 Concetta Lotti1†
Concetta Lotti1† Anna Paola Minervini2†
Anna Paola Minervini2† Chiara Delvento2
Chiara Delvento2 Pasquale Losciale2
Pasquale Losciale2 Liliana Gaeta3
Liliana Gaeta3 Raquel Sánchez-Pérez4
Raquel Sánchez-Pérez4 Luigi Ricciardi2
Luigi Ricciardi2 Stefano Pavan2*
Stefano Pavan2*- 1Department of Agriculture, Food, Natural Resources and Engineering, University of Foggia, Foggia, Italy
- 2Department of Soil, Plant and Food Sciences, Section of Plant Genetics and Breeding, University of Bari Aldo Moro, Bari, Italy
- 3Council for Agricultural Research and Economics-Research Centre for Agriculture and Environment (CREA-AA), Bari, Italy
- 4Plant Breeding Department, Fruit Breeding Group, CEBAS-CSIC, Campus Universitario de Espinardo, Espinardo, Spain
Almond [Prunus dulcis Miller (D. A. Webb), syn. Prunus amygdalus L.)] is the major tree nut crop worldwide in terms of production and cultivated area. Almond domestication was enabled by the selection of individuals bearing sweet kernels, which do not accumulate high levels of the toxic cyanogenic glucoside amygdalin. Previously, we showed that the Sweet kernel (Sk) gene, controlling the kernel taste in almond, encodes a basic helix loop helix (bHLH) transcription factor regulating the amygdalin biosynthetic pathway. In addition, we characterized a dominant allele of this gene, further referred to as Sk-1, which originates from a C1036→T missense mutation and confers the sweet kernel phenotype. Here we provide evidence indicating that the allele further referred to as Sk-2, originally detected in the cultivar “Atocha” and arising from a T989→G missense mutation, is also dominantly inherited and confers the sweet kernel phenotype in almond cultivated germplasm. The use of single nucleotide polymorphism (SNP) data from genotyping by sequencing (GBS) for population structure and hierarchical clustering analyses indicated that Sk-2 occurs in a group of related genotypes, including the widespread cultivar “Texas”, descending from the same ancestral population. KASP and dual label functional markers were developed for the accurate and high-throughput selection of the Sk-1 and Sk-2 alleles, and the genotyping of a panel of 134 almond cultivars. Overall, our results provide further insights on the understanding of the almond cultivation history. In addition, molecular marker assays and genotypic data presented in this study are expected to be of major interest for the conduction of almond breeding programs, which often need to select sweet kernel individuals in segregant populations.
1 Introduction
Almond [Prunus dulcis Miller (D.A. Webb), syn. Prunus amygdalus L.] is the tree nut species with the highest cultivated area and production worldwide (FAOSTAT data, 2021). U.S. accounts for about 80% of the almond global yield (1.7 million tonnes of shelled product), followed by Australia (6%), and Spain (5%) (FAOSTAT data, 2021; Pérez de los Cobos et al., 2021).
The kernels of wild almond relatives are bitter and highly toxic to humans, as they accumulate the cyanogenic glucoside amygdalin. Almond domestication, which took place in the Fertile Crescent about 12,000 years ago, was therefore signed by the selection of sweet kernel mutants (Sánchez-Pérez et al., 2019). Genetic studies indicated that the kernel taste is controlled by a single gene located on the almond chromosome 5, referred to as Sweet kernel (Sk) (Dicenta and García, 1993; Sánchez-Pérez et al., 2010). More recently, we showed that the Sk gene encodes a basic helix-loop-helix (bHLH) transcription factor. In addition, we characterized a dominant allele of this gene, further referred to as Sk-1, conferring the sweet kernel phenotype and originating from a C1036→T substitution in the gene coding sequence. This leads to a nonsynonymous change (Leu346→Phe) in the protein helix loop helix (HLH) domain which impairs the expression of the first two genes of the amygdalin biosynthetic pathway, encoding the P450 cytochromes PdCYP79D16 and PdCYP71AN24 (Thodberg et al., 2018; Sánchez-Pérez et al., 2019).
Resequencing of the Sk gene in a panel of 30 sweet kernel cultivars showed the occurrence of the Sk-1 allele in all cases but one, represented by the Spanish cultivar “Atocha”. However, “Atocha” displayed, at the heterozygous state, another nucleotide substitution (T989→G), also leading to a nonsynonymous change (Leu330→Arg) in the HLH domain (Sánchez-Pérez et al., 2019).
Almond breeding programs often involve the cross between cultivars heterozygous for the wild-type recessive sk allele. This implies that bitter kernel individuals must be discarded in segregating progenies. Phenotypic selection for kernel taste is costly and time consuming, as the almond juvenile phase prevents the evaluation of fruit traits prior to 3-4 years. Therefore, several studies were addressed to the identification of DNA markers suitable for assisted selection (Sánchez-Pérez et al., 2007; Sánchez-Pérez et al., 2010; Ricciardi et al., 2018).
Restriction fragment length polymorphism (RFLP), simple sequence repeat (SSR), and cleaved amplified length polymorphism (CAPS) molecular markers, located at variable genetic distance from the Sk locus, are currently available for the selection of sweet kernel genotypes (Sánchez-Pérez et al., 2007; Sánchez-Pérez et al., 2010; Ricciardi et al., 2018). Although they can be obtained with relatively inexpensive laboratory equipment, they are not ideal for large scale genotyping, as they require restriction enzyme digestion and/or manual scoring of electrophoretic profiles. In contrast, the Kompetitive Allele Specific PCR (KASP) technology, developed at LGC Genomics and based on competitive allele-specific PCR amplification of target sequences and endpoint fluorescence (Smith and Maughan, 2015), provides an automatized, high-throughput and cost-effective solution for genotyping and marker-assisted selection (Semagn et al., 2014; Nadeem et al., 2018; Ayalew et al., 2019; Li et al., 2022). In addition, dual label assays, based on fluorescently tagged and allele specific probes, are also suitable for high-throughput genotyping and used in plant breeding (Broccanello et al., 2018).
