
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 24 July 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1170004
This article is part of the Research TopicAdvances on Genomics and Genetics of Horticultural Crops and their Contribution to Breeding Efforts, volume IIView all 12 articles
 Shuai Yuan1,2†
Shuai Yuan1,2† Cong Nie1†
Cong Nie1† Shangang Jia3
Shangang Jia3 Tianqi Liu1
Tianqi Liu1 Junming Zhao1
Junming Zhao1 Jinghan Peng1
Jinghan Peng1 Weixia Kong1
Weixia Kong1 Wei Liu1Wenlong Gou2
Wei Liu1Wenlong Gou2 Xiong Lei2
Xiong Lei2 Yi Xiong1
Yi Xiong1 Yanli Xiong1
Yanli Xiong1 Qingqing Yu1
Qingqing Yu1 Yao Ling1*
Yao Ling1* Xiao Ma1*
Xiao Ma1*Hordeum L. is widely distributed in mountain or plateau of subtropical and warm temperate regions around the world. Three wild perennial Hordeum species, including H. bogdanii, H. brevisubulatum, and H. violaceum, have been used as forage and for grassland ecological restoration in high-altitude areas in recent years. To date, the degree of interspecies sequence variation in the three Hordeum species within existing gene pools is still not well-defined. Herein, we sequenced and assembled chloroplast (cp) genomes of the three species. The results revealed that the cp genome of H. bogdanii showed certain sequence variations compared with the cp genomes of the other two species (H. brevisubulatum and H. violaceum), and the latter two were characterized by a higher relative affinity. Parity rule 2 plot (PR2) analysis illuminated that most genes of all ten Hordeum species were concentrated in nucleotide T and G. Numerous single nucleotide polymorphism (SNP) and insertion/deletion (In/Del) events were detected in the three Hordeum species. A series of hotspots regions (tRNA-GGU ~ tRNA-GCA, tRNA-UGU ~ ndhJ, psbE ~ rps18, ndhF ~ tRNA-UAG, etc.) were identified by mVISTA procedures, and the five highly polymorphic genes (tRNA-UGC, tRNA-UAA, tRNA-UUU, tRNA-UAC, and ndhA) were proved by the nucleotide diversity (Pi). Although the distribution and existence of cp simple sequence repeats (cpSSRs) were predicted in the three Hordeum cp genomes, no rearrangement was found between them. A similar phenomenon has been found in the cp genome of the other seven Hordeum species, which has been published so far. In addition, evolutionary relationships were reappraised based on the currently reported cp genome of Hordeum L. This study offers a framework for gaining a better understanding of the evolutionary history of Hordeum species through the re-examination of their cp genomes, and by identifying highly polymorphic genes and hotspot regions that could provide important insights into the genetic diversity and differentiation of these species.
As secretory organs and active metabolic centers, chloroplasts (cp) are considered the source of energy that drives the evolution of early life (Liu et al., 2018). Although most of the genetic information is provided by the nuclear genome, the cp genome is used to perform variation analysis due to its small size and matrilineal inheritance without gene recombination interference (Gumeni et al., 2017; Shen et al., 2018). Therefore, sequence variation in cp genomes plays a key role in studying plant evolution, and genetic diversity (Xiong et al., 2020a). With the advent of high-throughput sequencing technologies, especially Illumina sequencing, sequence and structure information obtained from the whole cp genome has been elucidated in some vital species (Ogihara et al., 2000; Sajjad et al., 2017). Cp genomes contain several functional genes, such as photosynthesis-related genes, expression-related genes, and biosynthesis-related genes (Bailey et al., 2020). Differential gene detection and phylogeny analysis among genera or families using cp genome sequences is another effective method for studying evolutionary patterns due to the conservative property of cp DNA, mainly in content and arrangement mode. Generally, the structure of the cp genome is quadrantal, containing two inverted repeat (IR) sequences divided by a large single-copy (LSC) region and a small single-copy (SSC) region (Wu et al., 2021). However, four specific Hordeum species, H. pubiflorum, H. murinum, H. marinum, and H. bulbosum, were a noticeable exception to this typical structure with IR loss or missing introns (Bernhardt et al., 2017). It is noteworthy that this phenomenon was rarely reported in plants in the Poaceae family but it was often found in plants in the Leguminaceae family (Xue et al., 2019).
Derived from the Triticeae tribe of the Gramineae family, Hordeum L. is composed of approximately 45 species or subspecies, which are distributed in the southern and northern hemispheres, with four species diversity centers, including Southwest Asia, Central Asia, North America, and Southern America (Brassac and Blattner, 2015; and Reinert et al., 2019). The genus Hordeum consists of one cultivated species, namely H. vulgare, and abundant wild species, such as H. vulgare subsp. spontaneum, H. bogdanii, H. brevisubulatum, H.violaceum (H. roshevilzii), etc. Wild species — which gradually undergo environmental selection — often possess favorable genes such as disease resistance and insect resistance genes and thus are considered important germplasms for genetic improvement (Alyr et al., 2020). Investigation of the genetic diversity and kinship between wild and cultivated species may provide a perspective for the development and utilization of advantageous genes and extension of the genetic basis of cultivars. Previous studies have explored the phylogenetic relationships between wild and cultivated and annual and perennial Hordeum species, which mainly depended on the mitochondrial genome sequences (Hisano et al., 2016) or partial nuclear single-copy genome sequence analysis (Jonathan and Blattner, 2015). However, there are relatively few reports on the phylogenetic relationships using complete cp genomes of the genus Hordeum. Particularly, large-scale phylogenetic analysis of wild perennial species originating from North Central Asia (H. bogdanii, H. brevisubulatum, and H. violaceum) and those distributed elsewhere is still insufficient. Therefore, performing complete cp genome sequencing of these three wild perennial Hordeum species to identify some plastid key genes in interspecific genetic differentiation between the wild and cultivated and/or perennial and annual Hordeum species is of great significance, to further improve the phylogenetic relationships and genome structure of the genus Hordeum.
Here, complete cp genomes of three wild perennial Hordeum species, H. bogdanii, H. brevisubulatum, and H. violaceum, were sequenced and annotated, to determine the cp genome size, nucleotide diversity (Pi), repeat sequences, insertions/deletions (In/Dels), single nucleotide polymorphisms (SNPs). Sequence synteny, relative synonymous codon usage, Parity rule 2 (PR2) analysis, rearrangements, and IR expansions or contractions were evaluated among 10 Hordeum species (H. bogdanii, H. brevisubulatum, H. violaceum, H. jubatum, H. bulbosum, H. marinum, H. murinum, H. pubiflorum, H. vulgare subsp. spontaneum, and H. vulgare). In addition, phylogenetic relationships of the sequenced Hordeum species from other whole sequenced Poaceae species were revealed. Meanwhile, the degree of variation between wild and cultivated and annual and perennial Hordeum species was further evaluated. This study contributes to the expansion of the cp genome database.
Three Hordeum species, H. bogdanii, H. brevisubulatum, and H. violaceum, were from NPGS (National Plant Germplasm System of the United States; Supplementary Table 1). In total, 100 mg leaves were harvested at the three-leaf stage, and then total genomic DNA was extracted using the plant DNA Extraction Kit (Tiangen, Beijing, China) as per manufacture’s instruction. DNA concentration was quantified using 0.1% agarose gel, libraries were established, and DNA with good quality was selected and sequenced using the Illumina NovaSeq platform with a read length of PE150.
The complete circular genome sequence cannot be directly obtained by one-time splicing because of the characteristics of next-generation sequencing (NGS), genomic repeats, a specific structure of the genome, and related factors. Therefore, a different complicated strategy was performed: The kernel modules were assembled using the SPAdes v3.10.1 (Saint Petersburg State University, Saint, Russia) (Safonova et al., 2014) software for the cp genome of three species, which is not dependent on the reference genome. The contig was obtained using the kmer iterative extend seed. The SSPACE v2.0 procedure was used (BaseClear BV, Einsteinweg, Leiden, The Netherlands) (Boetzer et al., 2011) to acquire scaffolds by connecting contig sequences. The gap of scaffolds sequence was constructed using Gapfiller V2.1.1 procedure (BaseClear BV, Einsteinweg, Leiden, The Netherlands) to assemble a complete pseudo sequence (Boetzer and Pirovano, 2012). The alignment-correction method was used to align the sequencing sequence into the pseudo genome, which was later rearranged according to the cp structure of the three species, thereby obtaining a complete cp circular genome sequence.
Cp gene structure annotation plays an important role in cp genome sequencing. Blast v2.2.25 (U.S. National Library of Medicine 8600 Rockville Pike, Bethesda MD, 20894 USA) (Kent and Brumbaugh, 2002) was used to align CDS sequences of cp genome in NCBI. The gene annotation results of cp genomes for three Hordeum species were acquired using a manual correction. Moreover, to obtain gene annotation, rRNA and tRNA sequences of cp genomes were aligned in NCBI (https://www.ncbi.nlm.nih.gov/) database using HMMER v3.1b2 (HHMI/Harvard University, Boston, USA; The European Bioinformatics Institute, Cambridge, UK) (Finn et al., 2011) and Aragorn v1.2.38 programs (Murdoch University, Western Australia, Australia; Lund University, Lund, Sweden) (Dean and Bjorn, 2004). In addition, H. vulgare subsp. spontaneum (KC912688.1) was used as a reference sequence for quality control of the cp genome after assembly.
The Simple Sequence Repeats (SSRs) markers are a class of tandem repeats with motifs consisting of several nucleotides group (usually 1~6) as repeating units. The SSR marker is called cpSSR marker on cp genomes. CpSSR were identified and analyzed using the software MISA v1.0 (Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) Gatersleben, Corrensstr. 3, 06466 Seeland, Germany) (Beier et al., 2017). CpSSR parameters were described as A-B, with A representing the number of repetitions and B representing the total number of the base unit in a sequence. For example, 1-8 indicates more than 8 repetitions of a single-base, 2-5 indicates more than 5 repetitions of a double-base, 3-3 more than three repetitions of triple-base, 4-3, 5-3, 6-3 and so on. Furthermore, the interspersed repeats sequences, which are a different kind of repetitive sequences from tandem repeats and have both forward and palindromic repeats (including reverse and complementary) with a minimum size of 15 bp, sequence coherence of more than 90% and are distributed throughout the genome, were identified using the Vmatch v2.3.0 (http://www.vmatch.de/) program.
The degeneracy of codons show that each amino acid has one to six codons. The heterogeneity of synonymous codon usage is called Relative Synonymous Codon Usage (RSCU). To highlight the relative biasness between amino acids and codons, the RSCU was analyzed using the MEGA v10.1.8 program (Kumar et al., 2008).
The complete cp genomes of the three Hordeum species sequenced in this study and seven other Hordeum species (H. bulbosum, H. jubatum, H. marinum, H. murinum, H. pubiflorum, H. vulgare subsp. spontaneum, H. vulgare) were downloaded from the NCBI database and used for PR2 analysis to evaluate nucleotide usage bias in the coding genes of them (Wei et al., 2014). Base A, T, C and G content at the third site of synonymous codons were calculated using the MEGA v10.1.8 software.
SNP (Single Nucleotide Polymorphism) refers to the DNA sequence polymorphism caused by the variation (insertions or deletions (In/Dels)) of a single nucleotide at the genomic level and accounts for more than 90% of known polymorphisms. The cp genomes of three Hordeum materials were aligned using MAFFT program, version v7.310 (https://mafft. cbrc. jp/alignment/software/) (Standley, 2013) to identify SNP and In/Dels. In addition, the nucleotide diversity (Pi) and Ka/Ks in this study were calculated using the conjunct genes and protein-coding genes of the three Hordeum materials detected. Base mutation, including non-Synonymous mutations (Ka) and synonymous mutations (Ks) causes changes in amino acids, which ratios > 1 is called a positive selection effect and < 1 is named a purified selection effect. Pi is considered an important tool that able to reveal the variation of size of nucleic acid sequences, and a range of potential molecular markers can be provided based on the regions of high variability for population genetics (Meng et al., 2018). The Ka/Ks and Pi values were calculated using KaKs_Calculator v2.0 (https://sourceforge.net/projects/kakscalculator2/) (Zhang et al., 2006) and VCFTOOLS (Danecek et al., 2011), respectively. Nevertheless, before achieving the above tasks, the CDS sequences of the conjunct genes in each species were globally aligned using MAFFT software.
