 Sanjib Sapkota1
Sanjib Sapkota1 Rishi R. Burlakoti
Rishi R. Burlakoti Kurt Lamour
Kurt Lamour
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 29 June 2023
Sec. Plant Pathogen Interactions
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1161864
This article is part of the Research TopicNew Insights in Small Fruit DiseasesView all 5 articles
Phytophthora rubi is a primary causal agent of Phytophthora root rot and wilting of raspberry (Rubus idaeus L.) worldwide. The disease is a major concern for raspberry growers in Canada and USA. To date, no information is available on genomic diversity of P. rubi population from raspberry in Canada. Using a PCR-free library prep with dual-indexing for an Illumina HiSEQX running a 2x150 bp configuration, we generated whole genome sequence data of P. rubi isolates (n = 25) recovered during 2018 to 2020 from nine fields, four locations and four cultivars of raspberry growing areas of British Columbia, Canada. The assembled genome of 24 isolates of P. rubi averaged 8,541 scaffolds, 309× coverage, and 65,960,000 bp. We exploited single nucleotide polymorphisms (SNPs) obtained from whole genome sequence data to analyze the genome structure and genetic diversity of the P. rubi isolates. Low heterozygosity among the 72% of pathogen isolates and standardized index of association revealed that those isolates were clonal. Principal component analysis, discriminant analysis of principal component, and phylogenetic tree revealed that P. rubi isolates clustered with the raspberry specific cultivars. This study provides novel resources and insight into genome structure, genetic diversity, and reproductive biology of P rubi isolated from red raspberry. The availability of the P. rubi genomes also provides valuable resources for future comparative genomic and evolutionary studies for oomycetes pathogens.
Phytophthora rubi, the primary causal agent of Phytophthora root rot and wilting (PRRW), is the most destructive pathogen of red raspberry (Rubus idaeus L.) in the Pacific Northwest regions of Canada (Sapkota et al., 2022a; Sapkota et al., 2022b) and the United States (Stewart et al., 2014; Weiland et al., 2018) and other raspberry producing areas worldwide (Duncan et al., 1987; Wilcox et al., 1993; Wilcox and Latorre, 2002; Jennings et al., 2003; Sapkota et al., 2022b). This pathogen poses a serious economic impacts to raspberry producers by reducing plant vigor, lifespan, and yield (Burlakoti and Dossett, 2020; Sapkota et al., 2022b). In addition with P. rubi, other species of Phytophthora such as, P. gonapodyides, P. cryptogea, P. citricola, and P. megasperma were also sporadically reported to infect raspberry (Wilcox, 1989; Wilcox and Latorre, 2002; Sapkota et al., 2022b; Burlakoti et al., 2023).
Phytophthora rubi is considered as a homothallic species and produces both sexual and asexual spores (Tabima et al., 2018; Abad et al., 2023). Mycelia and oospores in infected tissue and soil are major sources of inocula. The mycelia and sporangia can directly infect the basal stem and root tissues of plants. Sporangia also produces motile zoospores for infection in presence of high soil moisture. Sporangia and zoospores can travel in soil via water allowing P. rubi to migrate within and among fields. The dispersal and germination of these spores are favored by high soil moisture due to either rain or irrigation and average temperatures of 15 to 20°C (Graham et al., 2021). The pathogen spores colonizes root tissues, interferes water and nutrient movement in vascular system and develop dark-reddish brown lesions on roots and crown. As the disease progresses, foliage symptoms, such as necrosis, scorching, and chlorosis of leaves, and wilting of branches appears (Sapkota et al., 2022b).
In the past decades, genomes of multiple species of Phytophthora, such as P. infestans (Haas et al., 2009), P. sojae, P. ramorum (Tyler et al., 2006), P. capsici (Lamour et al., 2012), and P. fragariae (Gao et al., 2015) were sequenced. Recently, a draft genome of one isolate of P. rubi of raspberry was reported from the western USA (Tabima et al., 2017). However, whole genome sequencing resources of isolates of P. rubi from Canada are not available. The genetic structure of P. rubi isolated from raspberry has been analyzed in western USA using amplified fragment length polymorphism, and genotyping by sequencing (Stewart et al., 2014; Tabima et al., 2018). Both studies showed a low genetic diversity and low differentiation among P. rubi population. In addition, single nucleotide polymorphisms (SNPs) obtained from high throughput sequencing have recently been utilized to infer population structure, and its reproductive strategies in many species of Phytophthora such as P. capsici (Castro-Rocha et al., 2016; Siegenthaler et al., 2022), P. pluvialis (Brar et al., 2018), and P. lateralis (Quinn et al., 2013). High throughput sequencing is progressively becoming less expensive and data provide useful tools to explore genetic diversity and potentially aid in identifying the areas of diversity driving host varietal preference.
Although P. rubi has been reported as a major causal agent of PRRW in raspberry growing areas in Canada (Sapkota et al., 2022a; Sapkota et al., 2022b), no information is available on genetic structure and diversity of P. rubi population. Therefore, the objectives of this study were to: i) generate genomic resources of P. rubi using whole genome sequencing, and ii) study the genome structure and genetic diversity of P. rubi isolates from raspberry.
Isolates in this study were selected from the large culture collection from our previous study on raspberry root rot and wilting complex of raspberry on BC (Sapkota et al., 2022a). These isolates (n = 25) were collected over multiple years (2018 to 2020) from nine fields, and four cultivars of raspberry (Table 1). The detail of sampling, isolation and identification of the pathogen was described in (Sapkota et al., 2022a). In brief, pathogen isolates were obtained from infected root and cane tissues of raspberry using CMA-PARPH (corn meal agar medium amended with pimaricin, ampicillin, rifampicin, pentachloronitrobenzene and hymexazol). The hyphal tips of each isolate grown on CMA-PARPH were transferred to 20% clarified V8PAR for further morphological characterization. Colony and sporangia morphology was used for morphological characterization. For the molecular identification, isolates were grown on V8PAR media with cellophane on the surface for 10 to 13 days. Mycelia were then collected, freeze-dried, and ground using a magnetic MixerMill bead beating device (Qiagen). High quality genomic DNA was extracted using the MagMAX kit (ThermoFisher Scientific Inc.) following the manufacturer’s instructions. Pathogens were identified using multiplex targeted-sequencing with degenerate primers of three nuclear genes: heat shock protein90, elongation factor 1 alpha and beta tubulin as described in (Sapkota et al., 2022a).
