 Wujun Zhang
Wujun Zhang Zhaolei Zhang
Zhaolei Zhang Baocai Liu
Baocai Liu Jingying Chen1*
Jingying Chen1*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 22 June 2023
Sec. Plant Bioinformatics
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1163325
This article is part of the Research TopicStructural Variation of the Chloroplast Genome and Related Bioinformatics ToolsView all 11 articles
Pseudostellaria heterophylla (Miq.) Pax is a well-known medicinal and ecologically important plant. Effectively distinguishing its different genetic resources is essential for its breeding. Plant chloroplast genomes can provide much more information than traditional molecular markers and provide higher-resolution genetic analyses to distinguish closely related planting materials. Here, seventeen P. heterophylla samples from Anhui, Fujian, Guizhou, Hebei, Hunan, Jiangsu, and Shandong provinces were collected, and a genome skimming strategy was employed to obtain their chloroplast genomes. The P. heterophylla chloroplast genomes ranged from 149,356 bp to 149,592 bp in length, and a total of 111 unique genes were annotated, including 77 protein-coding genes, 30 tRNA genes, and four rRNA genes. Codon usage analysis showed that leucine had the highest frequency, while UUU (encoding phenylalanine) and UGC (encoding cysteine) were identified as the most and least frequently used codons, respectively. A total of 75–84 SSRs, 16–21 short tandem repeats, and 27–32 long repeat structures were identified in these chloroplast genomes. Then, four primer pairs were revealed for identifying SSR polymorphisms. Palindromes are the dominant type, accounting for an average of 47.86% of all long repeat sequences. Gene orders were highly collinear, and IR regions were highly conserved. Genome alignment indicated that there were four intergenic regions (psaI-ycf4, ycf3-trnS, ndhC-trnV, and ndhI-ndhG) and three coding genes (ndhJ, ycf1, and rpl20) that were highly variable among different P. heterophylla samples. Moreover, 10 SNP/MNP sites with high polymorphism were selected for further study. Phylogenetic analysis showed that populations of Chinese were clustered into a monophyletic group, in which the non-flowering variety formed a separate subclade with high statistical support. In this study, the comparative analysis of complete chloroplast genomes revealed intraspecific variations in P. heterophylla and further supported the idea that chloroplast genomes could elucidate relatedness among closely related cultivation materials.
Pseudostellaria heterophylla (Miq.) Pax (tai-zi-shen or hai-er-shen) is a well-known traditional medicinal plant of the Caryophyllaceae family. It is commonly used for the treatment of fatigue, spleen asthenia, anorexia, asthenia after severe illness, and cough due to lung dryness either in China (Commission, 2015) or in Korea (Liu et al., 2017). Recent pharmacologic research has indicated that P. heterophylla has anti-diabetes (Liu et al., 2017), immune enhancement (Yang et al., 2020), and anti-oxidant properties (Ng et al., 2004) due to its composition containing numerous active compounds such as cyclic peptides (pseudostellarin), polysaccharides, amino acids, saponins, and sapogenins (Wang et al., 2013). P. heterophylla is mainly distributed in the Fujian, Guizhou, Shandong, Anhui, and Jiangsu provinces of China (Kang et al., 2016), Japan, Korea, and the Russian Far East (Choi and Pak, 2000). P. heterophylla has been cultivated in China for over 100 years with abundant germplasm resources (Xiao et al., 2015), represented by significant variability in leaf length, leaf width, number of main stems, total biomass, and number of above-ground stem nodes. Currently, the breeding of P. heterophylla is progressing slowly since the introduction of varieties is not standardized and the genetic background of the cultivated populations cannot be traced. Moreover, there are few sexually reproduced varieties. However, long-term clonal reproduction is the main means of propagation in various regions, which leads to the erosion of the species genetic variability and restricts the development of utilization and applications (Wu et al., 2016). Therefore, finding a method that can distinguish different germplasm resources in P. heterophylla is urgent.
Previously, the chloroplast genome rbcL and matK regions, the Internal Transcribed Spacers (ITS) of the nuclear ribosomal DNA, sequence-related amplified polymorphism (SRAP), inter simple sequence repeat (ISSR), and expressed sequence tag-simple sequence repeat (ESR-SSR) have been used to characterize the genetic diversity of P. heterophylla germplasm (Yi et al., 2013; Xiao et al., 2014; Xu et al., 2023). Yi et al. (2013) found that the ITS sequences of different P. heterophylla varieties had several specific single nucleotide mutation sites and could be used to identify and distinguish samples from nine different producing areas. Xiao et al. (2014) used ISSR to analyze the diversity of 12 P. heterophylla cultivars. A total of 73 polymorphic bands were identified, accounting for 89.02% of the total amplified bands, which revealed the clustering of these 12 cultivars into three clades.
With the development of high-throughput sequencing technologies and the decrease in sequencing costs, complete chloroplast genomes assembled from shotgun genomic DNA sequencing provide a more convenient and higher -resolution means to study the relationship among plant cultivated varieties (Straub et al., 2012). The chloroplast genome length is usually between 115 kb and 165 kb, and the length differences are mostly due to inverted repeat (IR) expansion/contraction (Zhu et al., 2016) or gene losses (Lei et al., 2016). As the second-largest plant genome, the chloroplast genome contains rich genetic information for species identification, phylogenetic analysis, and population genetic studies (Palmer, 1991). Dong et al. (2014) employed a chloroplast genomic strategy to design taxon-specific DNA mini-barcodes and applied them to species identification in the ginsengs. Liu et al. (2022) obtained chloroplast genome sequences of 24 plant samples in the genus Atractylodes and provided a new understanding of their phylogenetic relationship. Utilizing massively parallel sequencing technology for chloroplast genome sequencing in plants can facilitate a better understanding and discrimination of low-level systematic relationships among different taxa in plant classification (Parks et al., 2009). The first P. heterophylla chloroplast genome sequence distributed in Korea was reported and indicated that the P. heterophylla chloroplast genome has a double-stranded, circular, typically four-segment structure (Kim et al., 2019). However, there is still a lack of population genetic analyses in P. heterophylla using chloroplast genomes.
Here, we collected 17 P. heterophylla plant samples with remarkable phenotypic characteristics and obtained their chloroplast genome sequences using next-generation sequencing. This study aimed to (1) elucidate the conservation and diversity of P. heterophylla chloroplast genomes through comparative genomic approaches; (2) identify the most variable chloroplast genome regions to utilize them as markers for further germplasm conservation and genetic improvement; and (3) determine the relationships between genotypes using the chloroplast genome sequence data.
In this study, 17 samples of P. heterophylla were collected from seven provinces and represented dominant cultivars in China (Table 1). Zheshen No. 1 has an erect growth habit, is unbranched and short, and its leaves are ovate. Its flowers are white, and its roots are spindle -shaped. It is moderately susceptible to leaf spot disease. Zheshen No. 2 has four to six upright branches (more than the Zheshen No. 1), ovate-lanceolate thick leaves, carrot-shaped root tubers, and moderate resistance to leaf spot disease. It does not flower. Zheshen No. 3 is a tetraploid P. heterophylla genotype induced by Zheshen No. 1. Zheshen No. 3 has oval, large, thick, dark green leaves, a low seed setting rate, and high-yielding roots. The Minxuan No. 6 and Minxuan No. 7 biotypes have long, oval, and thick leaves, flowering, large root tubers, and are more resistant to viral diseases. The Zherong Datiao was introduced from Guizhou and has characteristically large root tubers. Shitai No. 1 is a variety obtained using a mixed breeding approach. Its plants are upright, tall, and flowering, with round to long oval leaves and long spindle roots. The Guizhou cultivar plants are upright and tall, with oblong-ovate leaves and long spindle roots. The Jurong cultivar is a native cultivated variety with oval and thick leaves and high-yielding roots. The Hunan cultivar plants are upright, tall, and flowering, with long, ovate leaves and fusiform root tubers. The Xuancheng cultivar plants are upright and multi-branched and have tall plants with oblong-ovate leaves and large root tubers. The Shandong cultivar has been domesticated from a wild population. Its plants are tall with branches, and its leaves are oval-lanceolate and thin. The root tuber of the Shandong cultivar is long, spindle-shaped, and thin, and yields for this cultivar are high. The Hebei cultivar was introduced from Shandong and has morphological characteristics like the Shandong cultivar. These samples were identified by Prof. Jingying Chen from the Fujian Academy of Agricultural Sciences.
