,3*‡ Tom Ruttink1,2‡
Tom Ruttink1,2‡ Elia Lacchini2,3‡
Elia Lacchini2,3‡ Stephane Rombauts2,3‡
Stephane Rombauts2,3‡ Annelies Haegeman1‡
Annelies Haegeman1‡ Ellen De Keyser1‡
Ellen De Keyser1‡ Christof Van Poucke4‡Sandrien Desmet5‡
Christof Van Poucke4‡Sandrien Desmet5‡ Thomas B. Jacobs2,3‡
Thomas B. Jacobs2,3‡ Tom Eeckhaut1‡
Tom Eeckhaut1‡ Alain Goossens2,3*†‡
Alain Goossens2,3*†‡ Katrijn Van Laere1*†‡
Katrijn Van Laere1*†‡Industrial chicory (Cichorium intybus var. sativum) and witloof (C. intybus var. foliosum) are crops with an important economic value, mainly cultivated for inulin production and as a leafy vegetable, respectively. Both crops are rich in nutritionally relevant specialized metabolites with beneficial effects for human health. However, their bitter taste, caused by the sesquiterpene lactones (SLs) produced in leaves and taproot, limits wider applications in the food industry. Changing the bitterness would thus create new opportunities with a great economic impact. Known genes encoding enzymes involved in the SL biosynthetic pathway are GERMACRENE A SYNTHASE (GAS), GERMACRENE A OXIDASE (GAO), COSTUNOLIDE SYNTHASE (COS) and KAUNIOLIDE SYNTHASE (KLS). In this study, we integrated genome and transcriptome mining to further unravel SL biosynthesis. We found that C. intybus SL biosynthesis is controlled by the phytohormone methyl jasmonate (MeJA). Gene family annotation and MeJA inducibility enabled the pinpointing of candidate genes related with the SL biosynthetic pathway. We specifically focused on members of subclade CYP71 of the cytochrome P450 family. We verified the biochemical activity of 14 C. intybus CYP71 enzymes transiently produced in Nicotiana benthamiana and identified several functional paralogs for each of the GAO, COS and KLS genes, pointing to redundancy in and robustness of the SL biosynthetic pathway. Gene functionality was further analyzed using CRISPR/Cas9 genome editing in C. intybus. Metabolite profiling of mutant C. intybus lines demonstrated a successful reduction in SL metabolite production. Together, this study increases our insights into the C. intybus SL biosynthetic pathway and paves the way for the engineering of C. intybus bitterness.
The Asteraceae plant family comprises many economically relevant crops, including chicory, of which three cultivated groups are distinguished: root chicory, leaf chicory and witloof (Kiers et al., 2000; Barcaccia et al., 2016; Raulier et al., 2016). Root chicory, known as industrial chicory (Cichorium intybus var. sativum), is mainly grown for the extraction of inulin-type fructan carbohydrates, an important health-promoting dietary fiber (Chi et al., 2011), from the taproot. The industrial chicory root has further potential as a raw material to make a gluten-free, nutritionally beneficial chicory flour for use in the food industry. Within the leaf chicory group, Sugarloaf (C. intybus var. porphyreum), Radicchio (C. intybus var. latifolium), and Catalogne (C. intybus var. sylvestre) can be distinguished as subgroups, all consisting of leafy vegetables. Witloof (C. intybus var. foliosum), also referred to as witlof, Belgian endive or chicon, composes a vegetable of tightly packed, etiolated leaves. Cichorium intybus crops are all characterized by a bitter taste (Drewnowski and Gomez-Carneros, 2000; Meyer and Blaauwhoed, 2009), attributed to the production of sesquiterpene lactones (SLs) as specialized metabolites. SLs also have a role in plant defense (Padilla-Gonzalez et al., 2016) and are responsible for many of the health benefits of chicory, including antifungal, anti-inflammatory, antitumor and anticytotoxic activity, and are therefore of economic importance (Barrero et al., 2000; Bischoff et al., 2004; Chadwick et al., 2013; Häkkinen et al., 2021; Meng et al., 2022). Better knowledge of the metabolic pathway and the functionality of the genes involved would enable modification of SL content and open new markets for Cichorium use and consumption.
The majority of SLs are thought to be derived from the germacranolides (De Kraker et al., 2001) with the guaianolides being the most important with regard to bitterness (De Kraker et al., 1998). The principal guaianolide SLs reported in Cichorium species are lactucin, 8-deoxylactucin, lactucopicrin and their derivatives, such as 11,13-dihydro-analogs (Van Beek et al., 1990). While multiple SL extraction, isolation, identification and quantification methods were developed (Ivanescu et al., 2015), a total of 16 guaianolide SL metabolites could be detected in leaf extracts of C. intybus using ultra-high performance liquid chromatography (UHPLC) – high-resolution mass spectrometry (HRMS) (Kips, 2017), and more recently one 12,8-guaianolide, four 12,6-guaianolides, and 16 analogs were isolated from C. intybus roots (Meng et al., 2022). Some guaianolide SL metabolites are present both in the free forms and as their oxalates or glycosides (bound to carbohydrates) (Ferioli et al., 2015; Kips, 2017) (Figure 1).
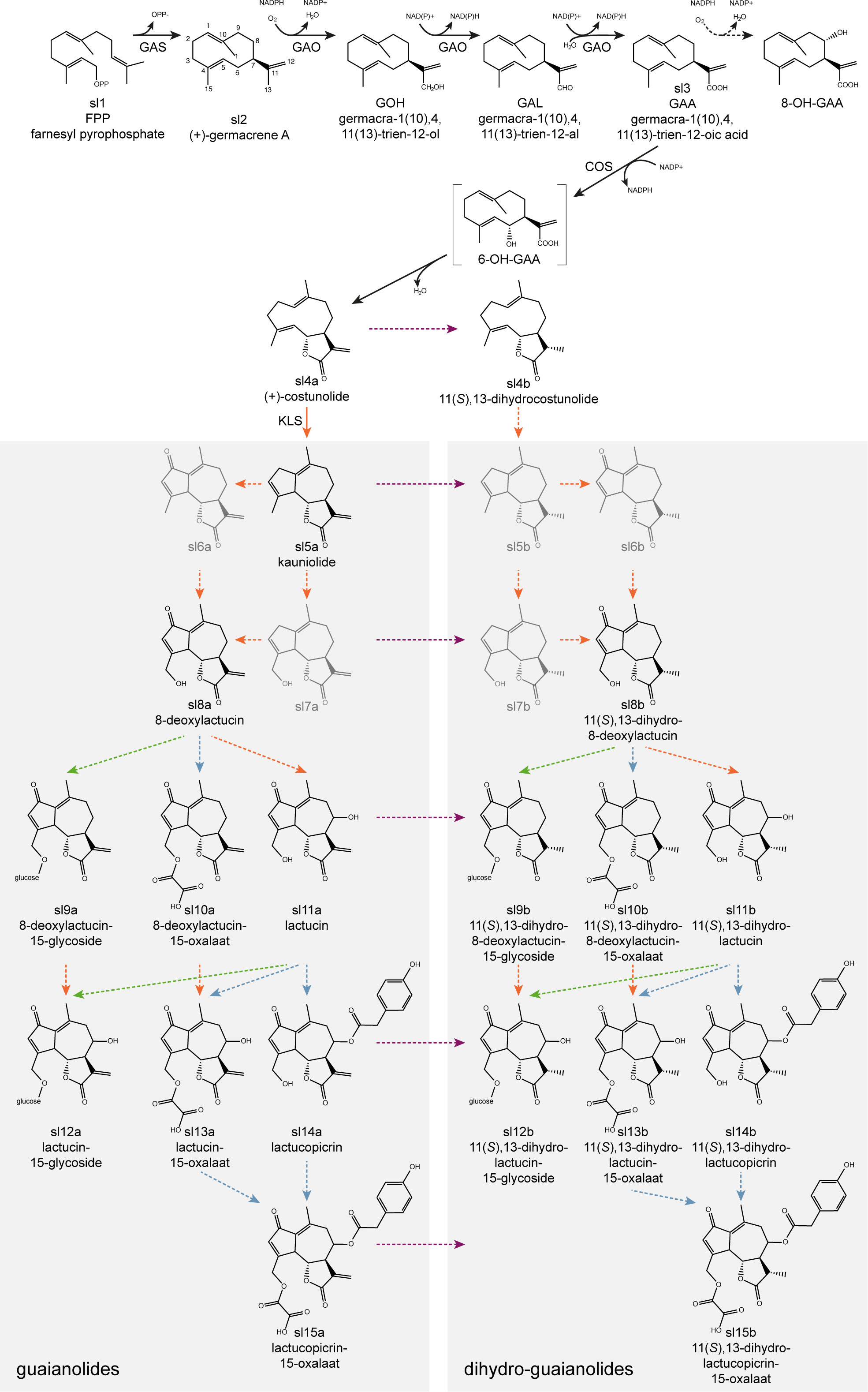
Figure 1 Proposed putative biosynthetic pathway for the formation of guaianolide SL metabolites. It starts with the cyclization of farnesyl pyrophosphate (FPP) to (+)-germacrene A by GERMACRENE A SYNTHASE (GAS), followed by the conversion into germacrene acid by GERMACRENE A OXIDASE (GAO), into (+)-costunolide by COSTUNOLIDE SYNTHASE (COS), and into kauniolide by KAUNIOLIDE SYNTHASE (KLS). Conversion of costunolide into 11(S),13-dihydrocostunolide occurs by an unknown enzyme. Both kauniolide and 11(S),13-dihydrocostunolide could be further converted into undetected intermediates (5b, 6a, 6b, 7a, 7b) necessary for subsequent conversions into guaianolide- (left) and dihydro-guaianolide- (right) SL metabolites, catalyzed by yet unknown enzymes. Arrows indicate putative conversions with enzymes belonging to oxidase (orange), reductase (purple), acyltransferase (blue) and glycosylase (green) gene families. Dashed arrows indicate putative pathway branches for which no enzymes have been identified yet. The point(s) of conversion from SL metabolites to dihydro-SL metabolites (on the right side of the purple dotted line) is (are) unknown.
The biosynthetic pathway that forms bitter guaianolide SL metabolites has been studied in a number of plants of the Asteraceae family: C. intybus (Bouwmeester et al., 2002; Nguyen et al., 2010; Cankar et al., 2011; Liu et al., 2011; Bogdanović et al., 2019; Bogdanović et al., 2020; Cankar et al., 2021; Cankar et al., 2022), C. endiva (Zhang et al., 2022), Helianthus annuus and Lactuca sativa (Nguyen et al., 2010; Ikezawa et al., 2011), Tanacetum parthenium (Liu et al., 2014; Liu et al., 2018), Artemisia annua (Majdi et al., 2016), Saussurea lappa and Barnadesia spinosa (Nguyen et al., 2010), among others. The biosynthesis starts with the cyclization of farnesyl pyrophosphate (FPP) by the terpene synthase (TPS) GERMACRENE A SYNTHASE (GAS) to form germacrene A (De Kraker et al., 1998; Bouwmeester et al., 2002) (Figure 1). Four paralogous GERMACRENE A SYNTHASE genes (CiGAS) have previously been identified in C. intybus (Bogdanović et al., 2019), of which two genes CiGASl and CiGASs were functionally characterized by Bouwmeester et al. (2002). In addition, CRISPR/Cas knock-out mutations in the four CiGAS paralogous genes reduce the SL metabolite levels (Cankar et al., 2021). Three-step hydroxylation/oxidation of the methyl group at C-12 of germacrene A by the cytochrome P450 (CYP) GERMACRENE A OXIDASE (GAO) then leads to the formation of germacrene A acid (Nguyen et al., 2010) (Figure 1). Subsequent hydroxylation at the C6- (or C8-) position of germacrene A acid by the CYP COSTUNOLIDE SYNTHASE (COS) produces an intermediate that undergoes spontaneous non-enzymatic lactonization to yield costunolide, the first SL metabolite (De Kraker et al., 2002; Ikezawa et al., 2011; Liu et al., 2011) (Figure 1). Two CiGAO genes and one CiCOS gene, isolated from C. intybus, have previously been functionally characterized (Nguyen et al., 2010; Cankar et al., 2011; Liu et al., 2011). The further conversion from the germacranolide SL substrate costunolide to guaianolide SLs was first established by the isolation and characterization of a CYP from feverfew (T. parthenium), KAUNIOLIDE SYNTHASE (KLS), which catalyzes the formation of the guaianolide kauniolide (Liu et al., 2018) (Figure 1). Recently, three CiKLS genes were also identified in chicory, and their catalytic activity was confirmed using a yeast microsome assay (Cankar et al., 2022). Inactivation of these CiKLS genes by CRISPR/Cas genome editing resulted in the interruption of SL biosynthesis in chicory leaves and taproots. Genes encoding enzymes involved further downstream of kauniolide have not been identified yet. Furthermore, alternative pathways leading to the formation of germacranolide and guaianolide SLs may exist, as it is currently unclear at which point in the pathway hydroxylation occurs to form the lactone, how dihydro-derivates are formed and at which point(s) glycosylation occurs (De Kraker et al., 1998; De Kraker et al., 2002).
