
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 09 May 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1179009
This article is part of the Research TopicStructural Variation of the Chloroplast Genome and Related Bioinformatics ToolsView all 11 articles
 Xiaomin Wang1,2,3*
Xiaomin Wang1,2,3* Shengyi Bai1
Shengyi Bai1 Zhaolei Zhang4
Zhaolei Zhang4 Fushun Zheng1Lina Song1Lu Wen1
Fushun Zheng1Lina Song1Lu Wen1 Meng Guo1,2,3Guoxin Cheng1,2,3Wenkong Yao1,2,3Yanming Gao1,2,3Jianshe Li1,2,3*
Meng Guo1,2,3Guoxin Cheng1,2,3Wenkong Yao1,2,3Yanming Gao1,2,3Jianshe Li1,2,3*In order to compare and analyze the chloroplast (cp) genomes of tomato germplasms and understand their phylogenetic relationships, the cp genomes of 29 tomato germplasms were sequenced and analyzed in this study. The results showed highly conserved characteristics in structure, number of gene and intron, inverted repeat regions, and repeat sequences among the 29 cp genomes. Moreover, single-nucleotide polymorphism (SNP) loci with high polymorphism located at 17 fragments were selected as candidate SNP markers for future studies. In the phylogenetic tree, the cp genomes of tomatoes were clustered into two major clades, and the genetic relationship between S. pimpinellifolium and S. lycopersicum was very close. In addition, only rps15 showed the highest average KA/KS ratio in the analysis of adaptive evolution, which was strongly positively selected. It may be very important for the study of adaptive evolution and breeding of tomato. In general, this study provides valuable information for further study of phylogenetic relationships, evolution, germplasm identification, and molecular marker-assisted selection breeding of tomato.
Cultivated tomato (Solanum lycopersicum) is an annual or perennial herb, which is a model system for Solanaceae and fruiting vegetables. The origin center of tomato is the Andes Mountains of South America, which is native to Peru, Ecuador, and other places in South America (Aoki et al., 2010). It has become one of the major cultivated vegetables in the world and contains rich nutrients. Significant differences in the nutrient composition of different varieties were detected by using high-performance liquid chromatography and electrochemical methods (Wang et al., 2023). Previous studies have shown that S. pimpinellifolium is the ancestor of cultivated tomatoes (Lin et al., 2014b), and S. habrochaites is an important wild relative of cultivated tomato, which has a variety of excellent disease resistance and stress resistance traits (Safari et al., 2021). Wild germplasm resources are widely used in modern tomato breeding. Therefore, it is necessary to identify the phylogenetic relationships among tomato germplasms by chloroplast (cp) genome sequencing, assembly, and annotation.
Chloroplast is the main place for energy conversion and photosynthesis of plants (Sadali et al., 2019). Compared with the large nuclear genome, the cp genome is smaller, and the copy number is more (Ashworth and Vanessa, 2017). The genome of cp is generally maternal inheritance; there is no problem of gene recombination (Li et al., 2014; Zhao et al., 2015). In recent years, the cp genome has been mainly used in phylogenetic, population genetics, and phylogeography studies (Wang et al., 2022b), in which plant phylogenetic analysis is the most basic cp genome analysis, which has been widely used in the phylogenetic study of the plant kingdom or one of the groups and to identify the relationships of angiosperm order, family, genus, interspecies, and intraspecies (Sun et al., 2020; Guo et al., 2021; Li et al., 2021; Xie et al., 2021; Liu et al., 2022; Raman et al., 2022; Wang et al., 2022a). The cp genome has unique advantages in plant phylogenetic studies. It had been favored in the study of plant molecular systematics because of its distinctive differences of molecular evolution rates in different regions, moderate nucleic acid replacement rates, and easily accessible sequences. The cp genome contains a large number of functional genes related to photosynthesis, gene expression, and other biosynthesis. Most of the cytoplasmically inherited traits are maternally inherited, and the development of cp molecular markers associated with maternally inherited traits has important applications in molecular marker-assisted selection breeding. Studies have shown that yellowing is the most common mutant phenotype in Chinese cabbage (Brassica campestris ssp. pekinensis), which is mostly inherited maternally. The mutated gene is the cp 16S small subunit protein gene rps4, and the presence of a single-nucleotide polymorphism (SNP) (A–C) with a mutation rate higher than 99% in the coding region of rps4 resulted in the conversion of the RPS4 protein’s 193rd amino acid Val to Gly (Tang et al., 2018). Li et al. (2011) hypothesized that the upregulated expression of the RPS15a gene may be associated with the occurrence of the multi-ovary in wheat. Therefore, comparing cp genomes can identify some important variations in the evolution of species and provide a theoretical basis for the study of species relatedness and interspecific identification, while more cp molecular markers associated with maternal genetic traits can be developed for molecular marker-assisted selection breeding.
As an important family of plants, Solanaceae plants include many edible and medicinal plants (Martins da Silva et al., 2014), but there are only a few reports on the study of tomato cp genome. Daniell et al. (2006) conducted a comparative analysis of the complete cp genomes of wild potato (Solanum bulbocastanum), tomato (S. lycopersicum), tobacco (Nicotiana tabacum), and Atropa (Atropa belladonna). The results showed that deletions or insertions within some intergenic spacer regions result in less than 25% sequence identity. They can be used as an effective chloroplast marker in low-level phylogenetic studies. Chung et al. (2006) sequenced the cp genome of a cultivated potato (Solanum tuberosum) and compared it with the cp genome of six other Solanaceae plants, including S. lycopersicum. The results showed that there was a 241-bp deletion in the large single copy region (LSC) of cultivated potato, which could be used as a new method to identify cultivated potato and wild potato. Rachele et al. (2020) carried out a cp genome analysis with tomato as main material at first time. They sequenced seven cp genomes of cultivated accessions from Southern Italy and two wild species among the closest (S. pimpinellifolium) and most distantly related (S. neorickii) species to cultivated tomatoes. In total, 11 tomato cp genome sequences were retrieved in GenBank for comparative analysis with the abovementioned set. Finally, they found that S. pimpinellifolium was the nearest ancestor of all cultivated tomatoes. The local materials were closely related to other cultivated tomatoes. However, the SNP loci that could be used for future research were not screened, and adaptive evolution analysis was not carried out in the abovementioned study. Moreover, the research on the complete genome sequencing and analysis of tomato core germplasms from China and wild tomatoes’ cp genome has not been reported widely.
In view of the lack of classification and phylogenetic relationship between tomato interspecies and intraspecies, 29 tomato germplasms with different genetic backgrounds at home and abroad were screened from the core germplasms by our research group, and six wild resources were collected from Tomato Genetic Resource Center. The second-generation high-throughput sequencing technology was used to sequence the complete cp genome of tomato germplasms as well as assemble and annotate. Then, bioinformatics analysis was performed; a high-definition map of cp genome and a phylogenetic tree were constructed to clarify the phylogenetic relationship of tomato germplasms. Moreover, SNP loci with high polymorphism were selected as candidate SNP markers, and adaptive evolution analysis was conducted. This study will provide valuable information for further phylogenetic relationships, evolution, germplasm identification, and molecular marker-assisted selection breeding of tomato.
In this study, 29 tomato core collections were selected and cultivated in the experimental farm of Ningxia University (Table 1). The fresh and healthy leaves of 29 tomato germplasms were collected, and then the leaf tissue samples were frozen fresh at -80°C until DNA extraction. The total genomic DNA was extracted using the New Plant Genome Extraction Kit (DP320) (Tiangen, Beijing, China) according to the manufacturer’s instructions. The purity and integrity of the genomic DNA samples were identified using 1% agarose gel electrophoresis. The concentration of genomic DNA samples was measured using a NanoDrop 2000C spectrophotometer (Thermo Scientific; Waltham, MA, USA).