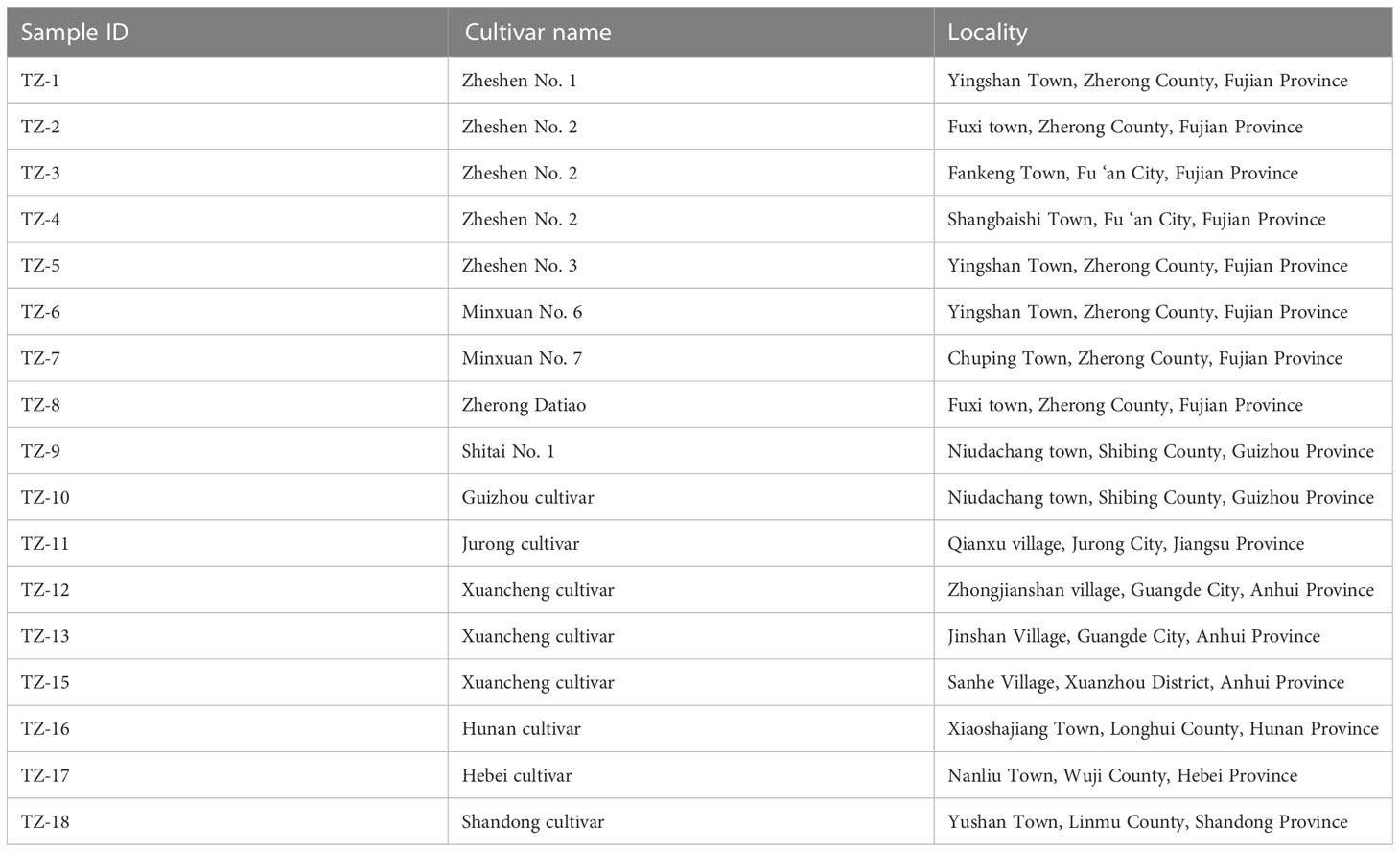
Table 1 Collection information of 17 P. heterophylla samples.
The total genomic DNA from P. heterophylla leaf tissues was extracted using a modified CTAB method. DNA quantity and quality were determined using Qubit4.0 (Thermo Fisher Scientific Inc., USA). Subsequently, the genomic DNA was purified and fragmented to construct sequencing libraries (350 bp) using the TruSeq DNA PCR-Free High Throughput Library Prep Kit (Illumina, San Diego, CA). High-throughput sequencing (2 × 150 bp) was performed with the NovaSeq 6000 sequencer (Illumina, San Diego, CA).
The PCR-free sequencing data were used to assemble the chloroplast genome sequences of P. heterophylla using the GetOrganelle pipeline (Jin et al., 2020). Gene annotation of the chloroplast genome sequences was performed using CpGAVAS2 (Shi et al., 2019) and then manually evaluated and corrected. Graphical maps of P. heterophylla chloroplast genome sequences were drawn using OrganellarGenomeDRAW (OGDRAW) (Greiner et al., 2019).
The REPuter (Kurtz et al., 2001) software was used to recognize four types of sequence repeats, including forward (F), reverse (R), complementary (C), and palindromic (P). The minimum repeat size of oligonucleotide repeats was set at 30 bp, and the Hamming distance was set at 3. Tandem repeats were analyzed using the Tandem Repeats Finder (TRF) software (Benson, 1999) with default parameters. Simple sequence repeats (SSRs) were detected using the MIcroSAtellite identification tool (MISA) (Beier et al., 2017). The minimum repeat thresholds of mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs were set as 10, 6, 5, 5, 5, and 5, respectively. Primers for SSRs were designed with Primer 3.0 software (Untergasser et al., 2012).
The mVISTA program with the Shuffle-Lagan model (Frazer et al., 2004) was employed to compare the chloroplast genome sequences of P. heterophylla. IRscope (Amiryousefi et al., 2018) was used to visualize the contraction and extension of IR boundaries between the four parts of the genome (LSC/IRb/SSC/IRa). Gene rearrangements were observed using the co-linear blocks obtained by the Mauve alignment algorithm (Darling et al., 2004).
ParaAT2.0 software (Zhang et al., 2012) was used to align protein sequences derived from specific protein-encoded DNA sequences extracted from 17 P. heterophylla chloroplast genomes. The nucleic acid alignment corresponding to the codon was translated back according to the protein alignment result. KaKs_Calculator 3.0 software (Zhang, 2022) was then used to calculate synonymous (Ks), nonsynonymous (Ka), and Ka/Ks ratios after homologous sequence alignment.
The concatenated protein-coding gene sequences of the 17 Pseudostellaria chloroplast genomes were used for phylogenetic analysis, with Cerastium arvense, Gymnocarpos przewalskii, and Dianthus caryophyllus as outgroup species. A maximum likelihood (ML) phylogenetic tree of 1,000 bootstrap replications was constructed using RAxML v8.2.12 (Stamatakis, 2014).