Here, we describe a discovery workflow for genes involved in the SL biosynthetic pathway in C. intybus. First, a comprehensive genome-wide screen and gene family annotation were carried out and combined with phylogenetic and transcriptome analyses. We used an annotated reference genome of C. intybus var. sativum (Waegneer et al., 2023) to mine for all the corresponding genes of the two most important SL biosynthesis gene families, namely TPSs and CYPs. Using protein alignment and phylogenetic analysis, gene clades were delineated and clades were identified containing known members (GAS, GAO, COS, KLS) with a previously demonstrated biochemical activity in C. intybus or closely related plant species. Further, we performed a comprehensive transcriptome analysis of methyl jasmonate (MeJA)-treated samples of three species, C. intybus var. sativum, C. intybus var. foliosum and L. sativa, to confirm that SL biosynthesis is induced by MeJA and to further select TPS and CYP gene family members putatively involved in the SL biosynthetic pathway. Subsequent functional analysis by heterologous expression in Nicotiana benthamiana leaves established the enzymatic activity of several of the cloned CYP genes and determined their possible role in the SL biosynthetic pathway. CRISPR/Cas9 genome editing (De Bruyn et al., 2020) was used to create GAO, COS, and KLS mutants in C. intybus and provide insights into the in planta role of the CYP genes in the formation of the guaianolide SL metabolites.
An in-house generated C. intybus var. sativum reference genome sequence (https://bioinformatics.psb.ugent.be/orcae/overview/Cicin) (Waegneer et al., 2023) was used for genome-wide identification of gene family members of the selected TPS and CYP gene families. First, the corresponding homology groups (HOMgroups) with all known orthologs of multiple species were extracted from the PLAZA dicots 4.0 database (https://bioinformatics.psb.ugent.be/plaza) (Van Bel et al., 2018). The coding DNA sequences (CDS) and protein sequences were used as queries to identify the C. intybus homologs using BLASTn and BLASTp searches (e-value <1e-80), respectively, against the CDS and protein sequences of all 53,507 predicted C. intybus genes. ORCAE (Sterck et al., 2012) was used for manual curation of the structural gene annotation. Each predicted gene was manually curated, using available supporting data in ORCAE (such as RNA-sequencing (RNA-seq) read alignments, splicing sites, gapped alignments of de novo assembled transcripts, and blast hits of orthologs from closely related species). Predicted C. intybus proteins were further validated via multiple protein sequence alignment (MUSCLE) and predicted TPS and CYP C. intybus genes leading to proteins smaller than 350 amino acids or without a C-terminus were classified as pseudogenes. A tBLASTx was performed between the CDS of C. intybus and L. sativa (V8, 2016-01-20, id28333, https://lgr.genomecenter.ucdavis.edu/) (Reyes-Chin-Wo et al., 2017) in both directions (e-value <1e-80) to identify the corresponding L. sativa orthologs (as L. sativa is the closest relative to Cichorium containing also a fully sequenced and annotated genome and produces guaianolide SL metabolites). L. sativa genes were not manually curated but genes leading to proteins smaller than 350 amino acids were classified as pseudogenes. Proteins encoded by C. intybus and L. sativa genes (excluding the pseudogenes), were used for phylogenetic analysis (UPGMA tree) in Geneious 10.2.6 (http://www.geneious.com). The C. intybus CYP protein sequences were further classified by David R. Nelson (University of Tennessee, USA).
C. intybus var. foliosum ‘Topmodel’ and ‘Van Tongelen’, C. intybus var. sativum ‘OBOE’ and ‘VL70’ and L. sativa ‘ZORBA’ seeds were sterilized in 70% EtOH (1 min) and 1.5% NaOCl + 0.02% Tween20 (15 min). Eight sterilized seeds were transferred to sterile plastic containers (145 mm x 100 mm x 60 mm; Eco2nv, Zottegem, Belgium) filled with growth medium (4.4 g.L-1 Murashige and Skoog (MS) + vitamins (Murashige and Skoog, 1962), 20 g.L-1 sucrose, 7 g.L-1 agar, pH 5.8). Seeds were topped with an autoclaved 145 mm x 100 mm 100 µM mesh (Prosep bvba, Zaventem, Belgium) and containers were kept in a growth chamber for 2 weeks at 23 ± 2°C under a 16-h/8-h (light/dark) photoperiod at 40 µmol.m-2.s-1 photosynthetic active radiation (PAR). After two weeks of growth, the number of seedlings was reduced to four well-grown seedlings per container and the mesh containing the seedlings was transferred to mock growth medium (4.4 g.L-1 MS + vitamins, 20 g.L-1 sucrose, 7 g.L-1 agar, 0.02% Tween20, pH 5.8) or growth medium supplemented with 100 µM MeJA. After 2 h, 6 h or 24 h of treatment, the four seedlings in each container were pooled and frozen in liquid nitrogen. A total of 120 samples (five plant varieties, two treatments, three time points, four biological replicates each) were harvested.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was performed to validate MeJA induction of CiGASl (AF497999.1), CiGASs (AF498000.1), CiGAO (GU256644.1) and CiCOS (JF816041.1) candidate genes and to select the most suitable samples for RNA-seq. RNA was extracted from 150 to 200 mg ground plant material, using the 3% CTAB RNA extraction protocol (Luypaert et al., 2017). RNA extraction, quantification and RT-qPCR were performed as previously described (De Keyser et al. (2020). Eleven reference genes were selected for industrial chicory and witloof (Delporte et al., 2015) and ten for lettuce (Borowski et al., 2014; Sgamma et al., 2016). The geNorm (Vandesompele et al., 2002) module in qbase+ (Hellemans et al., 2007) (CellCarta) was used for reference gene selection. RT-qPCR with the reference genes was performed in a LightCycler480 (Roche). RT-qPCR primers targeting CiGASl, CiGASs, CiGAO and CiCOS were developed (Supplementary Table 1) to amplify at least one paralogous gene copy. RT-qPCR analysis was done on the entire sample set, all no-RT samples, including no template controls (NTC) using both target and selected reference genes [TIP41 and PP2AA2 for industrial chicory and witloof (M-value = 0.124; CV-value = 0.043) and TIP41 and PP2AA3 for lettuce (M-value = 0.423; CV-value = 0.146)].
A total of 18 RNA samples were selected (three species, i.e. C. intybus var. intybus, C. intybus var. foliosum, L. sativa, three biological replicates each per MeJA treatment versus mock) for RNA-seq analysis. Libraries were prepared and sequenced by the VIB Nucleomics Core facility (Leuven, Belgium) with SE-75 reads on an Illumina NextSeq500 instrument (NCBI SRA Bioproject PRJNA738883). Adapters and low quality 3’ ends were trimmed from the RNA-seq reads (with a quality threshold of 20) using Cutadapt (Martin, 2011). Next, reads were dereplicated using prinseq-lite (Schmieder and Edwards, 2011). Industrial chicory and witloof reads were mapped on the CDS of an assembled and annotated reference genome of C. intybus var. sativum (Waegneer et al., 2023), while lettuce reads were mapped on the CDS of the annotated reference genome of L. sativa (Reyes-Chin-Wo et al., 2017), using BWA-MEM (Li and Durbin, 2009) with default settings. Mapped reads were sorted using SAMtools (Li et al., 2009) and reads with a mapping quality below 20 were discarded. Subsequent data analysis was done in R v.3.4.3 (R Core Team, 2017) using the package edgeR (Robinson et al., 2010). Genes that occurred in less than two samples with less than four counts per million reads mapped (CPM) were discarded. Normalization to scale the raw library sizes was performed using the trimmed mean of M-values (to the reference) as proposed by Robinson and Oshlack (2010). To identify differentially expressed genes (DEGs), a negative binomial generalized log-linear model was fitted to the read counts for each gene and subsequently likelihood ratio tests were performed between the MeJA treatment and the mock per cultivar. Thresholds for further filtering of the DEGs were set at minimum 2-fold expression change (log2-fold change of 1) and an adjusted p-value (Benjamini-Hochberg multiple testing correction) cutoff of 0.05 was used to be further considered as statistically significant DEGs between MeJA and mock treatment. A heatmap was constructed visualizing the gene expression ratio (log2-fold change) of MeJA- over mock-treated samples of industrial chicory, witloof and lettuce.
Full-length CDS (flCDS) of the CiGAO, CiCOS, CiKLS and CYP71BZ candidate genes were amplified from cDNA with Q5 High Fidelity Polymerase (New England Biolabs) using gene-specific primers (Supplementary Table 2). PCR amplification products were used in an additional PCR with Q5 High Fidelity Polymerase using the gene-specific primers flanked by AttB sites for Gateway cloning. The PCR amplification products were run on a 1.5% agarose gel, extracted and purified with the GeneJET Gel Extraction Kit (Thermo Fisher Scientific). Previously identified CiGASs (AF498000.1) (Bouwmeester et al., 2002), CiGAO (GU256644.1) (Nguyen et al., 2010), CiCOS (JF816041.1) (Liu et al., 2011) and TpKLS (MF197559.1) (Liu et al., 2018) gene coding sequences were synthesized by Twist Bioscience (San Francisco, US). All amplified or synthesized sequences were cloned into the Gateway donor vector pDONR207 (Invitrogen) using Gateway™ BP clonase™ (Thermo Fisher Scientific) and sequence-verified by Sanger sequencing (Eurofins) prior to insertion into the N. benthamiana binary expression vector pEAQ-HT-DEST1 (Sainsbury et al., 2009) using Gateway™ LR clonase™ (Thermo Fisher Scientific).
The recombinant N. benthamiana expression vectors were individually transformed into the Agrobacterium tumefaciens strain C58C1, carrying the pMP90 helper plasmid, by electroporation. Transformed A. tumefaciens were grown and used for infiltration in N. benthamiana (Moses et al., 2015). Each vector was infiltrated to the abaxial side of three fully expanded leaves of 3- to 4-week-old N. benthamiana plants using 1-mL syringes. Infiltration with A. tumefaciens containing an empty pEAQ-HT-DEST1 vector was used as a negative control. The agroinfiltrated plants were incubated for 3 days in a plant growth chamber maintained at 25°C in a 14-h/10-h (light/dark) photoperiod.
Agroinfiltrated N. benthamiana leaves were harvested and ground to a fine powder in liquid nitrogen. Around 100 mg of leaf material was used for extraction of the SL metabolites. Leaf material was first vortexed with 1 mL of hexane for 10 min, followed by centrifuging for 15 min at 3,000 rpm. The resulting organic extract was evaporated to dryness under vacuum. For GC-MS analysis, the residue obtained from metabolite extraction was trimethylsilylated using 10 µL of pyridine and 50 µL of N-methyl-N-(trimethylsilyl)trifluoroacetamine. GC-MS analysis was performed on a 7890B GC system and a 7250 GC/QTOF (Agilent). Analyses were performed on a VF-5ms capillary column (30m x 0.25 mm x 0.25 µm; Varian CP9013; Agilent) at a constant helium flow of 1.2 mL/min, using a 1-µL aliquot, which was injected in splitless mode. After injection, the oven was held at 70°C for 2 min, ramped to 210°C at a rate of 5°C/min, held at 210°C for 5 min, ramped to 320°C at a rate of 20°C/min, held at 320°C for 5 min, and finally cooled to 70°C at a rate of 50°C/min at the end of the run. The injector, the MS transfer line, the MS ion source, and the quadrupole were set to 280°C, 280°C, 230°C and 150°C, respectively. Full electron ion (EI)-MS spectra were generated for each sample by scanning the m/z range of 50 to 800 with a solvent delay of 15 min. Electron ionization energy was 70 eV. For relative quantification, the peak areas were integrated using Agilent Masshunter Quantitative Analysis software (version 10.0). Identification of costunolide in the agroinfiltrated N. benthamiana leaves was performed by comparison with the retention time and mass spectrum of a costunolide standard (Sigma-Aldrich).
