 Mingwei Guan1,2,3
Mingwei Guan1,2,3 Xiangtian Shi1,2,3Si Chen1,2,3Yuanyuan Wan1,2,3Yunshan Tang1,2,3Tian Zhao1,2,3Lei Gao1,2,3Fujun Sun1,2,3
Xiangtian Shi1,2,3Si Chen1,2,3Yuanyuan Wan1,2,3Yunshan Tang1,2,3Tian Zhao1,2,3Lei Gao1,2,3Fujun Sun1,2,3 Nengwen Yin1,2,3Huiyan Zhao1,2,3
Nengwen Yin1,2,3Huiyan Zhao1,2,3 Kun Lu1,2,3
Kun Lu1,2,3 Jiana Li1,2,3*
Jiana Li1,2,3* Cunmin Qu1,2,3*
Cunmin Qu1,2,3*- 1Integrative Science Center of Germplasm Creation in Western China (CHONGQING) Science City and Southwest University, College of Agronomy and Biotechnology and Academy of Agricultural Sciences, Southwest University, Chongqing, China
- 2Academy of Agricultural Sciences, Southwest University, Chongqing, China
- 3Affiliation Engineering Research Center of South Upland Agriculture, Ministry of Education, Chongqing, China
Yellow seed coat in rapeseed (Brassica napus) is a desirable trait that can be targeted to improve the quality of this oilseed crop. To better understand the inheritance mechanism of the yellow-seeded trait, we performed transcriptome profiling of developing seeds in yellow- and black-seeded rapeseed with different backgrounds. The differentially expressed genes (DEGs) during seed development showed significant characteristics, these genes were mainly enriched for the Gene Ontology (GO) terms carbohydrate metabolic process, lipid metabolic process, photosynthesis, and embryo development. Moreover, 1206 and 276 DEGs, which represent candidates to be involved in seed coat color, were identified between yellow- and black-seeded rapeseed during the middle and late stages of seed development, respectively. Based on gene annotation, GO enrichment analysis, and protein–protein interaction network analysis, the downregulated DEGs were primarily enriched for the phenylpropanoid and flavonoid biosynthesis pathways. Notably, 25 transcription factors (TFs) involved in regulating flavonoid biosynthesis pathway, including known (e.g., KNAT7, NAC2, TTG2 and STK) and predicted TFs (e.g., C2H2-like, bZIP44, SHP1, and GBF6), were identified using integrated gene regulatory network (iGRN) and weight gene co-expression networks analysis (WGCNA). These candidate TF genes had differential expression profiles between yellow- and black-seeded rapeseed, suggesting they might function in seed color formation by regulating genes in the flavonoid biosynthesis pathway. Thus, our results provide in-depth insights that facilitate the exploration of candidate gene function in seed development. In addition, our data lay the foundation for revealing the roles of genes involved in the yellow-seeded trait in rapeseed.
Introduction
Rapeseed (Brassica napus L., AACC, 2n = 38) is an important oilseed crop worldwide, serving as a source of edible vegetable oil and feed meal (Saeidnia and Gohari, 2012; Carruthers et al., 2017; Kaur et al., 2020). Increasing seed oil content is an important breeding objective in rapeseed. Improving the seed oil content of rapeseed by 1% is equivalent to a 2.3–2.5% increase in seed yield (Wang, 2004). The vegetable oil from rapeseed is excellent for human health (Lu et al., 2011; Liu et al., 2016). Therefore, the demand for high-quality rapeseed has risen sharply. Yellow-seeded rapeseed has many favorable qualities over black-seeded varieties, such as higher oil and protein contents, lower fiber content, and fewer pigments and polyphenols (Tang et al., 2010; Chao et al., 2022). Therefore, the yellow-seeded trait is a major breeding objective for rapeseed.
Seeds are specific sites of oil accumulation in B. napus. Seed development in rapeseed, like the model plant Arabidopsis (Arabidopsis thaliana), can divided into two phases: embryogenesis and seed maturation (Ruuska et al., 2002; Locascio et al., 2014). Carbohydrates, proteins, triacylglycerol (TAG), and pigments are important reserves in developing seeds. Many genes involved in the biosynthetic pathways of these compounds have been identified in Arabidopsis (White et al., 2001; Zhang M. et al., 2009; Lu et al., 2020; Seok et al., 2021). Among these pathways, the flavonoid biosynthesis pathway is a ‘model’ for secondary metabolite production. Flavonoids perform different functions in plants, including color formation and resistance to abiotic and biotic stresses (Nishihara and Nakatsuka, 2011; Chezem and Clay, 2015; Davies et al., 2018). Much effort has focused on revealing the flavonoid biosynthesis pathway involved in seed color formation using Arabidopsis TRANSPARENT TESTA (TT) mutants, such as mutants of TT1-TT19, TTG1, and TTG2 (Albert et al., 1997; Dong et al., 2001; Bharti and Khurana, 2003; Kerhoas et al., 2006; Fu et al., 2007; Stracke et al., 2007; Yoshida et al., 2008; Cutanda-Perez et al., 2009; Baudry et al., 2010; Appelhagen et al., 2011; Chen et al., 2013; Pang et al., 2013; Stein et al., 2013; Zhang et al., 2013; Gesell et al., 2014; Ichino et al., 2015; Gonzalez et al., 2016; Zhang et al., 2017). Numerous genes involved in the formation of seed coat color have also been identified in Brassica species. For example, TRANSPARENT TESTA GLABRA 1 (TTG1) shares the same functions in B. rapa as its Arabidopsis counterpart, controlling both hairiness and seed coat color traits in this crop (Zhang J.F. et al., 2009). The bHLH transcription factor (TF) gene TT8 regulates the accumulation of proanthocyanidins (PAs) and controls seed coat color in Brassica species (Li et al., 2012); silencing of TT1 genes and knockout of TT2 homologs altered seed coat color in B. napus (Lian et al., 2017; Xie et al., 2020). In B. napus, flavonoid biosynthesis pathway genes, including the P-type H+-ATPase genes (AHA10), TT10, TT12, and TTG1, are located within the quantitative trait locus (QTL) region for seed coat color on chromosome A09 (Liu et al., 2005; Badani et al., 2006; Fu et al., 2007; Xiao et al., 2007; Yan et al., 2009; Zhang et al., 2011; Stein et al., 2013; Zhang et al., 2013; Qu et al., 2015). In addition, several candidate genes (e.g., MYB2, MYB3, MYB4, TT8, and MYBL2.1) involved in PA accumulation have been identified in Brassica species using transcriptomic analysis, providing important information for identifying candidate genes for seed coat color (Hong et al., 2017; Jiang et al., 2019; He et al., 2022). However, the molecular mechanisms controlling the yellow-seeded trait in rapeseed remain largely unexplored.
To explore the dynamic regulation of seed coat color formation, we comprehensively investigated the dynamic changes in gene expression during seed development in yellow- and black-seeded rapeseed by performing multistage comparative transcriptomic analysis. Many candidate genes were uncovered, such as KNOTTED-like homeobox of Arabidopsis thaliana 7 (KNAT7), SEPALLATA 2 (SEP2), G-box binding factor 6 (GBF6), SEEDSTICK (STK), and TTG2, which play important roles in regulating seed development and seed coat color formation in rapeseed. Notably, some candidate genes, including BnaA02g17180D, BnaC06g22430D, BnaA07g21710D, BnaA05g00070D, and BnaA04g01810D, were differentially expressed in black-seeded vs. yellow-seeded B. napus during seed development. These results increase our understanding of the gene networks involved in yellow trait formation during seed development in rapeseed, which will facilitate breeding efforts for this trait.
Materials and methods
Plant materials
Seven inbred rapeseed (Brassica napus) lines, four yellow-seeded lines (GH06, 16G15, 16G16, and 16G47), and three black-seeded lines (ZY821, 16G48, and 17G56) were grown in experimental field in Beibei (106.38°E, 29.84°N), Chongqing, China. Inbred lines GH06 (Y1) and ZY821 (B1) are typical yellow- and black-seeded B. napus lines (Qu et al., 2013), respectively. Two groups of near-isogenic lines developed by Chongqing Rapeseed Technology Research Center (CRTRC) were planted during 2019–2020 in the field in CRTRC in Beibei, Chongqing, China: 16G15 (Y2), 16G16 (Y3) and 16G48 (B2); and 17G56 (B3), and 16G47 (Y4). At the flower initiation stage, individual flowers were marked with different colored wool to ensure that seed development could be dated exactly. Subsequently, seeds were sampled at 15, 30, and 50 DAP (days after pollination). At each stage, samples from five individual plants were pooled as a biological replicate, immediately frozen in liquid nitrogen, and stored at −80°C until total RNA extraction.
RNA extraction, cDNA library construction, and sequencing
For each sample, 3 μg RNA was used as input material for cDNA library preparation. Total RNA was extracted from the samples using an RNAprep Pure Plant Kit (Tiangen Biotech, Beijing, China) following the manufacturer’s instructions, and index codes were added to attribute sequences to each sample. First-strand cDNA was synthesized using random hexamer primers. Second-strand cDNA synthesis was then performed using DNA Polymerase I and RNase H. The remaining overhangs were converted into blunt ends using a MicroPoly (A) Purist Kit (Ambion, USA). The library fragments were purified with an AMPure XP system (Beckman Coulter, Beverly, MA, USA). Size-selected, adaptor-ligated cDNA was digested with 3 μl USER Enzyme (NEB, USA) at 37°C for 15 min and then at 95°C for 5 min. The fragments were amplified by PCR using Phusion High-Fidelity DNA Polymerase, Universal PCR Primers, and Index (X) Primer. Following quality checking, the RNA was used for library construction using a NEB Next Ultra RNA Library Prep Kit for Illumina with an insert size of 300 bp. The Illumina HiSeq X was used, which generates short reads of 150 bp in PE mode.
Identification of differential gene expression (DEGs)
After removing low-quality reads, Illumina sequencing reads were mapped to the B. napus Darmor-bzh reference genome (http://www.genoscope.cns.fr/brassicanapus) (Chalhoub et al., 2014) using HISAT with default settings (Kim et al., 2015). The bam files of uniquely mapped reads were used as inputs for HTseq, and FPKM (fragments per kilobase of transcript per million mapped reads) values were calculated to measure the expression levels of genes (Roberts et al., 2011; Anders et al., 2015). The Pearson correlation coefficient between biological replicates was calculated based on the normalized expression levels of log2(FPKM + 1). DEGs were recognized by Cuffdiff with a cut-off of log2 fold change (FC) ≥ 1 and a false discovery rate (FDR) ≤ 0.05 (Wang et al., 2009; Love et al., 2014).
Enrichment analysis of DEGs
Clustering analysis was performed using the TCseq clustering package, and PCA was performed using the Pheatmap package. Transformed and normalized gene expression values with log2 (FPKM + 1) were used for hierarchical clustering.
GOseq was used to obtain significant GO terms for the DEGs in the significant modules (Young et al., 2010), and the clusterProfiler package in R was used to perform KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis of DEGs in the modules: P < 0.02 was selected as the cut-off value.
Identification of hub genes and sub-networks associated with seed color
Hub genes, which are highly interconnected with nodes in a module, are functionally significant. The top 25% of genes evaluated by Cytoscape_v3.7.2 with the CytoNCA (BC, Betweenness centrality; CC, Closeness centrality; DC, Degree centrality) application of the STRING database (http://www.string-db.org) were considered to be hub genes. Hub genes were identified among DEGs between black- and yellow-seeded rapeseed. Protein–protein interaction (PPI) networks were established with STRING, with a cut-off confidence score > 0.4 (medium confidence). Cytoscape_v3.7.2 was then used to calculate the sub-modules of the DEG interaction network. Hub genes were identified according to BC, CC, and DC (Shannon et al., 2003; Chin et al., 2014). The top 25% of genes were ultimately identified as crucial genes based on the centrality values of genes in the PPI network.
Weight gene co-expression networks analysis (WGCNA) and integrative gene regulatory network (iGRN) analysis to identify target genes
The RNA-seq data were analyzed to construct gene co-expression networks using the R package WGCNA (Langfelder and Horvath, 2008). To reduce noise, genes with criterion FPKM ≤ 3 in each sample were excluded. In total, 21 samples were used for analysis, including 7 lines each at the early, middle, and late stages of seed development. The following parameters were used to identify each gene module: weighted network, unsigned; hierarchical clustering tree, dynamic hybrid tree cut algorithm; power, 30; and minimum module size, 250.