The P. heterophylla chloroplast genome sequence length ranged from 149,356 bp to 149,592 bp, with a variation of 236 bp among the different samples. Each chloroplast genome had the typical quadripartite structure, with a large single copy (LSC) region (80,994–81,144 bp), a small single copy (SSC) region (16,860 to 17,154 bp), and a pair of IR regions (IRa and IRb) (25,650 to 25,732 bp). The chloroplast genome GC content in all samples ranged from 36.50% to 36.52%, and the GC content in the IR region (approximately 42%) was significantly higher compared to the LSC region and SSC region (approximately 34% and 29%). A total of 111 unique genes were annotated in the P. heterophylla chloroplast genomes sequenced, including 77 protein-coding genes, 30 tRNA genes, and four rRNA genes (rrn23S, rrn16S, rrn5S, and rrn4.5S). Among these genes, 46 were related to photosynthesis, and 58 were involved in chloroplast transcription and translation activities. Fifteen genes were in the IR region with two copies, including four protein-coding genes, seven tRNA genes, and four rRNA genes. Seventeen genes contained introns, of which 14 genes (eight protein-coding genes and six tRNA genes) contained one intron, and three genes (rps12, ycf3, and clpP) contained two introns. Small exons were also identified in the petB, petD, and rpl16 genes, with lengths of 6 bp, 8 bp, and 9 bp, respectively. In addition, rps12 was identified as a trans-splicing gene. Further detailed chloroplast genome information is presented in Tables 2, S1 and Figure 1.
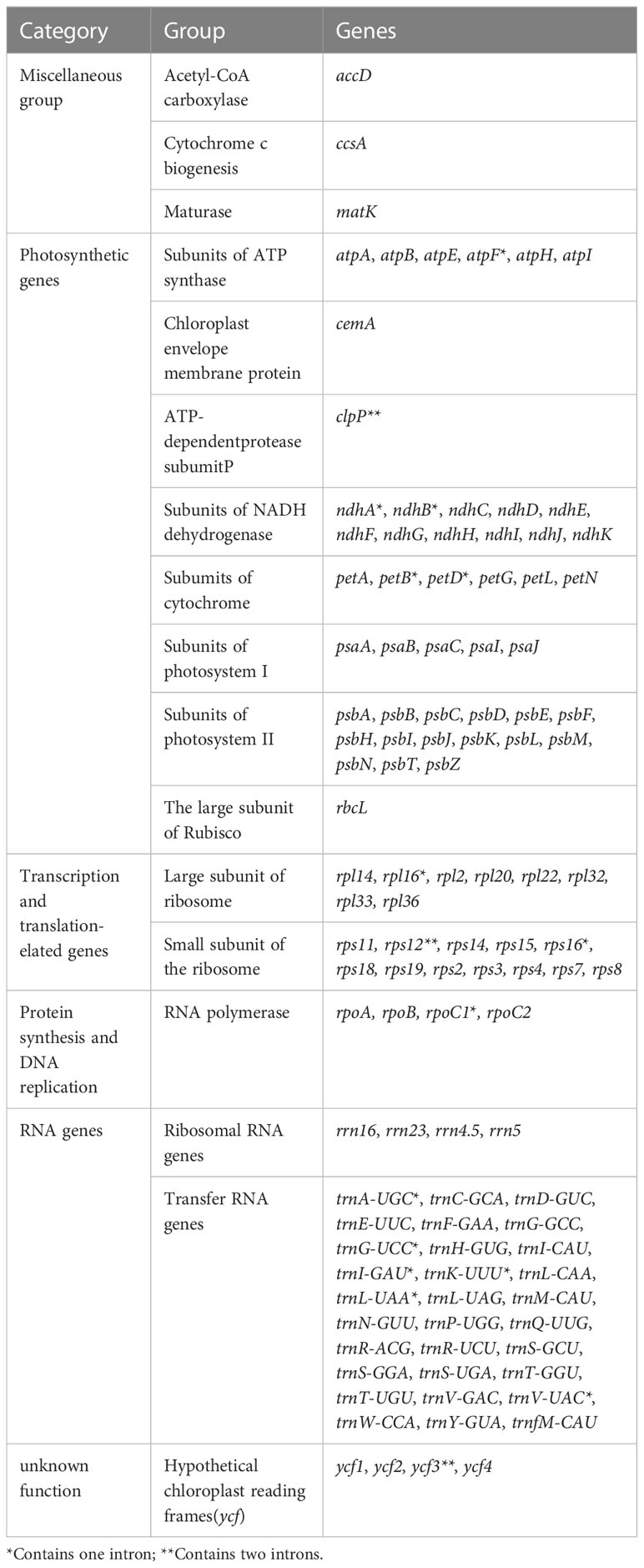
Table 2 Genes in the chloroplast genome of P. heterophylla.
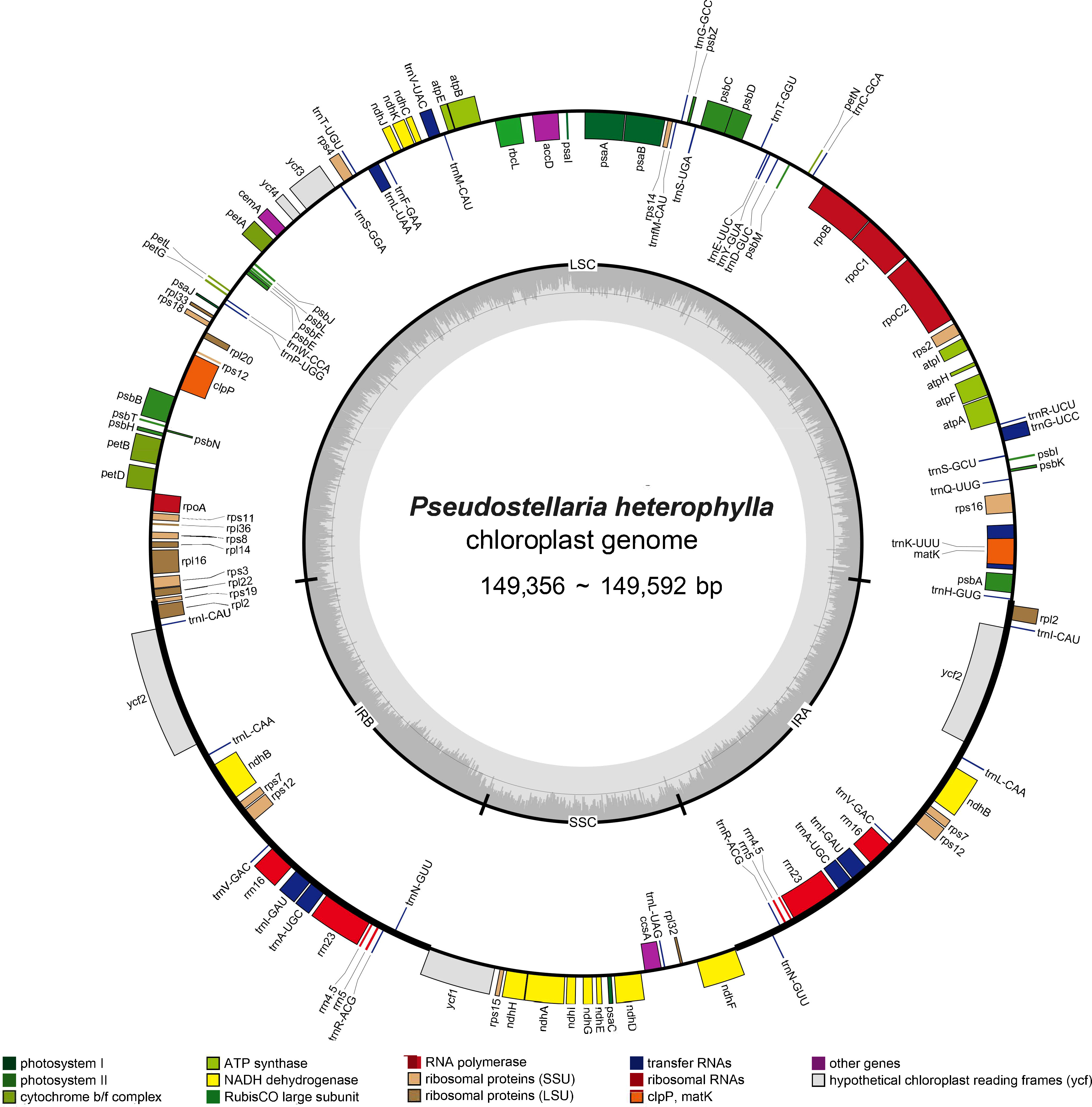
Figure 1 Circular chloroplast genome map of P. heterophylla. Genes drawn outside the circle are transcribed clockwise, and those inside are counterclockwise. Genes belonging to different functional groups are color-coded. The dark gray area in the inner circle denotes GC content. LSC, large single copy; SSC, small single copy; and IR, inverted repeat.