Alignment and collinearity of 10 Hordeum species complete cp genomes, H. bogdanii, H. brevisubulatum, H. violaceum, H. jubatum, H. bulbosum, H. marinum, H. murinum, H. pubiflorum, H. vulgare subsp. spontaneum, and H. vulgare, was analyzed using Mauve (Darling et al., 2004) and Mvista tools (http://genome.lbl.gov/vista/mvista/submit.html). The IRSCOPE online software (https://irscope.shinyapps.io/irapp/) was used to evaluate the expansion or contraction of IR and SC regions boundary for six species (H. bogdanii, H. brevisubulatum, H. violaceum, H. jubatum, H. vulgare subsp. spontaneum, and H. vulgare).
A total of 28 Poaceae species published in NCBI (Supplementary Table 2), and three hordeum species (H. bogdanii (CNS0491101), H. brevisubulatum (CNS0491102), H. violaceum (CNS0491103)) that in this study were sequenced to establish the phylogenetic tree. Saccharum spontaneum (LN896360.1) and Sorghum bicolor (NC008602.1) were the outgroups. MAFFT and RAxML v8.2.10 software (https://cme. h-its. org/exelixis/software. html) that follow GTR model and Hill Climbing algorithm were carried out to achieve the multi-sequence alignment and construction of the phylogenetic tree for different species, respectively.
Due to the loss of the IR region in the cp genomes of H. pubiflorum, H. murinum, H. marinum, and H. bulbosum, cp genome characteristics of only six Hordeum species, H. bogdanii, H. brevisubulatum, H. violaceum, H. jubatum, H. vulgare subsp. spontaneum, and H. vulgare were selected for comparison of cp genome characteristics (Figure 1). This comparison also included IR expansion and contraction. H. vulgare had the smallest cp genome size (136,462 bp) compared with that of the other five species (H. bogdanii (137,141 bp), H. brevisubulatum (137,002 bp), H. violaceum (137,032 bp), and H. spontaneum (136,536 bp), H. jubatum (136,826 bp), while it also had the highest GC content and total number of genes. Illumina paired-end sequencing yielded 26,262,890, 25,330,242, and 25,890,515 ReadSum (pair-end reads) from H. bogdanii, H. brevisubulatum, and H. violaceum, respectively. Q20 and Q30 (the percentage of bases with a mass value ≥20 and ≥30, respectively) were both more than 85%. The three perennial species (H. bogdanii, H. brevisubulatum, and H. violaceum) belonged to a typical quadrantal model, consisted of two copies of IR regions (IRs 21,573-21,587 bp), and were separated by LSC (81,128-81,169 bp) and SSC (12,728-12,798 bp) regions, which are the common feature of the majority of plants in the Poaceae family (Figure 1, Table 1). The overall GC content in the cp genomes of H. bogdanii, H. brevisubulatum, and H. violaceum was 38.23, 28.28, and 38.27%, respectively, and the percentage distributed in the IR regions was the highest than that in LSC and SSC regions. A total of 129, 131, and 131 genes were located in the complete cp genomes of H. bogdanii, H. brevisubulatum, and H. violaceum, respectively. Thirty-eight ribosomal RNA (rRNA) genes, 8 transfer RNA (tRNA) genes, and 85 messenger RNA (mRNA) genes were distributed in both H. brevisubulatum and H. violaceum. Interestingly, the annual cultivated species (H. vulgare) had the largest number of genes compared with the other five species, but these genes these genes were all attributed to tRNA.
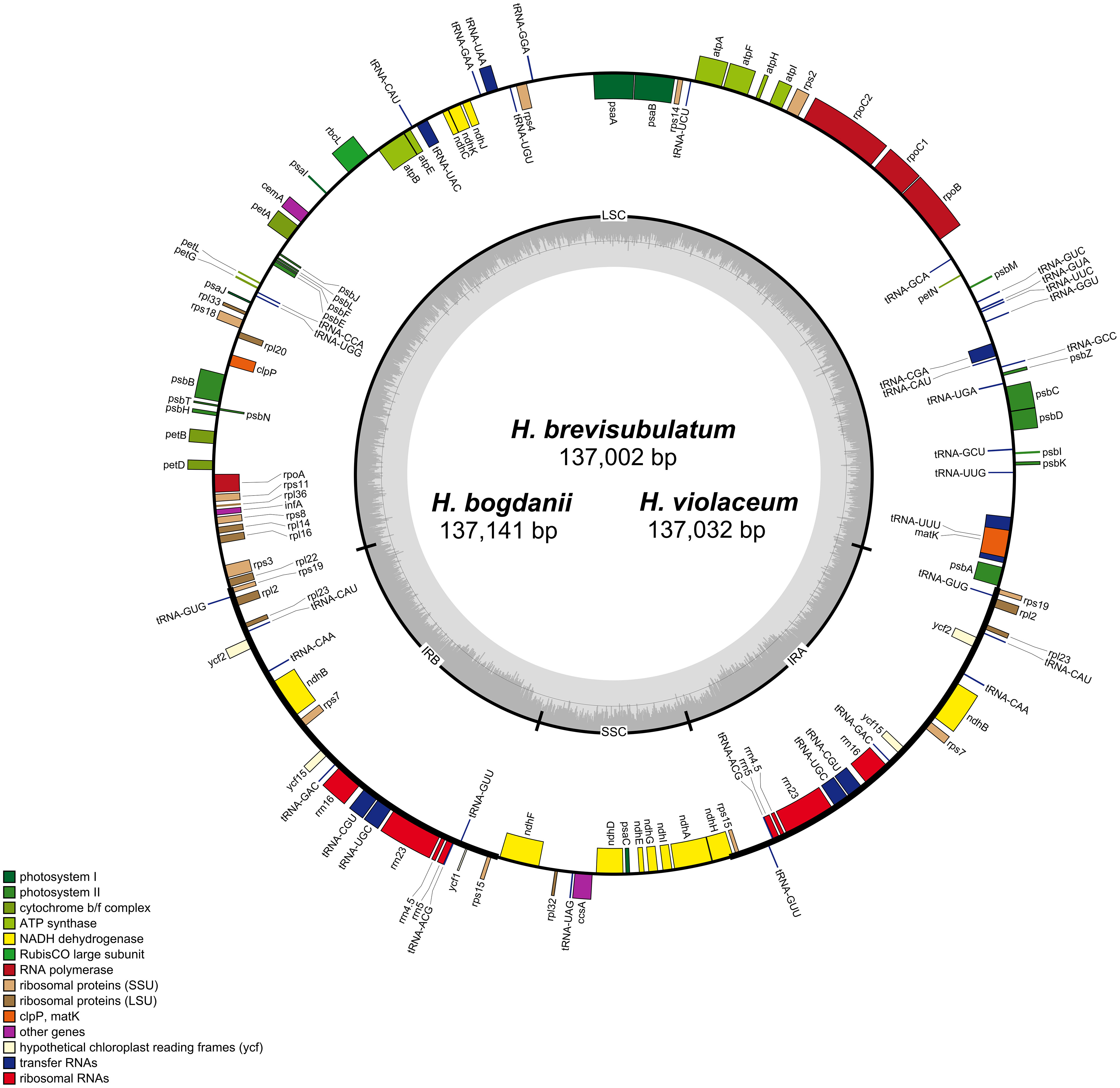
Figure 1 Gene maps of H. bogdanii, H. brevisubulatum, and H. violaceum cp genomes. Genes inside and outside the circle undergo clockwise and counterclockwise transcription in the gene map. Dark gray and light gray color represent guanine and cytosine (GC) content and adenine and thymine (AT) content, respectively.
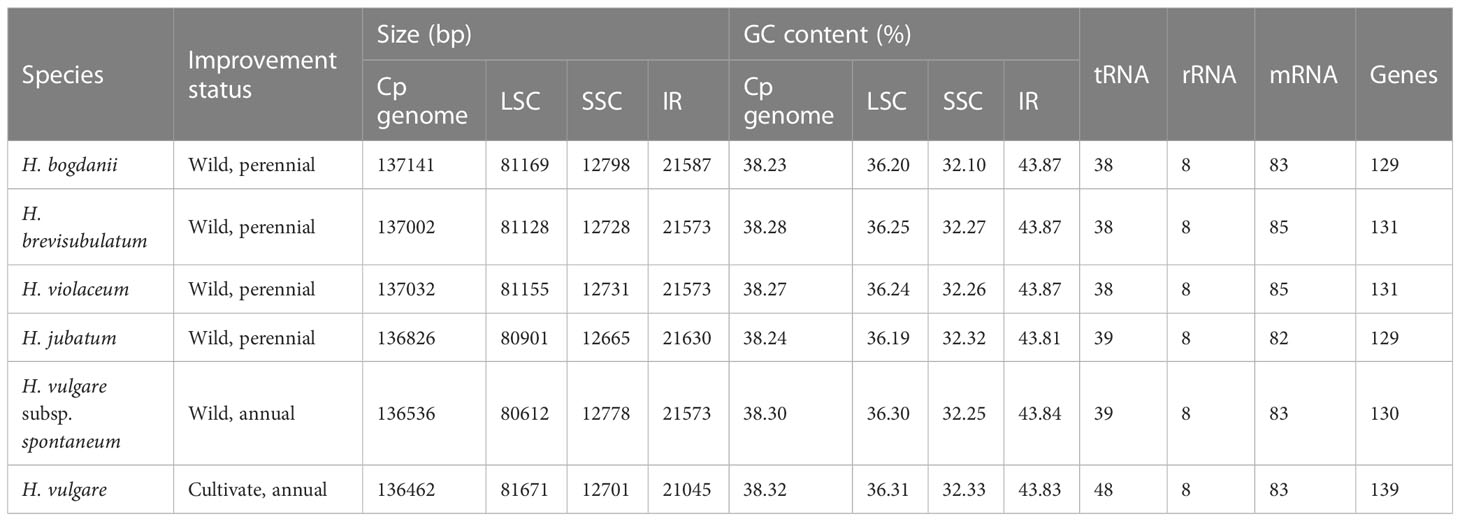
Table 1 Comparison of the six Hordeum chloroplast genomes.
Out of the 113 genes were shared by the five cp Hordeum genomes (H. bogdanii, H. brevisubulatum, H. violaceum, H. vulgare subsp. spontaneum, and H. vulgare) (Table 2). 46 were annotated to photosynthesis-related genes such as the large subunit of rubisco, a subunit of photosystem I, a subunit of photosystem II, a subunit of ATP synthase, cytochrome b/f complex, c-type cytochrome synthesis, and subunit of NADH dehydrogenase. Thirty-four genes were involved in self-replication, of which 30 genes and 4 genes were related to tRNA and rRNA, respectively. In addition, 12 genes encoding ribosomal proteins, as well as 14 genes were assembled into transcription. Interestingly, trnI-GAU, trnG-UCC, rps12, and rps16 genes were unique to two annual species (H. vulgare subsp. spontaneum and H. vulgare), while trnT-CGU and trnS-CGA genes were specific to three perennial species (H. bogdanii, H. brevisubulatum, and H. violaceum). More mutations may accumulate in introns because they are less constrained by natural selection than exons (Xiong et al., 2020b). Ten genes that contained a single intron in three cp genomes were collected (Supplementary Table 3).