Gene regulation is a dynamic process in which TFs play an important role in controlling spatiotemporal gene expression (De Clercq et al., 2021). To identify TFs closely related to seed color, iGRN was used to predict candidate genes. iGRN is used to determine the enrichment of TFs associated with (i.e., regulating) a set of input genes. Enrichment statistics were computed using hypergeometric distribution combined with Benjamini–Hochberg correction for multiple hypotheses testing (with a q-value cut-off of 1e-3). Hub genes confirmed by PPI and Cytoscape_v3.7.2 were used to predict candidate TFs. The results of analysis, combined with the classification of hub genes by WGCNA, were jointly used to predict TFs that are closely associated with the formation of seed color in rapeseed. The expression values of the TFs were calculated using FPKM of materials with the same seed color. A heatmap was generated using the R package Pheatmap (scale = “row”, cluster_row = T) (Diao et al., 2018).
Results
Overview of sequencing data analysis
Through library construction and sequencing, 158.95 million reads in yellow- and black-seeded B. napus during seed development were generated using the Illumina sequencing platform, and deposited in the NCBI database (Accession No., PRJNA931458). An average of 89.26% of the reads were mapped to the B. napus Darmor-bzh reference genome (version 4.1, http://www.genoscope.cns.fr/brassicanapus/data/; Chalhoub et al., 2014) using HTseq version 2.2, and the expression profiles of the genes were quantified in terms of FPKM. Of these mapped reads, an average of 83.32% were uniquely mapped and used to calculate normalized gene expression levels. Detailed information about the transcriptome sequencing data is given in Table 1. Based on the B. napus reference genome (http://www.genoscope.cns.fr/brassicanapus), 89,136 expressed genes were detected during seed development. After eliminating 31,089 genes with lower expression levels (FPKM < 1) in all samples, the 58,047 remaining genes were used for further analysis.
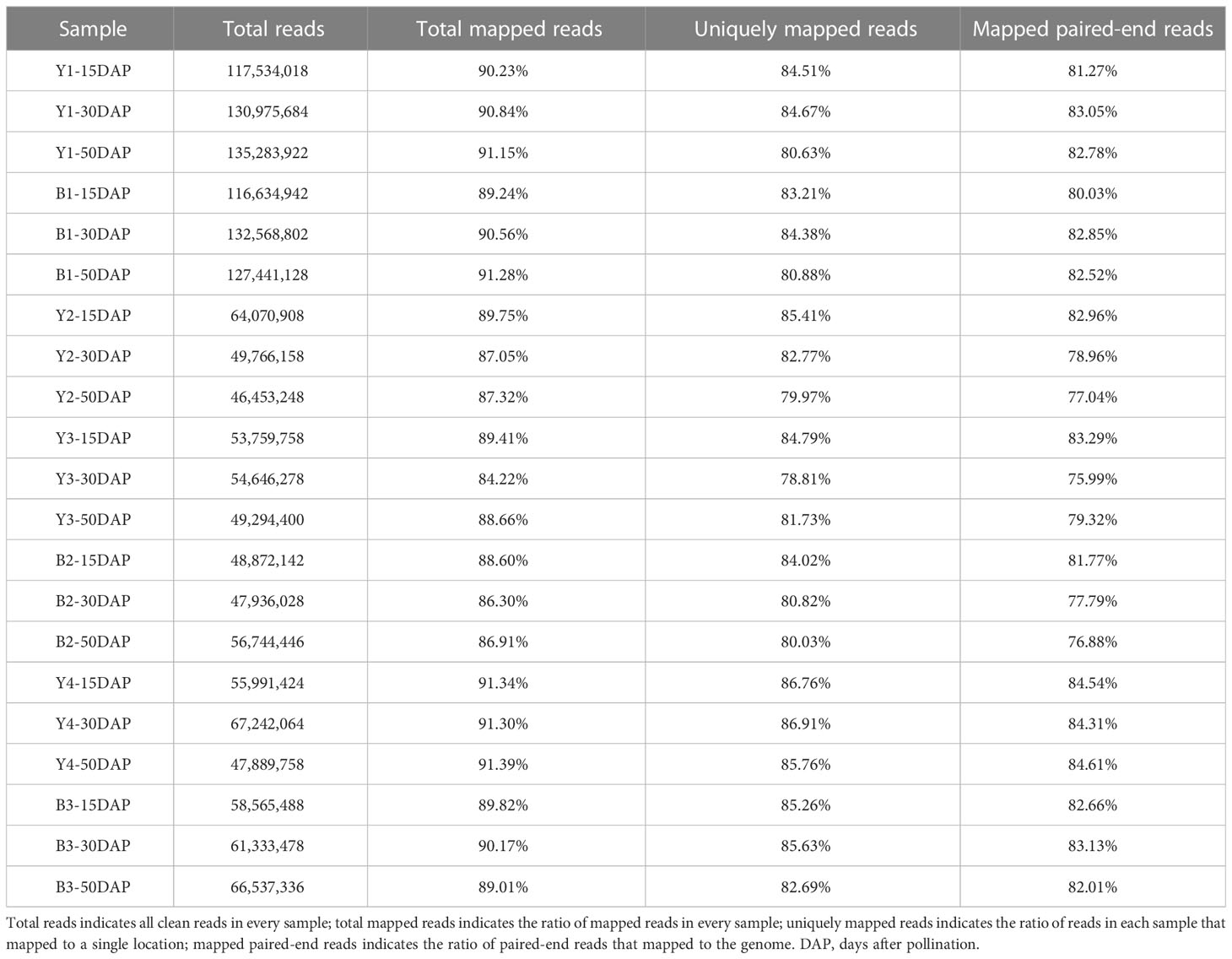
Table 1 Summary statistics of RNA-Seq results during seed development in B. napus.
Identification and annotation of differentially expressed genes during seed development
In this study, 31,843, 32,538, and 24,944 genes with high expression levels (FPKM ≥ 1) were identified at 15, 30, and 50 days after pollination (DAP), respectively (Figure 1A). Of these genes, 21,563 were expressed throughout seed development, and 3,120, 2,321, and 1,427 were specifically expressed at 15, 30, and 50 DAP, respectively. Based on the clustering analysis and principal component analysis (PCA) of the differentially expressed genes (DEGs), these DEGs were divided into three groups, which are consistent with the stages of seed development: early (E, 15 DAP), middle (M, 30 DAP), and late (L, 50 DAP) stages (Figure 1B).
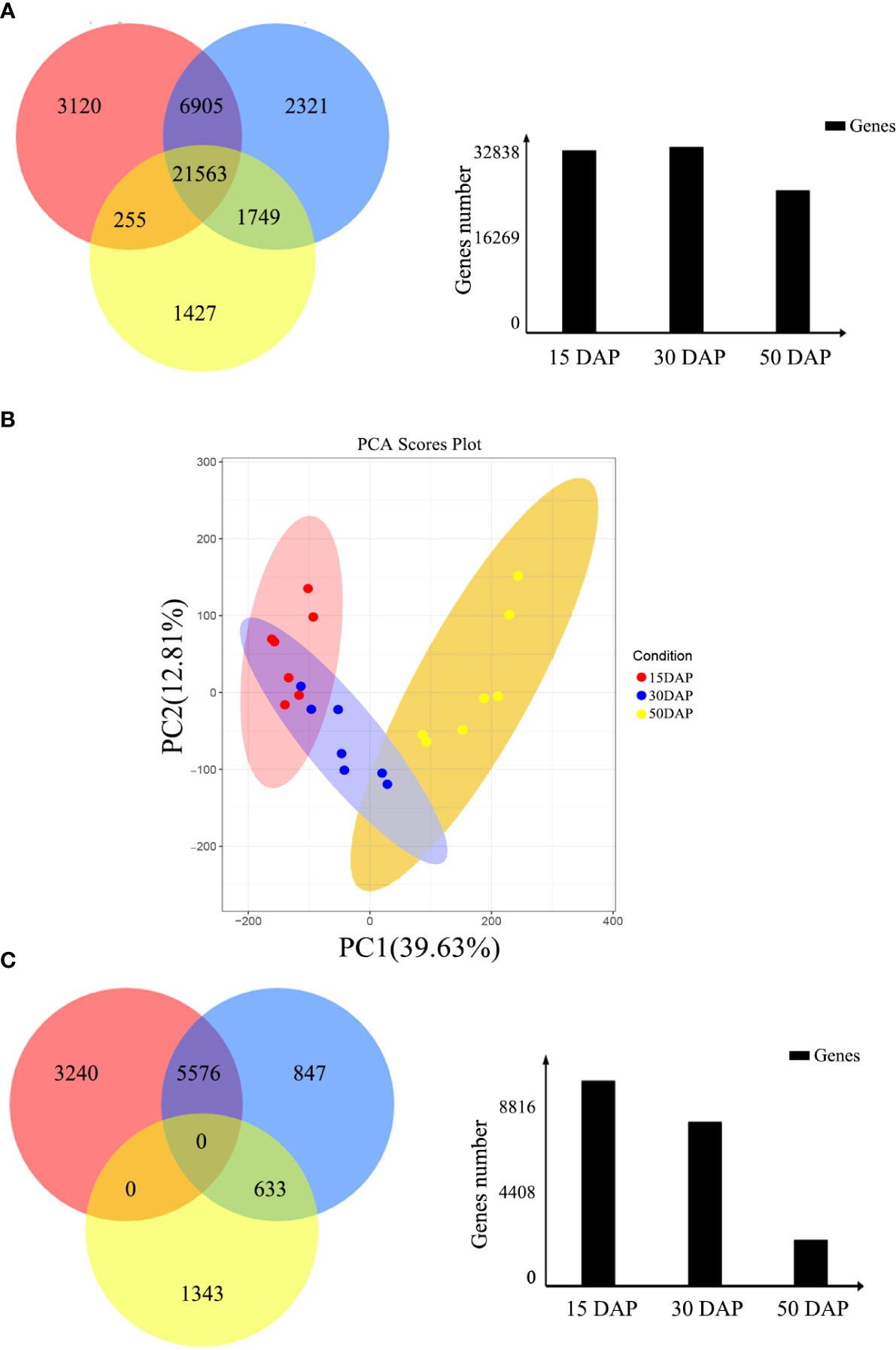
Figure 1 Identification of differentially expressed genes during seed development. (A) Venn diagram of the number of genes expressed during seed development. The red, blue, and yellow circles represent the early, middle, and late stages of seed development, respectively. The numbers represent the number of genes. The bar chart shows the total number of genes expressed during the early (15 DAP), middle (30 DAP), and late (50 DAP) stages of seed development. (B) Cluster dendrogram showing three distinct developmental stages: the early, middle, and late stages of seed development. (C) Venn diagram of DEGs expressed during seed development. The red, blue, and yellow circles represent the early, middle, and late stages of seed development, respectively. The numbers represent the number of DEGs. The bar chart shows the total number of DEGs expressed during the early (15 DAP), middle (30 DAP), and late (50 DAP) stages of seed development.
Moreover, the expression levels of the DEGs during the three stages of seed development were investigated via pairwise comparisons. Using DEGseq and DESeq, the DEGs with adjusted log2 fold change (FC) > 1 and q-value ≤ 0.05 were further investigated. The results of DEG analysis between materials are shown in Table S1. The results showed that 3,240, 847, and 1,343 DEGs were specifically expressed during the early, middle, and late stages of seed development, respectively, in all materials (Figure 1C). However, other genes did not show stable differences in expression among materials during seed development. In addition, a total 1,224 DEGs (FPKM ≥ 1), including 438 genes with high expression levels (FPKM ≥ 10) during seed development, had stable expression levels throughout development. These genes could be core genes for seed development in rapeseed. These DEGs were grouped into six clusters by K-means clustering (TCseq package; Figure 2). The expression of DEGs in clusters 1 and 5 peaked during early seed development, while the expression of DEGs in clusters 2 and 6 peaked during late seed development, and genes in the other clusters were expressed at their highest levels during the middle stage of seed development (Figure 2).