The amino acid frequencies, the number of codons, and the relative synonymous codon usage (RSCU) in P. heterophylla chloroplast genomes are shown in Table S2. The average RSCU value was 63.97, and the number of codons ranged from 22,012 (TZ-3) to 22,017 (TZ-5). Among the codons, leucine was the amino acid with the most abundant codons. UUU (encoding phenylalanine) and UGC (encoding cysteine) were the most and least used codons, respectively. Almost all amino acids had more than one synonymous codon, except for methionine and tryptophan. Four start codon types were identified in the 77 protein-coding genes. Among them, 73 genes possessed ATG as their start codon, while two genes (ndhD and psbL) had ACG, one gene (rps19) had GTG, and one gene (ycf1) had TTG as their start codon. All the samples had the same three stop codon types (TAA, TAG, and TGA). The most used stop codon was TAA (60.98%), followed by TGA (21.95%) and TAG (17.07%).
For the SSR analysis, 75–84 SSR loci were detected in the P. heterophylla chloroplast genomes (Figure 2), among which polyadenine (poly-A) (54.78%, 41–47) and polythymine (poly-T) (38.75%, 29–32) represented the most abundant simple sequence repeats. SSRs and their 500 bp upstream and downstream sequences were extracted, and 69 primer pairs were designed using Primer 3.0 software. After electronic amplification evaluation allowing for two mismatches, four pairs of SSR primers targeting highly polymorphic SSR regions were obtained (Table S3). Sixteen to 21 short tandem repeats were found in the P. heterophylla chloroplast genomes (Table S4), ranging in length from 11 to 32 bp, with most located in the intergenic space (IGS) regions. Twenty-seven to 32 long repeat structures were identified in the P. heterophylla chloroplast genomes, including forward, palindromic, reverse, and complement repeats (Table S5). Palindromic was the most common repeat sequence type, accounting for an average of 47.86% of all repeat sequences, followed by forward (40.12%), reverse (11.21%), and complement (0.81%).
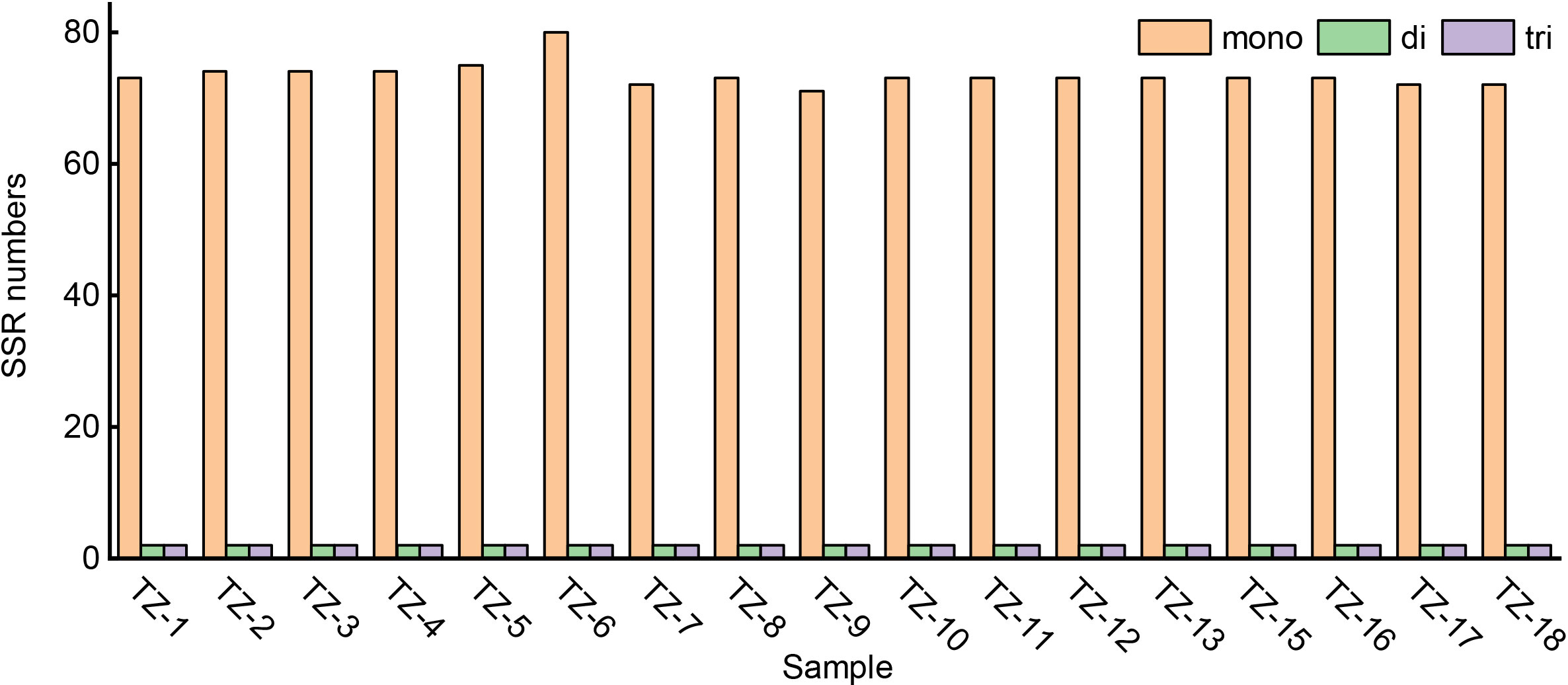
Figure 2 Simple sequence repeats (SSRs) in the chloroplast genome of P. heterophylla.
The P. heterophylla chloroplast genome exhibits four boundaries between the IRs and LSC/SSC regions: LSC-IRb, IRb-SSC, SSC-IRa, and IRa-LSC (Figure S1). The LSC-IRb, IRb-SSC, SSC-IRa, and IRa-LSC boundaries in all samples were located at rps19, ycf1, ndhF, and rpl2-trnH, respectively. The P. heterophylla chloroplast genomes from the Chinese populations were highly conserved. The nucleotide lengths of rps19 and ycf1 located in the IRb region were 195 bp and 105 bp, of ndhF located in the IRa region was 56 bp, and of trnH from the IRa-LSC boundary was 29 bp.