Functional or diagnostic markers are defined as those markers designed on the DNA polymorphism affecting phenotypic variation (Andersen and Lübberstedt, 2003; Ma et al., 2017). Functional markers are in complete linkage with functional motifs, and thus are fully predictive of the phenotype; in addition, they overcome issues related to other marker types, such as the lack of polymorphism between the parental genotypes of breeding populations, and the need of phenotypic validation of selected individuals (Pavan et al., 2013; Salgotra and Stewart, 2020). Thus, functional markers were indicated as major contributors to the development of precision plant breeding (Salgotra and Stewart, 2020).
Here, we performed allele mining on the Sk gene, indicating that two different alleles associated with the almond sweet kernel phenotype were selected with domestication and occur in cultivated germplasm. We characterized the distribution of the two Sk alleles in a large almond germplasm collection, including cultivars worldwide used in breeding programs. Finally, we developed KASP and dual label functional markers targeting the Sk DNA polymorphisms.
2 Materials and methods
2.1 Plant material and genomic DNA isolation
Plant material considered in this study was sampled from the ex situ collections of CREA-AA (Council for Agricultural Research and Analysis of Agricultural Economics—Research Centre for Agriculture and Environment), Bari, Italy, and CEBAS-CSIC (Spanish National Research Council—Center for Edaphology and Applied Biology of the Segura River), Murcia, Spain. In total, 144 individuals were considered, including 134 sweet kernel cultivars originating from main almond-growing countries (Supplementary Table 1) and 10 bitter kernel individuals selected from the segregant “R1000” x “Desmayo Largueta” segregant population previously described by Ricciardi et al. (2018) and Sánchez-Pérez et al. (2019). Genomic DNA was extracted by young leaf material, using the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. DNA quality and concentration were checked using 0.8% agarose gel electrophoresis and the NanoDrop 1000 spectrophotometer (ThermoScientific, Pittsburgh, PA, U.S.).
2.2 Marker development
Molecular markers assays were designed on the C1036T and the T989G single nucleotide polymorphisms (SNPs) occurring within the coding sequence of the Sk gene, based on the KASP and the dual label technologies. For KASP assays, three primers were designed on the sequence flanking the SNP position: two allele-specific forward primers, marked with the FAM and HEX fluorescence dyes, and a common reverse primer (Table 1). PCR reactions were performed at LGC genomics Service Lab (Hoddesdon, U.K.) according to standard protocols. Output fluorescence result tables were converted into scatter plots using the ggplot2 R package (Wickham, 2016).
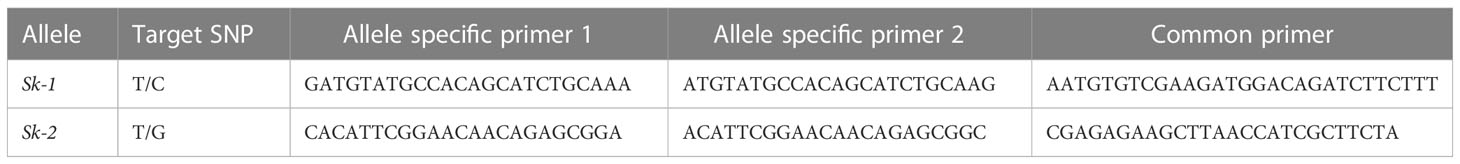
Table 1 Information on KASP assays designed in this study to detect the Sk-1 and Sk-2 alleles.
The ddPCR mutation detection assay, adapted to the CFX 96 Real-Time PCR detecting system (Bio-Rad Laboratories, Hercules, CA, U.S.) was used as dual label strategy. Sequences extending at both sides of the SNP position for 61 base pairs were used to design primers and probes, whose sequences were not provided by the supplier. PCR mixes were prepared in 10 µl reaction containing 1x SsoAdvanced Universal Probes Supermix, 1x target (FAM) and wild-type (HEX) primers/probes, and about 50 ng of genomic DNA. PCR reactions were carried with the following conditions: 95° C for 10 min, 40 cycles at 94° C for 30 s followed by 58°C for 30s. Genotypic calls were obtained through the allelic discrimination function available in the Bio-Rad CFX Manager v.3.1. software. Fluorescence result tables were exported and converted into scatter plots using the ggplot2 R package (Wickham, 2016).
Validation of genotypic calls on 23 selected individuals was performed by the inspection of electropherograms resulting from Sanger sequencing. This was performed at Eurofins Genomics (Ebersberg, Germany) using the primer combination SkFw (5’-GGTGCTTGAACTGGCTTCTC-3’)/SkRev (5’-CTTCCGATCCCAAAATCT-3’), flanking the Sk coding sequence (NCBI accession number MK041092). In addition, the occurrence of the T989→G mutation in the cultivar “Texas” was confirmed blasting the Sk coding sequence against the “Texas” reference genome sequence (Alioto et al., 2020).
2.3 Search of genomic variation data and quality control
The raw variant call format (VCF) SNP file reported by (Pavan et al., 2021) (doi: 10.6084/m9.figshare.12205652), which refers to the application of genotyping by sequencing (GBS) (Elshire et al., 2011) on an almond diversity panel, was used. Data relative to the cultivars genotyped in this study for the Sk locus were extracted. Then, quality control was performed by filtering for biallelic SNP loci with minor allele frequency >0.05 and missing rate <0.3, and for cultivars with missing data <0.4, using Tassel 5 (Bradbury et al., 2007). Finally, SNPs were pruned for linkage disequilibrium using the flag indep-pairwise 50 5 0.5 available in PLINK v.1.90 (Purcell et al., 2007).