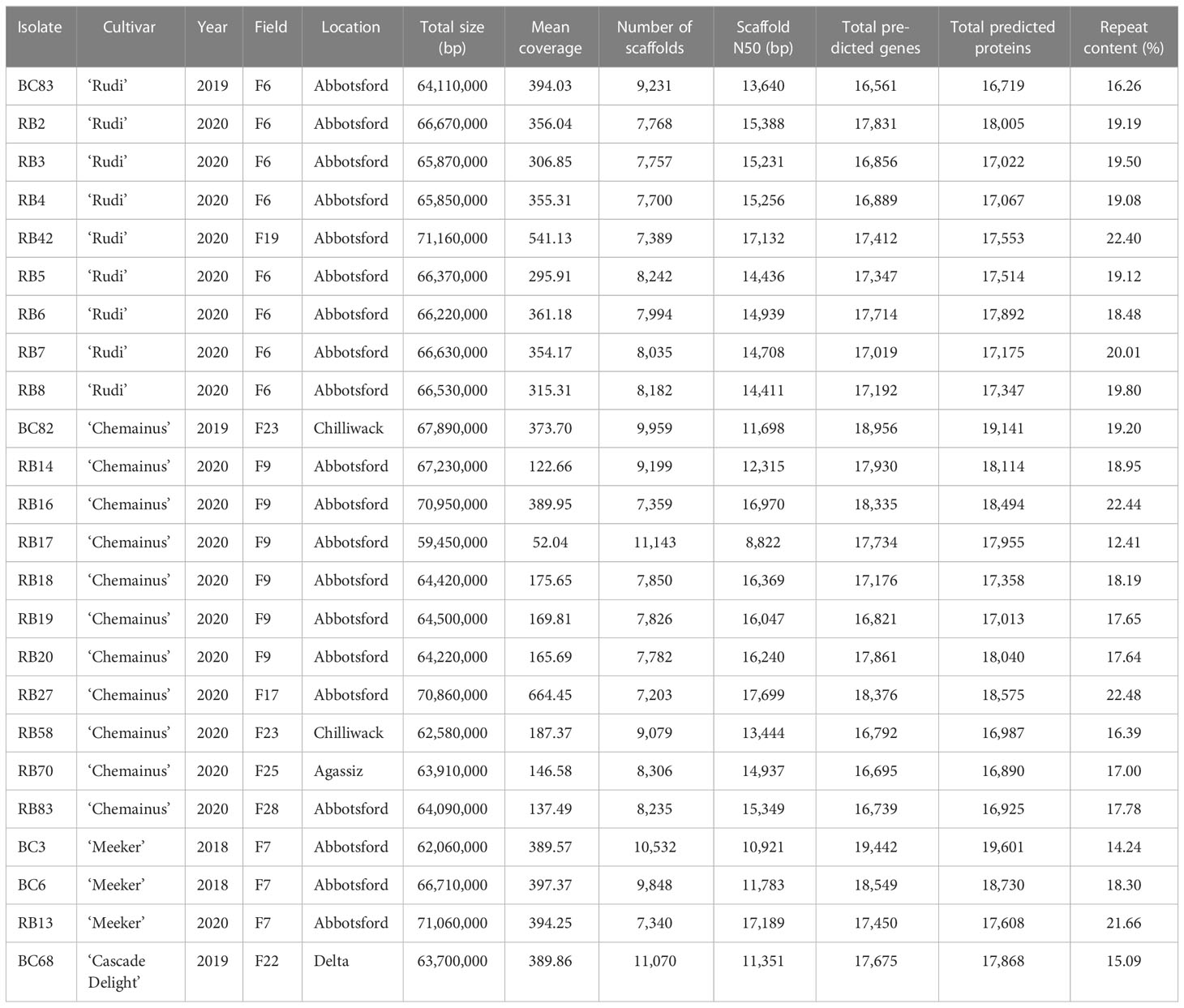
Table 1 Summary of comparative assembly statistics and gene predictions for genome resequencing of 24 isolates of Phytophthora rubi.
Genomic DNA of the pathogen was extracted as described in the previous section. Approximately, one µg of high quality genomic DNA was sheared to a size in between 250 and 500 bp with a Covaris M220 focused ultrasonicator (Covaris, Inc., Woburn, MA, U.S.A.). After shearing, fragmented DNA was ligated with dual-indices using a KAPA Hyperprep PCR-free library kit (Roche) for Illumina according to the manufacturer’s instructions. Libraries were loaded and sequenced on an Illumina HiSEQX next-generation sequencing device running a 2 x 150 bp paired-end configuration at Admera Health LLC (Plainfield, NJ) according to the manufacturer’s directions. Raw paired-end sequence data for each sample are publicly available for download in NCBI BioProject accession number PRJNA856328.
Raw sequence data was quality checked using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Paired-end reads were trimmed for the presence of adapters and low-quality sequences with BBDuk (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/). To identify SNPs, paired-end reads were randomly subsampled to 50 million read pairs with reformat.sh (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/) and then aligned to P. rubi reference genome (Tabima et al., 2017) using BWA-mem2 (Vasimuddin et al., 2019). SAMtools software package was used to sort, validate, and filter aligned reads (Li et al., 2009). SNPs or variants were called using Freebayes (Garrison and Marth, 2012), then filtered using VCFtools (Danecek et al., 2011) with minimum mapping quality score of 20, minimum depth of 10, and maximum depth of 300.
For whole genome assembly and annotation, a workflow was developed in the Nextflow workflow management system (Di Tommaso et al., 2017). Briefly, raw reads were quality and adapter trimmed by BBduk, then mapped to masked human and plant genomes using BBMap to remove host and contamination DNA, followed by normalization and error correction with BBNorm and Tadpole (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/). The processed reads were assembled using assemblers abyss-pe v2.3.5 (Jackman et al., 2017) and Spades v3.15 (Prjibelski et al., 2020), and the assembly with the highest N50 value was selected for annotation. Assemblies were filtered with Tiara (Karlicki et al., 2022) (minimum length 1000bp) to identify potential non-eukaryotic co-isolates, and any sequences identified as bacteria or archaea were excluded from annotation. Filtered assemblies were then repeat masked with EDTA (Ou et al., 2019), and annotated with Funannotate v1.8.13 (https://github.com/nextgenusfs/funannotate), with RNAseq data SRR10207404 (Adams et al., 2020) used as transcript evidence. Gene annotation was performed using Interproscan (Jones et al., 2014) and SignalP v6 (Teufel et al., 2022), and integrated into the assembly using Funannotate. Genome completeness was evaluated using BUSCO v5.4.4 in ‘protein’ mode with the Eukaryota Odb10 gene set (Simão et al., 2015) on the predicted proteins from each genome.
