 Weijie Xu
Weijie Xu Haiying Ren1,3,4*†
Haiying Ren1,3,4*† Xingjiang Qi
Xingjiang Qi Jianbo Xie
Jianbo Xie
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 14 April 2023
Sec. Plant Abiotic Stress
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1155504
This article is part of the Research Topic Heat Stress: Response, Mitigation, and Tolerance in Plants View all 14 articles
Stress response in plant is regulated by a large number of genes co-operating in diverse networks that serve multiple adaptive process. To understand how gene regulatory networks (GRNs) modulating abiotic stress responses, we compare the GRNs underlying drought and cold stresses using samples collected at 4 or 6 h intervals within 48 h in Chinese bayberry (Myrica rubra). We detected 7,583 and 8,840 differentially expressed genes (DEGs) under drought and cold stress respectively, which might be responsive to environmental stresses. Drought- and cold-responsive GRNs, which have been built according to the timing of transcription under both abiotic stresses, have a conserved trans-regulator and a common regulatory network. In both GRNs, basic helix-loop-helix family transcription factor (bHLH) serve as central nodes. MrbHLHp10 transcripts exhibited continuous increase in the two abiotic stresses and acts upstream regulator of ASCORBATE PEROXIDASE (APX) gene. To examine the potential biological functions of MrbHLH10, we generated a transgenic Arabidopsis plant that constitutively overexpresses the MrbHLH10 gene. Compared to wild-type (WT) plants, overexpressing transgenic Arabidopsis plants maintained higher APX activity and biomass accumulation under drought and cold stress. Consistently, RNAi plants had elevated susceptibility to both stresses. Taken together, these results suggested that MrbHLH10 mitigates abiotic stresses through the modulation of ROS scavenging.
A disturbance in the environment triggers rapid and global reprogramming of cells, which requires the spatial and temporal coordination of multiple TFs (Zhang et al., 2019; Zhou et al., 2021). Transcriptional regulation occurs on a multitude of time scales, from minutes to days, making temporally dynamic patterns possible (Wang et al., 2021). The regulation of phytohormone signaling pathways, light-signaling pathways, circadian clock regulation, and reactive oxygen species homeostasis at the transcriptional, epigenetic, and post-translational levels have been identified during environmental stress including heat, drought and cold stress (Ohama et al., 2017; Li et al., 2018; Ding et al., 2020).
In the presence of excess ROS, for example due to environmental stress, the cells are subjected to oxidative conditions that are detrimental to them (Wang et al., 2008; Kuge et al., 2010). H2O2, as ROS, is an important indicator of the generation of ROS (Bhattacharjee, 2012; De la Garma et al., 2015; He et al., 2018). Thus, the maintenance of steady state ROS by scavenging routes is necessary in order to prevent oxidative damage due to adverse environmental stress (Mittler et al., 2004; Miller et al., 2010; Jiang and Jie, 2002). The AsA-GSH cycle and Superoxide Dismutase (SOD) are both essential for scavenging ROS (Shi et al., 2013; Hernández et al., 2015). Superoxide dismutase (SOD) is the primary defense mechanism against ROS in plants, and it converts superoxide to oxygen (O2) and H2O2 at the molecular level (Huang et al., 2016; He et al., 2018; Wang et al., 2021). In the AsA-GSH cycle, APX uses AsA to reduce H2O2 to H2O (Hernández et al., 2015; Xing et al., 2018). In previous studies, APX is present in many organelles as well as in the cytosol. It has a unique ability to adapt to stress in various environments. (He et al., 2018; Xing et al., 2018). For example, it has been shown that overexpression of APX2 gene increases tolerance to exogenous hydrogen peroxide and assists in ROS detoxification under both stress and normal conditions in A. thaliana (Mittler et al., 2006; Rossel et al., 2007).
bHLH superfamily, a TF family with a large number of members, is prevalent throughout eukaryotes (Pires and Dolan, 2010; Sánchez-Pérez et al., 2019; Guo et al., 2021). In general, bHLH is composed of about 60 amino acids and has two functional areas: a base area with 13 to 17 predominantly base amino acids for DNA binding, and an HLH area capable of forming a homodimer or heterodimer with one or more partners. (Tian et al., 2019; Guo et al., 2021). bHLH TFs are important regulators in controlling responses to environmental stresses, development processes (Sun et al., 2020), and the biosynthesis of secondary metabolites (Groszmann et al., 2010; Zhao et al., 2020) by targeting dehydration and cold responsive genes (Guo et al., 2021). In Arabidopsis, AtAIB (ABA-inducible bHLH-type transcription factor), which enhance drought tolerance, control stomatal closure by modulating stomatal movement associated with H2O2 signalling (Cui et al., 2015; Li et al., 2017). In wheat, CBF (ICE1, bHLH116), a MYC-type bHLH TFs, was upregulated by cold stress and the knock-down bHLH116 seedling displayed reduced cold stress tolerance accompanied with increased ROS levels and reduced antioxidant enzyme activities (Guo et al., 2021). Extensive studies uncovered several downstream genes of bHLH. For example, bHLH122, can directly bind to the promoters of CYP707A3 gene, repressing its expression and increasing the APX content. AtbHLH68, AtbHLH112 and AtbHLH122, have been reported to control abiotic stress responses by regulating the APX and ABA signaling pathway genes in A. thaliana. These results indicate that bHLH genes play important roles in plant abiotic stress tolerance through crossing with phytohormone ABA and ROS scavenging pathway. However, the functional importance of bHLH gene in controlling plant response to multiple abiotic stresses remains to be investigated and the downstream genes extending the bHLH pathway remain unclear.
The Chinese bayberry (Myrica rubra Sieb. and Zucc.), which is widespread in tropical and subtropics, is one of the most important sub-tropical fruit crops. It is also a good candidate for studying fruit quality (Ren et al., 2019; Ren et al., 2021). M. rubra species are sensitive to drought and cold stress, which will directly decrease the yield and quality of M. rubra (Larcheveque et al., 2011; Windt et al., 2011). Moreover, the whole genome of M. rubra will be helpful to study the function genome and improve its genetics. (Ren et al., 2019). In this study, we performed a time-course transcriptome investigation in response to drought and cold stress, in an effort to identify the potential multifunctional TFs involved abiotic stress in M. rubra.
The 1-year-old M. rubra seedlings were grown in a greenhouse at Lanxi, Zhejiang Province, China (29.22 N, 119.45 E) (15.0 h light, 22–25°C, 75% humidity). We watered the seedlings once a day to maintain the optimum level of moisture in the field. In order to carry out the cold-treatment, the plants in the treatment group were transferred to the growing room (Sanyo) where they grew under the same conditions as those of the control (He et al., 2018). Three biological replicates were used for each group of seedlings, with each group exposed to 4°C for 0, 30 min, 1, 3, 6, 9, 12, 24, 36, or 48 h in growth chambers (Sanyo). Treatment groups were moved to Sanyo growth chambers with the same growth conditions as those in the control group when performing drought treatments. The seedlings were also divided into ten groups with three biological replicates each, and seedlings from each group were treated with 8% polyethylene glycol (PEG) for either 0, 30 min, 1, 3, 6, 9, 12, 24, 36 or 48 h in growth chambers (Sanyo) at 23°C. Each of the three to five fully expanded leaves of the seedling were measured.
The pigment content and enzyme activity of M. rubra leaves were determined immediately after freezing in liquid nitrogen at the same time. In few words, 0.1 g of the leaf sample was frozen in liquid nitrogen, homogenized in cold 0.01M phosphate buffer (1.5 mL, pH 7.2),and centrifuged at 14,000 grams for 10 minutes at 4°C for 10 minutes, and then Plant superoxide dismutase (SOD) Assay Kit (Nanjing Jiancheng Bioengineering Institute, Jiangsu Province, China) was used to measure the activity of SOD. First, 1.5 mL reaction buffer (6.5 × 10-6 M riboflavin, 0.013 M met, 6.3 × 10-6 M NBT, 1 × 10-4 M Ethylene Diamine Tetraacetic Acid (EDTA), 0.05 M phosphate buffer, pH=7.8) was added to the supernatant followed by incubation at 25°C for 30 min, and absorbance at 560 nm was measured with a spectrophotometer. The activity of malondialdehyde (MDA) was measured in accordance with the manufacturer’s specification using Catalase Assay Kit (Nanjing Jiancheng Bioengineering Institute). Using a plastic pestle and an ice-cold 0.01 M phosphate buffer (pH 7.2) with 1.13 mg dithiothreitol, the leaf specimens were crushed in a micro-centrifuge. A centrifuge was used for 10 minutes at 4°C, weighing 14,000 g. We assayed the supernatant for MDA activity by measuring the linear rate of decrease in absorbance at 240 nm with a spectrophotometer. For APX and peroxidase (POD) activity, 0.1 g samples of leaves were homogenized in 1.5 mL ice-cold 0.01 M phosphate buffer (pH 7.2) for 30 min and centrifuged at 14,000 g for 10 min at 4°C. A Plant APX and POD Assay Kit (Nanjing Jiancheng Bioengineering Institute) were used to measure APX and POD activity in the supernatant. The supernatant was added to a mixture of 0.5 mL 0.1 M phosphate buffer, 0.5 mL 0.1 M guaiacol buffer and incubated at 30°C for 8 min. The absorbance of the sample at 470 nm was measured with an optical spectrophotometer.
In the case of drought and cold-stress, the 3rd to 5th leaves of M. rubra were picked up, then frozen in liquid nitrogen, and kept at -80 degrees Celsius until application. According to the manufacturer’s guidelines, RNeasy Kit (Qiagen) was used to extract the total RNA. NanoDrop ND-2000 (A260/A280 1.9-2.1) and Agilent 2100 bioanalyzer (28S/18S 1.8-2.0) were used for the determination of RNA quality. A strand-specific RNA-seq library was constructed on an Illumina HiSeq 4000 platform according to the manufacturer’s instructions and index codes. The Beijing Novogene Technologies performed the construction of the libraries and the paired-end sequencing. Following quality control and removal of adapter-and poly(N)-containing reads, clean reads acquired after mapping on the reference genome of M. rubra (http://www.bayberrybase.cn/) were analyzed (http://www.bayberrybase.cn/) as previously described (Ren et al., 2019) with TopHat (v. 2.0.0) with default parameters (Trapnell et al., 2009). Transcript levels were normalized based on FPKM with Cufflinks (v. 2.1.1) with default options (Trapnell et al., 2012). It was considered significant that genes with P-value<0.05 (adjusted for the false discovery rate, Q-value< 0.05) and >2-fold change were differentially expressed.