2.4 Population structure analysis and hierarchical clustering
The analysis of genetic structure was performed by the software ADMIXTURE (Alexander et al., 2009), assuming from 1 to 10 ancestral populations and running 1,000 bootstrap replicates to estimate parameter standard errors. The most suitable number of ancestral populations was identified in correspondence with the lowest cross‐validation (CV) error. In accordance with the previous study by Pavan et al. (2021), individuals were assigned to one of the ancestral populations when their membership coefficient (qi) for that population was higher than 0.6, otherwise they were considered of admixed ancestry. Hierarchical clustering was performed using the AWclust R package (Gao and Starmer, 2008), based on the allele-sharing distance and the Ward’s minimum variance clustering algorithm.
3 Results
3.1 Development of KASP and dual label functional markers for the selection of the Sk-1 allele
Two KASP and dual label assays were designed to detect the Sk-1 allele, originating from the C1036→T mutation in the gene coding sequence (Sánchez-Pérez et al., 2019). When tested on a panel of 30 individuals, each of the two assays yielded three fluorescence clusters (Figure 1). In addition, genotypic calls resulting from the two assays were fully consistent. As expected, the Sk-1 allele was not detected in ten bitter kernel individuals selected from a previously described F1 segregant population (Ricciardi et al., 2018), which were therefore assigned to the C1036:C1036 genotypic cluster. Sweet kernel cultivars were assigned to two genotypic clusters, predicted to have one (T1036:C 1036) or two (T1036:T1036) copies of the Sk-1 allele. With no exception, Sanger sequencing confirmed the correct call of the T1036:C 1036 and T1036:T1036 genotypic clusters.
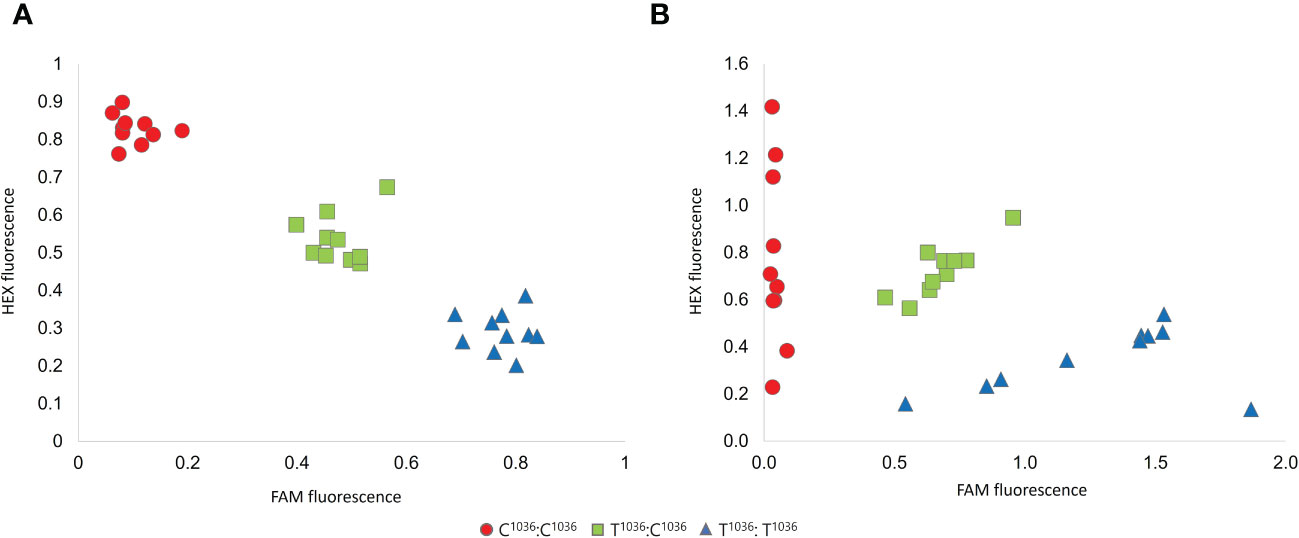
Figure 1 Validation of the KASP (A) and dual label (B) assays designed on the Sk-1 allele. Red circles, green squares and blue triangles indicate individuals belonging to the genotypic clusters associated with zero (C1036:C1036), one (T1036:C1036) or two (T1036T1036) Sk-1 copies, respectively.
Once performed assay validation, we genotyped additional 114 sweet kernel cultivars, originating from the main almond-growing countries. Overall, the KASP and dual label assays yielded a call rate of 97% and 99.2%, respectively. All the genotypic calls, reported in Supplementary Table 1, were consistent between the two assays.
3.2 The Sk-2 allele is also associated with the almond sweet kernel phenotype
Although at least one copy of the Sk-1 allele was detected in most cultivars, exceptions were found, represented by the cultivars “Texas”, “Pizzuta d’Avola”, and “Mollar de Tarragona” (Supplementary Table 1). Sanger sequencing confirmed the C1036:C1036 genotype of these cultivars.
Sánchez-Pérez et al. (2019) reported, for the Sk gene of the Spanish cultivar “Atocha”, a C1036:C1036 genotype and, at the same time, a heterozygous point mutation (T989→G) leading to a nonsynonymous change (Leu330→Arg) in the HLH protein domain. Notably, according to Sanger sequencing, the C1036:C1036 cultivars also carried at least one copy of the G989 mutation. Indeed, “Texas” and “Pizzuta D’Avola” were scored as heterozygous (T989:G989), whereas “Mollar de Tarragona” was scored as homozygous (G989:G989). The occurrence of the T989→G mutation in “Texas” was further confirmed by BLAST search against the newly released reference genome of the same cultivar (Alioto et al., 2020).