The filtered VCF files were analyzed using the R packages vcfR (Knaus and Grünwald, 2017), poppr (Kamvar et al., 2014), data.table (Dowle et al., 2019), and adegenet (Jombart et al., 2010). To determine genetic diversity of the P rubi isolates, Simpson’s diversity index, λ, (Simpson, 1949), Shannon-Weiner’s genotypic diversity, H, (Shannon, 2001 and review), and Nei’s unbiased gene diversity, Hexp, (Nei, 1978) were calculated. The heterozygous variants per kb were determined to investigate the distribution of variants in pathogen population. To test the hypothesis of clonal reproduction, we estimated the standardized index of association (r̄d). Statistical significance of r̄d test was calculated with 999 permutations of the random test. The r̄d values would be expected to be zero, if isolates are mating randomly (i.e. freely recombining) (Agapow and Burt, 2001). We carried out discriminant analysis of principal components (DAPC) in the R package adegenet to show clustering among genetically related populations (Jombart et al., 2010). The advantage of the DAPC approach is that it does not make assumptions on population models or data structure. Principal component analysis (PCA) was also performed using the glpca command to display major genetic variations. A phylogenetic tree was constructed based on Bruvo’s (Bruvo et al., 2004) distance to calculate the genetic relatedness among the population of P. rubi. These results were plotted and visualized using the R package “ggplot2” (Wickham, 2016). We excluded one isolate (RB15) in the genome assembly since we could not identify a clear reasons for size difference of this isolates with remaining 24 isolates.
In total, whole genomes of 25 P. rubi isolates collected from diverse cultivars and locations were sequenced. The summary of total genome size, number of scaffolds, scaffold N50, total predicted genes, total predicted proteins, and percent repeat content of these isolates is shown in Table 1. The average genome size was 65,960,000 bp (range: 59,450,000 to 71,160,000 bp). The mean coverage ranged from 52 to 664×, with an average of 309×. The assembled genomes averaged 8,541 scaffolds (range: 7,203 to 11,143) with an average N50 of 14,426 (range: 8,822 to 17,668). The total number of predicted genes and protein ranged between 16,561 to 18,956 and 16,719 to 19,141 respectively. The repetitive content in P. rubi genome assemblies ranged from 12.41 to 22.48%. In addition, BUSCO completeness scores ranged from 92.2 to 94.2%, with an average of 93.33% (Supplementary Table 1).
Genotypic diversity estimates of overall population of P. rubi was high (H = 3.22 and λ = 0.96) (Table 2). H and λ values of isolates grouped by cultivar was also high. Low genetic diversity (Hexp = 0.02) was observed among these isolates. The Hexp was lowest (0.011) in the isolates from ‘Meeker’ and was the highest (0.019) in the isolates from ‘Chemainus’ (Table 2). The SNPs per kb for isolates grouped by cultivar ranged from 0.60 to 1.70. Low heterozygous variants per kb were observed in 72% of pathogen isolates and the alternate allele coverage fell into a distribution of near 100% or 0% in these isolates (Figure 1A). Seven isolates, recovered from ‘Rudi’, showed signs of increased heterozygosity (SNPs per kb: 1.57 to 1.70; Figure 1B). The r̄d values for the overall population and oomycetes isolates grouped by cultivar were significantly deviated from hypothesis of random mating among isolates of P. rubi from BC (Table 2).
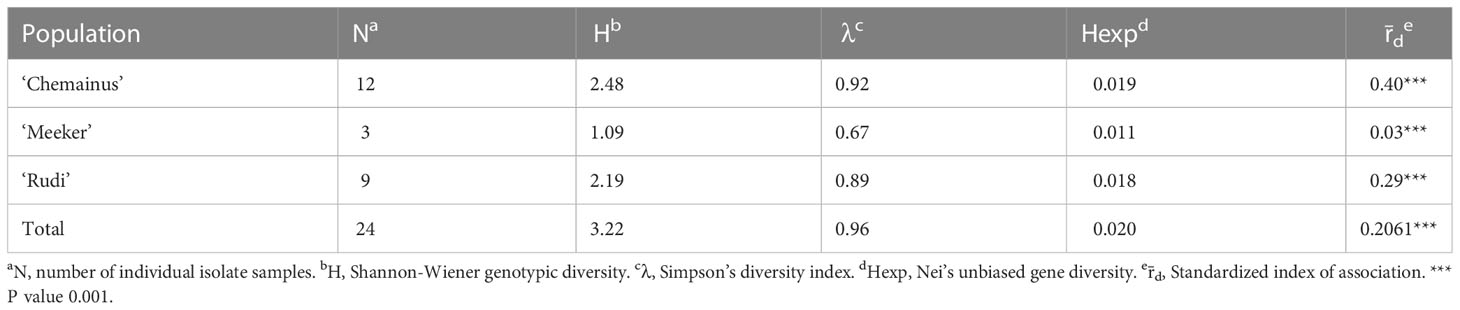
Table 2 Indices of genotypic diversity for Phytophthora rubi from cultivar population based on SNPs generated by whole genome resequencing.