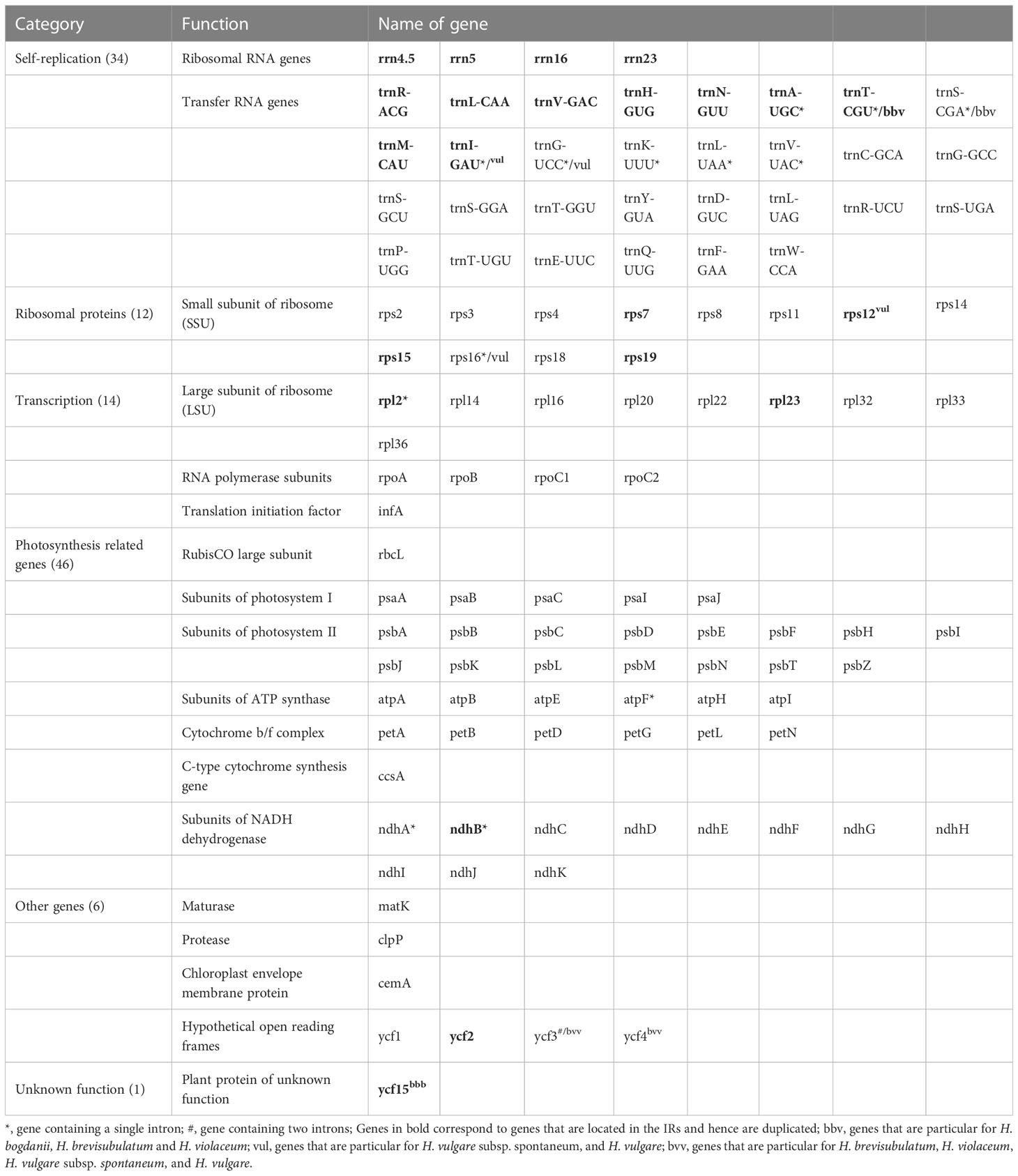
Table 2 List of genes annotated in the plastomes of the three wild perennial Hordeum species (H. bogdanii, H. brevisubulatum, and H. violaceum) from Central Asia and two annual species (H. vulgare subsp. spontaneum and H. vulgare).
Two different types of repeat sequences, which includes scattered repetitive sequences (palindrome repeats and direct repeats) and simple sequence repeats (SSR), were carefully analyzed Using MISA v1.0 and Vmatch v2.3.0, respectively. A total of 231 (forward type, 125 and palindromic type, 106), 220 (forward type 115 and palindromic type, 105), and 218 (forward type,115 and palindromic type 103) scattered repetitive sequences were predicted in H. bogdanii, H. brevisubulatum, and H. violaceum, respectively (Figure 2A). Their common characteristic is the number of repeats reached the peak at a repeat length of 15 bp (Figure 2C). SSR, a tandem repeat sequence of dozens of nucleotides generally composed of a series of repeat units (1-6 bp in length), was distributed throughout the genome. A total of 182 SSR in the cp genome of H. bogdanii was detected, which was greater than that of H. brevisubulatum (178) and H. violaceum (176) (Figure 2B). The number of mononucleotides (primarily poly-A or poly-T) accounted for the largest proportion of total SSR, which was above 59% (Figure 2B). Interestingly, trinucleotide (AGC) and tetranucleotide (AACA and AGAA) SSR were found only in H. bogdanii, and other types of SSR nucleotides in the cp genome of the three wild perennial Hordeum species were predicted with a fixed distribution (Figure 2D), which warrants further investigation in the future. The mononucleotide T was repeated 13 times and was unique to H. brevisubulatum and H. violaceum (Figure 2D). Furthermore, the majority of SSR were distributed in the LSC region, of which the proportion of H. bogdanii was 75.6%, slightly lower than that of H. brevisubulatum (76%) and H. violaceum (76%) (Figure 2E).
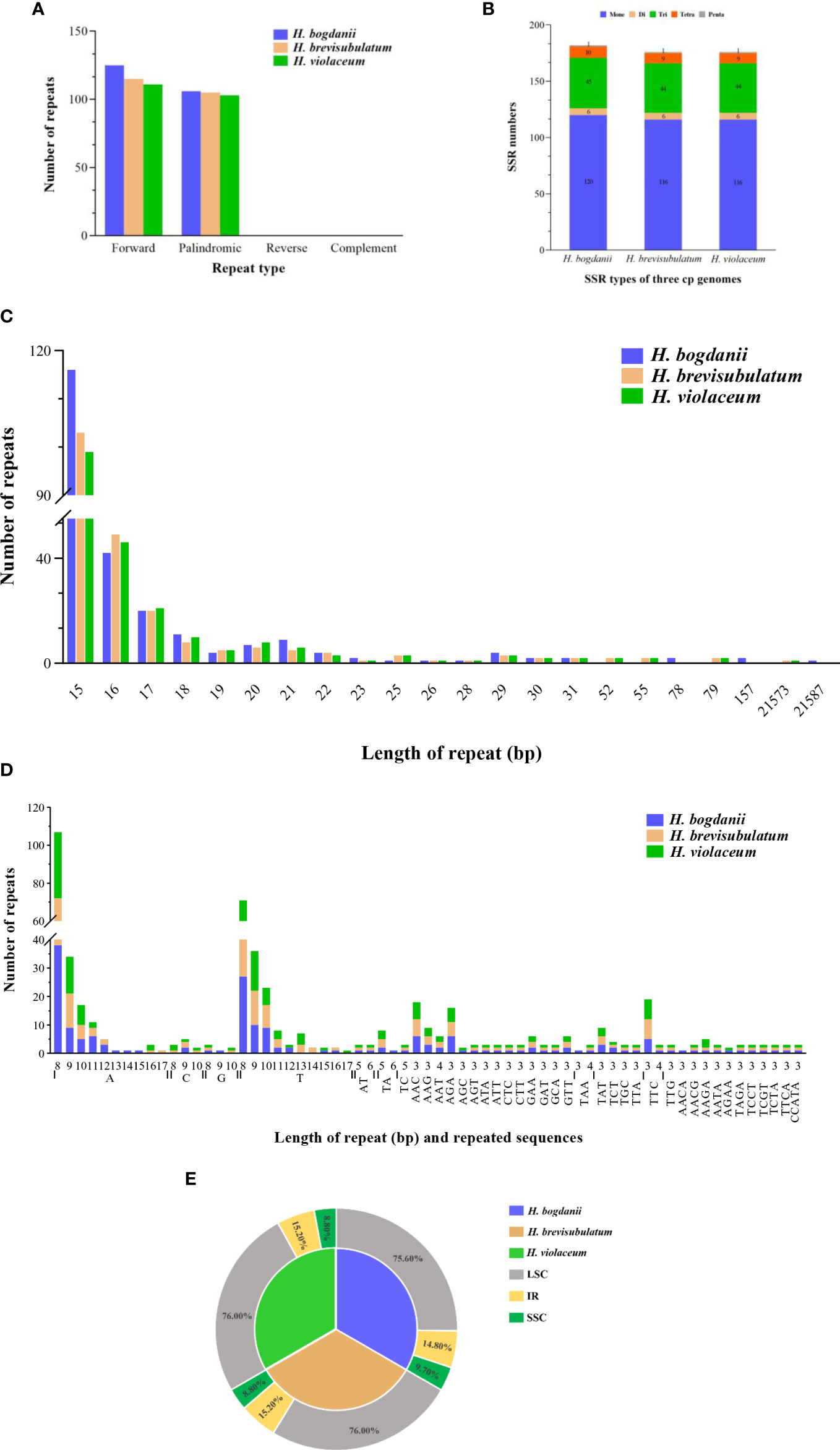
Figure 2 Simple sequence repeats (SSRs) and scattered repetitive sequences in the three Hordeum cp genomes. (A) frequency of repeat types; (B) compare of the number of SSR type in the three Hordeum cp genomes; (C) frequency of repeats length; (D) motifs in the cp genome of Hordeum; (E) Distribution region of repeating sequences of three Hordeum cp genome. IR, inverted repeat; LSC, large single-copy; SSC, small single-copy.
RSCU, which is caused by the unequal usage of a synonymous codon, was further analyzed (Figure 3). Each amino acid corresponds to at least one codon and at most six codons owing to the redundancy of codons. RSCU values for the initial codon (AUG) were 1.987, 1.983, and 1.987 in H. bogdanii, H. brevisubulatum, and H. violaceum, respectively. RSCU values for termination codons, UAA, UAG, and UGA, were 1.771, 0.651, and 0.578 in H. bogdanii, 1.730, 0.671, and 0.600 in H. brevisubulatum, and 1.730, 0.671, and 0.600 in H. violaceum, respectively. Codons with RSCU values >1, which are usually considered to be preferred codons, accounted for 51.61% (32/62) of codons, and the third nucleotide of most codons was biased towards either A or U. Notably, only one codon, UGG (corresponding to tryptophan), showed no bias in the three Hordeum species, and its RSCU was 1.00.