According to the comparative analysis of the whole chloroplast genome of P. heterophylla using the LAGAN program, several regions were variable and were able to distinguish different populations (Figure S2). In terms of genes, the most variable coding genes were ndhJ, ycf1, and rpl20, and the most variable intergenic regions were psaI-ycf4, ycf3-trnS, ndhC-trnV, and ndhI-ndhG (Figure 3). Among these genes and intergenic regions, ycf1 and ndhI-ndhG contained a higher number of SNP and MNP polymorphic loci. Particularly, 10 highly polymorphic SNP/MNP loci were identified, which could be used as candidate SNP/MNP markers to distinguish different populations (Table 3). Then, the Mauve algorithm was used to identify the local collinear blocks (LCBs) of the P. heterophylla chloroplast genomes, with NC_044183 selected as the reference genome (Figure S3). Among all the chloroplast genomes of the samples, the collinear blocks, including the LSC, SSC, and IR regions, showed relatively high levels of conservation and no gene rearrangements. Thirty-two protein-coding genes with polymorphic sites were used to analyze the synonymous (Ks) and non-synonymous (Ka) substitution rates (Table S6). The average Ka value of the 15 genes was higher than 0.001 (Figure S4), with rps15, rpoC2, and rpl20 exhibiting the highest Ka values. Meanwhile, the average Ks value of 17 genes, such as rps19, rps18, and rpl14, was higher than 0.001. The Ka/Ks ratio of all these 32 protein-coding genes ranged from 0.001 to 49.884, with an average value of 19.244. The Ka/Ks ratio of 15 genes was higher than 1, and the gene with the highest Ka/Ks ratio was rps15 (49.88).
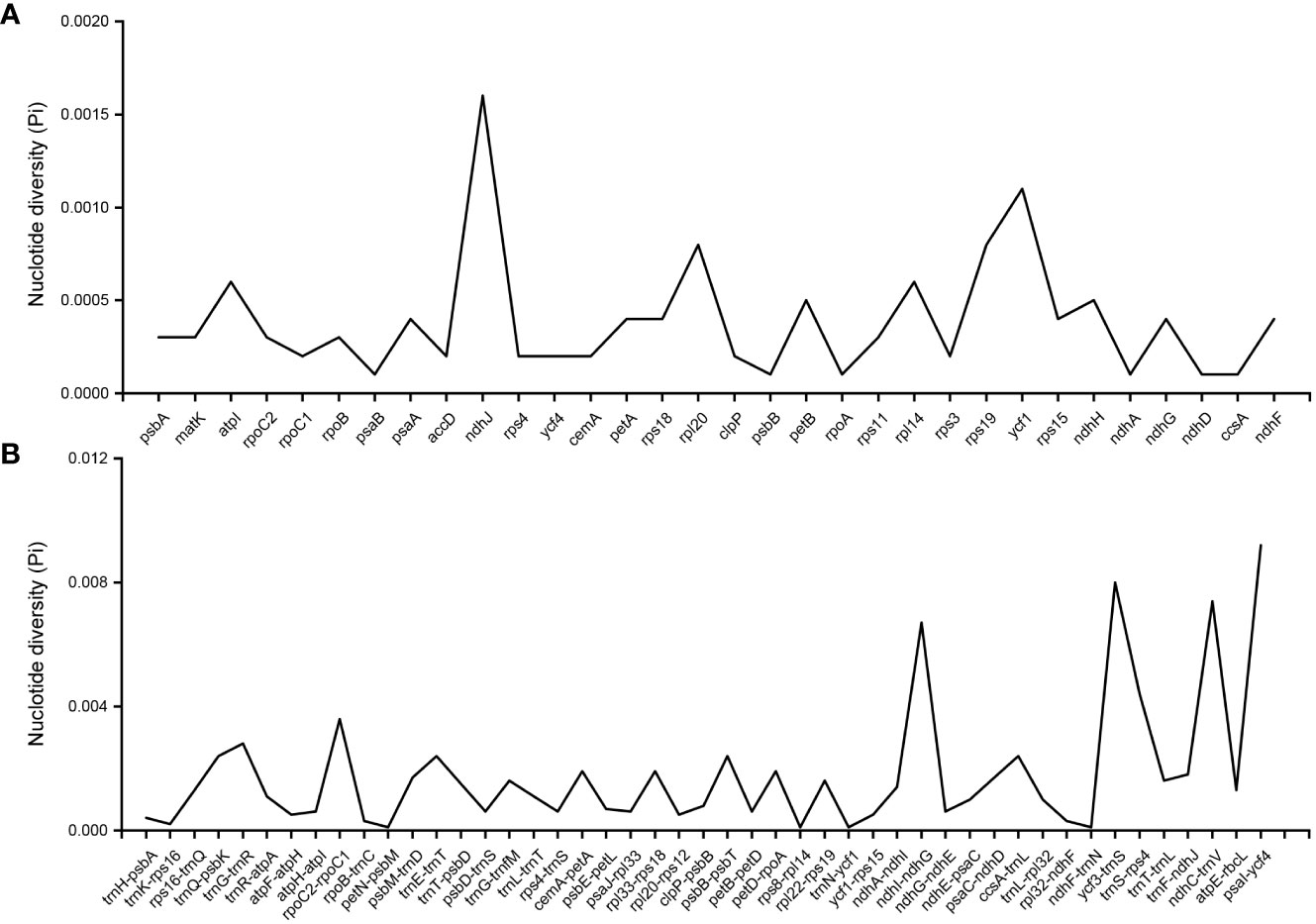
Figure 3 The nucleotide diversity of genes and intergenic regions in the P. heterophylla chloroplast genomes. (A) Coding region; (B) Noncoding region.
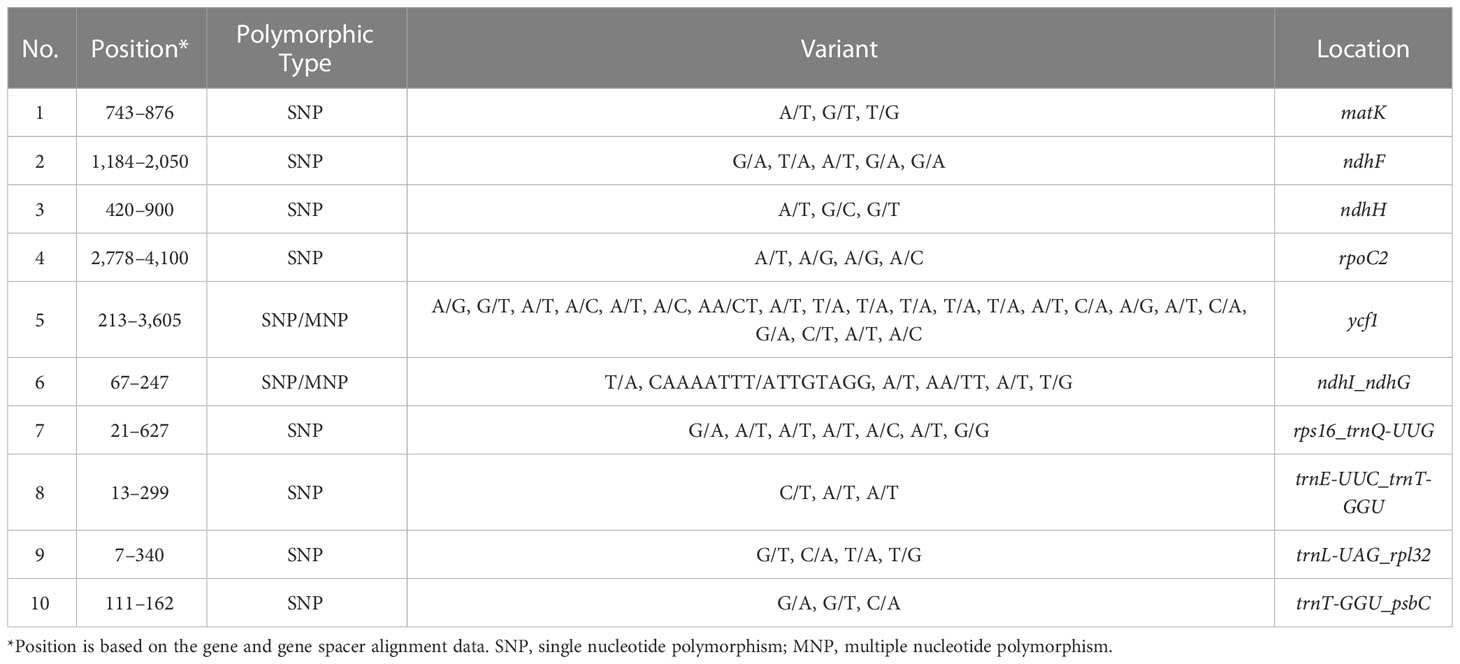
Table 3 Candidate polymorphic DNA markers from the chloroplast genome of P. heterophylla.
