 Hyo Ju Lee
Hyo Ju Lee Yi Lee
Yi Lee Sang-Choon Lee
Sang-Choon Lee Chang-Kug Kim
Chang-Kug Kim Ji-Nam Kang
Ji-Nam Kang Soo-Jin Kwon1
Soo-Jin Kwon1 Sang-Ho Kang
Sang-Ho Kang- 1Genomics Division, National Institute of Agricultural Sciences, Jeonju, Republic of Korea
- 2Department of Industrial Plant Science and Technology, Chungbuk National University, Cheongju, Republic of Korea
- 3Phyzen Genomics Institute, Seongnam, Republic of Korea
The family Schisandraceae is a basal angiosperm plant group distributed in East and Southeast Asia and includes many medicinal plant species such as Schisandra chinensis. In this study, mitochondrial genomes (mitogenomes) of two species, Schisandra repanda and Kadsura japonica, in the family were characterized through de novo assembly using sequencing data obtained with Oxford Nanopore and Illumina sequencing technologies. The mitogenomes of S. repanda were assembled into one circular contig (571,107 bp) and four linear contigs (10,898–607,430 bp), with a total of 60 genes: 38 protein-coding genes (PCGs), 19 tRNA genes, and 3 rRNA genes. The mitogenomes of K. japonica were assembled into five circular contigs (211,474–973,503 bp) and three linear contigs (8,010–72,712 bp), with a total of 66 genes: 44 PCGs, 19 tRNA genes, and 3 rRNA genes. The mitogenomes of the two species had complex structural features with high repeat numbers and chloroplast-derived sequences, as observed in other plant mitogenomes. Phylogenetic analysis based on PCGs revealed the taxonomical relationships of S. repanda and K. japonica with other species from Schisandraceae. Finally, molecular markers were developed to distinguish between S. repanda, K. japonica, and S. chinensis on the basis of InDel polymorphisms present in the mitogenomes. The mitogenomes of S. repanda and K. japonica will be valuable resources for molecular and taxonomic studies of plant species that belong to the family Schisandraceae.
1 Introduction
Mitochondria are essential organelles that play an important role in energy production through aerobic respiration in most eukaryotic cells (Newton, 1988). They also regulate vital activities in vivo by participating in metabolic processes such as cell differentiation, apoptosis, cell growth, and cell division (Kroemer and Reed, 2000; van Loo et al., 2002; Bonora and Pinton, 2014). Mitochondria arose from endosymbiotic α-proteobacteria in archaea-derived host cells and eventually came to possess an independent mitochondrial genome (mitogenome) via their evolution into eukaryotic organelles (Gray et al., 1999; Lang et al., 1999).
The mitogenomes of plants are larger and more complicated than those of other eukaryotes. They contain varied master circular, linear, and branching structures (Backert et al., 1997; Rice et al., 2013; Gualberto et al., 2014). Since mitogenomes of Marchantia polymorpha and Arabidopsis thaliana were first mapped, a total of hundreds land plants mitogenomes have been deposited in the Organelle Genome Resources of NCBI. The mitogenome sizes in land plants vary greatly, from 66 kb (Viscum scurruloideum; Skippington et al., 2015) to 11.3 Mb (Silene conica; Sloan et al., 2012). Similarly, the number of protein-coding genes (PCGs) ranges from 19 (Viscum scurruloideum) to 79 (Ammopiptanthus mongolicus; unpublished accession NC_039660). Mitogenome size also differs between related species (Sloan et al., 2012; Gualberto and Newton, 2017). It has diversified because of foreign DNA insertion, nuclear or chloroplast sequence insertion, and recombination activity in non-coding regions such as repeat and intron sequences (Alverson et al., 2011; Gandini and Sanchez-Puerta, 2017; Gualberto and Newton, 2017; Cui et al., 2021).
In land plants such as Cucurbita pepo (Alverson et al., 2010), Cycas taitungensis (Chaw et al., 2008), Garcinia mangostana (Wee et al., 2022), and Oryza sativa (Notsu et al., 2002), short repeats of less than 100 bp or large repeats of 1 kb or more contribute significantly to mitogenome size (Arrieta-Montiel et al., 2009; Wynn and Christensen, 2019). At least 300 MYA, when plants first evolved into seed plants, chloroplast DNA containing PCGs and tRNAs frequently transferred into the mitogenome (Wang et al., 2007). Such transfer of non-functional plastid-derived sequences to the mitogenome can introduce new chimeric genes, tRNAs, or promoters (Nakazono et al., 1996; Miyata et al., 1998; Hao and Palmer, 2009; Wang et al., 2012). Furthermore, introns can be acquired by the mitogenome via gene transfer (Palmer et al., 2000), and AT contents can be increased even in species with relatively abundant GCs in the genome (Sloan and Wu, 2014). These characteristics provide useful information for the study of plant evolution and phylogeny (Wallace et al., 1988; Cameron et al., 2007; Mower et al., 2012).
The family Schisandraceae is composed of basal angiosperm plants of the order Austrobaileyales and branches out after Amborellales and Nymphaeales (Qiu et al., 1999; Leitch and Hanson, 2002). Schisandraceae plants that belong to the genera Schisandra and Kadsura are distributed throughout Korea, China, and Japan and are valuable economic and medicinal resources (Liu et al., 2014). The fruits of Schisandraceae have anticancer, antioxidant, and anti-inflammatory properties (Li et al., 2018; Zhou et al., 2021) and contain lignans, polysaccharides, flavonoids, and organic acids (Jiang et al., 2015; Zhou et al., 2021). These features have driven interest in Schisandraceae plants (Liu et al., 2022), and research has been conducted on the extraction of fruit-derived substances (Lee and Kim, 2010; Nam et al., 2014; Yi et al., 2016), individual breeding techniques (Kim et al., 2014; Boo and Kim, 2020), flower growth, and sex determination (Liu et al., 2022). In addition, genes involved in compound biosynthesis have been recently identified using transcriptome and metabolome analyses (Chun-Yu et al., 2020; Hong et al., 2022; Li et al., 2022). However, it is difficult to distinguish between Schisandra species because the dried mature fruits are morphologically similar (Lee et al., 2013). Furthermore, Schisandraceae plant classification is complex and has changed several times over the last few decades (Li et al., 2018).
In this study, we aimed to gain a better understanding of mitogenome characteristics, structure, evolution, and function. We therefore assembled the mitogenomes of Schisandra repanda and Kadsura japonica using Nanopore and Illumina sequencing. We used the resulting assembled mitogenomes to conduct taxonomic studies and investigate the relationships between highly utilized Schisandraceae species. Our results will help protect these species and develop their genetic resources.
2 Materials and methods
2.1 DNA extraction and sequencing
Fresh leaf samples were collected from single S. repanda and K. japonica plant growing at Jeju Agricultural Research Field, and genomic DNA was extracted. For Illumina sequencing, 200 ng genomic DNA was sonicated to a fragment size of 350 bp using a Covaris S2 system (Covaris, USA) and processed with the Illumina TruSeq Nano Sample Prep kit (Illumina, USA) according to the manufacturer’s instructions. The constructed library was quantified using digital PCR and Taqman Probe (Thermo Fisher Scientific, USA) and sequenced using an Illumina NovaSeq 6000 platform. For Oxford Nanopore sequencing, 10 µg unfragmented genomic DNA was processed using the Quick Ligation Kit (NEB, USA) and SQK-LSK109 Ligation kit (ONT, UK) according to the manufacturers’ instructions. The purified library was loaded into a MinION Flow Cell (ONT, UK) and sequenced for 72 h.
2.2 Mitogenome assembly and annotation
The quality trim tool (Phred score > 20) in the CLC Assembly Cell package (ver. 4.2.1, Qiagen, Denmark) and Porechop software (ver. 0.2.3; https://github.com/rrwick/Porechop) with default parameters were used to trim the Illumina and Nanopore raw sequencing data and remove adaptor and chimeric sequences.
The trimmed Nanopore data were assembled using the NextDenovo program (ver. 2.3.1; https://github.com/Nextomics/NextDenovo) with 1-kb read cutoff and 100-Mb genome size as the parameters. The total assembled contigs were aligned with mitochondrial genome sequence of Schisandra chinensis (MK860624) using nucmer (ver. 4.0.0beta2; https://github.com/mummer4/mummer) with default parameters and then contigs that were aligned with the mitochondrial genome sequence were selected as mitochondrial contigs. The selected contigs were merged, gap-filled, and error-corrected by a series of mapping the trimmed Illumina data using clc_ref_assemble and clc_mapping_viewer with default parameters in CLC Assembly Cell package ver. 4.2.1 (QIAGEN, Denmark) (Kim et al., 2015).
The assembled mitogenome sequences were validated by read mapping of Nanopore and Illumina sequencing data. The trimmed reads were mapped to the assembled mitogenome sequences, and the consistency and connectivity of the mapped reads on the mitogenome sequences and junctions between contigs were confirmed using clc_ref_assemble, clc_mapping_viewer, and clc_mapping_info with default parameters in CLC Assembly Cell package ver. 4.2.1 (QIAGEN, Denmark).
The mitogenome sequences were annotated using Artemis (Carver et al., 2012) and GeSeq (Tillich et al., 2017) programs on the basis of similarity with the mitochondrial reference genome (S. chinensis chromosome 1 mitochondrion, complete sequence [MK860624]) (Baek et al., 2019). In addition, manual curation with BLAST was used to pinpoint particular gene areas.
Circular maps of the mitogenome were created with the annotated data using OGDRAW (http://ogdraw.mpimp-golm.mpg.de; Lohse et al., 2007).
2.3 Repeat sequence analysis
Repeat sequences, such as forward and reverse repeats, in mitogenomes were identified using the REPuter program (ver. 2.3.0), with maximum size of gaps between repeat instances of 30 (Kurtz et al., 2001). Simple sequence repeats (SSRs) were detected using the MISA program (ver. 2.1; https://webblast.ipk-gatersleben.de/misa/), with mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide repeat parameters set as 10, 5, 3, 4, 3, and 3, respectively (Beier et al., 2017). Tandem repeats were found using the Tandem Repeats Finder program (ver. 4.09; https://tandem.bu.edu/trf/trf.html), with min match = 2, min mismatch = 7, min score = 50, and max period = 2,000 (Benson, 1999).
2.4 Identification plastid-derived sequences in mitochondrial DNA
The mitogenome sequences of S. repanda and K. japonica were examined for the presence of plastid-derived sequences using BLASTn with megaBlast parameters and a cutoff e-value of 1e–5 against the S. chinensis chloroplast genome sequence (NC_034908).
2.5 RNA-editing analysis
PREPACT (ver. 3.12.0; http://www.prepact.de/prepact-main.php) with default options was utilized to predict RNA editing sites within PCGs of S. repanda and K. japonica mitogenomes. The cutoff value was set to 0.001 to ensure prediction accuracy (Lenz et al., 2010; Lenz and Knoop, 2013; Lenz et al., 2018).
2.6 Ka/Ks analysis
The multiple sequence alignments of 33 commonly found genes from four species were performed using MAFFT v7.305b and the alignments were corrected with Gblocks v0.91b94, and then we used ParaAT v2.0 was used to estimate the Ka/Ks ratio (Castresana, 2000; Zhang et al., 2012; Katoh and Standley, 2013). The plot was drawn using in-house Python and R scripts.
