 Naama Geraffi1‡
Naama Geraffi1‡ Priya Gupta
Priya Gupta Naama Wagner
Naama Wagner Isaac Barash
Isaac Barash Tal Pupko
Tal Pupko Guido Sessa
Guido Sessa
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 31 July 2023
Sec. Plant Pathogen Interactions
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1198160
This article is part of the Research Topic Molecular Interactions Between Bacterial Pathogens and Plants: Selected Contributions to the 14th International Conference on Plant Pathogenic Bacteria (14th ICPPB) View all 10 articles
Acquisition of the pathogenicity plasmid pPATH that encodes a type III secretion system (T3SS) and effectors (T3Es) has likely led to the transition of a non-pathogenic bacterium into the tumorigenic pathogen Pantoea agglomerans. P. agglomerans pv. gypsophilae (Pag) forms galls on gypsophila (Gypsophila paniculata) and triggers immunity on sugar beet (Beta vulgaris), while P. agglomerans pv. betae (Pab) causes galls on both gypsophila and sugar beet. Draft sequences of the Pag and Pab genomes were previously generated using the MiSeq Illumina technology and used to determine partial T3E inventories of Pab and Pag. Here, we fully assembled the Pab and Pag genomes following sequencing with PacBio technology and carried out a comparative sequence analysis of the Pab and Pag pathogenicity plasmids pPATHpag and pPATHpab. Assembly of Pab and Pag genomes revealed a ~4 Mbp chromosome with a 55% GC content, and three and four plasmids in Pab and Pag, respectively. pPATHpag and pPATHpab share 97% identity within a 74% coverage, and a similar GC content (51%); they are ~156 kb and ~131 kb in size and consist of 198 and 155 coding sequences (CDSs), respectively. In both plasmids, we confirmed the presence of highly similar gene clusters encoding a T3SS, as well as auxin and cytokinins biosynthetic enzymes. Three putative novel T3Es were identified in Pab and one in Pag. Among T3SS-associated proteins encoded by Pag and Pab, we identified two novel chaperons of the ShcV and CesT families that are present in both pathovars with high similarity. We also identified insertion sequences (ISs) and transposons (Tns) that may have contributed to the evolution of the two pathovars. These include seven shared IS elements, and three ISs and two transposons unique to Pab. Finally, comparative sequence analysis revealed plasmid regions and CDSs that are present only in pPATHpab or in pPATHpag. The high similarity and common features of the pPATH plasmids support the hypothesis that the two strains recently evolved into host-specific pathogens.
Pantoea agglomerans is a Gram‐negative facultative anaerobic bacterium of the Erwineaceae family (Adeolu et al., 2016). It is widespread in nature and found in association with many plant species as an epiphyte and endophyte (Manulis and Barash, 2003; Sulja et al., 2022). Strains of P. agglomerans have evolved into tumorigenic pathogens displaying host specificity on various plants by acquiring a pathogenicity plasmid, which is designated as pPATH. Two P. agglomerans pathogenic pathovars can be distinguished: P. agglomerans pv. gypsophilae (Pag), which induces galls on gypsophila and triggers an immune response on sugar beet, and P. agglomerans pv. betae (Pab), which causes galls on both beet and gypsophila (Weinthal et al., 2007; Barash and Manulis-Sasson, 2009). Pathogenicity of both pathovars is dependent on a type III secretion system (T3SS) and effectors (T3Es), and on auxin and cytokinins biosynthetic pathways that are all encoded in the pathogenicity plasmids pPATHpag, in Pag, and pPATHpab, in Pab (Manulis and Barash, 2003). The extensively characterized pPATHpag plasmid has a size of ~131 kb and contains a pathogenicity island (PAI) of ~75 kb that harbors genes encoding T3SS structural, regulatory and effector proteins, and plasmid maintenance determinants, and carries multiple insertion sequences (IS) (Lichter et al., 1996; Guo et al., 2002; Weinthal et al., 2007). The PAI structure, composition and location on the plasmid support a recent evolution of pathogenesis (Manulis and Barash, 2003).
The inventory of T3Es in Pab and Pag bacteria was previously determined based on draft genome sequences in combination with a machine-learning approach and translocation assays into beet roots, where eight and nine plasmid-borne effectors were identified in Pab and Pag strains, respectively (Nissan et al., 2018). Five of them (DspA/E, HopX2, HopAY1, HopAF1, and HrpK) are in common between Pag and Pab, and shared with other phytopathogenic bacteria (Lindeberg et al., 2005; Petnicki-Ocwieja et al., 2005; Boureau et al., 2006; Washington et al., 2016; Saint-Vincent et al., 2020). HopD1 was also reported in other bacteria (Block et al., 2014) but it is only present in Pag. Conversely, four T3Es (HsvB, HsvG, PthG and PseB) were only identified in Pag and Pab strains. HsvG and HsvB are putative transcription factors which may contribute to host specificity in gypsophila and beet, respectively (Valinsky et al., 1998; Nissan et al., 2006; Nissan et al., 2012). PthG is present only in Pag and triggers an immune response in beet species, while PseB is present only in Pab and its function is still unknown (Nissan et al., 2018). The small repertoire and plasmid location of T3Es in the two pathovars are consistent with recent evolution of P. agglomerans pathogenesis and limited functional redundancy between effectors. Remarkably, transformation of HsvG and PthG or HsvB and PseB was found to convert nonpathogenic bacteria into host-specific gall-forming pathogens on gypsophila and beet, respectively (Nissan et al., 2019).
Draft genome sequences of the Pab 4188 and Pag 824-1 strains were previously generated using MiSeq second-generation sequencing technology and partially assembled into 79 and 55 contigs for Pab and Pag, respectively (Nissan et al., 2018). In this study, we employed Pacific Biosciences (PacBio) third-generation sequencing technology, which provides longer reads than MiSeq (Bachall, 2009), to sequence and completely assemble the Pab and Pag genomes. Comparative sequence analysis of the newly assembled pPATHpag and pPATHpab pathogenicity plasmids identified common and unique genes involved in plasmid housekeeping and bacterial virulence that may have shaped the evolution of the Pag and Pab pathogenic pathovars.
The bacterial strains used are Pantoea agglomerans pv. betae strain 4188 (Pab) (Burr et al., 1991) and Pantoea agglomerans pv. gypsophilae strain 824-1 (Pag) (Manulis et al., 1991). These strains were grown at 28°C in Lysogeny Broth (LB) medium supplemented with Rifampicin (100 µg/ml). The same strains were sequenced before (Nissan et al., 2018). The strains used for both sequencing efforts (MiSeq, PacBio) were drawn from the same stock, which was kept frozen in -80°C. Thus, it is unlikely that mutations have accumulated between the two sequencing efforts.
Bacteria were grown overnight in LB liquid medium, and bacterial genomic DNA was isolated as described by Chen and Kuo (1993). The DNA was sent to Macrogen (Seoul, South Korea) for sequencing. PacBio/single-molecule real-time (SMRT) sequencing was used to sequence the genome of the Pag and Pab strains. Samples were prepared according to standard instructions for SMRTbell templates for sequencing on the PacBio RS System, and were sequenced using SMRT® sequencing. In Pag, the sequencing yielded 82,397 reads (692,414,886 read bases). The read N50 was 12,640 bp and the average read length was 8,748 bp. In Pab, the sequencing yielded 81,985 reads (706,785,250 read bases). The read N50 was 12,688 bp and the average read length was 8,906 bp.
The PacBio reads were used to complete the assembly of the bacterial genomes. These reads are long, and thus allow achieving longer contigs, and in bacterial genomes even the full chromosome sequence. Nevertheless, they are prone to more errors than Illumina reads. In order to obtain a more accurate assembly, previously published draft genome sequences from MiSeq data (Nissan et al., 2018) were used to correct the assembly done using PacBio reads. The PacBio reads were used as input to Canu v1.7 (Koren et al., 2017) to generate the draft complete assembly, with the following parameters: -pacbio-raw corMhapSensitivity=high genomeSize= 5m. The average coverage was assessed by mapping corrected and trimmed reads obtained by Canu v1.7 against the assembly using BWA v0.7.17 (Li and Durbin, 2009; Li and Durbin, 2010), calculating the alignment depth using SAMtools v1.3.3 (Danecek et al., 2021), and the average depth per molecule using awk. Next, we used Circlator (Hunt et al., 2015) to convert the linear contigs into a circular sequence. To run Circlator, the following additional programs were used: BWA v0.7.17 (Li and Durbin, 2009; Li and Durbin, 2010), Prodigal v2.6.3 (Hyatt et al., 2010), Canu v1.7 (Koren et al., 2017), SAMtools v1.3.3 (Danecek et al., 2021), and MUMmer v3.23 (Kurtz et al., 2004). Following this step, we used the abovementioned Illumina reads to polish the assembly using Pilon v1.22 (Walker et al., 2014). To this end, we mapped the Illumina reads to the draft genome using BWA v0.7.17 (Li and Durbin, 2009; Li and Durbin, 2010), converted the output SAM file to BAM file and sorted it using SAMtools v1.3.3 (Danecek et al., 2021), and finally used it to correct the assembly using Pilon with the default parameters values and including –changes to keep track of the corrections done in the assembly. We repeated this process until no further corrections were introduced to the assembly. Two and three rounds were required to fully correct Pab and Pag assemblies, respectively. The average coverage of the Illumina reads was assessed in the same manner as assessed for the PacBio reads.
Genomes were annotated using two different programs: (i) Prokka v1.13.3 (Seemann, 2014) with default parameter values; (ii) RAST: a webserver that was used with default settings (Aziz et al., 2008; Overbeek et al., 2014; Brettin et al., 2015). Finally, ISFinder (Siguier et al., 2006) was used to find and locate ISs and Tns in the plasmids. Whole genome alignment was performed using Mugsy-1.2.2. (Angiuoli and Salzberg, 2011). CDSs were aligned using Emboss Needle global alignment (Rice et al., 2000).
Draft genome sequences of the Pab 4188 and Pag 824-1 strains (~5 Mb) (NCBI accession no. ASM166202v1 and ASM166198v1) were previously generated by MiSeq second-generation sequencing technologies and partially assembled into 79 contigs for Pab and 55 for Pag (Nissan et al., 2018). In this study, PacBio third-generation sequencing technology, which provides longer reads than MiSeq (Bachall, 2009), was employed to sequence the Pab and Pag genomes. The newly sequenced data (NCBI accession no.: ASM166202v2 and ASM166198v2), as well as the previously sequenced MiSeq sequencing data, were used to assemble the genome, aiming that the short-read data would correct errors introduced to the assemblies using the long-read data. Both the sequencing data and the final assemblies were deposited to NCBI and can be found under BioProject PRJNA320975.
Assembly of the Pab and Pag PacBio reads revealed four and five circular contigs respectively, representing the chromosome for each strain, three plasmids for Pab, and four plasmids for Pag (Table 1). The chromosomes have a similar length of ~4 Mb with a 55% GC content and each consists of ~4,000 CDSs. Among the plasmids, the previously identified pPATH pathogenicity plasmids Pab and Pag (pPATHpab and pPATHpag; Manulis and Barash, 2003), have a length of ~156 kb and ~131 kb, respectively, a 51% GC content, and consist of 163 and 138 CDSs, respectively. Two other homologous plasmids were identified in the two pathovars: Plasmid 02 with a length of ∼540 kb in Pab and ∼580 kb in Pag, and Plasmid 03 with a length of ~180 kb in Pab and ∼140 kb in Pag. Plasmid 02 and 03 have a GC content ranging between 52% and 54%, and they consist of ~600 and ~200 CDSs, respectively. An additional ~79 kb plasmid, Plasmid 04, was detected in Pag. It has a 52% GC content and consists of ~80 CDSs. In a BLASTn search, Plasmid 04 was found to be homologous to plasmid pAR1aD of the P. agglomerans strain AR1a (accession no. CP059087) with 67% coverage and 99.8% identity, and to the pEM02 plasmid of Erwinia spp. (accession no. LN907829) with 44% coverage and 98% identity.
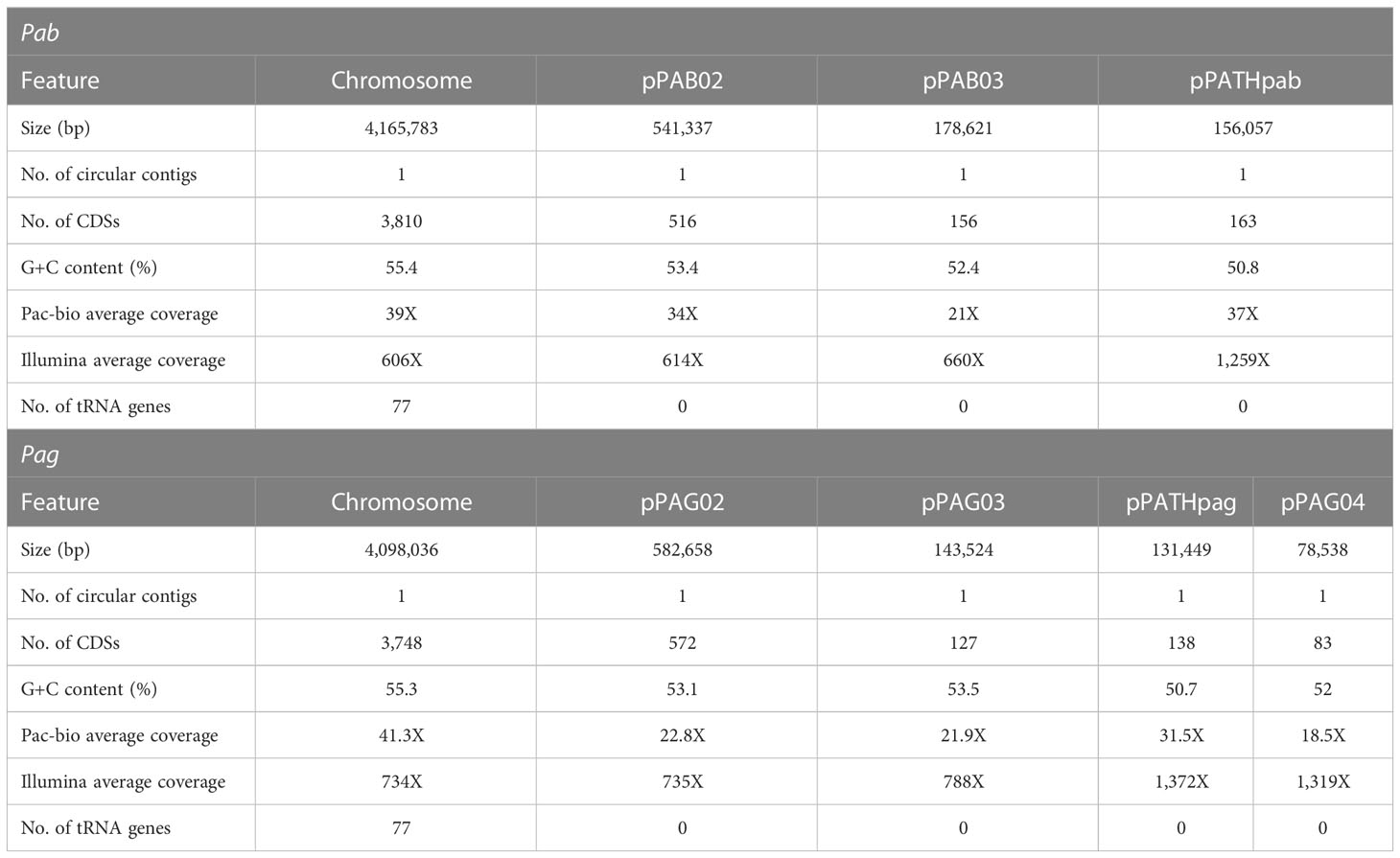
Table 1 Features of Pab and Pag genomes following sequencing with PacBio, assembly with Canu, polishing with Pilon, and annotation with PGAP.
