 Chun-Jiao Li
Chun-Jiao Li Xin-Tong Xie1
Xin-Tong Xie1 Ruo-Nan Wang
Ruo-Nan Wang De-Zhu Li
De-Zhu Li- 1College of Life Science, Shenyang Normal University, Shenyang, Liaoning, China
- 2Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, Yunnan, China
- 3School of Life Sciences, Sun Yat-Sen University, Guangzhou, China
Lobelia species, as rich source of the alkaloid lobeline which has been shown to have important biological activity, have been used in folk medicine throughout East Asia to treat various diseases. However, Lobelia is a complex and varied genus in East Asia and is thus difficult to identify. Genomic resources would aid identification, however the availability of such information is poor, preventing a clear understanding of their evolutionary history from being established. To close this gap in the available genomic data, in this study, 17 plastomes of East Asian lobelias were newly sequenced and assembled. Although the plastomes of Lobelia sect. Hypsela, L. sect. Speirema, and L. sect. Rhynchopetalum shared the gene structure, the inverted repeat (IR)/large single copy (LSC) boundaries, genome size, and the number of repeats were variable, indicating the non-conservative nature of plastome evolution within these sections. However, the genomes of the Lobelia sect. Delostemon and L. sect. Stenotium showed rearrangements, revealing that these two sections might have undergone different evolutionary histories. We assessed nine hotspot genes and 27-51 simple sequence repeat motifs, which will also serve as valuable DNA barcode regions in future population genetics studies and for the delineation of plant species. Our phylogenetic analysis resolved the evolutionary positions of the five sections in agreement with previous evolutionary trees based on morphological features. Although phylogenetic reconstruction of Lobelioideae based on the rpoC2 gene has rarely been performed, our results indicated that it contains a considerable amount of phylogenetic information and offers great promise for further phylogenetic analysis of Lobelioideae. Our site-specific model identified 173 sites under highly positive selections. The branch-site model exhibited 11 positive selection sites involving four genes in the East Asian branches. These four genes may play critical roles in the adaptation of East Asian taxa to diverse environments. Our study is the first to detect plastome organization, phylogenetic utility, and signatures of positive selection in the plastomes of East Asian lobelias, which will help to further advance taxonomic and evolutionary studies and the utilization of medicinal plant resources.
1 Introduction
Valuable medicines with anti-inflammatory, antioxidative, antiviral, and antiepileptic functions can be made from lobeline and its derivatives (Tamboli et al., 2012; Thawabteh et al., 2019). Lobeline is predominantly derived from plants of the Lobelia (Lammers, 2007b; Zheng et al., 2021). Lobelia L. is the largest genus among all the 29 genera in the subfamily Lobelioideae (Campanulaceae) and has an almost cosmopolitan distribution mainly in tropical to warm-temperate climate regions (Lammers, 2007a; Lammers, 2011). Approximately 12% of Lobelia species are found in Asia, with 27 of them being found in East Asia alone (Lammers, 2007a; Hong et al., 2011). East Asian lobelias are widely distributed in subtropical areas, with Lobelia sessilifolia extending north into eastern Siberia (Lammers, 2011). The lobelias, which have colonized East Asia, are classified into five sections based on morphological traits: Lobelia sect. Delostemon, L. sect. Stenotium, L. sect. Hypsela, L. sect. Speirema, and L. sect. Rhynchopetalum (Hong et al., 2011; Lammers, 2011).
In East Asia, Lobelia is a complex and varied genus, making it challenging to delineate because species and infra-specific taxa exhibit many morphological similarities. The distinctive features are confined to a few morphological traits, which are frequently ineffective for species identification, such as minor differences in plant size, plant color, and indumentum type (Li et al., 2017). For example, L. erectiuscula is distinct from L. davidii due to differences of corolla size between purple-blue corollas (1.3–1.9 cm long) and purple-red or red-purple corollas (1.1–2.8 cm long) (Li et al., 2017). Furthermore, the investigation of the herbarium field specimens revealed several intermediates and various forms. Macroscopic morphological similarity led to a high degree of misidentification between closely related taxa. Our previous study demonstrated that the leaf epidermal characteristics helped differentiate species of Lobelia (Li et al., 2017). However, no molecular characterization or identification research has been conducted by this group.