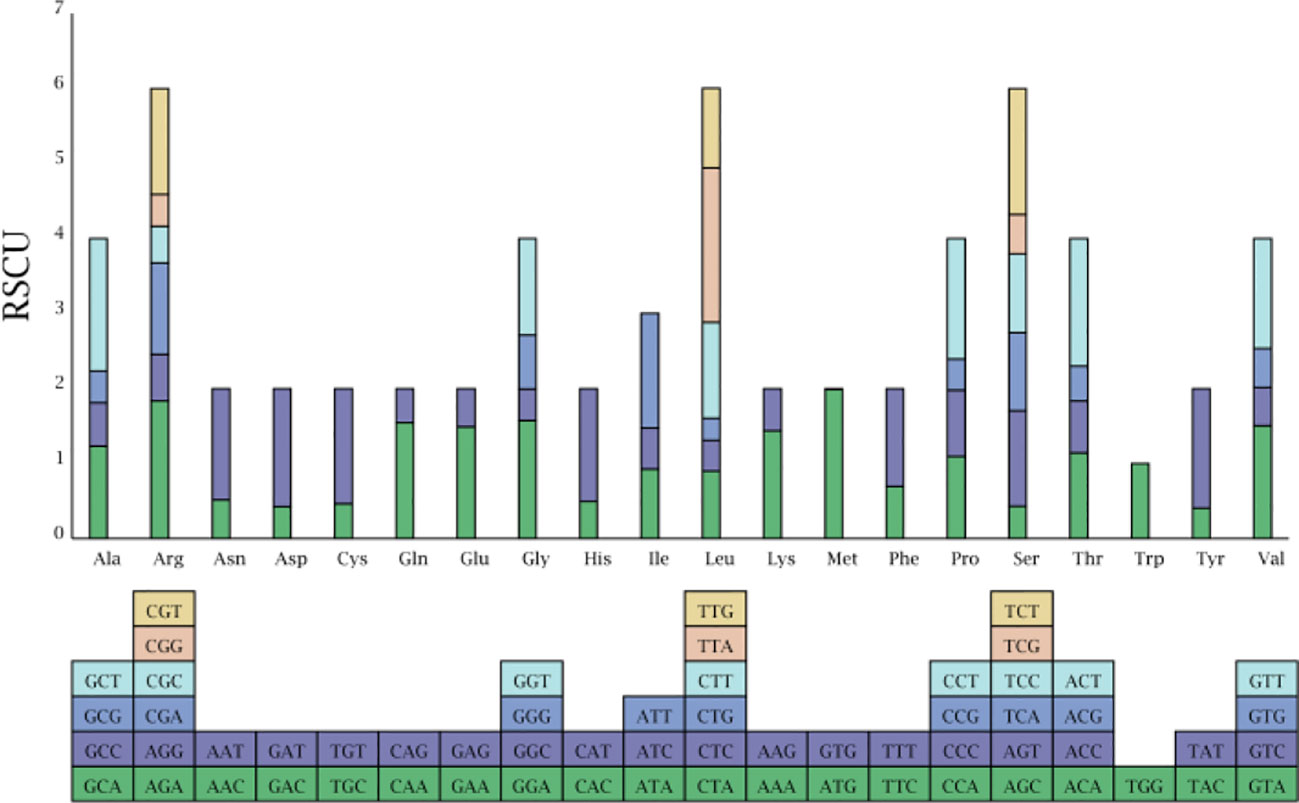
Figure 3 Relative frequency of synonymous codon for the twenty amino acids in the three Hordeum species chloroplast genomes.
Forty-four coding sequences (CDS, ≥300 bp long) containing start (ATG) and stop (TAG, TGA, TAA) codons were collected from the 10 cp genomes, to carry out PR2-plot analysis to further understand codon bias (Figure 4). The results showed that the 44 genes of the 10 species were not evenly distributed within the four regions, but mainly in G3/(G3+C3) > 0.5 and A3/(A3+T3) < 0.5 regions. This suggests that there may be a bias towards G and T bases at the third position of synonymous codons, which needs further investigation.
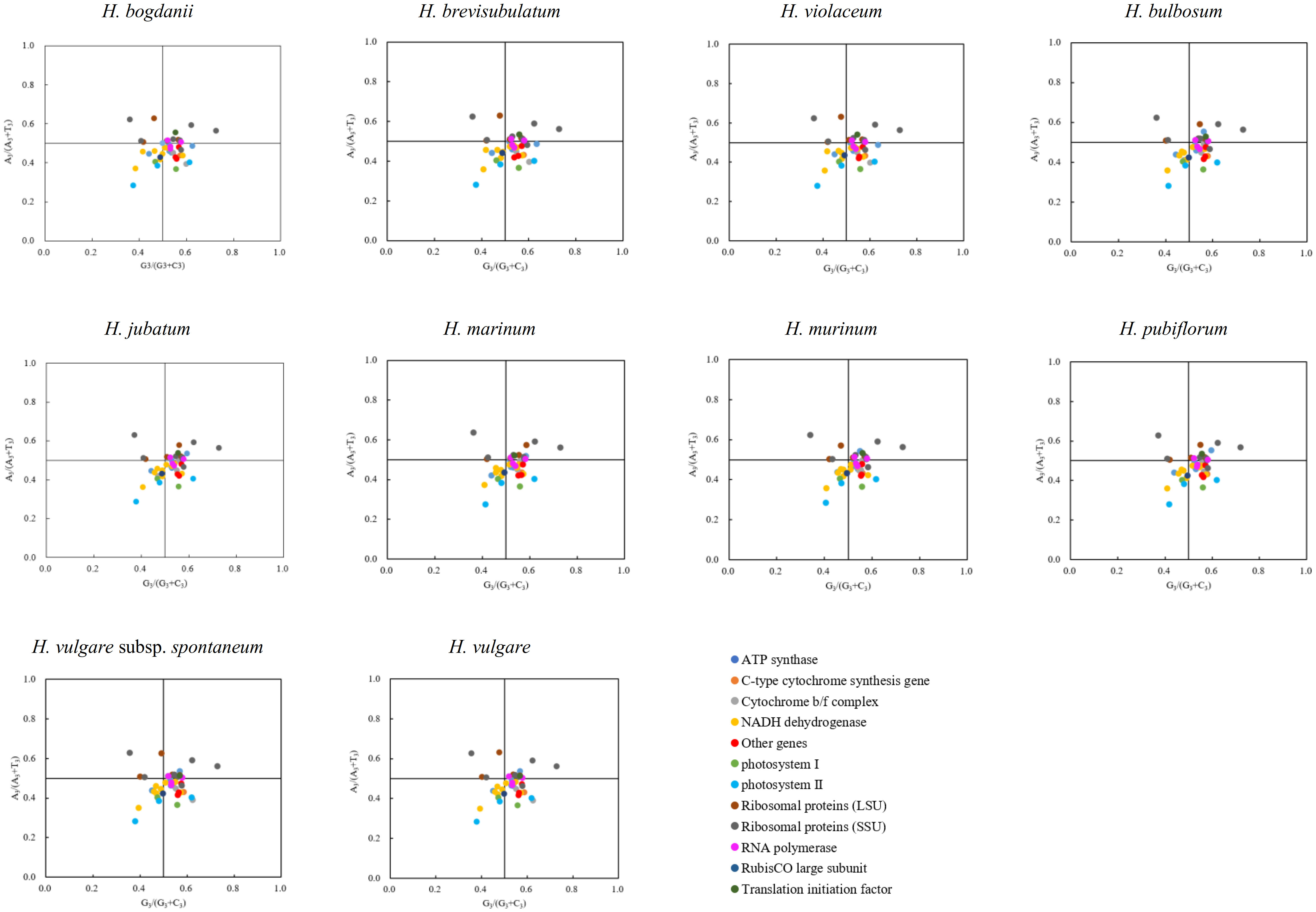
Figure 4 PR2-plot analysis of cp genomes ten Hordeum species. Base A, T, C and G content at the third site of synonymous codons were replaced through A3, T3, C3 and G3, respectively.
InDels and SNPs (mainly containing Tn (transition) and Tv (transversion)) were detected among the three Hordeum cp genomes using MAFFT software (Standley, 2013). A total of 109, 112, and 33 In/Dels were identified in H. bogdanii vs H. brevisubulatum, H. bogdanii vs H. violaceum, and H. brevisubulatum vs. H. violaceum, respectively, in which 4 InDels were discovered in the coding sequence (Supplementary Table 4). There were similar quantities of Tn and Tv in both H. bogdanii vs H. brevisubulatum (Tn = 61, Tv = 304) and H. bogdanii vs H. violaceum (Tn = 66, Tv = 298), most of which were encoded in the noncoding sequence. However, 19 Tn (2 coding, 17 noncoding) and 60 Tv (23 coding, 37 noncoding) were detected during H. brevisubulatum vs H. violaceum. Interestingly, we found that both InDels and SNPs were mainly concentrated in LSC and the intergenic region for each pairwise comparison, while InDels did not occurred in the IR region of H. brevisubulatum vs H. violaceum (Figure 5).
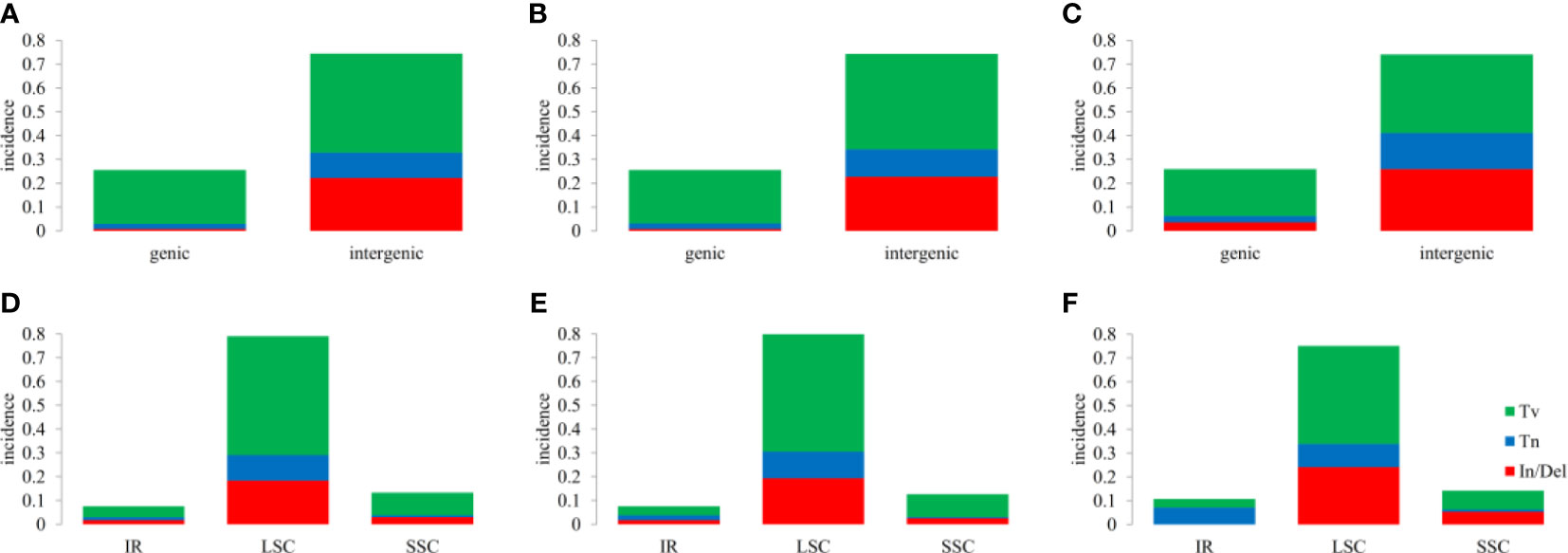
Figure 5 Overview of single nucleotide polymorphisms (SNPs) and Insertions/Deletions (In/Dels). (A, B), (C, D), and (E, F) the differences between H. bogdanii vs H. brevisubulatum, H. bogdanii vs H. violaceum and Hordeum brevisubulatum vs Hordeum. violaceum. Tv, transversion; Tn, transition; In/Del, insertion/deletion; IR, inverted repeat; LSC, large single-copy; SSC, small single-copy.
The non-synonymous/Synonymous mutation ratio (Ka/Ks) ratio of 83 common protein-coding genes in cp genomes of the three Hordeum species was calculated using Ka/Ks Calculator software (Zhang et al., 2006) (Supplementary Table 3). Ka/Ks values of H. bogdanii vs H. brevisubulatum, H. bogdanii vs H. violaceum, and H. brevisubulatum vs H. violaceum were 16, 19, and 2, respectively. In addition, the Ka/Ks values of some genes (ropB, atpI, psaB, etc.) could not be computed because Ka or/and Ka was 0, which suggests that these genes were relatively conservative without any Ka or Ks nucleotide substitution. Pi values were calculated using VCFTOOLS software. A total of 101 common genes in the three wild perennial Hordeum species were examined, whose Pi values ranged between 0 to 0.1674 (Figure 6). It is noteworthy that relatively higher Pi values (Pi ≥ 0.1) were detected in five genes, including tRNA-UGC, tRNA-UAA, tRNA-UUU, tRNA-UAC, and ndhA. Meanwhile, these genes were also among those with Ka/Ks > 1. Moreover, other genes with a Pi ≥ 0.1 were found in single-copy (SC) rather than IR regions, except for tRNA-UGC.