2.7 Phylogenetic analysis
Phylogenetic analysis was performed using 14 conserved coding sequences (CDSs; atp1, atp6, atp8, atp9, cob, cox2, mttB, nad1, nad2, nad3, nad5, nad6, rpl5, and rps12) in 17 species. Concatenated CDSs were aligned using the MAFFT program (ver. 7) with default parameters and used as input data for the phylogenetic analysis (Katoh et al., 2019). The RAxML program (ver. 8.2.12) was used for the phylogenetic analysis, with the substitution model GTR + I + G, and 1,000 bootstrap replicates (Stamatakis, 2014). The FigTree program (ver. 1.4.3; http://tree.bio.ed.ac.uk/software/figtree/) was used to visualize the trees.
2.8 Gene clusters conserved in the mitogenomes
Gene orders in the mitogenomes of S. repanda and K. japonica were compared with those in the mitogenomes of 15 other species. Two nearby genes with no intervening genes and less than 5 kb of intergenic sequences between them were defined as clusters. A gene cluster was considered conserved when the two flanking genes in the S. repanda and K. japonica mitogenomes were also present in other mitogenomes.
2.9 Identification of InDel polymorphisms and PCR amplification
The mitogenomes of S. repanda, K. japonica, and S. chinensis were compared reciprocally using megaBLAST with a cutoff e-value of 1e–5 and minimum length of 10 kb. Based on the megaBLAST results, homologous regions of >10 kb among the three mitogenomes were selected and InDel polymorphisms were identified. InDels of >5 bp were selected and used to design InDel primers. The flanking sequences ( ± 300 bp) of the selected InDels were extracted from S. chinensis mitogenome sequences and used to design primer sets for PCR amplification of the InDel sites. The PCR primers were designed using Primer 3 software with modified parameters: primer size of 17–25 mer, temperature of 50–60°C, GC of 45–55, and amplicon size of 300–500 bp (Untergasser et al., 2012). The specificity of the designed primers was confirmed using BLASTN against S. chinensis mitogenome sequences. PCR conditions were 5 min at 95°C for pre-denaturation; 40 cycles of 30 s each at 95°C, 58°C, and 72°C; and a final extension for 5 min at 72°C. Gel electrophoresis on 4% agarose gels in TAE buffer with loading dye was used to analyze the PCR products.
3 Results
3.1 Whole-genome sequencing and assembly of the S. repanda and K. japonica mitogenomes
The mitogenomes of S. repanda and K. japonica were assembled using sequencing data obtained from the third- and next-generation sequencing platforms of Oxford Nanopore and Illumina technology, respectively. Additionally, we confirmed the assembly sequences through read mapping and checking read depth (Supplementary figures 1, 2). For S. repanda, 10.79 and 2.69 Gb were obtained by trimming 10.82 Gb of raw Nanopore data and 3.02 Gb of raw Illumina data, respectively. For K. japonica, 9.80 and 1.99 Gb were obtained by trimming 9.84 Gb of raw Nanopore data and 2.27 Gb of raw Illumina data, respectively (Supplementary table 1). The trimmed sequencing data were then de novo assembled to characterize the mitogenomes of the two species.
Five and eight mitochondrial contigs with varying genome sizes and gene contents were assembled from S. repanda and K. japonica, respectively. The S. repanda mitogenome had one circular and four linear contigs: (circular-1: 571,107 bp; linear-1: 607,403 bp; linear-2: 215,128 bp; linear-3: 42,796 bp; and linear-4: 10,898 bp; Supplementary figure 3). We investigated gene content (PCGs, tRNA, and rRNA) in these five S. repanda mitochondrial contigs: S. repanda circular-1 included 34 genes (18 PCGs, 13 tRNAs; 3 rRNAs), linear-1 contained 34 genes (20 PCGs, 11 tRNAs, 3 rRNAs), linear-2 contained 11 genes (4 PCGs, 4 tRNAs, 3 rRNA), linear-3 contained 4 PCGs, and linear-5 contained 2 PCGs (Supplementary table 2). In all, 60 genes were identified, comprising 38 protein-coding (23 unique core genes except nad9 and 15 variable genes), 19 tRNA, and 3 rRNA genes (Table 1).
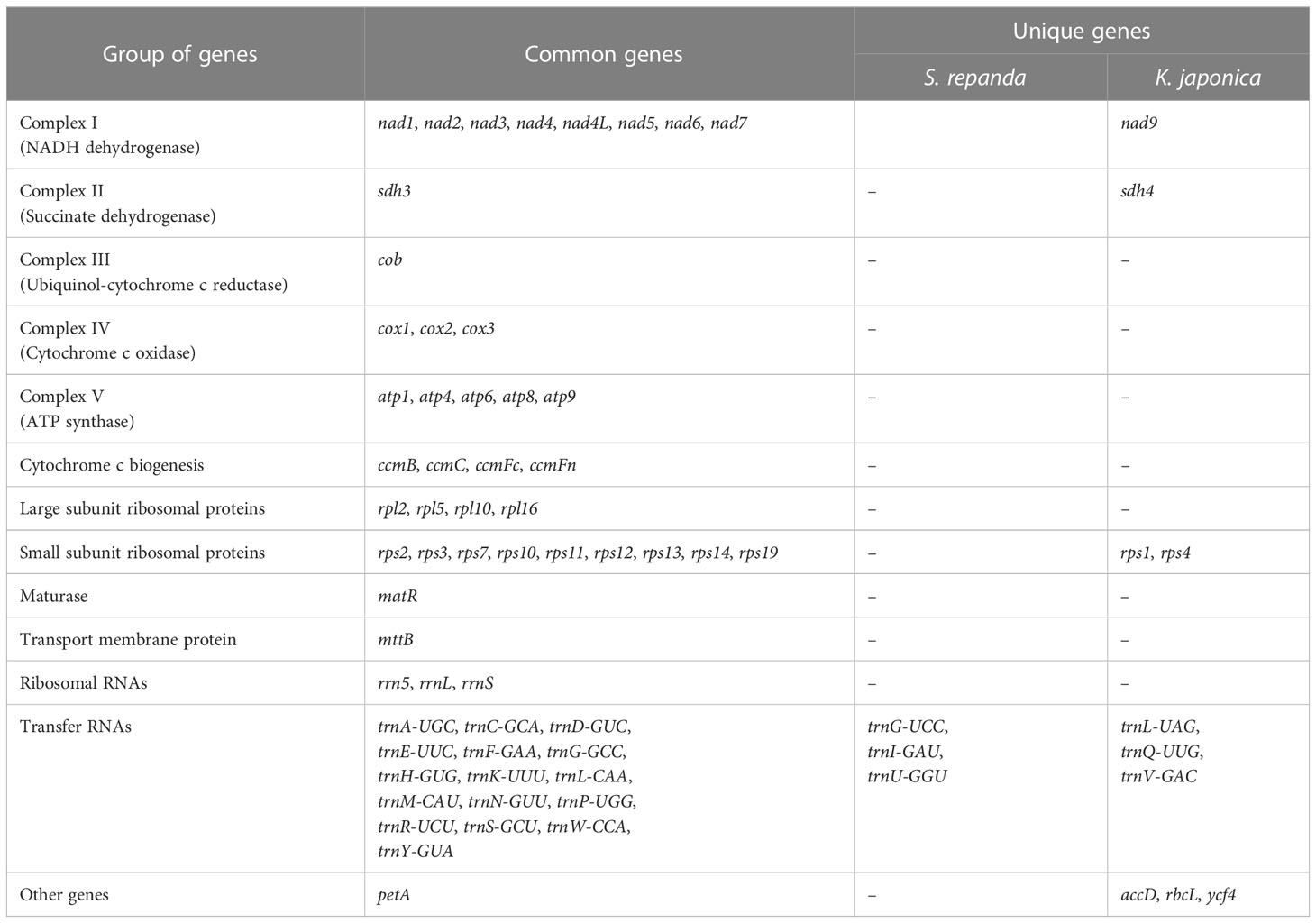
Table 1 Annotated genes in the mitogenomes of S. repanda and K. japonica.
The mitogenome of K. japonica had five circular and three linear contigs: (circular-1: 973,503 bp; circular-2: 897,204 bp; circular-3: 848,837 bp; circular-4: 261,590 bp; circular-5: 211,474 bp; linear-1: 72,712 bp; linear-2: 68,176 bp; and linear-3: 8,010 bp; Supplementary figure 4). K. japonica circular-1 included 45 genes (29 PCGs, 13 tRNAs, 3 rRNAs), circular-2 included 43 genes (28 PCGs, 12 tRNAs, 3 rRNAs), circular-3 included 43 genes (27 PCGs, 13 tRNAs, 3 rRNAs), circular-4 included 14 genes (7 PCGs, 7 tRNAs), circular-5 included 9 genes (7 PCGs, 2 tRNA), linear-1 contains 3 genes (1 PCG, 2 tRNAs), linear-2 contained 6 genes (5 PCGs, 1 tRNA), and linear-3 contained only 1 PCG (Supplementary table 2). A total of K. japonica 66 genes were identified, including 44 protein-coding (24 unique core genes and 20 variable genes), 19 tRNA, and 3 rRNA genes (Table 1).
Based on the assembled contig sequences, we performed a BLASTN search to look for regions of similarity between contigs. As a result, 18 (1,054-43,663 bp) and 72 (1,041-819,626 bp) similar sequences were identified in S. repanda and K. japonica, respectively (Supplementary table 3; Supplementary figure 5 and 6). In particular, the K. japonica circular -1 to -3 sequences 815,430 bp, 815,483 bp, and 819,626 bp were identified as the longest similar sequences. Many of the ends of the linear contigs are homologous to other genomic regions (Supplementary figure 5 and 6), suggesting a multipartite structure of these genomes. The mitogenomes of five contigs of S. repanda and eight contigs of K. japonica have been deposited in NCBI under GenBank Accession Numbers OK077167–OK077171 and OK077159–OK077166, respectively. S. repanda circular-1 (Accession No. OK077168) and K. japonica circular-1 (Accession No. OK077159) were used in all further analyses (repeat sequence analysis and DNA migration from chloroplasts to mitochondria), except prediction of RNA-editing, Ka/Ks, phylogenetic analysis, gene clusters, and identification of InDel, as they are the largest circular and contain the most genes (Figure 1).