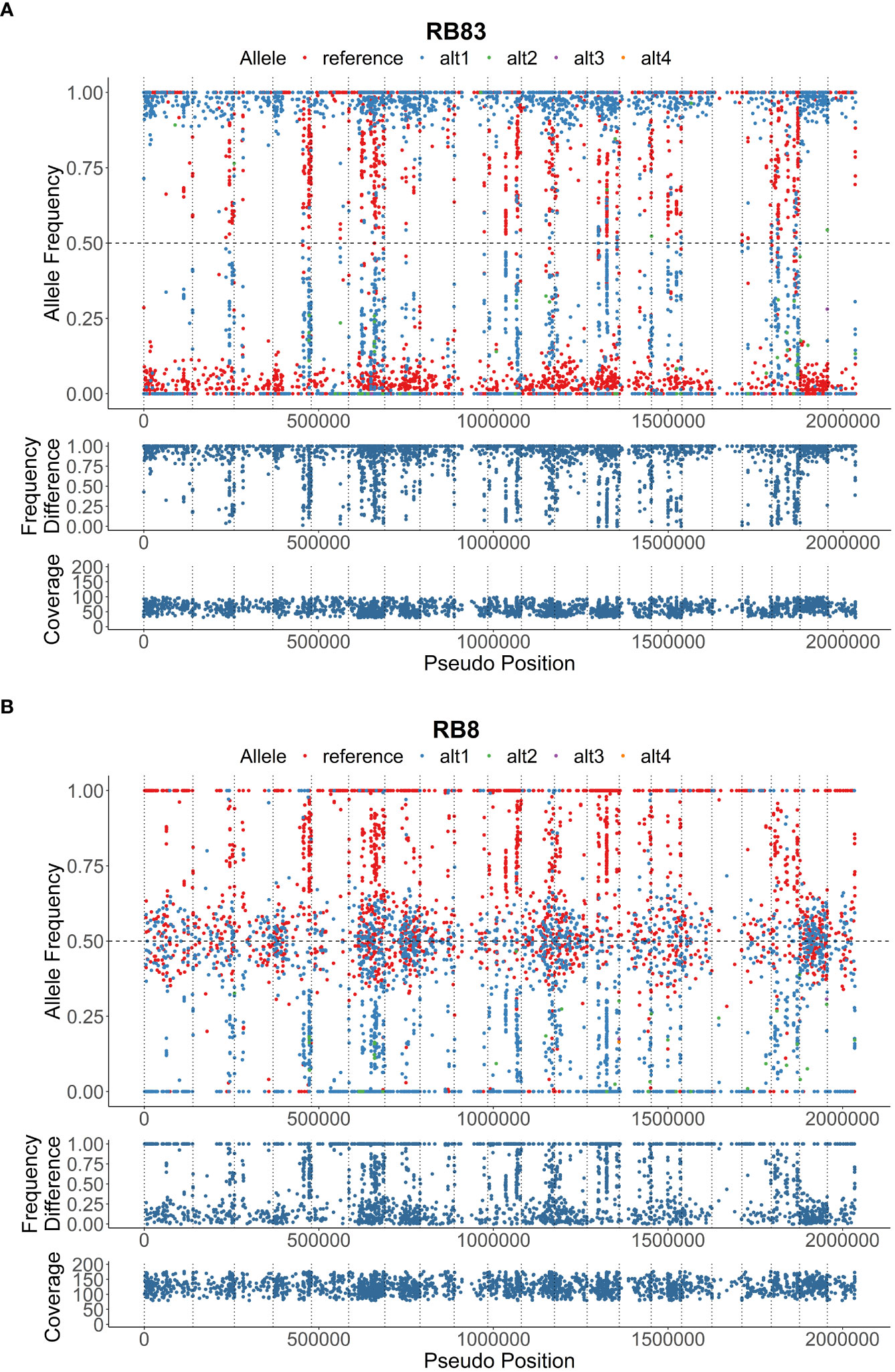
Figure 1 Heterozygous allele frequencies plots across 2 Mbp of the reference genome; (A), isolate RB83 exhibiting low heterozygosity, and (B), isolate RB8 exhibiting moderate heterozygosity. The allele frequency, frequency difference, and coverage are denoted on below the visualizations of heterozygous allele frequencies.
DAPC indicated the presence of unique genetic clusters of P rubi isolates corresponding to raspberry cultivars (Figure 2A). Analysis of isolates using PCA based on Bruvo’s distance also showed the major grouping of isolates based on cultivars (Figure 2B). A phylogenetic tree also supported the results obtained from DAPC and PCA analyses (Figure 2C). Among the isolates of P. rubi, eight isolates from ‘Rudi’ formed one cluster and 11 isolates from ‘Chemainus’ and three isolates from ‘Meeker’ formed a distinct cluster.
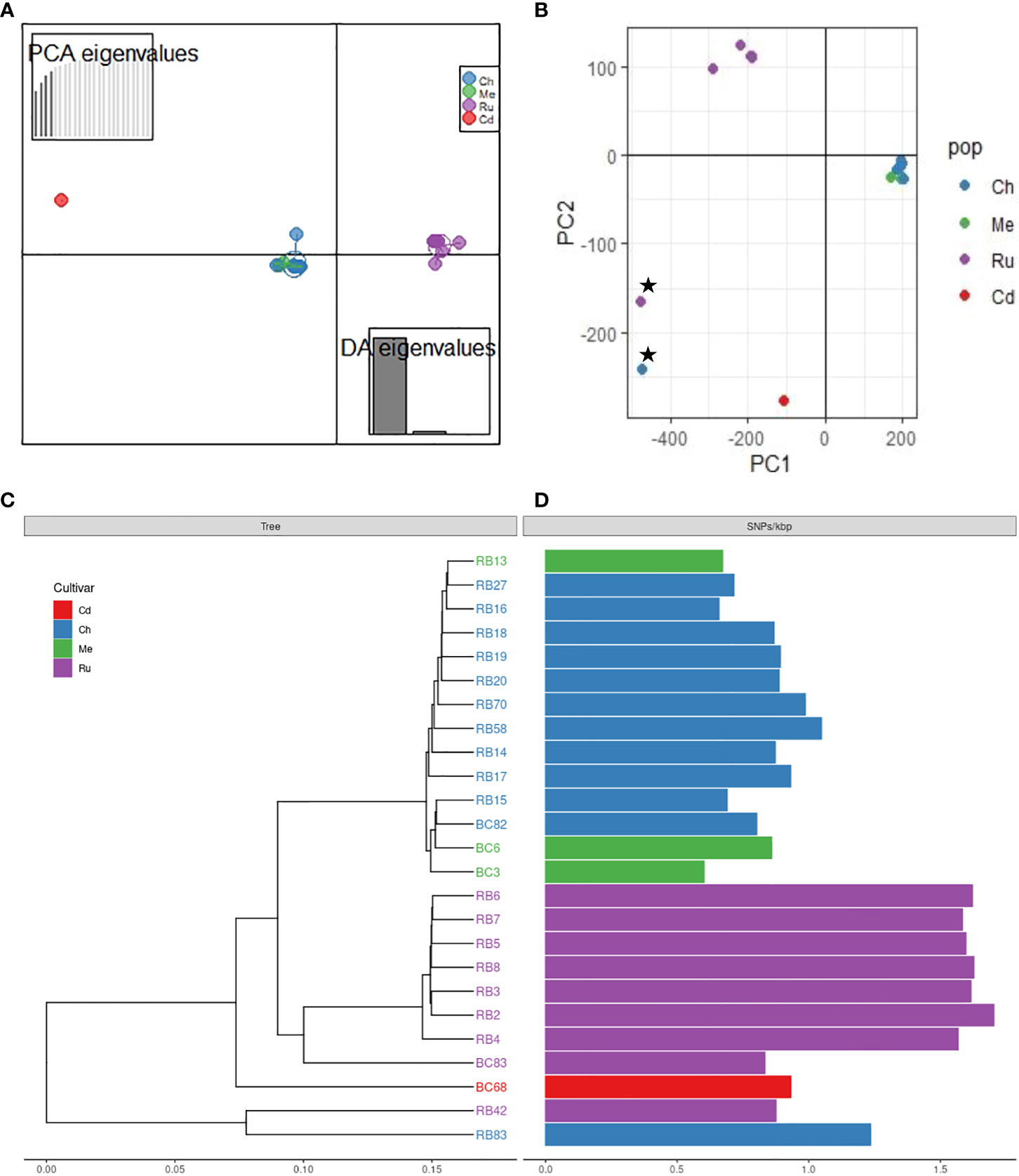
Figure 2 Population structure of Phytophthora rubi isolates from different cultivars of raspberry (cultivar abbreviation: Ru = ‘Rudi’, Ch = ‘Chemainus’, Me = ‘Meeker’, Cd = ‘Cascade Delight’) based on SNPs generated by whole genome resequencing; (A) discriminant analysis of principal components (DAPC) plot, (B) principal component analysis (PCA) plot; star symbols indicate outlier isolates RB83 and RB42, recovered from cultivar ‘Chemainus’ and ‘Rudi’, (C) genetic distance tree, (D) single nucleotide polymorphisms (SNPs) per Kbp.