The results above mentioned indicated that, besides Sk-1, another dominant allele conferring the sweet kernel phenotype, arising from a T989→G mutation, was selected with domestication and occurs within almond cultivated germplasm. We further named this allele Sk-2 and designed new KASP and dual label assays for its specific detection, which were tested on the same panel of cultivars above mentioned. As shown in Figure 2, both assays resulted in three fluorescence clusters. The call rates were 98.5% and 100% for the KASP and the dual label assay, respectively. In addition, genotypic calls were again fully consistent. “Mollar de Tarragona” was genotyped as homozygous for the Sk-2 allele (G989:G989), whereas “Texas” and “Pizzuta D’Avola” were genotyped as heterozygous (T989:G989), in accordance with the results of Sanger Sequencing. In addition, the cultivars “Nikitsky”, “Butte” and “Del Cid” were also genotyped as heterozygous (Supplementary Table 1). Overall, the relative frequencies of the Sk-1 and Sk-2 alleles in the panel of almond cultivars subjected to genotyping were 96.5% and 3.5%, respectively.
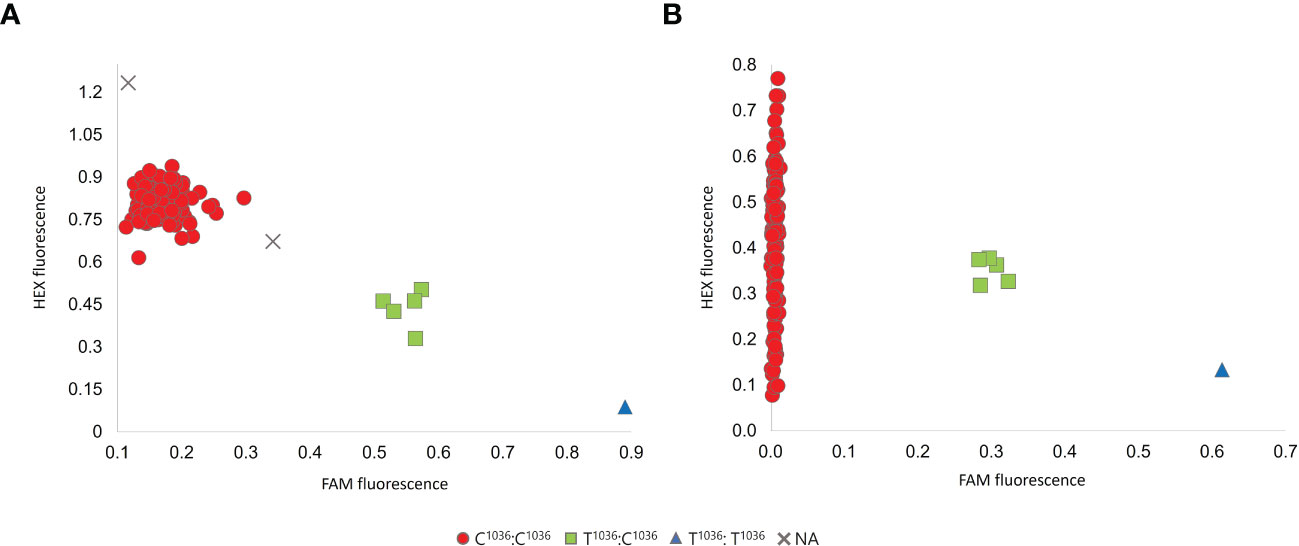
Figure 2 Results of the KASP (A) and dual label (B) assays designed on the Sk-2 allele. Red circles, green squares and blue triangles indicate individuals belonging to the genotypic clusters associated with zero (T989:T989), one (T989:G989) or two (G989:G989) Sk-2 copies, respectively. Grey crosses indicate individuals associated with missing genotypic call.
3.3 Genetic relationships among cultivars carrying the two Sk alleles
Aiming to investigate the origin of the two Sk alleles, we exploited the availability of DNA polymorphism data for most of the cultivars genotyped in this study, including “Mollar de Tarragona”, “Nikisky”, “Texas” and “Pizzuta D’Avola”, carrying the Sk-2 allele (Pavan et al., 2021). The application of quality control procedure resulted in a dataset of 114 cultivars and 44,638 SNPs.
Population structure analysis indicated a model with four ancestral populations (named from K1 to K4) as suitable to explain data (Supplementary Figure 1). With a few exceptions, cultivars assigned to the ancestral populations K1, K2 and K3 originated from Italy, whereas cultivars assigned to the ancestral population K4 originated from U.S., except for the Ukranian cultivar “Nikitsky” (Figure 3; Table 2). Concerning the four cultivars in the panel bearing the Sk-2 allele, two of them (“Texas” and “Nikitsky”) were assigned to K4, and two (“Mollar de Tarragona” and “Pizzuta D’Avola) to the admixed group. The inspection of membership coefficients (qi) returned by the ADMIXTURE parametric model, indicating the estimated percentage of the genome deriving from each ancestry, revealed that the K4 ancestry was the only one associated with all the four cultivars bearing Sk-2 (Table 2), suggesting that the Sk-2 mutation arose or was selected in the ancestral population K4. Hierarchical clustering resulted in the identification of two main clusters (named C1 and C2), with C1 including the four cultivars mentioned above carrying the Sk-2 allele (Figure 4).