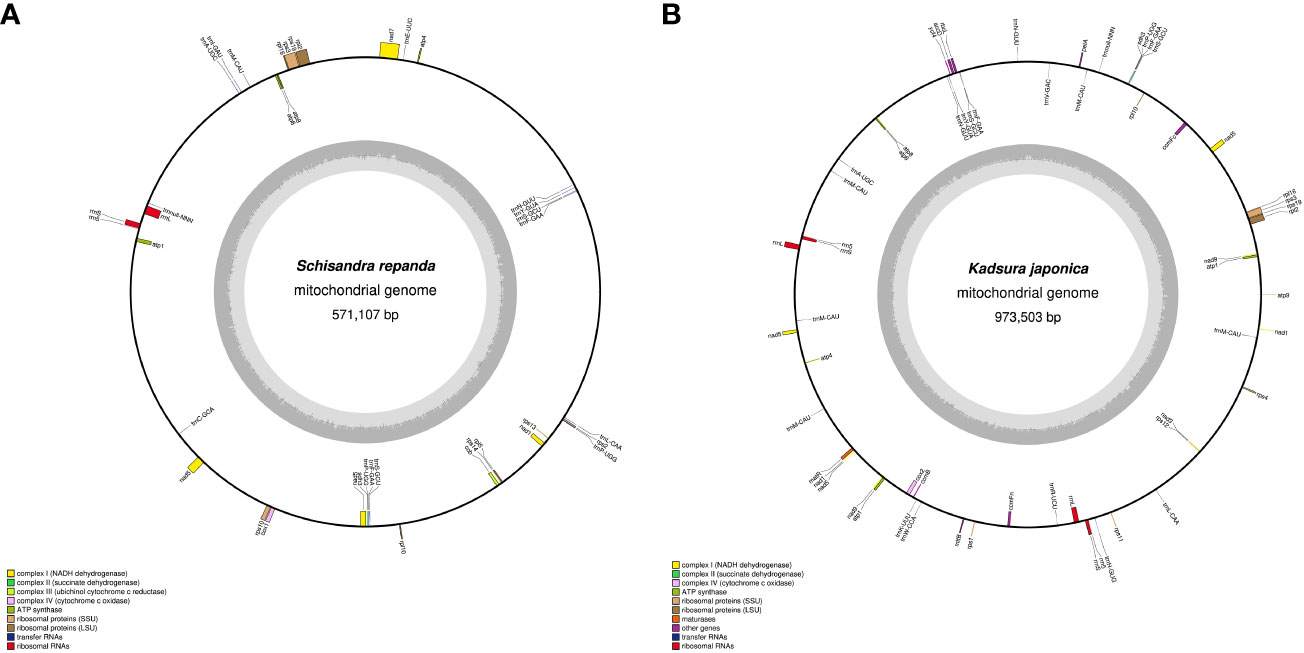
Figure 1 Maps of S. repanda and K. japonica circular-1 mitogenomes. (A) The S. repanda mitogenome has a total length of 571,107 bp. (B) The K. japonica mitogenome has a total length of 973,503 bp. Mitochondrial gene functional groups are represented by the same color.
3.2 Repeat sequence analysis
SSRs are DNA sequences in eukaryotic genomes that typically consist of 1- to 6-bp nucleotides (Lovin et al., 2009). SSR frequency varies among plant species, although SSRs are uniformly dispersed throughout the mitogenome (Zhang et al., 2020). MISA program analysis revealed that the SSR distribution ratios of S. repanda and K. japonica were highly comparable (Figure 2A; Supplementary table 4). S. repanda and K. japonica had 200 and 320 SSRs, respectively; the proportion of tetranucleotide repeats was the highest at 36.5% and 32.7%, and the proportion of hexanucleotide repeats was the lowest at 4.5% and 3.4%, respectively.
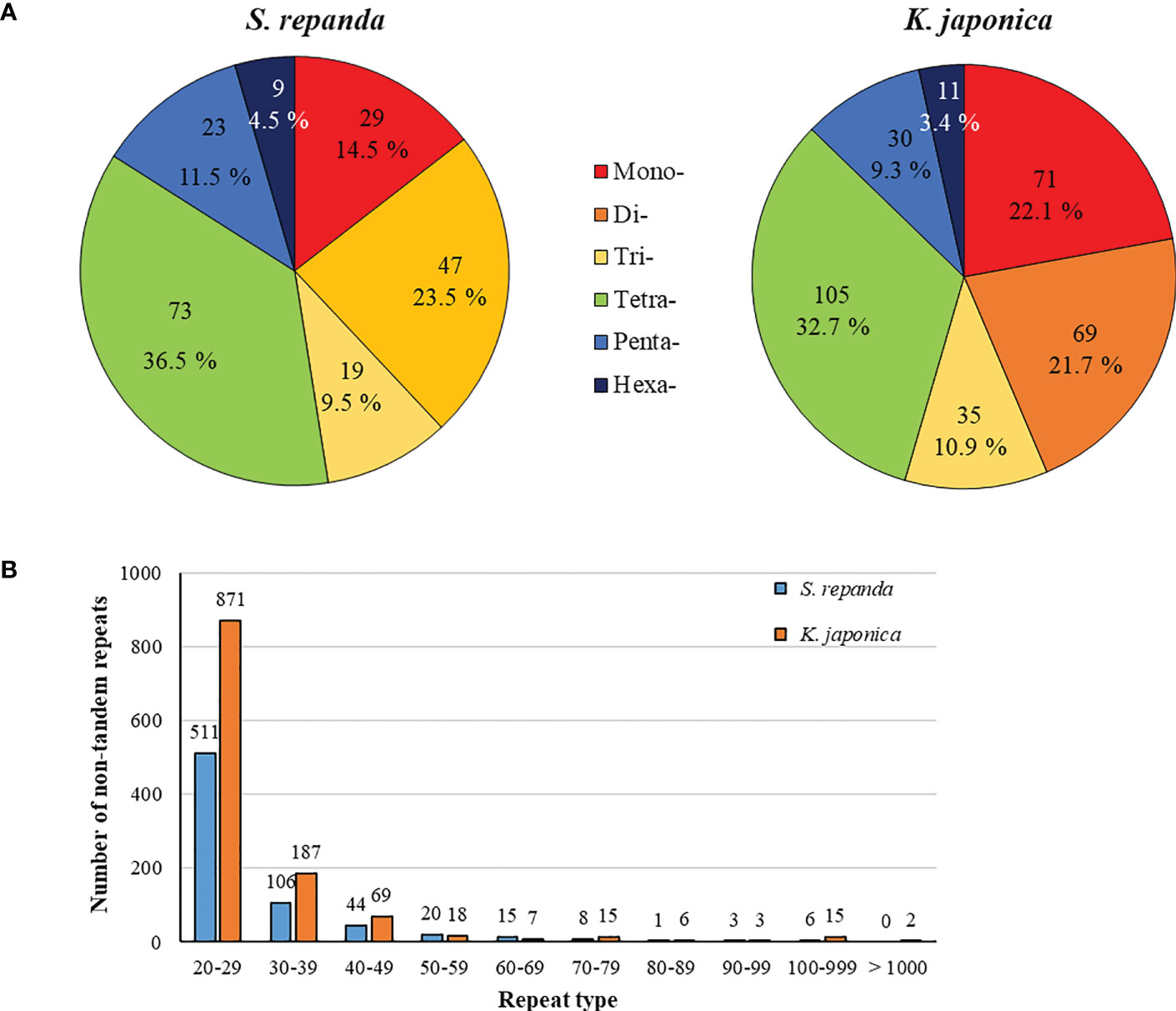
Figure 2 Frequency distribution of SSRs and non-tandem repeats in S. repanda and K. japonica mitogenomes. (A) Number of SSRs. Different types of SSRs are represented by different colors. (B) Number of non-tandem repeats.
Analysis of pentanucleotide and hexanucleotide locations revealed four SSRs in S. repanda located in nad5, nad7, and rpl5 and two SSRs in K. japonica located in rpl2 and rps3 (Supplementary table 5). The Tandem Repeats Finder program was used to analyze perfect tandem repeats, with 11 and 24 identified in S. repanda and K. japonica, respectively (Supplementary table 6). Among them, rpl2, rrnL, and nad5 in S. repanda had three tandem repeats, and rrnL in K. japonica had two tandem repeats. The SSRs and tandem repeat sequences were mostly in the intergenic spacers.
Using the REPuter program to analyze non-tandem repeats other than SSRs and tandem repeats, 714 (total length, 21,695 bp; 3.8%) and 1,193 (total length, 53,852 bp; 5.53%) repeats were detected in S. repanda and K. japonica, respectively (Supplementary table 7). The most abundant repeats in S. repanda and K. japonica were 511 and 871 repeats in the 20- to 29-bp range, with the longest repeats measuring 539 and 12,605 bp, respectively (Figure 2B). Non-tandem repeats were similarly distributed in the two species, and only K. japonica had two long repeat sequences of more than 1 kb. According to a repeat analysis, K. japonica had longer repeats than S. repanda.
3.3 DNA migration from chloroplasts to mitochondria
Intergenomic gene transfer is the movement of DNA sequences between mitochondria, chloroplasts, and the nucleus, as opposed to DNA sequence movement by inheritance, and it is an important cause of mitogenome expansion and evolution (Adams et al., 2001; Renner and Bellot, 2012; Hao et al., 2022). We compared the complete chloroplast sequence of S. chinensis (NC_034908.1) to the S. repanda and K. japonica mitogenomes and identified 16 (32–2,807 bp, 3.39%) and 37 (39–7,515 bp, 4.34%) fragments with high similarities to chloroplast genome, respectively (Figure 3; Supplementary table 8). There were eight complete tRNAs (trnH-GTG, trnL-CAA, trnM-ATG, trnN-GUU, trnP-TGG, trnR-TCT, trnV-GAC, and trnW-CCA) and three partial tRNAs (trnH-GTG, trnN-GUU, and trnR-TCT). The chloroplast-derived sequences also included two partial rRNAs (rrn16s and rrn23S) and 29 partial PCGs (accD, atp1, atpA, atpE, atpF, atpH, cemA, infA, ndhB, ndhI, petA, petD, psaA, psaI, psaJ, psbE, rbcL, rpl20, rpl33, rpl36, rpoA, rpoC1, rpoC2, rps2, rps7, rps8, ycf1, ycf2, and ycf4).
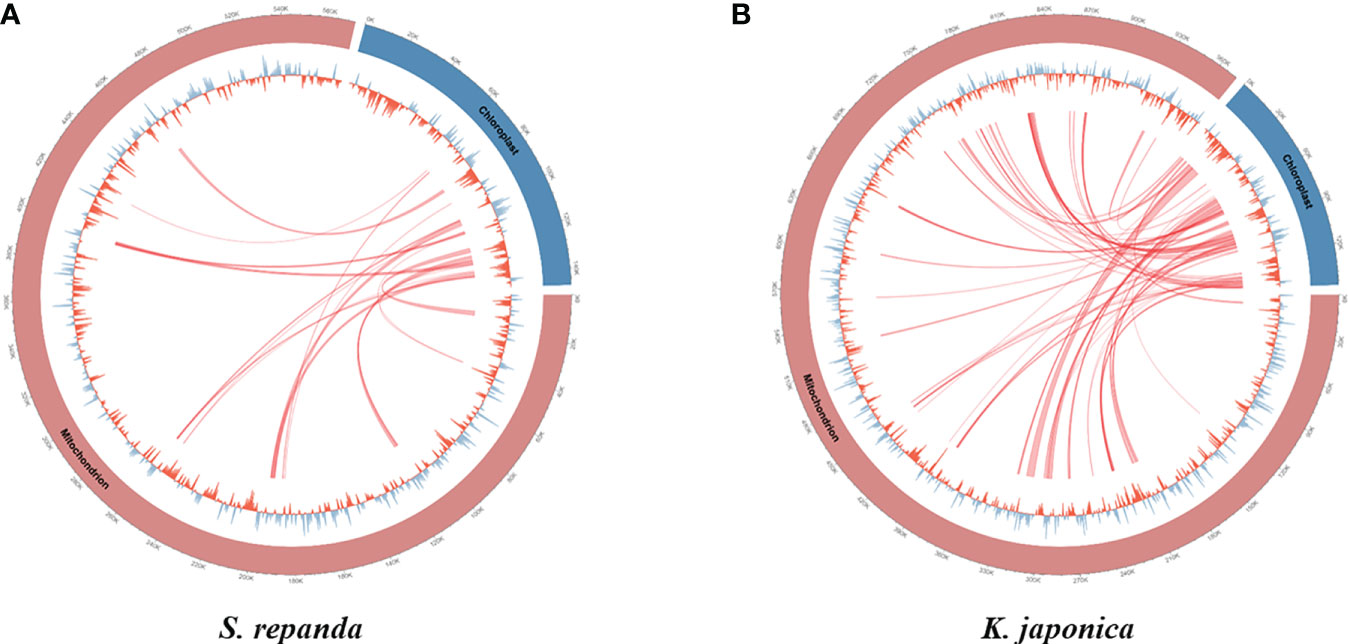
Figure 3 Representation of gene transfers between mitochondrial and chloroplast genomes for S. repanda and K. japonica. Regions of chloroplast sequences that have been inserted into the mitogenome are indicated by red lines. (A) S. repanda. (B) K. japonica.