This is the first genome-level study of P. rubi isolates collected from diverse cultivars and locations of raspberry growing areas in BC, Canada. Genome assemblies of 24 P. rubi isolates were generated along with gene predictions and annotation. BUSCO analysis found at least 92% of single-copy eukaryotic genes in the predicted proteins from each of the sequenced genomes, indicating that the gene prediction has captured a relatively complete representation of the gene content of each isolate. BUSCO values from our study are similar to results for the Eukarya dataset for previous Phytophthora genome assemblies studies produced from both short-read and long-read sequences (Cui et al., 2019; Engelbrecht et al., 2021; Kronmiller et al., 2023). Previous researchers sequenced only one (Tabima et al., 2017) or three isolates (Adams et al., 2020) of P. rubi resulting in assemblies of 74.65 and 77.84 Mbp in 9,434 and 13,882 scaffolds, respectively. The genome assemblies and scaffolds of P. rubi isolates generated in our study ranged from 59.45 to 71.16 Mbp and 7,203 to 11,143, respectively (Table 1), indicating that the genome structure in our study varied slightly than the previous studies, which is likely due to the inclusion of multiple isolates of P. rubi collected from diverse cultivars and locations in our study compared to either one or few isolates in previous studies.
We found a high genotypic diversity and low gene diversity among isolates of P. rubi from BC. We hypothesize that genotypic variability could be associated with three possible reasons (Milgroom, 2015; Zhan, 2016; Brugman et al., 2022). First, the continuous availability of host to pathogen populations is likely to promote rapid coevolution between the P. rubi pathogen and its raspberry host. Second, high disease pressure of PRRW in the field as a result of multiple infection events could increase diversity. Third, the mutation and sexual reproduction (through self-fertilization) can affect genotypic variability. Previous studies also reported the high genotypic diversity in clonal populations of P. infestans in Denmark (Maurice et al., 2019), and P. palmivora in Indonesia (Brugman et al., 2022). In contrast, genetic structure study of P. rubi population in the western USA using amplified fragment length polymorphism (Stewart et al., 2014) and using genotyping by sequencing (Tabima et al., 2018) showed low genetic diversity among isolates.
Phytophthora rubi is thought to be homothallic like other Phytopthora species (Tabima et al., 2018; Abad et al., 2023), with sexual reproduction through selfing and rare outcrossing. Selfing is expected to dramatically reduce heterozygosity and may reduce genetic diversity and thus considerably decrease the level of heterozygosity relative to sexual populations. Thus, we hypothesized that in P. rubi population very low heterozygous sites per Kb should be present in the genome. Our findings reveal the significant deficit of heterozygosity in the majority of sequenced genomes (18 of 25 isolates = 72%) indicating that the P. rubi population from BC are predominantly clonal (Figure 2D). However, the remaining seven isolates showed signs of increased heterozygosity, and these isolates were recovered from the raspberry cultivar ‘Rudi’. It is likely due to the accumulation of mutations during the successive asexual reproduction but may be due to a rare outcrossing event (Balloux et al., 2003). Nevertheless, our findings of clonality is congruent with the study in western USA by (Tabima et al., 2018) who also reported that P. rubi from raspberry are predominantly clonal and/or selfing. Previous studies also revealed low heterozygosity among populations of other homothallic Phytopthhora species, such as P. plurivora (Schoebel et al., 2014), and P. sojae (Stewart et al., 2016).
The production of oospores by pathogen does not always refer to sexual reproduction or its contribution to epidemics, hence it is important to test the null hypothesis of random mating when interpreting population structure of P. rubi. The r̄d test showed the presence of clonality among isolates of P. rubi from BC. Past studies also used r̄d to refer to clonality in populations of P. pluvialis (Brar et al., 2018), and P. plurivora (Schoebel et al., 2014). It is important to remember that the pathogen population dynamics would change over time and continuous monitoring of structure of pathogen populations is required to provide valuable information for breeders to identify resistant plant materials (McDonald, 2015; Milgroom, 2015). For example, Engelbrecht et al. (2017) reported clonal mode of reproduction in P. cinnamomi, a causal agent of Avocado root rot, populations in South Africa; however after a few years an evidence of random mating among pathogen populations was reported (Engelbrecht et al., 2022).
DAPC, PCA, and phylogenetic analyses revealed the presence of distinct clusters of P. rubi isolates with raspberry cultivars. The isolate from ‘Rudi’ formed one cluster while isolates from ‘Chemainus’ and ‘Meeker’ formed a further distinct cluster. This clustering may suggest that the genetic diversity in cultivars could pose varying levels of selection pressure on P. rubi populations (Moore and Daubeny, 1993; Moore, 2004; Kempler et al., 2006). We further speculate that the ability of variety of isolates of P. rubi to cause PRRW disease on a different raspberry cultivars might have led for cultivar specificity among isolates. Studies of 11 isolates of P. fragariae var rubi, (recently separated into a distinct species P. rubi) on roots of six raspberry genotypes showed that the existence of three different races, suggesting a significant cultivar/isolate interaction (Kennedy and Duncan, 1993). The presence of physiological races among pathogen populations was reported from other Phytophthora spp. such as P. capsici (Jiang et al., 2015; Barchenger et al., 2018) and P. sojae (Xue et al., 2015; Ping et al., 2016). Further research would be required to fully understand the pathogen races status of P. rubi isolates from BC using cultivars with varying levels of resistance.