Next, detailed comparative analysis was carried out for proteins encoded in the pPATHpab and pPATHpag pathogenicity plasmids (Barash and Manulis-Sasson, 2009). This analysis detected proteins that are involved in plasmid housekeeping and plant pathogenicity, including proteins required for plasmid maintenance, structural and regulatory proteins of the T3SS, T3Es, Type 3 chaperons (T3Cs), harpins, and enzymes of biosynthetic pathways of plant growth hormones. Homologous proteins encoded in the pPATHpab and pPATHpag plasmids were compared and their closest homolog in other bacteria was determined. The obtained data were used to generate an updated map of pPATHpag (Figure 1) and the first map of pPATHpab (Figure 2).
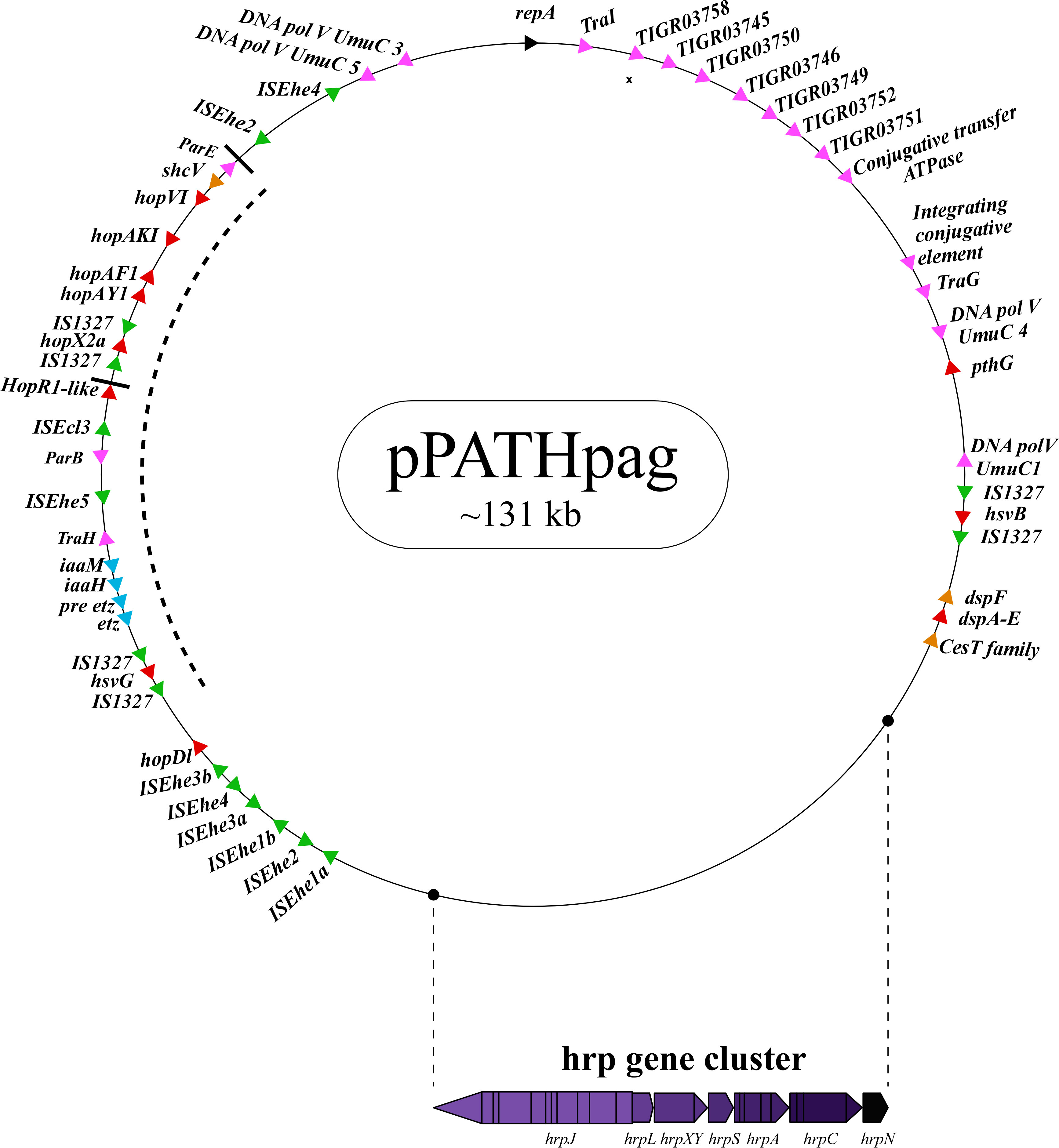
Figure 1 Schematic representation of the pPATHpag plasmid. The plasmid contains the hrp gene cluster, genes encoding validated and putative T3SS effector proteins (red), a gene cluster encoding indole-3-acetic acid (IAA) and cytokinins (CK) biosynthetic genes (blue), insertion sequence (IS) elements (green), plasmid maintenance genes (pink), T3C (orange) and the repA gene (black). Arrows indicate gene orientation. A cluster of effector genes is marked by thick bars, and an inversion fragment by a dotted line.
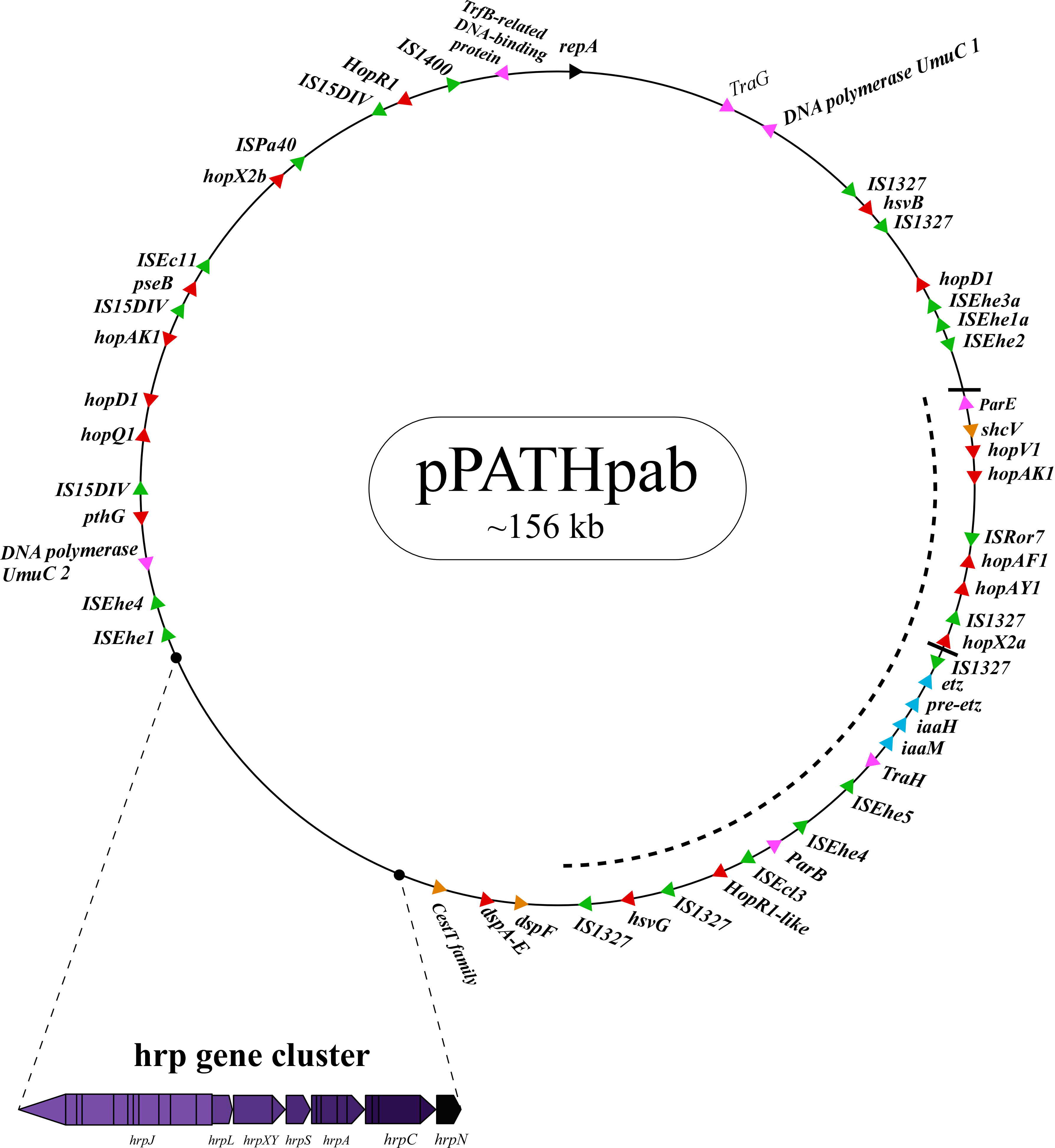
Figure 2 Schematic representation of the pPATHpab plasmid. The plasmid contains the hrp\hrc gene cluster, genes encoding validated and putative T3SS effector proteins (red), a gene cluster encoding indole-3-acetic acid (IAA) and cytokinins (CK) biosynthetic genes (blue), insertion sequence (IS) elements (green), plasmid maintenance genes (pink), T3C (orange) and the repA gene (black). Arrows indicate gene orientation. A cluster of effector genes is marked by thick bars, and an inversion fragment by a dotted line.
To examine if other bacterial strains have plasmids with similar structures as observed in pPATHpab and pPATHpag, we conducted a small-scale comparative genomics analysis with publicly available P. agglomerans genomes. Specifically, we searched for genes related to the T3SS, which in Pantoea are known to be encoded on plasmids. We have blasted (tblastn) the protein sequences of the T3SS regulators and components listed in Tables 2, 3 versus all the fully assembled genomes of P. agglomerans available in NCBI. To consider the presence of each of the components, an E-value lower than 10-10 and percentage of identical matches higher than 50% were required. Interestingly, a full cluster was found in one genome – P. agglomerans strain DAPP-PG734, on plasmid P2. This cluster was not identified in any other genome. In addition to the T3SS, we also searched for the effectors HsvB and HsvG using tblastn. These effectors were found only on pPATH plasmid of Pag and Pab. These results suggest that the presence of a T3SS and associated effectors is a derived state that characterizes a few specific strains rather than an ancestral state that characterizes the entire P. agglomerans species.
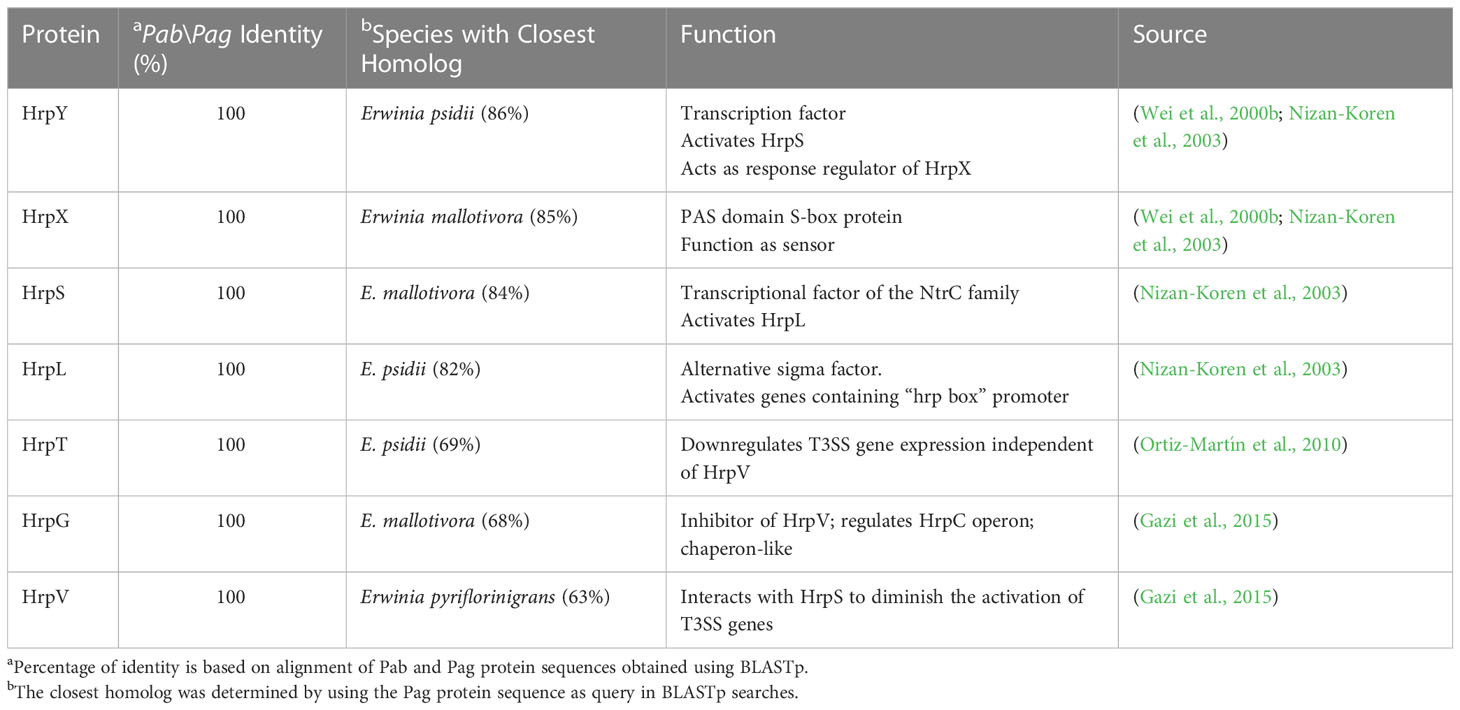
Table 2 Pab and Pag regulatory Hrp proteins.