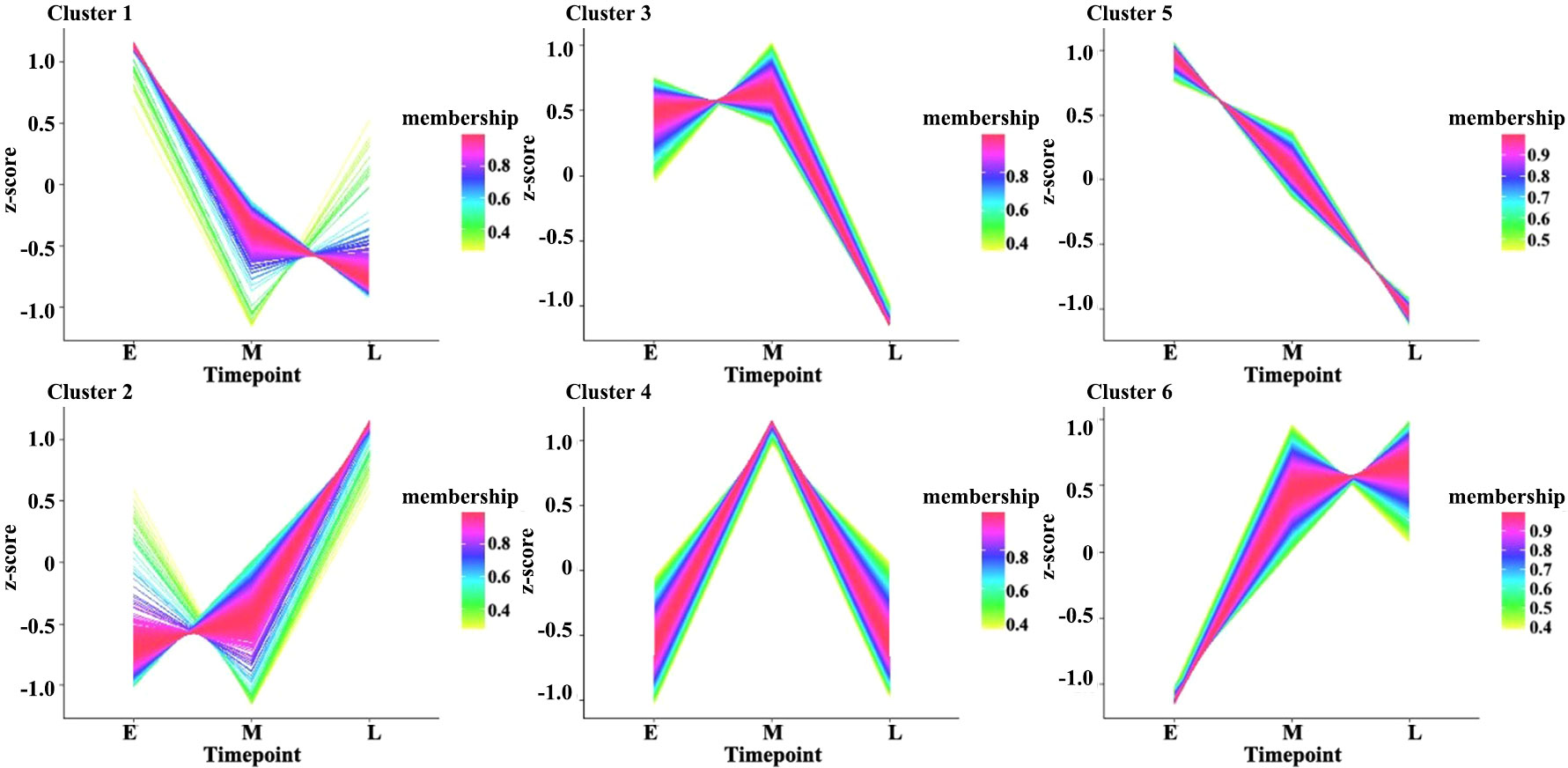
Figure 2 Clustered gene expression profiles in developing seeds. The clusters were defined based on the temporal expression profiles of genes using K-means clustering in R. The Y-axis represents the standardized FPKM values of genes, and E, M, and L on the X-axis represent seed samples at 15, 30, and 50 DAP, respectively.
Besides, the functions of these DEGs were predicted using Gene Ontology (GO) and KEGG enrichment analysis. The GO terms were divided into the biological process (BP), cellular component (CC), and molecular function (MF) categories (Figure 3). As shown in Figure 3, the DEGs were enriched in different GO terms during the three stages of development. For example, the most highly enriched terms during early seed development included carbohydrate metabolic process in the BP category and catalytic activity and enzyme inhibitor activity in the MF category. During the middle stage of seed development, the most highly enriched GO terms included photosynthesis in the BP category and photosynthetic membrane and photosystem in the CC category. During late seed development, the most highly enriched GO terms included embryo development in the BP category, lipid droplet and monolayer-surrounded lipid storage body in the CC category, and nutrient reservoir activity in the MF category (Table S2). KEGG enrichment result showed that the pathways involved mainly carbohydrate, starch and sucrose metabolism in the early seed development; photosynthesis proteins and photosynthesis metabolism in the middle stage; and cutin, suberine and wax biosynthesis, transporters, cytochrome P450 and lipid in the late stage, etc. (Table S3). These results suggest that these DEGs play different roles during seed development in B. napus.
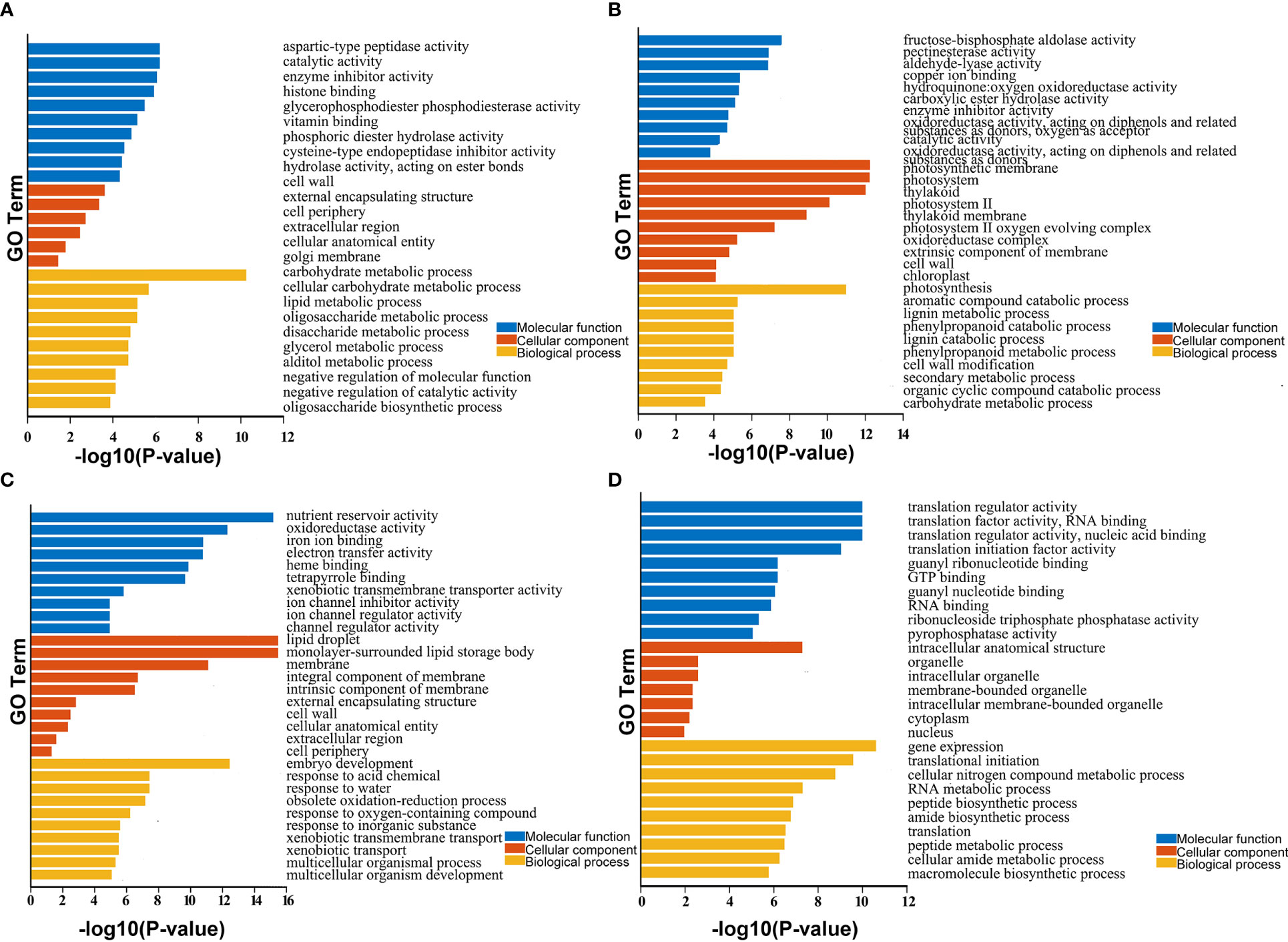
Figure 3 GO enrichment analysis of identified DEGs and core genes in developing seeds. (A–D) show enriched GO terms in seeds at 15, 30, and 50 DAP and 438 core genes with stable, high expression levels during seed development. Only significant categories (top 10 sorted by P-value in MF, CC, and BP) are displayed.
GO enrichment analysis of the 438 core genes with stable, high expression levels during seed development revealed that these genes were clustered into 89 GO terms. The top three terms were translation regulator activity, translation factor activity, and RNA binding and translation regulator activity in the MF category; intracellular anatomical structure, organelle, and intracellular organelle in the CC category; and gene expression, translational initiation, and cellular nitrogen compound metabolic process in the BP category (Table S2). KEGG enrichment result showed the most highly enriched in the pathway of translation factors, protein families: genetic information processing and spliceosome, etc. (Table S3). These results suggest that these DEGs may play crucial roles in translation and the regulation of seed development in B. napus.
Identification of DEGs between the yellow- and black-seeded rapeseed
To identify changes in gene expression related to the yellow-seeded trait, the expression patterns of DEGs were further investigated between yellow- and black-seeded lines during seed development (15, 30, and 50 DAP). In total, 516, 1,206, and 276 DEGs between yellow- and black-seeded rapeseed were obtained in the early, middle, and late stages of seed development, respectively (Figure 4). The highest number of DEGs was observed in the middle stage of seed development (1,206 DEGs, including 982 upregulated and 224 downregulated DEGs in yellow-seeded materials), while fewer DEGs were detected in the late stage of seed development (276 DEGs, including 96 upregulated and 180 downregulated DEGs in yellow-seeded materials). The DEGs in the middle stage of seed development may play important roles in seed color formation. In addition, we identified 47 common DEGs during all three stages of seed development, including 27 upregulated and 20 downregulated genes (Table S4).