To explore the relationships among P. heterophylla cultivars, a maximum likelihood (ML) phylogenetic tree was constructed, and C. arvense, G. przewalskii, and D. caryophyllus were selected as out group species (Figure 4). The samples belonging to the Korean P. heterophylla population formed a separate cluster from the samples from the Chinese population. In terms of the Chinese P. heterophylla population samples, TZ-1, TZ-8, TZ-10–TZ-13, TZ-15, and TZ-16 were clustered into a big branch, which may be due to the mutual introduction of P. heterophylla from Fujian, Jiangsu, Anhui, Hunan, Guizhou, and other locations, resulting in a high similarity of the germplasm resources. TZ-17, TZ-18, and TZ-7 were clustered into a smaller branch that is related to Shandong P. heterophylla sources. Three samples of Zheshen No. 2 (TZ-2, TZ-3, and TZ-4) from different places were clustered into a separate branch. TZ-5, TZ-6, and TZ-9 were in separate branches that were located towards the edges of the phylogenetic tree. TZ-6 was selected for its virus resistance. The above results indicated that chloroplast genome sequence analyses could provide useful information for assessing the genetic background of a species. They could be used to assist breeding and provide a molecular –biological basis for cultivar identification.
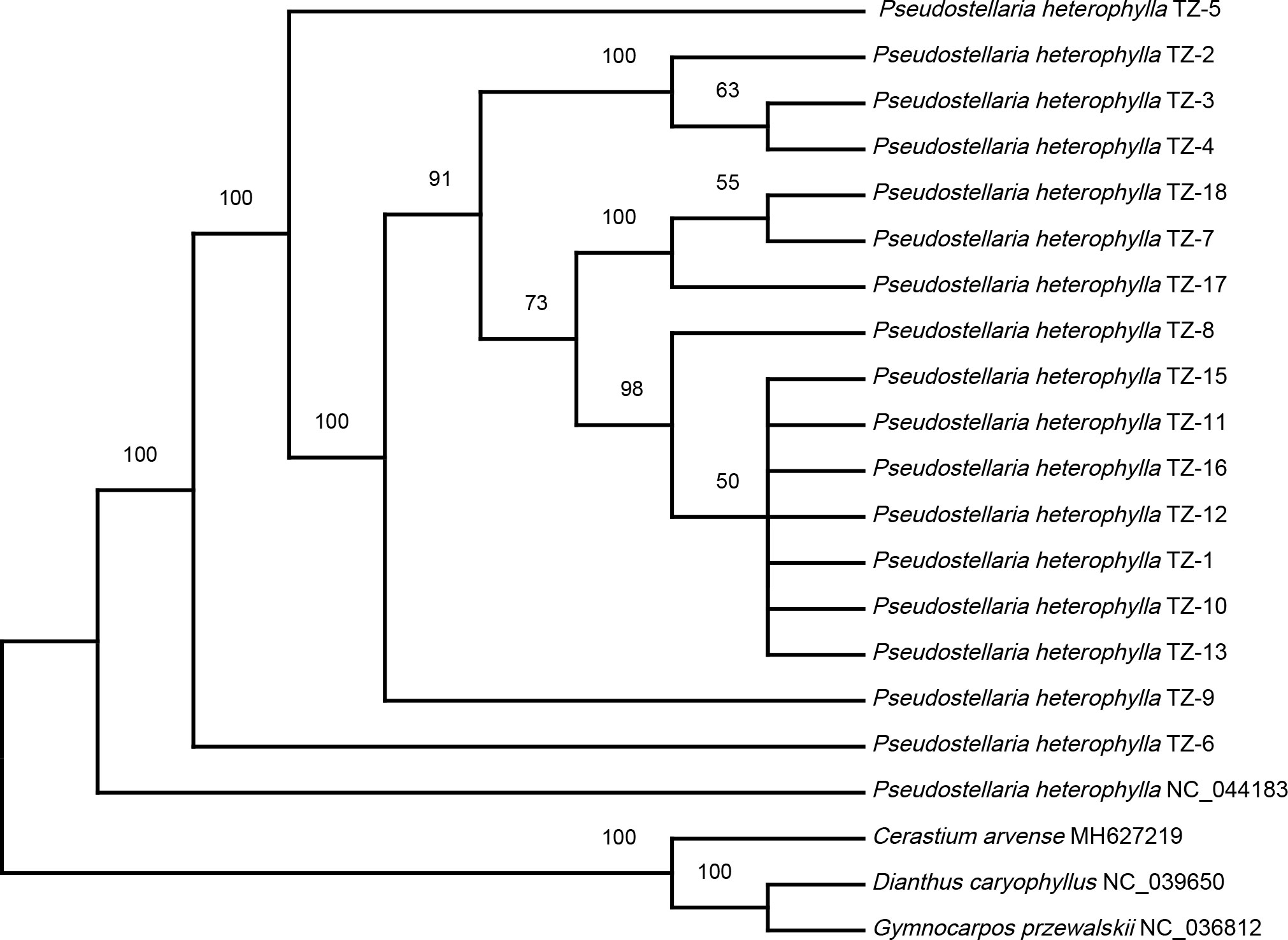
Figure 4 Maximum likelihood (ML) phylogenetic tree of P. heterophylla inferred from chloroplast genome sequences. Bootstrap values are shown at each node.
Distinguishing germplasm resources is essential for plant breeding. Traditional breeding efforts in P. heterophylla have usually used plant morphological characteristics, such as leaf size, shape, and thickness; rhizome length, diameter, and texture; plant height; and the number of flowers, to distinguish varieties. However, phenotypes are easily affected by cultivation methods and environmental factors and require long-term observation (Chen et al., 2010). In addition, the irregular introduction of P. heterophylla has also impacted the distribution of P. heterophylla genetic resources, which affected the uniform collection, classification, and identification of germplasm resources (Xu et al., 2023). P. heterophylla cultivation has a history of more than 180 years, and at its earliest stage of cultivation, P. heterophylla germplasm resources were mainly derived from wild populations. Since the 1960s, Fujian has successively introduced resources from Jiangsu, Anhui, Zhejiang, Shandong, and other places that formed novel germplasm resources, such as Zheshen No. 1 and Zheshen No. 2. Guizhou Province has no wild P. heterophylla populations, and P. heterophylla was introduced from Fujian for cultivation in the 1990s. Wild resources of P. heterophylla are highly abundant in Jiangsu Province, where there are rarely germplasm introductions from other locations. Due to the use of seeds to raise seedlings, P. heterophylla in Jiansu Province is less affected by viral diseases and achieves a higher yield. Interestingly, phylogenetic analysis in this study has provided clues to trace the breeding history of these resources and further verified that the chloroplast genome can provide useful information for analyzing the genetic background of this species. The Korean population was located on the outermost part of the phylogenetic tree as an outgroup. Previous studies reported that P. heterophylla is distributed throughout the mountains of Korea, and the morphological characteristics of the Korean population are different from those distributed in China (Choi and Pak, 2000). Therefore, it is possible to obtain new genetic resources or special breeding materials by hybridizing the Korean population with Chinese populations based on the theory of distant hybridization.