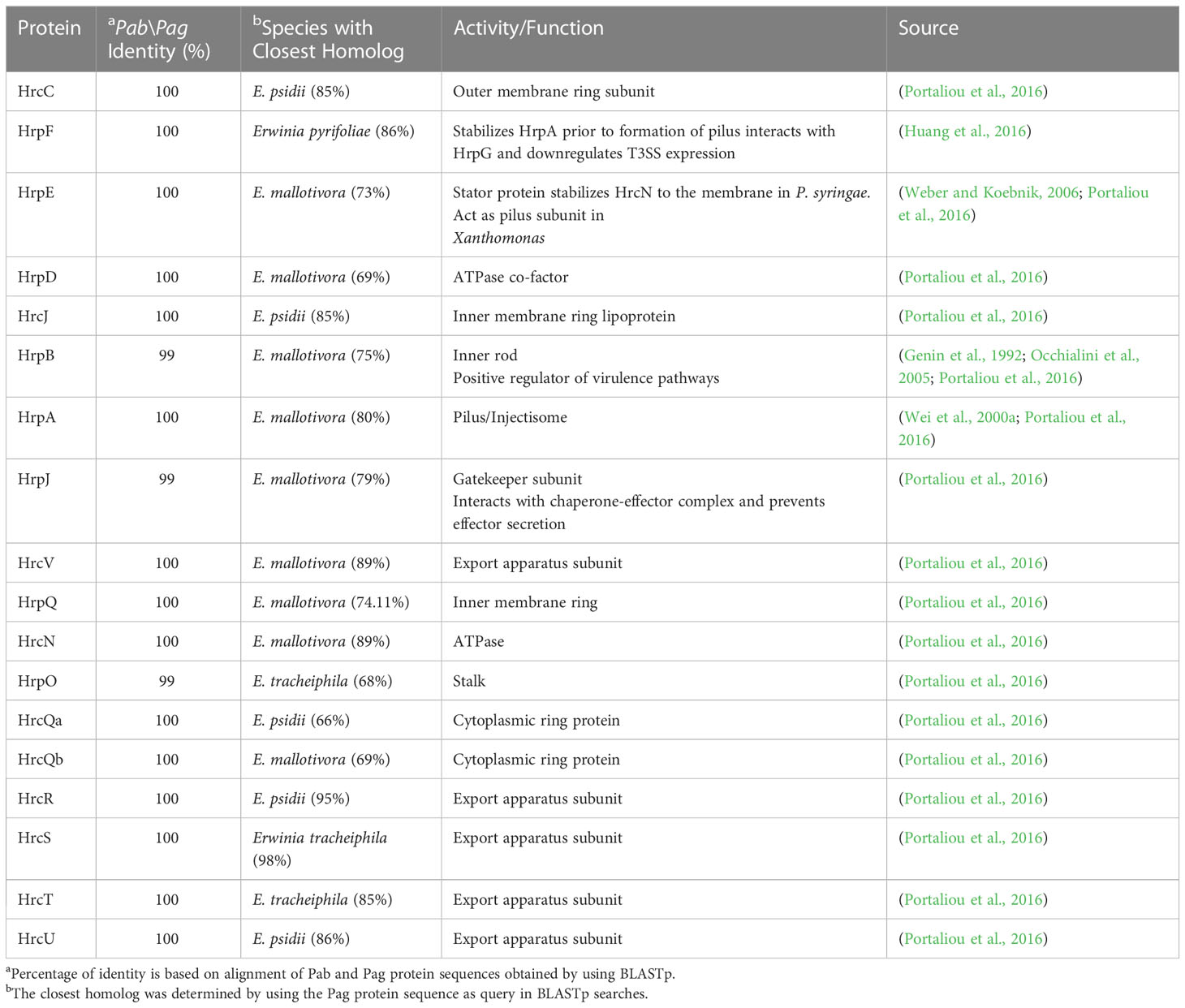
Table 3 Pab and Pag structural Hrp\Hrc proteins.
Proteins involved in plasmid maintenance, replication, and transfer were found to be encoded in the pPATHpab and pPATHpag plasmids (Table 4). Within this group of proteins is RepA that in Pseudomonas was shown to initiate plasmid replication by binding to the origin of replication (Díaz-López et al., 2003; Weinthal et al., 2007), which is yet to be determined in pPATHpab and pPATHpag. Several proteins of the partition system that assures equal segregation of chromosome and plasmids were also detected (Bignell and Thomas, 2001; Funnell, 2016). We also found a transcriptional repressor and toxin which are involved in plasmid maintenance. These include a TrfB-related DNA binding protein (transcriptional repressor of genes involved in plasmid inheritance) (Thomas and Smith, 1986), and ParA and ParB partitioning proteins (Bignell and Thomas, 2001; Funnell, 2016). The ParE toxin is predicted to inhibit DNA gyrase to stop replication during stress conditions (Jiang et al., 2002), and to be involved in plasmid maintenance via post segregation killing of plasmid free daughter cells using toxin antitoxin systems (Engelberg-Kulka and Glaser, 1999). ParA is only present in pPATHpag, while ParB and ParE are present in both pathovars. In addition, we found two and four copies of polymerase V in Pab and Pag, respectively. Polymerase V participates in DNA repair (Wang, 2001). Interestingly, proteins involved in the conjugative transfer of integrative conjugative elements (ICEs) are present in pPATHpag. ICEs are self-transmissible mobile genetic elements that encode the machinery for conjugation, as well as regulatory systems to control their excision and conjugative transfer (Baltrus et al., 2022; Daveri et al., 2023). They include six integrating conjugative element proteins, a conjugal transfer protein, a conjugal transfer lipoprotein, and a conjugative transfer ATPase. In addition, we found two proteins, TraH (contains an ATP binding motif) and TraG (NTPase), encoded in pPATH of both pathovars, whose homologs in other bacteria participate in pilus synthesis and assembly (Zatyka and Thomas, 1998). Finally, a TraI domain containing protein, which plays a putative function as DNA helicase/relaxase, was detected in pPATHpag in the proximity of RepA and can be a part of relaxosome that facilitates plasmid transfer (Matson and Ragonese, 2005).
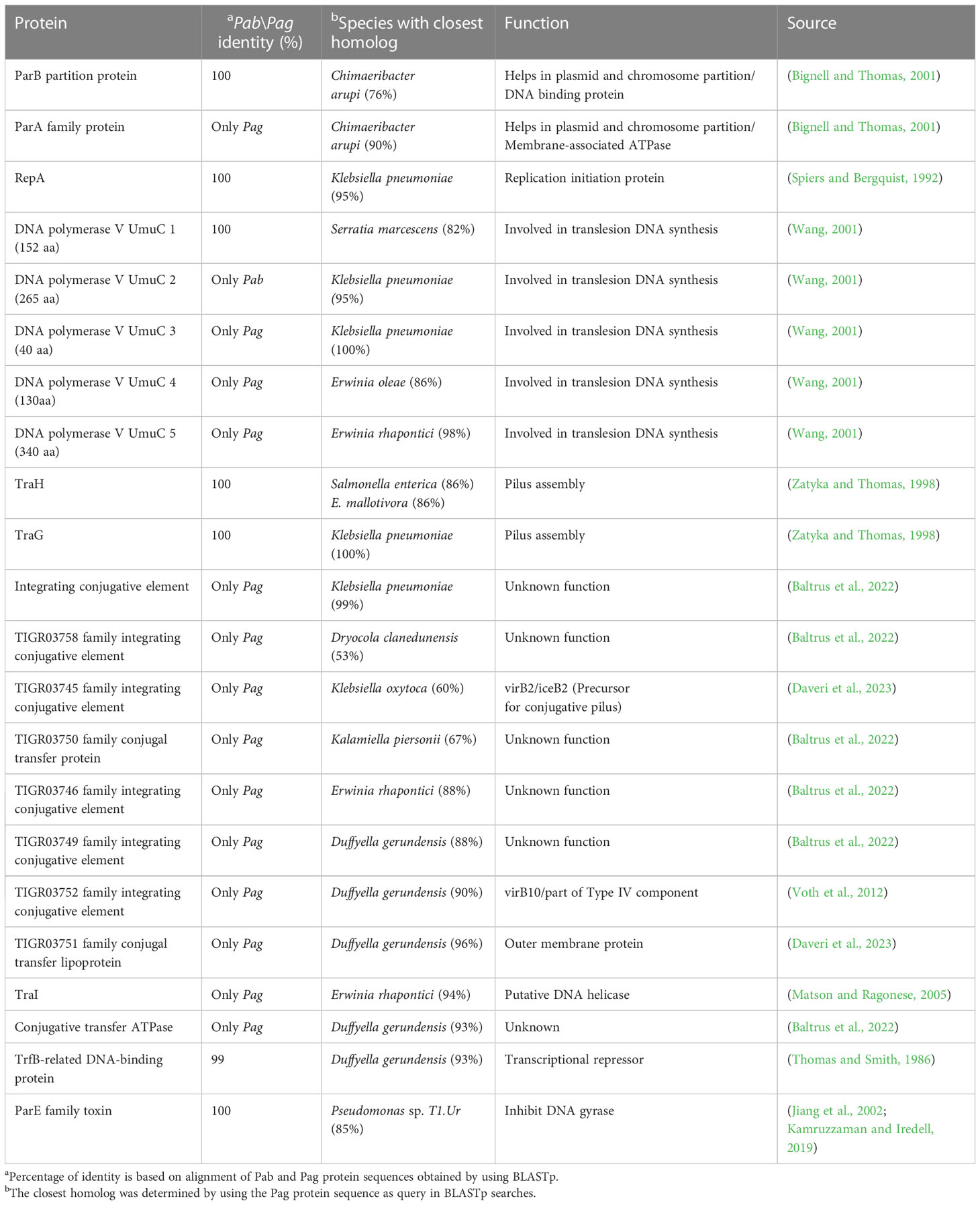
Table 4 Housekeeping proteins.
The T3SS is a syringe-like structure that delivers effector proteins inside the plant cell (Galán and Collmer, 1999). It is a complex of proteins encoded by hrp (hypersensitive response and pathogenicity) and hrc (hypersensitive response and conserved) genes (Alfano and Collmer, 1997). Structure and function of the Pag T3SS were extensively characterized in previous studies (Nizan et al., 1997; Mor et al., 2001). Here, we identified and compared structural and regulatory T3SS proteins of pPATHpab and pPATHpag and determined their closest homologs in other bacteria (Tables 2, 3). All Hrp\Hrc proteins are identical in the two pathovars. They display 63%-98% sequence similarity to proteins in different Erwinia spp., most commonly Erwinia mallotivora and Erwinia psidii. As schematically shown in Figure 3, the hrp\hrc gene cluster consists of four operons: hrpJ, hrpA, hrpC, hrpXY, and three single genes: hrpL, hrpS and hrpN. The genetic arrangement of these operons was found to be the same as in Pag (Mor et al., 2001). Operons hrpJ, hrpA and hrpC mainly encode T3SS structural components, while the hrpXY operon encodes regulatory proteins (Tables 2, 3). The hrpJ operon is the largest and consists of 11 genes (hrpJ, hrcV, hrpQ, hrcN, hrpO, hrcQa, hrcQb, hrcR, hrcS, hrcT and hrcU), all encoding structural proteins of the T3SS basal body, except HrcN which is an ATPase and HrpJ that acts as a gatekeeper protein that regulates translocator and effector secretion (Portaliou et al., 2016). hrpA is a smaller operon and consists of five genes (hrpA, hrpB, hrcJ, hrpD and hrpE) encoding pilus/injectisome (HrpA, HrpB and HrcJ) components, an ATPase cofactor (HrpD) and a stator protein (HrpE) to stabilize HrcN. The last structural operon is hrpC consisting of five genes (hrpF, hrpG, hrcC, hrpT and hrpV) with different functions. Homologs of hrpT, hrpV and hrpG in Pseudomonas syringae and Erwinia amylovora were shown to have a regulatory role (Ortiz-Martín et al., 2010; Gazi et al., 2015). They act in concert to control hrp/hrc gene expression which should be coupled with the assembly and function of the T3SS under inducing condition (Ortiz-Martín et al., 2010). The hrpXY two-gene operon together with hrpS and hrpL is responsible for regulation of T3SS genes that contain a hrp box in their promoter (Tables 2, 3).