Plant material of industrial chicory C. intybus var. sativum ‘L9001’ was provided by COSUCRA (Belgium), and witloof C. intybus var. foliosum ‘Van Hamme’ was provided by Nationale Proeftuin voor Witloof (Herent, Belgium) and subcultured in vitro as previously described (De Bruyn et al. (2020). To target multiple putative paralogous CiGAO, CiCOS, CiKLS or CYP71BZ genes, the identified CiGAO, CiCOS, CiKLS and CYP71BZ clades were further divided into groups GAO_A, GAO_B, COS, KLS, BZ-II_A, BZ-II_B and BZ-III using nucleotide alignments and phylogenetic clustering (Supplementary Figure 1). Multiple gRNAs were designed to target the putative paralogous SL biosynthesis genes of each subclade (see Supplementary Table 3 for the corresponding primers). CRISPR/Cas9 vector construction, protoplast isolation, transfection and regeneration were performed as previously described (De Bruyn et al. (2020) with minor changes. Protoplasts were cotransfected with up to four CRISPR/Cas9 vectors, each containing a unique gRNA (grouped per subclade), using a total of 20 µg vector. The CRISPR/Cas9 gene target sequences and primers used for HiPlex amplicon sequencing are shown in Supplementary Figure 1 for each gene (see Supplementary Table 4 for the corresponding primer sequences). HiPlex amplicon sequencing, ploidy level and mutation analyses were performed as described in De Bruyn et al. (2020). Mutant types were defined as carriers of unique mutations (i.e., a discrete set of non-reference haplotype alleles identified by SMAP haplotype-window (Schaumont et al., 2022)). Mutant types were further aggregated based on the predicted consequences of the mutation(s) on the encoded protein functionality. By assuming loss of function (LOF) in case of out-of-frame indels or large in-frame indels (>9 amino acids), M types were grouped into L types if the functional consequences were similar, despite small differences in the actual mutated sequence. L types were defined by the homozygous or heterozygous LOF state per locus, for a given set of mutated genes (Figure 2 and Supplementary Table 5).
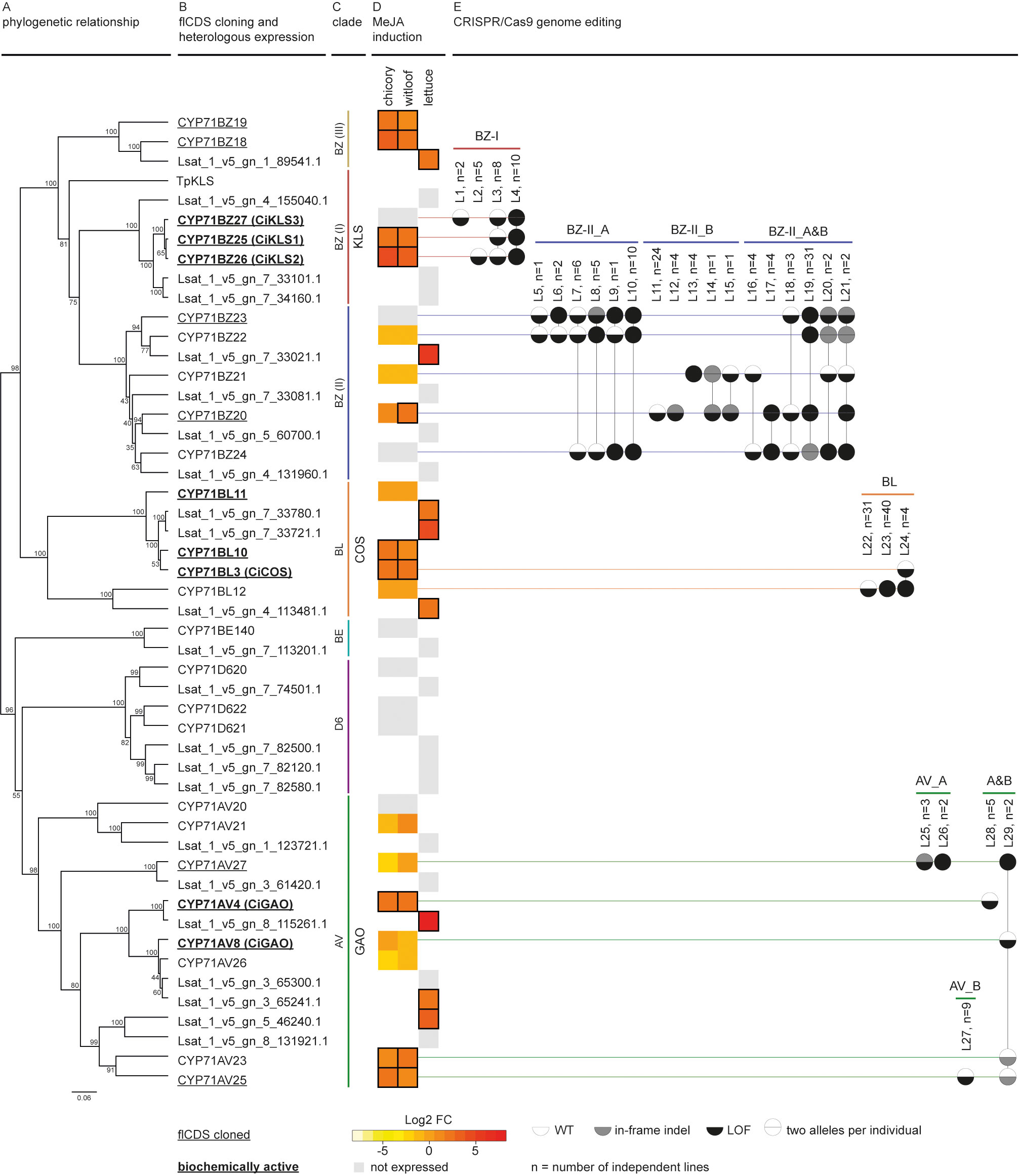
Figure 2 Discovery workflow of C. intybus CYP71 genes. (A) Phylogenetic analysis of C. intybus and corresponding L. sativa orthologous CYP71 genes. The phylogenetic tree was inferred from the protein alignment using the UPGMA method. Bootstrap values (shown at the branching points) are based on 1,000 replicates. (B) Full-length (fl)CDS were cloned from 14 genes (underlined), and functionally tested by heterologous expression in N. benthamiana (bold=biochemically active). KLS from T. parthenium (TpKLS) (AXG24152.1) was included for phylogenetic analysis. CYP71AV4/CYP71AV8 and CYP71BL3 were previously identified as functional CiGAO (ADF43080.1) and CiCOS (AEG79727.1), respectively. Also CiKLS1, CiKLS2, CiKLS3 were previously described as functional. (C) The various clades are indicated by vertical lines for GAO (green), COS (orange) and KLS (red). (D) Heatmap of gene expression response to MeJA treatment (log2-fold change in gene expression versus mock treatment as measured via RNA-seq) in industrial chicory, witloof and lettuce. Gray boxes indicate non-expressed genes while MeJA-inducible genes with log2-fold change ≥ 1 are marked with a black outline. (E) CRISPR/Cas9 genome editing was applied by targeting groups of genes (GAO_A, GAO_B, COS, KLS, BZ-II_A, BZ-II_B, and BZ-III). Mutants (listed by mutation type in Supplementary Table 5) are shown as two alleles per gene per circle, colored by wild type (WT), in-frame indel, or loss-of-function (LOF). LOF types are labeled L1-L29 and aggregated number of plants (n) are given.
For metabolite profiling of CRISPR/Cas9 genome-edited plants and wild-type control plants, for each plant, around 3 g of inner leaves were freshly harvested after between 2 and 6 months of growth and used for three technical replicates. Only for regenerant M3 and the two M6 genotypes material could be harvested for only one analysis without replicates. Each plant sample was immediately frozen and ground in liquid nitrogen, freeze-dried and stored under vacuum conditions at -20°C till analysis. Given that chicory bitterness is mostly attributed to the SLs downstream of compounds sl7a and sl7b (Figure 1), we performed metabolite profiling of the genome-edited plants only by UHPLC-HRMS analysis. To this end, extraction, separation and detection were done according to the method developed by Kips (2017). All compounds (sl08a: deoxylactucin, sl08b: dihydrodeoxylactucin, sl09a: deoxylactucin glycoside, sl09b: dihydrodeoxylactucin glycoside, sl10a: deoxylactucin oxalate, sl10b: dihydrodeoxylactucin oxalate, sl11a: lactucin, sl11b: dihydrolactucin, sl12a: lactucin glycoside, sl12b: dihydrolactucin glycoside, sl13a: lactucin oxalate, sl13b: dihydrolactucin oxalate, sl14a: lactucopicrin, sl14b: dihydrolactucopicrin, sl15a: lactucopicrin oxalate and sl15b: dihydrolactucopicrin oxalate) were identified based on the accurate mass and fragmentation pattern and reported as relative peak areas (area compound/area internal standard (santonine, Sigma-Aldrich)). Four compounds were quantified with reference standards: lactucin (sl11a), dihydrolactucin (sl11b), lactucopicrin (sl14a) and dihydrolactucopicrin (sl14b) (all from Extrasynthese, Genay, France), while no standards were available for the other compounds. Data recording was achieved with MassLynx™ (v.4.1) while the integration was performed with TargetLynx™ (v. 4.1) (Waters). Data analysis of metabolite profiles was done in R v.4.0.4 (R Core Team, 2017). Mean and standard error values were calculated for each mutation type of the relative peak areas of all 16 guaianolide SL metabolites extracted from the plants containing mutations in (multiple) paralogous CiGAO, CiCOS or CiKLS genes. Significant SL metabolite changes between the wild-type plants and the mutants were analyzed using a Pairwise Wilcoxon Rank Sum Test (p < 0.05).
Given their reported involvement in SL biosynthesis, our C. intybus var. sativum reference genome sequence (https://bioinformatics.psb.ugent.be/orcae/overview/Cicin) (Waegneer et al., 2023) was used for genome-wide identification of members from the TPS and CYP gene families. In total, 447 C. intybus annotated genes were manually curated, respectively 51 TPSs and 396 CYPs, of which 87 (19%) were classified as pseudogenes. Identification of the corresponding L. sativa orthologs resulted in 52 TPSs and 391 CYPs, with 42 (9%) pseudogenes. In summary, excluding pseudogenes, 40 TPS and 320 CYP gene family members were identified in C. intybus and 47 TPSs and 354 CYPs in L. sativa. Both C. intybus and L. sativa protein models were used for the creation of the TPS (Supplementary Figure 2) and CYP (Supplementary Figure 3) phylogenetic trees and clade classification.
Jasmonates (JAs) are widely known as elicitors of plant specialized metabolism, including many terpene biosynthetic pathways (Wasternack and Strnad, 2019; Lacchini and Goossens, 2020; Nguyen et al., 2022). Notably, transcript profiling studies have demonstrated that treatment with JA or MeJA induces the expression of genes encoding enzymes involved in the biosynthesis of artemisinin, the SL from the medicinal plant Artemisia annua (Yu et al., 2012). Yet, potential responsiveness to JAs has to the best of our knowledge not been investigated for guaianolide SL production in C. intybus or any other species. Therefore, we performed a comprehensive transcriptome analysis of seedlings of three species, C. intybus var. sativum, C. intybus var. foliosum and L. sativa, treated with MeJA over 24 h. First, we assessed the MeJA responsiveness of the known SL biosynthetic pathway genes CiGASl, CiGASs, CiGAO, and CiCOS by RT-qPCR analysis. This analysis confirmed the MeJA responsiveness of the SL pathway in all three species (Figure 3). As such, it became plausible that MeJA inducibility could be relevant to select candidate CYP gene family members potentially involved in C. intybus SL biosynthesis. Strongest upregulation was observed after a 6-h MeJA treatment in all three species. Thus, samples of industrial chicory ‘OBOE’, witloof ‘Topmodel’ and lettuce ‘ZORBA’, after 6 h of mock or MeJA treatment were used for subsequent RNA-seq analysis. The industrial chicory and witloof reads were mapped to the CDS of C. intybus var. sativum, containing 53,507 annotated genes, of which 38% and 37% were expressed in industrial chicory and witloof, respectively. The lettuce reads were mapped to the CDS of L. sativa, containing 37,829 annotated genes (Reyes-Chin-Wo et al., 2017), of which 48% were expressed (Supplementary Table 6). Hereof, 3,384 industrial chicory genes, 2,696 witloof genes and 1,395 lettuce genes were significantly differentially expressed between MeJA and mock treatment (padj<0.05) (Supplementary Table 6). Comparison between industrial chicory and witloof gene sets identified a total of 1,930 genes that were commonly differentially expressed upon MeJA treatment. As anticipated, these included known SL biosynthesis genes. In industrial chicory, witloof, and lettuce, the number of downregulated genes (MeJA vs. mock) was 1,489, 1,076 and 586, respectively, and the number of upregulated genes (MeJA vs. mock) was 1,895, 1,620 and 809, respectively (Supplementary Table 6).