Molecular markers derived from the chloroplast genome, such as rbcL, matK, and psbA-trnH, are effective for species identification and phylogenetic resolution (Shi et al., 2011; Liu et al., 2014), and several DNA barcode libraries have been established (Liu et al., 2017). However, species or biotype identification with molecular markers still faces many challenges, especially for closely related species and different populations within species. Previous studies demonstrated that the identification efficiency of DNA barcode markers in specific regions for closely related species was only about 80% (Chen et al., 2014). Several highly informative DNA barcode markers for specific taxa have been developed using comparative analyses of c hloroplast genomes (Zhou et al., 2022). After a comparative analysis of the Rheum palmatum, R. tanguticum, and R. officinale chloroplast genomes, five hypervariable regions (rps16-trnQ, psaA-ycf3, psbE-petL, ndhF-rpl32, and trnT-trnL) were identified and used as specific DNA barcodes for the identification of 42 samples among R. tanguticum, R. officinale, and R. palmatum (Li et al., 2022). The trnl-GAU intron region was detected to be highly variable and will be useful for future evolutionary studies, although the data from four widely distributed varieties were highly conserved (Wang et al., 2018). The chloroplast genome comparison of Gentiana species revealed that the six most InDel-variable loci could be selected as regions for DNA barcode genotyping, confirming that chloroplast genomes could improve the discriminatory capacity for species identification (Zhou et al., 2018). Seven regions (rpl32-ccsA, rpl20-clpP, trnC-rpoB, ycf1b, accD-ycf4, ycf1a, and psbK-accD) were identified from the Pterocarpus chloroplast genome by quantifying nucleotide diversity and had remarkably higher variability compared to the plant universal barcodes (rbcL, matK, and trnH-psbA) (Jiao et al., 2019). The comparison of the rose chloroplast genome revealed that 15 cpSSRs and 150 flanking single nucleotide variations (SNVs) exhibited higher divergence and stronger power for the genotyping of rose varieties (Li et al., 2020). Moreover, the chloroplast genome can also be used as a super-barcode for phylogenetic and closely related taxon identification studies (Chen et al., 2018).
Using high-throughput sequencing approaches, we obtained the complete chloroplast genome sequences of seventeen P. heterophylla varieties. The gene contents and gene orders of the chloroplast genomes were highly conserved. Among these cultivars, 75–84 SSRs, 16–21 short tandem repeats, and 27–32 long repeat structures were detected. Four primer pairs were designed to target highly polymorphic SSR loci. Gene orders were collinear, and IR regions were conserved. Four intergenic regions and three coding genes were found to be highly variable, and ten SNP/MNP sites with polymorphisms were identified and selected for further study. Phylogenetic analysis showed that Chinese populations were clustered into a monophyletic group, in which the non-flowering varieties formed a separate subclade. This study verified that chloroplast genomes could elucidate the relationship among closely related cultivated materials and provide useful information for developing new, highly polymorphic, and informative molecular makers.
The original contributions presented in the study are publicly available. This data can be found here: NCBI, PRJNA932041. The GenBank numbers provided are: OQ405025.1~OQ405039.1, OK643505.1, and OK643506.1.
WZ and JC conceived and designed the experiments. WZ and BL performed the experiments. WZ, ZZ, YZ, and YH analyzed and interpreted the data. WZ and ZZ wrote the manuscript. JC revised and approved the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was supported by the Natural Science Foundation of Fujian Province, China (Grant No. 2022J01485), the High-Quality Agricultural Development “5511 “ Collaborative Innovation Project Special Topic of Fujian Provincial People’s Government & Chinese Academy of Agricultural Sciences (Grant No. XTCXGC2021003), the Scientific and Technological Innovation Team of Fujian Academy of Agricultural Sciences (Grant No. CXTD2021014-2), the Construction of “ The Belt and Road “ National Traditional Herbal Medicine Physical Database and Picture Information Database (Grant No. 2018FY100702), the Public Welfare Scientific Research of Fujian Province (Grant No. 2021R1034006), and the Fujian Medicinal Plant Germplasm Resource Nursery (Grant No. ZYBHDWZX202203).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1163325/full#supplementary-material
Supplementary Table 1 | Genome features of P. heterophylla chloroplast genomes.
Supplementary Table 2 | Codon usage of P. heterophylla chloroplast genomes.
Supplementary Table 3 | Primer pairs for SSRs.
Supplementary Table 4 | Tandem repeats in P. heterophylla chloroplast genomes.
Supplementary Table 5 | Repeat structures in P. heterophylla chloroplast genomes.
Supplementary Table 6 | Ka/Ks analysis of genes with variable sites.
Supplementary Figure 1 | The comparison of the P. heterophylla chloroplast genome junction boundaries. JLB, junction of LSC and IRb; JLA, junction of LSC and IRb.
Supplementary Figure 2 | The comparative analysis with LAGAN program of the whole-chloroplast genome of P. heterophylla. The x-axis represents the coordinate in the chloroplast genome.
Supplementary Figure 3 | A comparison of the whole plastid genomes of P. heterophylla using the Mauve algorithm. The red LCBs indicate syntenic regions, while the histograms within each block represent the degree of sequence similarity. rRNA, protein-coding, and tRNA gene annotations are denoted by red, white, and green boxes, respectively.
Supplementary Figure 4 | Ka/Ks analysis of P. heterophylla chloroplast gnomes. (A) Ka, rate of nonsynonymous substitution; (B) Ks, rate of synonymous substitution; (C) Ka/Ks, rate of non-synonymous vs. synonymous substitutions.
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Chen, S., Pang, X., Song, J., Shi, L., Yao, H., Han, J., et al. (2014). A renaissance in herbal medicine identification: from morphology to DNA. Biotechnol. Adv. 32, 1237–1244. doi: 10.1016/j.biotechadv.2014.07.004
Chen, S., Yao, H., Han, J., Liu, C., Song, J., Shi, L., et al. (2010). Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PloS One 5, 8613. doi: 10.1371/journal.pone.0008613
Chen, X., Zhou, J., Cui, Y., Wang, Y., Duan, B., Yao, H. (2018). Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 9. doi: 10.3389/fphar.2018.00695
Choi, K., Pak, J. H. (2000). A natural hybrid between Pseudostellaria heterophylla and P. palibiniana (Caryophyllaceae). Acta Phytotaxon. Geobot. 50, 161–171. doi: 10.18942/bunruichiri.KJ00001077422
Commission, C. P. (2015). People’s republic of China pharmacopoeia. 2015 Edition (Beijing, China: China Medical Science and Technology Press).