Overall, this study provided the genome sequence resources of economically important raspberry pathogen P. rubi and investigated the genome structure and genetic diversity among these isolates. Knowledge gained from the genome sequences can provide insight into pathogen genomic structure, biology, comparative genomic and evolutionary studies among oomycetes. More recently, researcher have been using whole genome sequence data in examining effector biology and in comparative genomic analysis of different Phytophthora species including P. rubi and P. fragariae (Adams et al., 2020). The genome structure and diversity of pathogen populations could be associated with origin, dispersal of pathogen, and reproductive strategies (Milgroom and Peever, 2003; McDonald, 2015; Milgroom, 2015; Grünwald et al., 2017). This information could be useful for screening and developing resistant cultivars, and deploying disease management strategies. For example, pathogen microorganisms showing sexual or mixed reproduction could pose a high risk by emerging new genotypes during the sexual cycle (McDonald and Linde, 2002). Larger sampling and analysis of population within and outside production regions is warrant to understand overall population diversity of P. rubi populations both regionally and globally.
The datasets presented in this study can be found in online repositories. NCBI BioProject accession number is PRJNA856328. The variant data file (VCF) is available in the European Variation Archive (https://www.ebi.ac.uk/eva/?Home) and accessions associated with the submission are Project: PRJEB62133 and Analyses: ERZ18306854.
SS, RB, and KL conceived and designed the study. RB supervised the first author to conduct the experiment and drafting the manuscript. SS, RB, and KL curated the data. SS and ML performed all bioinformatics analyses. SS and RB wrote a first draft of the manuscript. All authors read and approved the submitted version.
This research was supported by funding from the Canadian Agricultural Partnership (CAP)-AgriScience Program, the BC Raspberry Industry Development Council and the BC Lower Mainland Horticultural Improvement Association (Project ID: CAP–AgriScience Project No: ASP-007)”.
We thank Floodlight Genomics LLC for conducting the sequencing work as part of their Educational and Research Outreach Program (EROP) at low or no cost. We thank all raspberry growers in Fraser Valley areas of BC who allowed us to monitor their fields. We thank Amy Novinscak, Taysia Nikaido-Landry, Carol Koch, and Rebecca Kehtler for technical support. We also thank the Storkan-Hanes-McCaslin research foundation for providing a research award support to SS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1161864/full#supplementary-material
Abad, Z. G., Burgess, T., Redford, A. J., Bienapfl, J. C., Mathew, R., Srivastava, S. K., et al. (2023). IDphy: an international online resource for molecular and morphological identification of Phytophthora. Plant Dis. 107, 987–998. doi: 10.1094/PDIS-02-22-0448-FE
Adams, T. M., Armitage, A. D., Sobczyk, M. K., Bates, H. J., Tabima, J. F., Kronmiller, B. A., et al. (2020). Genomic investigation of the strawberry pathogen Phytophthora fragariae indicates pathogenicity is associated with transcriptional variation in three key races. Front. Microbiol. 11, 490. doi: 10.3389/fmicb.2020.00490
Agapow, P. M., Burt, A. (2001). Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes. 1, 101–102. doi: 10.1046/j.1471-8278.2000.00014.x
Balloux, F., Lehmann, L., De Meeûs, T. (2003). The population genetics of clonal and partially clonal diploids. Genetics 164, 1635–1644. doi: 10.1093/genetics/164.4.1635
Barchenger, D. W., Sheu, Z. M., Kumar, S., Lin, S. W., Burlakoti, R. R., Bosland, P. W. (2018). Race characterization of phytophthora root rot on capsicum in Taiwan as a basis for anticipatory resistance breeding. Phytopathology 108, 964–971. doi: 10.1094/PHYTO-08-17-0289-R
Brar, S., Tabima, J., Mcdougal, R., Dupont, P. Y., Feau, N., Hamelin, R., et al. (2018). Genetic diversity of Phytophthora pluvialis, a pathogen of conifers, in new Zealand and the west coast of the united states of America. Plant Pathol. 67, 1131–1139. doi: 10.1111/ppa.12812
Brugman, E., Wibowo, A., Widiastuti, A. (2022). Phytophthora palmivora from sulawesi and Java islands, Indonesia, reveals high genotypic diversity and lack of population structure. Fungal Biol. 126, 267–276. doi: 10.1016/j.funbio.2022.02.004
Bruvo, R., Michiels, N. K., D’souza, T. G., Schulenburg, H. (2004). A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol. Ecol. 13, 2101–2106. doi: 10.1111/j.1365-294X.2004.02209.x
Burlakoti, R., Dossett, M. (2020). Past efforts and future perspectives of managing major diseases of red raspberries in British Columbia. Acta Hortic. 1277, 397–402. doi: 10.17660/ActaHortic.2020.1277.56
Burlakoti, R. R., Sapkota, S., Lubberts, M., Lamour, K. (2023). First report of phytophthora gonapodyides causing root rot on raspberry in Canada. Plant Dis. doi: 10.1094/PDIS-08-22-1940-PDN
Castro-Rocha, A., Shrestha, S., Lyon, B., Grimaldo-Pantoja, G. L., Flores-Marges, J. P., Valero-Galván, J., et al. (2016). An initial assessment of genetic diversity for Phytophthora capsici in northern and central Mexico. Mycol. Prog. 15, 1–12. doi: 10.1007/s11557-016-1157-0
Cui, C., Herlihy, J. H., Bombarely, A., McDowell, J. M., Haak, D. C. (2019). Draft assembly of Phytophthora capsici from long-read sequencing uncovers complexity. Mol. Plant Microbe Interact. 32, 1559–1563. doi: 10.1094/MPMI-04-19-0103-TA
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., Depristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Di Tommaso, P., Chatzou, M., Floden, E. W., Barja, P. P., Palumbo, E., Notredame, C. (2017). Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319. doi: 10.1038/nbt.3820
Dowle, M., Srinivasan, A., Gorecki, J., Chirico, M., Stetsenko, P., Short, T., et al. (2019). Package ‘data. table’. Extension of ‘data. frame’. [Computer software]. Vol. 596.