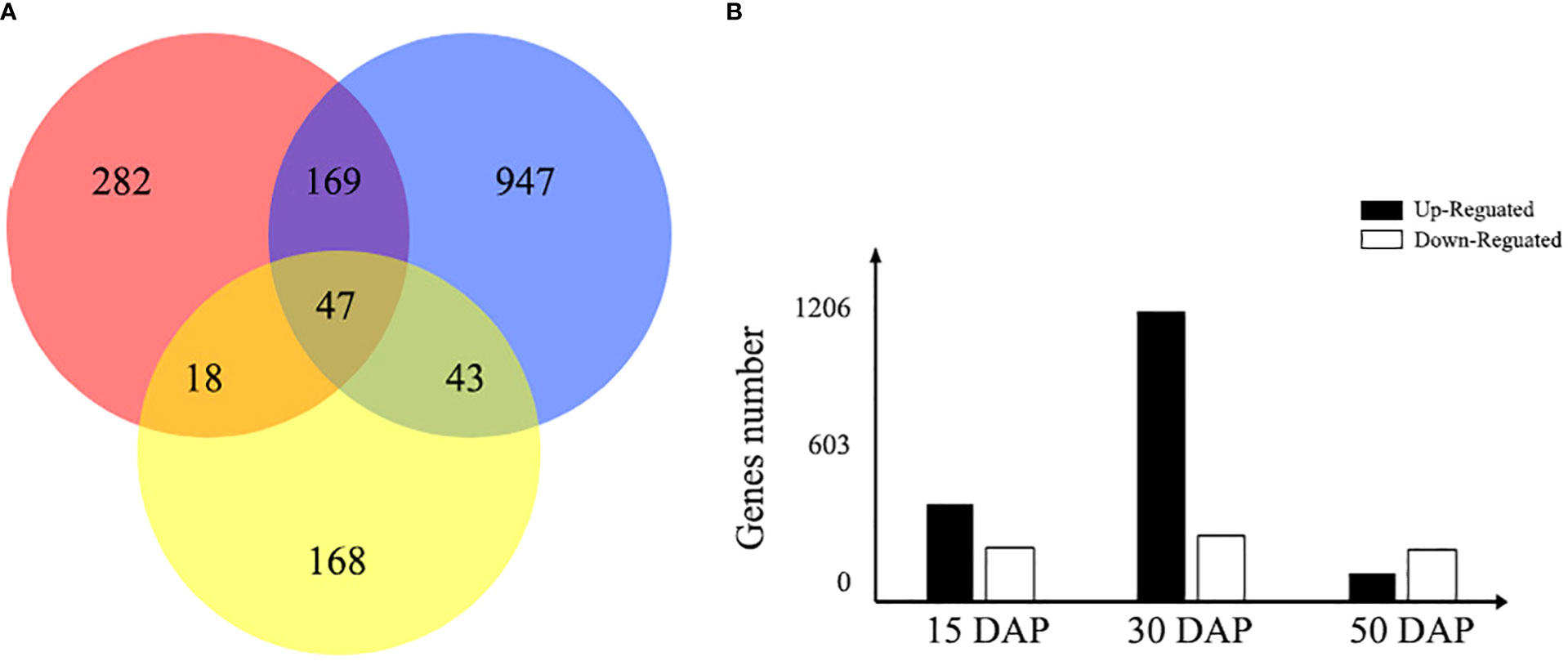
Figure 4 Venn diagram of the number of DEGs expressed during seed development. (A) The red, blue, and yellow circles represent the early, middle, and late stages of seed development, respectively. The number represents the number of DEGs between yellow- and black-seeded rapeseed. (B) The bar chart shows the total number of DEGs expressed during early (15 DAP), middle (30 DAP), and late (50 DAP) seed development. Black bars indicate upregulated DEGs in yellow seeds, and white bars indicate downregulated DEGs in yellow seeds.
Identification of hub genes associated with seed color via PPI network analysis
To explore the molecular mechanism of seed color formation in B. napus, protein–protein interaction (PPI) networks were constructed using the STRING online database, with parameters including a minimum required interaction score > 0.4 (medium confidence). The upregulated and downregulated DEGs in yellow-seeded B. napus seeds at three stages of development were submitted to the STRING database for predicting PPIs and construct PPI networks (Figure S1; Table S5). Strikingly, the PPI networks of downregulated DEGs were highly consistent, especially during the middle and late stages of seed development; these genes are mainly associated with the proanthocyanidin and flavonoid biosynthesis pathways (Table S5). To further elucidate the interactions of these genes, these downregulated and repeatedly detected DEGs in the middle and late stages of seed development were submitted to the STRING database and reconstructed the PPI networks. We then selected the top 25% of genes as hub genes following evaluation by Cytoscape_v3.7.2 with the CytoNCA (BC, CC, DC) application (Figure 5). These hub genes, including TT (TT1, TT4, TT8, TT12, and TT16), BANYULS (BAN), Cinnamate-4-hydroxylase (C4H), AHA10, Leucoanthocyanidin dioxygenase (LDOX), MYB-LIKE 2 (MYBL2), and Phenylalanine ammonia-lyase (PAL), were downregulated in yellow- vs. black-seeded rapeseed (Table S5), suggesting they might play important roles in seed color formation in rapeseed.
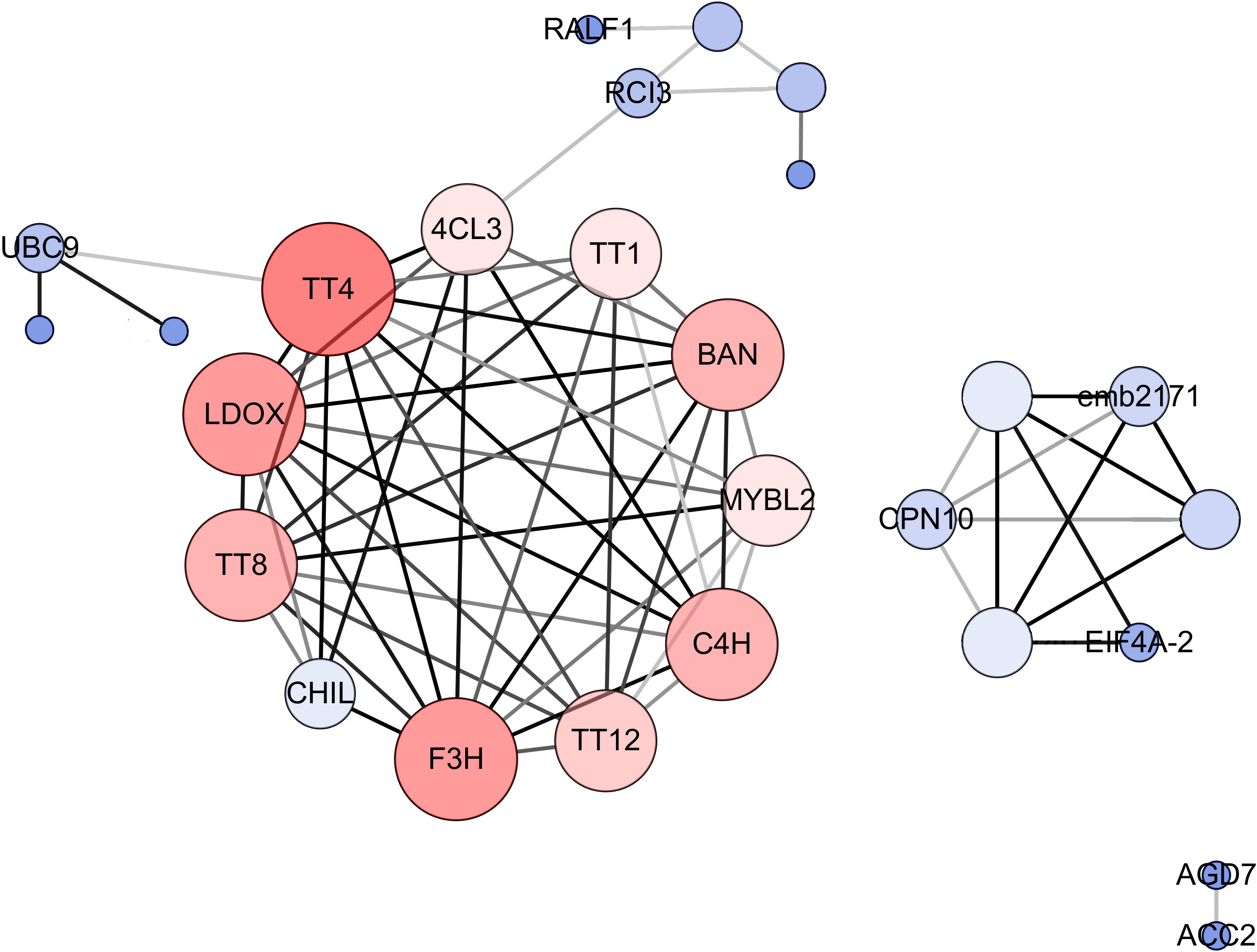
Figure 5 PPI networks for hub genes involved in B. napus seed color. The size and color depth of the circle represent the degree of center, and the color depth of the line represents the degree of correlation. Crucial genes were identified based on eigenvector centrality (EGC), degree centrality (DC), and closeness centrality (CC), which were calculated using the Cytoscape_v3.7.2 plug-in CytoNCA.
Weight gene co-expression network analysis (WGCNA) to identify DEGs for seed color in rapeseed
To further identify the genes/functional pathways associated with seed color, the weighted gene co-expression networks of yellow- and black-seeded rapeseed were constructed using RNA-seq data in this study. To reduce noise, genes with FPKM ≤ 3 in each sample were excluded. The co-expression networks were constructed based on pairwise correlations (>0.8) and minModuleSize = 250. A total of 15 distinct modules labeled with different colors were identified, with the number of genes per module ranging from 258 (orange) to 13,402 (gray) (Figure 6).
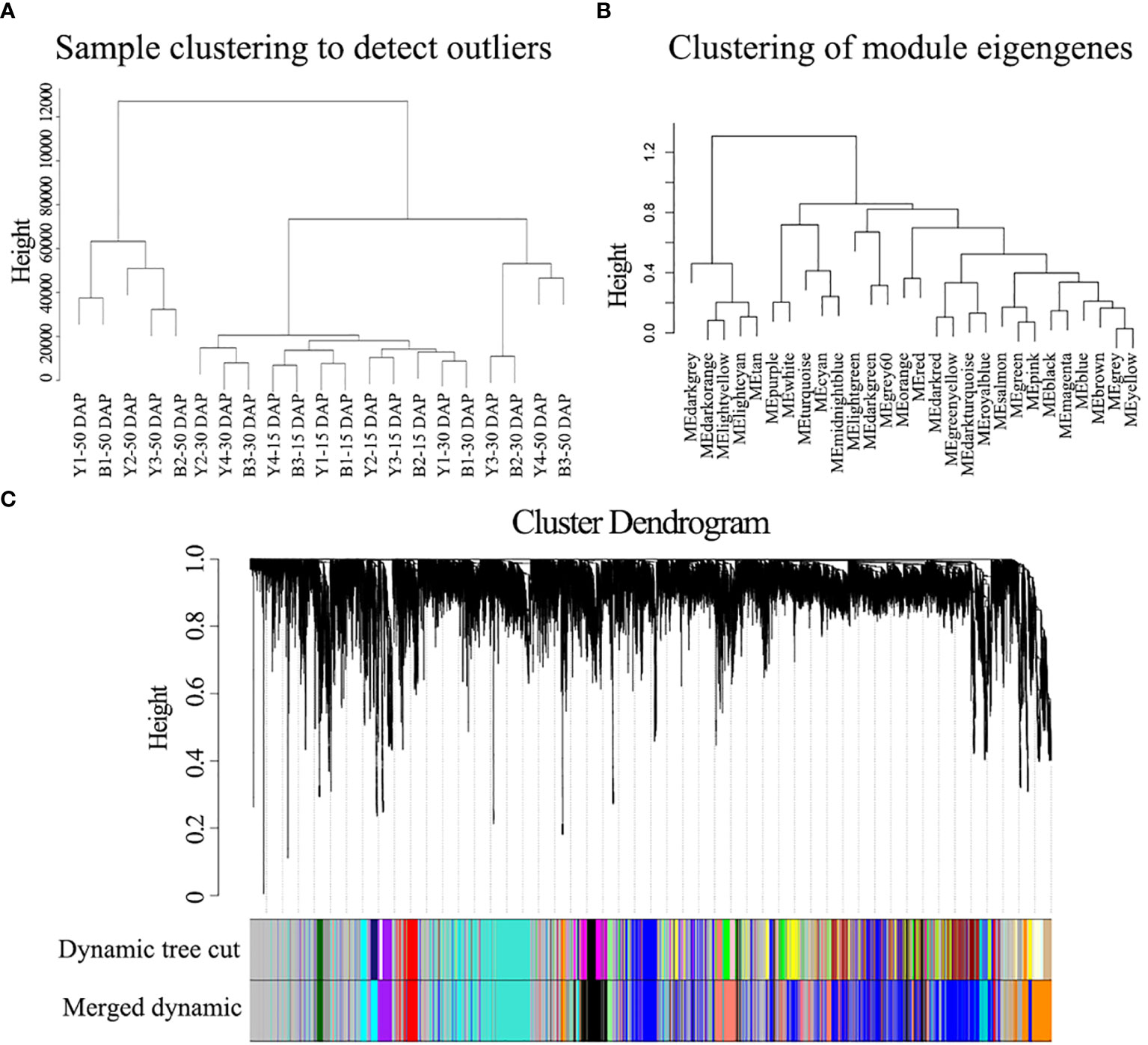
Figure 6 WGCNA of the gene expression matrix in seeds. (A) Clustering tree analysis of different samples. (B) Clustering tree analysis of different modules. (C) Clustering dendrogram of the average network adjacency for the identification of gene co-expression modules.