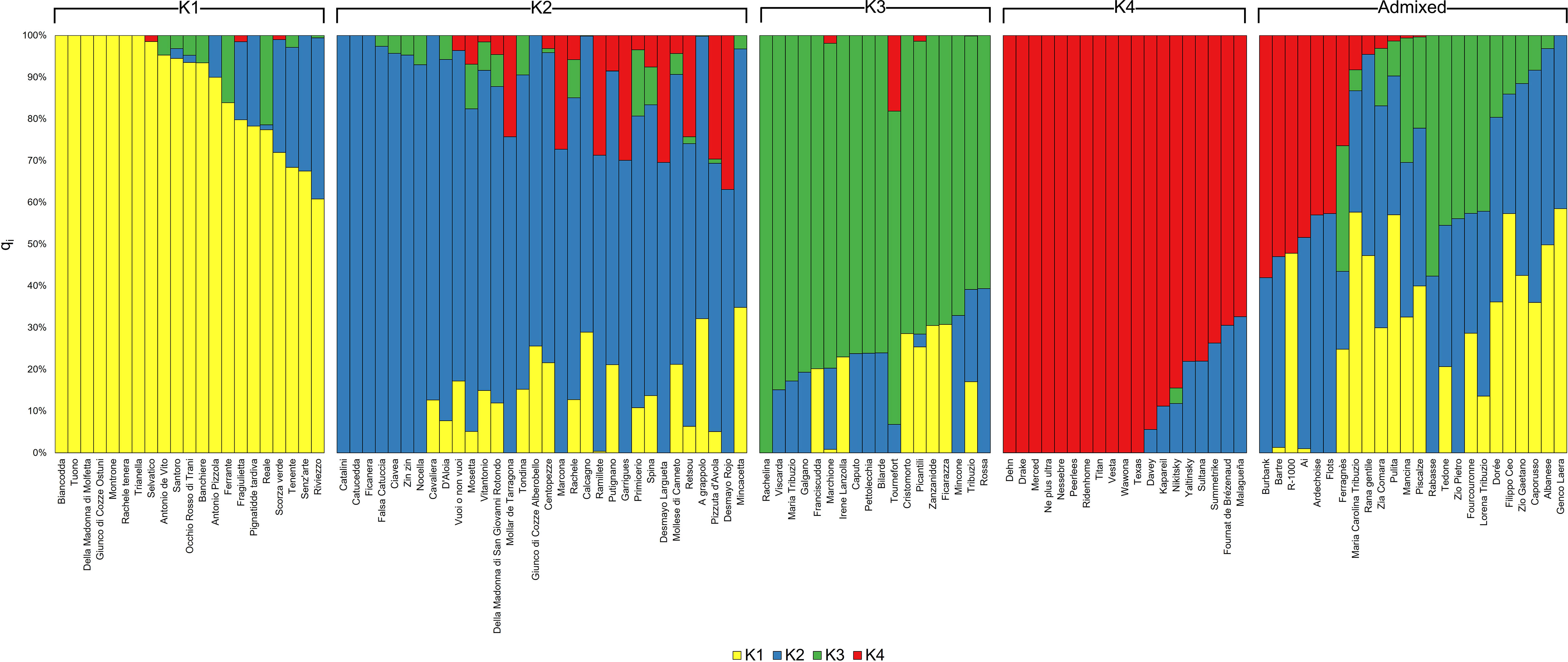
Figure 3 Results of ADMIXTURE population structure analysis for a model with four ancestral populations, named from K1 to K4. Each cultivar is represented by a vertical bar. In turn, each bar is divided into segments whose length and color represent the proportion of the genome (qi) contributed by each ancestral population. Cultivars assigned to one of the ancestral populations have a membership coefficient (qi) for that population >0.6. The remaining cultivars are assigned to the admixed group.
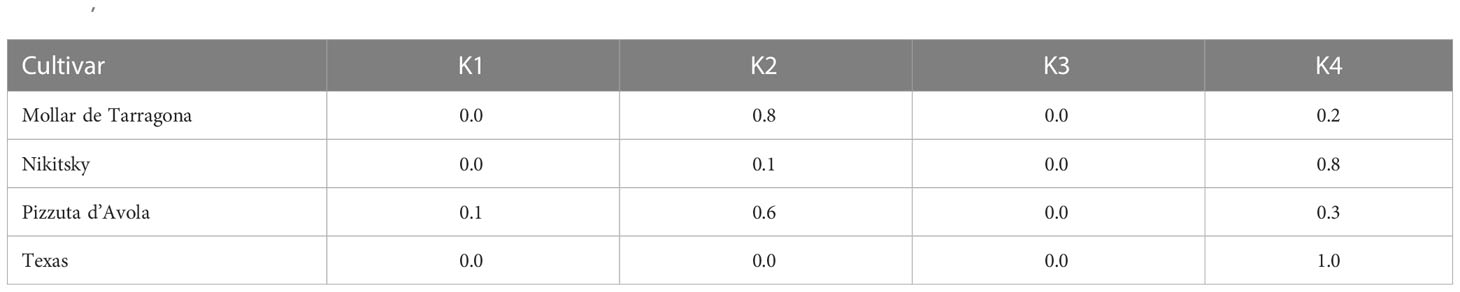
Table 2 ADMIXTURE membership coefficient for the four ancestral populations K1, K2, K3 and K4, relative to four cultivars (“Mollar de Tarragona”, “Nikisky”, “Texas” and “Pizzuta D’Avola”) carrying at least one copy of the Sk-2 allele.