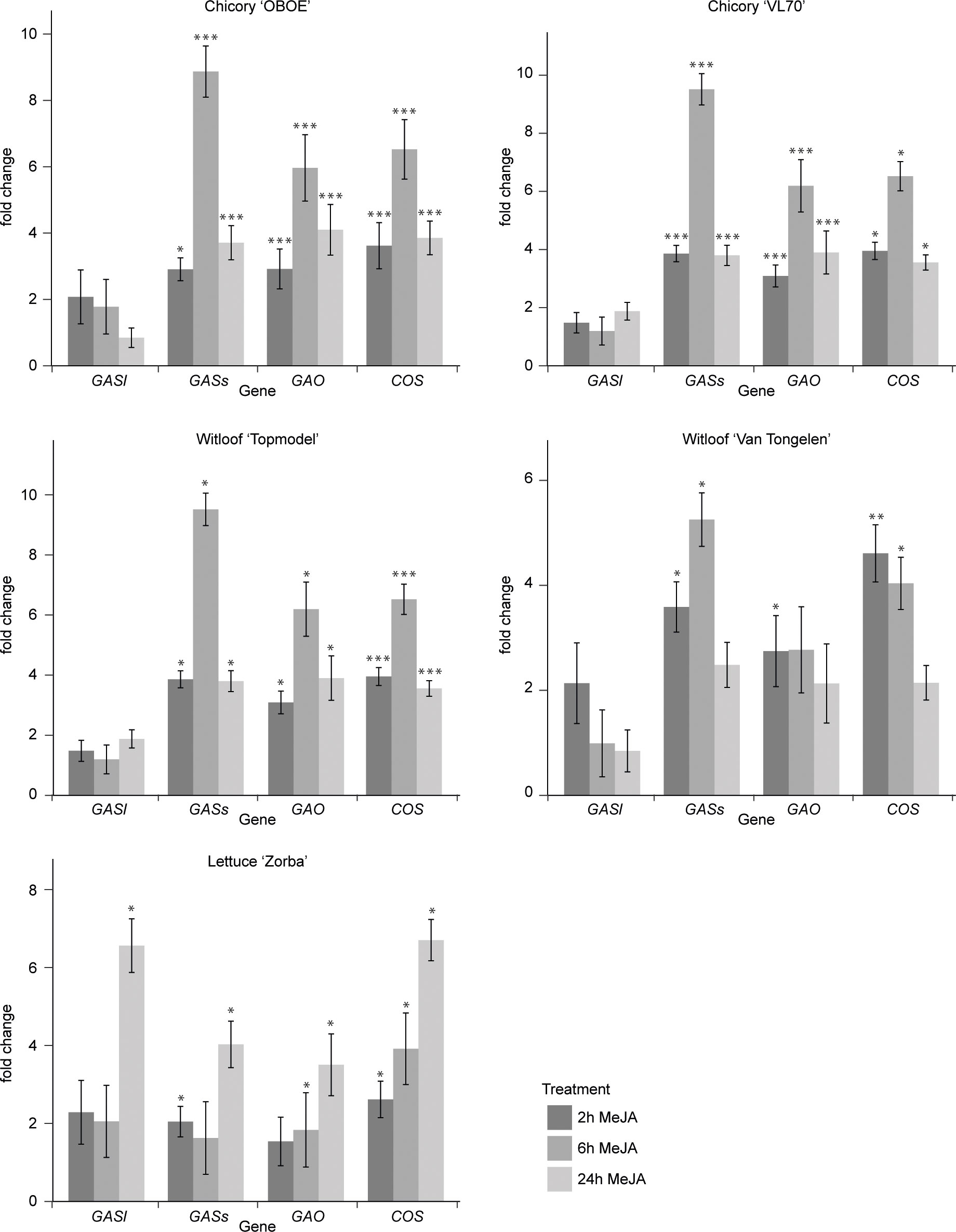
Figure 3 Fold changes of SL biosynthesis gene expression. Steady-state levels of transcripts corresponding to CiGASl, CiGASs, CiGAO (CYP71AV4) and CiCOS (CYP71B3)) were determined by RT-qPCR in seedlings of five plant varieties after 2 h, 6 h and 24 h of mock or MeJA treatment. Columns reflect the increase in transcript levels caused by MeJA treatment. The values in the y-axis correspond to the mean ± SE (n = 4) of the ratio of the transcript levels in MeJA-treated samples versus those in mock-treated samples at each timepoint. Statistical differences relative to the respective mock treatments were determined by Kruskal-Wallis rank sum test at p<0.05, using Pairwise Wilcoxon rank sum test (*p<0.05, **p<0.01 and ***p<0.001.).
A total of 40 and 47 TPS gene family members were identified in C. intybus and L. sativa, respectively, of which 15 industrial chicory, 14 witloof and 16 lettuce genes were MeJA-inducible (Supplementary Table 6; Supplementary Figure 2). Protein alignment of all TPS genes showed that the previously described CiGAS-L, the tandem duplicated genes CiGAS-S1 and CiGAS-S2, and CiGAS-S3 (Bouwmeester et al., 2002; Bogdanović et al., 2019; Cankar et al., 2021) corresponded to cicin09g10370, cicin03g50310 and cicin03g50280, and cicin06g42820, respectively, thus identifying the clade with functional CiGAS proteins (Supplementary Figure 2). Two additional CiGAS paralogs belonging to the same clade were identified: cicin06g42850 and cicin02g48520 (Supplementary Figure 2 and Supplementary Figure 4). The six paralogs were located on four different chromosomes, with two tandem duplicated genes on CiChr3 and two tandem duplicated genes on CiChr6. Of these, cicin03g50310, cicin03g50280 and cicin06g42820 were MeJA-inducible in both industrial chicory and witloof (Supplementary Figure 2). In the same clade, four lettuce orthologs were identified, of which only Lsat_1_v5_gn_8_116340.1 was MeJA-inducible. Based on the protein alignment and phylogenetic clustering of the TPS genes with the previously described CiGAS genes, a total of six genes in the genome of C. intybus could potentially encode GAS enzymes, more specifically cicin03g50280, cicin03g50310, cicin06g42850, cicin06g42820, cicin09g10370, and cicin02g48520 (Supplementary Figure 2; GAS clade indicated by the blue vertical bar).
A total of 320 and 354 genes were identified in the CYP superfamily in C. intybus and L. sativa, respectively, of which 49 industrial chicory, 51 witloof and 33 lettuce genes were MeJA-inducible (Supplementary Table 6; Supplementary Figure 3). Phylogenetic analysis identified the CYP71 clade with the previously described CiGAO, CiCOS and CiKLS genes (Liu et al., 2018; Cankar et al., 2022), the clade on which we further focused in this study. This CYP71 clade contains a total of 26 C. intybus and 23 L. sativa genes (Figures 2A, B), of which 9 industrial chicory, 10 witloof and 8 lettuce genes were MeJA-inducible (Figure 2D). Phylogenetic analysis further identified three subclades that each contained multiple putative paralogs of CiGAO, CiCOS and CiKLS genes (Figure 2C).
Two functional CiGAO genes (CYP71AV4 and CYP71AV8) were previously described in C. intybus (Nguyen et al., 2010; Cankar et al., 2011; Liu et al., 2011). Our analysis revealed eight potential paralogs in the CYP71AV CiGAO clade (Figures 2A–C and Supplementary Figure 4): CYP71AV4 located on CiChr3, CYP71AV27 located on CiChr7, tandem duplicated genes CYP71AV8 and CYP71AV26 located on CiChr7, CYP71AV23 and CYP71AV25 located on CiChr4, and the slightly more distantly related CYP71AV20 and CYP71AV21 located on CiChr2. CYP71AV4, CYP71AV23, and CYP71AV25 were MeJA-inducible in both industrial chicory and witloof. In the same clade, seven lettuce orthologs were identified, of which three genes (Lsat_1_v5_gn_8_115261.1, Lsat_1_v5_gn_3_65241.1 and Lsat_1_v5_gn_5_46240.1) were MeJA-inducible (Figure 2D).
Four potential paralogs were identified in the CYP71BL CiCOS clade: CYP71BL11, CYP71BL10, CYP71BL3 and CYP71BL12 (Figures 2A–C and Supplementary Figure 4), which were all located on CiChr5 in proximity of each other. CYP71BL3 revealed 99.8% nucleotide identity (one SNP difference) to the previously described functional CiCOS gene (Liu et al., 2011). CYP71BL10 and CYP71BL3 were MeJA-inducible in both industrial chicory and witloof (Figure 2D). Within the same clade, three lettuce orthologs were identified, which were all MeJA-inducible (Lsat_1_v5_gn_7_33780.1, Lsat_1_v5_gn_7_33721.1 and Lsat_1_v5_gn_4_113481.1).
The CYP71BZ clade containing CiKLS and TpKLS revealed a total of ten potential paralogs in C. intybus (Figures 2A–C and Supplementary Figure 4), divided in three subclades (here named I to III). The three tandem duplicated genes, CYP71BZ25, CYP71BZ26, and CYP71BZ27 of subclade I, corresponding to CiKLS1, CiKLS2, and CiKLS3 (Cankar et al., 2022), respectively, were located on CiChr5. Of these, CiKLS1 and CiKLS2 were MeJA-inducible, while their lettuce orthologs (Lsat_1_v5_gn_4_155040.1, Lsat_1_v5_gn_7_33101.1, Lsat_1_v5_gn_7_34160.1) were not expressed. CYP71BZ subclade II contained five genes; CYP71BZ22, CYP71BZ23 and CYP71BZ24 were tandem duplicated genes located on CiChr5, while CYP71BZ21 and CYP71BZ20 were tandem duplicated genes located on CiChr4, and only CYP71BZ20 was MeJA-inducible in witloof. Within that subclade, a total of four lettuce orthologs were identified, of which only Lsat_1_v5_gn_7_33021.1 was MeJA-inducible. In subclade III, the tandem duplicated genes CYP81BZ19 and CYP71BZ18 located on CiChr2 were both MeJA-inducible, as was their lettuce ortholog (Lsat_1_v5_gn_1_89541.1).
Finally, two other CYP71 subclades were identified: CYP71BE with one gene of chicory and lettuce each (CYP71BE140, Lsat_1_v5_gn_7_113201.1), and CYP71D6 with three genes of chicory (CYP71D620, CYP71D621, and CYP71D622), and four orthologs of lettuce (Lsat_1_v5_gn_7_74501.1, Lsat_1_v5_gn_7_82500.1, Lsat_1_v5_gn_7_82120.1, Lsat_1_v5_gn_7_82580.1), respectively, but none of these genes were MeJA-inducible.
To investigate which of the candidate CYP71 paralogous genes described above are involved in SL biosynthesis, we first reconstructed the C. intybus SL biosynthesis pathway up to costunolide in N. benthamiana leaves by agroinfiltration (Figure 4). The SL-specific candidate genes were co-expressed with a gene encoding a truncated, feedback-free version of the 3-HYDROXY-3-METHYLGLUTARYL-COA REDUCTASE (tHMGR) enzyme (Reed et al., 2017), to boost production of the precursor of all sesquiterpenes. First, the successfully cloned flCDS of four putative paralogous CiGAO genes were transformed in N. benthamiana leaves by co-agroinfiltration with CiGASs (cicin03g50280; also described as CiGAS-S2 in (Cankar et al., 2021)) and CiCOS (CYP71BL3) to assess GAO functionality and/or relative catalytic efficiency (Supplementary Table 7). Co-agroinfiltration of CiGASs, CiGAO and CiCOS was used as a positive control for the production of costunolide, as described previously (Liu et al., 2018). Costunolide was detected in N. benthamiana leaves transformed with CiGAO (CYP71AV4) and CYP71AV8, but not CYP71AV27 or CYP71AV25 (Figures 4B, C). Notably, the activity of CYP71AV8, which is non-MeJA-inducible (Figure 2D), was relatively higher than that of CYP71AV4.