A GO analysis was performed on DEGs and a GO annotation was obtained from M. rubra (http://www.bayberrybase.cn/) (Ren et al., 2019). The result of the GO enrichment analysis along with the P-value modified using the Benjamini and Hochberg (1995) FDR method were input into OmicShare platform (https://www.omicshare.com/tools), and created a visual tree map of the outcome of the GO analysis. Significant enrichment of GO terms with corrected P value < 0.05 and Q value < 0.05 was observed.
The tissue specificity score was calculated as previously described (Liao and Zhang, 2006). The tissue specificity score was generated as follows in order to further quantify the tissue specificity of gene expression: n represents the sum of tissues, aij is the mean expression of the gene i in tissue j, and the tissue specificity of the gene i is defined as:
Using PlantPAN v.2.0, it was determined that cis-regulatory elements were found in the 2 kb promoter region of the candidate genes (Chow et al., 2016; Xu et al., 2022). It was predicted that these motifs are TF target sites based on 80% confidence values. Then, a backward elimination random forest (BWERF) algorithm is employed to construct ML-hGRNs, which uses the genes and TFs encoding transport, photosynthesis and oxidoreductases (Deng et al., 2017; Xu et al., 2022).
ExPASy (http://web.expasy.org/computepi/) was used for the analysis of the isoelectric point (pI) and the molecular weight of the bHLH protein (Kumar et al., 2018). In order to analyze genetic variation and phylogenetic relationships, a plurality of sequence alignment of bHLH protein sequences was performed in MEGA 7 (Kumar et al., 2018). To deal with the gaps and lack of data, we chose a partial deletion with an 80% coverage limit. Therefore, Jones-Taylor-Thornton (JTT) + (G) + (F) was chosen as the optimum amino acid replacement model. The Maximum Likelihood (ML) method in MEGA 7 was utilized to construct a phylogenetic tree of protein sequences with 1000 bootstrapping replicas (Kumar et al., 2022). For all positions, 90 per cent of the site coverage was removed; in other words, no more than 10 per cent of the alignment space, no data, and no clear basis were permitted. A phylogenetic tree was visualized using Figtree software (http://tree.bio.ed.ac.uk/software/figtree/).
Generic transformation, cloning and expression analysis were carried out on A. thaliana seedlings at long time (16 hours/8 hours darkness) to produce overexpression and RNAi lines (Quan et al., 2021; Zhang et al., 2022). With the help of gene-specific primers, we cloned the full-length coding region of MrbHLH10 gene from a M. rubra clone template cDNA. During the process of cloning the full-length gene, the coding region of MrbHLH10 gene was cloned into pDONR222 vector. Subsequently, LR reactions were employed to re-sequence the encoding area and target vector pGWB405 in accordance with Nakagawa et al. (2007), verified by sequencing. Two kinds of vector were introduced to Agrobacterium GV3101 via Agrobacterium-mediated transformation together with gene silencing inhibitor P19, and then transformed into Nicotiana benthamiana based on previous study (Zhang et al., 2022). The Agrobacterium-mediated floral dipping approach was used to create transgenic A. thaliana seedlings (Wang et al., 2022). Transgenic plants were confirmed by PCR analysis using vector- and gene-specific primers in Table S1.
On a plate containing 1% sucrose and ½×MS medium (0.1 mM MgSO4, 0.1 mM CaCl2, 0.6 mM NaCl, and 0.3 mM ZnSO4), seeds of WT and transgenic A. thaliana were grown. The germination count was calculated up to the 5 days (d) after stratification (nearly emerged radicle). Based on the prior research, we obtained the germination rate (GR) by the number of seeds on each side of WT A. thaliana and transgenic plants (Quan et al., 2020; Zhang et al., 2022). Leaf physiological traits were measured in mature leaves of WT and transgenic A. thaliana seedlings at 20 days after germination (DAG). Seedling length of A. thaliana was calculated by ImageJ software (https://imagej.en.softonic.com/) (Banugopan et al., 2012).
The statistical significance of treatment differences was assessed with either one way or twoway analysis of variance, with SPSS 17.0 (IBM, Chicago, IL, USA) and Excel 2013 (Microsoft Corp., Redmond, WA) on the basis of prior research (Xu et al., 2022). For the calculation of P values, the Students’t test (* P < 0.05, * * P < 0.01) was adopted. After normalization, all samples had a normal distribution with respect to variance homogeneity (Xu et al., 2022).
In order to determine the effects of drought and cold stress on physiological activity, we first examined the activity of SOD, POD, MDA, APX, CAT and H2O2 under drought and cold stress. As expected, the activity of SOD, POD, MDA, APX, CAT and H2O2 showed an overall increase during the drought and cold treatment (Figures 1A–H). For example, under drought stress, the activities of SOD, MDA, APX and CAT and H2O2 increased significantly from 6 to 36 h (Figures 1B–D); the activities of SOD, MDA and APX peaked at 36 h, 36 h and 12 h, respectively (Figures 1B, C). By contrast, the activities of SOD and APX peaked at 12 h after cold treatment (Figures 1B, D). Interestingly, the activities of APX and MDA were positively correlated with each other (P>0.05, R>0.6) under the two stresses. Totally, the anti-oxidant enzyme activities were strongly induced under drought and cold stress.
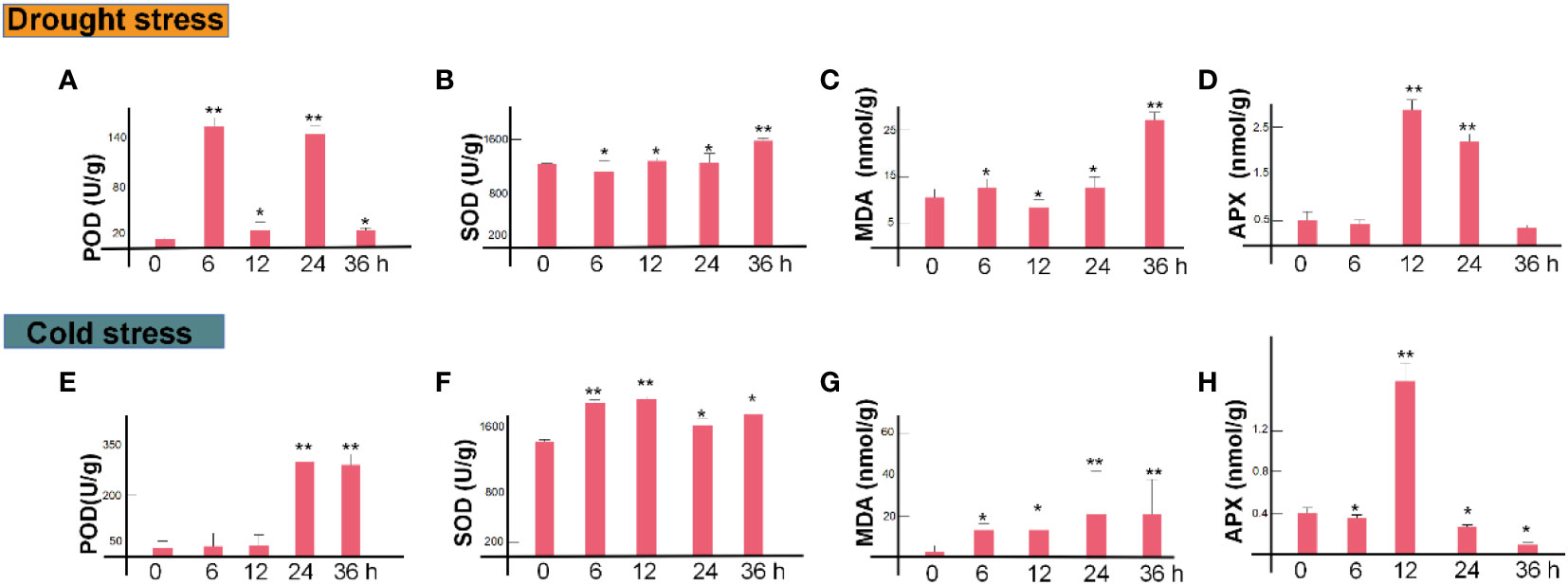
Figure 1 Temporal dynamics of M. rubra physiological characteristics during drought and cold treatment. Peroxidase (POD) activity (A), superoxide dismutase (SOD) activity (B), MDA (C) and APX content (D), in M. rubra under drought stress. Peroxidase (POD) activity (E), superoxide dismutase (SOD) activity (F), MDA content (G) and APX content (H), in M. rubra under cold stress. (*P < 0.05, **P < 0.01, Student’s t-test).
To identify the stress-responsive genes under both stresses, we sequenced the total RNAs of the leaf tissues across all the time points. A total of 7,583 and 8,840 differentially expressed genes (DEGs) were obtained under drought stress and cold stress, respectively (Fold Change>2 and FDR < 0.01) (Figure 2A; Table S1). To gain insight into the transcriptome dynamics under the two stresses, we performed principal component analysis (PCA; Figures 2B, C). As a result, the transcriptome data can be generally divided into four and two clusters under drought and cold stress, respectively (Figures 2B, C). Consistent with this, a previous study reported that abiotic stress response in higher plants occurs in different phases (Wu et al., 2021). Notably, the cold-responsive transcription profile was clearly divided into two phases, and the majority of cold-responsive DEGs were induced in the second phase timepoint from 24 to 48 h (Figure 2D). By contrast, there were only <1500 DEGs in the first phase (0 to 24 h. The fewer DEGs within the early response might be reminiscent of M. rubra species that lived in freezing environment (under 4°C) with consistent cold pressure (Ren et al., 2019).