3.4 Prediction of RNA editing sites
RNA editing, i.e., the post-transcriptional addition, deletion, or conversion of nucleotides in the coding region of transcribed RNA, occurs frequently in the plant mitogenome (Gray, 2003; Yang et al., 2017). In the PCGs of S. repanda and K. japonica mitogenomes, we detected 639 and 684 RNA editing sites, respectively (Supplementary table 9). The first base position of the codon had 224 and 237 RNA editing sites, and the second base position had 415 and 447, respectively, in S. repanda and K. japonica. No RNA-editing sites were predicted at the third base position (Supplementary table 10). To date, the RNA-editing prediction programs cannot reliably predict RNA-editing at third base positions due to technical limitation. After RNA editing, 28.79% and 29.39% of the amino acids remained hydrophobic, whereas 13.3% remained hydrophilic in S. repanda and K. japonica. However, 47.42% and 47.37% of the amino acids were converted from hydrophilic to hydrophobic, and 9.86% and 9.36% were converted from hydrophobic to hydrophilic in S. repanda and K. japonica, respectively; only four of them were changed to stop codons in the S. repanda and K. japonica mitogenome. After RNA editing, leucine was the predominant amino acid in 48.67% of S. repanda and 41.96% of K. japonica, respectively.
3.5 The substitution rates of protein-coding genes
The number of non-synonymous substitutions (Ka) and synonymous substitutions (Ks) is important for phylogenetic reconstruction of related species and for understanding the evolutionary dynamics of protein coding sequences (Fay and Wu, 2003). Ka/Ks values are used to determine whether certain protein-coding genes have undergone selection pressure during evolution. Ka/Ks=1 indicates neutral evolution, Ka/Ks > 1 indicates positive selection, and Ka/Ks <1 indicates negative selection (Zhang et al., 2006). In this study, all of the 33 PCGs of S. repanda, K. japonica, S. chinensis, and S. sphenanthera mitogenome were used to calculate the Ka and Ks substitution rates. As a result, due to the limitation of the ParaAT, Ka/Ks cannot be calculated when sequences are identical between species. In the comparison of the four species, the Ka/Ks of rps13 was the highest at 2.26 to 2.42, and appeared to be under positive selection along with genes such as atp9 (Figure 4A; Supplementary table 11). Genes such as ccmFc, cob, cox2, matR, nad6, sdh3, RPL and RPS showed Ka/Ks values less than 1, indicating that they were under negative selection (Figure 4B-G). The cox1 and cox3 exhibited the lowest Ka/Ks ratio, suggesting that these genes had fewer changes and well conserved between species.
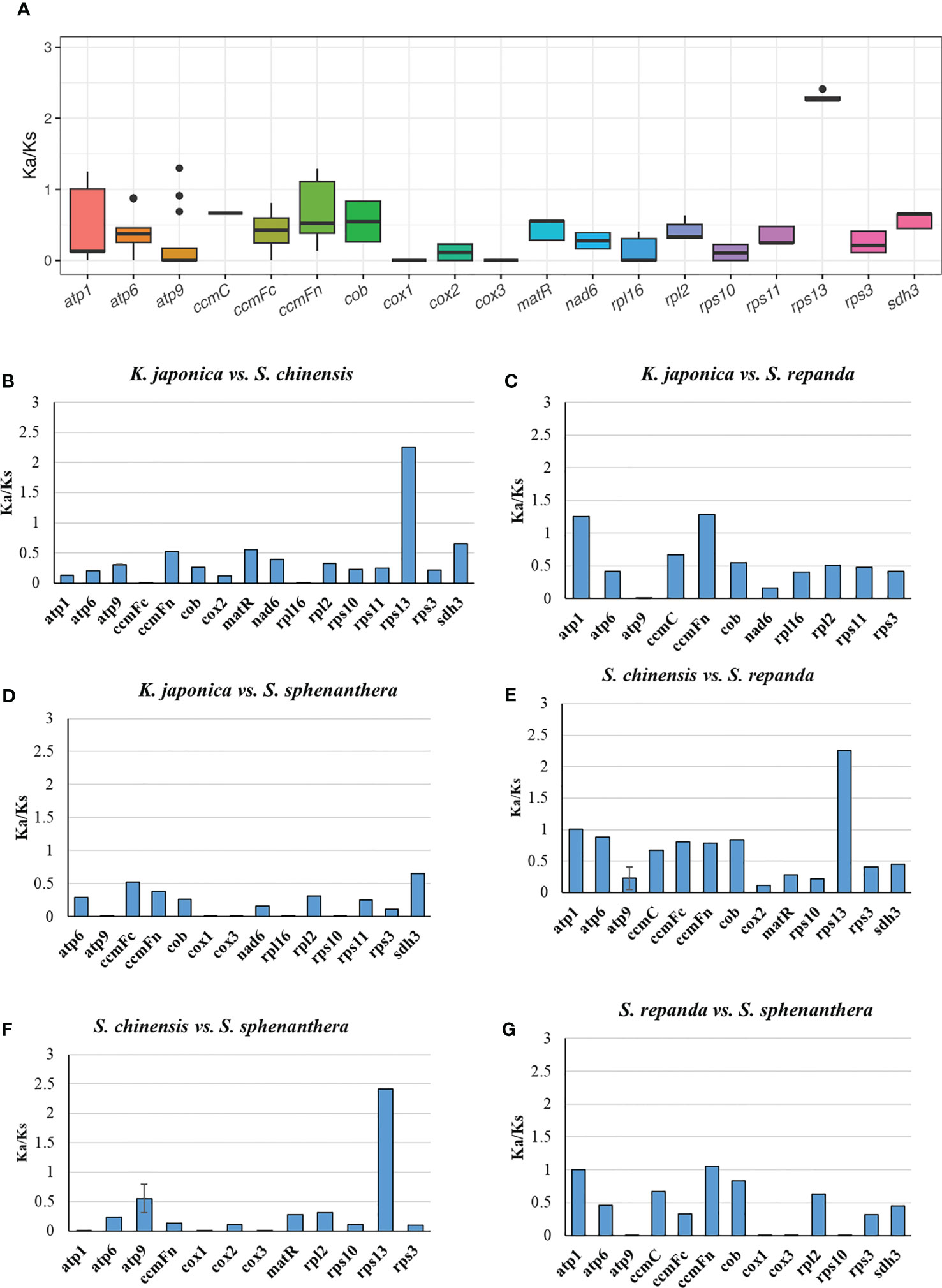
Figure 4 The Ka/Ks values of 33 PCGs of S. repanda, K. japonica, S. chinensis, and S. sphenanthera. (A) Box plot for pairwise divergence of Ka/Ks ratio (mean ± SD, and range). (B-G) Bar graph for pairwise divergence of Ka/Ks ratio (error bars representing either the mean ± SD). The missing values, such as atp1 in S. repanda vs S. sphenanthera, could be due to the limitation of the ParaAT, Ka/Ks cannot be calculated when sequences are identical between species.
3.6 Phylogenetic analysis
Phylogenetic analysis using the maximum likelihood (ML) method was performed using a dataset of 14 PCGs from the following 17 plant taxa (including four species from Schisandraceae): Amborella trichopoda, Nymphaea colorata, S. chinensis, S. repanda, K. japonica, Schisandra sphenanthera, Liriodendron tulipifera, Magnolia biondii, Spirodela polyrhiza, Nelumbo nucifera, Vitis vinifera, Bombax ceiba, Hibiscus cannabinus, Carica papaya, Cercis canadensis, Senna occidentalis, and Senna tora (Figure 5). The ML tree contained 15 nodes, of which 14 had at least 90% of the support value with 100% for 10 and 85% for only one; S. repanda and K. japonica were classified into one clade with bootstrap support values of 100 and 94. The four plants of the family Schisandraceae were placed quite close to each other, with S. repanda being the closest to S. chinensis. Finally, Schisandraceae plants were classified as basal angiosperms along with Amborella and the Nymphaeales, and the placement indicated that the family evolved from mesangiosperms into monocots and eudicots.
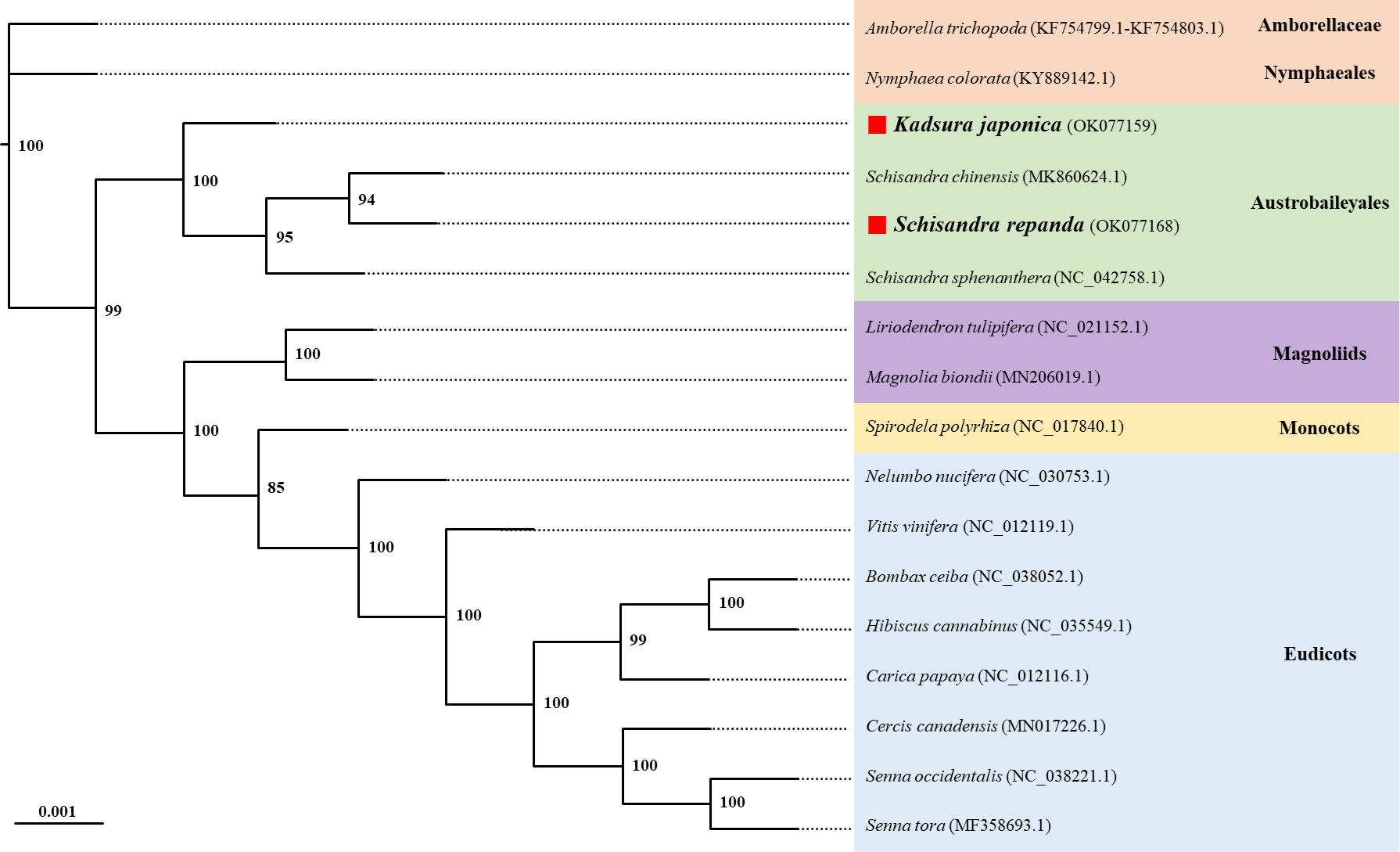
Figure 5 Phylogenetic relationships of S. repanda and K. japonica with other 15 plant species. The numbers next to the branches indicate bootstrap values of maximum likelihood phylogenetic trees. GenBank accession numbers are listed after the scientific names. Positions of S. repanda and K. japonica are indicated in bold.