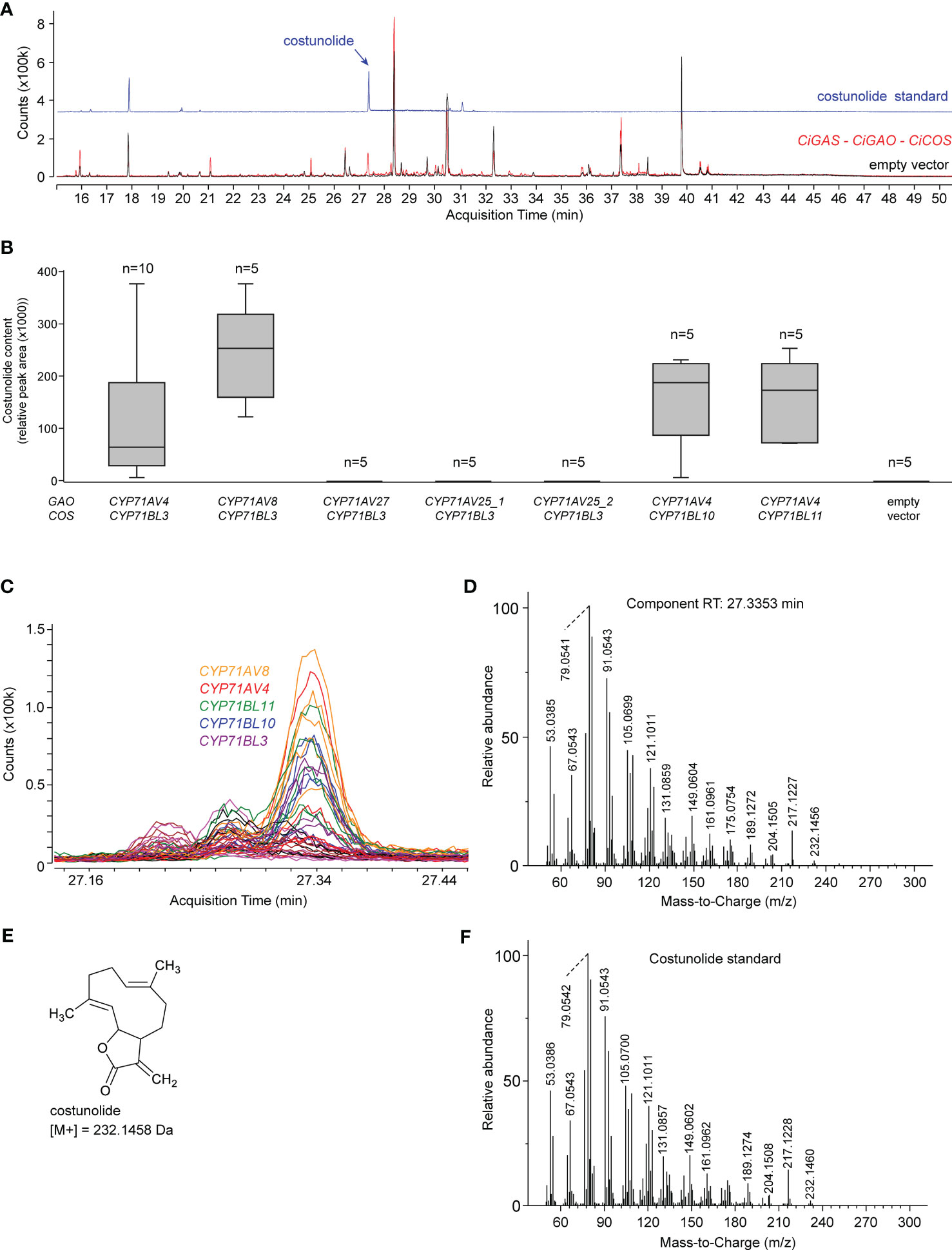
Figure 4 Costunolide production in agroinfiltrated N. benthamiana leaves. (A) Overlay of the EIC GC-MS chromatograms of the costunolide standard (blue) and N. benthamiana extracts co-agroinfiltrated with CiGASs, CYP71AV4 (CiGAO) and CYP71BL3 (CiCOS) (red) or empty vector controls (black). The peak at ±27.3 min (indicated with a black arrow) corresponds to costunolide. (B) Boxplot of costunolide production (relative peak area) in N. benthamiana leaves (n = 3) co-agroinfiltrated with CiGASs and various combinations of potential CiGAO and CiCOS genes, including CYP71AV4 (CiGAO), CYP71AV8 or paralogous candidate CiGAO genes (CYP71AV27, CYP71AV25), and CYP71BL3 (CiCOS) or paralogous candidate CiCOS genes (CYP71BL10, CYP71BL11). Labels _1, _2, etc. indicate different allelic variants of the corresponding CYP71 genes cloned from the different C. intybus varieties used. (C) Overlay of the EIC (232.15 ± 0.05 Da; 27.1 – 27.5 min) GC-MS chromatograms of all samples (three replicates of all assays in Supplementary Table 10). The peak at 27.34 min corresponds to the blue peak in panel (B) The assays displayed indicate which agroinfiltrated N. benthamiana extracts produce this peak. The peak was not present in all other assays, indicated by all colored horizontal lines. (D) Deconvoluted EI-MS spectrum of the peak eluting at 27.34 min. (E) Structure of costunolide with the corresponding molecular weight. (F) Deconvoluted EI-MS spectrum of the costunolide standard.
The successfully cloned flCDS of three putative paralogous CiCOS genes were then transformed in N. benthamiana leaves by co-agroinfiltration with CiGASs (cicin03g50280) and CiGAO (CYP71AV4) (Supplementary Table 7). Costunolide could be detected in N. benthamiana leaves transformed with each of these CiCOS paralogs (Figures 4B, C). As with CiGAO (CYP71AV4), this included MeJA-inducible (CYP71BL3 and CYP71BL10) and non-MeJA-inducible (CYP71BL11) genes (Figure 2D). No marked differences in the catalytic performance of the three paralogs was apparent.
In conclusion, a total of two paralogous CiGAO genes (CYP71AV8 and CYP71AV4) and three paralogous CiCOS genes (CYP71BL10, CYP71BL11 and CYP71BL3) displayed catalytic activity in the synthetic costunolide biosynthetic pathway in N. benthamiana.
Successfully cloned flCDS of seven CiKLS subclade genes were transformed in N. benthamiana leaves by co-agroinfiltration with CiGASs, CiGAO (CYP71AV4) and CiCOS (CYP71BL3) (Supplementary Table 8). Co-agroinfiltration of CiGASs, CiGAO and CiCOS with TpKLS was used as positive control for the potential production of kauniolide (Figure 5), as previously described (Liu et al., 2018). Kauniolide was detected in N. benthamiana leaves transformed with TpKLS as well as with CiKLS1 (CYP71BZ25), CiKLS2 (CYP71BZ26) and CiKLS3 (CYP71BZ27) (Figures 5A–C). No kauniolide production was observed in any of the other combinations (CYP71BZ18 and CYP71BZ19 (subclade III); CYP71BZ20 and CYP71BZ23 (subclade II)). As with CiGAO and CiCOS, the functional CiKLS gene set included MeJA-inducible (CYP71BZ25 and CYP71BZ26) and non-MeJA-inducible (CYP71BZ27) genes (Figure 2D).
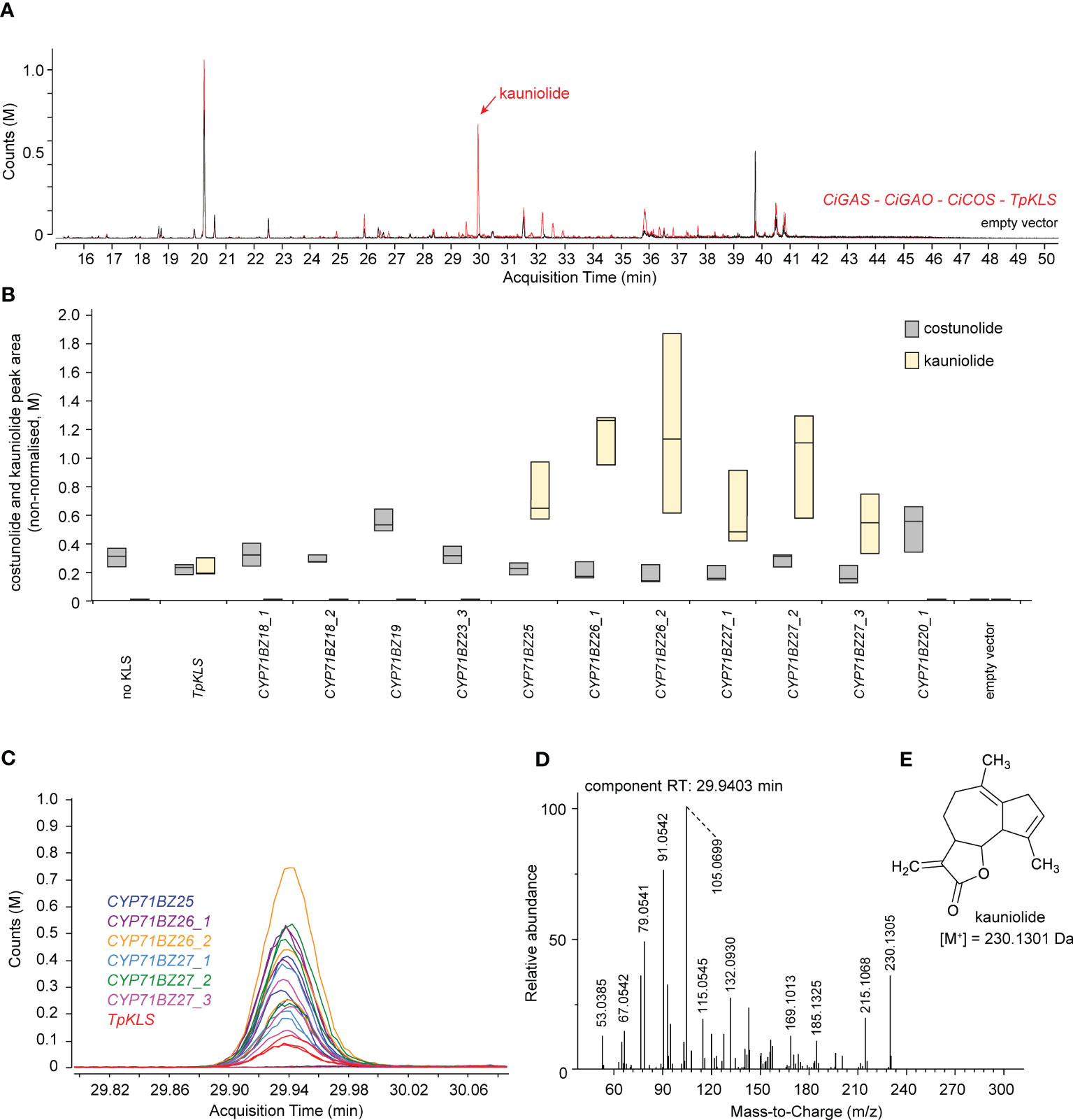
Figure 5 Kauniolide production in agroinfiltrated N. benthamiana leaves. (A) Overlay of the EIC GC-MS chromatogram of N. benthamiana extracts co-agroinfiltrated with CiGASs, CYP71AV4 (CiGAO), CYP71BL3 (CiCOS), and TpKLS (red chromatogram) or empty vector controls (black chromatogram). The red peak at ±30 min (indicated with a black arrow) is present in the N. benthamiana extracts co-agroinfiltrated with both CYP71BL3 and TpKLS and absent in the N. benthamiana extracts agroinfiltrated with only CYP71BL3. (B) Boxplot of kauniolide production (relative peak area) in N. benthamiana leaves (n = 5) co-agroinfiltrated with CiGASs, CYP71AV4 (CiGAO), CYP71BL3 (CiCOS), and differential potential KLS genes, including TpKLS, CiKLS or paralogous candidate CiKLS genes. Labels _1, _2, etc. indicate different allelic variants of the corresponding CYP71 genes cloned from the different C. intybus varieties used. (C) Overlay of the EIC (230.13 ± 0.05 Da; 29.8 – 30.1 min) GC-MS chromatograms of all samples (three replicates of all assays in Supplementary Table 10). The peak at 29.94 min corresponds to the red peak in panel (B) The assays displayed indicate which agroinfiltrated N. benthamiana extracts produce this peak. The peak was not present in all other assays, indicated by all colored horizontal lines. (D) Deconvoluted EI-MS spectrum of the peak eluting at 29.94 min. (E) Structure of kauniolide with the corresponding molecular weight.