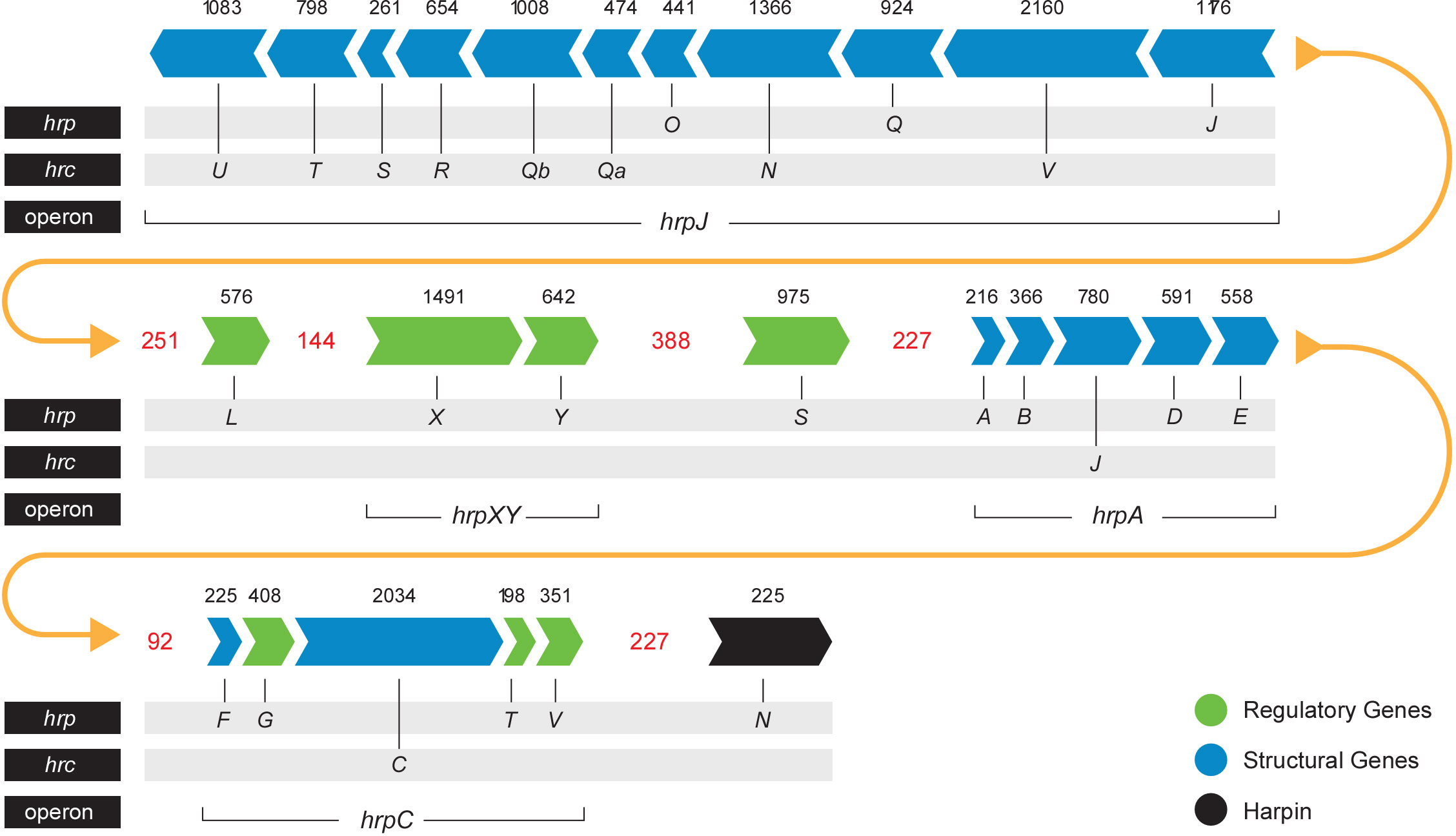
Figure 3 Pab and Pag hrp\hrc gene cluster. Arrows indicate gene orientation. Black numbers denote the gene size (base pairs). Red numbers denote the distance (base pairs) between operons\genes. Yellow arrows represent the continuity of the gene cluster.
T3Es are secreted through the T3SS directly inside the plant cell and manipulate host cellular processes to promote bacterial growth in the apoplast (Macho, 2016). Previous reports identified nine effectors in Pag (HsvG, HsvB, DspA/E, HopAY1, HopX2, HopAF1, HrpK, PthG, and HopD1) and eight in Pab (HsvG, HsvB, PseB, DspA/E, HopAY1, HopX2, HopAF1 and HrpK) (Nissan et al., 2018). Truncated forms of PthG and HopD1 were found in Pab, and a truncated HopAY1 was found in Pag. In addition, homologs of HopV1 and HopR1 effectors, which are known to be functional in other bacteria (Wei et al., 2007), were found, but their translocation was not assessed by secretion assays (Nissan et al., 2018). Notably, in our analysis, HrpK was not retrieved in any of the pathovar assemblies. The composition of the Pab and Pag T3E pools was further refined by the identification of three new candidate effectors in pPATHpab and one in pPATHpag. One of these candidate T3Es was named HopR1-like, based on its similarity to HopR1 of Pab (47%). HopR1-like is present and identical in the two pathovars and represents a new member of the AvrE-family of T3Es displaying 96% similarity to a transducer protein in E. psidii (Table 5). Two additional newly identified candidate effectors present in Pab are HopQ1 and HopX2b. HopQ1 displays high sequence similarity (98%) to HopQ1 of P. syringae pv. tomato DC3000 and to XopQ of Xanthomonas euvesicatoria (63%) (Giska et al., 2013; Teper et al., 2014). HopX2a (previously reported as HopX2) is present in both pathovars. Another CDS (HopX2b) which is more closely related (98%) to HopX2 of P. syringae, was found only in Pab (Table 5). HopX2b displays 70% identity to HopX2a and they both belong to XopE/AvrPphe family. However, translocation of HopR1-like, HopX2b and HopQ1 into plant cells and their contribution to bacterial virulence is yet to be determined.
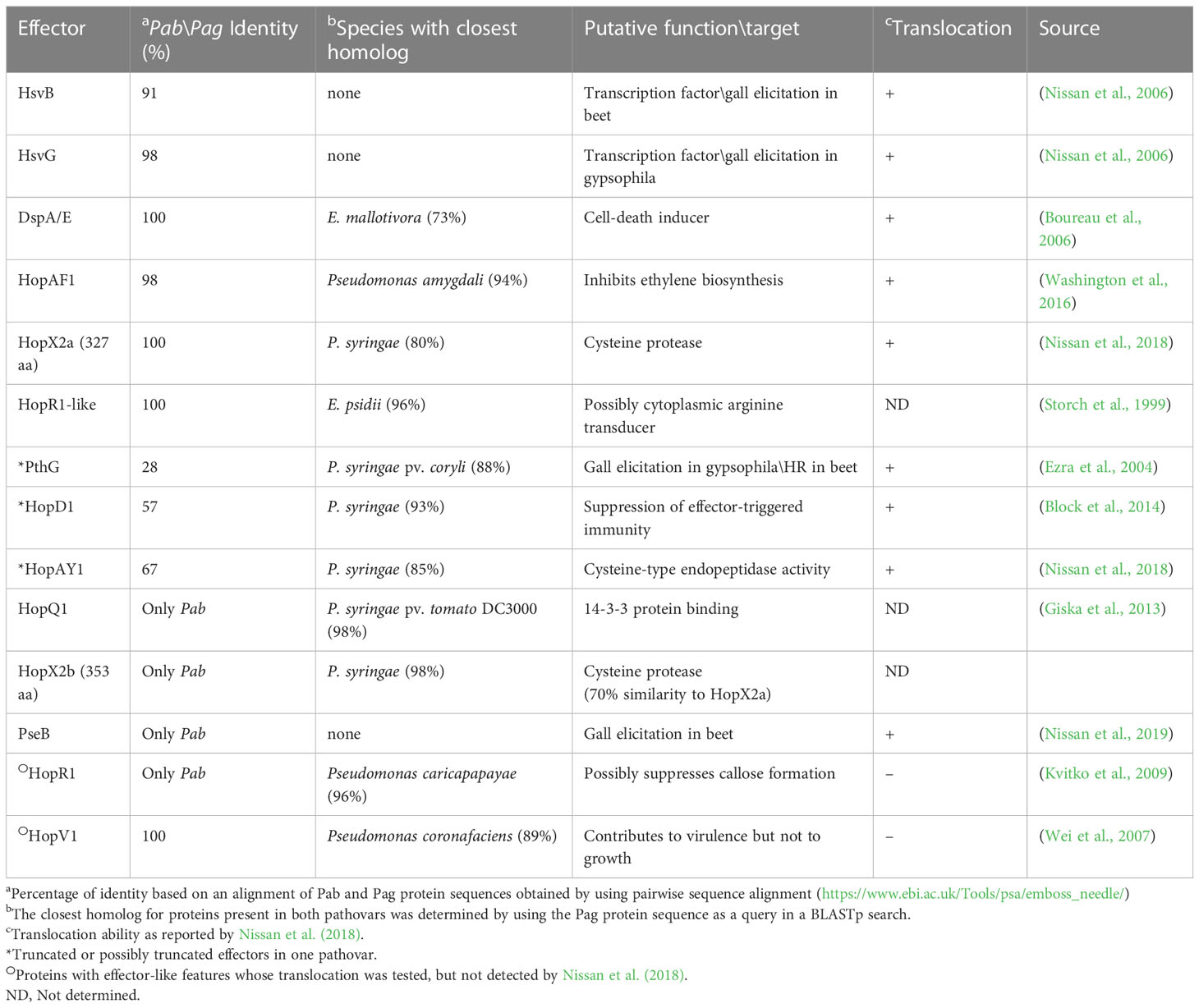
Table 5 Pab and Pag T3Es.
T3Es of the two pathovars, either in full-length or truncated, display a high degree of sequence similarity (91%-100%). The majority of them display high sequence similarity to effectors of other bacteria, mainly of Pseudomonas spp. (73%-97%) (Table 5). Remarkably, in this study we found a homolog in P. syringae pv. coryli (87.91%) for PthG that, along with HsvB, HsvG and PseB, has not been previously detected in any other bacteria. Most of the effector genes are distributed throughout the pPATHpab and pPATHpag plasmids, with the exception of a gene cluster including the HopV1, HopAF1, HopAY1, and HopX2a effector genes, and the HopAKI harpin (Figures 1, 2). Putative functions of the effectors are listed in Table 5.
Harpins represent a class of proteins secreted through the T3SS that facilitate translocation of T3Es into plant cells (Li et al., 2019a). Our analysis confirmed the presence of the previously reported harpins HrpN and HopAK1 in both pathovars (Nissan et al., 2018), and identified an additional homolog of HopAK1 in pPATHpab (HopAK1-1). Sequence comparison revealed that HrpN of Pab and Pag are almost identical (99%), while HopAK1 homologs of the two pathovars display 91% similarity. Closest homologs of HopAK1 and HrpN were found in P. syringae (86%) and E. psidii (65%), respectively. In terms of location of the genes within the pPATH plasmids, hrpN is at the edge of the hrp\hrc cluster in both pathovars, while hopAK1 is located within a cluster of effector genes (Figures 1, 2). Pab hopAK1-1 encodes a harpin, which has its closest homolog in P. syringae (64%) and is located upstream of the PseB effector gene.
T3Cs are small (15-20 kDa), cytoplasmic, and acidic proteins that play roles in T3Es secretion, such as prevention of T3E premature aggregation and cytoplasmic proteolysis (Lohou et al., 2013). Our analysis detected three T3Cs that are encoded in both pPATH plasmids: DspF, ShcV and CesT (Table 6). DspF was previously reported to be present in Pag (Mor et al., 2001) and shares relatively high sequence similarity to DspF of E. piriflorinigrans (74%). In E. amylovora it was shown to facilitate translocation of the DspA/E T3E by interacting with its N-terminus through a predicted β-sheet helix-binding groove (Gaudriault et al., 2002; Triplett et al., 2009). ShcV displays the highest similarity to its Pseudomonas coronafaciens homolog (88%). ShcV was reported to interact with and assist the translocation of HopPtoV effector in P. syringae. This effector-chaperon interaction is also supported by the genomic location of these two proteins: the CDS for the ShcV T3C and the HopPtoV T3E are adjacent to each other (Wehling et al., 2004). In pPATHpab and pPATHpag, ShcV and DspF are encoded by CDS adjacent to the HopV1 and DspA/E T3E genes, respectively (Figures 1, 2) in support of the hypothesis that they play a function as chaperones of the encoded T3Es. An additional T3C encoded in both Pab and Pag is a member of the CesT family of chaperons that were shown to assist in the recruitment of multiple T3Es to the T3SS (Thomas et al., 2005). It shares a relatively low sequence similarity to a protein in E. psidii (53%) and its location upstream to the DspA/E CDS suggests its involvement in folding and\or secretion of this effector.

Table 6 Type III chaperones.
Galls formation may be caused by interference of the bacteria with the hormone balance of the plant, in particular with the ratio between auxin and cytokinin concentrations. We identified four plant hormone biosynthetic genes (iaaM, iaaH, etz and pre-etz) in both the pPATH plasmids, as previously reported for pPATHpag (Table 7) (Lichter et al., 1995; Manulis et al., 1998). IaaM and IaaH are enzymes participating in auxin synthesis through the indole-3-acetamide pathway (Morris, 1986). The operon for cytokinin biosynthesis consists of two genes: pre-etz and etz. The function of pre-etz is unknown, while etz encodes the enzyme isopentenyl transferase (Guo et al., 2001). The similarity of these genes in the two pathovars is high (96%-97%), and all enzymes, except pre-Etz, are very similar to homologs in Erwinia spp. No putative homologs have been found for pre-Etz. All four genes are clustered together in the pPATHpag and pPATHpab plasmids (Figures 1, 2).

Table 7 Biosynthetic enzymes of plant hormones.
TEs, including ISs and Tns, are major determinants in the evolution of pathogenic bacteria (Siguier et al., 2014; Nicolas et al., 2015). Tns differ from ISs because in addition to the transposase, they carry passenger/cargo genes, which are not involved in catalysis or regulation of the TE movement (Siguier et al., 2014). ISs belong to diverse families and groups based on the type of transposase, number of CDSs, size, conserved terminal base pairs at the end, number of base pairs present in direct repeats produced at the target site after transposition, and mechanism of transposition (Mahillon and Chandler, 1998). Previous studies detected the presence of ISEhe1, ISEhe2, ISEhe3, ISEhe4, ISEhe5, IS1327 (six copies) in pPATHpag (Lichter et al., 1996; Guo et al., 2002). Information about the presence of ISs and Tns in Pab was not reported earlier.
In this study, we used the ISFinder tool to retrieve TE sequences in pPATHpab and pPATHpag, and sequences with the highest significance were analyzed for their location in the plasmid and number of copies. This analysis identified ten types of ISs and two Tns (ISPa40 and ISRor7) in pPATHpab and seven ISs in pPATHpag. Seven of all the identified ISs are common to both pathovars (ISEhe1, ISEhe2, ISEhe3, ISEhe4, ISEhe5, IS1327, ISEcl3) and belong to diverse IS families: IS1, IS3, IS5, IS6 and IS630. Exclusively present in Pab are the ISs IS1400, ISEcl1 and IS15DIV and the Tns ISRor7 and ISPa40. In Pag we found an additional copy of ISEhe2 and ISEhe4, and new IS ISEcl3 (Table 8). The presence of such diverse ISs indicates massive horizontal gene transfer (HGT) (Barash and Manulis-Sasson, 2009).