The hub genes detected by PPI network analysis belonged to four modules (Table 2). Most hub genes (23/28) were classified into the orange module, which were significantly enriched in the GO terms sulfur compound biosynthetic process, aromatic amino acid family metabolic process, and sulfur compound metabolic process. The blue and red modules each contained two hub genes, which were strongly enriched in the GO terms nuclear processes and macromolecule biosynthetic process, respectively. The salmon module, with one hub gene, was significantly enriched in protein transport, cellular macromolecule localization, and protein localization (Table S6). These results indicate that the modules containing hub genes are most strongly correlated with seed color, thus representing suites of interconnected genes underlying the regulation of seed color formation.
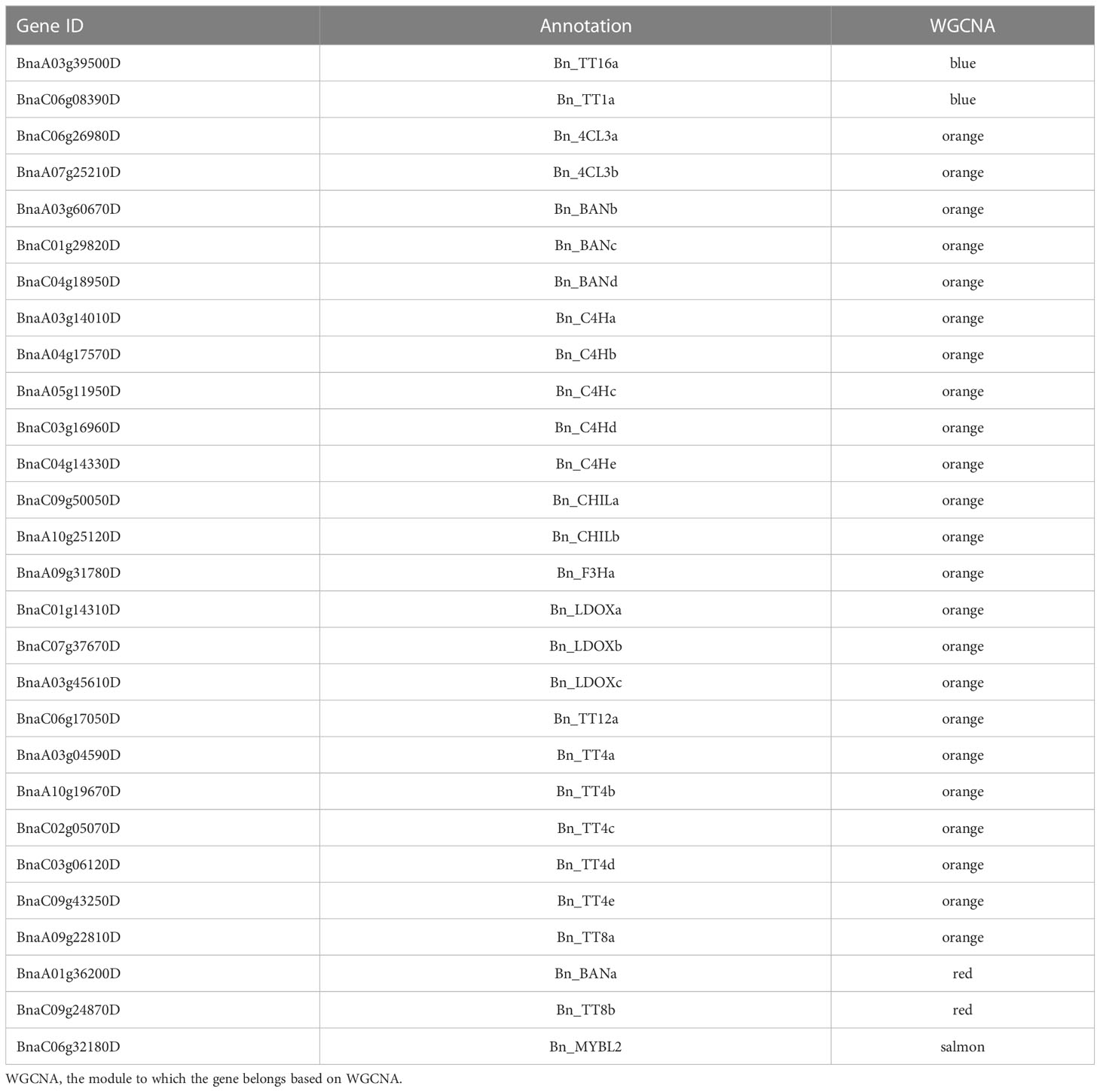
Table 2 WGCNA of DEGs associated with the flavonoid biosynthesis pathway in rapeseed.
The integrated gene regulatory network is linked to the flavonoid biosynthesis pathway in rapeseed
Flavonoids play important roles in seed color formation and are much more complex in Brassica species than in the model plant Arabidopsis (Qu et al., 2016). To enhance our understanding of the regulatory gene network of the flavonoid biosynthesis pathway, an iGRN associated with B. napus seed color were constructed using the DEGs between the yellow- and black-seeded rapeseed. Twenty-five TFs highly interacted with hub genes involved in the flavonoid biosynthesis pathway (Table 3). Among genes known to be involved in the flavonoid biosynthesis pathway, the homologous genes of KNAT7 (BnaA09g52990D), NAC2 (NAC domain containing protein 2, BnaC08g43050D and BnaCnng64100D), STK (BnaA03g24210D, BnaAnng39120D and BnaCnng46740D), and SEP2 (BnaC05g48320D, BnaA01g33070D and BnaC09g42060D) were expressed at higher levels in black-seeded materials, whereas TTG2 (BnaA03g17120D and BnaC03g20650D) showed the opposite pattern (Figure 7A). We also predicted novel TF genes, including Integrase-type DNA-binding superfamily (BnaA10g00620D, BnaC05g00680D, and BnaCnng08620D), bZIP44 (basic leucine-zipper 44; BnaA02g17180D, BnaC06g22430D, and BnaA07g21710D), Exonuclease family (BnaA05g00070D), SHP1 (BnaA04g01810D, and BnaA07g18050D), which showed differential expression levels between yellow- and black-seeded rapeseed (Figure 7A). Notably, five TF genes that were categorized in the orange and red modules by WGCNA (Figure 7B) had similar expression trends to most hub genes, which were closely related to the flavonoid pathway or proanthocyanidin pathway. Six TFs in the salmon module regulate the expression of the hub genes via Bn_MYBL2 (BnaC06g32180D), and other TFs in the blue module play important roles in regulating the flavonoid biosynthesis pathway via Bn_TT1a (BnaC06g08390D) and Bn_TT16a (BnaA03g39500D) (Figure 7). These findings suggest that these TFs might be involved in regulating flavonoid pathways, thus affecting seed color in rapeseed.
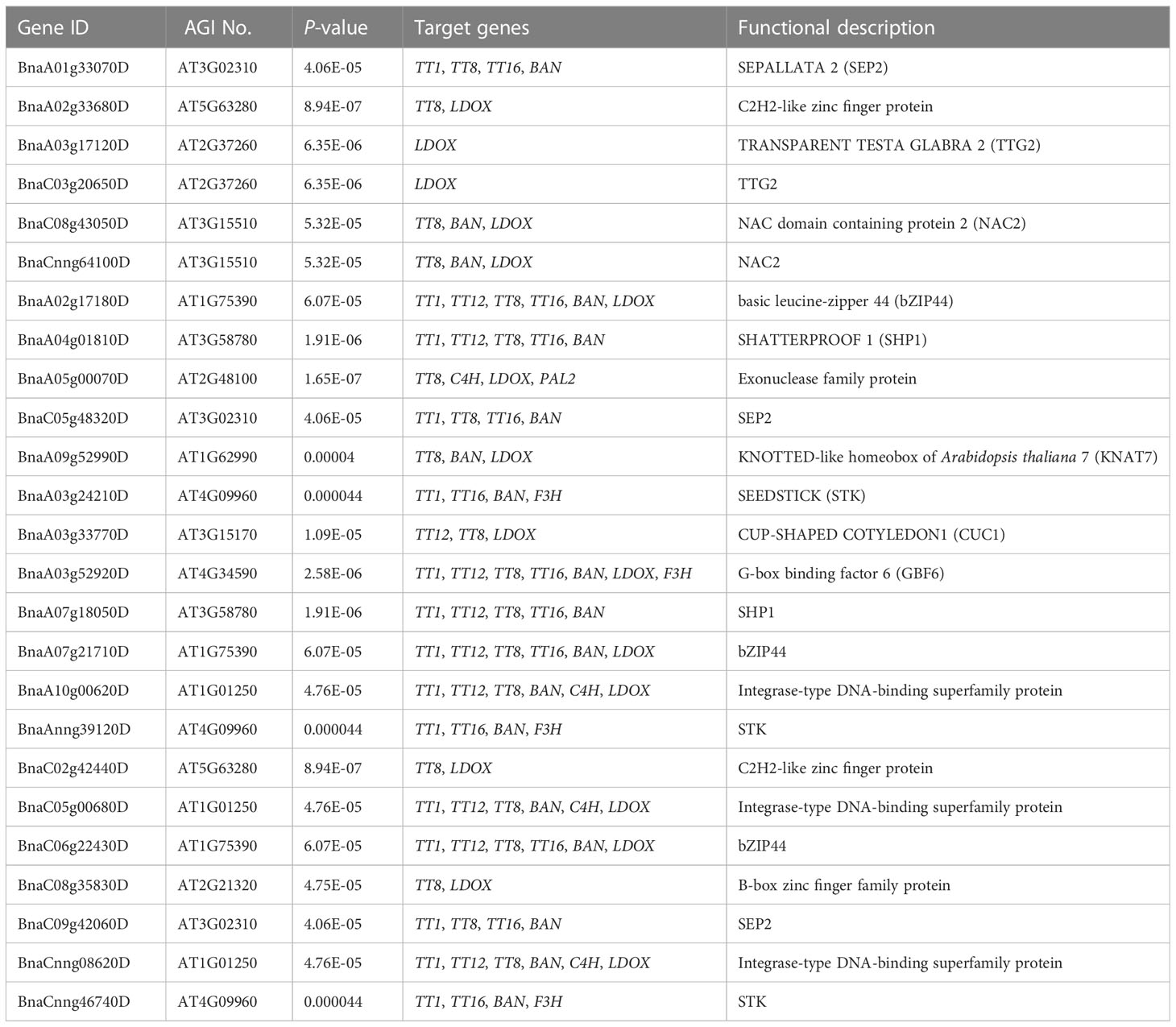
Table 3 The predicted 25 transcription factors by iGRN.