In conclusion, a total of three paralogous CiKLS genes displayed catalytic activity in the synthetic kauniolide biosynthetic pathway in N. benthamiana. Thus, these results correlated well with those published by Cankar et al. (2022), who demonstrated the KLS activity of these three genes in a yeast microsome assay.
To investigate whether any of the hitherto non-functional CYP71BZ genes of subclades II and III could act in SL biosynthesis, the flCDS of all remaining successfully cloned, non-functional CYP71 genes were transformed in N. benthamiana leaves by co-agroinfiltration with CiGASs, CiGAO, CiCOS and CiKLS2 (Supplementary Table 9). None of the tested combinations led to further conversion of kauniolide to putative downstream SLs, suggesting that these CYP71 family members may not be involved in SL biosynthesis, accept costunolide or kauniolide as a substrate, or be expressed or functional in N. benthamiana.
CRISPR/Cas9 genome editing was used to simultaneously target multiple CYP71 genes in protoplasts of C. intybus, followed by regeneration of plants. For this experiment, the identified CiGAO, CiCOS and CiKLS (sub)clades were further divided into groups (GAO_A, GAO_B, COS, KLS, BZ-II_A, BZ-II_B and BZ-III; Figure 2E) and multiple gRNAs were designed to target the putative paralogous SL biosynthesis genes within each group (Supplementary Figure 1). A series of CRISPR/Cas9 protoplast co-transfection and regeneration experiments using up to four CRISPR/Cas9 vectors (combined per group) were performed to create a broad spectrum of plant genotypes containing different (LOF) mutations in multiple genes. A total of 939 regenerated plants were acclimatized to the greenhouse and characterized by HiPlex amplicon sequencing at all target loci, 46 plants targeted in the CiGAO paralogous candidate genes, 109 plants targeted in the CiCOS paralogous candidate genes, and 784 plants targeted in the CiKLS or CYP71BZ paralogous candidate genes., respectively. Mutation analysis revealed 225 plants with at least one mutated allele in one SL biosynthesis candidate gene (Figure 2E), which were grouped into 46 unique mutation types (Supplementary Table 5), and further aggregated into LOF types. An overall plant mutation frequency of 23.9% was obtained, creating a broad spectrum of mutants. Ploidy levels were determined for 368 acclimatized plants, including 46 control plants and 322 CRISPR/Cas9-edited plants, resulting in 48.1% diploid plants and 51.9% tetraploid plants. However, the tetraploid mutated plants did not contain more complex editing patterns (no more than two unique alleles detected per locus), suggesting that the ploidy change occurred after the mutation was induced.
36 CRISPR/Cas9 mutants, each containing mutations in one or more paralogous CYP71 genes, were sampled for SL metabolite profiling (4 GAO mutants, 13 COS mutants, 21 KLS or CYP71BZ mutants; Supplementary Table 5), corresponding to different 23 mutation types. The production of 14 out of 16 SL metabolites was successfully eliminated in one mutant type (M6) (Supplementary Table 10), containing a homozygous LOF mutation in the three paralogous CiKLS genes, which were functionally characterized here and in Cankar et al. (2022). These results confirm that the three paralogous CiKLS genes are indeed important SL biosynthesis genes for the production of the guaianolide SL metabolites in chicory. Unfortunately, the plants with LOF alleles in the three CiKLS genes were slow-growing and not persistent, making it impossible to include biological and technical replicates. Therefore, no statistical analysis could be performed on the SL metabolite values of these mutants compared to wild-type control plants. Besides mutation type M6, no other mutation type that could be analyzed showed such a consistent noticeable modulation in SL levels (Supplementary Table 10). None of the mutant lines with LOF alleles for the genes encoding the CYP71 proteins that were non-functional in N. benthamiana (subclade BZ-II) showed any detectable alteration in SL levels, further indicating that these genes are not involved in SL biosynthesis in C. intybus. Nonetheless, it should be noted that for genes from subclade BZ-III in the CYP71 phylogenetic tree (e.g. CYP71BZ18 and CYP71BZ19) that were non-functional in N. benthamiana, we did not generate homozygous LOF mutations, hence their possible involvement cannot be excluded unambiguously yet.
It should further be noted that most SL-profiled plants (also the wild type) were tetraploid plants. Comparing the SL metabolite content of the four quantitatively measurable (ng/mg dry weight) SL metabolites, lactucin, lactucopicrin, dihydrolactucin and dihydrolactucopicrin, in the analyzed diploid and tetraploid plants (Supplementary Table 10; see M3, M46 and WT) showed an SL metabolite concentration increase in these tetraploid plants, suggesting that the plant’s ploidy level has an influence on SL metabolite production.
Modifying the bitterness of C. intybus could open new markets for this crop in addition to the existing inulin market. Not only reducing the bitterness in Cichorium can be of value, but also increasing the production of SL metabolites can improve market options, such as the extraction of bioactive SLs as natural compounds for the pharmaceutical and cosmetic industries (Häkkinen et al., 2021; Matos et al., 2021; Rolnik and Olas, 2021). Although there is some natural variation in the SL content among Cichorium species (Foster et al., 2011; Testone et al., 2016), variants with a considerably low SL content have not yet been identified. Better knowledge of the pathway and the functionality of the genes involved would open opportunities for modification of SL biosynthesis.
Delineation of the putative biochemical functions involved in the SL biosynthetic pathway based on a hypothetical SL pathway connecting the conversions of FPP to the detectable guaianolide SL metabolites (Kips, 2017) and our current knowledge of this pathway drove us to select two specific enzyme families, TPS and CYP. The functional diversity of the TPS enzymes underlies the generation of diverse terpenoids across the plant kingdom (Karunanithi and Zerbe, 2019). In this study, we found 40 genes encoding TPSs in the genome of C. intybus, including cicin03g50310, cicin03g50280, cicin06g42820, and cicin09g10370, which correspond to the previously described CiGAS-S1, CiGAS-S2, CiGAS-S3 and CiGAS-L, respectively (Bouwmeester et al., 2002; Bogdanović et al., 2019; Cankar et al., 2021). The CYP superfamily is one of the largest enzyme families in plants and has also been widely identified in animals, fungi, protists, archaea, bacteria, insects and viruses (Xu et al., 2015). In this study, 320 genes were found to potentially encode functional CYPs in the genome of C. intybus, including CYP71AV4/CYP71AV8 and CYP71BL3, which were previously functionally characterized as CiGAO and CiCOS, respectively (Nguyen et al., 2010; Cankar et al., 2011; Liu et al., 2011), as well as the three recently identified CiKLS genes (Cankar et al., 2022). Several phylogenetic clades were identified in the two gene families, and anchored to CiGAS, CiGAO, CiCOS, and CiKLS. Notably, each clade contains multiple putative paralogous genes, often derived from tandem duplication, which generally display high levels of amino-acid sequence similarity, suggesting similar protein folding structures and putatively conserved biochemical activities (e.g. CiKLS1, CiKLS2, CiKLS3). Such a comprehensive genome-wide overview of paralogs and the underlying mechanisms that drive gene family expansion and gene evolution (including non-, sub- and neo-functionalization) is important in the frame of pathway elucidation. For instance, it defines how many and which genes (redundantly) encode enzymes at each subsequent step of the pathway, which regulatory elements are shared or unique across paralogs (leading to co-expression networks and/or control of pathway activity by alternative regulatory factors or signaling pathways), and whether knockouts, e.g. by genome editing, will lead to observable phenotypes (e.g. will LOF mutations be required in multiple genes to overcome functional redundancy).
Given the reported importance of members of the CYP71 gene family in C. intybus SL biosynthesis, we performed an in-depth characterization of this gene family. First, the CYP71 genes were functionally characterized using an N. benthamiana heterologous expression assay. In addition to confirming the two previously described CiGAO genes (Nguyen et al., 2010; Cankar et al., 2011), one CiCOS (Liu et al., 2011), and the three CiKLS genes (Cankar et al., 2022), two additional functional CiCOS genes were identified. Conversely, no biochemical activity was observed for CYP71AV25, CYP71AV27, CYP71BZ18, CYP71BZ19, CYP71BZ20, and CYP71BZ23 in this system, suggesting that these candidate genes may either be non-functional in the guaianolide SL biosynthetic pathway, or may act further downstream.
The role of the putative paralogous CYP71 genes in SL biosynthesis was also analyzed in C. intybus by the creation of LOF mutants. A broad spectrum of mutants was created containing mutations in single or multiple paralogous CiGAO, CiCOS, CiKLS and CYP71BZ genes and their guaianolide SL metabolite levels were determined. Because many of the CiGAO, CiCOS, CiKLS or CYP71BZ mutated plant genotypes only contained one heterozygous LOF allele in one paralog of a specific SL biosynthesis gene (CiGAO, CiCOS, CiKLS or CYP71BZ), the mutation effect on guaianolide SL metabolite quantities and composition was in most cases most likely masked due to the presence of at least one functional allele and/or functional paralog. There was one notable exception: mutation type M6 that contained a LOF mutation in the three paralogous CiKLS genes eliminated the accumulation of nearly all SL metabolites detectable in wild-type plants. These results further underscore that the enzymes encoded by these three CiKLS genes are necessary for in planta SL biosynthesis, more particularly for the production of the kauniolide intermediate, thereby corroborating similar observations reported by Cankar et al. (2022). Furthermore, our results also suggest that successful LOF mutations in all alleles of multiple paralogs of a specific SL biosynthesis gene are required to significantly decrease guaianolide SL metabolite production levels.
In this study, we also showed that the C. intybus SL pathway is controlled by the phytohormone MeJA, a known elicitor of plant specialized metabolism. Interestingly, when we integrated the outcome of the comprehensive RNA-seq analysis of MeJA-elicited C. intybus seedlings with that of the phylogenetic and functional analysis of the CYP71 gene family, we discovered that all the functional clades, i.e. CiGAO, CiCOS, and CiKLS, contained both MeJA-inducible and non-MeJA-inducible paralogs, suggesting the existence of multiple regulatory routes controlling C. intybus SL biosynthesis. Furthermore, MeJA inducibility was often conserved between industrial chicory and witloof orthologs and even some orthologous L. sativa genes, indicating that the control of SL biosynthesis by this hormone evolved before the species diversification. Conversely, some genes were only MeJA-inducible in one C. intybus variety (industrial chicory or witloof) or L. sativa, which appoints them as plausible candidates for contributing to SL differentiation between these species (Testone et al., 2016).
Reducing SL metabolite production in C. intybus has also been achieved via targeting of the CiGAS genes. Bogdanović et al. (2020) used gene silencing via artificial microRNAs to downregulate the expression of CiGASs and CiGASl genes. This approach resulted in only partial gene silencing, leading to variable levels of SL metabolite production. It should also be noted that the authors only evaluated the guaianolide oxalate SL metabolites, because these SL metabolites are often present in high amounts in wild-type Cichorium plants (Sessa et al., 2000). Cankar et al. (2021) used CRISPR/Cas9 genome editing to target the four previously identified CiGAS paralogs (Bogdanović et al., 2019), resulting in a significant decrease of all six measured SL metabolites but with a corresponding increase of squalene and phenolic compounds. Because FPP is the precursor for sesquiterpene and sterol biosynthesis, it is not surprising that inactivation of CiGAS leads to changes in the metabolic flux from FPP to other compounds. Indeed, accumulation of FPP has been shown upon silencing of the amorpha-4,11-diene synthase genes in A. annua (Catania et al., 2018) while silencing of terpene synthase genes (TPS9 and TPS12) in tomato led to the upregulation of several genes involved in flavonoid biosynthesis (Coppola et al., 2018). The changes in these metabolic pathways with FPP as precursor should be considered when evaluating the bitter taste in plants containing mutations in the guaianolide SL biosynthesis genes.