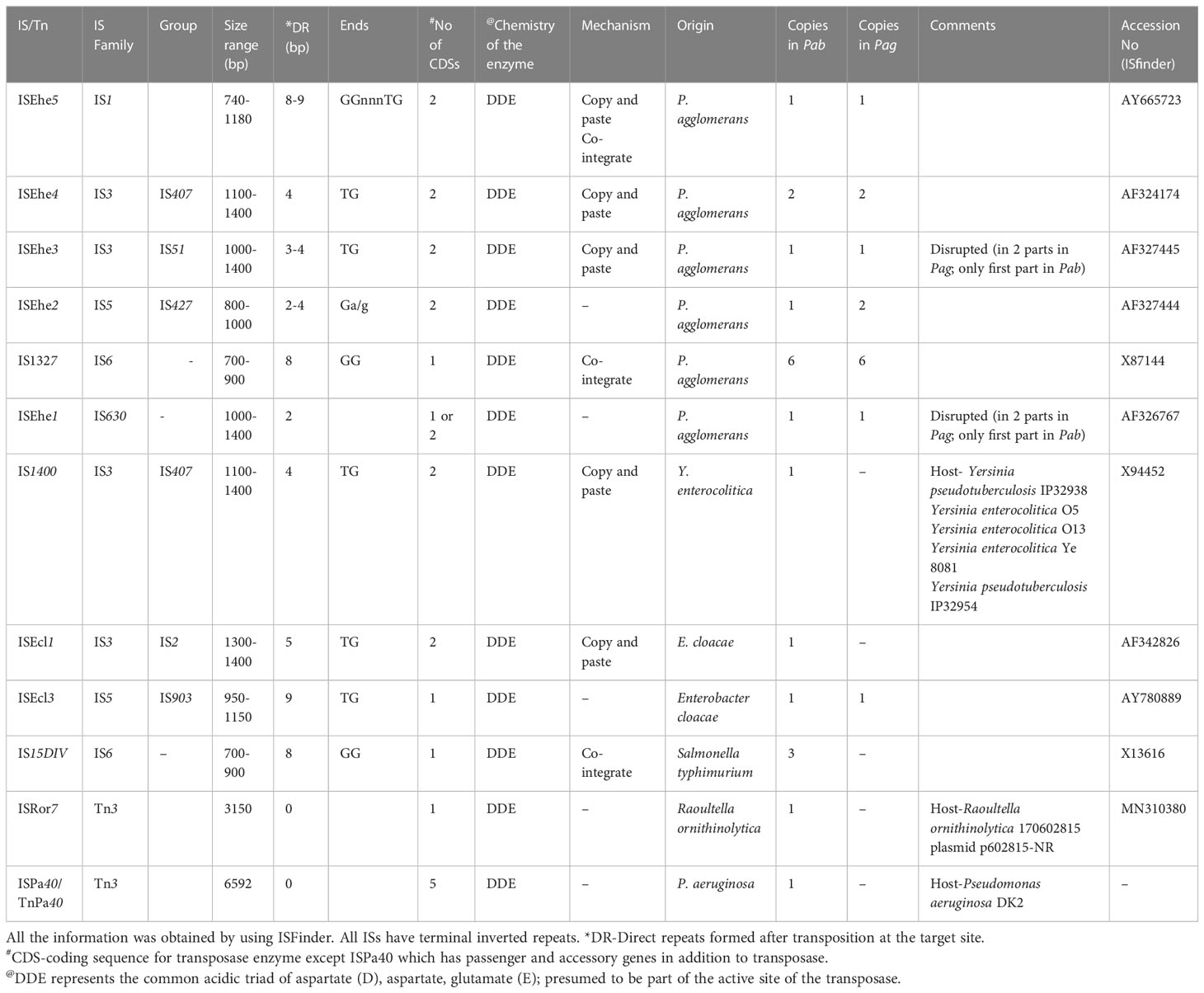
Table 8 Insertional sequences and transposons present in pPATHpab and pPATHpag.
Homologs of ISPa40, IS1400 and IS15DIV were found in plant and animal pathogenic bacteria, such as P. aeruginosa, Yersinia enterocolitica and Salmonella typhimurium. In contrast, ISEhe1, 3, 5 and IS1327, display very little similarity to proteins in other bacteria (Lichter et al., 1996; Guo et al., 2002). As previously reported by Guo et al. (2002), ISEhe3 and ISEhe1 are separated into two parts in Pag due to the insertion of ISEhe4 and ISEhe2, respectively. Conversely in Pab, we found only the first fragment of ISEhe3 and ISEhe1. It is likely that the second fragment of ISEhe3 and ISEhe1 has been lost during evolution of pPATHpab (Figures 1, 2). All ISs are quite dispersed throughout the pPATH plasmid in both pathovars, though in pPATHpag there is a typical clustering of ISEhe1-4 downstream to the T3SS cluster (Figures 1, 2).
To investigate differences between pPATHpab and pPATHpag, we aligned the two plasmids and analyzed the CDSs in all the unaligned fragments. Sequences that are present in pPATHpag, but not in pPATHpab, are indicated in Figure 4 (fragments 1-5). Three unique sequences are located downstream to the repA gene: the first (~15 kb) contains a CDS encoding an antirestriction protein, DUF1281 with unknown function, six integrating conjugative element protein, TIGR03750 family conjugal transfer protein, and TIGR03751 family conjugal transfer lipoprotein (Figure 4, fragment 1). The second fragment (261 bp) encodes a 3’-5’ exonuclease (Figure 4, fragment 2). The third one (1,524 bp) contains a CDS that encodes a polymerase V (Figure 4, fragment 3). Fragment 4 (2,131 bp) contains CDSs encoding two ISs (ISEhe2 and ISEhe4), a membrane-associated ATPase (ParA family protein), and two polymerase V (Figure 4). Fragment 5 of the unique area (5,930 bp) ends closely to repA. It contains seven CDSs that are unique to Pag and encode: MFS (major facilitator superfamily) transporter, which has a role in resistance to toxic compounds, epoxide hydrolase, DUF1697, a protein containing a MEKHLA domain, tyrosine recombinase XerC that is involved in transposition, polymerase V, and peptidase (Figure 4). The vast majority of these CDSs display high sequence similarity to genes present in genomes of Erwinia spp. (85%-96%) (Table 9).
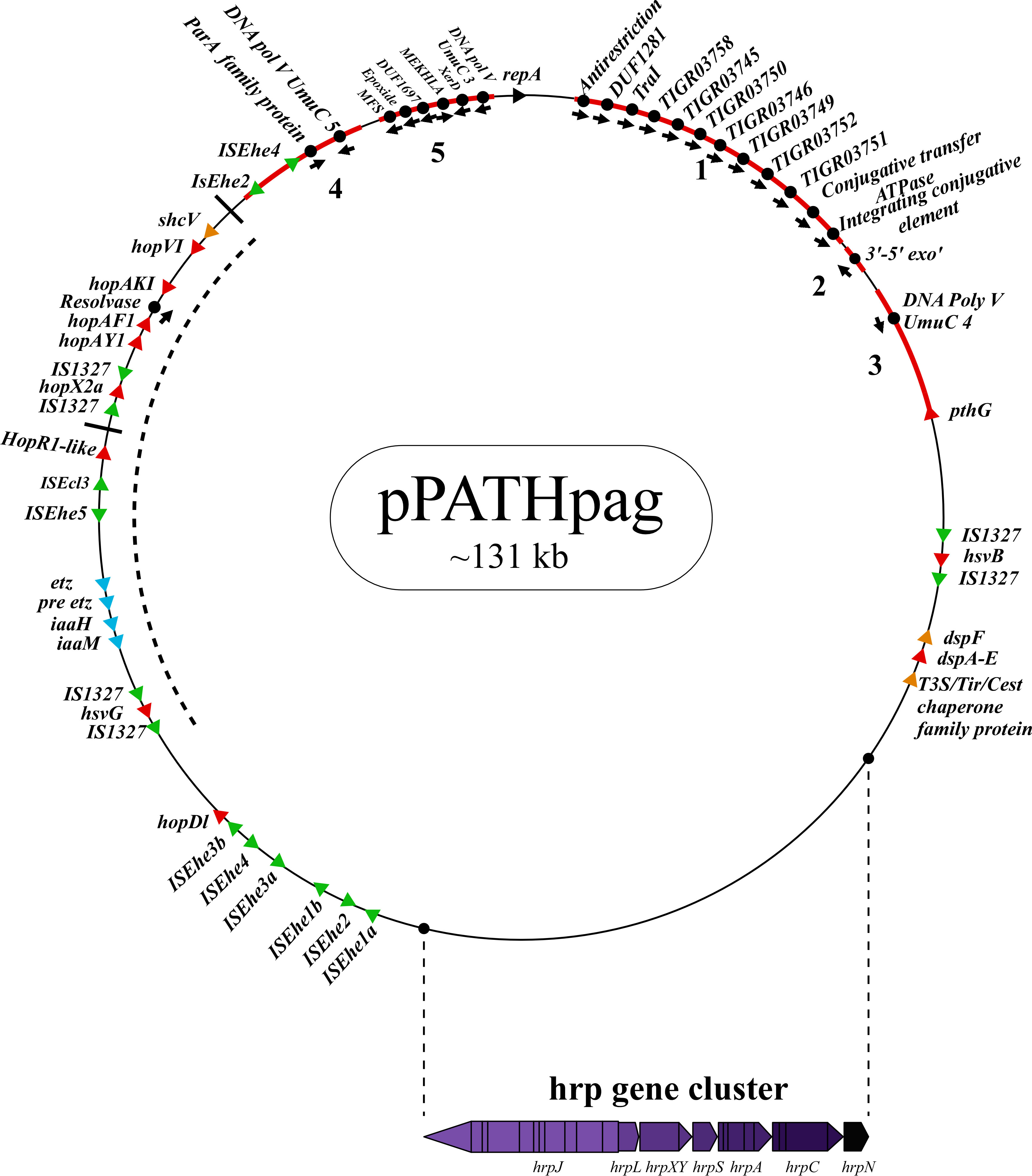
Figure 4 Unique sequences of the pPATHpag plasmid. Sequences that are present in pPATHpag, but not in pPATHpab, are marked in red and numbered from 1 to 5. Genes located on these unique sequences and present in pPATHpag, but not in pPATHpab, are indicated with black dots. Color code for arrows for rest of the genes are similar to Figure 1.
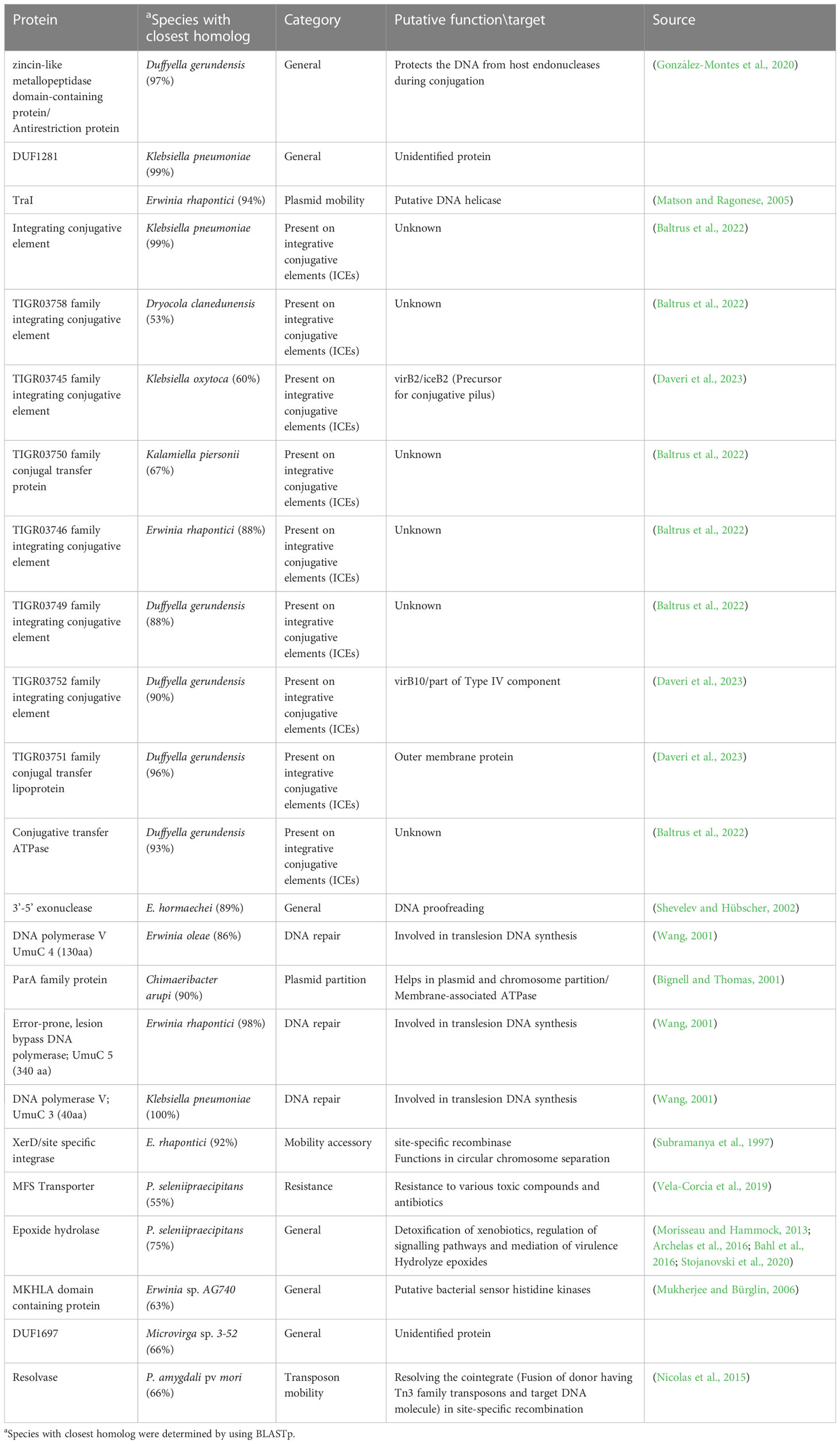
Table 9 Unique CDS in Pag.
Sequences that are present in pPATHpab but not in pPATHpag are indicated in Figure 5. There are two consecutive unique sequences in the first ~16 kb downstream to repA (Figure 5, fragments 1 and 2). Fragment 1 contains four CDSs encoding ArdC-like ssDNA-binding domain-containing protein/DUF1738 (antirestriction protein), STY4534 family ICE replication protein/DUF3577 (unknown function), DUF4160 (unknown function) and a pilL protein involved in pilus assembly. Fragment 2 encodes a resolvase I gene involved in recombination processes, and an ATPase gene involved in the zeta toxin\antitoxin system. An additional unique sequence of ~5,400 bp is located within the T3E cluster (Figure 4, fragment 3); it includes a resolvase II gene and the transposon ISRor7, which belongs to the Tn3 family. There is also a ~20 kb unique sequence upstream to repA (Figure 5, fragment 4) that includes a copy of HopD1 and HopAKI, and CDSs encoding four effectors present only in pPATHpab (HopQ1, PseB, HopX2b and HopR1). In addition, throughout this region, there are the ISs IS15DIV (three copies), ISEcl1, IS1400 and the Tn ISPa40. Other unique CDSs within fragment 4 encode proteins involved in type II toxin\antitoxin two components system, which enhances bacteria fitness, antibiotics resistance and maintenance. A CDS next to the truncated PthG encoding polymerase is also unique to Pab (Table 10).