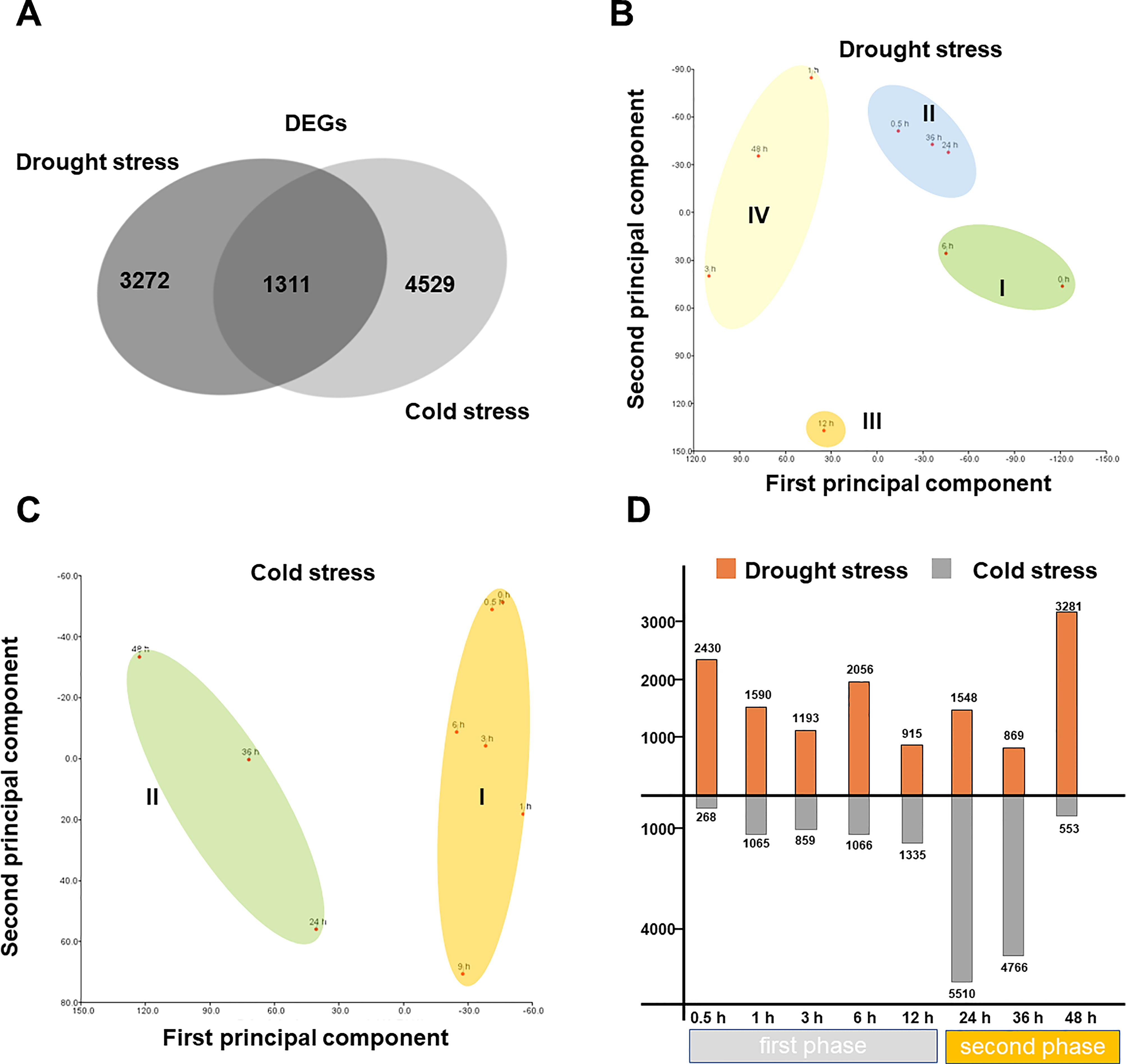
Figure 2 Temporal dynamics of M. rubra transcriptome during drought and cold treatment. (A) Venn diagrams of DEGs overlapping between drought stress and cold stress. DEG indicates dfferentially expressed gene. (B) PCA of the transcriptomes of the 8 time point samples under drought stress and (C) cold stress. (D) Number of drought-responsive (upper bars) and cold-responsive (lower bars) genes at each time point compared to the control group (0 h).
The GO analysis of the drought and cold-response genes showed that there were 251 and 227 significant enrichment terms (P<0.001). The upregulated DEGs under drought stress were enriched for terms including “response to hormone-mediated signaling pathway,” “oxidoreductase activity,” “photosynthesis,” “transcription factor activity,” “abscisic acid biosynthetic process,” and “ABA signal transduction pathway” (Table S3). By contrast, upregulated cold-responsive DEGs were enriched for terms of photosynthesis, response to “oxidoreductase activity,” “photosynthesis,” “external biotic stimulus,” “transcription factor activity,” “transcription regulation,” “calcium-binding,” “DNA binding,” and “serine/threonine kinase activity” (Table S3). These indicates that the patterns of the functional shifts were consistent with the physiological changes under the two stresses.
To examine the shared and unique transcriptomic network under drought and cold stresses, DEGs were clustered into 20 dynamic groups by using K-means clustering algorithm (Wu et al, 2021). The ten largest dynamic groups in response to drought and cold stress contained most of the stress-responsive DEGs—83.4% (3,142 drought-responsive genes) and 88.7% (4,108 cold-responsive genes) respectively (Figure 3A). The genes in the D2, D3, C3, C9, and C10 groups were continuously down- or up-regulated under both stresses (Figure 3A). On the other hand, the D1 and C2 groups were observed to have transient changes in gene expression at earlier timepoints, presumably in response to early stress. Thus, the groups with similar expression dynamics probably kept similar biological processes regardless of the stress they were exposed to. Under the two types of stress, the D4 and C4 groups with transient gene expression peaks at 36 hours showed a higher level of photosynthetic gene accumulation (Figures 3A, B). Likewise, the D3 and C7 groups that showed an increasing tendency to continue showed oxidoreductase activity, as well as the genes associated with abiotic stress reaction (Figures 3A, B). These GO terms are consistent with the early and late physiological changes, which were observed in plant cells under drought and cold stress (Muns et al., 2007; Guo et al., 2021). This similarity in biological functions indicates that abiotic stress-response genes have maintained conserved expression dynamics.
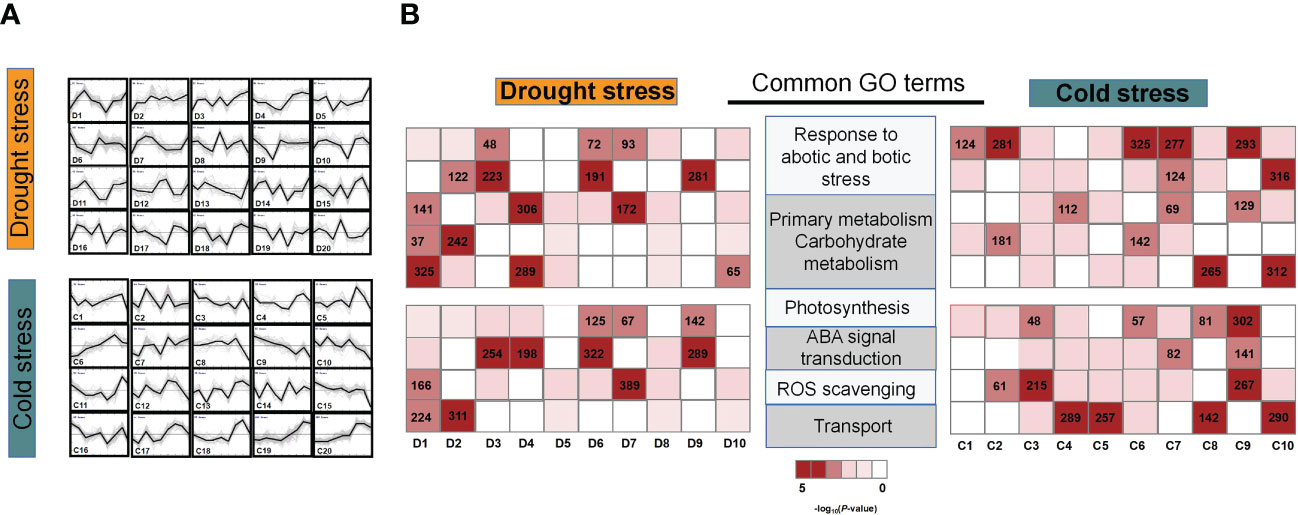
Figure 3 Temporal dynamics of M. rubra transcriptome during drought and cold treatment. (A) Expression dynamics of the 20 largest gene clusters over time under drought (D1–D20) and cold stress (C1–C20). (B) GO terms enriched in DEGs, with red indicating more significant enrichment (–log10(P-value)). The heatmaps show enriched GO terms detected in temporally dynamic groups of both drought and cold stress from 1 to 48 h.
Our objective was to gain insight into the regulatory mechanism underpinning drought and cold stress by using hierarchical clustering (Figures S1, S2). Under drought and cold stress response, the hierarchical clustering network consist of differentially expressed genes with 6 cluster (DI-VI) and 4 cluster (CI-IV) (Figures S1, S2), respectively. Cluster enrichment analyses revealed that, despite the presence of multiple metabolic pathways, some patterns could be discerned in a single cluster. For example, in drought stress response, genes involved in “membrane,” “abiotic stress stimulus,” “oxidoreductase activity,” “response to abiotic stress,” “response to stress,” “ADP metabolic process,” and “oxidation-reduction process,” were mainly enriched in cluster D-III, which exhibited an increasing trend from 12 to 36 h, indicating that membrane biosynthesis, ADP metabolic process and oxidoreductase activity pathways are enhanced in response to drought stress (Figure S1). In cold stress response, genes involved in “oxidoreductase activity,” “abiotic stress stimulus,” “antioxidant activity,” “photosynthesis,” and “plant-pathogen interactions” were enriched in cluster C-IV and exhibited an increasing trend from 0 to 12 h, suggesting active ROS scavenging. DEGs involved in “response to abiotic stimulus” and “organic substance biosynthetic process,” and “response to cold,” were enriched in cluster C-II and showed continuously decreasing trends to 48 h (Figure S2). As a result of drought and cold stress, M. rubra exhibits early and late physiological changes that are consistent with these two GO terms.
Expression dynamics are conserved, indicating that some regulators are common to both early and late responses in two abiotic stresses (Wu et al., 2021). Gene expression profiles under abiotic stress were sample-specific for TFs, which dominated the network rather than other genes (Yin et al., 2019; Wu et al., 2021). In our current research, a total of 1,923 and 1,731 stress-responsive TFs were identified under drought and cold stress, respectively, 26.6% (548) of which are stress-conserved TFs (Figure 4A). TFs had more sample-specific expression profiles under different abiotic stresses (Wu et al., 2021), and the number of stress-responsive TFs increased significantly across the samples in our study (Figure 4B). Specially, TF families of bHLH, MYB, ethylene responsive factor (ERF), MYB, WRKY, and NAM, ATAF, and CUC (NAC) showed one of the highest enrichments under drought and cold stress (Figure 4B). The bHLHs proteins, for instance, form a large family of plant-specific TFs that play an important role in plant defense and biotic and abiotic stresses (Yoda et al., 2002; Journot-Catalino et al., 2006; Zheng et al., 2006; Liu et al., 2007). Interestingly, the transcription of MrbHLH18, MrbHLH31, MrbHLH10 and MrbHLH75 increased significantly under both drought and cold stress, which are grouped in cluster D3 and C7 related to response to abiotic stimulus and oxidoreductase activity in both abiotic stresses.