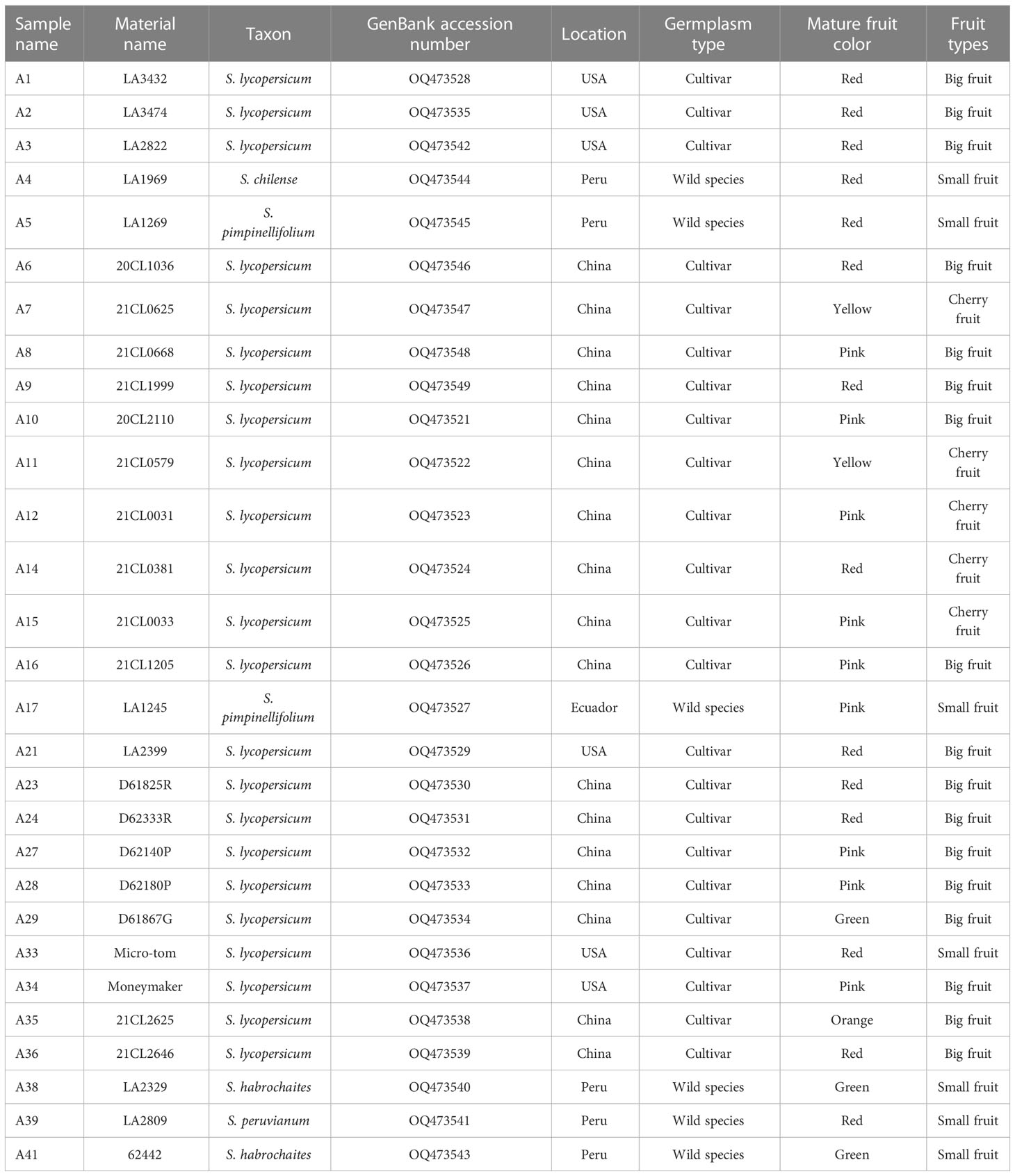
Table 1 Material sources of 29 tomato germplasms.
The high-throughput sequencing of 29 tomato germplasms was completed by Berry Genomics Co., Ltd. After the DNA samples were qualified, the genomic DNA was randomly cut into 350-bp fragments by enzyme digestion. After terminal repair and poly A addition, the sequencing adapters were connected at both ends of the fragments. Lastly, the libraries were analyzed for size distribution using agarose gels and were quantified using real-time PCR. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using Novaseq 6000 S4 Reagent Kit (Illumina) according to the manufacturer’s instructions. After cluster generation, the DNA libraries were sequenced on Illumina NovaSeq 6000 platform, and 150-bp paired-end reads were generated.
Repetitive sequences in the cp genome play a critical role in genome evolution and rearrangements. The simple sequence repeats (SSR) motifs were analyzed in the cp genome of 29 tomato germplasms using MISA v2.1 (Raman et al., 2022). The minimum repeat thresholds of 10, six, five, five, five, and five are for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs, respectively. Tandem repeats were analyzed using the TRF (Benson, 1999) software with default parameters (Liu et al., 2022). In addition, oligonucleotide repeat analysis of four types of repeats in the cp genome was carried out. The forward, reverse, complement, and palindromic repeats were detected using REPuter online software (Kurtz et al., 2001) with a minimum repeat size of 30 bp and 90% sequence identity (Hamming distance of 3).
The connecting regions of IR-LSC and IR-SSC in the cp genomes of 29 tomato germplasms were compared by using IRscope online software (https://irscope.shinyapps.io/irapp/) (Amiryousefi et al., 2018). The mVISTA online software (http://genome.lbl.gov/vista/mvista/submit.shtml) was used to compare the cp genomics of 29 tomato germplasms (Frazer et al., 2004). The comparative analysis was carried out by using the shuffle-LAGAN mode in mVISTA, and the sequence alignment was visualized in an mVISTA plot. The size of the sliding window is set to 100, and the default values for minimum and maximum Y are 50% to 100%. Mauve V2.4.0 was used to compare the cp genomes of 29 tomato germplasms to determine the collinearity of cp genome structure and identify possible rearrangements (Katoh et al., 2019). We also calculated the nucleotide diversity (Pi) of 80 protein-coding genes and intergenic spacer regions among the 29 tomato germplasms (Raman et al., 2022).
The coding sequences, intergenomic sequences, and the complete cp genomes of 29 tomato germplasms were selected to construct a phylogenetic tree, and Solanum bulbocastanum DQ347958 was selected as the outgroup. To analyze the phylogenetic relationship of tomato, alignments were used to construct the phylogenetic trees using the maximum likelihood (ML) method implemented in RAxML v8.2.12 with 1,000 bootstrap replicates (Nguyen et al., 2015).
In this study, the complete cp genomes of 29 tomato germplasms were compared. By extracting the same specific protein-encoded DNA sequence and translating it into a protein sequence, protein sequence alignment was performed using ParaAT3.0 software, and then the nucleic acid alignment result corresponding to a codon was translated back according to the protein alignment result. After the homologous sequence alignment, KaKs_Calculator 3.0 software was used to calculate the synonymous (KS) and nonsynonymous (KA) substitution rates and KA/KS ratios (Zhang, 2022).
The raw data were deposited in the Sequence Read Archive under BioProject accession number PRJNA936910. In this study, the cp genomes of 29 tomato germplasms were sequenced and characterized. The final cp genomes were assembled, annotated, and submitted to GenBank. The complete cp genomes of the 29 tomato germplasms ranged from 155,257 to 155,461 bp in length. Each cp genome was made up of three distinct regions (Figure 1). The length of the IR region ranged from 25,594 to 25,612 bp. The length of the LSC region ranged from 85,688 bp (A39 and A41) to 85,875 bp (A4), and the length of the SSC region ranged from 18,355 bp (A38, A39, and A41) to 18,375 bp (A4). Among 29 tomato germplasms, the total GC content of the cp genomes of A38 was the lowest (37.84%), while that of A5 was the highest (37.87%). The total GC content of the cp genome of each of the remaining 27 germplasms was 37.86% (Supplementary Table S1).