While many botanists have attempted to understand the phylogenetic relationships of lobelias worldwide (Antonelli, 2008; Lagomarsino et al., 2014; Knox and Li, 2017), East Asian lobelias are much less well-studied. Only three sections of East Asian lobelias have been sequenced, with genomic coverage being much lower for the L. sect. Delostemon and L. sect. Stenotium (Antonelli, 2008; Antonelli et al., 2010; Knox, 2014; Kagame et al., 2021). Previous phylogenetic studies have failed to reconstruct a well-supported phylogenetic tree forming the backbone of the East Asian clades. Many discordances between the phylogenetic relationships of East Asian lobelias inferred from nuclear ribosomal ITS (nrITS) and those inferred from a small number of plastid gene regions have been noted (Chen et al., 2016; Kagame et al., 2021). Plastid markers have been widely used for reconstructing phylogenetic relationships of plants because of their high copy numbers, conserved structure, and uniparental inheritance (Wicke et al., 2011; Gitzendanner et al., 2018). Nevertheless, the utilization of only a few plastid genes is often inadequate to determine the general or species-level phylogeny because of the low mutation rate (Hernandez-Leon et al., 2013; Shaw et al., 2014; Zhou et al., 2019). The plastid genome can be easily obtained cost-effectively using next-generation sequencing technologies, which have been successfully and extensively applied to resolve complex phylogenetic problems from deep to the shallow taxonomic levels of angiosperms (Heckenhauer et al., 2019; Thode et al., 2020). Knox and Li (2017), based on the plastid genomes of several East Asian lobelias, inferred ancestral lineages of the Hawaiian and African giant lobelias, which existed in both South Africa and Eastern Asia but subsequently underwent extinction. However, the possibility remains that they might not be sampled. Therefore, the plastid phylogenomic approach, as opposed to the previous marker-based approach, may be a better choice for elucidating the phylogenetic relationships of East Asian lobelias. Moreover, expanding the collection of fully-sequenced East Asian lobelias will allow the origin of giant lobelias to be reassessed.
Angiosperm plastid genomes are generally highly conserved in their organization, gene order, and content. However, plastid genomes from Lobelioideae are unique due to their unusual structural characteristics (Knox, 2014), showing unprecedented variations in content and gene order (Knox, 2014; Knox and Li, 2017; Li et al., 2020). Lobelioideae plastid genomes generally contain dozens of large open reading frames (ORFs, putative protein-coding genes), an uncommon feature in other angiosperms (Knox, 2014). Previous studies have also revealed extensive genome rearrangements and gene/intron loss throughout the subfamily, as well as severe reduction of inverted repeats in the genera of Downingia and Isotoma (Knox and Li, 2017). This diversity makes the plastomes of Lobelioideae an excellent basis for studying genomic evolution.
In this study, we sequenced and assembled 17 plastomes from 15 species covering all five sections of East Asian lobelias. We evaluated the sequences using a comparative genomics approach to investigate structural variation in the plastomes of East Asian lobelias. We reconstructed a phylogenomic tree to detect the phylogenetic position of East Asian Lobelia species within Lobelioideae and examined highly divergent regions as genetic markers. We also tested how informative the plastid protein-coding genes (PCGs) were in resolving the phylogenetic relationships between the Lobelioideae genera and sections. The nucleotide substitution rates in plastid genomes were also explored to reveal mechanisms related to the adaptation of Asian Lobelioideae species to diverse environments, especially alpine environments. This study is the first survey of plastome organization, phylogenetic utility, and the signatures of positive selection in plastids of the genus Lobelia, with the findings being of medicinal importance in East Asia.