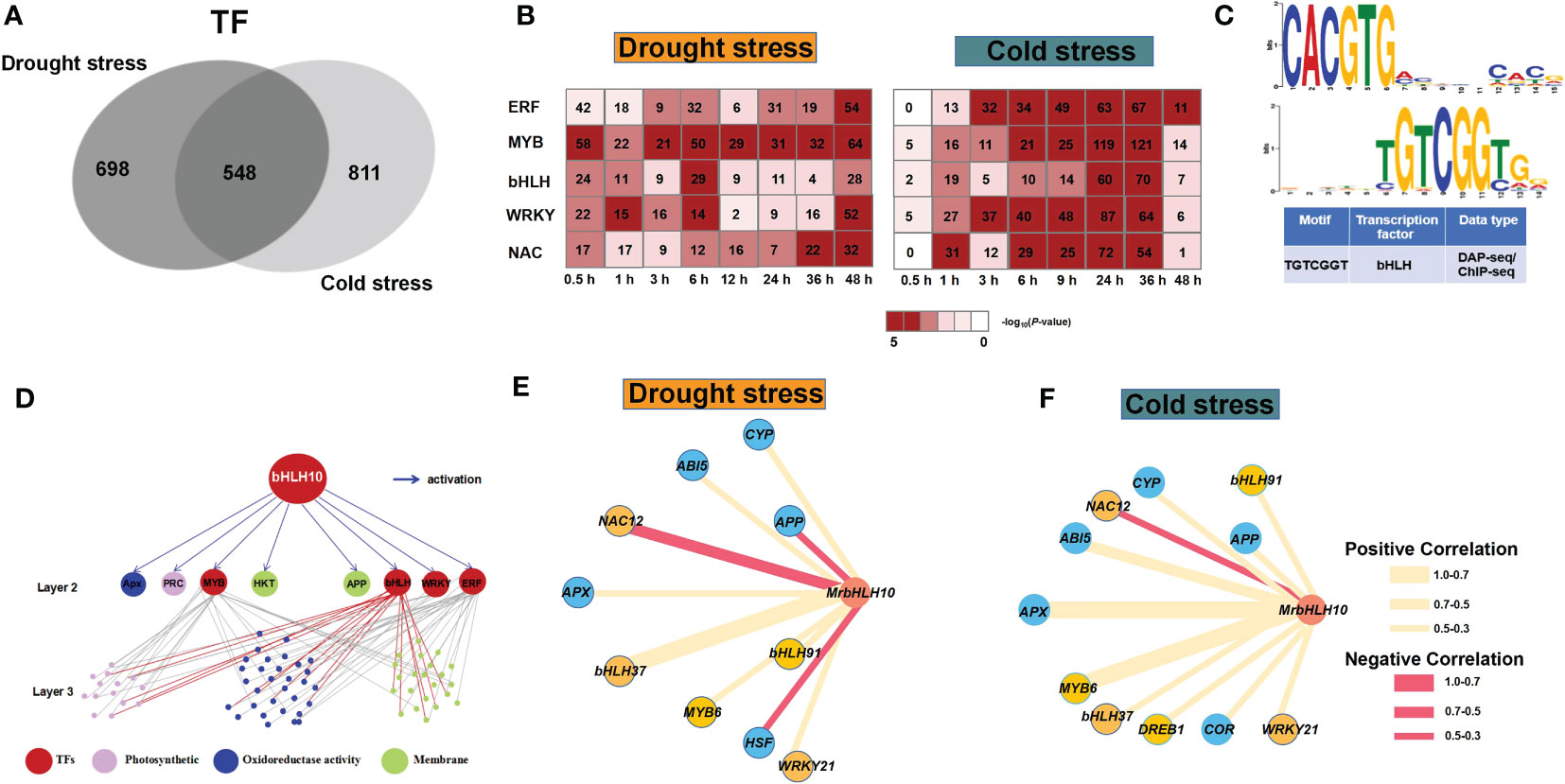
Figure 4 Gene regulatory networks of drought and cold-responsive transcriptional modulators. (A) Venn diagrams of TFs overlapping between drought stress and cold stress. (B) Enrichment (–log10 (P-value)) of TF families in timepoint (0–48 h) under drought stress (left) and cold stress (right). The number represents the number of differentially expressed TF genes. The heatmap presents TF families enriched in at least one of 20 groups. (C) Bioinformatic analysis of bHLH Binding Site (TFBS) Motifs based on PlantTFDB database (http://planttfdb.gao-lab.org/). (D) Gene regulatory network of photosynthesis, oxidoreductase activity and membrane regulation under drought stress and cold stress. (E) Coexpression network of genes that are differentially expressed under drought stress and cold stress (F) treatment in M. rubra at different timepoints.
To further examine the potential regulatory relationships, we constructed gene regulatory network (GRN) that interlinks TFs with their potential target genes (PTGs) based on the expression data and examined the presence of MrbHLH10 potential TF binding site (TFBS) to verify the GRN (Figure 4C). As a result, totally 227 TF-PTG pairs were identified, and we observed that MYBs, WRKYs, bHLHs and ERFs which targeted large numbers of genes (twenty or more target genes from each TF as indicated by solid or arrows line, Figure 4D). Notably, MrbHLH10 regulated 39 downstream ROS signaling pathway genes, and clear functional associations were observed in both stresses—MrbHLH10 were connected to APX (GENE_006840) gene (Figures 4E, F; R > 0.6; P <0.05), while multiple previously stress-responsive gene, such as WRKY21 (Zhang et al., 2017) and MYB6 (Li et al., 2020) were also associated with MrbHLH10 (Figures 4E), indicating a role as the master regulators under the cold and drought stress.
The bHLH superfamily, one of the largest TF families, is widespread in eukaryotes (Pires & Dolan, 2010). Among the amino acid sequences of the MrbHLH proteins, there is a high conservation domain comprising 60 amino acids with two distinct functional areas (Figure 5A). The base region, which is located at the N-terminal end of the domain, participates in DNA binding, is composed of 15 amino acids with many base residues (Tian et al., 2019). The MrbHLH10 coding sequencing (CDS) is 1,483 bp long, encoding 493 amino acids and having a molecular mass of ~54 kD and an isoelectric point of 5.42 (Table S6). The multiple amino acid sequence alignment and a phylogenetic tree indicated that the bHLH protein sequences from multiple species consist of the same conserved domains (Guo et al., 2021) (Figures 5B, C). Therefore, MrbHLH10 and ATbHLH10 might have the same biological function, which has been demonstrated to respond to various abiotic stresses (Guo et al., 2021).
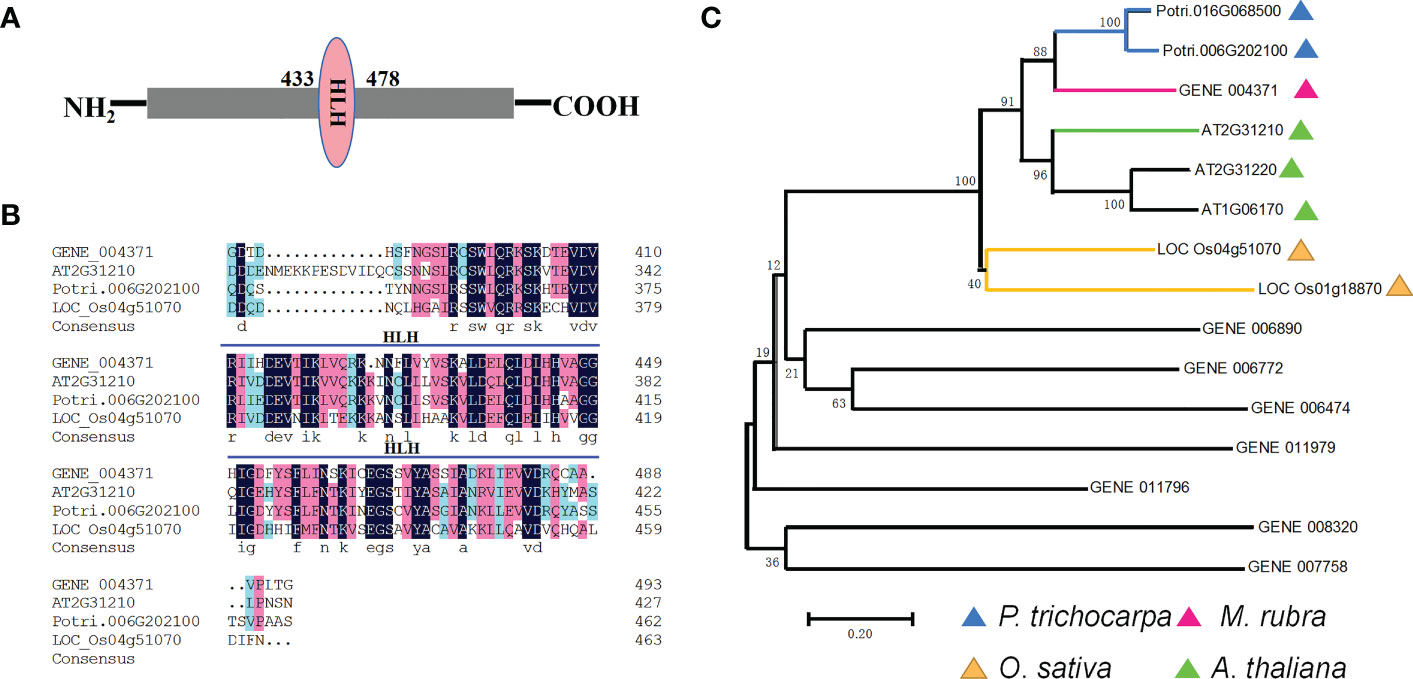
Figure 5 Bioinformatics analysis of bHLH gene (A) Domain architecture analysis of MrbHLH protein. (B) Multiple amino acid sequence alignment of bHLH10 from M. rubra, O. sativa, P. trichocarpa and A. thaliana. (C) Phylogenetic analysis of bHLH homologs from M. rubra, O. sativa, P. trichocarpa and A. thaliana. Scale bars, 0.2.
In order to probe into the potential biology function of MrbHLH10 gene in M. rubra, we made a deep analysis on the expression mode of bHLH. Notably, a total of 241 MrbHLH members are detected in M. rubra, which show four major expression patterns (Figure S3). MrbHLH10 showed different expression patterns in 12 of the selected organs or tissues (Figure S3), for instance, the expression profiles of bHLH10 gene in the branch, xylem and mature leaf show similar trends, and expression patterns in the young leaf, phloem and petiole grouped in one cluster (Figure S3), indicating that bHLH10 gene regulate plant growth and stress response in M. rubra.