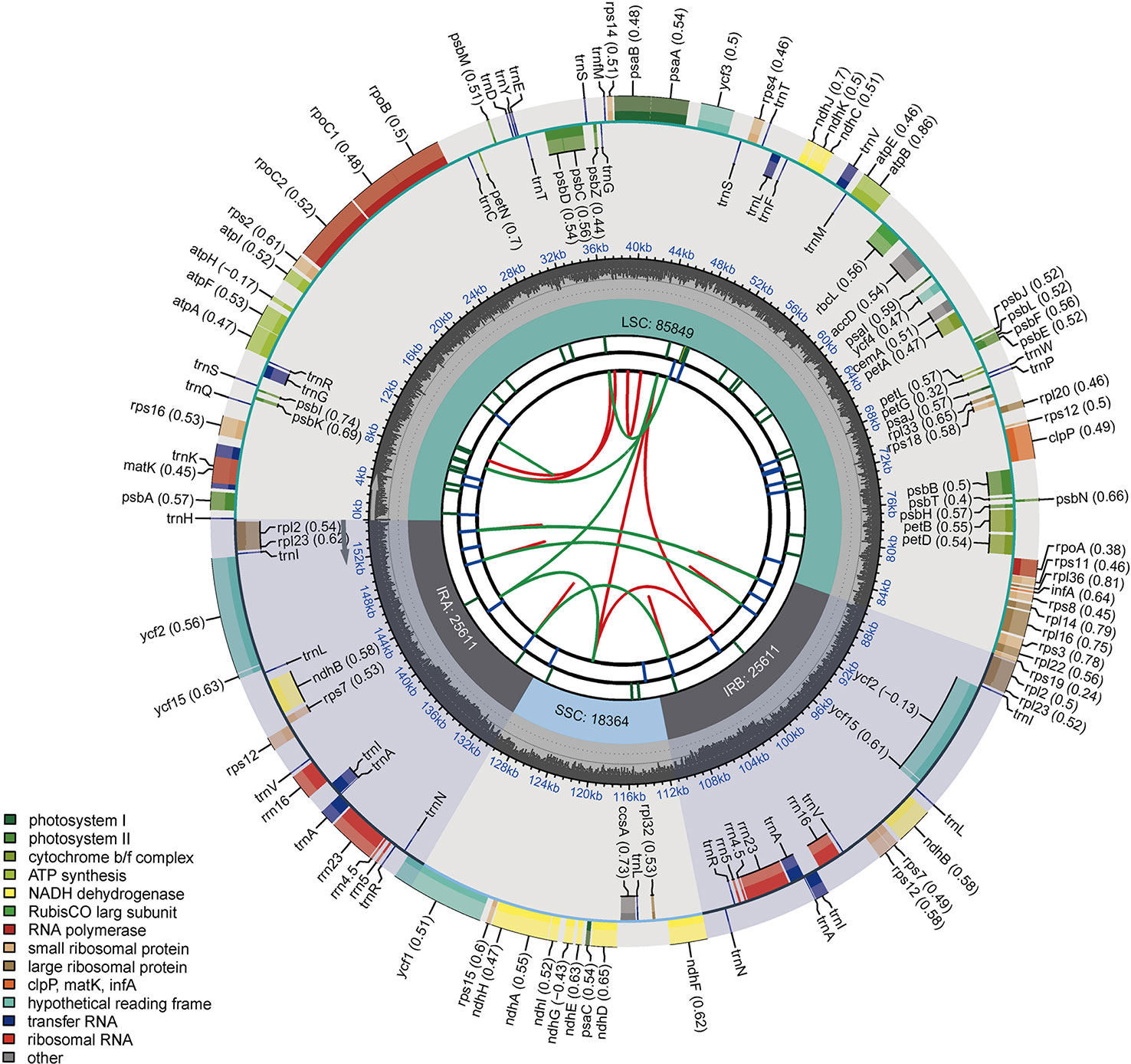
Figure 1 Circular chloroplast genome map of tomato. Genes drawn outside the circle are transcribed clockwise, and those inside are counterclockwise. Genes belonging to different functional groups are color-coded. The darker gray in the inner circle shows the GC content, while the lighter gray shows the AT content.
In addition, the number of genes and introns were highly conserved, and the same suite of protein-coding genes, ribosomal RNA (rRNA) genes, and transfer RNA (tRNA) genes was found in all taxa. Each cp genome included 113 unique genes, which contained 80 protein-coding genes, 29 tRNA genes, and four rRNA genes (Supplementary Table S2). In 113 genes, 18 genes with introns were identified. Among them, nine protein-coding (atpF, ndhA, ndhB, petB, petD, rpl16, rpl2, rpoC1, and rps16) and six tRNA genes (trnA-UGC, trnG-GCC, trnI-GAU, trnK-UUU, trnL-UAA and trnV-UAC) contained one intron, whereas clpP, rps12, and ycf3 contained two introns.
There were four borders between LSC, IRb, SSC, and IRa in the cp genome: LSC/IRb border, IRb/SSC border, SSC/IRa border, and IRa/LSC border. The borders of the 29 tomato germplasm cp genomes were compared (Figure 2). The four borders were conservative. The rpl22 gene was present in the LSC region, and rpl2 gene existed entirely in the IR region. Additionally, the rps19 gene straddled the boundary of the LSC/IRb regions. The ndhF gene was located at the IRb/SSC border, and the distance between ndhF and the JSB line was 17 bp. The ycf1 gene was observed at the JSA line, which straddled the boundary of the SSC/IRa regions. The trnH noncoding gene was located on the right side of the JLA line with a distance of 1 bp. In addition, the psbA gene existed in the LSC.

Figure 2 Comparison of the borders of large single copy (LSC), small single copy (SSC), and inverted repeat (IR) regions among 29 tomato germplasm chloroplast genomes. JLB line indicates the border between LSC and IRb, JSB line indicates the border between SSC and IRb, JSA line indicates the border between SSC and IRa, and JLA line indicates the border between LSC and IRa.
The amino acid frequency, codon usage, and relative synonymous codon usage (RSCU) of 80 protein-coding regions in 29 tomato germplasms were analyzed using Codon W. The RSCU values ranged from 63.99 to 64.03, the number of codons ranged from 23,010 to 23,016 in 29 tomato germplasms, and the number of amino acids ranged from 22,930 to 22,936. Of these amino acids, leucine (2,439–2,445 codons) was the most abundant amino acid, with a frequency of 10.63%–10.66%, while the frequency of cysteine (257 codons) was 1.12%. However, the most often used codon was ATT (encoding isoleucine), and the least used was TGA (termination codon). Almost all the amino acids had more than one synonymous codon; the exceptions were methionine and tryptophan. Furthermore, 62 codons displayed RSCU values exceeding 1.00. ATG and TGG, encoding methionine and tryptophan separately, exhibited no bias (RSCU = 1.00) (Supplementary Table S3). Moreover, three types of starting codons (ATG, GTG, and ACG) were detected in 80 protein-coding genes. Most genes used ATG as the starting codon. TAA, TAG, and TGA were present as stop codons in these genes. The most often used stop codon was TAA at 52.5%, followed by TAG (26.25%) and TGA (21.25%).
In this study, the repeat sequences of the cp genomes of 29 tomato germplasms were analyzed. The distribution of SSRs, tandem repeats, and dispersed repeats differs among the 29 tomato germplasms (Figure 3A). Furthermore, we identified the total number of SSRs per cp genome that ranges from 36 to 42. Mononucleotides were the most frequent in the SSRs, with a distribution of 97.36%, followed by dinucleotide and trinucleotides at 2.47% and 0.17%, respectively, in the 29 tomato germplasms (Figure 3B). There was a dinucleotide with a predominant motif of TA per cp genome in 29 tomato germplasms. Trinucleotides were absent in the cp genome of most tomato germplasms, except for a TTA motif in A4 and a TAA motif in A38. Moreover, the distribution of tandem repeats in the tomato cp genomes ranges from 26 to 30.