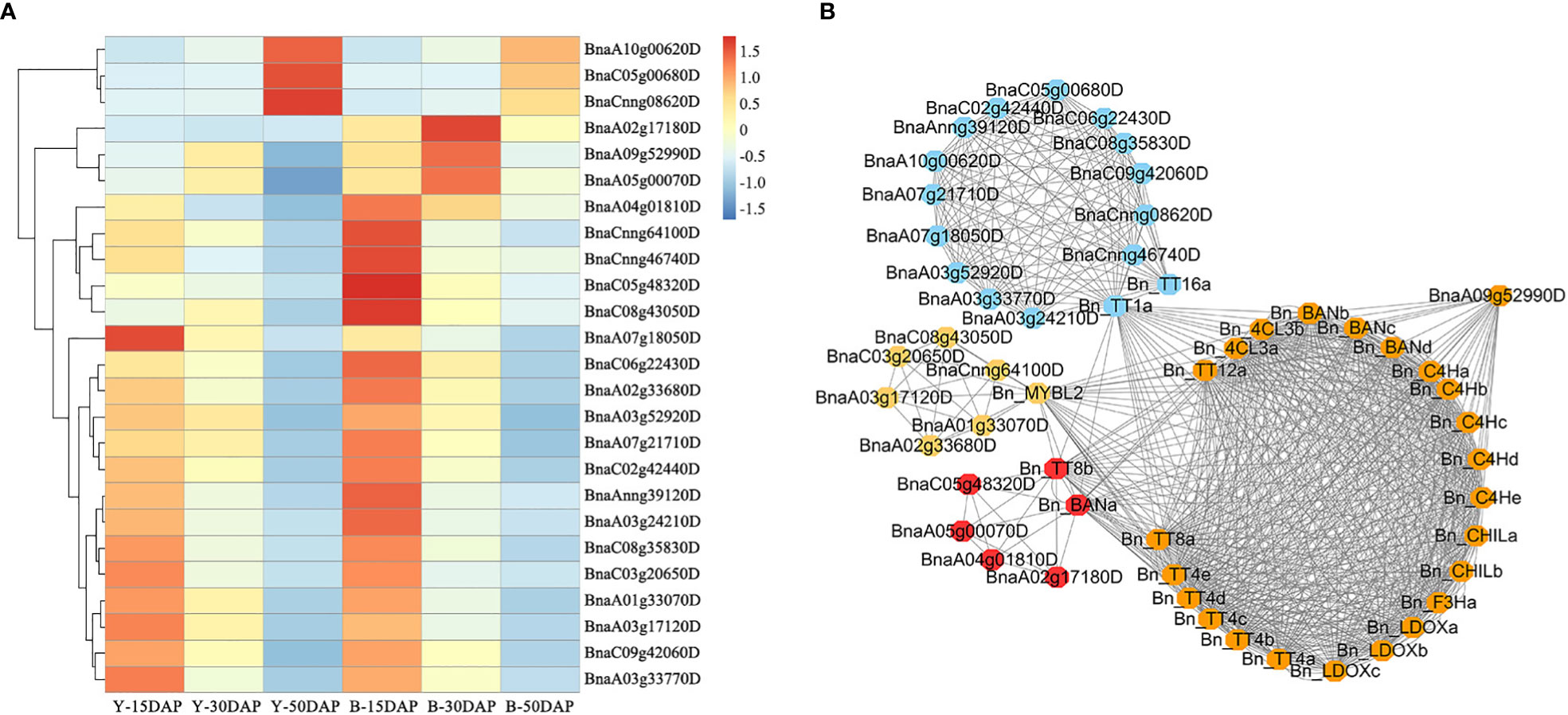
Figure 7 Expression profiles of 25 candidate TF genes. (A) Comparative expression analysis of 25 TF genes associated with seed color between yellow- and black-seeded rapeseed. The expression levels of the TF genes were calculated using FPKM values of materials with the same seed color. The heatmap was generated using the R package Pheatmap (scale = “row”, cluster_row = T). (B) Co-expression network of 25 TFs and hub genes. The same color indicates that genes are located in the same module, as determined by WGCNA.
Discussion
In rapeseed, the yellow-seeded trait is desirable because it offers many advantages, including higher oil and protein content, lower pigment and polyphenol contents, and lower fiber content, compared to black-seeded varieties (Abraham and Bhatia, 1986; Simbaya et al., 1995; Tang et al., 2010). To date most yellow-seeded varieties were developed via the interspecific hybridization of Brassica species (Chen and Heneen., 1992; Liu et al., 2010; Qu et al., 2013), but the genetic stability and underlying mechanism remain unclear. As known, seed color is considered to be a typical quantitative trait. Despite numerous studies involving QTL analysis, GWAS, and candidate gene identification have been performed to elucidate the molecular mechanism underlying yellow seed coat formation in rapeseed (Liu et al., 2005; Badani et al., 2006; Xiao et al., 2007; Yan et al., 2009; Zhang et al., 2011; Stein et al., 2013; Zhang et al., 2013; Lian et al., 2017; Xie et al., 2020; Chao et al., 2022), few major QTL and candidate genes involved in seed color have been successfully cloned in rapeseed. Recently, a major yellow-seed QTL on chromosome A09 was reported to control seed color, oil content and fiber content, which includes important candidate genes (e.g., BnaA09.JAZ1, BnaA09.GH3.3, and BnaA09.LOX3) (Chao et al., 2022).
Numerous studies have revealed that the phenolic compounds cyanidin and procyanidins play dominant roles in seed color formation in rapeseed (Auger et al., 2009; Zhai et al., 2019). The flavonoid biosynthesis pathway is a major factor determining seed color in rapeseed. In the current study, GO enrichment and PPI analyses showed that down-regulated DEGs in yellow-seeded rapeseed were mainly associated with the proanthocyanidin biosynthetic pathway and the flavonoid biosynthetic pathway. The hub genes TT16, BAN, TT4, LDOX, CHIL, F3H, TT12, 4CL, and C4H genes were located in four modules (Table 2) and were expressed at higher levels in black- vs. yellow-seeded rapeseed, especially during the middle and late stages of seed development (Figure 5). BnTT12 was previously identified as a candidate gene involved in seed color formation (Chai et al., 2009) and was detected within QTL intervals for seed color in rapeseed (Wang et al., 2017); TT16 is involved in proanthocyanidin accumulation and is expressed in endothelial cells (Chen et al., 2013). However, compared to black-seeded rapeseed, photosynthesis-related genes were more active in yellow-seeded rapeseed, which were clustered into the GO terms xenobiotic transmembrane transport (GO:0006855), xenobiotic transport (GO:0042908), transport (GO:0006810), and amino acid metabolism (GO:1901605) (Table S7). It appears that yellow-seeded rapeseed has more advantageous traits compared to black-seeded varieties, such as higher oil and protein contents; this notion requires further study.
The MYB-bHLH-WD40 (MBW) complex plays crucial roles in determining seed color by regulating the flavonoid biosynthesis pathway in various plant species (Appelhagen et al., 2010; Xu et al., 2014; Xu et al., 2015), but little is known about this process in rapeseed. In this study, BnTT1 and BnTT8 were identified as hub genes (Table 2). Indeed, the recently generated rapeseed mutant tt8 and tt2 (created by CRISPR/Cas9-mediated gene editing) has yellow seeds (Zhai et al., 2019; Xie et al., 2020). In addition, downregulating BnTT1 via RNA interference (RNAi) reduced the accumulation of flavonoids in seeds (Lian et al., 2017). Moreover, the ternary MBW complex (TT2-TT8-TTG1) was previously shown to function in the flavonoid biosynthesis pathway by regulating DFR, LDOX/TT18, BAN, and TT12 expression, leading to changes in seed color (Lepiniec et al., 2006; Hichri et al., 2011; Xu et al., 2015). We also determined that BAN, LDOX, and TT12 were more highly expressed in black- vs. yellow-seeded rapeseed. These genes shared similar expression patterns with BnTT8 (Figure 7; Table 2), suggesting they might function together to regulate the flavonoid biosynthesis pathway.
With the development of high-throughput technologies such as genomic, transcriptomic, and proteomic profiling, numerous databases have been used to elucidate the complex networks involved in plant development and molecular responses to environmental cues. The release of B. napus datasets (Shen et al., 2015; Song et al., 2020) has made it possible to reveal the flavonoid biosynthetic pathway in rapeseed at the genome-wide level. iGRN analysis is helpful for uncovering the regulatory mechanisms of TFs in the flavonoid biosynthetic pathway in rapeseed using a network-based approach based on supervised learning for large-scale functional data integration (De Clercq et al., 2021). In the current study, 25 TF genes were identified that could be involved in regulating the genes associated with the flavonoid biosynthesis pathway based on iGRN (Figure 7; Table 3). Of these genes, KNAT7, NAC2, and SEP2 are associated with the development of secondary cell walls in the seed coat (Grallert et al., 1997; Kunieda et al., 2008; Li et al., 2011), KNAT7 is related to anthocyanin and proanthocyanidin biosynthesis (Bhargava et al., 2013), and SEP2 is co-expressed with multiple MYB- and bHLH-related genes (Ahmad et al., 2022). Furthermore, SHP1 and CUC1 are involved in seed development (Kusumanjali et al., 2012; Mizzotti et al., 2012; Cucinotta et al., 2018). In addition, GBF6, STK, and TTG2 are candidate genes involved in seed coat color. The expression pattern of GBF6 is similar to that of PAP1, which regulates the anthocyanin pathway in Arabidopsis (Gonzalez et al., 2008; Adamiec et al., 2011). STK is a key gene involved in proanthocyanidin production (Mizzotti et al., 2014), and TTG2 regulates genes in the PA biosynthetic branch of the flavonoid pathway (Gonzalez et al., 2016). Therefore, we demonstrated that iGRN can provide accurate and reliable information about protein structure and function.
In addition, some novel regulators could be involved in seed coat color, including BnbZIP44 (BnaA02g17180D, BnaC06g22430D, and BnaA07g21710D), Exonuclease family protein (BnaA05g00070D), and SHP1 (BnaA04g01810D), which were more highly expressed in black-vs. yellow-seeded B. napus (Figure 7). bZIP44 is closely related to external environmental stimuli; its knockout lines showed slower germination during seed coat rupture (Weltmeier et al., 2009; Iglesias-Fernandez et al., 2013). Recently, bZIP44 was considered as key candidate transcription factors associated with anthocyanin biosynthesis in C. maximowiczii fruits (Zhang et al., 2023) SHP1 is a key gene that regulates inner seed coat development in Arabidopsis (Ehlers et al., 2016). Therefore, these identified TFs can be the targets for further molecular characterization.
In conclusion, the present study is the comprehensive report on transcriptome profiling of developing seeds in yellow- and black-seeded rapeseed. A total of 1206 and 276 DEGs involved in seed coat color could be identified in the middle and late stages, and the downregulated DEGs are mainly enriched for the phenylpropanoid and flavonoid biosynthesis pathways. Moreover, twenty-five TFs involved in seed color are predicted by iGRN and WGCNA that are associated with regulating flavonoid biosynthesis pathway. Our findings provide many clues for elucidating the regulatory networks of the flavonoid biosynthesis pathway and for better understanding the molecular mechanism of the yellow-seeded trait in rapeseed.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI Bioproject PRJNA931458. (Pending release on acceptance).