To investigate the potential function of bHLH10 in the response to enviromental stresses, we generated transgenic A. thaliana seedling overexpressing (OE) and RNAi bHLH10 via Agrobacterium tumefaciens-mediated transformation (Figure 6A) (Zhang et al., 2022). Compared to the WT and RNAi line, the OE line had a significantly longer root length and heavier fresh weight increased by 22.9% and 62.8% respectively under normal condition (P<0.05; Figure S3), indicating that MrbHLH10 gene is implicated in plant growth (Figures 6A–C).
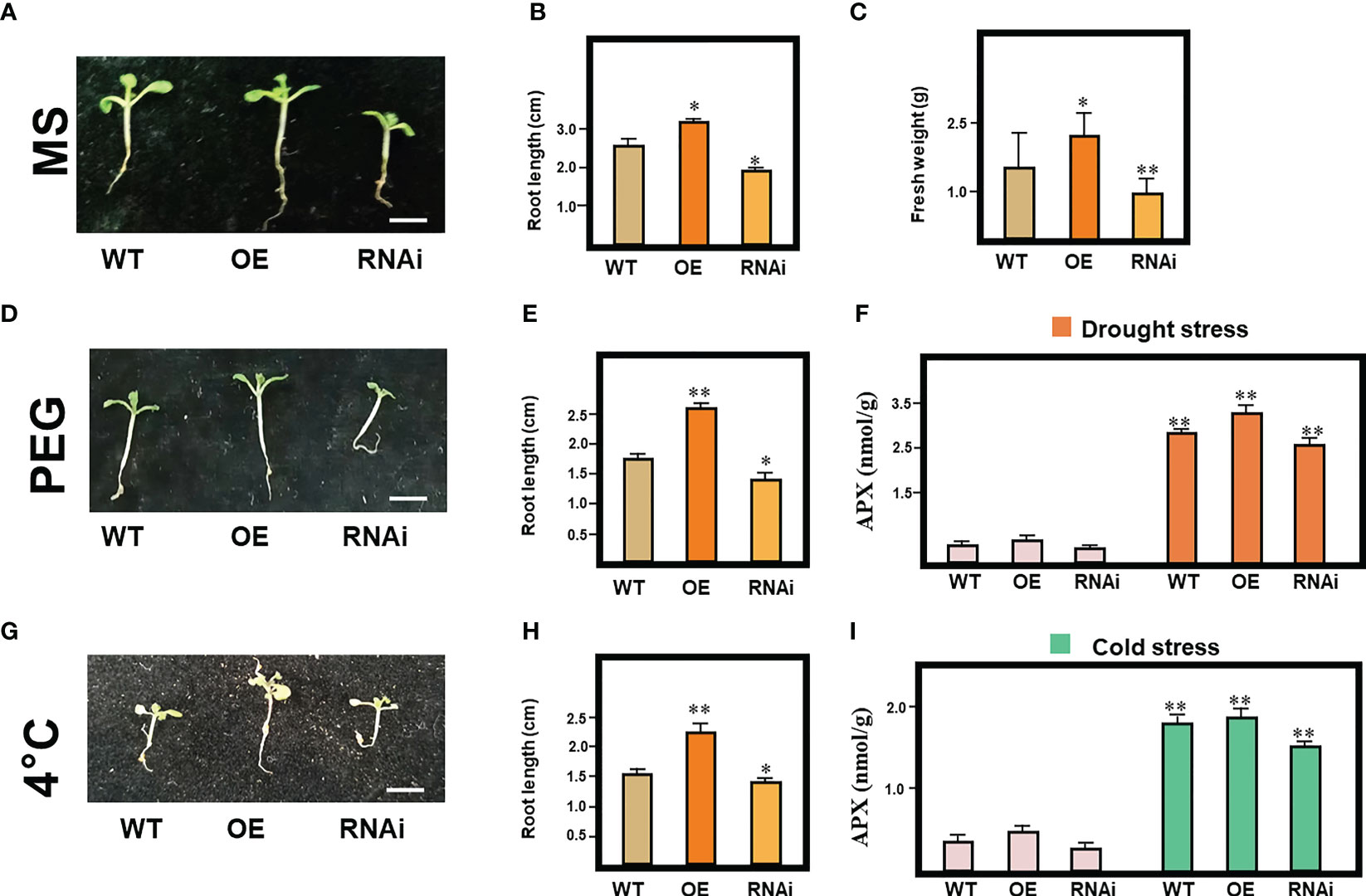
Figure 6 MrbHLH10 expression in response to drought and cold stress. (A) Time course of MrbHLH10 expression in A. thaliana exposed to drought and cold stress on standard ½ MS medium. (B) Root length and (C) fresh weight of wild-type (WT), MrbHLH10-OE and MrbHLH10-RNAi lines grown on standard ½ MS medium for 1 weeks. (D) Time course of MrbHLH10 expression in A. thaliana exposed to drought stress on standard ½ MS medium with 8% PEG for 1 weeks. (E) Root length of wild-type (WT), MrbHLH10-OE and MrbHLH10-RNAi lines grown on standard ½ MS medium with 8% PEG for 1 weeks. (F) APX content in leaves of wild-type (WT), MrbHLH10-OE and MrbHLH10-RNAi lines grown on standard ½ MS medium with 8% PEG for 1 weeks. (G) Time course of MrbHLH10 expression in A. thaliana exposed to drought stress on standard ½ MS medium with 4 °C for 1 weeks. (H) Root length of wild-type (WT), MrbHLH10-OE and MrbHLH10-RNAi lines grown on standard ½ MS medium, with 4 °C for 1 weeks. (I) APX content of wild-type (WT), MrbHLH10-OE and MrbHLH10-RNAi lines grown on standard ½ MS medium, with 4 °C for 1 weeks. Data are presented as means ± SD (n = 3). (*P < 0.05, **P < 0.01, Student’s t-test).
Compared with WT and RNAi lines, the primary root length of OE was obviously increased (P<0.05; Figure 6D). After 7 days of treatment at 4 °C, the WT and RNAi lines showed a significant increase in the primary root length of OE line compared with WT and RNAi (P<0.05; Figure 6G), indicating that bHLH10 can positively increase the growth of plants under drought and cold stress. Ascorbate peroxidase (APX) maintain cellular ROS homeostasis and is major ROS scavengers (Xing et al., 2018; Wang et al., 2021). APX activity were measured in OE, RNAi and WT line. Total APX activity was strongly induced, and OE line showed distinctly APX activity higher than WT plants and RNAi line respectively under drought and cold stress (P<0.05; Figures 6F, I). Thus, bHLH10 could be used as an indirect way to improve drought and cold-tolerance by keeping ROS in a stable state.
The improvement of the plant’s ability to withstand many kinds of stress is one of the fundamental objectives of breeding. It is of great importance to enhance the comprehensive resistance of plants by means of core functional genes in molecular breeding. (Hu et al., 2017; Xu et al., 2023). In this study, we constructed a high-temporal-resolution dynamic transcriptome landscape of drought and cold stress responses using nine time points. The dynamic transcriptome profiles were clealy grouped into four and two stages within the drought and cold stress response respectively, indicating the early-responsive and late-responsive phase (Hickman, 2017; Wu et al., 2021). We found there were 7,453 and 9,614 genes mainly expressed at the stages of early- and late-responsive phase respectively. Biological processes in the K-Means groups with similar expression dynamics were probably maintained across the two types of abiotic stresses. It is also possible that there are common regulators of early and late responses in both stressors due to the conserved expression dynamics. Specifically, we have identified 2,311 stress-conserved genes, among them 548 TFs, which will undoubtedly become the goal of functional genomics in the future. Because of this vast array of genes, we will be able to carry out further functional research, which will significantly improve our knowledge of the genetic mechanisms that govern the response to environmental stresses.
The TFs of bHLH, MYB, NAC, WRKY and ERF are known to regulate stress-responsive genes (He et al., 2018; Wu et al., 2021). Some TFBSs, which were enriched in the promoter, were related to the up-regulation of TF genes. The bHLH TF gene and some bHLH binding sites were significantly enriched (Guo et al., 2019; Yu et al., 2021). Therefore, transcriptomic regulation during salt stress in M. rubra is mediated by dynamic regulation of bHLHs and other TFs. In A. thaliana, these TFs are also associated with a cold stress response (Xu et al., 2014; Guo et al., 2021; Yu et al., 2021). In addition, bHLH genes function in leaf formation and growth as well as heat and drought stress responses (Khadiza et al., 2017; Zhao et al., 2021), indicating that in the two main dicotyledons and probably in other plants, bHLH orthologues are thought to confer a stress-tolerance mechanism.
To adapt to abiotic stress conditions including extreme temperatures (heat and freezing), plants regulate a series of genes, so as to form a GRN in previous study (Guo et al., 2021; Jia et al., 2022). In the absence of a central trans-regulator gene, a GRN could be deleted completely or network connectivity could be reorganized (Wu et al., 2021; Xu et al., 2022). However, there is still little investigation on the construction of GRNs in response to abiotic stresses combination (Reményi et al., 2004; Song et al., 2016; Vihervaara et al., 2018).
Systemic signaling in plants during abiotic stress combination (Zandalinas et al., 2020). In the bHLH10-mediated GRN, direct target genes including MYB6, WRKY41, TCP4, CYP1, STZ and APX1 were identified. In O. sativa, overexpression of OsbHLH148 gene increases the plant’s drought tolerance by regulating the JA pathway and the expression of OsJAZ protein (jasmonate ZIM domain) (Seo et al., 2011). With the expression of MfbHLH38 gene from resurrection plants (Myrothamnus flabellifolius), transgenic Arabidopsis has improved water retention and drought tolerance, as well as the increase of their oxidative stress tolerance and osmotic regulatory ability, which is associated with the ABA response and elevated ABA content. In our study, multiple target genes of bHLH10, such as APX1 and WRKY41 were known to function in ROS scavenging and ABA signal processes (Reményi et al., 2004; Song et al., 2016; Vihervaara et al., 2018). In addition, recent studies showed that AtSTZ, a downstream transcription repressor, enhances abiotic stress tolerance after growth delay in Arabidopsis (Park et al., 2015; Sakamoto, 2004), may be regulated by MAP kinases in Arabidopsis (Nguyen et al., 2016). CYP1 mediate the last steps of auxin biosynthesis, as well as root growth inhibition in response to stress (Rodríguez et al., 2010).