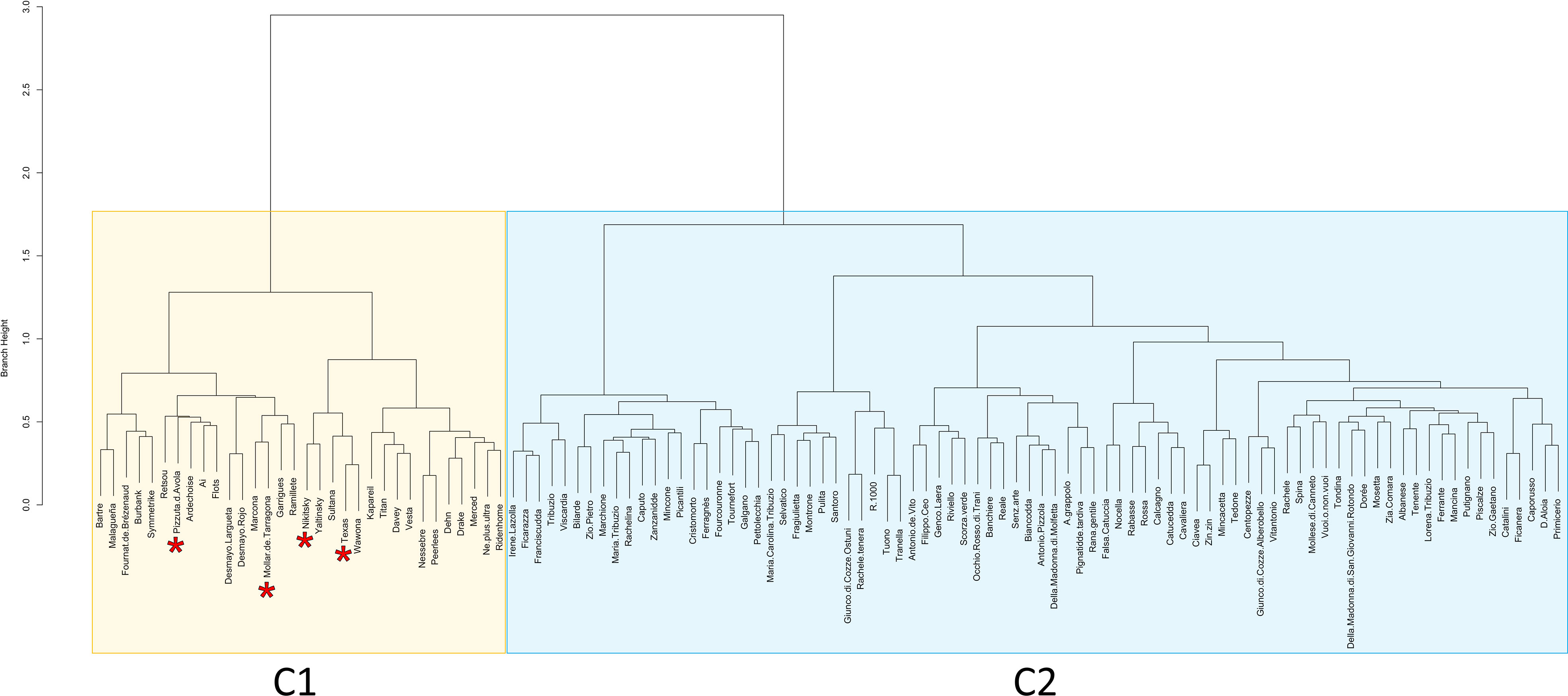
Figure 4 Results of hierarchical clustering with the AWclust algorithm. The two main clusters of the dendrogram (C1 and C2) are shaded with different colors. The names of the cultivars “Mollar de Tarragona”, “Nikisky”, “Texas” and “Pizzuta D’Avola”, harboring at least one copy of the Sk-2 allele, are highlighted with an asterisk.
4 Discussion
We recently reported the isolation of the almond Sk gene and the identification of its dominant C1036→T mutant allele, re-named here as Sk-1, which confers the sweet kernel phenotype (Sánchez-Pérez et al., 2019). In this study, allele mining indicated that a second Sk mutant allele, originating from a T989→G mutation and named Sk-2, also occurs in almond cultivated germplasm. The presence of at least one copy of Sk-2 in all the sweet kernel cultivars lacking Sk-1 indicates that Sk-2 is also a dominant allele functionally related to the sweet kernel phenotype. In addition, it suggests that Sk-1-and Sk-2 are the only mutations leading to the sweet kernel taste that occur in almond cultivated germplasm. Both the Sk-1 and Sk-2 mutations lead to nonsynonymous changes in the protein HLH domain, which is known to play a major role in the dimerization of bHLH transcription factors (Murre, 2019).
Sweet kernel cultivars exhibit trace amounts of amygdalin, and variation in amygdalin concentration in sweet kernel cultivars are of commercial importance, as they have an influence on the kernel taste (Arrázola et al., 2012; Lee et al., 2013). Thus, it would be interesting to investigate whether the different genotypic combinations harboring the Sk-1 and Sk-2 alleles are associated with significantly different amygdalin concentrations.
The lower frequency of Sk-2 with respect to Sk-1 in almond cultivated germplasm, together with the occurrence of Sk-2 in specific genetic groups (Figures 3, 4), suggest that Sk-1 was the first allele to be selected with domestication, and that the Sk-2 mutation arose or was selected in a later stage during the almond cultivation history. Population structure analysis identified the ancestral population K4, significantly contributing to the differentiation of Spanish, French, and U.S. cultivars, as the one in which Sk-2 first appeared. Importantly, although literature indicates that U.S. almond breeding originates from the introduction of French germplasm in late XIX century, genotyping failed to detect the Sk-2 allele, occurring in the U.S. cultivars “Texas” and “Butte”, in French germplasm. Conversely, the occurrence of Sk-2 in several Spanish cultivars (“Del Cid”, “Atocha”, “Mollar de Tarragona”) suggests that at least part of the U.S. almond germplasm might originate from Spanish introductions.