Author contributions
JL and CQ conceived the project and designed the experiment plans; MG, XS, SC, YW, YT, TZ, LG, and FS conducted the experiments; MG, HZ, KL, and NY analyzed the data and wrote the article; MG, HZ, NY, JL and CQ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Science Foundation of China (32272150, 31830067), Natural Science Foundation of Chongqing (CSTB2022NSCQ-LZX0034), China Agriculture Research System of MOF and MARA, Innovation and Entrepreneurship Training Program for graduates (CYS21124), the 111 Project (B12006).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1154208/full#supplementary-material
References
Abraham, V., Bhatia, C. R. (1986). Development of strains with yellow-seed coat in Indian mustard (Brassica juncea czern & coss). Plant Breed. 97, 86–88. doi: 10.1111/j.1439-0523.1986.tb01307.x
Adamiec, M., Luciński, R., Jackowski, G. (2011). The irradiance dependent transcriptional regulation of AtCLPB3 expression. Plant Sci. 181, 449–456. doi: 10.1016/j.plantsci.2011.07.004
Ahmad, S., Chen, J. L., Chen, G. Z., Huang, J., Hao, Y., Shi, X. L., et al. (2022). Transcriptional proposition for uniquely developed protocorm flowering in three orchid species: Resources for innovative breeding. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.942591
Albert, S., Delseny, M., Devic, M. (1997). BANYULS, a novel negative regulator of flavonoid biosynthesis in the Arabidopsis seed coat. Plant J. 11, 289–299. doi: 10.1046/j.1365-313X.1997.11020289.x
Anders, S., Pyl, P. T., Huber, W. (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1101/002824
Appelhagen, I., Huep, G., Lu, G. H., Strompen, G., Weisshaar, B., Sagasser, M. (2010). Weird fingers: Functional analysis of WIP domain proteins. FEBS Lett. 584, 3116–3122. doi: 10.1016/j.febslet.2010.06.007
Appelhagen, I., Lu, G. H., Huep, G., Schmelzer, E., Weisshaar, B., Sagasser., M. (2011). TRANSPARENT TESTA 1 interacts with R2R3-MYB factors and affects early and late steps of flavonoid biosynthesis in the endothelium of Arabidopsis thaliana seeds. Plant J. 67, 406–419. doi: 10.1111/j.1365-313X.2011.04603.x
Auger, B., Baron, C., Lucas, M. O., Vautrin, S., Berges, H., Chalhoub, B., et al. (2009). Brassica orthologs from BANYULS belong to a small multigene family, which is involved in procyanidin accumulation in the seed. Planta 230, 1167–1183. doi: 10.1007/s00425-009-1017-0
Badani, A. G., Snowdon, R. J., Wittkop, B., Lipsa, F. D., Baetzel, R., Horn, R., et al. (2006). Colocalization of a partially dominant gene for yellow seed colour with a major QTL influencing acid detergent fibre (ADF) content in different crosses of oilseed rape (Brassica napus). Genome 49, 1499–1509. doi: 10.1139/G06-091
Baudry, A., Heim, M. A., Dubreucq, B., Caboche, M., Weisshaar, B., Lepiniec, L. (2010). TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 39, 366–380. doi: 10.1111/j.1365-313X.2004.02138.x
Bhargava, A., Ahad, A., Wang, S. C., Mansfield, S. D., Haughn, G. W., Douglas, C. J., et al. (2013). The interacting MYB75 and KNAT7 transcription factors modulate secondary cell wall deposition both in stems and seed coat in Arabidopsis. Planta 237, 1199–1211. doi: 10.1007/s00425-012-1821-9
Bharti, A. K., Khurana, J. P. (2003). Molecular characterization of transparent testa (tt) mutants of Arabidopsis thaliana (ecotype estland) impaired in flavonoid biosynthetic pathway. Plant Sci. 165, 1321–1332. doi: 10.1016/S0168-9452(03)00344-3
Carruthers, J. M., Cook, S. M., Wright, G. A., Osborne, J. L., Clark, S. J., Swain, J. L., et al. (2017). Oilseed rape (Brassica napus) as a resource for farmland insect pollinators: quantifying floral traits in conventional varieties and breeding systems. Global Change Biol. Bioenergy 9, 1370–1379. doi: 10.1111/gcbb.12438
Chai, Y. R., Lei, B., Huang, H. L., Li, J. N., Yin, J. M., Tang, Z. L., et al. (2009). TRANSPARENT TESTA 12 genes from brassica napus and parental species: cloning, evolution, and differential involvement in yellow seed trait. Mol. Genet. Genomics 281, 109–123. doi: 10.1007/s00438-008-0399-1
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H. B., Wang, X. Y., et al. (2014). Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chao, H., Guo, L., Zhao, W., Li, H., Li, M. (2022). A major yellow-seed QTL on chromosome A09 significantly increases the oil content and reduces the fiber content of seed in Brassica napus. Theor. Appl. Genet. 135, 1293–1305. doi: 10.1007/s00122-022-04031-0
Chen, G. Q., Deng, W., Peng, F., Truksa, M., Singer, S., Snyder, C. L., et al. (2013). Brassica napus TT16 homologs with different genomic origins and expression levels encode proteins that regulate a broad range of endothelium-associated genes at the transcriptional level. Plant J. 74, 663–677. doi: 10.1111/tpj.12151
Chen, B. Y., Heneen, W. K. (1992). Inheritance of seed colour in Brassica campestris l. and breeding for yellow-seeded B. napus l. Euphytica 59, 157–163. doi: 10.1007/BF00041268
Chezem, W. R., Clay, N. K. (2015). “Regulatory and biosynthetic mechanisms underlying plant chemical defense responses to biotic stresses,” in R. A. E, Laitinen Ed. Molecular mechanisms in plant adaptation. (John Wiley & Sons, Inc). doi: 10.1002/9781118860526.ch5
Chin, C. H., Chen, S. H., Wu, H. H., Ho, C. W., Ko, M. T., Lin, C. T. (2014). cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, S11. doi: 10.1186/1752-0509-8-S4-S11
Cucinotta, M., Manrique, S., Cuesta, C., Benkova, E., Novak, O., Colombo, L. (2018). CUP-SHAPED COTYLEDON1 (CUC1) and CUC2 regulate cytokinin homeostasis to determine ovule number in Arabidopsis. J. Exp. Bot. 69, 5169–5176. doi: 10.1093/jxb/ery281
Cutanda-Perez, M. C., Ageorges, A., Gomez, C., Vialet, S., Terrier, N., Romieu, C., et al. (2009). Ectopic expression of VlmybA1 in grapevine activates a narrow set of genes involved in anthocyanin synthesis and transport. Plant Mol. Biol. 69, 633–648. doi: 10.1007/s11103-008-9446-x
Davies, K. M., Albert, N. W., Zhou, Y., Schwinn, K. E. (2018). Functions of flavonoid and betalain pigments in abiotic stress tolerance in plants. Am. Cancer Soc. 1, 21–61. doi: 10.1002/9781119312994.apr0604
De Clercq, I., Van de Velde, J., Luo, X., Liu, L., Storme, V., Van Bel, M., et al. (2021). Integrative inference of transcriptional networks in arabidopsis yields novel ROS signalling regulators. Nat. Plants 7, 1–14. doi: 10.1038/s41477-021-00894-1
Diao, C., Xi, Y., Xiao, T. (2018). Identification and analysis of key genes inosteosarcoma using bioinformatics. Oncol. Lett. 15, 2789–2794. doi: 10.3892/ol.2017.7649
Dong, X. Y., Braun, E. L., Grotewold, E. (2001). Functional conservation of plant secondary metabolic enzymes revealed by complementation of Arabidopsis flavonoid mutants with maize genes. Plant Physiol. 127, 46–57. doi: 10.1104/pp.127.1.46
Ehlers, K., Bhide, A. S., Tekleyohans, D. G., Wittkop, B., Snowdon, R. J., Becker, A. (2016). The MADS box genes ABS, SHP1, and SHP2 are essential for the coordination of cell divisions in ovule and seed coat development and for endosperm formation in Arabidopsis thaliana. PloS One 11, e0165075. doi: 10.1371/journal.pone.0165075
Fu, F. Y., Liu, L. Z., Chai, Y. R., Chen, L., Yang, T., Jin, M. Y., et al. (2007). Localization of QTLs for seed color using recombinant inbred lines of Brassica napus in different environments. Genome 50, 840–854. doi: 10.1139/G07-068
Gesell, A., Yoshida, K., Tran, L. T., Constabel, C. P. (2014). Characterization of an apple TT2-type R2R3 MYB transcription factor functionally similar to the poplar proanthocyanidin regulator PtMYB134. Planta 240, 497–511. doi: 10.1007/s00425-014-2098-y
Gonzalez, A., Brown, M., Hatlestad, G., Akhavan, N., Smith, T., Hembd, A., et al. (2016). TTG2 controls the developmental regulation of seed coat tannins in Arabidopsis by regulating vacuolar transport steps in the proanthocyanidin pathway. Dev. Biol. 419, 54–63. doi: 10.1016/j.ydbio.2016.03.031
Gonzalez, A., Zhao, M., Leavitt, J. M., Lloyd, A. M. (2008). Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in arabidopsis seedlings. Plant J. 53, 814–827. doi: 10.1111/j.1365-313X.2007.03373.x
Grallert, A., Miklos, I., Sipiczki, M. (1997). Division-site selection, cell separation, and formation of anucleate minicells in schizosaccharomyces pombe mutants resistant to cell-wall lytic enzymes. Protoplasma 198, 218–229. doi: 10.1007/BF01287571
He, Q., Xue, Y. H., Wang, Y. X., Zhang, N. A., Zhang, L. G. (2022). Metabolic profiling and transcriptomic data providing critical flavonoid biosynthesis mechanisms disclose color differences of purple heading Chinese cabbages (Brassica rapa l.). Lebensmittel-Wissenschaft. Und-Technol. 168, 113885. doi: 10.1016/j.lwt.2022.113885
Hichri, I., Barrieu, F., Bogs, J., Kappel, C., Delrot, S., Lauvergeat, V. (2011). Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 62, 2465–2483. doi: 10.1093/jxb/erq442
Hong, M. Y., Hu, K. N., Tian, T. T., Li, X., Chen, L., Zhang, Y., et al. (2017). Transcriptomic analysis of seed coats in yellow-seeded Brassica napus reveals novel genes that influence proanthocyanidin biosynthesis. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01674
Ichino, T., Fuji, K., Ueda, H., Takahashi, H., Koumoto, Y., Takagi, J., et al. (2015). GFS9/TT9 contributes to intracellular membrane trafficking and flavonoid accumulation in Arabidopsis thaliana. Plant J. 80, 410–423. doi: 10.1111/tpj.12637
Iglesias-Fernandez, R., Barrero-Sicilia, C., Carrillo-Barral, N., Onate-Sanchez, L., Carbonero, P. (2013). Arabidopsis thaliana bZIP44: A transcription factor affecting seed germination and expression of the mannanase-encoding gene AtMAN7. Plant J. 74, 767–780. doi: 10.1111/tpj.12162
Jiang, J., Zhu, S., Yuan, Y., Wang, Y., Zeng, L., Batley, J., et al. (2019). Transcriptomic comparison between developing seeds of yellow- and black-seeded Brassica napus reveals that genes influence seed quality. BMC Plant Biol. 19, 203. doi: 10.1186/s12870-019-1821-z
Kaur, H., Wang, L. H., Stawniak, N., Sloan, R., van Erp, H., Eastmond, P. (2020). The impact of reducing fatty acid desaturation on the composition and thermal stability of rapeseed oil. Plant Biotechnol. J. 18, 983–991. doi: 10.1111/pbi.13263
Kerhoas, L., Aouak, D., Cingoz, A., Routaboul, J. M., Lepiniec, L., Einhorn, J. (2006). Structural characterization of the major flavonoid glycosides from Arabidopsis thaliana seeds. J. Agric. Food Chem. 54, 6603–6612. doi: 10.1021/jf061043n
Kim, D., Landmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kunieda, T., Mitsuda, N., Ohme-Takagi, M., Takeda, S., Aida, M., Tasaka, M., et al. (2008). NAC family proteins NARS1/NAC2 and NARS2/NAM in the outer integument regulate embryogenesis in Arabidopsis. Plant Cell 20, 2631–2642. doi: 10.1105/tpc.108.060160
Kusumanjali, K., Kumari, G., Srivastava, P. S., Das, S. (2012). Sequence conservation and divergence in miR164C1 and its target CUC1 in Brassica species. Plant Biotechnol. Rep. 6, 149–163. doi: 10.1007/s11816-011-0208-x
Langfelder, P., Horvath, S. (2008). WGCNA: An r package for weighted correlation network analysis. BMC Bioinf. 9, 559–571. doi: 10.1186/1471-2105-9-559
Lepiniec, L., Debeaujon, I., Routaboul, J. M., Baudry, A., Pourcel, L., Nesi, N., et al. (2006). Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 57, 405–430. doi: 10.1146/annurev.arplant.57.032905.105252
Li, X., Chen, L., Hong, M. Y., Zhang, Y., Zu, F., Wen, J., et al. (2012). A Large insertion in bHLH transcription factor BrTT8 resulting in yellow seed coat in Brassica rapa. PloS One 7, e44145. doi: 10.1371/journal.pone.0044145