Van Beek et al. (1990) were the first to show that the bitterness in Cichorium plants is related to their guaianolide SL metabolite concentration and composition. However, not all guaianolide SL metabolites that can be successfully measured today have been considered regarding bitterness perception. Almost all studies measuring guaianolide SL metabolite profiles focus on measuring six guaianolide SL metabolites: lactucin, 8-deoxylactucin, lactucopicrin, 11(S),13-dihydro-8-deoxylactucin, 11(S),13-dihydrolactucin and 11(S),13-dihydrolactucopicrin (Van Beek et al., 1990; Sessa et al., 2000; Foster et al., 2011; Beharav et al., 2015; D’antuono et al., 2016; Yanagisawa and Misaka, 2021). The oxalate and glycoside metabolite forms of guaianolide SL are not taken into account (Kips (2017), resulting in the unknown perception of their bitter taste. Still, previous studies analyzing these six guaianolide SL metabolites have shown that 11(S),13-dihydrolactucopicrin is perceived as extremely bitter, while lactucopicrin, 8-deoxylactucin, 11(S),13-dihydro-8-deoxylactucin, 11(S),13-dihydrolactucin and lactucin are perceived as less bitter. Furthermore, previous research on the quantitative analysis of these six guaianolide SL metabolites showed that the bitterness responses were dose-dependent and that the concentration at which the bitterness is perceived is different for each metabolite (Yanagisawa and Misaka, 2021). Moreover, the perception of bitterness seems not only to be related to the guaianolide SL metabolite concentrations and composition. A previous study strongly correlated glucose and sucrose concentrations in industrial chicory leaves with crunchiness and bitterness, while fructose concentrations were correlated with sweetness (François et al., 2008). Another study indicated that the balance between different SL metabolites and phenolic compounds affects the bitterness in a rather complex manner (D’antuono et al., 2016). Therefore, SL metabolites are possibly not the only metabolites that should be considered when studying bitterness in plant tissues. Cellular assays and human sensory tests can be used to evaluate the link between SL metabolite concentration and composition, and bitterness (Yanagisawa and Misaka, 2021).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/; PRJNA738883.
CDB, AG, TR, and KVL conceived and designed the research and supervised the experiments; CDB, SR, CVP, SD, and EL performed the experiments; SR designed and supervised genomic DNA extraction and sequencing, did assemblies and gene prediction; CDB, TR, SR, AH, CVP, SD, EL, TJ, TE, AG, and KVL designed the experiments and analyzed the data; CDB, TR, AG, and KVL wrote the article. All authors contributed to the article and approved the submitted version.
The authors gratefully acknowledge the financial support of the Research Foundation Flanders (FWO SB Grant No. 1S01520N to CDB) and the European Union’s Horizon 2020 research and innovation program under Grant Agreement No 825730 (Endoscape) to AG.
The authors gratefully acknowledge Annick Bleys for critical reading of the manuscript, Ronald Van den Oord (ILVO) for technical support in plant tissue culture, Nancy Mergan and Carina Pardon (ILVO) for technical support in DNA-extraction, Veerle Buysens and Sophie Carbonelle (ILVO) for technical support in SL metabolite extraction, Laurence Desmet for technical support in RT-qPCR analysis, Rina Vanhaecke and Roger Dobbelaere (ILVO) for technical support in the greenhouse, Liesbeth De Milde (VIB-UGent) for technical support in N. benthamiana leaf infiltrations, and Geert Goeminne from the VIB Metabolomics Core - Ghent for the costunolide and kauniolide profiling.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1200253/full#supplementary-material
Barcaccia, G., Ghedina, A., Lucchin, M. (2016). Current advances in genomics and breeding of leaf chicory (Cichorium intybus l.). Agriculture 6, 50. doi: 10.3390/agriculture6040050
Barrero, A. F., Oltra, J. E., Álvarez, M., Raslan, D. S., Saúde, D. A., Akssira, M. (2000). New sources and antifungal activity of sesquiterpene lactones. Fitoterapia 71, 60–64. doi: 10.1016/s0367-326x(99)00122-7
Beharav, A., Stojakowska, A., Ben-David, R., Malarz, J., Michalska, K., Kisiel, W. (2015). Variation of sesquiterpene lactone contents in Lactuca georgica natural populations from Armenia. Genet. Resour. Crop Evol. 62, 431–441. doi: 10.1007/s10722-014-0171-9
Bischoff, T. A., Kelley, C. J., Karchesy, Y., Laurantos, M., Nguyen-Dinh, P., Arefi, A. G. (2004). Antimalarial activity of lactucin and lactucopicrin: sesquiterpene lactones isolated from Cichorium intybus l. J. Ethnopharmacol. 95, 455–457. doi: 10.1016/j.jep.2004.06.031
Bogdanović, M., Cankar, K., Dragićević, M., Bouwmeester, H., Beekwilder, J., Simonović, A., et al. (2020). Silencing of germacrene a synthase genes reduces guaianolide oxalate content in Cichorium intybus l. GM Crops Food 11, 54–66. doi: 10.1080/21645698.2019.1681868
Bogdanović, M., Cankar, K., Todorović, S., Dragicević, M., Simonović, A., Van Houwelingen, A., et al. (2019). Tissue specific expression and genomic organization of bitter sesquiterpene lactone biosynthesis in Cichorium intybus l. (Asteraceae). Ind. Crop Prod. 129, 253–260. doi: 10.1016/j.indcrop.2018.12.011
Borowski, J. M., Galli, V., Messias Rda, S., Perin, E. C., Buss, J. H., Delmar Dos Anjos E Silva, S., et al. (2014). Selection of candidate reference genes for real-time PCR studies in lettuce under abiotic stresses. Planta 239, 1187–1200. doi: 10.1007/s00425-014-2041-2
Bouwmeester, H. J., Kodde, J., Verstappen, F. W. A., Altug, I. G., De Kraker, J.-W., Wallaart, T. E. (2002). Isolation and characterization of two germacrene a synthase cDNA clones from chicory. Plant Physiol. 129, 134–144. doi: 10.1104/pp.001024
Cankar, K., Bundock, P., Sevenier, R., Häkkinen, S. T., Hakkert, J. C., Beekwilder, J., et al. (2021). Inactivation of the germacrene a synthase genes by CRISPR/Cas9 eliminates the biosynthesis of sesquiterpene lactones in Cichorium intybus l. Plant Biotechnol. J. 19, 2442–2453. doi: 10.1111/pbi.13670
Cankar, K., Hakkert, J. C., Sevenier, R., Campo, E., Schipper, B., Papastolopoulou, C., et al. (2022). CRISPR/Cas9 targeted inactivation of the kauniolide synthase in chicory results in accumulation of costunolide and its conjugates in taproots. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.940003
Cankar, K., Van Houwelingen, A., Bosch, D., Sonke, T., Bouwmeester, H., Beekwilder, J. (2011). A chicory cytochrome P450 mono-oxygenase CYP71AV8 for the oxidation of (+)-valencene. FEBS Lett. 585, 178–182. doi: 10.1016/j.febslet.2010.11.040
Catania, T. M., Branigan, C. A., Stawniak, N., Hodson, J., Harvey, D., Larson, T. R., et al. (2018). Silencing amorpha-4,11-diene synthase genes in Artemisia annua leads to FPP accumulation. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00547
Chadwick, M., Trewin, H., Gawthrop, F., Wagstaff, C. (2013). Sesquiterpenoids lactones: benefits to plants and people. Int. J. Mol. Sci. 14, 12780–12805. doi: 10.3390/ijms140612780
Chi, Z.-M., Zhang, T., Cao, T.-S., Liu, X.-Y., Cui, W., Zhao, C.-H. (2011). Biotechnological potential of inulin for bioprocesses. Bioresour Technol. 102, 4295–4303. doi: 10.1016/j.biortech.2010.12.086
Coppola, M., Cascone, P., Bossi, S., Corrado, G., Garonna, A. P., Maffei, M., et al. (2018). TPS genes silencing alters constitutive indirect and direct defense in tomato. Int. J. Mol. Sci. 19, 2748. doi: 10.3390/ijms19092748
D'antuono, L. F., Ferioli, F., Manco, M. A. (2016). The impact of sesquiterpene lactones and phenolics on sensory attributes: an investigation of a curly endive and escarole germplasm collection. Food Chem. 199, 238–245. doi: 10.1016/j.foodchem.2015.12.002
De Bruyn, C., Ruttink, T., Eeckhaut, T., Jacobs, T., De Keyser, E., Goossens, A., et al. (2020). Establishment of CRISPR/Cas9 genome editing in witloof (Cichorium intybus var. foliosum). Front. Genome Ed. 2. doi: 10.3389/fgeed.2020.604876
De Keyser, E., Desmet, L., Losschaert, M., De Riek, J. (2020). A general protocol for accurate gene expression analysis in plants. Methods Mol. Biol. 2065, 105–118. doi: 10.1007/978-1-4939-9833-3_9
De Kraker, J.-W., Franssen, M. C. R., Dalm, M. C. F., De Groot, A., Bouwmeester, H. J. (2001). Biosynthesis of germacrene a carboxylic acid in chicory roots. demonstration of a cytochrome P450 (+)-germacrene a hydroxylase and NADP+-dependent sesquiterpenoid dehydrogenase(s) involved in sesquiterpene lactone biosynthesis. Plant Physiol. 125, 1930–1940. doi: 10.1104/pp.125.4.1930
De Kraker, J.-W., Franssen, M. C. R., De Groot, A., KoüNig, W. A., Bouwmeester, H. J. (1998). (+)-germacrene a biosynthesis: the committed step in the biosynthesis of bitter sesquiterpene lactones in chicory. Plant Physiol. 117, 1381–1392. doi: 10.1104/pp.117.4.1381
De Kraker, J.-W., Franssen, M. C. R., Joerink, M., De Groot, A., Bouwmeester, H. J. (2002). Biosynthesis of costunolide, dihydrocostunolide, and leucodin. demonstration of cytochrome p450-catalyzed formation of the lactone ring present in sesquiterpene lactones of chicory. Plant Physiol. 129, 257–268. doi: 10.1104/pp.010957
Delporte, M., Legrand, G., Hilbert, J.-L., Gagneul, D. (2015). Selection and validation of reference genes for quantitative real-time PCR analysis of gene expression in Cichorium intybus. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00651
Drewnowski, A., Gomez-Carneros, C. (2000). Bitter taste, phytonutrients, and the consumer: a review. Am. J. Clin. Nutr. 72, 1424–1435. doi: 10.1093/ajcn/72.6.1424
Ferioli, F., Manco, M. A., D’antuono, L. F. (2015). Variation of sesquiterpene lactones and phenolics in chicory and endive germplasm. J. Food Compos Anal. 39, 77–86. doi: 10.1016/j.jfca.2014.11.014
Foster, J. G., Cassida, K. A., Sanderson, M. A. (2011). Seasonal variation in sesquiterpene lactone concentration and composition of forage chicory (Cichorium intybus l.) cultivars. Grass Forage Sci. 66, 424–433. doi: 10.1111/j.1365-2494.2011.00801.x
François, I. M., Wins, H., Buysens, S., Godts, C., Van Pee, E., Nicolaï, B., et al. (2008). Predicting sensory attributes of different chicory hybrids using physico-chemical measurements and visible/near infrared spectroscopy. Postharvest Biol. Technol. 49, 366–373. doi: 10.1016/j.postharvbio.2008.02.011
Häkkinen, S. T., Soković, M., Nohynek, L., Ćirić, A., Ivanov, M., Stojković, D., et al. (2021). Chicory extracts and sesquiterpene lactones show potent activity against bacterial and fungal pathogens. Pharmaceuticals 14, 941. doi: 10.3390/ph14090941
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F., Vandesompele, J. (2007). qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19. doi: 10.1186/gb-2007-8-2-r19
Ikezawa, N., Göpfert, J. C., Nguyen, D. T., Kim, S.-U., O'maille, P. E., Spring, O., et al. (2011). Lettuce costunolide synthase (CYP71BL2) and its homolog (CYP71BL1) from sunflower catalyze distinct regio- and stereoselective hydroxylations in sesquiterpene lactone metabolism. J. Biol. Chem. 286, 21601–21611. doi: 10.1074/jbc.M110.216804
Ivanescu, B., Miron, A., Corciova, A. (2015). Sesquiterpene lactones from Artemisia genus: biological activities and methods of analysis. J. Anal. Methods Chem. 2015, 247685. doi: 10.1155/2015/247685
Karunanithi, P. S., Zerbe, P. (2019). Terpene synthases as metabolic gatekeepers in the evolution of plant terpenoid chemical diversity. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01166
Kiers, A. M., Mes, T. H. M., van der Meijden, R., Bachmann, K. (2000). A search for diagnostic AFLP markers in Cichorium species with emphasis on endive and chicory cultivar groups. Genome 43, 470–476. doi: 10.1139/g00-024
Kips, L. (2017). Characterization and processing of horticultural byproducts: a case study of tomato and Belgian endive roots (Faculty of Bioscience Engineering, Ghent University, Belgium: PhD-dissertation).