Duncan, J. M., Kennedy, D. M., Seemuller, E. (1987). Identities and pathogenicities of Phytophthora spp. causing root rot of red raspberry. Plant Pathol. 36, 276–289. doi: 10.1111/j.1365-3059.1987.tb02235.x
Engelbrecht, J., Duong, T., Berg, N. V. (2017). New microsatellite markers for population studies of Phytophthora cinnamomi, an important global pathogen. Sci. Rep. 7, 1–10. doi: 10.1038/s41598-017-17799-9
Engelbrecht, J., Duong, T. A., Paap, T., Hubert, J. M., Hanneman, J. J., Van Den Berg, N. (2022). Population genetic analyses of Phytophthora cinnamomi reveals three lineages and movement between natural vegetation and avocado orchards in south Africa. Phytopathology 112, 1568–1574. doi: 10.1094/PHYTO-10-21-0414-R
Engelbrecht, J., Duong, T. A., Prabhu, S. A., Seedat, M., Van den Berg, N. (2021). Genome of the destructive oomycete Phytophthora cinnamomi provides insights into its pathogenicity and adaptive potential. BMC Genom. 22, 1–15. doi: 10.1186/s12864-021-07552-y
Gao, R., Cheng, Y., Wang, Y., Wang, Y., Guo, L., Zhang, G. (2015). Genome sequence of Phytophthora fragariae var. fragariae, a quarantine plant-pathogenic fungus. Genome Announc. 3, e00034–e00015. doi: 10.1128/genomeA.00034-15
Garrison, E., Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv. 1207, 3907. doi: 10.48550/arXiv.1207.3907
Graham, K. A., Beck, B. R., Zasada, I. A., Scagel, C. F., Weiland, J. E. (2021). Growth, sporulation, and pathogenicity of the raspberry pathogen Phytophthora rubi under different temperature and moisture regimes. Plant Dis. 105, 1791–1797. doi: 10.1094/PDIS-09-20-1916-RE
Grünwald, N. J., Everhart, S. E., Knaus, B., Kamvar, Z. N. (2017). Best practices for population genetic analyses. Phytopathology 107, 1000–1010. doi: 10.1094/PHYTO-12-16-0425-RVW
Haas, B. J., Kamoun, S., Zody, M. C., Jiang, R. H., Handsaker, R. E., Cano, L. M., et al. (2009). Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461, 393–398. doi: 10.1038/nature08358
Jackman, S. D., Vandervalk, B. P., Mohamadi, H., Chu, J., Yeo, S., Hammond, S. A., et al. (2017). ABySS 2.0: resource-efficient assembly of large genomes using a bloom filter. Genome Res. 27, 768–777. doi: 10.1101/gr.214346.116
Jennings, S. N., Brennan, R., Gordon, S. C. (2003). Commercial breeding for pest and disease resistance in cane and bush fruits. IOBC WPRS Bull. 26, 67–72.
Jiang, L., Sanogo, S., Bosland, P. W. (2015). Using recombinant inbred lines to monitor changes in the race structure of Phytophthora capsici in chile pepper in new Mexico. Plant Health Prog. 16, 235–240. doi: 10.1094/PHP-RS-15-0034
Jombart, T., Devillard, S., Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 1–15. doi: 10.1186/1471-2156-11-94
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., Mcanulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kamvar, Z. N., Tabima, J. F., Grünwald, N. J. (2014). Poppr: an r package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281. doi: 10.7717/peerj.281
Karlicki, M., Antonowicz, S., Karnkowska, A. (2022). Tiara: deep learning-based classification system for eukaryotic sequences. Bioinformatics 38, 344–350. doi: 10.1093/bioinformatics/btab672
Kempler, C., Daubeny, H. A., Frey, L., Walters, T. (2006). ‘Chemainus’ red raspberry. HortScience 41, 1364–1366. doi: 10.21273/HORTSCI.41.5.1364
Kennedy, D., Duncan, J. (1993). Occurrence of races in Phytophthora fragariae var rubi on raspberry. Acta Hortic. 352, 555–562. doi: 10.17660/ActaHortic.1993.352.81
Knaus, B. J., Grünwald, N. J. (2017). Vcfr: a package to manipulate and visualize variant call format data in r. Mol. Ecol. Resour. 17, 44–53. doi: 10.1111/1755-0998.12549
Kronmiller, B. A., Feau, N., Shen, D., Tabima, J. F., Ali, S. S., Armitage, A. D., et al. (2023). Comparative genomic analysis of 31 Phytophthora genomes reveals genome plasticity and horizontal gene transfer. Mol. Plant Microbe Interact. 36, 26–46. doi: 10.1094/MPMI-06-22-0133-R
Lamour, K. H., Mudge, J., Gobena, D., Hurtado-Gonzales, O. P., Schmutz, J., Kuo, A., et al. (2012). Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici. Mol. Plant Microbe Int. 25, 1350–1360. doi: 10.1094/MPMI-02-12-0028-R
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Maurice, S., Montes, M. S., Nielsen, B. J., Bødker, L., Martin, M. D., Jønck, C. G., et al. (2019). Population genomics of an outbreak of the potato late blight pathogen, Phytophthora infestans, reveals both clonality and high genotypic diversity. Mol. Plant Pathol. 20, 1134–1146. doi: 10.1111/mpp.12819
McDonald, B. A. (2015). How can research on pathogen population biology suggest disease management strategies? the example of barley scald (Rhynchosporium commune). Plant Pathol. 64, 1005–1013. doi: 10.1111/ppa.12415
McDonald, B. A., Linde, C. J. (2002). Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 40, 349–379. doi: 10.1146/annurev.phyto.40.120501.101443
Milgroom, M. G. (2015). Population biology of plant pathogens: genetics, ecology, and evolution (MN, USA: APS Press, The American Phytopathological Society St. Paul).