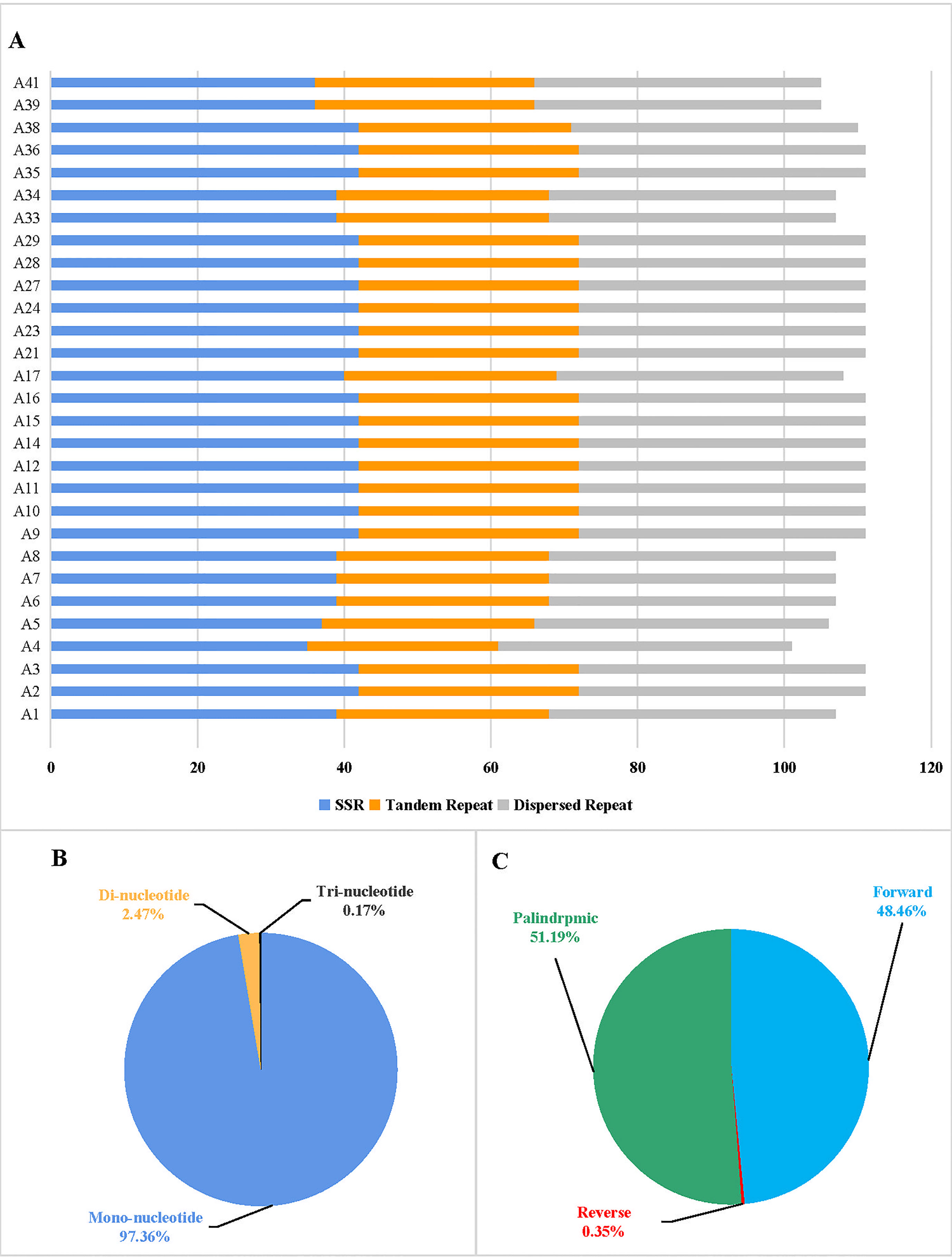
Figure 3 Histogram showing the number of repeats in 29 tomato germplasm chloroplast genomes. (A) Distribution of simple sequence repeats (SSRs), tandem repeats, and dispersed repeats in 29 tomato germplasms. (B) Proportion of different SSR repeat types. (C) Proportion of different dispersed repeat types.
In addition to SSRs and tandem repeats, a dispersed repeat analysis of four types of repeats in the cp genome, including forward (F), reverse (R), palindromic (P), and complementary (C), was performed using REPuter. The total number of 39 dispersed repeats was identified in each cp genome of most tomato germplasms, while A4 and A5 had 40 dispersed repeats. Among the tomato cp genomes, forward repeats and palindromic repeats were the most common, accounting for 48.46% and 51.19%, respectively (Figure 3C). Only A4, A5, A39, and A41 had one reverse repeat, respectively. Complement repeats were not observed in the cp genomes of 29 tomato germplasms (Supplementary Table S4).
Multiple alignments of 29 tomato cp genomes were conducted using the online software platform mVISTA. A comparison of overall sequence variation showed that the tomato cp genome is highly conserved. Only ycf1 open reading frame had a divergence in the coding region (Supplementary Figure S1). Subsequently, Mauve was used to identify the local collinear blocks (LCBs) of 29 tomato germplasm cp genomes (Supplementary Figure S2). These germplasms showed a consistent sequential order in all genes. The LCBs of all cp genomes showed a relatively high conservation with no gene rearrangement.
In addition, the nucleotide diversity (Pi) of 80 protein genes and intergenic spacer regions in the tomato cp genomes was calculated. A total of 80 genes were related to transcription, translation, and photosynthetic processes, with low variation (Figure 4A). Among these 80 genes, petL had the highest Pi value (0.0035), followed by rps15 (0.0024) and ycf1 (0.0015), showing obvious divergences. Meanwhile, 32 genes had a Pi value of 0. However, obvious divergences were detected in the intergenic spacer regions of psbI-trnS-GCU, rpl36-infA, infA-rps8, trnR-ACG-trnN-GUU, ccsA-ndhD, ndhH-rps15, rps15-ycf1, and trnN-GUU-trnR-ACG (Figure 4B). The Pi value of these six genes was above 0.003. In addition, SNP/MNP loci with high polymorphism located at 17 fragments were selected as candidate SNP markers for future studies (Table 2). Among the screened SNP markers, those localized to fragments of the ndhH gene and the ndhK-ndhC-trnV-UAC intergenic spacer regions could be used for interspecific identification.