The identification of molecular markers suitable for the selection of sweet kernel individuals has been for a long time one of the main research aims for almond breeding. Differently from the molecular markers reported so far, which are located at various genetic distances from the Sk gene (Sánchez-Pérez et al., 2007; Sánchez-Pérez et al., 2010; Ricciardi et al., 2018), those identified in this study directly target Sk polymorphisms causing kernel taste variation, and thus fall within the definition of functional markers provided by Andersen and Lübberstedt (2003). Functional markers are considered optimal to assist selection, as they overcome issues related to recombination and allow precision breeding (Lau et al., 2015). Both KASP and dual label assays used in this study represent state-of-the-art and high-throughput technologies for marker-assisted selection, which are widely used in plant breeding (Broccanello et al., 2018; Radanović et al., 2022). KASP technology is known to provide a lower cost per data point, however it provides a less efficient separation of genotypic clusters and requires higher amount of DNA template (Ayalew et al., 2019). In our study, KASP assays resulted in slightly lower call rates than dual label assays.
In conclusion, we provide evidence indicating that two mutations of the same transcription factors were selected with domestication to make almonds palatable. In addition, we describe functional marker assays allowing the accurate and high-throughput selection of sweet kernel individuals in almond breeding programs. Finally, we report genotypic data of a large germplasm collection, including cosmopolitan cultivars and the four most widely used founding clones for almond breeding programs worldwide, i.e. “Cristomorto”, “Tuono”, “Nonpareil” and “Texas” (Pérez de los Cobos et al., 2021), which are expected to be of great relevance for the almond breeding community.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
SP conceived the study. LG, PL and RS-P assembled the genetic materials. LR and SP provided funds to the research. CL and AM performed lab work. CD and AM performed data analyses. SP wrote the manuscript draft. All authors contributed to the article and approved the submitted version.
Funding
This study was carried out within the framework of: 1) the Agritech National Research Center, receiving funding from the European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) – MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4 – D.D. 1032 17/06/2022, CN00000022); 2) The project “Recupero e valorizzazione del germoplasma frutticolo pugliese” “Re.Ge.Frup.2.1”, receiving funds from “PSR Puglia 2014-2020. Misura 10 - Sottomis. 10.2 - Operazione 10.2.1 - Progetti per la conservazione e valorizzazione delle risorse genetiche in agricoltura”; 3) the project “ALmond ADaptation in NOvel Environments: Multiscale Approach from Genome to Function PrediCtion: ALADINO MAGIC” (MINECO-Spain).
This manuscript reflects only the authors’ views and opinions, neither the European Union nor the European Commission can be considered responsible for them.
Acknowledgments
We are grateful to Dr. Zheng Zheng (Chinese Academy of Agricultural Science, Zhengzhou, China) for technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1171195/full#supplementary-material
References
Alexander, D. H., Novembre, J., Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Alioto, T., Alexiou, K. G., Bardil, A., Barteri, F., Castanera, R., Cruz, F., et al. (2020). Transposons played a major role in the diversification between the closely related almond and peach genomes: results from the almond genome sequence. Plant J. 101, 455–472. doi: 10.1111/tpj.14538
Andersen, J. R., Lübberstedt, T. (2003). Functional markers in plants. Trends Plant Sci. 8, 554–560. doi: 10.1016/j.tplants.2003.09.010
Arrázola, G., Sánchez-Pérez, R., Dicenta, F., Grané, N. (2012). Content of the cyanogenic glucoside amygdalin in almond seeds related to the bitterness genotype. Agron. Colomb. 30, 260–265.
Ayalew, H., Tsang, P. W., Chu, C., Wang, J., Liu, S., Chen, C., et al. (2019). Comparison of TaqMan, KASP and rhAmp SNP genotyping platforms in hexaploid wheat. PloS One 14, e0217222. doi: 10.1371/journal.pone.0217222
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Broccanello, C., Chiodi, C., Funk, A., McGrath, J. M., Panella, L., Stevanato, P. (2018). Comparison of three PCR-based assays for SNP genotyping in plants. Plant Methods 14, 28. doi: 10.1186/s13007-018-0295-6
Dicenta, F., García, J. E. (1993). Reciprocal crosses in almond. Plant Breed. 110, 77–80. doi: 10.1111/j.1439-0523.1993.tb00571.x
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-Sequencing (GBS) approach for high diversity species. PloS One 6, e19379. doi: 10.1371/journal.pone.0019379
FAOSTAT data (2021) Available at: http://www.fao.org/faostat/ [Accessed January 2023].
FAOSTAT. Food and Agriculture Organization of the United Nations (2022) FAOSTAT database: Data. Available at: https://www.fao.org/faostat/en/#data/QCL.