Li, E., Wang, S., Liu, Y., Chen, J. G., Douglas, C. (2011). OVATE FAMILY PROTEIN 4 (OFP4) interaction with KNAT7 regulates secondary cell wall formation in arabidopsis thaliana. Plant J. Cell Mol. Biol. 67, 328–341. doi: 10.1111/j.1365-313X.2011.04595.x
Lian, J. P., Lu, X. C., Yin, N. W., Ma, L. J., Lu, J., Liu, X., et al. (2017). Silencing of BnTT1 family genes affects seed flavonoid biosynthesis and alters seed fatty acid composition in Brassica napus. Plant Sci. 254, 32–47. doi: 10.1016/j.plantsci.2016.10.012
Liu, S., Fan, C. C., Li, J. N., Cai, G. Q., Yang, Q. Y., Wu, J., et al. (2016). A genome-wide association study reveals novel elite allelic variations in seed oil content of Brassica napus. Theor. Appl. Genet. 129, 1203–1215. doi: 10.1007/s00122-016-2697-z
Liu, Z. W., Fu, T. D., Tu, J. X., Chen, B. Y. (2005). Inheritance of seed colour and identification of RAPD and AFLP markers linked to the seed colour gene in rapeseed (Brassica napus l.). Theor. Appl. Genet. 110, 303–310. doi: 10.1007/s00122-004-1835-1
Liu, X. P., Tu, J. X., Chen, B. Y., Fu, T. D. (2010). Identification and inheritance of a partially dominant gene for yellow seed colour in Brassica napus. Plant Breed. 124, 9–12. doi: 10.1111/j.1439-0523.2004.01051.x
Locascio, A., Roig-Villanova, I., Bernardi, J., Varotto, S. (2014). Current perspectives on the hormonal control of seed development in Arabidopsis and maize: A focus on auxin. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00412
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/PREACCEPT-8897612761307401
Lu, C. F., Napier, J. A., Clemente, T. E., Cahoon, E. B. (2011). New frontiers in oilseed biotechnology: Meeting the global demand for vegetable oils for food, feed, biofuel, and industrial applications. Curr. Opin. Biotechnol. 22, 252–259. doi: 10.1016/j.copbio.2010.11.006
Lu, C. Q., Xie, Z. J., Feng, Y. (2020). Mitochondrial ribosomal protein s9m is involved in male gametogenesis and seed development in Arabidopsis. Plant Biol. 22, 655–667. doi: 10.1111/plb.13108
Mizzotti, C., Ezquer, I., Paolo, D., Rueda-Romero, P., Guerra, R. F., Battaglia, R., et al. (2014). SEEDSTICK is a master regulator of development and metabolism in the Arabidopsis seed coat. PloS Genet. 10, e1004856. doi: 10.1371/journal.pgen.1004856
Mizzotti, C., Mendes, M. A., Caporali, E., Schnittger, A., Kater, M. M., Battaglia, R., et al. (2012). The MADS box genes SEEDSTICK and ARABIDOPSIS bsister play a maternal role in fertilization and seed development. Plant J. 70, 409–420. doi: 10.1111/j.1365-313X.2011.04878.x
Nishihara, M., Nakatsuka, T. (2011). Genetic engineering of flavonoid pigments to modify flower color in floricultural plants. Biotechnol. Lett. 33, 433–441. doi: 10.1007/s10529-010-0461-z
Pang, Y. Z., Cheng, X. F., Huhman, D. V., Ma, J. Y., Peel, G. J., Yonekura-Sakakibara, K., et al. (2013). Medicago glucosyltransferase UGT72L1: Potential roles in proanthocyanidin biosynthesis. Planta 238, 139–154. doi: 10.1007/s00425-013-1879-z
Qu, C. M., Fu, F. Y., Lu, K., Zhang, K., Wang, R., Xu, X. F., et al. (2013). Differential accumulation of phenolic compounds and expression of related genes in black- and yellow-seeded Brassica napus. J. Exp. Bot. 64, 2885–2898. doi: 10.1093/jxb/ert148
Qu, C. M., Hasan, M., Lu, K., Liu, L. Z., Zhang, K., Fu, F. Y., et al. (2015). Identification of QTL for seed coat colour and oil content in Brassica napus by association mapping using SSR markers. Can. J. Plant Sci. 95, 387–395. doi: 10.4141/CJPS2013-411
Qu, C. M., Zhao, H. Y., Fu, F. Y., Wang, Z., Zhang, K., Zhou, Y., et al. (2016). Genome-wide survey of flavonoid biosynthesis genes and gene expression analysis between black- and yellow-seeded Brassica napus. Front. Plant Sci. 7, 1775. doi: 10.3389/fpls.2016.01755
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. (2011). Improving RNA-seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22. doi: 10.1186/gb-2011-12-3-r22
Ruuska, S., Girke, A. T., Ohlrogge, J. B. (2002). Contrapuntal networks of gene expression during arabidopsis seed filling. Plant Cell 14, 1191–1206. doi: 10.1105/tpc.000877
Saeidnia, S., Gohari, A. R. (2012). Importance of Brassica napus as a medicinal food plant. J. Med. Plants Res. 6, 2700–2703. doi: 10.5897/JMPR11.1103
Seok, H. Y., Bae, H., Kim, T., Mehdi, S. M. M., Nguyen, L. V., Lee, S. Y., et al. (2021). Non-TZF protein AtC3H59/ZFWD3 is involved in seed germination, seedling development, and seed development, interacting with PPPDE family protein Desi1 in Arabidopsis. Int. J. Mol. Sci. 22, 4738. doi: 10.3390/ijms22094738
Shannon, P., Markiel, A., Ozier, O., Baliga, N., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecularinteraction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shen, E. H., Zou, J., Behrens, F. H., Chen, L., Ye, C. Y., Dai, S. T., et al. (2015). Identification, evolution, and expression partitioning of miRNAs in allopolyploid Brassica napus. J. Exp. Bot. 66, 7241–7253. doi: 10.1093/jxb/erv420
Simbaya, J., Slominski, B., Rakow, G., Campbell, L. D., Downey, R. K., Bell, J. M. (1995). Quality characteristics of yellow-seeded brassica seed meals: Protein, carbohydrate, and dietary fiber components. J. Agric. Food Chem. 43, 2062–2066. doi: 10.1021/jf00056a020
Song, J. M., Liu, D. X., Xie, W. Z., Yang, Z. Q., Guo, L., Liu, K., et al. (2020). BnPIR: Brassica napus pan-genome information resource for 1,689 accessions. Plant Biotechnol. J. 19, 412–414. doi: 10.1111/pbi.13491
Stein, A., Wittkop, B., Liu, L. Z., Obermeier, C., Friedt, W., Snowdon, R. J. (2013). Dissection of a major QTL for seed colour and fibre content in Brassica napus reveals colocalization with candidate genes for phenylpropanoid biosynthesis and flavonoid deposition. Plant Breed. 132, 382–389. doi: 10.1111/pbr.12073
Stracke, R., Ishihara, H., Barsch, G. H. A., Mehrtens, F., Niehaus, K., Weisshaar, B. (2007). Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 50, 660–677. doi: 10.1111/j.1365-313X.2007.03078.x
Tang, Z. L., Li, J. N., Zhang, X. K., Chen, L., Wang, R. (2010). Genetic variation of yellow-seeded rapeseed lines (Brassica napus l.) from different genetic sources. Plant Breed. 116, 471–474. doi: 10.1111/j.1439-0523.1997.tb01033.x
Wang, H. Z. (2004). Strategy for rapeseed genetic improvement in China in the coming fifteen years. Chin. J. Oil Crop Sci. 26, 98–101. doi: 10.3321/j.issn:1007-9084.2004.02.025
Wang, Z., Gerstein, M., Snyder, M. (2009). RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wang, J., Xian, X. H., Xu, X. F., Qu, C. M., Lu, K., Li, J. N., et al. (2017). Genome-wide association mapping of seed coat color in Brassica napus. J. Agric. Food Chem. 65, 5229–5237. doi: 10.1021/acs.jafc.7b01226
Weltmeier, F., Rahmani, F., Ehlert, A., Dietrich, K., Schutze, K., Wang, X., et al. (2009). Expression patterns within the Arabidopsis C/S1 bZIP transcription factor network: Availability of heterodimerization partners controls gene expression during stress response and development. Plant Mol. Biol. 69, 107–119. doi: 10.1007/s11103-008-9410-9
White, J. A., Todd, T., Newman, T., Focks, N., Girke, T., de Ilarduya, O. M., et al. (2001). A new set of arabidopsis expressed sequence tags from developing seeds. the metabolic pathway from carbohydrates to seed oil. Plant Physiol. 124, 1582–1594. doi: 10.1104/pp.124.4.1582
Xiao, S. S., Xu, J. S., Li, Y., Zhang, L., Shi, S. J., Shi, S. W., et al. (2007). Generation and mapping of SCAR and CAPS markers linked to the seed coat color gene in Brassica napus using a genome-walking technique. Genome 50, 611–618. doi: 10.1139/g07-044
Xie, T., Chen, X., Guo, T. L., Rong, H., Chen, Z. Y., Sun, Q. F., et al. (2020). Targeted knockout of BnTT2 homologues for yellow-seeded Brassica napus with reduced flavonoids and improved fatty acid composition. J. Agric. Food Chem. 68, 5676–5690. doi: 10.1021/acs.jafc.0c01126
Xu, W. J., Dubos, C., Lepiniec, L. (2015). Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 20, 176–185. doi: 10.1016/j.tplants.2014.12.001
Xu, W. J., Grain, D., Bobet, S., Le Gourrierec, J., Thevenin, J., Kelemen, Z., et al. (2014). Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB-bHLH-WDR complexes and their targets in Arabidopsis seed. New Phytol. 202, 132–144. doi: 10.1111/nph.12620
Yan, X. Y., Li, J. N., Fu, F. Y., Jin, M. Y., Chen, L., Liu, L. Z. (2009). Co-Location of seed oil content, seed hull content and seed coat color QTL in three different environments in Brassica napus l. Euphytica 170, 355–364. doi: 10.1007/s10681-009-0006-5
Yoshida, K., Iwasaka, R., Kaneko, T., Sato, S., Tabata, S., Sakuta, M. (2008). Functional differentiation of lotus japonicus TT2s, R2R3-MYB transcription factors comprising a multigene family. Plant Cell Physiol. 49, 157–169. doi: 10.1093/pcp/pcn009
Young, M. D., Wakefield., M. J., Smyth, G. K., Oshlack, A. (2010). Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 11, R14. doi: 10.1186/gb-2010-11-2-r14
Zhai, Y. G., Yu, K. D., Cai, S. L., Hu, L. M., Amoo, O., Xu, L., et al. (2019). Targeted mutagenesis of BnTT8 homologs controls yellow seed coat development for effective oil production in Brassica napus l. Plant Biotechnol. J. 18, 1153–1168. doi: 10.1111/pbi.13281
Zhang, X. B., Abrahan, C., Colquhoun, T. A., Liu, C. J. (2017). A proteolytic regulator controlling chalcone synthase stability and flavonoid biosynthesis in Arabidopsis. Plant Cell 29, 1157–1174. doi: 10.1105/tpc.16.00855
Zhang, M., Fan, J. L., Taylor, D. C., Ohlrogge, J. B. (2009). DGAT1 and PDAT1 acyltransferases have overlapping functions in Arabidopsis triacylglycerol biosynthesis and are essential for normal pollen and seed development. Plant Cell 21, 3885–3901. doi: 10.1105/tpc.109.071795
Zhang, Y., Li, X., Chen, W., Yi, B., Wen, J., Shen, J. X., et al. (2011). Identification of two major QTL for yellow seed color in two crosses of resynthesized Brassica napus line no. 212717. Mol. Breed. 28, 335–342. doi: 10.1007/s11032-010-9486-1
Zhang, K., Lu, K., Qu, C. M., Liang, Y., Wang, R., Chai, Y. R., et al. (2013). Gene silencing of BnTT10 family genes causes retarded pigmentation and lignin reduction in the seed coat of Brassica napus. PloS One 8, e61247. doi: 10.1371/journal.pone.0061247
Zhang, J. F., Lu, Y., Yuan, Y. X., Zhang, X. W., Geng, J. F., Chen, Y., et al. (2009). Map-based cloning and characterization of a gene controlling hairiness and seed coat color traits in Brassica rapa. Plant Mol. Biol. 69, 553–563. doi: 10.1007/s11103-008-9437-y
Keywords: Brassica napus L., yellow trait, transcriptome, flavonoid pathway, protein-protein interaction
Citation: Guan M, Shi X, Chen S, Wan Y, Tang Y, Zhao T, Gao L, Sun F, Yin N, Zhao H, Lu K, Li J and Qu C (2023) Comparative transcriptome analysis identifies candidate genes related to seed coat color in rapeseed. Front. Plant Sci. 14:1154208. doi: 10.3389/fpls.2023.1154208
Received: 30 January 2023; Accepted: 17 February 2023;
Published: 09 March 2023.
Edited by:
Hongbo Chao, Zhengzhou University, ChinaReviewed by:
Libin Zhang, Huazhong University of Science and Technology, ChinaYu Liang, Guangxi Normal University, China
Raheel Shahzad, Universitas Muhammadiyah Bandung, Indonesia
Copyright © 2023 Guan, Shi, Chen, Wan, Tang, Zhao, Gao, Sun, Yin, Zhao, Lu, Li and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiana Li, bGpuMTk1MEBzd3UuZWR1LmNu; Cunmin Qu, ZHJxdWN1bm1pbkBzd3UuZWR1LmNu