Milgroom, M. G., Peever, T. L. (2003). Population biology of plant pathogens: the synthesis of plant disease epidemiology and population genetics. Plant Dis. 87, 608–617. doi: 10.1094/PDIS.2003.87.6.608
Moore, P. P. (2004). ‘Cascade delight’ red raspberry. HortScience 39, 185–187. doi: 10.21273/HORTSCI.39.1.185
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590. doi: 10.1093/genetics/89.3.583
Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J. R., Hellinga, A. J., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 1–18. doi: 10.1186/s13059-019-1905-y
Ping, J., Fitzgerald, J. C., Zhang, C., Lin, F., Bai, Y., Wang, D., et al. (2016). Identification and molecular mapping of Rps11, a novel gene conferring resistance to Phytophthora sojae in soybean. Theor. Appl. Genet. 129, 445–451. doi: 10.1007/s00122-015-2638-2
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A., Korobeynikov, A. (2020). Using SPAdes de novo assembler. Curr. Protoc. Bioinf. 70, e102. doi: 10.1002/cpbi.102
Quinn, L., O'neill, P. A., Harrison, J., Paskiewicz, K. H., Mccracken, A. R., Cooke, L. R., et al. (2013). Genome-wide sequencing of Phytophthora lateralis reveals genetic variation among isolates from Lawson cypress (Chamaecyparis lawsoniana) in northern Ireland. FEMS Microbiol. Lett. 344, 179–185. doi: 10.1111/1574-6968.12179
Sapkota, S., Burlakoti, R. R., Lamour, K., Lubberts, M., Punja, Z. K. (2022a). Development and application of multiplex targeted-sequencing approaches to identify Phytophthora species associated with root rot and wilting complex of red raspberry. PloS One 17, e0275384. doi: 10.1371/journal.pone.0275384
Sapkota, S., Burlakoti, R. R., Punja, Z. K., Dossett, M., Gerbrandt, E. (2022b). Understanding the root rot and wilting complex of raspberry: current research advances and future perspectives. Can. J. Plant Pathol. 44, 323–344. doi: 10.1080/07060661.2021.2011420
Schoebel, C. N., Stewart, J., Gruenwald, N. J., Rigling, D., Prospero, S. (2014). Population history and pathways of spread of the plant pathogen Phytophthora plurivora. PloS One 9, e85368. doi: 10.1371/journal.pone.0085368
Shannon, C. E. (2001). A mathematical theory of communication. ACM SIGMOBILE mob. Comput. Commun. Rev. 5, 3–55. doi: 10.1145/584091.584093
Siegenthaler, T. B., Lamour, K., Hansen, Z. R. (2022). Population structure of Phytophthora capsici in the state of Tennessee. Mycol. Prog. 21, 159–166. doi: 10.1007/s11557-021-01769-7
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stewart, J. E., Kroese, D., Tabima, J. F., Larsen, M. M., Fieland, V. J., Press, C. M., et al. (2014). Pathogenicity, fungicide resistance, and genetic variability of Phytophthora rubi isolates from raspberry (Rubus idaeus) in the western united states. Plant Dis. 98, 1702–1708. doi: 10.1094/PDIS-11-13-1130-RE
Stewart, S., Robertson, A., Wickramasinghe, D., Draper, M., Michel, A., Dorrance, A. (2016). Population structure among and within Iowa, Missouri, Ohio, and south Dakota populations of Phytophthora sojae. Plant Dis. 100, 367–379. doi: 10.1094/PDIS-04-15-0437-RE
Tabima, J. F., Coffey, M. D., Zazada, I. A., Grünwald, N. J. (2018). Populations of phytophthora rubi show little differentiation and high rates of migration among states in the western united states. Mol. Plant-Microbe Int. 31, 614–622. doi: 10.1094/MPMI-10-17-0258-R
Tabima, J. F., Kronmiller, B. A., Press, C. M., Tyler, B. M., Zasada, I. A., Grünwald, N. J. (2017). Whole genome sequences of the raspberry and strawberry pathogens Phytophthora rubi and P. fragariae. Mol. Plant-Microbe Int. 30, 767–769. doi: 10.1094/MPMI-04-17-0081-A
Teufel, F., Almagro Armenteros, J. J., Johansen, A. R., Gíslason, M. H., Pihl, S. I., Tsirigos, K. D., et al. (2022). SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025. doi: 10.1038/s41587-021-01156-3
Tyler, B. M., Tripathy, S., Zhang, X., Dehal, P., Jiang, R. H., Aerts, A., et al. (2006). Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 313, 1261–1266. doi: 10.1126/science.1128796
Vasimuddin, M., Misra, S., Li, H., Aluru, S. (2019). “Efficient architecture-aware acceleration of BWA-MEM for multicore systems,” in 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). (Riode Janeiro, Brazil), 314–324.
Weiland, J. E., Benedict, C., Zasada, I. A., Scagel, C. R., Beck, B. R., Davis, A., et al. (2018). Late-summer disease symptoms in western washington red raspberry fields associated with co-occurrence of Phytophthora rubi, Verticillium dahliae, and Pratylenchus penetrans, but not raspberry bushy dwarf virus. Plant Dis. 102, 938–947. doi: 10.1094/PDIS-08-17-1293-RE
Wickham, H. (2016). Ggplot2: Elegant graphics for data analysis, (Houston, TX: Springer), 189–201. Available at: https://link.springer.com/book/10.1007/978-3-319-24277-4.
Wilcox, W. F. (1989). Identity virulence and isolation frequency of seven phytophthora spp. causing root rot of raspberry in new York USA. Phytopathology 79, 93–101. doi: 10.1094/Phyto-79-93
Wilcox, W. F., Latorre, B. A. (2002). Identities and geographic distributions of Phytophthora spp. causing root rot of red raspberry in Chile. Plant Dis. 86, 1357–1362. doi: 10.1094/PDIS.2002.86.12.1357
Wilcox, W. F., Scott, P. H., Hamm, P. B., Kennedy, D. M., Duncan, J. M., Brasier, C. M., et al. (1993). Identity of a Phytophthora species attacking raspberry in Europe and north America. Mycol. Res. 97, 817–831. doi: 10.1016/S0953-7562(09)81157-X
Xue, A. G., Marchand, G., Chen, Y., Zhang, S., Cober, E. R., Tenuta, A. (2015). Races of phytophthora sojae in Ontario, canada 2010–2012. Can. J. Plant Pathol. 37, 376–383. doi: 10.1080/07060661.2015.1052562
Keywords: clonal, genome, genetic diversity, Phytophthora rubi, raspberry, heterozygosity
Citation: Sapkota S, Burlakoti RR, Lubberts M and Lamour K (2023) Genome resources and whole genome resequencing of Phytophthora rubi isolates from red raspberry. Front. Plant Sci. 14:1161864. doi: 10.3389/fpls.2023.1161864
Received: 08 February 2023; Accepted: 15 May 2023;
Published: 29 June 2023.
Edited by:
Darko Jevremović, Fruit Research Institute, SerbiaReviewed by:
Alessandro Cestaro, Fondazione Edmund Mach, ItalyCopyright © 2023 Kurt Lamour and His Majesty the King in Right of Canada, as represented by the Minister of Agriculture and Agri-Food Canada for the contribution of Sanjib Sapkota, Rishi R. Burlakoti, and Mark Lubberts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rishi R. Burlakoti, cmlzaGkuYnVybGFrb3RpQGFnci5nYy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.