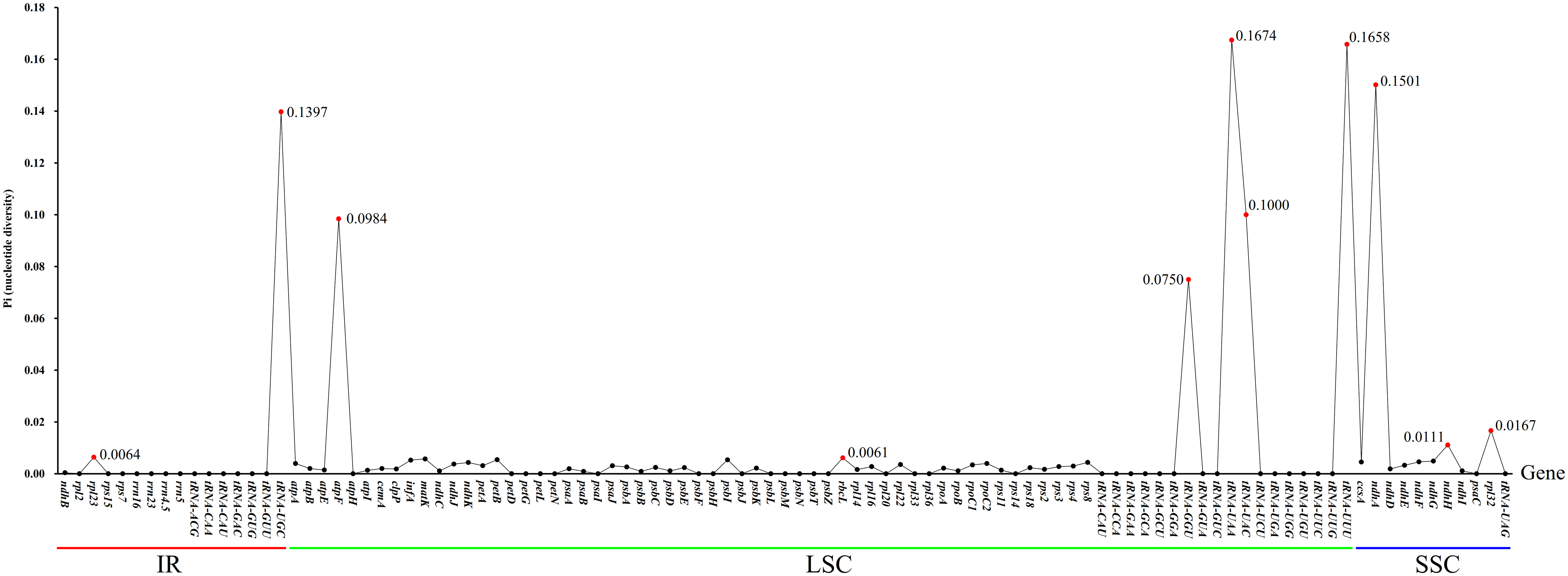
Figure 6 The nucleotide diversity (Pi) calculated by 101 genes shared in three wild perennial Hordeum species. Genes with Ka/Ks value > 1 are highlighted in red; The genes above the red line, green line and blue line were located in IR, LSC and SSR regions, respectively.
To understand the sequence divergence between wild and cultivated, as well as annual and perennial species in genus Hordeum, and elaborate further on the evolutionary events that occurred, including gene mutation, rearrangement and loss, we analyzed and compared the cp genomes of two annual species (one cultivated species, H. vulgare and one wild species, H. vulgare subsp. spontaneum), and eight perennial wild species (H. bogdanii, H. brevisubulatum, H. violaceum, H. bulbosum, H. jubatum, H. marinum, H. murinum, and H. pubiflorum) were compared and analyzed. It was found that the coding region is more conservative than the non-coding region, as well as the divergence frequency was higher in the LSC and SSC region than in IR region (Figure 7). The two annual species (especially H. vulgare) had many conserved regions compared with the other eight wild perennial species, this was the case in the CNS (Conserved Noncoding Sequences) of LSC and SSC regions. The highly variable regions are called hotspots regions, and these regions were mainly concentrated in small RNA molecules such as tRNA-GGU ~ tRNA-GCA, tRNA-UGU ~ ndhJ, psbE ~ rps18, ndhF ~ tRNA-UAG. Furthermore, MAUVE software revealed rearrangement events with scanty genes in the cp genomes of 10 species (Supplementary Figure 1).
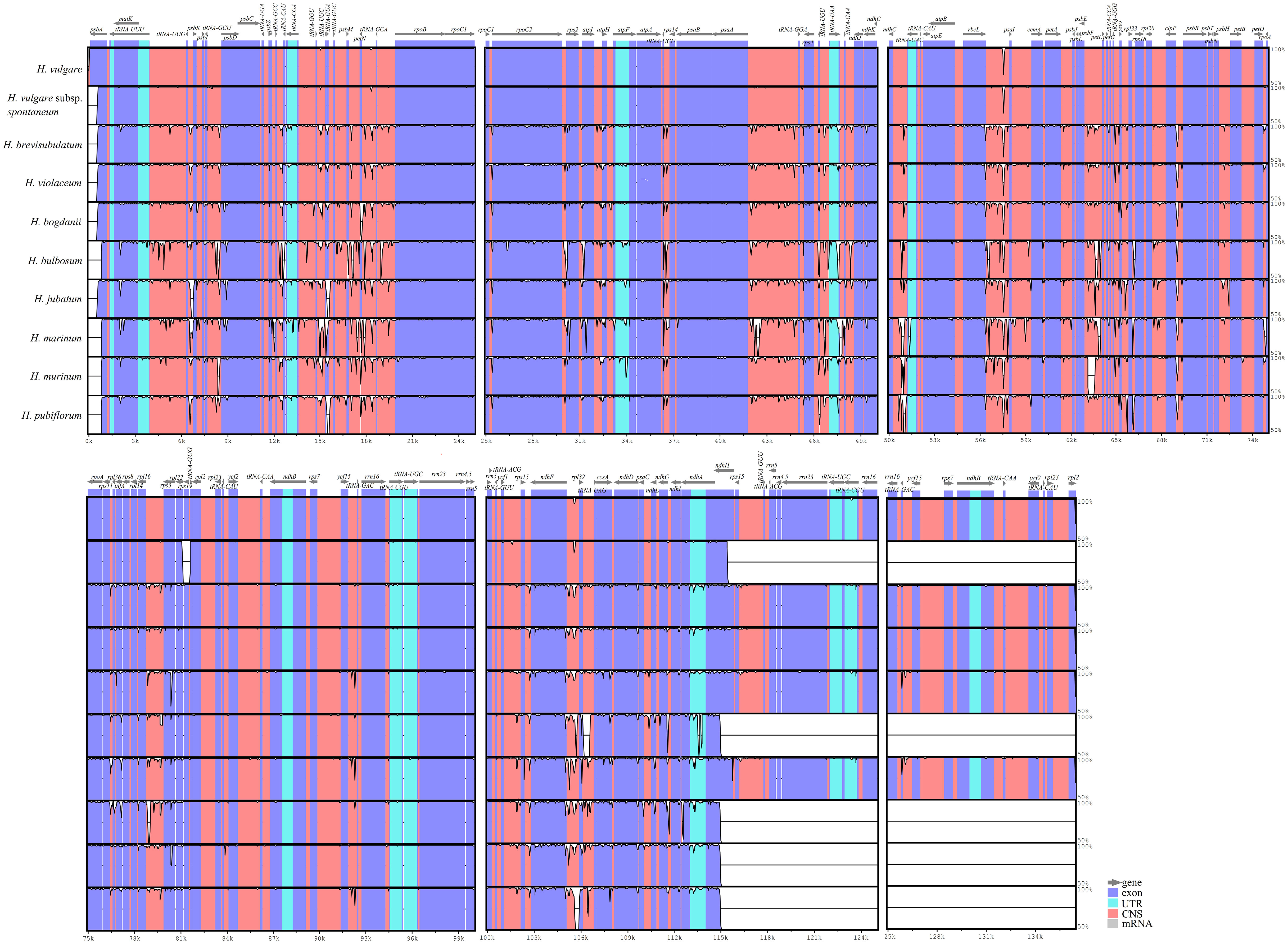
Figure 7 Alignment of the ten Hordeum species cp genome sequences. Exon, untranslated region (UTR), conserved noncoding sequences (CNS), and mRNA were marked by different colors. The x-axis and level a clinic columnar strip express the paratactic and sequences stability in the cp genome and the peaks represent hotspot regions.
Expansion and contraction of IR regions, recognized as an evolutionary event, are generally concentrated in the junction of IR/SSC or IR/LSC. Moreover, this phenomenon is the primary cause of the variation of cp genomes size. Therefore, the IR borders of six species in the Hordeum genus were compared to explore their differences. The species studied included two annuals (including one cultivated species, H. vulgare and one wild species, H. vulgare subsp. spontaneum), and four perennial wild species (H. bogdanii, H. brevisubulatum, H. violaceum, and H. jubatum) (Figure 8). The results showed significant differences in the junction sites between the annual and perennial species. The genes ndhF-ndhH and rpl2-trnH-psbA-rpl22-rps19 were found close in SSC/IR and LSC/IR boundaries, respectively. The ndhH genes of the other five species ranged from 207 (H. bogdanii, H. brevisubulatum, H. violaceum) to 216 (H. vulgare) bp in IRa region throughout the SSC/IRa junction, with the exception of H. vulgare subsp. spontaneum. Two genes, trnH and rpl2, were found near the junction of LSC/IR region in H. vulgare, whereas the genes around this junction region of the other five species were rpl22 and rps19 genes. Additionally, we observed that only e ndhH gene for H. vulgar was separated from SSC/IRb boundary with 1 bp.
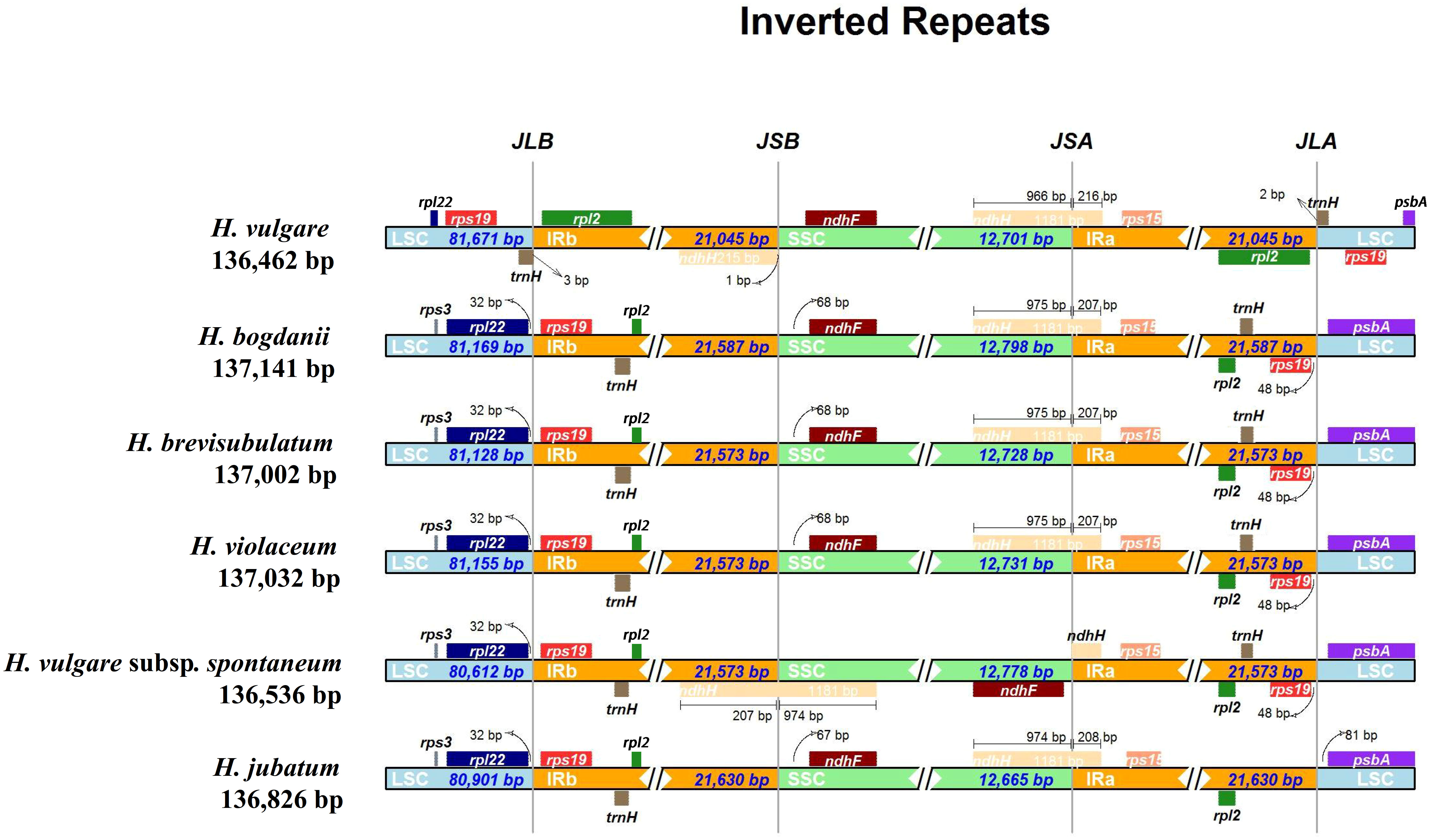
Figure 8 IRscope analysis of the six Hordeum cp genomes. JLB, JSB, JSA, and JLA represent the junction of LCS and IRb, SSC and IRb, SSC and IRa, and LSC and IRa region, respectively.