Gao, X., Starmer, J. D. (2008). AWclust: Point-and-click software for non-parametric population structure analysis. BMC Bioinf. 9, 77. doi: 10.1186/1471-2105-9-77
Lau, W. C. P., Rafii, M. Y., Ismail, M. R., Puteh, A., Latif, M. A., Ramli, A. (2015). Review of functional markers for improving cooking, eating, and the nutritional qualities of rice. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00832
Lee, J., Zhang, G., Wood, E., Rogel Castillo, C., Mitchell, A. E. (2013). Quantification of amygdalin in nonbitter, semibitter, and bitter almonds (Prunus dulcis) by UHPLC-(ESI)QqQ MS/MS. J. Agric. Food Chem. 61, 7754–7759. doi: 10.1021/jf402295u
Li, L., Sun, Z., Zhang, Y., Ke, H., Yang, J., Li, Z., et al. (2022). Development and utilization of functional kompetitive allele-specific PCR markers for key genes underpinning fiber length and strength in. Gossypium hirsutum L. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.853827
Ma, Y., Coyne, C. J., Main, D., Pavan, S., Sun, S., Zhu, Z., et al. (2017). Development and validation of breeder-friendly KASPar markers for er1, a powdery mildew resistance gene in pea (Pisum sativum l.). Mol. Breed. 37, 151. doi: 10.1007/s11032-017-0740-7
Murre, C. (2019). Helix-loop-helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev. 33, 6–25. doi: 10.1101/gad.320663.118
Nadeem, M. A., Nawaz, M. A., Shahid, M. Q., Doğan, Y., Comertpay, G., Yıldız, M., et al. (2018). DNA Molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol. Biotechnol. Equip. 32, 261–285. doi: 10.1080/13102818.2017.1400401
Pavan, S., Delvento, C., Mazzeo, R., Ricciardi, F., Losciale, P., Gaeta, L., et al. (2021). Almond diversity and homozygosity define structure, kinship, inbreeding, and linkage disequilibrium in cultivated germplasm, and reveal genomic associations with nut and seed weight. Hortic. Res. 8, 15. doi: 10.1038/s41438-020-00447-1
Pavan, S., Schiavulli, A., Appiano, M., Miacola, C., Visser, R. G. F., Bai, Y., et al. (2013). Identification of a complete set of functional markers for the selection of er1 powdery mildew resistance in Pisum sativum l. Mol. Breed. 31, 247–253. doi: 10.1007/s11032-012-9781-0
Pérez de los Cobos, F., Martínez-García, P. J., Romero, A., Miarnau, X., Eduardo, I., Howad, W., et al. (2021). Pedigree analysis of 220 almond genotypes reveals two world mainstream breeding lines based on only three different cultivars. Hortic. Res. 8, 11. doi: 10.1038/s41438-020-00444-4
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Radanović, A., Sprycha, Y., Jocković, M., Sundt, M., Miladinović, D., Jansen, C., et al. (2022). KASP markers specific for the fertility restorer locus Rf1 and application for genetic purity testing in sunflowers (Helianthus annuus l.). Genes (Basel). 13, 465. doi: 10.3390/genes13030465
Ricciardi, F., Del Cueto, J., Bardaro, N., Mazzeo, R., Ricciardi, L., Dicenta, F., et al. (2018). Synteny-based development of CAPS markers linked to the sweet kernel LOCUS, controlling amygdalin accumulation in almond (Prunus dulcis (Mill.) D.A.Webb). Genes (Basel). 9, 385. doi: 10.3390/genes9080385
Salgotra, R. K., Stewart, C. N., Jr (2020). Functional markers for precision plant breeding. Int. J. Mol. Sci. 21 (13), 4792. doi: 10.3390/ijms21134792
Sánchez-Pérez, R., Howad, W., Dicenta, F., Arús, P., Martínez-Gómez, P. (2007). Mapping major genes and quantitative trait loci controlling agronomic traits in almond. Plant Breed. 126, 310–318. doi: 10.1111/j.1439-0523.2007.01329.x
Sánchez-Pérez, R., Howad, W., Garcia-Mas, J., Arús, P., Martínez-Gómez, P., Dicenta, F. (2010). Molecular markers for kernel bitterness in almond. Tree Genet. Genomes 6, 237–245. doi: 10.1007/s11295-009-0244-7
Sánchez-Pérez, R., Pavan, S., Mazzeo, R., Moldovan, C., Aiese Cigliano, R., Del Cueto, J., et al. (2019). Mutation of a bHLH transcription factor allowed almond domestication. Science. 364, 1095–1098. doi: 10.1126/science.aav8197
Semagn, K., Babu, R., Hearne, S., Olsen, M. (2014). Single nucleotide polymorphism genotyping using kompetitive allele specific PCR (KASP): overview of the technology and its application in crop improvement. Mol. Breed. 33, 1–14. doi: 10.1007/s11032-013-9917-x
Smith, S. M., Maughan, P. J. (2015). SNP genotyping using KASPar assays. Methods Mol. Biol. 1245, 243–256. doi: 10.1007/978-1-4939-1966-6_18
Thodberg, S., Del Cueto, J., Mazzeo, R., Pavan, S., Lotti, C., Dicenta, F., et al. (2018). Elucidation of the amygdalin pathway reveals the metabolic basis of bitter and sweet almonds (Prunus dulcis). Plant Physiol. 178, 1096–1111. doi: 10.1104/pp.18.00922
Keywords: almond, kernel taste, allele mining, marker-assisted selection, breeding
Citation: Lotti C, Minervini AP, Delvento C, Losciale P, Gaeta L, Sánchez-Pérez R, Ricciardi L and Pavan S (2023) Detection and distribution of two dominant alleles associated with the sweet kernel phenotype in almond cultivated germplasm. Front. Plant Sci. 14:1171195. doi: 10.3389/fpls.2023.1171195
Received: 21 February 2023; Accepted: 23 March 2023;
Published: 14 April 2023.
Edited by:
Mohan Lal, North East Institute of Science and Technology (CSIR), IndiaReviewed by:
Mario Di Guardo, University of Catania, ItalyFabio Palumbo, University of Padua, Italy
Ezio Portis, University of Turin, Italy
Copyright © 2023 Lotti, Minervini, Delvento, Losciale, Gaeta, Sánchez-Pérez, Ricciardi and Pavan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Pavan, c3RlZmFuby5wYXZhbkB1bmliYS5pdA==
†These authors have contributed equally to this work and share first authorship