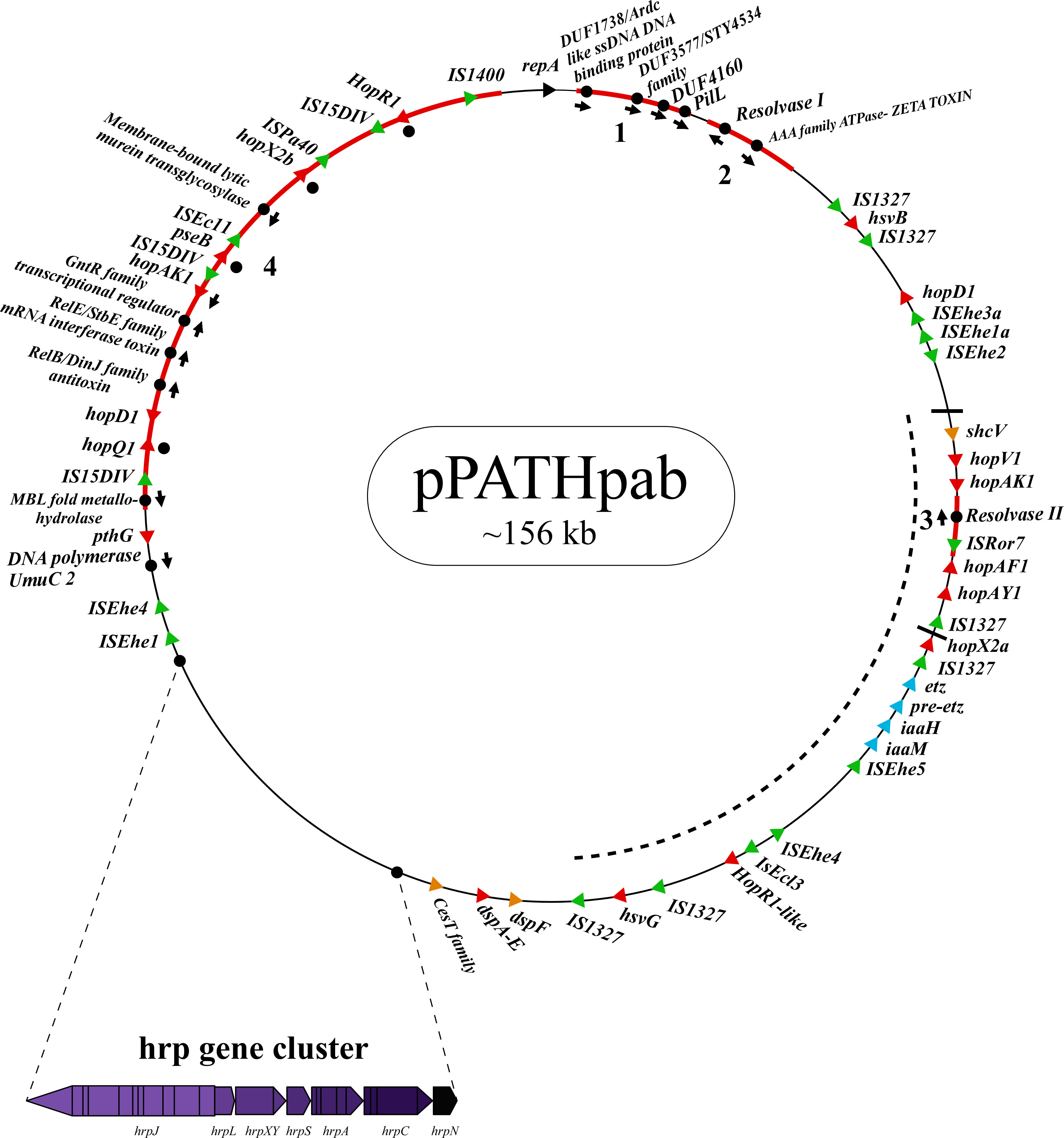
Figure 5 Unique sequences of the pPATHpab plasmid. Sequences that are present in pPATHpab, but not in pPATHpag, are marked in red and numbered from 1 to 4. Genes located on these unique sequences and present in pPATHpab, but not in pPATHpag, are indicated with black dots. Color code for arrows for rest of the genes are similar to Figure 2.
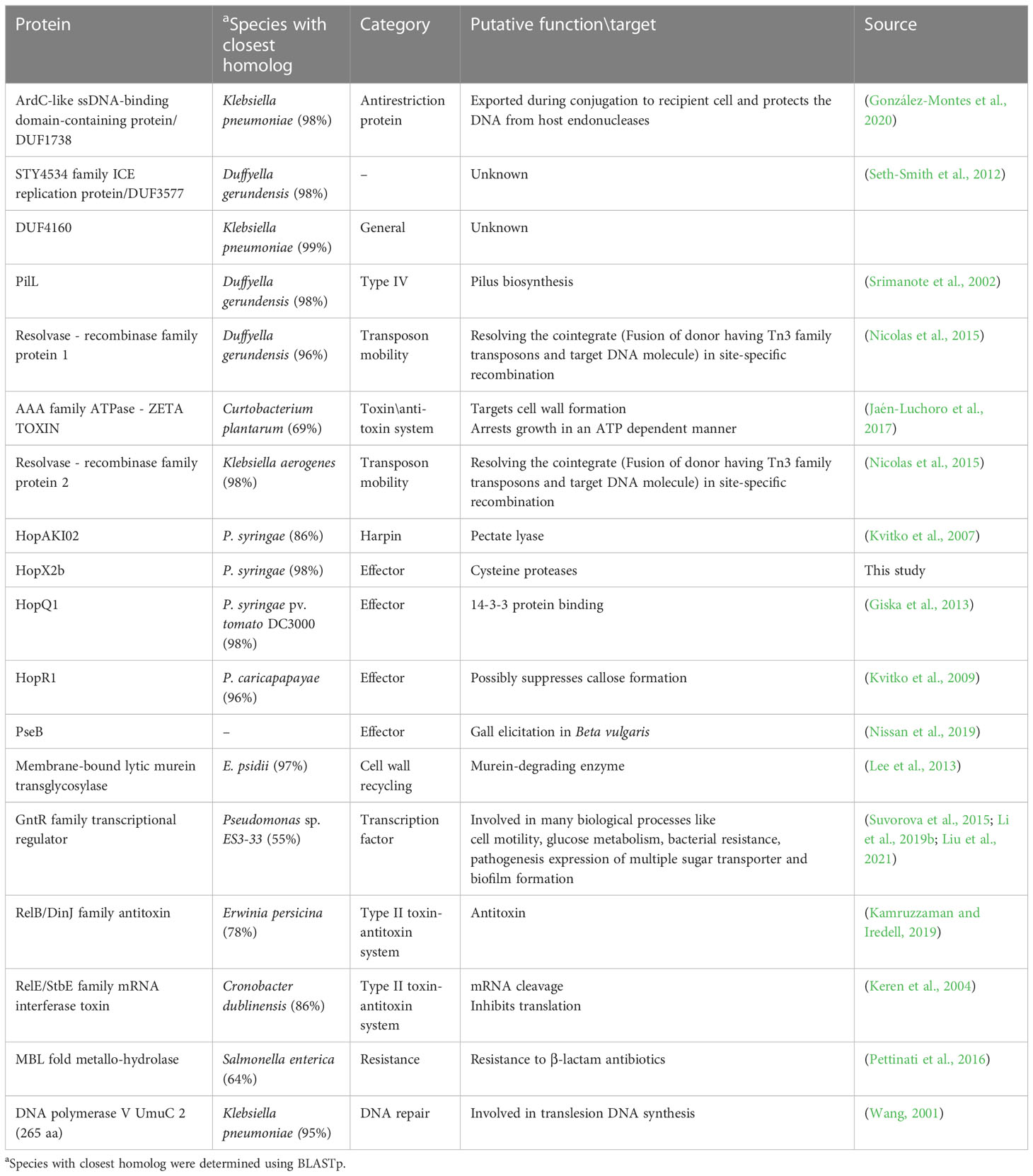
Table 10 Unique CDS in Pab.
Sequencing of the Pag and Pab genomes by PacBio technology allowed their complete assembly and disclosed the structure and composition of the pPATHpab and pPATHpag pathogenicity plasmids. Sequence analysis of pPATHpab and pPATHpag allows to formulate hypotheses about their evolutionary origin. The high similarity (97%) between pPATHpab and pPATHpag supports the notion that these plasmids evolved from a common ancestor plasmid. CDSs of the ~20 kb hrp\hrc gene cluster, which is highly conserved in Pab and Pag (>99% identity), display high similarity to hrp\hrc genes of Erwinia spp. This suggests that the ancestor P. agglomerans strain, which were possibly non-pathogenic, may have acquired the hrp\hrc gene cluster from a pathogenic Erwinia strain and thereby turned into a new pathogenic strain. In support of this hypothesis, copies of the T3E DspA/E are located in Pag and Pab at the edge of the hrp\hrc cluster, as similarly observed in Erwinia spp. (Siamer et al., 2011), and it is likely that DspA/E has been transferred from Erwinia to P. agglomerans along with the hrp\hrc cluster. Recently, a T3SS has also been reported in endophytic P. agglomerans DAPP‐PG 734 and, P. agglomerans BAV 2934 but it is distantly related to Pab and Pag T3SS suggesting different origin of T3SS in different Pantoea strains (Moretti et al., 2021; Sulja et al., 2022). It is possible that pPATH was introduced into a P. agglomerans population by a conjugative or mobilizable plasmid. P. agglomerans may have acquired the entire pPATH plasmid or the PAI was incorporated in a pre-existing plasmid (Barash and Manulis-Sasson, 2009). In either one of these cases, horizontal gene transfer (HGT) appears as a major evolutionary force that drove pPATH generation. Large mobile elements, such as Tns and ISs, are key players in HGT (Nicolas et al., 2015). The wide genetic interchange between P. agglomerans and other bacterial strains manifests itself in the large repertoire of IS elements occurring in pPATHpab and pPATHpag, and in the presence of T3E genes common to other phytopathogenic bacteria, and particularly widespread among P. syringae pathovars (Guo et al., 2002; Manulis and Barash, 2003).
Several lines of evidence indicate that P. agglomerans pathogenic strains are in an early stage of evolution. First, P. agglomerans pathovars have their T3SS gene cluster and effector genes in a plasmid, which suggests that the pPATH plasmids have been acquired recently, and the PAI has not been yet incorporated in the P. agglomerans chromosome, as observed in other pathogens (Hacker et al., 1997). In addition, comparison between corresponding plasmids of the two pathovars revealed a high identity (96%-97%) and similarity coverage (73%-74%) suggesting that pPATH, plasmid 02 and plasmid 03 were all present in the common ancestor strain before its splitting into two distinct pathovars. Finally, the repertoire of T3Es of the two pathovar is limited as compared to other pathogens. Based on our refined analysis, seven effectors are present in both pathovars (HsvG, HsvB, DspA/E, HopX2a, HopAF1, HopV1 and HopR1-like). In addition, HopD1 and PthG are present only in Pag, while PseB, HopQ1, HopAY1, HopR1 and HopX2b are exclusive to Pab and are located in a region of ~20 kb that is unique to pPATHpab (Figure 3).
Introduction of pathoadaptive mutations represents an important mechanism that may contribute to evolution of a new pathogen (Sokurenko et al., 1999; Bartoli et al., 2016). In support of the involvement of pathoadaptive mutations in the evolution of Pag and Pab, truncated variants of T3Es are present in the two pathovars: HopAY1 is truncated in Pag, while PthG and HopD1 are truncated in Pab. These genes acquired mutations that interrupted their CDSs, possibily contributing to the formation of the two distinct pathovars. Truncation of these effectors may have allowed bacteria to escape recognition by newly appeared resistance proteins of the host plant. Generation of PthG in Pag may be the result of pathoadaptive changes that occurred randomly and were preserved due to their beneficial effect. One possible scenario is that Pab evolved from Pag by a genetic modification that resulted in truncation of the PthG CDS and evasion of beet recognition and immunity (Ezra et al., 2000; Manulis and Barash, 2003; Ezra et al., 2004). Typically, these are mutations causing a functional modification or elimination of genes that confer enhanced pathogenicity to the bacteria (Sokurenko et al., 1999).
A 20 kb region which is perfectly mirrored in pPATHpab and pPATHpag is present in the two plasmids. This segment includes the cluster of plant hormone biosynthetic genes, the clustered effector genes, the hsvG gene, and the hopR1-like candidate effector gene. The inversion must have occurred sometime after the splitting into two pathovars and could have happened spontaneously or due to a replication-transcription conflict that resulted in DNA rearrangement (Merrikh et al., 2012; Merrikh and Merrikh, 2018). It has been known that head-on orientation genes can be beneficial to the bacteria due to their high mutation frequency (Merrikh and Merrikh, 2018). Altogether, we conclude that genetic rearrangements and mutations in the ancestor pathogenic plasmid supposedly shaped pPATHpag and pPATHpab resulting in the generation of two pathogenic strains with different host specificities.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, BioProject PRJNA320975.
Conceptualization, GS, IB and TP. Software, TP and NW. Formal Analysis, NG, PG and NW. Investigation, NG and PG. Data Curation, NG and PG. Writing – Original Draft Preparation, NG, PG, GS and IB. Writing – Review and Editing, TP and NW. Supervision, GS, TP and IB. Project Administration, GS. Funding Acquisition, GS and IB. All authors contributed to the article and approved the submitted version.