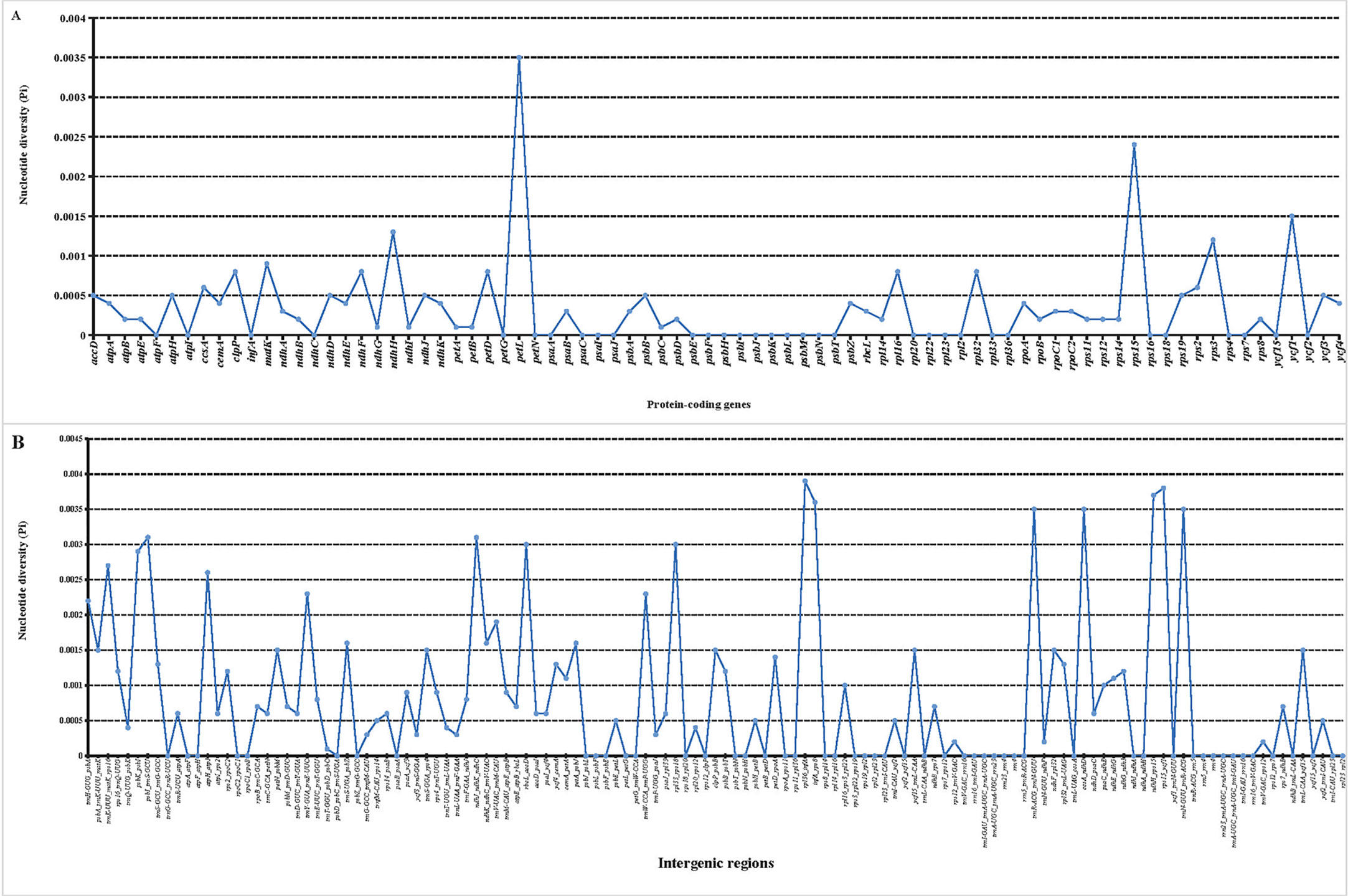
Figure 4 Nucleotide diversity (Pi) value in 29 tomato germplasm chloroplast genomes. (A) Pi value of protein-coding genes. (B) Pi value of intergenic spacer regions.
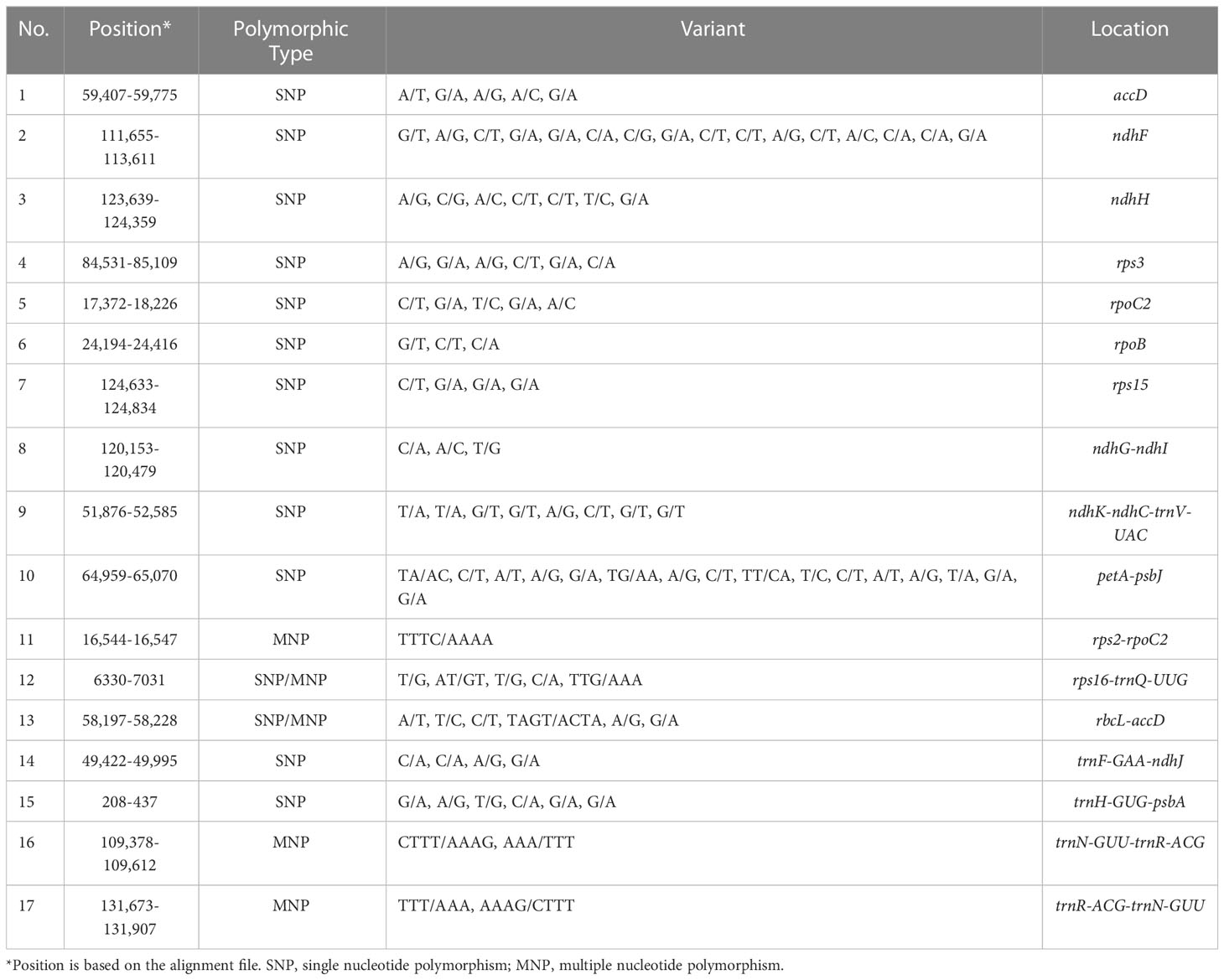
Table 2 Candidate polymorphic DNA markers among chloroplast genomes of 29 tomato germplasms.
In order to study the phylogenetic relationship among different tomato germplasms, ML phylogenetic trees were constructed using coding sequences, intergenomic sequences, and the complete cp genomes of 29 tomato germplasms (Figure 5). Solanum bulbocastanum DQ347958 selected as outgroups were retrieved from NCBI. In the phylogenetic tree, the tomatoes were clustered into two major clades based on coding sequences and the complete cp genomes, with one clade comprising all cultivated tomato and S. pimpinellifolium and the other clade containing the remaining four wild tomatoes. Furthermore, the first clade could be further divided into two minor clades, with all cultivated tomato clustered into the same minor clade and the other contained two S. pimpinellifolium, indicating a relatively closer interrelationship between cultivated tomato and S. pimpinellifolium. The second clade could also be divided into two minor clades, with one minor clade comprising only S. chilense and the other including S. habrochaites and S. peruvianum (Figures 5A, B). However, the phylogenetic results based on intergenomic sequences are different from the abovementioned results based on coding sequences and the complete cp genomes. Based on intergenomic sequences, the tomatoes were clustered into two major clades. The first can be further divided into two minor clades, with all cultivated tomatoes and two wild tomatoes (two S. pimpinellifolium and one S. chilense, respectively) clustered into the first minor clade and the other minor clade contained S. habrochaites A41 and S. peruvianum A39. S. habrochaites A38 was clustered separately as the second major clade. It indicated that S. chilense was more closely related to cultivated tomatoes than S. habrochaites and S. peruvianum.