The bHLH TFs are also involved in the course of development and growth, including the germination of seeds and the development of root, epidermis, xylem, carpels, anthers, fruits and stomata (Groszmann et al., 2010; Guo et al., 2021). In our study, root length significantly increased 131%, 114% and 78% in the MrbHLH10-OE line compared to the WT under drought, cold and normal condition respectively. Currently, there is evidence that bHLHs directly regulate cytokinin synthesis genes or cytokinin degradation genes such as CKXs (Hezhong et al., 2008; Hozain et al., 2012). bHLH10 may directly regulate MrCKX by GRN analysis, enhancing root development and cell division in root of OE plants, in agreement with prior reports (Hezhong et al., 2008). In contrast to other stress-tolerant genes like MYB6, DREB2C, WRKY45, and AtSAP5 (Lim et al., 2007; Hozain et al., 2012), overexpression of bHLH10 gene enhances plant growth and environmental stresses tolerance.
Excess ROS, for example, caused by environmental stresses, such as heat, salt, cold, and drought stress, results in oxidative conditions that are detrimental to plant cells (Wang et al., 2008; He et al., 2018). In response to environmental stresses, plants accumulate cryoprotectant molecules such as soluble sugars, sugar alcohols, and low-molecular-weight nitrogenous compounds (proline and glycinebetaine; Zhuo et al., 2017; He et al., 2018; Xing et al., 2018), and activated antioxidant defence systems which include MDA, CAT, SOD, POD and APX (He et al., 2018; Wang et al., 2021; Xie et al., 2018; Zhuo et al., 2017). These antioxidative enzymes can suppress ROS accumulation in plant cells due to environmental stress (Wang et al., 2021; Wu et al., 2018). The cycle of AsA-GSH and SOD is of great significance to the scavenging of ROS (He et al., 2018; Shi et al., 2014). SOD acts as a first defence mechanism for ROS, it catalyzes the conversion of oxygen ions (O2-) into oxygen (O2) and hydrogen peroxide (H2O2) (Huang et al., 2016; He et al., 2018), in the AsA-GSH cycle, AsA reduces H2O2 to water (H2O) (Xing et al., 2018). Our findings indicate that bHLH10 may activate stress-responsive gene including APX1 to modulate ROS accumulation under environmental stresses by GRN analysis. Further physiological analyses support this hypothesis, activities of APX increased in transgenic plants under normal, cold and drought stress conditions in our study. In previous study, APX subtypes are present in a variety of organelles as well as in the cytosol, whose adjustment modes differ under environmenta stress conditions (He et al., 2018; Xing et al., 2018). Recent studies shown that APX1 is induced by heat, cold, drought and H2O2 stresses, and APX genes confer tolerance to various abiotic stresses when overexpressed in transgenic plants (Wang et al., 2016; He et al., 2018; Wang et al., 2020). Absence of cytosolic APX1 in Wassilewskija background results in a breakdown of the H2O2-scavenging system in Arabidopsis chloroplasts, causing an increase in H2O2 and protein oxidation (Davletova et al., 2005). A knockout of APX1 also significantly inhibits Arabidopsis growth and development, leading to increase sensitivity to oxidative stress, stunted growth and delayed flowering (Davletova et al., 2005; Miller et al., 2007; He et al., 2018). In Populus, activated PeAPX1 promotes cytosolic APX that scavenges ROS under cold and heat stress, and transgenic Populus overexpressing PtAPX1 showed increased cold tolerance and led to lower MDA and H2O2 levels in leaf and roots, resulting in higher plant height and root biomass under cold stress, phenocopying the MrbHLH10 OE plants in our study (He et al., 2018; Zhou et al., 2020; Guo et al., 2021).
In this study, we present a systematic review of ROS scavenging regulation first, as well as the complex GRNs that accompany it, and such networks have an essential role to play in understanding plant responses to environmental stresses. Together, the results show that MrbHLH10, a versatile TF, can increase environmental stresses by regulating the expression of MrAPX1, a direct downstream gene, which in turn keeps ROS stable (Figure 7). These results indicate that over-expression of bHLH10 can enhance the antioxidative function of transgenic plants, which might be helpful to prevent hyperosmolar and excess ROS due to environmental stresses.
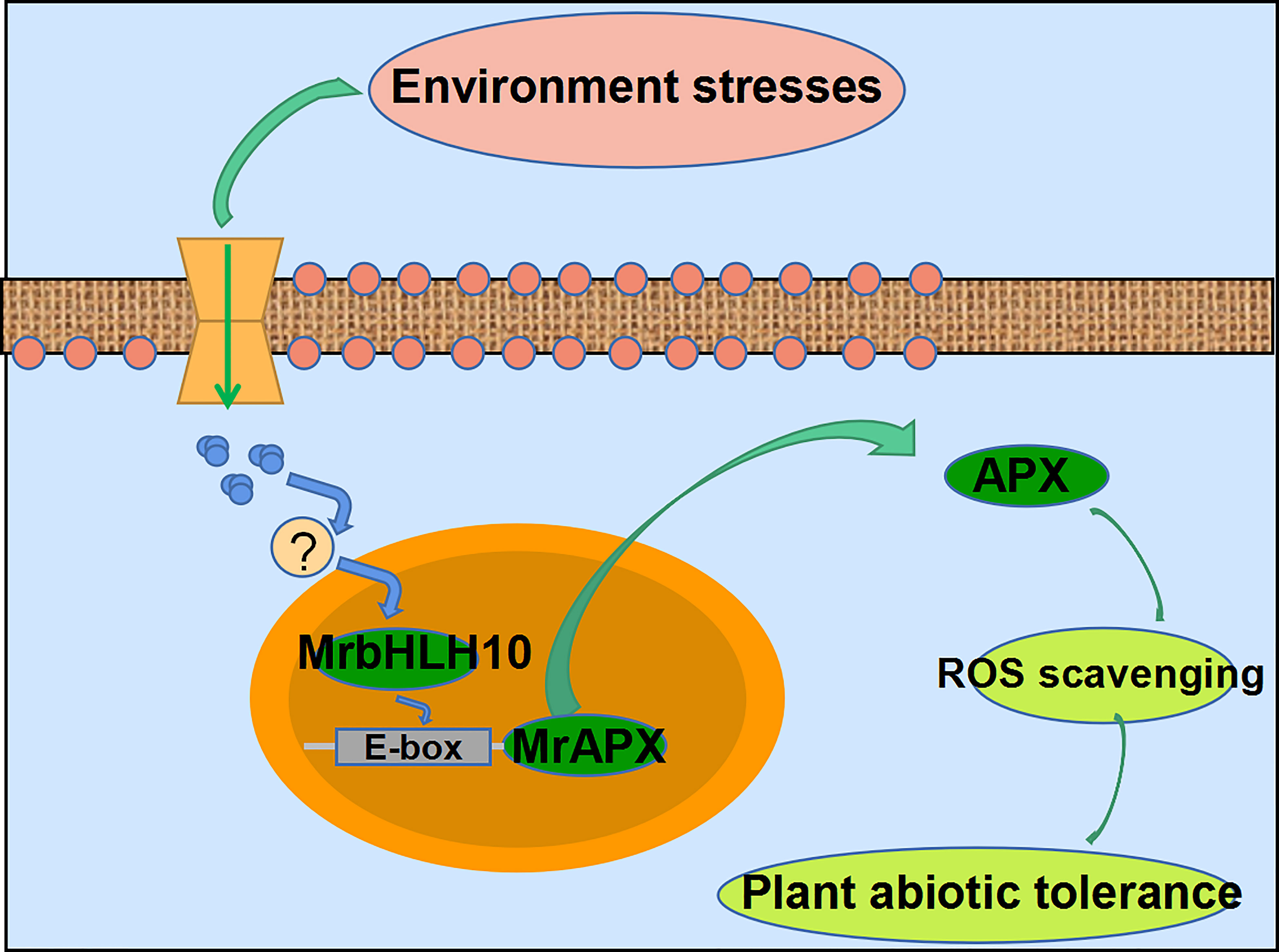
Figure 7 A potential working model for bHLH10 response to abiotic stresses in M. rubra. Under drought and cold stress, activating the environmental stresses signaling pathway and stimulating downstream stress signal transduction in plant cells, the upregulated bHLH10 acts upstream of APX and directly regulates its expression by binding to the E-box motif of its promoter. The activated APX then promotes cytosolic ascorbate peroxidase accumulation to scavenge ROS content under environmental stresses.
Raw data for RNA-seq are available at the BIGD Genome Sequence Archive (https://bigd.big.ac.cn) under accession number CRA006695
This study was completed within the laws of the People’s Republic of China. No specific permits were required for our field research. The study species is not included in the ‘List of Protected Plants in China.
HR and JX designed the conception and experiment; WX, HR, XQ, SZ and ZY performed the experiments; WX wrote the manuscript; JX and HR helped to analyze and assess the data; XQ and SZ provided the valuable suggestions on the manuscript; JX helped to revise the manuscript; HR and JX obtained the funding and were responsible for this manuscript; all authors read and approved the manuscript.
This study was supported by Breeding for Agricultural New Varieties in Zhejiang Province (2021C02066-2) and Key R&D Projects in Zhejiang Province (2019C02038, 2020C02001, and 2021C02009), State Key Laboratory for Managing Biotic and Chemical Threats to the Quality and Safety of Agro-products (2021DG700024-KF202306), the Key Laboratory of Horticultural Plant Genetic and Improvement of Jiangxi Province, the open project of the Key Laboratory of Horticultural Plant Genetic and Improvement of Jiangxi Province (2021KFJJ001), National Key R&D Program of China (No.2022YFD2201600; 2022YFD2200602), the Project of the National Natural Science Foundation of China (NOs., 32022057 and 31972954), Forestry and Grassland Science and Technology Innovation Youth Top Talent Project of China (No. 2020132607).
We thank Ms. Liju Yan, Mr. Qi Zhang, and Ms. Xiuzhu Guo for collecting samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1155504/full#supplementary-material
Benjamini, Y., Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. society: Ser. B (Methodological) 57 (1), 289–300.
Bhattacharjee, S. (2012). The language of reactive oxygen species signaling in plants. J. Bot. 2012, 1–22. doi: 10.1155/2012/985298
Chow, C. N., Zheng, H. Q., Wu, N. Y., Chien, C. H., Huang, H. D., Lee, T. Y., et al. (2016). PlantPAN 2.0: an update of plant promoter analysis navigator for reconstructing transcriptional regulatory networks in plants. Nucleic Acids Res. 44, D1154–D1160. doi: 10.1093/nar/gkv1035
Cui, L. G., Shan, J. X., Shi, M., Gao, J. P., Lin, H. X. (2015). DCA1 acts as a transcriptional co-activator of DST and contributes to drought and salt tolerance in rice. PloS Genet. 11, e1005617. doi: 10.1371/journal.pgen.1005617
Davletova, S., Rizhsky, L., Liang, H., Shengqiang, Z., Oliver, D. J., Coutu, J., et al. (2005). Cytosolic ascorbate peroxidase 1 is a central component of the reactive oxygen gene network of arabidopsis. Plant Cell 17 (1), 268–281.