The phylogenetic position of Triticeae was identified based on the cp genomes sequences of three studied Hordeum species and other 28 species downloaded from NCBI (Figure 9). The structure of this phylogenetic tree of these species conformed with the classical botanical classification. Twelve Hordeum species were divided into six sub-groups, among which H. brevisubulatum, and H. violaceum were in the same sub-groups, and H. bogdanii is further distant from them. Different accessions of the same species are placed in the same subgroup. In addition, genus Hordeum was more closely related to the species of Elymus, Aegilops, Triticum than to Agropyron.
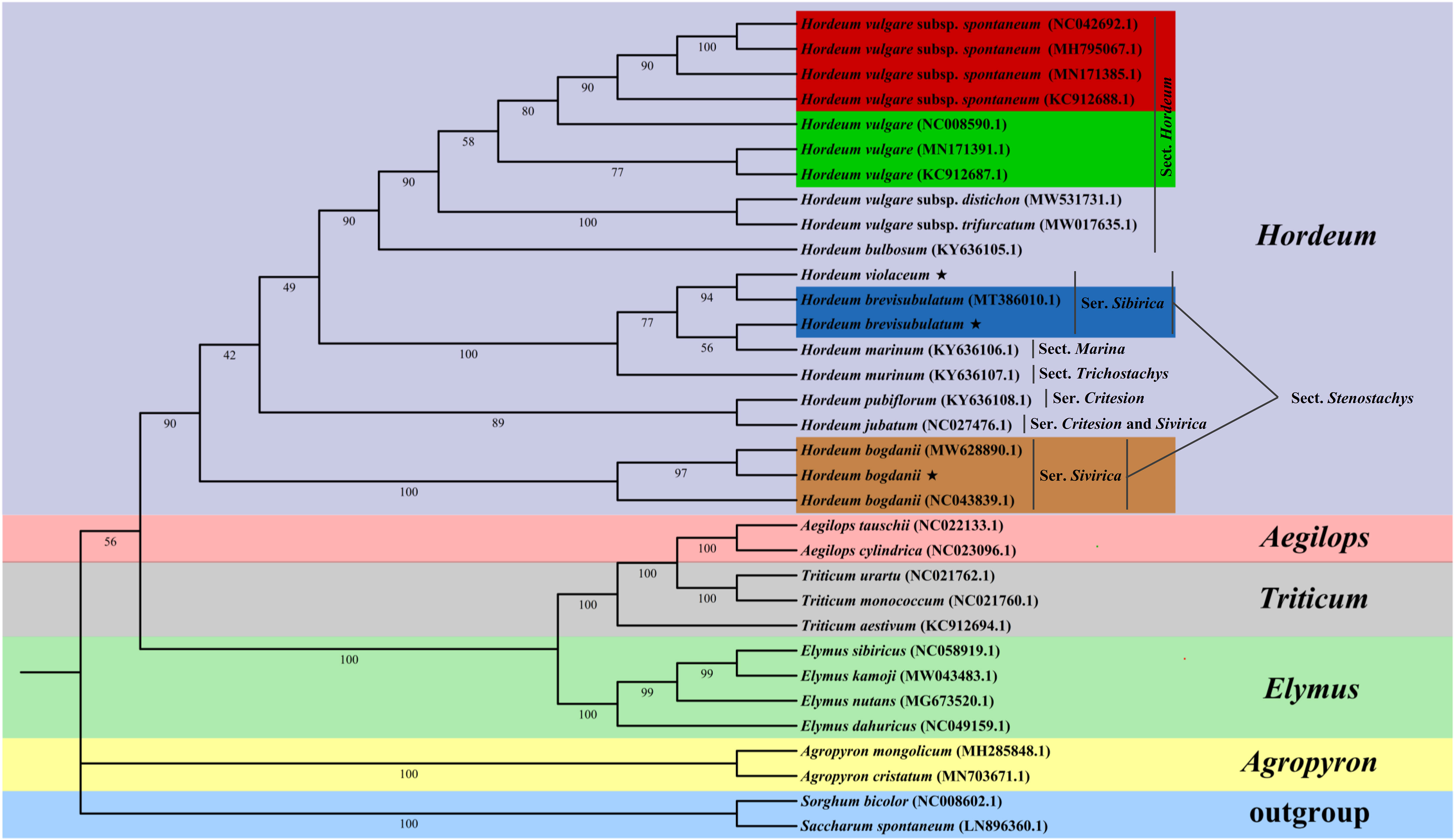
Figure 9 ML phylogenetic tree of 31 Poaceae species, with Saccharum spontaneum and Sorghum bicolor as outgroups. The bootstrap values are shown at the nodes; H. vulgare subsp. spontaneum, H. vulgare, H. brevisubulatum, and H. bogdanii species of different accession were represented by the base color of red, green, blue, and orange, respectively.
The total size and GC content of cp genomes were not significantly different among the three wild perennial Hordeum species (H. bogdanii, H. brevisubulatum, and H. violaceum). These results revealed that the cp genome size and GC content of Poaceae are highly conserved, and the occurrence of variation may help us to better understand the unique variation among species or subspecies (Liu et al., 2019). A total of 129, 131, and 131 genes were detected in the cp genomes of H. bogdanii, H. brevisubulatum, and H. violaceum, respectively. Notably, two mRNA genes, ycf3 and ycf4, which were detected in these transformants and have been shown to contribute to the unstable accumulation of photosystem I complexes in the thylakoid membranes (Boudreau et al., 1997), were not found in H. bogdanii. This may be because two genes were transferred from the cp genome of H. bogdanii to its nuclear genome during the evolution of the species (Xiong et al., 2020). Two transfer RNA genes (trnG-UUC and trnI-GAU) and two small subunit of ribosome genes (rps12 and rps16) were found to be unique to only two annual Hordeum species, including one wild species (H. vulgare subsp. spontaneum) and one cultivated species (H. vulgare). However, the functions of these four genes require further validation in the future. Genes specific for cultivated species (H. vulgare) in this study were not identified. This may be due to genetic changes may not exist in the cp genome but rather in the nuclear genome during plants domestication. Typically, cp genomes of Poaceae species are highly conserved in structure, which is a typical quadripartite (the IR region is separated by LSC and SSC). However, in some plants, cp genomes contain only one IR region (alfalfa) (Tao et al., 2016) or lack the IR region (algae) (Xue et al., 2019). H. bulbosum, H. marinum, H. murinum, and H. pubiflorum also fall into this category, with linear cp genomes without the IR region (Bernhardt et al., 2017). Therefore, the cp genome characteristics of these four Hordeum species were not analyzed and compared in the current study. However, cp genome characteristics of only two annual species (H. vulgare, and H. vulgare subsp. spontaneum) and four perennial species (H. bogdanii, H. brevisubulatum, H. violaceum, and H. jubatum) were analyzed and compared. The result demonstrated that the size and GC content of cp genomes of the six Hordeum species ranged from 136,462 to 137,141 bp and 38.23% to 38.32%, respectively, indicating that the cp genome length and GC content of synanthropic species were not significantly different, while the number of genes (139) in cultivated species were more abundant compared with that in wild species. The reason may be that natural selection has led to an accelerated rate of gene loss in wild species (Vishwakarma et al., 2017). It is well known that gene degradation and even loss occur because the cp genome of angiosperms evolves relatively fast (Lei et al., 2016). Our study found no significant difference in the total number of genes among the five wild Hordeum species, which ranged from 129 to 131 (Table 1), which was significantly lower than that of H. vulgare (139), with a maximum gap of 10 genes and a minimum of 8 genes, such as rps12, rps16, etc. There is evidence that these genes have been lost in Ulmus (Zuo et al., 2017) and Orchidaceae (Jing et al., 2014).
Introns, which are located in the non-coding region, typically have higher mutation rates than exons, as their functions are often more restricted (Gan et al., 2018). Nevertheless, it is noteworthy that introns play a crucial role in regulating gene expression (Ma et al., 2016). Nine genes, including atpF, ndhA, ndhB, tRNA-CGA, tRNA-CGU, tRNA-UAA, tRNA-UAC, tRNA-UGC, and tRNA-UUU, are shared by the three wild perennial Hordeum species and contain only one intron, while one gene, ycf3, contains two introns, which is unique to H. brevisubulatum and H. violaceum (Supplementary Table 3). In addition, the ycf3 gene in the cultivated Hordeum species contains two introns (Middleton et al., 2013). Therefore, we contemplated that the absence of ycf3 gene introns in H. bogdanii is unusual. Previous research has suggested that a species that a lack of gene introns in a species may indicate that it has taken on additional functions in diverse areas such as protease, RNA polymerase, and ribosomal pathways (Hakobyan et al., 2021).
Cp SSR in population genetics is considered a valuable molecular marker owing to its traits of matrilineal inheritance and low recombination frequency; gene insertion or deletion is also frequent in Cp SSRs (Xiao et al., 2019; Zong et al., 2019). Scattered repetitive sequences (SRS) and SSR of three wild perennial Hordeum species were analyzed and compared in the present study. The total number of SRS and cpSSRs in H. bogdanii, H. brevisubulatum, and H. violaceum were 231, 220, 218 and 182, 176, 176, respectively. H. bogdanii showed significantly different results from other two species, possibly due to their relatively close phylogenetic relatedness. In addition, the results of the study of Secale sylvestre (Skuza et al., 2022) and Spartina maritima (Rousseau-Gueutin et al., 2015) suggested that related species usually have similar SSR loci. Remarkably, most of the SSRs of the three Hordeum species are mononucleotides repeats dominated by poly-A or poly-T. This SSR phenomenon has not only been reported in the cp genomes of the Poaceae family (Phalaris arundinacea and P. aquatica) (Xiong et al., 2020) but also in other angiosperm families, such as Hibiscus rosa-sinensis (Abdullah et al., 2020), Firmiana (Abdullah et al., 2019), and Taenia (Yang et al., 2014).