Figure 5 Maximum likelihood phylogenetic tree of 29 tomato germplasms. (A) Based on protein-coding sequences. (B) Based on complete chloroplast genomes. (C) Based on intergenomic sequences. Values above the branches represent the maximum likelihood bootstrap value. .
In total, 49 protein-coding genes of all the 29 tomato cp genomes were used for the analysis of synonymous (KS) and non-synonymous (KA) substitution rates. The results showed that most protein-coding genes have relatively low average KS values (<0.008), except the petL genes (Figure 6B). In the same way, the average KA values of most protein-coding genes were comparatively low (<0.0015), except the rps15 and ycf1 genes (Figure 6A). The average KA/KS ratio of rps15 gene was the highest (2.41). Furthermore, the KA/KS ratios of all the protein-coding genes ranged from 0 to 2.41, with an average ratio of only 0.14 (Figure 6C).
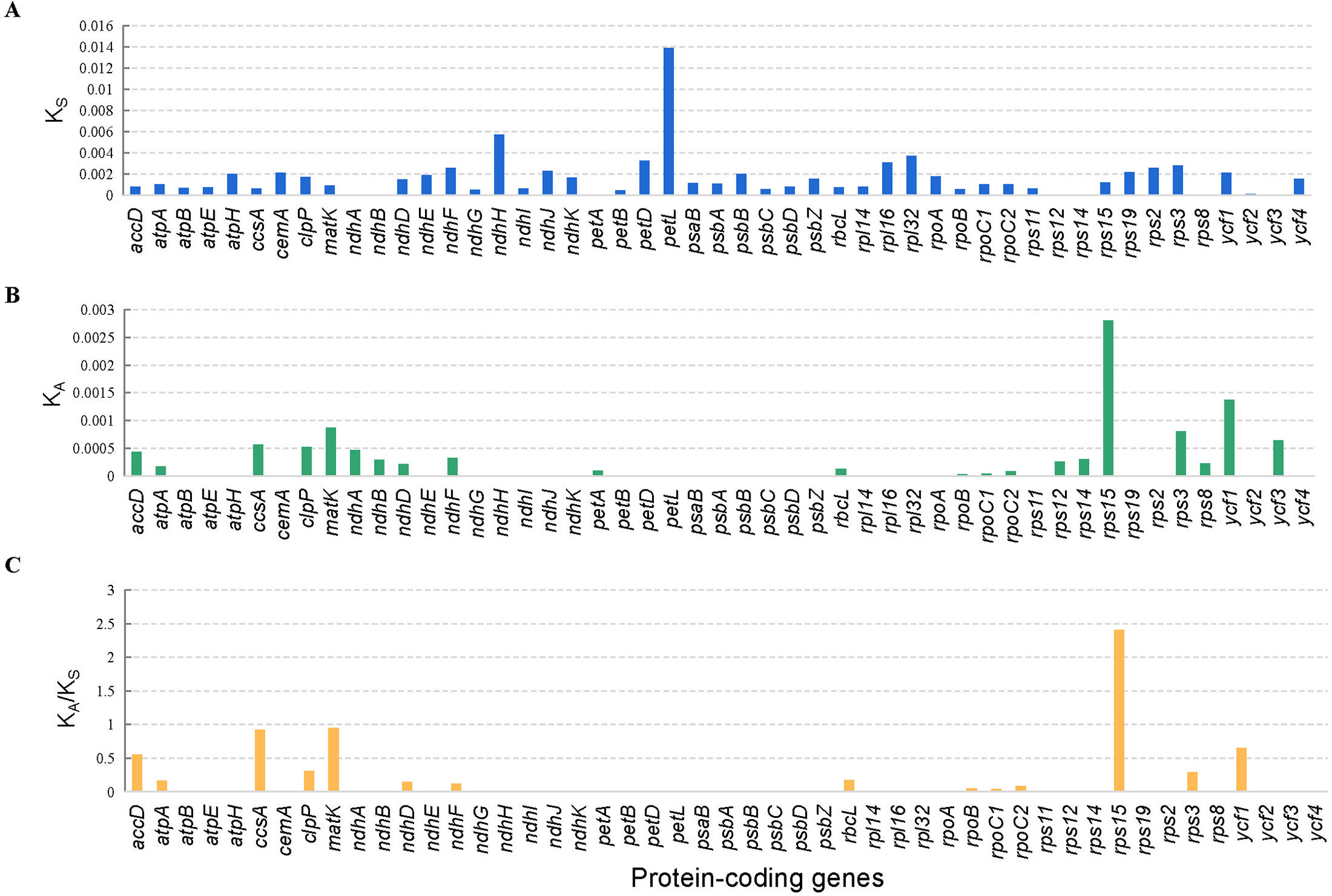
Figure 6 Selective pressure of 49 protein-coding genes in 29 tomato cp genomes. (A) KS, rate of synonymous substitution. (B) KA, rate of nonsynonymous substitution. (C) KA/KS, rate of non-synonymous vs. synonymous substitution.
Due to the slow evolution of plant cp genome, it has been used for plant classification and molecular evolution research. The study of species identification and phylogenetic evolution based on cp genome is a development trend of plant taxonomy biology, which has attracted more and more attention and recognition from researchers (Nock et al., 2015). The genetic background of tomato is more and more narrow due to the loss of genetic diversity, selfing characteristics, and long-term domestication in the process of tomato spreading from the origin to the world (Doebley et al., 2006). However, tomato wild relatives have a rich genetic diversity and are an inexhaustible gene library for the genetic improvement of tomato. So far, there are few reports on the comparative study of tomato cp genome. Rachele et al. (2020) compared and analyzed nine tomato cp genomes obtained by sequencing and 11 tomato cp genomes retrieved from GenBank. In this study, the cp genome sequences of 29 tomato germplasms were sequenced and compared, which also showed highly conserved characteristics in structure, number of gene and intron, inverted repeat regions, which was similar to the study of Rachele et al. (2020).
Repeats in the cp genome play an important role in genome evolution and rearrangements (Wang et al., 2022a). SSR can be used as a molecular marker and used in population genetics research because of its high polymorphism (Zhang et al., 2012; Zhao et al., 2015). However, among 29 tomato cp genomes, the number of mononucleotide repeats was the largest, accounting for the majority of all SSRs (97.36%). Therefore, polymorphisms existed in the SSRs of the tomato cp genome, but their repeat numbers were relatively conservative. Whether they could be used as molecular markers independently for population genetic analysis needs to be further validated. However, trinucleotide repeats were only present in A4 (S. chilense) and A38 (S. habrochaites) (TTA and TAA motifs, respectively), which may be used as SSR candidate markers for the study of subsequent germplasm identification. Moreover, the tandem repeats and dispersed repeats of tomato cp genome were relatively conservative. Previous studies have shown that repeats have a great influence on insertion and substitution, which can increase the genetic diversity of biological populations (Klein and O’Neill, 2018). The existence and abundance of cp repetitive sequences may also be related to multiple phylogenetic signals (Zhang et al., 2011; Wang et al., 2016), while the repetitive sequences on the tomato cp genome are relatively conservative, and whether it is related to phylogenetic signals needs further study.