3.7 Conservation of gene clusters
The sequences of rRNA and PCGs in plant mitogenomes are highly conserved, but the relative order of genes is frequently rearranged by homologous recombination. However, some gene clusters have been conserved during evolution (Palmer et al., 2000; Richardson et al., 2013; Gui et al., 2016). There were 47 and 46 conserved gene clusters in all S. repanda and K. japonica contigs, respectively (Figure 6). Three gene clusters (rpl16~rps3, rps3~rps19, and rps19~rpl2) can be traced back to mitochondrial origins in endosymbiotic bacterial ancestors (Takemura et al., 1992; Niu et al., 2022), two gene clusters (nad3~rps12 and cox1~rps10) could be found in gymnosperms, and the sdh3~trnP-UGG cluster arose during bryophyte evolution (Turmel et al., 2007). In addition, trnF-GAA~trnS-GCU and trnF-GAA~trnP-UGG arose during evolution into Schisandraceae and Mesangiospermae. However, sixteen gene clusters (blue box; rpl10~trnS-GCU, petA~trnM-CAU, trnK-UUU~trnW-CCA, rps2~trnP-UGG, nad1(exon4)~trnD-GUC, atp1~nad9, mttB~rps1, trnS-GCU~trnY-GUA, atp1~rrn5, rps2~trnL-CAA, trnD-GUC~trnM-CAU, accD~rbcL, accD~ycf4, nad5(exon1)~trnM-CAU, nad5(exon2)~petA, atp4~trnE-UUC) were regained in only Schisandraceae and were confirmed to be lost in the subsequent evolution process. The gene clusters of nad2(exon3)~trnY-GUA was not present in basal angiosperms, but they appeared to have been regained in the course of evolution in Mesangiospermae, except for Bombax ceiba.
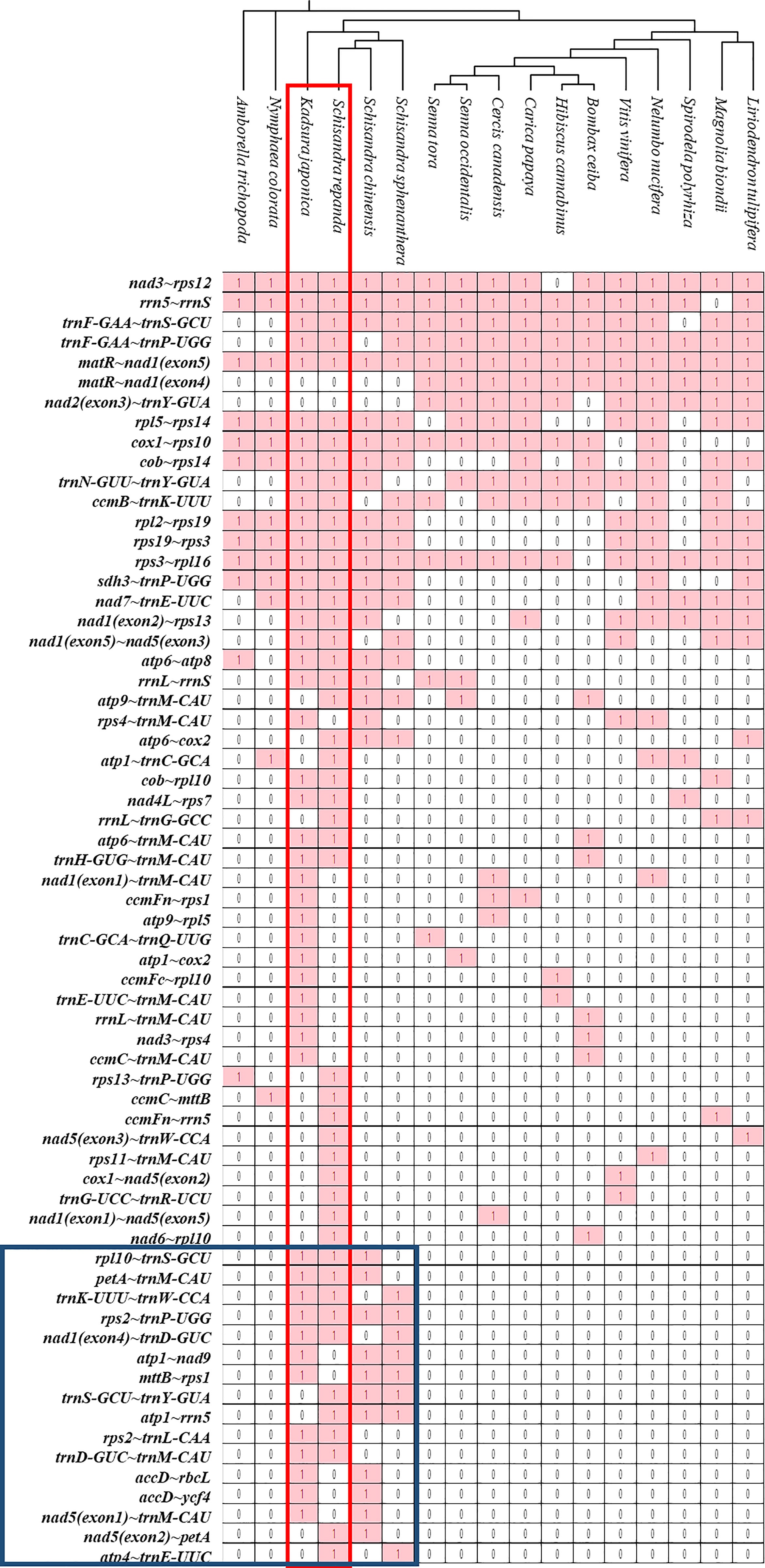
Figure 6 Gene clusters in the mitogenomes of 17 species. Gene sequences of S. repanda and K. japonica mitogenomes were compared with those of the other 15 species. The evolutionary relationships of the mitogenomes are indicated in the phylogenetic tree. ‘1’ indicates the presence of the gene in the mitogenome. ‘0’ indicateds the absence of the gene in the mitogenomes.
3.8 Species identification with InDel markers
InDel regions are widely utilized to create markers to distinguish between species because they can be easily detected (Sebbenn et al., 2019). However, Schisandra and Kadsura species have not been identified using this method on the basis of their mitogenomes. We used BLAST to search for InDel regions of the S. chinensis mitogenome in the S. repanda and K. japonica mitogenomes. InDel markers were designed for 95 of the 130 InDel regions confirmed to be longer than 5 bp (Supplementary table 12). We selected two InDel markers for PCR analysis (Figure 7A, B; Supplementary table 13). Notably, S. repanda, K. japonica, and S. chinensis could be distinguished using PCR amplification with the two primer sets. Based on the S. chinensis sequence, 6 and 11 bp were inserted into S. repanda and K. japonica, respectively, in the case of InDel_8_18_7873, and 5 bp were inserted to S. repanda and 6 bp were deleted from K. japonica in the case of InDel_1_21_11950 (Figure 7C, D). The three species can be distinguished readily using two InDel markers developed through this study.
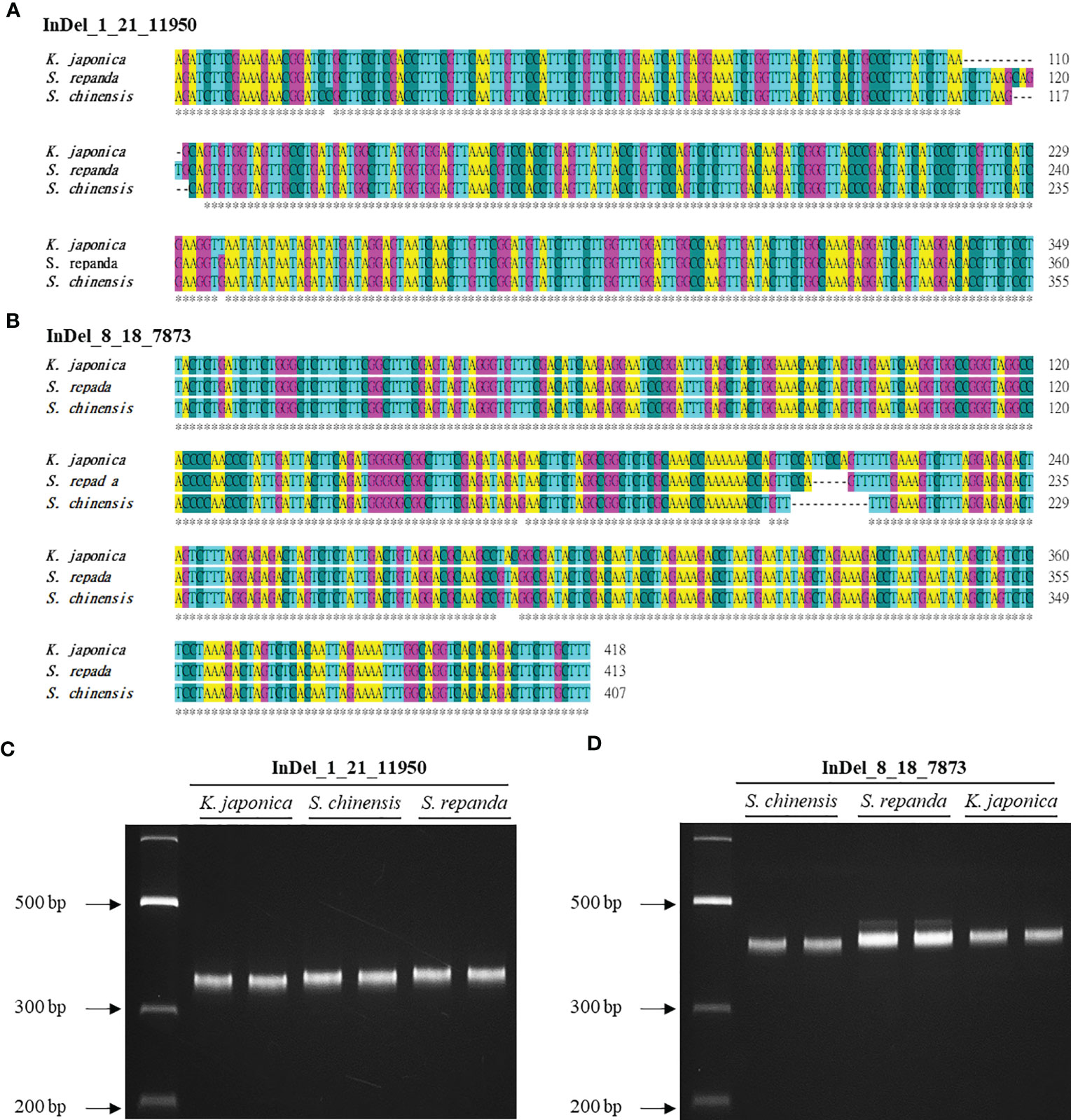
Figure 7 Development of InDel markers for S. repanda, K. japonica, and S. chinensis mitogenomes. (A, B) Alignment of the InDel regions. (C) PCR amplicons for K. japonica, S. chinensis, and S. repanda using InDel_1_21_11950 were 349, 355, and 360 bp, respectively. (D) The InDel_8_18_7873 primer set was used to obtain PCR results for S. chinensis, S. repanda, and K. japonica (407, 413, and 418 bp, respectively).