During the translation of mRNA into proteins, there is an uneven frequency of synonymous codon usage called RSCU (Tyagi et al., 2020). In our study, 90.62% of codons with RSCU > 1 preferentially select A/U as the third nucleotide site, which is much higher than those ending with G/C, with similar results in many angiosperms such as Nicotiana otophora (Asaf et al., 2016), Oryza minuta (Sajjad et al., 2017), and Medicago sativa (Tao et al., 2016). The preference for A/U-ending codons is a common feature among most angiosperms and may be associated with certain evolutionary processes (Wang et al., 2023). PR2-plot analysis is essential for exploring codon bias. If the values of G3/(G3+C3) and A3/(A3+T3) are equal to 1, codon bias is completely influenced due to base mutation pressure; if both values are equal to 0, it is entirely because of natural selection (Wen et al., 2016). The majority of genes in our study had G3/(G3+C3) values greater than 0.5 and A3/(A3+T3) values lower than 0.5, indicating a bias towards G and T nucleotides in the third codon position, possibly due to a combination of natural selection and base mutations (Chen et al., 2021).
In the process of natural mutation, the probability of point mutation (SNP) is normally greater than that of frameshift (In/Del) (Raes and Van de Peer, 2005). As previously stated, the results of the cp genomes of the three Hordeum demonstrated that most mutations supported this conclusion. Interestingly, these mutation sites were concentrated in the intergenic or LSC region. The number of SNPs and In/Dels was significantly higher between H. bogdanii vs H. brevisubulatum and H. bogdanii vs H. violaceum compared with H. brevisubulatum vs H. violaceum. The reason may be that H. bogdanii was phylogenetically more distant from H. brevisubulatum and H. violaceum. Notably, no In/Dels were detected in the IR regions of H. brevisubulatum vs H. violaceum, suggesting that IR regions were the most conservative in the four-part structure (LSC, SSC, and IRa/IRb) of the cp genome, which warrants further exploration (Ravi et al., 2008). Pi, which is one of the standards that estimate the degree of nucleotide sequence variation and provide greater insight into the genetic variation to reflect complex changeable selection pressures in species and population levels (Namgung et al., 2021). Five genes with relatively high Pi values (Pi ≥ 0.1) were identified in the cp genomes, including tRNA-UGC, tRNA-UAA, tRNA-UUU, tRNA-UAC, and ndhA. These mutation hotspots can serve as a basis for further development of barcode molecular markers and phylogenetic analysis of the genus Hordeum.
The cp genomes of the 10 Hordeum species were analyzed for sequence variant and collinearity of using mVISTA and MAUVE procedures, respectively. The results indicated that the cultivated species, H. vulgare, were relatively conservative compared with the other wild related species. The wild plants undergo rapid molecular evolution due to which they form hotspot regions more frequently that are mainly located in the non-coding region of the LSC (Peng et al., 2021). Similar observations have been reported with Morella rubra (Liu et al., 2017) and three Cardiocrinum species (Lu et al., 2016). Notably, a series of hotspots regions were discovered, which mainly concentrated on tRNA-GGU ~ tRNA-GCA, tRNA-UGU ~ ndhJ, psbE ~ rps18, ndhF ~ tRNA-UAG, etc. Repeated conversions of genes between IRa and IRb regions may be a key factor responsible for generating these hotspots (Park et al., 2019). Collinearity analysis is generally a crucial strategy to determine the degree of cp genome variation (Liu et al., 2018). Collinearity analysis demonstrated that no rearrangement was detected in the cp genomes of the ten Hordeum species. However, there were significant differences were observed based on the cp genomes size, genotype, and expansion or contraction of IR boundaries.
As plants continue to evolve, the IR boundary can expand or contract due to the insertion or deletion of certain genes in the IR or SC region, which are the main factors contributing to cp genome size variation (Li et al., 2020). Here, the junction sites of the IR/SC region of the six cp genomes were analyzed using an online IRSCOPE software. In addition to the two annual Hordeum species (H. vulgare and H. spontaneum), no significant gene expansion, contraction, or loss was detected in the LSC/IRs/SSC boundary of the remaining four wild perennial Hordeum species (H. bogdanii, H. brevisubulatum, H. violaceum, and H. jubatum). This could be related to the fact that annual species have a more rapid evolutionary rate compared to perennial species (Duchene and Bromham, 2013). The length of the SSC region of H. vulgare was relatively smaller, mainly because the ndhH gene spanned the SSC/IRa region with 966 bp, which was the smallest compared with the other four wild perennial Hordeum species, located in the SSC region. Furthermore, the sites of genes trnH and rps19 of H. vulgare changed significantly compared with those of the other Hordeum species. Besides, the rpl22 gene only existed in the LSC region of H. vulgare, suggesting that it was replicated. This phenomenon may be attributed to the continuous domestication of the cultivated species, H. vulgare, leading to genetic changes through natural selection (Suoi et al., 2016). Therefore, the variation of the IR boundary and can be useful for phylogenetic studies of Hordeum species.
The cp genome is quite conservative in sequence and structure, and the homology of molecular characters is easier to determine, thus it is a useful tool for constructing plant phylogeny (Yang et al., 2022). We conducted a phylogenetic analysis based on 31 Poaceae species (28 have been published and cp genomes of 3 Hordeum species were sequenced in the current study), with Saccharum spontaneum and Sorghum bicolor as the outgroups. The result showed that H. bogdanii has a further distance from H. brevisubulatum and H. violaceum. However, Jonathan et al. (Jonathan and Blattner, 2015) established a phylogenetic tree of these three Hordeum species based on the nuclear single-copy genome sequence analysis and demonstrated that they are clustered into a group. There may be two possible reasons for this difference. The first that the maternal ancestor of H. bogdanii is quite different from that of H. brevisubulatum and H. violaceum, and therefore it is hard to determine owing to relatively few reports on their matrilineal inheritance information. Another reason is the difference between the selected outgroups. In addition, although H. brevisubulatum (MT386010.1) has been published, the sequenced H. brevisubulatum in this study cannot be grouped into an identical subgroup. This may be because the former is a diploid or hexaploidy, while the latter is a tetraploid (Jakob and Blattner, 2006). Our findings provide valuable information for further investigation of the evolution trends of the cp genome in Hordeum species.
In summary, we sequenced and annotated the cp genomes of three Hordeum species (H. bogdanii, H. brevisubulatum, and H. violaceum) that exhibit a typical quadripartite structure. We then compared them to the cp genomes of two annual species, including one cultivated species (H. vulgare) and one wild species (H. vulgare subsp. spontaneum), as well as other five wild Hordeum species have been previously published. The results demonstrated that the cp genome of H. vulgare was more conserved although it contains a greater number of genes. Two mRNA genes, ycf3 and ycf4, were not identified in H. bogdanii, of which ycf3 contains two introns. Genes trnG-UUC, trnI-GAU, rps12, and rps16 that are specific to only two annual Hordeum (H. vulgare, and H. vulgare subsp. spontaneum) and may be closely related to the regulation of Hordeum growth. Five highly polymorphic genes (tRNA-UGC, tRNA-UAA, tRNA-UUU, tRNA-UAC, and ndhA) and a series of hotspot regions, which mainly concentrated on tRNA-GGU ~ tRNA-GCA, tRNA-UGU ~ ndhJ, psbE ~ rps18, ndhF ~ tRNA-UAG, etc., were identified. These findings lay the foundation for further development of barcode molecular markers and phylogenetic analysis of Hordeum L. In addition, based on the phylogenetic tree analysis, H. brevisubulatum and H. violaceum were classified into the same group and were found to be relatively close phylogenetic relatives as compared with H. bogdanii. Finally, the present study highlights the degree of variation between wild and cultivated, as well as annual and perennial Hordeum species, providing insights into phylogenetic evolution and population genetics in the genus Hordeum.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://db.cngb.org/, CNS0491101, https://db.cngb.org/, CNS0491102, https://db.cngb.org/, CNS0491103.
SY and CN: Conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review and editing, visualization. These authors contributed equally to this work and share the first authorship. YL and XM: Writing – review and editing, supervision, project administration, and funding acquisition. SJ, TL, JZ and JP: Investigation, resources, and writing – review and editing. WK and WL: Formal analysis, investigation, and data curation. YX and YLX: Methodology, software, validation, and formal analysis. XL and QY: Writing – review and editing. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (grant numbers 31570654), the Regional Innovation Cooperation Project of Sichuan Province (2022YFQ0076), the Project of Cooperation between Provincial School and Provincial College (2023YFSY0012). The Forage Position Expert Project of Sichuan Beef Cattle Innovation Team (SCCXTD-2020-13).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1170004/full#supplementary-material
Abdullah, S. I., Mehmood, F., Ali, Z., Waheed, M. T. (2019). Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 19, 100199. doi: 10.1016/j.plgene.2019.100199
Abdullah, S. I., Mehmood, F., Waseem, S., Mirza, B., Ahmed, I., Waheed, M. T. (2020). Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 112, 581–591. doi: 10.1016/j.ygeno.2019.04.010
Asaf, S., Khan, A. L., Khan, A. R., Waqas, M., Kang, S. M., Khan, M. A., et al. (2016). Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00843
Alyr, M. H., Pallu, J., Sambou, A., Nguepjop, J. R., Seye, M., Tossim, H. A., et al. (2020). Fine-mapping of a wild genomic region involved in pod and seed size reduction on chromosome A07 in Peanut (Arachis hypogaea L.). Genes 11, 1402. doi: 10.3390/genes11121402
Bailey, M., Ivanauskaite, A., Grimmer, J., Akintewe, O., Payne, A. C., Etherington, R., et al. (2020). The Arabidopsis NOT4A E3 ligase promotes PGR3 expression and regulates chloroplast translation. Nat. Commun. 2020, 21998. doi: 10.1038/s41467-020-20506-4
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bernhardt, N., Brassac, J., Kilian, B., Blattner, F. R. (2017). Dated tribe-wide whole chloroplast genome phylogeny indicates recurrent hybridizations within Triticeae. BMC Evol. Biol. 17, 141. doi: 10.1186/s12862-017-0989-9
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Boetzer, M., Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13, 1–9. doi: 10.1186/gb-2012-13-6-r56
Boudreau, E., Takahashi, Y., Lemieux, C., Turmel, M., Rochaix, J. D. (1997). The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO J. 16, 6095–6104. doi: 10.1093/emboj/16.20.6095
Brassac, J., Blattner, F. R. (2015). Species-level phylogeny and polyploid relationships in Hordeum (Poaceae) inferred by next-generation sequencing and In Silico cloning of multiple nuclear loci. Syst. Biol. 64, 792–808. doi: 10.1093/sysbio/syv035
Chen, S. Y., Zhang, H., Wang, X., Zhang, Y. H., Ruan, G. H., Ma, J.. (2021). Analysis of codon usage bias in the chloroplast genome of Helianthus annuus J-01. IOP Conf. Series: Earth Envir Sci. 792, 12006–12009. doi: 10.1088/1755-1315/792/1/012009