This research was supported by the Israel Science Foundation (ISF) under grant number 488/19.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adeolu, M., Alnajar, S., Naushad, S., Gupta, R. S. (2016). Genome-based phylogeny and taxonomy of the “Enterobacteriales”: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae. Int. J. Syst. Evol. Microbiol. 66, 5575–5599. doi: 10.1099/IJSEM.0.001485
Alfano, J. R., Collmer, A. (1997). The type III (Hrp) secretion pathway of plant pathogenic bacteria: trafficking harpins, Avr proteins, and death. J. Bacteriol. 179, 5655–5662. doi: 10.1128/jb.179.18.5655-5662.1997
Angiuoli, S. V., Salzberg, S. L. (2011). Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27, 334–342. doi: 10.1093/bioinformatics/btq665
Archelas, A., Iacazio, G., Kotik, M. (2016). “Epoxide hydrolases and their application in organic synthesis,” in Green Biocatalysis, 179–229. doi: 10.1002/9781118828083.ch8
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi: 10.1186/1471-2164-9-75
Bahl, C. D., Hvorecny, K. L., Morisseau, C., Gerber, S. A., Madden, D. R. (2016). Visualizing the mechanism of epoxide hydrolysis by the bacterial virulence enzyme Cif. Biochemistry 55, 788–797. doi: 10.1021/acs.biochem.5b01229
Baltrus, D. A., Feng, Q., Kvitko, B. H. (2022). Genome context influences evolutionary flexibility of nearly identical type III effectors in two phytopathogenic Pseudomonads. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.826365
Barash, I., Manulis-Sasson, S. (2009). Recent evolution of bacterial pathogens: the gall-forming Pantoea agglomerans case. Annu. Rev. Phytopathol. 47, 133–152. doi: 10.1146/annurev-phyto-080508-081803
Bartoli, C., Roux, F., Lamichhane, J. R. (2016). Molecular mechanisms underlying the emergence of bacterial pathogens: an ecological perspective. Mol. Plant Pathol. 17, 303–310. doi: 10.1111/mpp.12284
Bignell, C., Thomas, C. M. (2001). The bacterial ParA-ParB partitioning proteins. J. Biotechnol. 91, 1–34. doi: 10.1016/s0168-1656(01)00293-0
Block, A., Toruño, T. Y., Elowsky, C. G., Zhang, C., Steinbrenner, J., Beynon, J., et al. (2014). The Pseudomonas syringae type III effector HopD1 suppresses effector-triggered immunity, localizes to the endoplasmic reticulum, and targets the Arabidopsis transcription factor NTL9. New Phytol. 201, 1358–1370. doi: 10.1111/nph.12626
Boureau, T., ElMaarouf-Bouteau, H., Garnier, A., Brisset, M.-N., Perino, C., Pucheu, I., et al. (2006). DspA/E, a type III effector essential for Erwinia amylovora pathogenicity and growth in planta, induces cell death in host apple and nonhost tobacco plants. Mol. Plant Microbe Interact. 19, 16–24. doi: 10.1094/MPMI-19-0016
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 58365. doi: 10.1038/srep08365
Burr, T. J., Katz, B. H., Abawi, G. S., Crosier, D. (1991). Comparison of tumorigenic strains of Erwinia herbicola isolated from table beet with E. h. gypsophilae. Plant Dis. 75, 855–858. doi: 10.1094/PD-75-0855
Chen, W., Kuo, T. (1993). A simple and rapid method for the preparation of gram-negative bacterial genomic DNA. Nucleic Acids Res. 212260. doi: 10.1093/nar/21.9.2260
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10, giab008. doi: 10.1093/gigascience/giab008
Daveri, A., Benigno, V., van der Meer, J. R. (2023). Characterization of an atypical but widespread type IV secretion system for transfer of the integrative and conjugative element (ICEclc) in Pseudomonas putida. Nucleic Acids Res. 51, 2345–2362. doi: 10.1093/nar/gkad024
Díaz-López, T., Lages-Gonzalo, M., Serrano-López, A., Alfonso, C., Rivas, G., Díaz-Orejas, R., et al. (2003). Structural changes in RepA, a plasmid replication initiator, upon binding to origin DNA. J. Biol. Chem. 278, 18606–18616. doi: 10.1074/jbc.M212024200
Engelberg-Kulka, H., Glaser, G. (1999). Addiction modules and programmed cell death and antideath in bacterial cultures. Annu. Rev. Microbiol. 53, 43–70. doi: 10.1146/annurev.micro.53.1.43
Ezra, D., Barash, I., Valinsky, L., Manulis, S. (2000). The dual function in virulence and host range restriction of a gene isolated from the pPATH (Ehg) plasmid of Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Interact. 13, 683–692. doi: 10.1094/MPMI.2000.13.6.683
Ezra, D., Barash, I., Weinthal, D. M., Gaba, V., Manulis, S. (2004). pthG from Pantoea agglomerans pv. gypsophilae encodes an avirulence effector that determines incompatibility in multiple beet species. Mol. Plant Pathol. 5, 105–113. doi: 10.1111/j.1364-3703.2004.00211.x
Funnell, B. E. (2016). ParB partition proteins: complex formation and spreading at bacterial and plasmid centromeres. Front. Mol. Biosci. 3. doi: 10.3389/fmolb.2016.00044
Galán, J. E., Collmer, A. (1999). Type III secretion machines: bacterial devices for protein delivery into host cells. Science 284, 1322–1328. doi: 10.1126/science.284.5418.1322
Gaudriault, S., Paulin, J.-P., Barny, M.-A. (2002). The DspB/F protein of Erwinia amylovora is a type III secretion chaperone ensuring efficient intrabacterial production of the Hrp-secreted DspA/E pathogenicity factor. Mol. Plant Pathol. 3, 313–320. doi: 10.1046/j.1364-3703.2002.00124.x
Gazi, A. D., Charova, S., Aivaliotis, M., Panopoulos, N. J., Kokkinidis, M. (2015). HrpG and HrpV proteins from the type III secretion system of Erwinia amylovora form a stable heterodimer. FEMS Microbiol. Lett. 362, 1–8. doi: 10.1093/femsle/fnu011
Genin, S., Gough, C. L., Zischek, C., Boucher, C. A. (1992). Evidence that the hrpB gene encodes a positive regulator of pathogenicity genes from Pseudomonas solanacearum. Mol. Microbiol. 6, 3065–3076. doi: 10.1111/j.1365-2958.1992.tb01764.x
Giska, F., Lichocka, M., Piechocki, M., Dadlez, M., Schmelzer, E., Hennig, J., et al. (2013). Phosphorylation of HopQ1, a type III effector from Pseudomonas syringae, creates a binding site for host 14-3-3 proteins. Plant Physiol. 161, 2049–2061. doi: 10.1104/pp.112.209023
González-Montes, L., Del Campo, I., Garcillán-Barcia, M. P., de la Cruz, F., Moncalián, G. (2020). ArdC, a ssDNA-binding protein with a metalloprotease domain, overpasses the recipient hsdRMS restriction system broadening conjugation host range. PloS Genet. 16, e1008750–e1008750. doi: 10.1371/journal.pgen.1008750
Guo, M., Manulis, S., Barash, I., Lichter, A. (2001). The operon for cytokinin biosynthesis of Erwinia herbicola pv. gypsophilae contains two promoters and is plant induced. Can. J. Microbiol. 47, 1126–1131. doi: 10.1139/cjm-47-12-1126
Guo, M., Manulis, S., Mor, H., Barash, I. (2002). The presence of diverse IS elements and an avrPphD homologue that acts as a virulence factor on the pathogenicity plasmid of Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Interact. 15, 709–716. doi: 10.1094/MPMI.2002.15.7.709
Hacker, J., Blum-Oehler, G., Mühldorfer, I., Tschäpe, H. (1997). Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol. Microbiol. 23, 1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x
Huang, Y.-C., Lin, Y.-C., Wei, C.-F., Deng, W.-L., Huang, H.-C. (2016). The pathogenicity factor HrpF interacts with HrpA and HrpG to modulate type III secretion system (T3SS) function and T3SS expression in Pseudomonas syringae pv. averrhoi. Mol. Plant Pathol. 17, 1080–1094. doi: 10.1111/mpp.12349
Hunt, M., De Silva, N., Otto, T. D., Parkhill, J., Keane, J. A., Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16, 1–10. doi: 10.1186/S13059-015-0849-0
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Jaén-Luchoro, D., Aliaga-Lozano, F., Gomila, R. M., Gomila, M., Salvà-Serra, F., Lalucat, J., et al. (2017). First insights into a type II toxin-antitoxin system from the clinical isolate Mycobacterium sp. MHSD3, similar to epsilon/zeta systems. PloS One, 12, e0189459. doi: 10.1371/journal.pone.0189459
Jiang, Y., Pogliano, J., Helinski, D. R., Konieczny, I. (2002). ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 44, 971–979. doi: 10.1046/j.1365-2958.2002.02921.x
Kamruzzaman, M., Iredell, J. (2019). A ParDE-family toxin antitoxin system in major resistance plasmids of Enterobacteriaceae confers antibiotic and heat tolerance. Sci. Rep. 9, 9872. doi: 10.1038/s41598-019-46318-1
Keren, I., Shah, D., Spoering, A., Kaldalu, N., Lewis, K. (2004). Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/GR.215087.116
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5, 1–9. doi: 10.1186/GB-2004-5-2-R12
Kvitko, B. H., Park, D. H., Velásquez, A. C., Wei, C.-F., Russell, A. B., Martin, G. B., et al. (2009). Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 Type III secretion effector genes reveal functional overlap among Effectors. PloS Pathog. 5, e1000388. doi: 10.1371/journal.ppat.1000388
Kvitko, B. H., Ramos, A. R., Morello, J. E., Oh, H.-S., Collmer, A. (2007). Identification of harpins in Pseudomonas syringae pv. tomato DC3000, which are functionally similar to HrpK1 in promoting translocation of type III secretion system effectors. J. Bacteriol. 189, 8059–8072. doi: 10.1128/JB.01146-07
Lee, M., Hesek, D., Llarrull, L. I., Lastochkin, E., Pi, H., Boggess, B., et al. (2013). Reactions of all Escherichia coli lytic transglycosylases with bacterial cell wall. J. Am. Chem. Soc 135, 3311–3314. doi: 10.1021/ja309036q
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, Z., Xiang, Z., Zeng, J., Li, Y., Li, J. (2019b). A GntR family transcription factor in streptococcus mutans regulates biofilm formation and expression of multiple sugar transporter genes. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.03224
Li, P., Zhang, L., Mo, X., Ji, H., Bian, H., Hu, Y., et al. (2019a). Rice aquaporin PIP1;3 and harpin Hpa1 of bacterial blight pathogen cooperate in a type III effector translocation. J. Exp. Bot. 70, 3057–3073. doi: 10.1093/jxb/erz130
Lichter, A., Barash, I., Valinsky, L., Manulis, S. (1995). The genes involved in cytokinin biosynthesis in Erwinia herbicola pv. gypsophilae: characterization and role in gall formation. J. Bacteriol. 177, 4457–4465. doi: 10.1128/jb.177.15.4457-4465.1995
Lichter, A., Manulis, S., Valinsky, L., Karniol, B., Barash, I. (1996). IS1327, a new insertion-like element in the pathogenicity-associated plasmid of Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Interact. 9, 98–104. doi: 10.1094/MPMI-9-0098
Lindeberg, M., Stavrinides, J., Chang, J. H., Alfano, J. R., Collmer, A., Dangl, J. L., et al. (2005). Proposed guidelines for a unified nomenclature and phylogenetic analysis of type III hop effector proteins in the plant pathogen Pseudomonas syringae. Mol. Plant-Microbe Interact. 18, 275–282. doi: 10.1094/MPMI-18-0275
Liu, G. F., Wang, X. X., Su, H. Z., Lu, G. T. (2021). Progress on the GntR family transcription regulators in bacteria. Yi Chuan = Hered. 43, 66–73. doi: 10.16288/j.yczz.20-245
Lohou, D., Lonjon, F., Genin, S., Vailleau, F. (2013). Type III chaperones & Co in bacterial plant pathogens: a set of specialized bodyguards mediating effector delivery. Front. Plant Sci. 4. doi: 10.3389/fpls.2013.00435
Macho, A. P. (2016). Subversion of plant cellular functions by bacterial type-III effectors: beyond suppression of immunity. New Phytol. 210, 51–57. doi: 10.1111/nph.13605
Mahillon, J., Chandler, M. (1998). Insertion sequences. Microbiol. Mol. Biol. Rev. 62, 725–774. doi: 10.1128/MMBR.62.3.725-774.1998
Manulis, S., Barash, I. (2003). Pantoea agglomerans pvs. gypsophilae and betae, recently evolved pathogens? Mol. Plant Pathol. 4, 307–314. doi: 10.1046/j.1364-3703.2003.00178.x
Manulis, S., Gafni, Y., Clark, E., Zutra, D., Ophir, Y., Barash, I. (1991) Identification of a plasmid DNA probe for detection of strains of Erwinia herbicola pathogenic on Gypsophila paniculata Phytopathol. 81, 54–57. doi: 10.1094/Phyto-81-54
Manulis, S., Haviv-Chesner, A., Brandl, M. T., Lindow, S. E., Barash, I. (1998). Differential involvement of indole-3-acetic acid biosynthetic pathways in pathogenicity and epiphytic fitness of Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Interact. 11, 634–642. doi: 10.1094/MPMI.1998.11.7.634
Matson, S. W., Ragonese, H. (2005). The F-plasmid TraI protein contains three functional domains required for conjugative DNA strand transfer. J. Bacteriol. 187, 697–706. doi: 10.1128/JB.187.2.697-706.2005
Merrikh, C. N., Merrikh, H. (2018). Gene inversion potentiates bacterial evolvability and virulence. Nat. Commun. 9, 4662. doi: 10.1038/s41467-018-07110-3
Merrikh, H., Zhang, Y., Grossman, A. D., Wang, J. D. (2012). Replication-transcription conflicts in bacteria. Nat. Rev. Microbiol. 10, 449–458. doi: 10.1038/nrmicro2800
Mor, H., Manulis, S., Zuck, M., Nizan, R., Coplin, D. L., Barash, I. (2001). Genetic organization of the hrp gene cluster and dspAE/BF operon in Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Interact. 14, 431–436. doi: 10.1094/MPMI.2001.14.3.431