In addition, 17 SNP/MNP loci with high polymorphism were selected as candidate SNP markers in this study. SNP molecular markers play an important role in tomato breeding, mainly in the identification of genetic relationship and genetic diversity of germplasm resources, the construction of genetic linkage map, the localization of target genes, and the identification of variety purity and molecular marker-assisted selection breeding (Kim et al., 2021). At present, the development of chloroplast SNP markers has been applied to many plants—for example, Cesare et al. (2010) identified six cp markers containing both cp SSRs and SNPs, and these SNP markers can distinguish most Miscanthus species and detect intraspecific variations, which can be used for breeding purposes. Lin et al. (2014a) developed four cp SNP molecular markers to distinguish soybean male sterile lines and maintainer lines and also to distinguish hybrids and soybean maintainer lines. The highly polymorphic SNP loci screened in this study were located at the regions of ndhG-ndhI, ndhK-ndhC, petA-psbJ, rps2-rpoC2, rps16-trnQ-UUG, rbcL-accD, trnF-GAA-ndhJ, trnH-GUG-psbA, trnN-GUU-trnR-ACG, and trnR-ACG-trnN-GUU and in accD, ndhF, ndhH, rps3, rpoC2, rpoB, and rps15 genes. They will be developed as candidate cp genome SNP molecular markers for future studies. Among the screened SNP markers, those localized to segments of the ndhH gene and the ndhK-ndhC-trnV-UAC gene spacer region could be used for interspecific identification. The other developed SNP marker can be used to analyze genetic diversity and population structure at the cp genome level and to develop functional markers associated with traits such as male sterility, which has an important application value in tomato germplasm identification and molecular marker-assisted selection breeding.
The cultivated tomatoes were domesticated from wild tomatoes. The fruit weight of modern cultivated tomato is more than 100 times that of its ancestors. In order to reveal the secret of tomatoes from small to large, Lin et al. (2014b) used genome-wide variation group data to analyze the phylogeny and population structure of tomatoes, and they found that the tomato population was divided into three subgroups, namely, S. pimpinellifolium, S. lycopersicum var. cerasiforme, and large-fruit cultivated tomato (S. lycopersicum). Combined with abundant phenotypic data and population genetics analysis, it was proved that wild tomato (S. pimpinellifolium) evolved into cherry tomato (S. lycopersicum var. cerasiforme) and finally formed a two-step artificial selection process of large-fruit cultivated tomato—namely, domestication and improvement. The comparative analysis of the 29 tomato germplasm cp genomes sequenced in this work allowed the phylogenetic relationships among wild and cultivated germplasms to be defined and also indicated that the genetic relationship between S. pimpinellifolium and S. lycopersicum was very close—that is, S. pimpinellifolium may be the ancestor of cultivated tomatoes—which was consistent with the previous research results. Notably, the phylogenetic relationships based on coding sequences and the complete cp genomes were consistent with the results based on traditional botanical classification (Zhao, 2012). However, the phylogenetic relationships based on intergenomic sequences were different from the results based on coding sequences and the complete cp genomes and traditional botanical classification, which may be due to the fact that intergenomic sequences contain more variability. The phylogenetic relationships based on intergenomic sequences show that S. chilense is more closely related to cultivated tomatoes and S. pimpinellifolium than S. habrochaites and S. peruvianum.
The KA/KS ratio is associated with gene adaptive evolution, such as positive selection and purity selection (Raman et al., 2022). In general, non-synonymous substitutions can cause amino acid changes, resulting in changes in protein conformation and function. Therefore, it will cause adaptive evolution and bring the advantages or disadvantages of natural selection. Synonymous substitution does not change the composition of the protein, so it is not affected by natural selection; then, Ks can reflect the background base substitution rate of the evolutionary process (Hurst, 2002). The KA/KS ratio can explain the type of selection of this gene. When KA/KS <<1, the gene is selected by purification. The KA/KS of most genes is far less than 1 because generally non-synonymous substitutions bring evolutionary disadvantages, and only a few cases will result in evolutionary advantages. When KA/KS >>1, the genes are strongly positively selected, and these genes are rapidly evolving recently and are of great significance for the evolution of species. We can screen some genes for further functional studies according to the KA/KS ratio, which has been widely applied to the field of molecular evolution (Navarro and Barton, 2003). In this study, the analysis results of synonymous (KS) and non-synonymous (KA) substitution rates showed that petL genes have relatively high average KS values. The rps15 and ycf1 genes have relatively high average KA. Only rps15 showed the highest average KA/KS ratio of 2.41, which was strongly positively selected. It may be a gene that is rapidly evolving recently, and its function can be further studied. It may be very important for the study of adaptive evolution and breeding of tomato.
In this study, the latest sequencing results of the chloroplast genomes of 29 tomato germplasms were reported and compared. Genome annotation and comparative analysis showed that each chloroplast genome was a typical tetragonal structure. The 29 chloroplast genomes are highly conserved in terms of structure, gene and intron number, IR region, and repeat sequences. In addition, we had screened SNP/MNP loci with high polymorphism located at 17 fragments of the regions of ndhG-ndhI, ndhK-ndhC, petA-psbJ, rps2-rpoC2, rps16-trnQ-UUG, rbcL-accD, trnF-GAA-ndhJ, trnH-GUG-psbA, trnN-GUU-trnR-ACG, and trnR-ACG-trnN-GUU and in accD, ndhF, ndhH, rps3, rpoC2, rpoB, and rps15 genes in the cp genomes of 29 tomato germplasms, which will be used as candidate SNP markers in future studies. In the phylogenetic tree, the cp genomes of tomatoes were clustered into two major clades, and the genetic relationship between S. pimpinellifolium and S. lycopersicum was very close. Moreover, in the analysis of adaptive evolution, only rps15 showed the highest average KA/KS ratio of 2.4, which was strongly positively selected. In general, this study will provide valuable information for further study of phylogenetic relationships, germplasm identification, and molecular marker-assisted selection breeding of tomato.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
XW and JL conceived and designed the study. FZ, LS, and LW collected and identified the plant materials. SB and ZZ performed the experiments and analyzed the data. XW and SB wrote the manuscript. XW, WY, MG, GC, and YG revised the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by Ningxia Hui Autonomous Region Agricultural Special and Dominant Industry Breeding Project (NXNYYZ20200104).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1179009/full#supplementary-material
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Aoki, K., Yano, K., Suzuki, A., Kawamura, S., Sakurai, N., Suda, K., et al. (2010). Large-Scale analysis of full-length cDNAs from the tomato (Solanum lycopersicum) cultivar micro-tom, a reference system for the solanaceae genomics. BMC Genomics 11, 210. doi: 10.1186/1471-2164-11-210
Ashworth, Vanessa, E. T. M. (2017). Revisiting phylogenetic relationships in phoradendreae (viscaceae): utility of the trnl-f region of chloroplast dna and presence of a homoplasious inversion in the intergenic spacer. Botany 95 (3), 1–12. doi: 10.1139/cjb-2016-0241