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Darling, A., Mau, B., Blattner, F. R., Perna, A. (2004). Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dean, L., Bjorn, C. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucl. Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Duchene, D., Bromham, L. (2013). Rates of molecular evolution and diversification in plants: Chloroplast substitution rates correlate with species-richness in the Proteaceae. BMC Evol. Biol. 13, 65. doi: 10.1186/1471-2148-13-65
Finn, R. D., Clements, J., Eddy, S. R. (2011). HMMER web server: Interactive sequence similarity searching. Nucl. Acids Res. 39, 29–37. doi: 10.1093/nar/gkr367
Gan, K. A., Carrasco, P. S., Sewell, J. A., Fuxman, B. J. I. (2018). Identification of single nucleotide non-coding driver mutations in cancer. Front. Genet. 9. doi: 10.3389/fgene.2018.00016
Gumeni, S., Evangelakou, Z., Gorgoulis, V., Trougakos, I. (2017). Proteome stability as a key factor of genome integrity. Int. J. Mol. Sci. 18, 2036. doi: 10.3390/ijms18102036
Hakobyan, S., Loeffler-Wirth, H., Arakelyan, A., Binder, H., Kunz, M. (2021). A transcriptome-wide isoform landscape of melanocytic nevi and primary melanomas identifies gene isoforms associated with malignancy. Int. J. Mol. Sci. 22, 7165. doi: 10.3390/ijms22137165
Hisano, H., Tsujimura, M., Yoshida, H., Terachi, T., Sato, K. (2016). Mitochondrial genome sequences from wild and cultivated barley (Hordeum vulgare). BMC Genomics 17, 824. doi: 10.1186/s12864-016-3159-3
Jakob, S. S., Blattner, F. R. (2006). A chloroplast genealogy of Hordeum (Poaceae): Long-term persisting haplotypes, incomplete lineage sorting, regional extinction, and the consequences for phylogenetic inference. Mol. Bio Evol. 23, 1602–1612. doi: 10.1093/molbev/msl018
Jing, L., Hou, B. W., Niu, Z. T., Liu, W., Xue, Q. Y., Ding, X. Y. (2014). Comparative chloroplast genomes of photosynthetic orchids: insights into evolution of the Orchidaceae and development of molecular markers for phylogenetic applications. PloS One 9, e99016. doi: 10.1371/journal.pone.0099016
Jonathan, B., Blattner, F. R. (2015). Species-level phylogeny and polyploid relationships in Hordeum (Poaceae) inferred by next-generation sequencing and in silico cloning of multiple nuclear loci. Syst. Bio. 64, 792–808. doi: 10.1093/sysbio/syv035
Kent, W., Brumbaugh, H. (2002). BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kumar, S., Nei, M., Dudley, J., Tamura, K. (2008). MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Bri Bioinf 9, 299–306. doi: 10.1093/bib/bbn017
Lei, W., Ni, D., Wang, Y., Shao, J., Liu, C. (2016). Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci. Rep. 6, 21669. doi: 10.1038/srep21669
Li, D. M., Zhu, G. F., Xu, Y. C., Ye, Y. J., Liu, J. M. (2020). Complete chloroplast genomes of three medicinal Alpinia species: genome organization, comparative analyses and phylogenetic relationships in family Zingiberaceae. Plants 9, 286. doi: 10.3390/plants9020286
Liu, L. X., Li, R., Worth, J. R. P., Li, X., Li, P., et al. (2017). The complete chloroplast genome of Chinese bayberry (Morella rubra, myricaceae): implications for understanding the evolution of fagales. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00968
Liu, X., Li, Y., Yang, H., Zhou, B. (2018). Chloroplast genome of the folk medicine and vegetable plant Talinum paniculatum (Jacq.) Gaertn.: gene organization, comparative and phylogenetic analysis. Molecules 23, 857. doi: 10.3390/molecules23040857
Liu, H., Su, Z., Yu, S., Liu, J., Li, B. (2019). Genome comparison reveals mutation hotspots in the chloroplast genome and phylogenetic relationships of Ormosia species. BioMed. Res. Int. 2019, 1–11. doi: 10.1155/2019/7265030
Lu, R. S., Pan, L., Qiu, Y. X. (2016). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 72054. doi: 10.3389/fpls.2016.02054
Ma, J. E., Lang, Q. Q., Qiu, F. F., Li, Z., Li, X. G., Luo, W., et al. (2016). Negative glucocorticoid response-like element from the first intron of the chicken growth hormone gene represses gene expression in the rat pituitary tumor cell line. Int. J. Mol. Sci. 17, 1863. doi: 10.3390/ijms17111863
Meng, J., Li, X., Li, H., Yang, J., Wang, H., He, J. (2018). Comparative analysis of the complete chloroplast genomes of four Aconitum medicinal species. Molecules 23, 1015. doi: 10.3390/molecules23051015
Middleton, C. P., Senerchia, N., Stein, N., Akhunov, E. D., Keller, B., Wicker, T., et al. (2013). Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PloS One 9, e85761. doi: 10.1371/journal.pone.0085761
Namgung, J., Do, H. D. K., Kim, C., Choi, H. J., Kim, J. H. (2021). Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of Allioideae (Amaryllidaceae). Sci. Rep. 11, 1–3. doi: 10.1038/s41598-021-82692-5
Ogihara, Y., Isono, K., Kojima, T., Endo, A., Hanaoka, M., Shiina, T., et al. (2000). Chinese spring wheat (Triticum aestivum L.) chloroplast genome: Complete sequence and contig clones. Plant Mol. Bio Rep. 18, 243–253. doi: 10.1007/BF02823995
Park, I., Song, J. H., Yang, S., Kim, W. J., Moon, B. C. (2019). Cuscuta species identification based on the morphology of reproductive organs and complete chloroplast genome sequences. Int. J. Mol. Sci. 20, 2726. doi: 10.3390/ijms20112726
Peng, J., Zhao, Y. L., Dong, M., Liu, S. Q., Hu, Z. Y., Zhong, X. F., et al. (2021). Exploring evolution characteristic between cultivated tea and its wild relatives using complete chloroplast genomes. BMC Ecol. Evo. 21, 71. doi: 10.1186/s12862-021-01800-1
Raes, J., Van de Peer, Y. (2005). Functional divergence of proteins through frameshift mutations. Trends Genet. 21, 428–431. doi: 10.1016/j.tig.2005.05.013
Ravi, V., Khurana, J. P., Tyagi, A. K., Khurana, P. (2008). An update on chloroplast genomes. Plant Syst. Evol. 271, 101–122. doi: 10.1007/s00606-007-0608-0
Reinert, S., Osthoff, A., Léon, J., Naz, A. (2019). Population genetics revealed a new locus that underwent positive selection in barley. Int. J. Mol. Sci. 20, 202. doi: 10.3390/ijms20010202
Rousseau-Gueutin, M., Bellot, S., Martin, G. E., Boutte, J., Chelaifa, H., Lima, O., et al. (2015). The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): comparative analyses and molecular dating. Mol. Phyl Evol. 93, 5–16. doi: 10.1016/j.ympev.2015.06.013
Safonova, Y., Bankevich, A., Pevzner, P. A. (2014). DipSPAdes: assembler for highly polymorphic diploid genomes. Int. Conf. Res. Comput. Mol. Biol. 6, 528–545. doi: 10.1089/cmb.2014.0153
Sajjad, A., Waqas, M., Khan, A. L., Khan, M. A., Kang, S. M., Imran, Q. M., et al. (2017). The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00304
Shen, X. F., Guo, S., Yin, Y., Zhang, J. J., Yin, X. M., Liang, Z. W., et al. (2018). Complete chloroplast genome sequence and phylogenetic analysis of Aster tataricus. Molecules 23, 2426. doi: 10.3390/molecules23102426
Skuza, L., Gastineau, R., Sielska, A. (2022). The complete chloroplast genome of Secale sylvestre (Poaceae: Triticeae). J. Appl. Genet. 63, 115–117. doi: 10.1007/s13353-021-00656-x
Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Bio Evol. 30, 772. doi: 10.1093/molbev/mst010
Suoi, C. K., Gi, C. M., Seonjoo, P. (2016). The complete chloroplast genome sequences of three Veroniceae species (Plantaginaceae): comparative analysis and highly divergent regions. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00355
Tao, X., Ma, L., Zhang, Z., Liu, W., Liu, Z. (2016). Characterization of the complete chloroplast genome of alfalfa (Medicago sativa) (Leguminosae). Gene Rep. 6, 67–73. doi: 10.1016/j.genrep.2016.12.006
Tyagi, S., Jung, J. A., Kim, J. S., Won, S. Y. (2020). Comparative analysis of the complete chloroplast genome of mainland Aster spathulifolius and other Aster species. Plants 9, 568. doi: 10.3390/plants9050568
Vishwakarma, M. K., Kale, S. M., Manda, S., Talari, N., Yaduru, S., Garg, V., et al. (2017). Genome-wide discovery and deployment of insertions and deletions markers provided greater insights on species, genomes, and sections relationships in the genus Arachis. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.02064
Wang, Y. Z., Jiang, D. C., Guo, K. G., Zhao, L., Meng, F. F., Xiao, J. L., et al. (2023). Comparative analysis of codon usage patterns in chloroplast genomes of ten Epimedium species. BMC Geno Data 24, 3. doi: 10.1186/s12863-023-01104-x
Wei, L., He, J., Jia, X., Qi, Q., Liang, Z. S., Zheng, H., et al. (2014). Analysis of codon usage bias of mitochondrial genome in Bombyx mori and its relation to evolution. BMC Evol. Biol. 14, 1–12. doi: 10.1186/s12862-014-0262-4
Wen, Y., Zou, Z., Li, H., Xiang, Z., He, N. (2016). Analysis of codon usage patterns in Morus notabilis based on genome and transcriptome data. Genome 60, 473–484. doi: 10.1139/gen-2016-0129
Wu, L. W., Nie, L. P., Wang, Q., Xu, Z. C., Wang, Y., He, C. N., et al. (2021). Comparative and phylogenetic analyses of the chloroplast genomes of species of Paeoniaceae. Sci. Rep. 11, 14643. doi: 10.1038/s41598-021-94137-0
Xiao, C. W., Liu, Y., Wei, Q., Ji, Q. A., Li, K., Pan, L. J., et al. (2019). Inhibitory effects of berberine hydrochloride on Trichophyton mentagrophytes and the underlying mechanisms. Molecules 24, 742. doi: 10.3390/molecules24040742
Xiong, Y. L., Xiong, Y., He, J., Yu, Q. Q., Zhao, J. M., Lei, X., et al. (2020b). The complete chloroplast genome of two important annual Clover species, Trifolium alexandrinum and T. resupinatum: genome structure, comparative analyses and phylogenetic relationships with relatives in Leguminosae. Plants 9, 478. doi: 10.3390/plants9040478
Xiong, Y., Xiong, Y. L., Jia, S. G., Ma, X. (2020a). The complete chloroplast genome sequencing and comparative analysis of reed canary grass (Phalaris arundinacea) and Hardinggrass (P. aquatica). Plants 9, 748. doi: 10.3390/plants9060748
Xue, S., Shi, T., Luo, W., Ni, X., Gao, Z. (2019). Comparative analysis of the complete chloroplast genome among Prunus mume, P. Armeniaca, and P. salicina. Horticulture Res. 6, 89. doi: 10.1038/s41438-019-0171-1
Yang, X., Luo, X., Cai, X. (2014). Analysis of codon usage pattern in Taenia saginata based on a transcriptome dataset. Par Vect 7, 1–11. doi: 10.1186/s13071-014-0527-1
Yang, J. P., Zhang, F. W., Ge, Y. J., Yu, W. H., Xue, Q. Q., Wang, M. T., et al. (2022). Effects of geographic isolation on the Bulbophyllum chloroplast genomes. BMC Plant Bio 22, 1–14. doi: 10.1186/s12870-022-03592-y
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, K. S., Yu, J. (2006). KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genom. Prot. Bioinf 4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
Zong, D., Gan, P., Zhou, A., Li, J., He, C. (2019). Comparative analysis of the complete chloroplast genomes of seven Populus species: insights into alternative female parents of Populus tomentosa. PloS One 14, e218455. doi: 10.1371/journal.pone.0218455
Keywords: Hordeum, chloroplast genome, parity rule 2, repeated sequences, hotpot, phylogenic tree
Citation: Yuan S, Nie C, Jia S, Liu T, Zhao J, Peng J, Kong W, Liu W, Gou W, Lei X, Xiong Y, Xiong Y, Yu Q, Ling Y and Ma X (2023) Complete chloroplast genomes of three wild perennial Hordeum species from Central Asia: genome structure, mutation hotspot, phylogenetic relationships, and comparative analysis. Front. Plant Sci. 14:1170004. doi: 10.3389/fpls.2023.1170004
Received: 20 February 2023; Accepted: 05 July 2023;
Published: 24 July 2023.
Edited by:
Christos Bazakos, Max Planck Institute for Plant Breeding Research, GermanyReviewed by:
Peng-Fei Ma, Chinese Academy of Sciences (CAS), ChinaCopyright © 2023 Yuan, Nie, Jia, Liu, Zhao, Peng, Kong, Liu, Gou, Lei, Xiong, Xiong, Yu, Ling and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Ma, bWFyb2FyQDEyNi5jb20=; Yao Ling, bHk5NzI5NzUyQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.