4 Discussion
The mitogenome comprises repeated sequences and recombination regions, leading to a range of sizes and structures, whereas the plant chloroplast genome is made up of a single-ring structure. This difference in complexity has likely contributed to a lack of research into mitogenomes (Mower et al., 2012; Gualberto et al., 2014). Recently, mitogenome research has advanced because of the development of efficient, inexpensive, and precise genome sequencing tools (Han et al., 2022; Wee et al., 2022; Yang et al., 2022). In this study, S. repanda and K. japonica, two important medicinal plants, were examined for their mitogenome features. According to our sequencing data, S. repanda and K. japonica have five and eight mitogenome structures, respectively, which is not surprising given the various sizes and structures of the plant mitogenome. For example, five circular mitogenomes have been found in A. trichopoda (Rice et al., 2013), and fluorescence microscopy examination of lettuce revealed linear, branching, and circular mitogenome structures (Kozik et al., 2019).
Mitogenomes in angiosperms encode 24 core protein-coding genes, the majority of which are respiratory protein genes: atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFc, ccmFn, cob, cox1, cox2, cox3, matR, mttB, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9 (Adams et al., 2002). Although the full-length nad9 gene was not identified in the S. repanda mitogenome, some sequences similar to nad9 were observed in S. repanda circular-1, linear-1, and linear-2. Notably, we observed that the nad9 gene is similarly lacking from the reported mitogenomes (in GenBank) of plants including Pisum sativum subsp. elatius (MW160422), Camellia nitidissima (NC_067639.1 and ON645224), Kandelia obovata (NC_069222.1 and OP756530), and Ipomoea batatas (NC_068714.1 and OM808941). Thus, we conclude that nad9 might truly be absent from the S. repanda mitogenome. Future work will examine the S. repanda nuclear genome for evidence of a potential loss or shift of the nad9 gene.
Various repeat sequences, including short (<1 kb), long (>1 kb), and tandem repeats, have been found in the mitogenome sequence (Gualberto et al., 2014; Guo et al., 2017). These repeats not only expand the mitogenome but also cause genomic recombination (Dong et al., 2018; Wang et al., 2019). In this study, we identified 200 (2704 bp, 0.47%) and 321 (4,076 bp, 0.42%) SSRs in S. repanda and K. japonica, respectively, which can be used to develop important markers for species distinction, genetic diversity, and evolution studies (Powell et al., 1996; Morgante et al., 2002; Guang-Xin et al., 2019). The total lengths of perfect tandem repeats in S. repanda and K. japonica were 269 and 520 bp, respectively, and the longest non-tandem repeats were 539 and 12,605 bp, respectively. These lengths were greater in K. japonica (5.53%) than in S. repanda (3.8%). The repeat sequences may have increased the K. japonica mitogenome size relative to the S. repanda mitogenome size.
In the mitochondrial, chloroplast, and nuclear genomes, gene transfer is possible in both directions. (Martin et al., 1998; Cui et al., 2021). Nuclear and plastid DNA sequences can be transferred to the mitogenome, resulting in mitogenome size changes (Sloan et al., 2012). When a foreign gene is inserted into the mitogenome at this time, it is preferentially inserted into the intergenic region (Zhao et al., 2018). The plastid genome rarely integrates foreign genetic material (Zhao et al., 2018), whereas the mitogenome frequently integrates DNA sequences from the nucleus, chloroplast, and other mitogenomes (Rice et al., 2013; Zhao et al., 2018). The length of the DNA integrated into the mitogenome varies on the basis of plant species but is generally within the range of 1–12% of angiosperm plastome sequences (Mower et al., 2012), such as in G. mangostana (1.7%) (Wee et al., 2022) and C. pepo (11.6%) (Alverson et al., 2010). Similarly, 3.39% and 4.34% integrated DNA fragments were found in S. repanda and K. japonica, respectively, driving genome size and genetic and evolutionary diversity.
RNA editing is a post-transcriptional process required for plant development and stress response (Zhu et al., 2014; Shi et al., 2016; Tang and Luo, 2018). RNA editing, which occurs most frequently in mitochondria, causes reorganization of the mitochondrial protein structure by forming new start and stop codons via C-to-U conversion or altering the RNA structure via splicing site changes (Takenaka et al., 2013; Tang and Luo, 2018; Hao et al., 2021). RNA editing sites were first discovered in the wheat cytochrome c oxidase gene (Covello and Gray, 1989); subsequently, more than 400 sites have been found in the genes of many plants, such as Acer truncatum (Ma et al., 2022), Arabis alpina (Xu and Bi, 2018), A. thaliana (Unseld et al., 1997), O. sativa (Notsu et al., 2002), Suaeda glauca (Cheng et al., 2021), and Zea mays (Hoch et al., 1991). In the mitogenome of K. japonica, a start codon was formed in rpl2 and a stop codon was formed in ccmFC, ccmFN, and rps11. In the mitogenome of S. repanda, start codons were formed in atp8, rps10, and rpl10, but no stop codon was identified. The detection of RNA editing sites in these mitogenomes provides clues to predict gene functions, as different amino acids are produced (Cheng et al., 2021).
In most plant species, the PCGs of the mitogenome is relatively conserved compared to that of the animal species, so point mutations are very rare and contribute to maintaining function (Gualberto et al., 2014). Ka values for most genes are smaller than Ks values because deleterious mutations are eliminated during natural selection to maintain mitochondrial function (Hurst, 2002). Nevertheless, the sdh4 gene is only found in K. japonica, indicating that sdh4 is evolutionarily unstable and has frequently been deleted from the mitogenomes (Adams et al., 2001; Petersen et al., 2017).
PCG and rRNA gene sequences are highly conserved, but the relative order of genes often changes because of genome expansion or recombination (Palmer et al., 2000); nevertheless, a number of highly conserved gene clusters can be found (Richardson et al., 2013; Bi et al., 2016; Dong et al., 2018). The rps3~rpl16 gene cluster is highly conserved and can be traced to the mitochondrial origin of endosymbiotic bacterial ancestors (Takemura et al., 1992; Niu et al., 2022). Most of the gene clusters are shared by species with close evolutionary relationships (Niu et al., 2022). Eight gene clusters were not found in any other plants, except Schisandraceae. This implies significant genome rearrangements led to rapid degeneration of these gene clusters in other plants (Dong et al., 2018). These gene clusters may have been significant in the development of Schisandraceae plants, in contrast to Amborella and Nymphaeales.
Mitogenomes are ideal for lineage studies in seed plants because they evolve more slowly than nuclear and chloroplast genomes and undergo recombination less often (Drouin et al., 2008). Recently, genotyping of both plastomes and mitogenomes has enabled us to distinguish between different plant species (Sebbenn et al., 2019; Han et al., 2022; Ma et al., 2022). InDel markers have the advantage of simplifying plant species identification by comparing amplification product sizes from reference genomes with PCR, making them more cost-effective than single-nucleotide polymorphism or SSR markers (Sohn et al., 2017; Chun et al., 2019). To date, only nuclear and chloroplast genome markers have been reported for Schisandraceae plant identification (Kim et al., 2012; Lee et al., 2013; Jeong et al., 2021). Here, we designed markers based on two InDel regions in the mitogenome sequences of S. repanda and K. japonica compared to S. chinensis. These InDel markers could clearly distinguish between the three species of Schisandraceae. In addition, our phylogenetic analysis results will be helpful for future evolutionary research.
5 Conclusion
We assembled and analyzed the mitogenomes of S. repanda and K. japonica. Various forms of repeats and chloroplast-derived sequences in these mitogenome likely caused mitogenome expansion. In addition, we catalogued RNA editing sites, which will help predict novel gene activity that arises from changes in the protein structure. The sixteen gene clusters we identified as specific to Schisandraceae will be useful to study the evolution of this family. Also, S. repanda, K. japonica, and S. chinensis could be distinguished using two InDel markers that we designed. In conclusion, our work on the mitogenomes of S. repanda and K. japonica offers new avenues for investigating the evolution of Schisandraceae and for understanding the individual species.
Data availability statement
The datasets presented in this study can be found in online repositories at NCBI, accession numbers: OKO77167-OKO77171.
Author contributions
S-HK conceived and designed the study. HJL, YL, S-CL, C-KK, J-NK, S-JK prepared the materials and performed related analysis. HJL and S-HK interpreted the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Cooperative Research Program in the National Agricultural Genome Program (Grant No. PJ013490), Rural Development Administration (RDA), Republic of Korea.