De la Garma, J. G., Fernandez-Garcia, N., Bardisi, E., Pallol, B., Rubio-Asensio, J. S., Bru, R., et al. (2015). New insights into plant salt acclimation: the roles of vesicle trafficking and reactive oxygen species signalling in mitochondria and the endomembrane system. New Phytol. 205, 216–239. doi: 10.1111/nph.12997
Deng, W., Zhang, K., Busov, V., Wei, H. (2017). Recursive random forest algorithm for constructing multilayered hierarchical gene regulatory networks that govern biological pathways. PloS One 12, e0171532. doi: 10.1371/journal.pone.0171532
Ding, Y., Shi, Y., Yang, S. (2020). Molecular regulation of plant responses to environmental temperatures. Mol. Plant 13, 544–564. doi: 10.1016/j.molp.2020.02.004
Groszmann, M., Bylstra, Y., Lampugnani, E. R., Smyth, D. R. (2010). Regulation of tissue-specific expression of SPATULA, a bHLH gene involved in carpel development, seedling germination, and lateral organ growth in Arabidopsis. J. Exp. Bot. 61, 1495–1508. doi: 10.1093/jxb/erq015
Guo, J., Sun, B., He, H., Zhang, Y., Wang, B. (2021). Current understanding of bhlh transcription factors in plant abiotic stress tolerance. Int. J. Mol. Sci. 22 (9), 4921. doi: 10.3390/ijms22094921
Guo, J., Dong, X., Han, G., Wang, B. (2019). Salt-enhanced reproductive development of suaeda salsa l. coincided with ion transportergene upregulation in flflowers and increased pollen k+ content. Front. Plant Sci. 10, 333.
He, F., Wang, H. L., Li, H. G., Su, Y., Li, S., Yang, Y., et al. (2018). PeCHYR1, a ubiquitin eligase from Populus euphratica, enhances drought tolerance via ABA-induced stomatal closure by ROS production in Populus. Plant Biotechnol. J. 16, 1514–1528. doi: 10.1111/pbi.12893
Hernández, L. E., Sobrino-Plata, J., Montero-Palmero, M. B., Carrasco-Gil, S., Flores-Cáceres, M. L., Ortega-Villasante, C., et al. (2015). Contribution of glutathione to the control of cellular redox homeostasis under toxic metal and metalloid stress. J. Exp. Bot. 66, 2901–2911. doi: 10.1093/jxb/erv063
Hickman, R. (2017). Architecture and dynamics of the jasmonic acid gene regulatory network. Plant Cell 29, 2086–2105. doi: 10.1105/tpc.16.00958
Huang, S. B., Van Aken, O., Schwarzlander, M., Belt, K., Millar, A. H. (2016). The roles of mitochondrial reactive oxygen species in cellular signaling and stress response in plants. Plant Physiol. 171, 1551–1559. doi: 10.1104/pp.16.00166
Hezhong, J. (2008). Study on technique of extracting volatile oils of notopterygium incisum ting ex HT Chang by SFE-CO~ 2. J. Of Anhui Agric. Sci. 36 (5), 1740.
Hozain, M. D., Abdelmageed, H., Lee, J., Kang, M., Fokar, M., Allen, R. D., et al. (2012). Expression of AtSAP5 in cotton up-regulates putative stress-responsive genes and improves the tolerance to rapidly developing water deficit and moderate heat stress. J. Plant Physiol. 169 (13), 1261–1270.
Hu, Y., Jiang, Y., Han, X., et al. (2017). Jasmonate regulates leaf senescence and tolerance to cold stress: crosstalk with other phytohormones. J. Exp. Bot. 68, 1361–1369.
Jiang Jiye, L. (2002). The algorithm on Knowledge Reduction in incomplete information system. Int. J. Uncertainty Fuzziness Knowledge-Based Syst. 24 (1), 95–103.
Jia, Y. Q., Niu, Y. N., Zhao, H. M., Wang, Z. B. (2022). Hierarchical transcription factor and regulatory network for drought response in betula platyphylla. Hortic. Res. 9, 40. doi: 10.1093/hr/uhac040
Journot-Catalino, N., Somssich, I. E., Roby, D., Kroj, T. (2006). The transcription factors WRKY11 and WRKY17 act as negative regulators of basal resistance in arabidopsis thaliana. Plant Cell 18 (11), 3289–3302.
Khadija, A., Lahlou, Y., Chemlali, S., Kissa, J., Gharibi, A., Baite, M. (2017). Cone beam computed tomography study of intra-sinus calcifications. Glob J. Med. Res. 17 (1), 4–13.
Kesavaraju, B., Dickson, S. (2012). New technique to count mosquito adults: using imagej software to estimate number of mosquito adults in a trap. J. Am. Mosq. CONTR 28 (4), 330–333. doi: 10.2987/12-6254R.1
Kuge, Y., Takai, N., Ogawa, Y., Temma, T., Zhao, Y., Nishigori, K., et al. (2010). Imaging with radiolabelled anti-membrane type 1 matrix metalloproteinase (mt1-mmp) antibody: potentials for characterizing atherosclerotic plaques. Eur. J. Nucl. Med. Mol. Imaging 37 (11), 2093–2104.
Kumar, S., Thambiraja, T. S., Karuppanan, K., Subramaniam, G. (2022). Omicron and delta variant of SARS-CoV-2: a comparative computational study of spike protein. J. Med. Virol. 94 (4), 1641–1649.
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). Mega x: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 6, 6. doi: 10.1093/molbev/msy096
Larchevêque Maurel, M., Desrochers, A., Larocque, G. R. (2011). How does drought tolerance compare between two improved hybrids of balsam poplar and an unimproved native species?. Tree Physiology, 31(3), 240–249.
Li, B., Gao, K., Ren, H., Tang, W. (2018). Molecular mechanisms governing plant responses to high temperatures. J. Integr. Plant Biol. 60, 757–779. doi: 10.1111/jipb.12701
Li, H., Yang, Z., Zeng, Q., Wang, S., Luo, Y., Huang, Y., et al. (2020). Abnormal expression of bHLH3 disrupts a flflavonoid homeostasis network, causing differences in pigment composition among mulberry fruits. Hortic. Res. 7, 83.
Li, J. J., Li, Y., Yin, Z. G., Jiang, J. H., Zhang, M. H., Guo, X., et al. (2017). OsASR5 enhances drought tolerance through a stomatal closure pathway associated with ABA and H2O2 signalling in rice. Plant Biotechnol. J. 15, 183–196. doi: 10.1111/pbi.12601
Liao, B. Y., Zhang, J. (2006). Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol. Biol. Evol. 23, 530–540. doi: 10.1093/molbev/msj054
Liu, X. Q., Bai, X. Q., Wang, X. J., Chu, C. C. (2007). OsWRKY71, a rice transcription factor, is involved in rice defense response. J. Plant Physiol. 164 (8), 969–979.
Miller, G. E., Chen, E., Zhou, E. S. (2007). If it goes up, must it come down? chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. psychol. Bull. 133 (1), 25.
Miller, G., Suzuki, N., Ciftci-Yilmaz, S., Mittler, R. (2010). Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 33, 453–467. doi: 10.1111/j.1365-3040.2009.02041.x
Mittler, R. (2006). Abiotic stress, the field environment and stress combination. Trends Plant Sci. 11 (1), 15–19.
Mittler, R., Vanderauwera, S., Gollery, M., Breusegem, F. V. (2004). Reactive oxygen gene network of plants. Trends Plant Sci. 9, 490–498. doi: 10.1016/j.tplants.2004.08.009
Mun, S., Decker, E. A., McClements, D. J. (2007). Influence of emulsifier type on in vitro digestibility of lipid droplets by pancreatic lipase[J]. Food Res. Int. 40 (6), 770–781.
Nakagawa, T., Kurose, T., Hino, T., Tanaka, K., Kawamukai, M., Niwa, Y., et al. (2007). Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J. bioscience bioengineering 104 (1), 34–41.
Nguyen, P. K., Rhee, J. W., Wu, J. C. (2016). Adult stem cell therapy and heart failurTo 2016: a systematic review. JAMA Cardiol. 1 (7), 831–841.
Ohama, N., Sato, H., Shinozaki, K., Yamaguchi-Shinozaki, K. (2017). Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 22, 53–65. doi: 10.1016/j.tplants.2016.08.015
Park, S., Lee, C. M., Doherty, C. J., Gilmour, S. J., Kim, Y., Thomashow, M. F. (2015). Regulation of the Arabidopsis CBF regulon by a complex low-temperature regulatory network. Plant J. 82, 193–207. doi: 10.1111/tpj.12796
Pires, N., Dolan, L. (2010). Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol. 27, 862–874. doi: 10.1093/molbev/msp288
Quan, M., Liu, X., Xiao, L., Chen, P., Song, F., Lu, W., et al. (2021). Transcriptome analysis and association mapping reveal the genetic regulatory network response to cadmium stress in populus tomentosa[J]. J. Exp. Bot. 72 (2), 576–591.
Quan, M., Liu, X., Xiao, L., Chen, P., Song, F., Lu, W., et al. (2020). Transcriptome analysis and association mapping reveal the genetic regulatory network response to cadmium stress in populus tomentosa[J]. J. Exp. Bot 72 (2), 576-591. doi: 10.1093/jxb/eraa434
Reményi, A., Schöler, H. R., Wilmanns, M. (2004). Combinatorial control of gene expression. Nat. Struct. Mol. Biol. 11, 812–815. doi: 10.1038/nsmb820
Ren, H. Y., He, Y. H., Qi, X. J., Zheng, X. L., Zhang, S. W., Yu, Z. P., et al. (2021). The bayberry database: a multiomic database for myrica rubra, an important fruit tree with medicinal value. BMC Plant Biol. 21, 452. doi: 10.1186/s12870-021-03232-x
Ren, H., Yu, H., Zhang, S., Liang, S., Qi, X. (2019). Genome sequencing provides insights into the evolution and antioxidant activity of chinese bayberry. BMC Genomics 20 (1), 458. doi: 10.1186/s12864-019-5818-7
Rodríguez, P., Luna, A., Candela, I., Mujal, R., Teodorescu, R., Blaabjerg, F. (2010). Multiresonant frequency-locked loop for grid synchronization of power converters under distorted grid conditions. IEEE Trans. Ind. Electron. 58 (1), 127–138.