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Cesare, M. D., Hodkinson, T. R., Barth, S. (2010). Chloroplast DNA markers (cpSSRs, SNPs) for Miscanthus, Saccharum and related grasses (panicoideae, poaceae). Mol. Breeding 26 (3), 539–544. doi: 10.1007/s11032-010-9451-z
Chung, H. J., Jung, J. D., Park, H. W., Kim, J. H., Cha, H. W., Min, S. R., et al. (2006). The complete chloroplast genome sequences of solanum tuberosum and comparative analysis with solanaceae species identified the presence of a 241bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Rep. 25 (12), 1369–1379. doi: 10.1007/s00299-006-0196-4
Daniell, H., Lee, S. B., Grevich, J., Saski, C., Quesada-Vargas, T., Guda, C. (2006). Complete chloroplast genome sequences of solanum bulbocastanum, solanum lycopersicum and comparative analyses with other solanaceae genomes. Theor. Appl. Genet. 112 (8), 1503–1518. doi: 10.1007/s00122-006-0254-x
Doebley, J. F., Gaut, B. S., Smith, B. D. (2006). The molecular genetics of crop domestication. Cell. 127, 1309–1321. doi: 10.1016/j.cell.2006.12.006
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Inna, D. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Guo, X., Wang, Z., Zhang, Y., Wang, R. (2021). Chromosomal-level assembly of the Leptodermis oblonga (rubiaceae) genome and its phylogenetic implications. Genomics 113 (5), 3072–3082. doi: 10.1016/j.ygeno.2021.07.012
Hurst, L. D. (2002). The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18 (9), 486–487. doi: 10.1016/S0168-9525(02)02722-1
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kim, M., Jung, J. K., Shim, E. J., Chung, S. M., Park, Y., Lee, G. P., et al. (2021). Genome-wide SNP discovery and core marker sets for DNA barcoding and variety identification in commercial tomato cultivars. Scientia Horticulturae 276, 109734. doi: 10.1016/j.scienta.2020.109734
Klein, S. J., O’Neill, R. J. (2018). Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 26 (1), 5–23. doi: 10.1007/s10577-017-9569-5
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, X., Ma, S., Zhang, G., Niu, N. (2011). Cloning of ribosomal protein S15a gene (TaRPS15a) and its expression patterns based on temporal-spatial in multi-ovary line of wheat (Triticum aestivum). J. Agric. Biotechnol. 19 (2), 236–242. doi: 10.3969/j.issn.1674-7968.2011.02.006
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., Chen, S. (2014). Plant DNA barcoding: from gene to genome. Biol. Rev. 90 (1), 157–166. doi: 10.1111/brv.12104
Li, Q., Yu, Y., Zhang, Z., Wen, J. (2021). Comparison among the chloroplast genomes of five species of Chamaerhodos (rosaceae: potentilleae): phylogenetic implications. Nordic J. Botany 39 (6), e03121. doi: 10.1111/njb.03121
Lin, C., Zhang, C., Zhao, H., Xing, S., Wang, Y., Liu, X., et al. (2014a). Sequencing of the chloroplast genomes of cytoplasmic male-sterile and male-fertile lines of soybean and identification of polymorphic markers. Plant Science 229, 208–214. doi: 10.1016/j.plantsci.2014.09.005
Lin, T., Zhu, G., Zhang, J., Xu, X., Yu, Q., Zheng, Z., et al. (2014b). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226. doi: 10.1038/ng.3117
Liu, J., Shi, M., Zhang, Z., Xie, H., Kong, W., Wang, Q., et al. (2022). Phylogenomic analyses based on the plastid genome and concatenated nrDNA sequence data reveal cytonuclear discordance in genus Atractylodes (Asteraceae: carduoideae). Front. Plant Science 13. doi: 10.3389/fpls.2022.1045423
Martins da Silva, B. J., Rodrigues, A. P. D., Farias, L. H. S., Hage, A. A. P., Nascimento, J. L. M., Silva, E. O. (2014). Physalis angulata induces in vitro differentiation of murine bone marrow cells into macrophages. BMC Cell Biol. 15. doi: 10.1186/1471-2121-15-37
Navarro, A., Barton, N. H. (2003). Chromosomal speciation and molecular divergence-accelerated evolution in rearranged chromosomes. Science 300 (5617), 321–324. doi: 10.1126/science.1088277
Nguyen, L., Schmidt, H. A., Haeseler, A. V., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nock, C. J., Waters, D., Edwards, M. A., Bowen, S. G., Henry, R. J. (2015). Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9 (3), 328–333. doi: 10.1111/j.1467-7652.2010.00558.x
Rachele, T., Lorenza, S., Donata, C., Concita, C., Luigi, O., Teodoro, C., et al. (2020). Cultivated tomato (Solanum lycopersicum l.) suffered a severe cytoplasmic bottleneck during domestication: implications from chloroplast genomes. Plants 9 (11), 1443. doi: 10.3390/plants9111443
Raman, G., Nam, G.-H., Park, S. (2022). Extensive reorganization of the chloroplast genome of Corydalis platycarpa: a comparative analysis of their organization and evolution with other Corydalis plastomes. Front. Plant Science. 13. doi: 10.3389/fpls.2022.1043740
Sadali, N. M., Sowden, R. G., Ling, Q., Jarvis, R. P. (2019). Differentiation of chromoplasts and other plastids in plants. Plant Cell Rep. 38 (7), 803–818. doi: 10.1007/s00299-019-02420-2
Safari, Z. S., Ding, P., Nakasha, J. J., Yusoff, S. F. (2021). Controlling fusarium oxysporum tomato fruit rot under tropical condition using both chitosan and vanillin. Coatings 11, 367. doi: 10.3390/coatings11030367
Sun, C., Chen, F., Teng, N., Xu, Y., Dai, Z. (2020). Comparative analysis of the complete chloroplast genome of seven Nymphaea species. Aquat. Botany 170, 103353. doi: 10.21203/rs.3.rs-20050/v1
Tang, X., Wang, Y., Zhang, Y., Huang, S., Liu, Z., Fei, D., et al. (2018). A missense mutation of plastid RPS4 is associated with chlorophyll deficiency in Chinese cabbage (Brassica campestris ssp. pekinensis). BMC Plant Biol. 18 (1), 130. doi: 10.1186/s12870-018-1353-y
Wang, W., Chen, S., Zhang, X. (2016). Chloroplast genome evolution in actinidiaceae: clpP loss, heterogenous divergence and phylogenomic practice. PloS One 11 (9), e0162324. doi: 10.1371/journal.pone.0162324
Wang, J., Qian, J., Jiang, Y., Chen, X., Zheng, B., Chen, S., et al. (2022a). Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Polygonatum and tribe polygonateae. Front. Plant Science 13. doi: 10.3389/fpls.2022.882189
Wang, Y., Wen, F., Hong, X., Li, Z., Mi, Y., Zhao, B. (2022b). Comparative chloroplast genome analyses of Paraboea (Gesneriaceae): insights into adaptive evolution and phylogenetic analysis. Front. Plant Science 13. doi: 10.3389/fpls.2022.1019831
Wang, J., Xu, Q., Liu, J., Kong, W., Shi, L. (2023). Electrostatic self-assembly of MXene on ruthenium dioxide-modified carbon cloth for electrochemical detection of kaempferol. Small doi: 10.1002/smll.202301709
Xie, X., Huang, R., Li, F., Tian, E., Li, C., Chao, Z. (2021). Phylogenetic position of Bupleurum sikangense inferred from the complete chloroplast genome sequence. Gene. 798, 145801. doi: 10.1016/j.gene.2021.145801
Zhang, Z. (2022). Kaks_calculator 3.0: calculating selective pressure on coding and non-coding sequences. Genomics Proteomics Bioinf. 20 (3), 536–540. doi: 10.1016/j.gpb.2021.12.002
Zhang, Q., Li, J., Zhao, Y., Korban, S., Han, Y. (2012). Evaluation of genetic diversity in Chinese wild apple species along with apple cultivars using SSR markers. Plant Mol. Biol. Rep. 30 (3), 539–546. doi: 10.1007/s11105-011-0366-6
Zhang, Y., Ma, P., Li, D. (2011). High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: bambusoideae). PloS One 6 (5), e20596. doi: 10.1371/journal.pone.0020596
Keywords: tomato, germplasm, chloroplast genome, phylogenetic, adaptive evolution
Citation: Wang X, Bai S, Zhang Z, Zheng F, Song L, Wen L, Guo M, Cheng G, Yao W, Gao Y and Li J (2023) Comparative analysis of chloroplast genomes of 29 tomato germplasms: genome structures, phylogenetic relationships, and adaptive evolution. Front. Plant Sci. 14:1179009. doi: 10.3389/fpls.2023.1179009
Received: 03 March 2023; Accepted: 11 April 2023;
Published: 09 May 2023.
Edited by:
Linchun Shi, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Zhen-Hui Gong, Northwest A&F University, ChinaCopyright © 2023 Wang, Bai, Zhang, Zheng, Song, Wen, Guo, Cheng, Yao, Gao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaomin Wang, d2FuZ3hpYW9taW5fMTk4MUAxNjMuY29t; Jianshe Li, MTM3MDk1ODc4MDFAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.