Acknowledgments
We thank Dr. Hyun-Oh Lee and Kyunghyun Nam in Phyzen Co. for their assistance in mitogenomes assembly.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1183406/full#supplementary-material
References
Adams, K. L., Qiu, Y. L., Stoutemyer, M., Palmer, J. D. (2002). Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. 99, 9905–9912. doi: 10.1073/pnas.04269489
Adams, K. L., Rosenblueth, M., Qiu, Y. L., Palmer, J. D. (2001). Multiple losses and transfers to the nucleus of two mitochondrial succinate dehydrogenase genes during angiosperm evolution. Genetics 158, 1289–1300. doi: 10.1093/genetics/158.3.1289
Alverson, A. J., Rice, D. W., Dickinson, S., Barry, K., Palmer, J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 23, 2499–2513. doi: 10.1105/tpc.111.087189
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of citrullus lanatus and cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436–1448. doi: 10.1093/molbev/msq029
Arrieta-Montiel, M. P., Shedge, V., Davila, J., Christensen, A. C., Mackenzie, S. A. (2009). Diversity of the arabidopsis mitochondrial genome occurs via nuclear-controlled recombination activity. Genetics 183, 1261–1268. doi: 10.1534/genetics.109.108514
Backert, S., Nielsen, B. L., Börner, T. (1997). The mystery of the rings: structure and replication of mitochondrial genomes from higher plants. Trends Plant Sci. 2, 477–483. doi: 10.1016/S1360-1385(97)01148-5
Baek, J. H., Kim, H. J., Kang, S. H., Kwon, S. J., Kim, C. K. (2019). The complete mitochondrial genome sequence of Schisandra chinensis (Austrobaileyales: schisandraceae). Mitochondrial DNA Part B. 4, 2485–2486. doi: 10.1080/23802359.2019.1638326
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C., Paterson, A. H., Wang, X., Xu, Y., Wu, D., Qu, Y., et al. (2016). Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed. Res. Int. 2016, 5040598. doi: 10.1155/2016/5040598
Bonora, M., Pinton, P. (2014). The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front. Oncol. 4. doi: 10.3389/fonc.2014.00302
Boo, J. Y., Kim, J. S. (2020). A study on the native environment and cutting propagation for the black-berry magnolia vine [Schisandra repanda (Siebold & zucc.) radlk] in halla mountain. Korean J. Med. Crop Sci. 28, 354–359. doi: 10.7783/KJMCS.2020.28.5.354
Cameron, S. L., Lambkin, C. L., Barker, S. C., Whiting, M. F. (2007). A mitochondrial genome phylogeny of diptera: whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst. Entomol. 32, 40–59. doi: 10.1111/j.1365-3113.2006.00355.x
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., McQuillan, J. A. (2012). Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chaw, S. M., Chun-Chieh Shih, A., Wang, D., Wu, Y. W., Liu, S. M., Chou, T. Y. (2008). The mitochondrial genome of the gymnosperm Cycas taitungensis contains a novel family of short interspersed elements, bpu sequences, and abundant RNA editing sites. Mol. Biol. Evol. 25, 603–615. doi: 10.1093/molbev/msn009
Cheng, Y., He, X., Priyadarshani, S. V. G. N., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 22, 1–15. doi: 10.1186/s12864-021-07490-9
Chun, J., Jin, M., Jeong, N., Cho, C., Seo, M. S., Choi, M. S., et al. (2019). Genetic identification and phylogenic analysis of new varieties and 149 Korean cultivars using 27 InDel markers selected from dense variation blocks in soybean (Glycine max (L.) Merrill). Korean J. Plant Res. 32, 519–542. doi: 10.7732/kjpr.2019.32.5.519
Chun-Yu, C. H. E. N., Shu-Ying, L. I. U., Yan, Y. A. N., Le, Y. I. N., Peng, D. I., Hui-Min, L. I. U., et al. (2020). Candidate genes involved in the biosynthesis of lignan in Schisandra chinensis fruit based on transcriptome and metabolomes analysis. Chin. J. Nat. Med. 18, 684–695. doi: 10.1016/S1875-5364(20)60007-3
Covello, P. S., Gray, M. W. (1989). RNA Editing in plant mitochondria. Nature 341, 662–666. doi: 10.1038/341662a0
Cui, H., Ding, Z., Zhu, Q., Wu, Y., Qiu, B., Gao, P. (2021). Comparative analysis of nuclear, chloroplast, and mitochondrial genomes of watermelon and melon provides evidence of gene transfer. Sci. Rep. 11, 1–9. doi: 10.1038/s41598-020-80149-9
Dong, S., Zhao, C., Chen, F., Liu, Y., Zhang, S., Wu, H., et al. (2018). The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 19, 1–12. doi: 10.1186/s12864-018-4991-4
Drouin, G., Daoud, H., Xia, J. (2008). Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 49, 827–831. doi: 10.1016/j.ympev.2008.09.009
Fay, J. C., Wu, C. I. (2003). Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 4, 213–235. doi: 10.1146/annurev.genom.4.020303.162528
Gandini, C. L., Sanchez-Puerta, M. V. (2017). Foreign plastid sequences in plant mitochondria are frequently acquired via mitochondrion-to-mitochondrion horizontal transfer. Sci. Rep. 7, 1–8. doi: 10.1038/srep43402
Gray, M. (2003). Diversity and evolution of mitochondrial RNA editing systems. IUBMB Life 55, 227–233. doi: 10.1080/1521654031000119425
Gray, M. W., Burger, G., Lang, B. F. (1999). Mitochondrial evolution. Science 283, 1476–1481. doi: 10.1126/science.283.5407.1476
Gualberto, J. M., Mileshina, D., Wallet, C., Niazi, A. K., Weber-Lotfi, F., Dietrich, A. (2014). The plant mitochondrial genome: dynamics and maintenance. Biochimie 100, 107–120. doi: 10.1016/j.biochi.2013.09.016
Gualberto, J. M., Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
Guang-Xin, E., Hong, Q. H., Zhao, Y. J., Ma, Y. H., Chu, M. X., Zhu, L., et al. (2019). Genetic diversity estimation of yunnan indigenous goat breeds using microsatellite markers. Ecol. Evol. 9, 5916–5924. doi: 10.1002/ece3.5174
Gui, S., Wu, Z., Zhang, H., Zheng, Y., Zhu, Z., Liang, D., et al. (2016). The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci. Rep. 6, 1–11. doi: 10.1038/srep30158
Guo, W., Zhu, A., Fan, W., Mower, J. P. (2017). Complete mitochondrial genomes from the ferns Ophioglossum californicum and Psilotum nudum are highly repetitive with the largest organellar introns. New Phytol. 213, 391–403. doi: 10.1111/nph.14135
Han, F., Qu, Y., Chen, Y., Bi, C. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Salix wilsonii using PacBio HiFi sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1031769
Hao, J., Liang, Y., Su, Y., Wang, T. (2022). The complete mitochondrial genome of Ophioglossum vulgatum l. @ is with highly repetitive sequences: intergenomic fragment transfer and phylogenetic analysis. Genes 13, 1287. doi: 10.3390/genes13071287
Hao, W., Liu, G., Wang, W., Shen, W., Zhao, Y., Sun, J., et al. (2021). RNA Editing and its roles in plant organelles. Front. Genet. 1747. doi: 10.3389/fgene.2021.757109
Hao, W., Palmer, J. D. (2009). Fine-scale mergers of chloroplast and mitochondrial genes create functional, transcompartmentally chimeric mitochondrial genes. Proc. Natl. Acad. Sci. 106, 16728–16733. doi: 10.1073/pnas.0908766106
Hoch, B., Maier, R. M., Appel, K., Igloi, G. L., Kössel, H. (1991). Editing of a chloroplast mRNA by creation of an initiation codon. Nature 353, 178–180. doi: 10.1038/353178a0
Hong, C. P., Kim, C. K., Lee, D. J., Jeong, H. J., Lee, Y., Park, S. G., et al. (2022). Long-read transcriptome sequencing provides insight into lignan biosynthesis during fruit development in Schisandra chinensis. BMC Genom. 23, 1–14. doi: 10.1186/s12864-021-08253-2
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486–487. doi: 10.1016/S0168-9525(02)02722-1
Jeong, H. J., Lee, J. B., Gil, J. S., Hong, C. P., Kang, S. H., Kwon, S. J., et al. (2021). Development of chloroplast-based InDel markers for the discrimination of schisandraceae plant species. Korean J. Med. Crop Sci. 29, 11–16. doi: 10.7783/KJMCS.2021.29.1.11
Jiang, K., Song, Q. Y., Peng, S. J., Zhao, Q. Q., Li, G. D., Li, Y., et al. (2015). New lignans from the roots of Schisandra sphenanthera. Fitoterapia 103, 63–70. doi: 10.1016/j.fitote.2015.03.015
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinf. 20, 1160–1166. doi: 10.1093/bib/bbx108
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, J. S., Jang, H. W., Kim, J. S., Kim, H. J., Kim, J. H. (2012). Molecular identification of Schisandra chinensis and its allied species using multiplex PCR based on SNPs. Genes Genomics 34, 283–290. doi: 10.1007/s13258-011-0201-3
Kim, J. Y., Kim, C. S., You, D. H., Kim, D. W., Choi, D. C., Kim, J. M., et al. (2014). Cuttings for mass propagation affecting the impact of increasing reproductive efficiency of Schisandra chinensis. Korean J. Med. Crop Sci. 22, 231–236. doi: 10.7783/KJMCS.2014.22.3.231
Kim, K., Lee, S. C., Lee, J., Yu, Y., Yang, K., Choi, B. S., et al. (2015). Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 5, 1–13. doi: 10.1038/srep15655
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PloS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kroemer, G., Reed, J. C. (2000). Mitochondrial control of cell death. Nat. Med. 6, 513–519. doi: 10.1038/74994
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lang, B. F., Gray, M. W., Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397. doi: 10.1146/annurev.genet.33.1.351
Lee, H. J., Kim, C. Y. (2010). Simultaneous determination of nine lignans using pressurized liquid extraction and HPLC-DAD in the fruits of Schisandra chinensis. Food Chem. 120, 1224–1228. doi: 10.1016/j.foodchem.2009.11.068
Lee, Y. M., Moon, B. C., Ji, Y., Seo, H. S., Kim, H. K. (2013). Development of RAPD-derived SCAR markers and multiplex-PCR for authentication of the Schisandrae fructus. Korean J. Med. Crop Sci. 21, 165–173. doi: 10.7783/KJMCS.2013.21.3.165
Leitch, I. J., Hanson, L. (2002). DNA C-values in seven families fill phylogenetic gaps in the basal angiosperms. Bot. J. Linn. Soc 140, 175–179. doi: 10.1046/j.1095-8339.2002.00096.x
Lenz, H., Hein, A., Knoop, V. (2018). Plant organelle RNA editing and its specificity factors: enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinform. 19, 1–18. doi: 10.1186/s12859-018-2244-9
Lenz, H., Knoop, V. (2013). PREPACT 2.0: predicting c-to-U and U-to-C RNA editing in organelle genome sequences with multiple references and curated RNA editing annotation. Bioinform. Biol. Insights 7, BBI–S11059. doi: 10.4137/BBI.S1105
Lenz, H., Rüdinger, M., Volkmar, U., Fischer, S., Herres, S., Grewe, F., et al. (2010). Introducing the plant RNA editing prediction and analysis computer tool PREPACT and an update on RNA editing site nomenclature. Curr. Genet. 56, 189–201. doi: 10.1007/s00294-009-0283-5
Li, H., Fan, C., Liu, J., Wang, B., Li, H. (2022). Integration of full-length transcriptomes and anthocyanin metabolite analysis for understanding fruit coloration mechanism in Schisandra chinensis. Physiol. Mol. Biol. Plants, 28, 921–933. doi: 10.1007/s12298-022-01179-3
Li, Z., He, X., Liu, F., Wang, J., Feng, J. (2018). A review of polysaccharides from Schisandra chinensis and Schisandra sphenanthera: properties, functions and applications. Carbohydr. Polym. 184, 178–190. doi: 10.1016/j.carbpol.2017.12.058
Liu, J., Qi, Y., Lai, H., Zhang, J., Jia, X., Liu, H., et al. (2014). Genus Kadsura, a good source with considerable characteristic chemical constituents and potential bioactivities. Phytomedicine 21, 1092–1097. doi: 10.1016/j.phymed.2014.01.015
Liu, X., Zhang, L., Yang, S. (2022). Analysis of floral organ development and sex determination in Schisandra chinensis by scanning electron microscopy and RNA-sequencing. Life 12, 1260. doi: 10.3390/life12081260
Lohse, M., Drechsel, O., Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Lovin, D. D., Washington, K. O., deBruyn, B., Hemme, R. R., Mori, A., Epstein, S. R., et al. (2009). Genome-based polymorphic microsatellite development and validation in the mosquito Aedes aegypti and application to population genetics in Haiti. BMC Genom. 10, 1–9. doi: 10.1186/1471-2164-10-590
Ma, Q., Wang, Y., Li, S., Wen, J., Zhu, L., Yan, K., et al. (2022). Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 22, 1–17. doi: 10.1186/s12870-021-03416-5
Martin, W., Stoebe, B., Goremykin, V., Hansmann, S., Hasegawa, M., Kowallik, K. V. (1998). Gene transfer to the nucleus and the evolution of chloroplasts. Nature 393, 162–165. doi: 10.1038/30234
Miyata, S. I., Nakazono, M., Hirai, A. (1998). Transcription of plastid-derived tRNA genes in rice mitochondria. Curr. Genet. 34, 216–220. doi: 10.1007/s002940050389
Morgante, M., Hanafey, M., Powell, W. (2002). Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 30, 194–200. doi: 10.1038/ng822
Mower, J. P., Sloan, D. B., Alverson, A. J., Wendel, J. F. (2012). “Plant genome diversity,” in “Plant mitochondrial genome diversity: the genomics revolution,” (Vienna, Austria: Springer), 123–144.