Rossel, J. B., Wilson, P. B., Hussain, D., Woo, N. S., Gordon, M. J., Mewett, O. P., et al. (2007). Systemic and intracellular responses to photooxidative stress in arabidopsis. Plant Cell 19 (12), 4091–4110.
Sakamoto, Y., Suzuki, T., Kobayashi, M., Gao, Y., Fukai, Y., Inoue, Y., et al. (2004). Perfluoropentacene: high-performance p– n junctions and complementary circuits with pentacene. J. Am. Chem. Soc. 126 (26), 8138–8140.
Sánchez-Pérez, R., Pavan, S., Mazzeo, R., Moldovan, C. (2019). Mutation of a bHLH transcription factor allowed almond domestication. Science 364, 1095–1098. doi: 10.1126/science.aav8197
Seo, J. H., Gutacker, A., Sun, Y., Wu, H., Huang, F., Cao, Y., et al. (2011). Improved high-efficiency organic solar cells via incorporation of a conjugated polyelectrolyte interlayer. J. Am. Chem. Soc. 133 (22), 8416–8419.
Shi, H., Ye, T., Chen, F., Cheng, Z., Wang, Y., Yang, P., et al. (2013). Manipulation of arginase expression modulates abiotic stress tolerance in arabidopsis: effect on arginine metabolism and ROS accumulation. J. Exp. Bot. 64, 1367–1379.
Shi, H., Ye, T., Zhu, J. K., Chan, Z. (2014). Constitutive production of nitric oxide leads to enhanced drought stress resistance and extensivetranscriptional reprogramming in arabidopsis. J. Exp. Bot. 65, 4119–4131.
Song, L., Huang, S. S. C., Wise, A., Castanon, R., Nery, J. R., Chen, H., et al. (2016). A transcription factor hierarchy defines an environmental stress response network. Science 354, 1550. doi: 10.1126/science.aag1550
Sun, W., Jin, X., Ma, Z., Chen, H., Liu, M. (2020). Basic helix–loop–helix (bHLH) gene family in tartary buckwheat (Fagopyrum tataricum): Genome-wide identification, phylogeny, evolutionary expansion and expression analyses. Int. J. Biol. Macromol. 155, 1478–1490. doi: 10.1016/j.ijbiomac.2019.11.126
Tian, Q., Chen, J., Wang, D., Wang, H.-L., Liu, C., Wang, S., et al. (2016). Overexpression of a populus euphratica CBF4 gene in poplar confers tolerance to multiple stresses. Plant Cell Tissue Organ Culture 128, 391–407.
Trapnell, C., Pachter, L., Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25 (9), 1105–1111.
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat. Protoc. 7 (3), 562–578.
Vihervaara, A., Duarte, F. M., Lis, J. T. (2018). Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 19, 385–397. doi: 10.1038/s41576-018-0001-6
Wang, C., Liu, S., Dong, Y., Zhao, Y., Geng, A., Xia, X., et al. (2016). PdEPF1 regulates water-use efficiency and drought tolerance by modulating stomatal density in poplar. Plant Biotechnol. J. 14, 849–860. doi: 10.1111/pbi.12434
Wang, L. Q., Wen, S. S., Wang, R., Wang, C. (2021). Pagwox11/12a activates pagcyp736a12 gene that facilitates salt tolerance in poplar. Plant Biotechnol. J. 33, 128–139. doi: 10.1111/pbi.13653
Wang, Y. Y., Cao, Y. B., Liang, X. Y. (2022). A dirigent family protein confers variation of casparian strip thickness and salt tolerance in maize. Nat. Commun., 13.
Wang, R. G., Chen, S. L., Zhou, X. Y., Shen, X., Deng, L., Zhu, H. J., et al. (2008). Ionic homeostasis and reactive oxygen species control in leaves and xylem sap of two poplars subjected to NaCl stress. Tree Physiol. 28, 947–957.
Wang, M., Gong, J., Bhullar, ,. N. K. (2020). Iron defificiency triggered transcriptome changes in bread wheat. Comput. Struct. Biotechnol. J. 18, 2709–2722.
Windt, C. W., Soltner, H., Van Dusschoten, D., Blümler, P. (2011). A portable halbach magnet that can be opened and closed without force: The NMR-CUFF. J. Magnetic Resonance 208 (1), 27–33.
Wu, B. M., Li, L., Qiu, T. H., Zhang, X., Cui, S. X. (2018). Cytosolic APX2 is a pleiotropic protein involved in H2O2 homeostasis, chloroplast protection, plant architecture and fertility maintenance. Plant Cell Rep. 37, 833–848.
Wu, W., Li, J., Wang, Q., Lv, K., Du, K., Zhang, W., et al. (2021). Growth-regulating factor 5 (GRF5)-mediated gene regulatory network promotes leaf growth and expansion in poplar. New Phytol. 230 (2), 612–628.
Xing, C., Liu, Y., Zhao, L., Zhang, S., Huang, X. (2018). A novel MYB transcription factor regulates ascorbic acid synthesis and affects cold tolerance. Plant Cell Environ. 42, 830–841. doi: 10.1111/pce.13387
Xu, W. J., Wang, Y., Xie, J. B., Tan, S. X. (2022). Growth-regulating factor 15-mediated gene regulatory network enhances salt tolerance in poplar[J]. Plant Physiol. 1-18, kiac600. doi: 10.1093/plphys/kiac600
Xie, Y. P., Chen, P. X., Yan, Y., Bao, C. N., Li, X. W., Wang, L. P., et al. (2018). An atypical R2R3 MYB transcription factor increases cold hardiness by CBF-dependent and CBF-independent pathways in apple. New Phytol. 218, 201–218.
Xu, W., Wang, Y., Xie, J., Tan, S. X., Wang, H., Zhao, Y., et al. (2023). Growth-regulating factor 15-mediated gene regulatory network enhances salt tolerance in poplar. Plant Physiol. 00, 1–18.
Xu, W., Zhang, N., Jiao, Y., Li, R., Xiao, D., Wang, Z. (2014). The grapevine basic helix-loop-helix (bHLH) transcription factor positively modulates CBF-pathway and confers tolerance to cold-stress in arabidopsis. Mol. Biol. Rep. 41, 5329–5342. doi: 10.1007/s11033-014-3404-2
Yin, H., Li, M., Li, D., Khan, S. A, Hepworth, S. R, Wang, S. M, et al. (2019). Transcriptome analysis reveals regulatory framework for salt and osmotic tolerance in a succulent xerophyte. BMC Plant Biol. 19, 1–15. doi: 10.1186/s12870-019-1686-1
Yoda, H., Ogawa, M., Yamaguchi, Y., Koizumi, N., Kusano, T., Sano, H. (2002). Identification of early-responsive genes associated with the hypersensitive response to tobacco mosaic virus and characterization of a WRKY-type transcription factor in tobacco plants. Mol. Genet. Genomics 267, 154–161.
Yu, C., Yan, M., Dong, H., Luo, J., Ke, Y., Guo, A., et al. (2021). Maize bHLH55 functions positively in salt tolerance through modulation of AsA biosynthesis by directly regulating GDP-mannose pathway genes. Plant Sci. 302, 110676. doi: 10.1016/j.plantsci.2020.110676
Zandalinas, S. I., Fichman, Y., Devireddy, A. R., Sengupta, Mittler, R. (2020). Systemic signaling during abiotic stress combination in plants. PNAS. 117 (24), 202005077. doi: 10.1073/pnas.2005077117
Zhang, C., Wang, D., Yang, C., Kong, N., Shi, Z., Zhao, P., et al. (2017). Genome-wide identification of the potato WRKY transcription factor family[J]. PloS One 12 (7), e0181573.
Zhang, X., Zhang, P., Wang, G., Bao, Z., Ma, F. (2022). Chrysanthemum lavandulifolium homolog clmad1 modulates the floral transition during temperature shift. Environ. Exp. Bot. 194, 104720. doi: 10.1016/j.envexpbot.2021.104720
Zhao, Y., Xie, J., Wang, S., Zu, W., Chen, S., Song, X., et al. (2021). Synonymous mutation in growth regulating factor 15 of miR396a target sites enhances photosynthetic efficiency and heat tolerance in poplar[J]. J. Exp. Bot. 72 (12), 4502–4519.
Zhang, M., Liang, X., Wang, L., Cao, Y., Song, W., Shi, J., et al. (2019). A HAK family na+ transporter confers natural variation of salt tolerance in maize. Nat. Plants 5 (12), 1297–1308.
Zhao, R., Song, X., Yang, N., Chen, L., Xiang, L., Liu, X. Q., et al. (2020). Expression of the subgroup IIIf bHLH transcription factor CpbHLH1 from chimonanthus praecox (L.) in transgenic model plants inhibits anthocyanin accumulation. Plant Cell Rep. 39, 891–907. doi: 10.1007/s00299-020-02537-9
Zheng, Z. Y., Abu Qamar, S., Chen, Z. X., Mengiste, T. (2006). Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 48 (4), 592–605.
Zhou, J., Li, Z., Xiao, G., Zhai, M., Pan, X., Huang, R., et al. (2020). CYP71D8L is a key regulator involved in growth and stress responses.
Zhou, P., Enders, T. A., Myers, Z. A., Erika, M., Crisp, P. A., Noshay, J. M. (2021). Prediction of conserved and variable heat and cold stress response in maize using cis-regulatory information. Plant Cell. 1, 1. doi: 10.1093/plcell/koab267
Keywords: Chinese bayberry, ROS scavenging, abiotic stress tolerance, bHLH transcription factor, gene regulatory networks
Citation: Xu W, Ren H, Qi X, Zhang S, Yu Z and Xie J (2023) Conserved hierarchical gene regulatory networks for drought and cold stress response in Myrica rubra. Front. Plant Sci. 14:1155504. doi: 10.3389/fpls.2023.1155504
Received: 31 January 2023; Accepted: 20 March 2023;
Published: 14 April 2023.
Edited by:
Biao Jin, Yangzhou University, ChinaReviewed by:
Chongde Sun, Zhejiang University, ChinaCopyright © 2023 Xu, Ren, Qi, Zhang, Yu and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiying Ren, amJ4aWVAYmpmdS5lZHUuY24=; Jianbo Xie, amJ4aWVAYmpmdS5lZHUuY24=
†ORCID: Haiying Ren, orcid.org/0000-0001-5803-4738
Jianbo Xie, orcid.org/0000-0002-8650-7675
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.