Darling, A. C., Mau, B., Blattner, F. R., Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dong, W., Liu, H., Xu, C., Zuo, Y., Chen, Z., Zhou, S. (2014). A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 15, 138. doi: 10.1186/s12863-014-0138-z
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, 59–64. doi: 10.1093/nar/gkz238
Jiao, L., Lu, Y., He, T., Li, J., Yin, Y. (2019). A strategy for developing high-resolution DNA barcodes for species discrimination of wood specimens using the complete chloroplast genome of three Pterocarpus species. Planta 250, 95–104. doi: 10.1007/s00425-019-03150-1
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Kang, C. Z., Zhou, T., Jiang, W. K., Guo, L. P., Zhang, X. B., Xiao, C. H., et al. (2016). Research on quality regionalization of cultivated Pseudostellaria heterophylla based on climate factors. Zhongguo Zhong Yao Za Zhi 41, 2386–2390. doi: 10.4268/cjcmm20161303
Kim, Y., Xi, H., Park, J. (2019). The complete chloroplast genome of prince ginseng, Pseudostellaria heterophylla (Miq.) pax (Caryophyllaceae. Mitochondrial DNA Part B 4, 2251–2253. doi: 10.1080/23802359.2019.1623127
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lei, W., Ni, D., Wang, Y., Shao, J., Wang, X., Yang, D., et al. (2016). Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci. Rep. 6, 21669–21669. doi: 10.1038/srep21669
Li, R., Wu, L., Xin, T., Hai, L., Lin, Y. L., Hui, Y., et al. (2022). Analysis of chloroplast genomes and development of specific DNA barcodes for identifying the original species of rhei radix et rhizoma. Acta Pharm. Sin. 57, 1495–1505. doi: 10.1038/s41401-021-00781-7
Li, C., Zheng, Y., Huang, P. (2020). Molecular markers from the chloroplast genome of rose provide a complementary tool for variety discrimination and profiling. Sci. Rep. 10, 12188. doi: 10.1038/s41598-020-68092-1
Liu, J., Shi, L., Han, J., Li, G., Lu, H., Hou, J., et al. (2014). Identification of species in the angiosperm family apiaceae using DNA barcodes. Mol. Ecol. Resour. 14, 1231–1238. doi: 10.1111/1755-0998.12262
Liu, J., Shi, L., Song, J., Sun, W., Han, J., Liu, X., et al. (2017). BOKP: a DNA barcode reference library for monitoring herbal drugs in the Korean pharmacopeia. Front. Pharmacol. 8. doi: 10.3389/fphar.2017.00931
Liu, J., Shi, M., Zhang, Z., Xie, H., Kong, W., Wang, Q., et al (2022). Phylogenomic analyses based on the plastid genome and concatenated nrDNA sequence data reveal cytonuclear discordance in genus Atractylodes (Asteraceae: Carduoideae). Front. Plant Sci. 13. doi: 0.3389/fpls.2022.1045423
Ng, T., Liu, F., Wang, H. (2004). The antioxidant effects of aqueous and organic extracts of Panax quinquefolium, Panax notoginseng, Codonopsis pilosula, Pseudostellaria heterophylla and Glehnia littoralis. J. Ethnopharmacol. 93, 285–288. doi: 10.1016/j.jep.2004.03.040
Palmer, J. D. (1991). “Plastid chromosomes: structure and evolution,” in The molecular biology of plastids. Eds. Bogorad, L., Vasil, I. K. (San Diego: Academic Press), 5–53. doi: 10.1016/B978-0-12-715007-9.50009-8
Parks, M., Cronn, R., Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7, 84. doi: 10.1186/1741-7007-7-84
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, 65–73. doi: 10.1093/nar/gkz345
Shi, L. C., Zhang, J., Han, J. P., Song, J. Y., Yao, H., Zhu, Y. J., et al. (2011). Testing the potential of proposed DNA barcodes for species identification of zingiberaceae. J. Syst. Evol. 49, 261–266. doi: 10.1111/j.1759-6831.2011.00133.x
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Straub, S. C. K., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., Liston, A. (2012). Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am. J. Bot. 99, 349—364. doi: 10.3732/ajb.1100335
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3–new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115. doi: 10.1093/nar/gks596
Wang, J., Li, C., Yan, C., Zhao, X., Shan, S. (2018). A comparative analysis of the complete chloroplast genome sequences of four peanut botanical varieties. PeerJ 6, e5349. doi: 10.7717/peerj.5349
Wang, Z., Liao, S. G., He, Y., Li, J., Zhong, R. F., He, X., et al. (2013). Protective effects of fractions from Pseudostellaria heterophylla against cobalt chloride-induced hypoxic injury in H9c2 cell. J. Ethnopharmacol. 147, 540–545. doi: 10.1016/j.jep.2013.03.053
Wu, L., Chen, J., Wu, H., Qin, X., Wang, J., Wu, Y., et al. (2016). Insights into the regulation of rhizosphere bacterial communities by application of bio-organic fertilizer in Pseudostellaria heterophylla monoculture regime. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.01788
Xiao, C. H., Zhou, T., Jiang, W. K., Ai, Q., Yang, C. G., Xiong, H. X., et al. (2014). Genetic diversity and quality analysis of cultivated. Pesudostellaria heterophylla. 45, 1319–1325. doi: 10.7501/j.issn.0253-2670.2014.09.023
Xiao, C., Zhou, T., Jiang, W., Zhao, D., Kang, C., Liao, M. (2015). Analysis on genetic diversity of phenotypic traits in cultivated Pseudostellaria heterophylla. J. Chin. Med. Mater. 38, 1600–1606. doi: 10.13863/j.issn.1001-4454.2015.08.009
Xu, L., Li, P., Su, J., Wang, D., Kuang, Y., Ye, Z., et al. (2023). EST-SSR development and genetic diversity in the medicinal plant Pseudostellaria heterophylla (Miq.) pax. J. Appl. Res. Med. Aromat. Plants 33, 100450. doi: 10.1016/j.jarmap.2022.100450
Yang, Q., Cai, X., Huang, M., Wang, S. (2020). A specific peptide with immunomodulatory activity from Pseudostellaria heterophylla and the action mechanism. J. Funct. Foods 68, 103887. doi: 10.1016/j.jff.2020.103887
Yi, J., Liao, F. P., Zheng, W. W. (2013). Identification of Pseudostellaria heterophylla from different idioplasms by analysis of rDNA ITS sequences. Chin. Tradit. Herb. Drugs 44, 1318–1322. doi: 10.7501/j.issn.0253-2670.2013.10.022
Zhang, Z. (2022). KaKs_Calculator 3.0: calculating selective pressure on coding and non-coding sequences. Genomics Proteomics Bioinf. 20, 536–540. doi: 10.1016/j.gpb.2021.12.002
Zhang, Z., Xiao, J., Wu, J., Zhang, H., Liu, G., Wang, X., et al. (2012). ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 419, 779–781. doi: 10.1016/j.bbrc.2012.02.101
Zhou, T., Wang, J., Jia, Y., Li, W., Xu, F., Wang, X. (2018). Comparative chloroplast genome analyses of species in Gentiana section Cruciata (Gentianaceae) and the development of authentication markers. Int. J. Mol. Sci. 19, 1962. doi: 10.3390/ijms19071962
Zhou, Z., Wang, J., Pu, T., Dong, J., Guan, Q., Qian, J., et al. (2022). Comparative analysis of medicinal plant Isodon rubescens and its common adulterants based on chloroplast genome sequencing. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1036277
Keywords: Pseudostellaria heterophylla, chloroplast genome, comparative analysis, intraspecific variation, phylogenetic relationship
Citation: Zhang W, Zhang Z, Liu B, Chen J, Zhao Y and Huang Y (2023) Comparative analysis of 17 complete chloroplast genomes reveals intraspecific variation and relationships among Pseudostellaria heterophylla (Miq.) Pax populations. Front. Plant Sci. 14:1163325. doi: 10.3389/fpls.2023.1163325
Received: 10 February 2023; Accepted: 17 May 2023;
Published: 22 June 2023.
Edited by:
Weijun Kong, Capital Medical University, ChinaReviewed by:
Wei Gu, Nanjing University of Chinese Medicine, ChinaCopyright © 2023 Zhang, Zhang, Liu, Chen, Zhao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingying Chen, Y2p5NjYwMUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.