Nakazono, M., Nishiwaki, S., Tsutsumi, N., Hirai, A. (1996). A chloroplast-derived sequence is utilized as a source of promoter sequences for the gene for subunit 9 of NADH dehydrogenase (nad9) in rice mitochondria. Mol. Gen. Genet. 252, 371–378. doi: 10.1007/BF02173001
Nam, S. Y., Lee, J. Y., Ko, J. S., Kim, J. B., Jang, H. H., Kim, H. R., et al. (2014). Changes in antioxidant and antimicrobial activities of Schizandra chinensis baillon under different solvent extraction. Korean J. Int. Agric. 26, 513–518. doi: 10.12719/KSIA.2014.26.4.513
Newton, K. J. (1988). Plant mitochondrial genomes: organization, expression and variation. Annu. Rev. Plant Physiol. 39, 503–532. doi: 10.1146/annurev.pp.39.060188.002443
Niu, L., Zhang, Y., Yang, C., Yang, J., Ren, W., Zhong, X., et al. (2022). Complete mitochondrial genome sequence and comparative analysis of the cultivated yellow nutsedge. Plant Genome 15, e20239. doi: 10.1002/tpg2.20239
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (Oryza sativa l.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics 268, 434–445. doi: 10.1007/s00438-002-0767-1
Palmer, J. D., Adams, K. L., Cho, Y., Parkinson, C. L., Qiu, Y. L., Song, K. (2000). Dynamic evolution of plant mitochondrial genomes: mobile genes and introns and highly variable mutation rates. Proc. Natl. Acad. Sci. 97, 6960–6966. doi: 10.1073/pnas.97.13.6960
Petersen, G., Cuenca, A., Zervas, A., Ross, G. T., Graham, S. W., Barrett, C. F., et al. (2017). Mitochondrial genome evolution in alismatales: size reduction and extensive loss of ribosomal protein genes. PloS One 12, e0177606. doi: 10.1371/journal.pone.0177606
Powell, W., Morgante, M., Andre, C., Hanafey, M., Vogel, J., Tingey, S., et al. (1996). The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 2, 225–238. doi: 10.1007/BF00564200
Qiu, Y. L., Lee, J., Bernasconi-Quadroni, F., Soltis, D. E., Soltis, P. S., Zanis, M., et al. (1999). The earliest angiosperms: evidence from mitochondrial, plastid and nuclear genomes. Nature 402, 404–407. doi: 10.1038/46536
Renner, S. S., Bellot, S. (2012). Horizontal gene transfer in eukaryotes: fungi-to-plant and plant-to-plant transfers of organellar DNA. Genomics Chloroplasts Mitochondria, 223–235. doi: 10.1007/978-94-007-2920-9_10
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 342, 1468–1473. doi: 10.1126/science.1246275
Richardson, A. O., Rice, D. W., Young, G. J., Alverson, A. J., Palmer, J. D. (2013). The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11, 1–17. doi: 10.1186/1741-7007-11-29
Sebbenn, A. M., Blanc-Jolivet, C., Mader, M., Meyer-Sand, B. R., Paredes-Villanueva, K., Honorio Coronado, E. N., et al. (2019). Nuclear and plastidial SNP and INDEL markers for genetic tracking studies of Jacaranda copaia. Conserv. Genet. Resour. 11, 341–343. doi: 10.1007/s12686-019-01097-9
Shi, X., Germain, A., Hanson, M. R., Bentolila, S. (2016). RNA Recognition motif-containing protein ORRM4 broadly affects mitochondrial RNA editing and impacts plant development and flowering. Plant Physiol. 170, 294–309. doi: 10.1104/pp.15.01280
Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. United States America 112, E3515–E3524. doi: 10.1073/pnas.1504491112
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Sloan, D. B., Wu, Z. (2014). History of plastid DNA insertions reveals weak deletion and AT mutation biases in angiosperm mitochondrial genomes. Genome Biol. Evol. 6, 3210–3221. doi: 10.1093/gbe/evu253
Sohn, H. B., Kim, S. J., Hwang, T. Y., Park, H. M., Lee, Y. Y., Koo, B. C., et al. (2017). Chromosome reshuffling patterns of Korean soybean cultivars using genome-wide 202 InDel markers. Korean J. Breed. Sci. 49, 213–223. doi: 10.9787/KJBS.2017.49.3.213
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Takemura, M., Oda, K., Yamato, K., Ohta, E., Nakamura, Y., Nozato, N., et al. (1992). Gene clusters for ribosomal proteins in the mitochondrial genome of a liverwort, Marchantia polymorpha. Nucleic Acids Res. 20, 3199–3205. doi: 10.1093/nar/20.12.3199
Takenaka, M., Zehrmann, A., Verbitskiy, D., Härtel, B., Brennicke, A. (2013). RNA Editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352. doi: 10.1146/annurev-genet-111212-133519
Tang, W., Luo, C. (2018). Molecular and functional diversity of RNA editing in plant mitochondria. Mol. Biotechnol. 60, 935–945. doi: 10.1007/s12033-018-0126-z
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Turmel, M., Otis, C., Lemieux, C. (2007). An unexpectedly large and loosely packed mitochondrial genome in the charophycean green alga Chlorokybus atmophyticus. BMC Genom. 8, 1–12. doi: 10.1186/1471-2164-8-137
Unseld, M., Marienfeld, J. R., Brandt, P., Brennicke, A. (1997). The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 15, 57–61. doi: 10.1038/ng0197-57
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3–new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115. doi: 10.1093/nar/gks596
van Loo, G., Saelens, X., Van Gurp, M., MacFarlane, M., Martin, S. J., Vandenabeele, P. (2002). The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 9, 1031–1042. doi: 10.1038/sj.cdd.4401088
Wallace, D. C., Zheng, X., Lott, M. T., Shoffner, J. M., Hodge, J. A., Kelley, R. I., et al. (1988). Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 55, 601–610. doi: 10.1016/0092-8674(88)90218-8
Wang, S., Li, D., Yao, X., Song, Q., Wang, Z., Zhang, Q., et al. (2019). Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 11, 1192–1206. doi: 10.1093/gbe/evz063
Wang, D., Rousseau-Gueutin, M., Timmis, J. N. (2012). Plastid sequences contribute to some plant mitochondrial genes. Mol. Biol. Evol. 29, 1707–1711. doi: 10.1093/molbev/mss016
Wang, D., Wu, Y. W., Shih, A. C. C., Wu, C. S., Wang, Y. N., Chaw, S. M. (2007). Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Biol. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wee, C. C., Nor Muhammad, N. A., Subbiah, V. K., Arita, M., Nakamura, Y., Goh, H. H. (2022). Mitochondrial genome of Garcinia mangostana l. variety mesta. Sci. Rep. 12, 1–9. doi: 10.1038/s41598-022-13706-z
Wynn, E. L., Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3 9, 549–559. doi: 10.1534/g3.118.200948
Xu, Y., Bi, C. (2018). The complete mitochondrial genome sequence of an alpine plant Arabis alpina. Mitochondrial DNA Part B. 3, 725–727. doi: 10.1080/23802359.2018.1483758
Yang, J., Harding, T., Kamikawa, R., Simpson, A. G., Roger, A. J. (2017). Mitochondrial genome evolution and a novel RNA editing system in deep-branching heteroloboseids. Genome Biol. Evol. 9, 1161–1174. doi: 10.1093/gbe/evx086
Yang, Z., Ni, Y., Lin, Z., Yang, L., Chen, G., Nijiati, N., et al. (2022). De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas [L.] lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 22, 1–12. doi: 10.1186/s12870-022-03665-y
Yi, H., Chen, Y., Liu, J., Zhang, J., Guo, W., Xiao, W., et al. (2016). Extraction and separation of active ingredients in Schisandra chinensis (Turcz.) baill and the study of their antifungal effects. PloS One 11, e0154731. doi: 10.1371/journal.pone.0154731
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, G. K. S., Yu, J. (2006). KaKs_Calculator: calculating ka and ks through model selection and model averaging. Genomics Proteomics Bioinf. 4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
Zhang, Z., Xiao, J., Wu, J., Zhang, H., Liu, G., Wang, X., et al. (2012). ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 419, 779–781. doi: 10.1016/j.bbrc.2012.02.101
Zhang, D., Xing, Y., Xu, L., Zhao, R., Yang, Y., Zhang, T., et al. (2020). Comparative analysis of the mitochondrial genome sequences of two medicinal plants: Arctium lappa and A. tomentosum. Res. Square. doi: 10.21203/rs.3.rs-28443/v1
Zhao, N., Wang, Y., Hua, J. (2018). The roles of mitochondrion in intergenomic gene transfer in plants: a source and a pool. Int. J. Mol. Sci. 19, 547. doi: 10.3390/ijms19020547
Zhou, Y., Men, L., Sun, Y., Wei, M., Fan, X. (2021). Pharmacodynamic effects and molecular mechanisms of lignans from Schisandra chinensis Turcz.(Baill.), a current review. Eur. J. Pharmacol. 892, 173796. doi: 10.1016/j.ejphar.2020.173796
Zhu, Q., Dugardeyn, J., Zhang, C., Mühlenbock, P., Eastmond, P. J., Valcke, R., et al. (2014). The Arabidopsis thaliana RNA editing factor SLO2, which affects the mitochondrial electron transport chain, participates in multiple stress and hormone responses. Mol. Plant 7, 290–310. doi: 10.1093/mp/sst102
Keywords: Schisandra repanda, Kadsura japonica, mitogenome, genome assembly, InDel markers
Citation: Lee HJ, Lee Y, Lee S-C, Kim C-K, Kang J-N, Kwon S-J and Kang S-H (2023) Comparative analysis of mitochondrial genomes of Schisandra repanda and Kadsura japonica. Front. Plant Sci. 14:1183406. doi: 10.3389/fpls.2023.1183406
Received: 10 March 2023; Accepted: 09 June 2023;
Published: 03 July 2023.
Edited by:
Jeffrey P. Mower, University of Nebraska-Lincoln, United StatesReviewed by:
Xiaohua Jin, Institute of Botany, Chinese Academy of Sciences (CAS), ChinaBaosheng Liao, Guangzhou University of Chinese Medicine, China
Runxian Yu, Institute of Botany, Chinese Academy of Sciences (CAS), China
Copyright © 2023 Lee, Lee, Lee, Kim, Kang, Kwon and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sang-Ho Kang, aG9zYW5nOTNAa29yZWEua3I=