Moretti, C., Rezzonico, F., Orfei, B., Cortese, C., Moreno-Pérez, A., van den Burg, H. A., et al. (2021). Synergistic interaction between the type III secretion system of the endophytic bacterium Pantoea agglomerans DAPP-PG 734 and the virulence of the causal agent of olive knot Pseudomonas savastanoi pv. savastanoi DAPP-PG 722. Mol. Plant Pathol. 22, 1209–1225. doi: 10.1111/MPP.13105
Morisseau, C., Hammock, B. D. (2013). Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 53, 37–58. doi: 10.1146/annurev-pharmtox-011112-140244
Morris, R. O. (1986). Genes specifying auxin and cytokinin biosynthesis in phytopathogens. Annu. Rev. Plant Physiol. 37, 509–538. doi: 10.1146/annurev.pp.37.060186.002453
Mukherjee, K., Bürglin, T. R. (2006). MEKHLA, a novel domain with similarity to PAS domains, is fused to plant homeodomain-leucine zipper III proteins. Plant Physiol. 140, 1142–1150. doi: 10.1104/pp.105.073833
Nicolas, E., Lambin, M., Dandoy, D., Galloy, C., Nguyen, N., Oger, C. A., et al. (2015). The Tn3-family of replicative transposons. Microbiol. Spectr. 3. doi: 10.1128/microbiolspec.MDNA3-0060-2014
Nissan, G., Chalupowicz, L., Sessa, G., Manulis-Sasson, S., Barash, I. (2019). Two Pantoea agglomerans type III effectors can transform nonpathogenic and phytopathogenic bacteria into host-specific gall-forming pathogens. Mol. Plant Pathol. 20, 1582–1587. doi: 10.1111/mpp.12860
Nissan, G., Gershovits, M., Morozov, M., Chalupowicz, L., Sessa, G., Manulis-Sasson, S., et al. (2018). Revealing the inventory of type III effectors in Pantoea agglomerans gall-forming pathovars using draft genome sequences and a machine-learning approach. Mol. Plant Pathol. 19, 381–392. doi: 10.1111/mpp.12528
Nissan, G., Manulis-Sasson, S., Chalupowicz, L., Teper, D., Yeheskel, A., Pasmanik-Chor, M., et al. (2012). The type III effector HsvG of the gall-forming Pantoea agglomerans mediates expression of the host gene HSVGT. Mol. Plant Microbe Interact. 25, 231–240. doi: 10.1094/MPMI-06-11-0173
Nissan, G., Manulis-Sasson, S., Weinthal, D., Mor, H., Sessa, G., Barash, I. (2006). The type III effectors HsvG and HsvB of gall-forming Pantoea agglomerans determine host specificity and function as transcriptional activators. Mol. Microbiol. 61, 1118–1131. doi: 10.1111/j.1365-2958.2006.05301.x
Nizan, R., Barash, I., Valinsky, L., Lichter, A., Manulis, S. (1997). The presence of hrp genes on the pathogenicity-associated plasmid of the tumorigenic bacterium Erwinia herbicola pv. gypsophilae. Mol. Plant-Microbe Interact. 10, 677–682. doi: 10.1094/MPMI.1997.10.5.677
Nizan-Koren, R., Manulis, S., Mor, H., Iraki, N. M., Barash, I. (2003). The regulatory cascade that activates the Hrp regulon in Erwinia herbicola pv. gypsophilae. Mol. Plant Microbe Interact. 16, 249–260. doi: 10.1094/MPMI.2003.16.3.249
Occhialini, A., Cunnac, S., Reymond, N., Genin, S., Boucher, C. (2005). Genome-wide analysis of gene expression in Ralstonia solanacearum reveals that the hrpB gene acts as a regulatory switch controlling multiple virulence pathways. Mol. Plant Microbe Interact. 18, 938–949. doi: 10.1094/MPMI-18-0938
Ortiz-Martín, I., Thwaites, R., Mansfield, J. W., Beuzón, C. R. (2010). Negative regulation of the Hrp type III secretion system in Pseudomonas syringae pv. phaseolicola. Mol. Plant Microbe Interact. 23, 682–701. doi: 10.1094/MPMI-23-5-0682
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Petnicki-Ocwieja, T., Van Dijk, K., Alfano, J. R. (2005). The hrpK operon of Pseudomonas syringae pv. tomato DC3000 encodes two proteins secreted by the type III (Hrp) protein secretion system: HopB1 and HrpK, a putative type III translocator. J. Bacteriol. 187, 649–663. doi: 10.1128/JB.187.2.649-663.2005
Pettinati, I., Brem, J., Lee, S. Y., McHugh, P. J., Schofield, C. J. (2016). The chemical biology of human metallo-β-lactamase fold proteins. Trends Biochem. Sci. 41, 338–355. doi: 10.1016/j.tibs.2015.12.007
Portaliou, A. G., Tsolis, K. C., Loos, M. S., Zorzini, V., Economou, A. (2016). Type III secretion: building and operating a remarkable nanomachine. Trends Biochem. Sci. 41, 175–189. doi: 10.1016/j.tibs.2015.09.005
Rice, P., Longden, L., Bleasby, A. (2000). EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277. doi: 10.1016/S0168-9525(00)02024-2
Saint-Vincent, P. M. B., Ridout, M., Engle, N. L., Lawrence, T. J., Yeary, M. L., Tschaplinski, T. J., et al. (2020). Isolation, characterization, and pathogenicity of two Pseudomonas syringae pathovars from Populus trichocarpa seeds. mdpi.com 8, 1137. doi: 10.3390/microorganisms8081137
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seth-Smith, H. M. B., Fookes, M. C., Okoro, C. K., Baker, S., Harris, S. R., Scott, P., et al. (2012). Structure, diversity, and mobility of the Salmonella pathogenicity island 7 family of integrative and conjugative elements within Enterobacteriaceae. J. Bacteriol. 194, 1494–1504. doi: 10.1128/JB.06403-11
Shevelev, I. V., Hübscher, U. (2002). The 3’ 5’ exonucleases. Nat. Rev. Mol. Cell Biol. 3, 364–376. doi: 10.1038/nrm804
Siamer, S., Patrit, O., Fagard, M., Belgareh-Touzé, N., Barny, M.-A. (2011). Expressing the Erwinia amylovora type III effector DspA/E in the yeast Saccharomyces cerevisiae strongly alters cellular trafficking. FEBS Open Bio 1, 23–28. doi: 10.1016/j.fob.2011.11.001
Siguier, P., Gourbeyre, E., Chandler, M. (2014). Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol. Rev. 38, 865–891. doi: 10.1111/1574-6976.12067
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Sokurenko, E. V., Hasty, D. L., Dykhuizen, D. E. (1999). Pathoadaptive mutations: gene loss and variation in bacterial pathogens. Trends Microbiol. 7, 191–195. doi: 10.1016/S0966-842X(99)01493-6
Spiers, A. J., Bergquist, P. L. (1992). Expression and regulation of the RepA protein of the RepFIB replicon from plasmid P307. J. Bacteriol. 174, 7533–7541. doi: 10.1128/jb.174.23.7533-7541.1992
Srimanote, P., Paton, A. W., Paton, J. C. (2002). Characterization of a novel type IV pilus locus encoded on the large plasmid of locus of enterocyte effacement-negative Shiga-toxigenic Escherichia coli strains that are virulent for humans. Infect. Immun. 70, 3094–3100. doi: 10.1128/IAI.70.6.3094-3100.2002
Stojanovski, G., Dobrijevic, D., Hailes, H. C., Ward, J. M. (2020). Identification and catalytic properties of new epoxide hydrolases from the genomic data of soil bacteria. Enzyme Microb. Technol. 139, 109592. doi: 10.1016/j.enzmictec.2020.109592
Storch, K. F., Rudolph, J., Oesterhelt, D. (1999). Car: a cytoplasmic sensor responsible for arginine chemotaxis in the archaeon Halobacterium salinarum. EMBO J. 18, 1146–1158. doi: 10.1093/emboj/18.5.1146
Subramanya, H. S., Arciszewska, L. K., Baker, R. A., Bird, L. E., Sherratt, D. J., Wigley, D. B. (1997). Crystal structure of the site-specific recombinase, XerD. EMBO J. 16, 5178–5187. doi: 10.1093/emboj/16.17.5178
Sulja, A., Pothier, J. F., Blom, J., Moretti, C., Buonaurio, R., Rezzonico, F., et al. (2022). Comparative genomics to examine the endophytic potential of Pantoea agglomerans DAPP-PG 734. BMC Genomics 23, 1–15. doi: 10.1186/S12864-022-08966-Y
Suvorova, I. A., Korostelev, Y. D., Gelfand, M. S. (2015). GntR family of bacterial transcription factors and their DNA binding motifs: structure, positioning and co-evolution. PloS One 10, e0132618. doi: 10.1371/journal.pone.0132618
Teper, D., Salomon, D., Sunitha, S., Kim, J.-G., Mudgett, M. B., Sessa, G. (2014). Xanthomonas euvesicatoria type III effector XopQ interacts with tomato and pepper 14-3-3 isoforms to suppress effector-triggered immunity. Plant J. 77, 297–309. doi: 10.1111/tpj.12391
Thomas, N. A., Deng, W., Puente, J. L., Frey, E. A., Yip, C. K., Strynadka, N. C. J., et al. (2005). CesT is a multi-effector chaperone and recruitment factor required for the efficient type III secretion of both LEE- and non-LEE-encoded effectors of enteropathogenic Escherichia coli. Mol. Microbiol. 57, 1762–1779. doi: 10.1111/j.1365-2958.2005.04802.x
Thomas, C. M., Smith, C. A. (1986). The trfB region of broad host range plasmid RK2: the nucleotide sequence reveals incC and key regulatory gene trfB/korA/korD as overlapping genes. Nucleic Acids Res. 14, 4453–4469. doi: 10.1093/nar/14.11.4453
Triplett, L. R., Melotto, M., Sundin, G. W. (2009). Functional analysis of the N terminus of the Erwinia amylovora secreted effector DspA/E reveals features required for secretion, translocation, and binding to the chaperone DspB/F. Mol. Plant Microbe Interact. 22, 1282–1292. doi: 10.1094/MPMI-22-10-1282
Triplett, L. R., Wedemeyer, W. J., Sundin, G. W. (2010). Homology-based modeling of the Erwinia amylovora type III secretion chaperone DspF used to identify amino acids required for virulence and interaction with the effector DspE. Res. Microbiol. 161, 613–618. doi: 10.1016/j.resmic.2010.05.015
Valinsky, L., Manulis, S., Nizan, R., Ezra, D., Barash, I. (1998). A pathogenicity gene isolated from the pPATH plasmid of Erwinia herbicola pv. gypsophilae determines host specificity. Mol. Plant Microbe Interact. 11, 753–762. doi: 10.1094/MPMI.1998.11.8.753
Vela-Corcía, D., Aditya, D., Srivastava, Dafa-Berger, A., Rotem, N., Barda, O., Levy, M. (2019). MFS transporter from Botrytis cinerea provides tolerance to glucosinolate-breakdown products and is required for pathogenicity. Nat. Commun. 10, 2886. doi: 10.1038/s41467-019-10860-3
Voth, D. E., Broederdorf, L. J., Graham, J. G. (2012). Bacterial type IV secretion systems: versatile virulence machines. Future Microbiol. 7, 241–257. doi: 10.2217/fmb.11.150
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963. doi: 10.1371/JOURNAL.PONE.0112963
Wang, Z. (2001). Translesion synthesis by the UmuC family of DNA polymerases. Mutat. Res. 486, 59–70. doi: 10.1016/s0921-8777(01)00089-1
Washington, E. J., Mukhtar, M. S., Finkel, O. M., Wan, L., Banfield, M. J., Kieber, J. J., et al. (2016). Pseudomonas syringae type III effector HopAF1 suppresses plant immunity by targeting methionine recycling to block ethylene induction. Proc. Natl. Acad. Sci. U. S. A. 113, E3577–E3586. doi: 10.1073/pnas.1606322113
Weber, E., Koebnik, R. (2006). Positive selection of the Hrp pilin HrpE of the plant pathogen Xanthomonas. J. Bacteriol. 188, 1405–1410. doi: 10.1128/JB.188.4.1405-1410.2006
Wehling, M. D., Guo, M., Fu, Z. Q., Alfano, J. R. (2004). The Pseudomonas syringae HopPtoV protein is secreted in culture and translocated into plant cells via the type III protein secretion system in a manner dependent on the ShcV type III chaperone. J. Bacteriol. 186, 3621–3630. doi: 10.1128/JB.186.11.3621-3630.2004
Wei, Z., Kim, J. F., Beer, S. V. (2000b). Regulation of hrp genes and type III protein secretion in Erwinia amylovora by HrpX/HrpY, a novel two-component system, and HrpS. Mol. Plant Microbe Interact. 13, 1251–1262. doi: 10.1094/MPMI.2000.13.11.1251
Wei, C.-F., Kvitko, B. H., Shimizu, R., Crabill, E., Alfano, J. R., Lin, N.-C., et al. (2007). A Pseudomonas syringae pv. tomato DC3000 mutant lacking the type III effector HopQ1-1 is able to cause disease in the model plant Nicotiana benthamiana. Plant J. 51, 32–46. doi: 10.1111/j.1365-313X.2007.03126.x
Wei, W., Plovanich-Jones, A., Deng, W. L., Jin, Q. L., Collmer, A., Huang, H. C., et al. (2000a). The gene coding for the Hrp pilus structural protein is required for type III secretion of Hrp and Avr proteins in Pseudomonas syringae pv. tomato. Proc. Natl. Acad. Sci. U. S. A. 97, 2247–2252. doi: 10.1073/pnas.040570097
Weinthal, D. M., Barash, I., Panijel, M., Valinsky, L., Gaba, V., Manulis-Sasson, S. (2007). Distribution and replication of the pathogenicity plasmid pPATH in diverse populations of the gall-forming bacterium Pantoea agglomerans. Appl. Environ. Microbiol. 73, 7552–7561. doi: 10.1128/AEM.01511-07
Keywords: Pantoea agglomerans, sugar beet, gypsophila, type 3 secretion system, type 3 secreted effectors, plasmid, genome assembly, gall-forming
Citation: Geraffi N, Gupta P, Wagner N, Barash I, Pupko T and Sessa G (2023) Comparative sequence analysis of pPATH pathogenicity plasmids in Pantoea agglomerans gall-forming bacteria. Front. Plant Sci. 14:1198160. doi: 10.3389/fpls.2023.1198160
Received: 31 March 2023; Accepted: 10 July 2023;
Published: 31 July 2023.
Edited by:
Prem Lal Kashyap, Indian Institute of Wheat and Barley Research (ICAR), IndiaReviewed by:
Junjie Yue, Beijing Institute of Biotechnology, ChinaCopyright © 2023 Geraffi, Gupta, Wagner, Barash, Pupko and Sessa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Priya Gupta, Z3VwdGEucHJpeWE3MTlAZ21haWwuY29t
†Deceased
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.