Lacchini, E., Goossens, A. (2020). Combinatorial control of plant specialized metabolism: mechanisms, functions, and consequences. Annu. Rev. Cell Dev. Biol. 36, 291–313. doi: 10.1146/annurev-cellbio-011620-031429
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, Q., Beyraghdar Kashkooli, A., Manzano, D., Pateraki, I., Richard, L., Kolkman, P., et al. (2018). Kauniolide synthase is a P450 with unusual hydroxylation and cyclization-elimination activity. Nat. Commun. 9, 4657. doi: 10.1038/s41467-018-06565-8
Liu, Q., Majdi, M., Cankar, K., Goedbloed, M., Charnikhova, T., Verstappen, F. W. A., et al. (2011). Reconstitution of the costunolide biosynthetic pathway in yeast and Nicotiana benthamiana. PloS One 6, e23255. doi: 10.1371/journal.pone.0023255
Liu, Q., Manzano, D., Tanić, N., Pesic, M., Bankovic, J., Pateraki, I., et al. (2014). Elucidation and in planta reconstitution of the parthenolide biosynthetic pathway. Metab. Eng 23, 145–153. doi: 10.1016/j.ymben.2014.03.005
Luypaert, G., Witters, J., Van Huylenbroeck, J., De Clercq, P., De Riek, J., De Keyser, E. (2017). Induced expression of selected plant defence related genes in pot azalea, Rhododendron simsii hybrid. Euphytica 213, 227. doi: 10.1007/s10681-017-2010-5
Majdi, M., Ashengroph, M., Abdollahi, M. R. (2016). Sesquiterpene lactone engineering in microbial and plant platforms: parthenolide and artemisinin as case studies. Appl. Microbiol. Biotechnol. 100, 1041–1059. doi: 10.1007/s00253-015-7128-6
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Matos, M. S., Anastácio, J. D., Nunes Dos Santos, C. (2021). Sesquiterpene lactones: promising natural compounds to fight inflammation. Pharmaceutics 13, 991. doi: 10.3390/pharmaceutics13070991
Meng, X.-H., Lv, H., Ding, X.-Q., Jian, T.-Y., Feng, X.-J., Ren, B.-R., et al. (2022). Sesquiterpene lactones with anti-inflammatory and cytotoxic activities from the roots of Cichorium intybus. Phytochemistry 203, 113377. doi: 10.1016/j.phytochem.2022.113377
Meyer, D., Blaauwhoed, J.-P. (2009). “Inulin,” in Handbook of hydrocolloids. Eds. Phillips, G. O., Williams, P. A. (New York: CRC Press), 829–848.
Moses, T., Pollier, J., Shen, Q., Soetaert, S., Reed, J., Erffelinck, M.-L., et al. (2015). OSC2 and CYP716A14v2 catalyze the biosynthesis of triterpenoids for the cuticle of aerial organs of Artemisia annua. Plant Cell 27, 286–301. doi: 10.1105/tpc.114.134486
Murashige, T., Skoog, F. (1962). A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant 15, 473–497. doi: 10.1111/j.1399-3054.1962.tb08052.x
Nguyen, T. H., Goossens, A., Lacchini, E. (2022). Jasmonate: a hormone of primary importance for plant metabolism. Curr. Opin. Plant Biol. 67, 102197. doi: 10.1016/j.pbi.2022.102197
Nguyen, D. T., Göpfert, J. C., Ikezawa, N., Macnevin, G., Kathiresan, M., Conrad, J., et al. (2010). Biochemical conservation and evolution of germacrene a oxidase in asteraceae. J. Biol. Chem. 285, 16588–16598. doi: 10.1074/jbc.M110.111757
Padilla-Gonzalez, G. F., Dos Santos, F. A., Da Costa, F. B. (2016). Sesquiterpene lactones: more than protective plant compounds with high toxicity. Crit. Rev. Plant Sci. 35, 18–37. doi: 10.1080/07352689.2016.1145956
Raulier, P., Maudoux, O., Notté, C., Draye, X., Bertin, P. (2016). Exploration of genetic diversity within Cichorium endivia and Cichorium intybus with focus on the gene pool of industrial chicory. Genet. Resour. Crop Evol. 63, 243–259. doi: 10.1007/s10722-015-0244-4
R Core Team (2017). R: a language and environment for statistical computing (Vienna, Austria: Foundation for Statistical Computing). Available at: http://www.R-project.org/.
Reed, J., Stephenson, M. J., Miettinen, K., Brouwer, B., Leveau, A., Brett, P., et al. (2017). A translational synthetic biology platform for rapid access to gram-scale quantities of novel drug-like molecules. Metab. Eng 42, 185–193. doi: 10.1016/j.ymben.2017.06.012
Reyes-Chin-Wo, S., Wang, Z., Yang, X., Kozik, A., Arikit, S., Song, C., et al. (2017). Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nat. Commun. 8, 14953. doi: 10.1038/ncomms14953
Robinson, M. D., Mccarthy, D. J., Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Robinson, M. D., Oshlack, A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. doi: 10.1186/gb-2010-11-3-r25
Rolnik, A., Olas, B. (2021). The plants of the Asteraceae family as agents in the protection of human health. Int. J. Mol. Sci. 22, 3009. doi: 10.3390/ijms22063009
Sainsbury, F., Thuenemann, E. C., Lomonossoff, G. P. (2009). pEAQ: versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol. J. 7, 682–693. doi: 10.1111/j.1467-7652.2009.00434.x
Schaumont, D., Veeckman, E., van der Jeugt, F., Haegeman, A., Van Glabeke, S., Bawin, Y., et al. (2022). Stack mapping anchor points (SMAP): a versatile suite of tools for read-backed haplotyping. bioRxiv, 483555. doi: 10.1101/2022.03.10.483555
Schmieder, R., Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Sessa, R. A., Bennett, M. H., Lewis, M. J., Mansfield, J. W., Beale, M. H. (2000). Metabolite profiling of sesquiterpene lactones from Lactuca species: major latex components are novel oxalate and sulfate conjugates of lactucin and its derivatives. J. Biol. Chem. 275, 26877–26884. doi: 10.1074/jbc.M000244200
Sgamma, T., Pape, J., Massiah, A., Jackson, S. (2016). Selection of reference genes for diurnal and developmental time-course real-time PCR expression analyses in lettuce. Plant Methods 12, 21. doi: 10.1186/s13007-016-0121-y
Sterck, L., Billiau, K., Abeel, T., Rouzé, P., Van De Peer, Y. (2012). ORCAE: online resource for community annotation of eukaryotes. Nat. Methods 9, 1041. doi: 10.1038/nmeth.2242
Testone, G., Mele, G., Di Giacomo, E., Gonnella, M., Renna, M., Tenore, G. C., et al. (2016). Insights into the sesquiterpenoid pathway by metabolic profiling and de novo transcriptome assembly of stem-chicory (Cichorium intybus cultigroup “Catalogna”). Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01676
Van Beek, T. A., Maas, P., King, B. M., Leclercq, E., Voragen, A. G. J., De Groot, A. (1990). Bitter sesquiterpene lactones from chicory roots. J. Agric. Food Chem. 38, 1035–1038. doi: 10.1021/jf00094a026
Van Bel, M., Diels, T., Vancaester, E., Kreft, L., Botzki, A., Van de Peer, Y., et al. (2018). PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 46, D1190–D1196. doi: 10.1093/nar/gkx1002
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034–research0034.0011. doi: 10.1186/gb-2002-3-7-research0034
Waegneer, E., Rombauts, S. M., Baert, J., Dauchot, N., De Keyser, A., Eeckhaut, T., et al. (2023). Industrial chicory genome gives insight into the molecular time table of anther development and male sterility. Front. Plant Sci. 14, 1181529. doi: 10.3389/fpls.2023.1181529
Wasternack, C., Strnad, M. (2019). Jasmonates are signals in the biosynthesis of secondary metabolites [[/amp]]mdash; pathways, transcription factors and applied aspects [[/amp]]mdash; a brief review. New Biotechnol. 48, 1–11. doi: 10.1016/j.nbt.2017.09.007
Xu, J., Wang, X.-Y., Guo, W.-Z. (2015). The cytochrome P450 superfamily: key players in plant development and defense. J. Integr. Agric. 14, 1673–1686. doi: 10.1016/S2095-3119(14)60980-1
Yanagisawa, T., Misaka, T. (2021). Characterization of the human bitter taste receptor response to sesquiterpene lactones from edible asteraceae species and suppression of bitterness through ph control. ACS Omega 6, 4401–4407. doi: 10.1021/acsomega.0c05599
Yu, Z.-X., Li, J.-X., Yang, C.-Q., Hu, W.-L., Wang, L.-J., Chen, X.-Y. (2012). The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua l. Mol. Plant 5, 353–365. doi: 10.1093/mp/ssr087
Keywords: chicory, sesquiterpene lactones, guaianolides, cytochrome P450, terpene synthase, jasmonate, Nicotiana benthamiana, CRISPR/Cas9 genome editing
Citation: De Bruyn C, Ruttink T, Lacchini E, Rombauts S, Haegeman A, De Keyser E, Van Poucke C, Desmet S, Jacobs TB, Eeckhaut T, Goossens A and Van Laere K (2023) Identification and characterization of CYP71 subclade cytochrome P450 enzymes involved in the biosynthesis of bitterness compounds in Cichorium intybus. Front. Plant Sci. 14:1200253. doi: 10.3389/fpls.2023.1200253
Received: 04 April 2023; Accepted: 06 June 2023;
Published: 22 June 2023.
Edited by:
Chang-Jun Liu, Brookhaven National Laboratory (DOE), United StatesReviewed by:
Pan Liao, Hong Kong Baptist University, Hong Kong SAR, ChinaCopyright © 2023 De Bruyn, Ruttink, Lacchini, Rombauts, Haegeman, De Keyser, Van Poucke, Desmet, Jacobs, Eeckhaut, Goossens and Van Laere. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alain Goossens, YWxhaW4uZ29vc3NlbnNAcHNiLXZpYi51Z2VudC5iZQ==; Katrijn Van Laere, a2F0cmlqbi52YW5sYWVyZUBpbHZvLnZsYWFuZGVyZW4uYmU=; Charlotte De Bruyn, Y2hhcmxvdHRlLWRlLWJydXluQGhvdG1haWwuY29t
†These authors have contributed equally to this work and share last and senior authorship
‡ORCID: Charlotte De Bruyn, orcid.org/0000-0002-7979-3079
Tom Ruttink, orcid.org/0000-0002-1012-9399
Elia Lacchini, orcid.org/0000-0002-1598-8950
Stephane Rombauts, orcid.org/0000-0002-3985-4981
Annelies Haegeman, orcid.org/0000-0002-8192-5368
Ellen De Keyser, orcid.org/0000-0002-6171-1311
Christof Van Poucke, orcid.org/0000-0001-7566-925X
Sandrien Desmet, orcid.org/0000-0002-0957-708X
Thomas B. Jacobs, orcid.org/0000-0002-5408-492X
Tom Eeckhaut, orcid.org/0000-0001-5473-0314
Alain Goossens, orcid.org/0000-0002-1599-551X
Katrijn Van Laere, orcid.org/0000-0001-5398-5489
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.