2 Materials and methods
2.1 Sampling, sequencing, and assembly
A total of 82 Lobelioideae plastomes, representing 23 genera and 13 sections within the genera Lobelia, were obtained from previous studies (Lammers, 2007a; Knox, 2014; Crowl et al., 2016; Knox and Li, 2017). Seventeen plastomes of East Asian Lobelia species were newly sequenced and assembled. Trachelium caeruleum (Campanuloideae) and Cyhia elata var. gerrardii (Cyphioideae), two representatives of the closely related subfamilies of Lobelioideae, were selected as the outgroups. Table S1 provides the GenBank accession numbers of all taxa used in the present study and information about newly sequenced species. Total genomic DNA was isolated from leaves of herbarium specimens using the CTAB method (Doyle, 1987). Voucher specimens were deposited in the Herbarium of the Kunming Institute of Botany, Chinese Academy of Sciences (KUN). We used agarose gel electrophoresis with Thermo Scientific NanoDrop Products to estimate the DNA quality. Total DNA was randomly fragmented into 400-600 bp fragments using an ultrasonicator, according to the Illumina Manufacturer Manual. The NEBNext® Ultra™ II DNA Library Prep Kit for Illumina was used to establish the DNA libraries of 500 bp. The Illumina paired-end sequencing (2×150 bp) was performed on the Illumina HiSeq platform at the Germplasm Bank of Wild Species in Southwest China (KUN).
2.2 Plastome assembly and annotation
The raw reads were filtered, and the clean reads were assembled into a circular plastome using GetOrganelle (Jin et al., 2020), with a complete plastome from Lobelia chinensis (NC_035370) as the reference sequence. We used Bandage software (Wick et al., 2015) to view and examine the final assembly graph of the plastomes. We used Sequencher 5.4.6 (Gene Codes Corporation) to identify the junctions between the inverted repeats (IRs) and large single-copy regions (LSCs). A Plastid Genome Annotator (PGA) (Qu et al., 2019) was used to automatically annotate the plastid genome. We utilized Geneious 8.0.2 (Kearse et al., 2012) to adjust the final plastome assembly. The OGDRAW tool (Lohse et al., 2013) was used to draw a circular plastid map using default settings. We investigated the ORF(>400 bp) of the large single-copy region of each East Asian taxon using Geneious software.
Four junctions between the IRs and the small single copy (SSC)/LSC regions were validated using the sequences obtained from the Sanger sequencing of polymerase chain reaction (PCR) products. To amplify these junctions using PCR, we developed primers based on a reference sequence (L. chinensis) through the website application of Primer3 (http://frodo.wi.mit.edu/primer3/) by using standard options. Table S2 shows the PCR procedure. Sanger sequencing of PCR products was performed by the Shanghai Biosune Biotechnology Company. We used Geneious 8.0.2 (Kearse et al., 2012) to align the sequences to check for significant differences.
2.3 Phylogenetic analyses
Seventy-four protein-coding genes were extracted from 82 Lobelioideae plastomes and outgroups and aligned individually using MAFFT (Katoh and Standley, 2014) with default options. Alignments were then adjusted manually in Geneious. We excluded ycf1 and ycf2 because the uncertain homology assessment could generate potentially spurious and misleading results (Braukmann et al., 2017). The absence of genes and gaps was treated as missing or ambiguous data. The optimal partitioning strategy and evolution models were selected using PartitionFinder (Lanfear et al., 2012) for phylogenetic reconstruction. Maximum likelihood (ML) trees were reconstructed with RAxML using a standard bootstrapping (BS) approach with 1000 replicates (Stamatakis, 2006) under the GTR+I+G model, which was selected by PartitionFinder as the optimal model (Table S4). We employed the JModelTest 2.0 program (Darriba et al., 2012) to ascertain the most appropriate model dataset given the Akaike Information Criterion. In addition, the Bayesian inference (BI) tree was reconstructed in MrBayes version 3.2 on the concatenated genes and partitioned based on the best model as determined with Partition Finder. The Markov chain Monte Carlo (MCMC) analysis was performed over 100 million steps and was completed every 10,000 generations. We discarded the first ten million of all the sampled trees as burn-in and determined the consensus tree from the remnant trees. Effective sample size (ESS) values (>200) were used to examine stationarity using Tracer 1.6 (Rambaut et al., 2014). We used the Figtree v.1.4.0 program (Rambaut, 2012) to visualize and annotate trees.
2.4 Comparative plastome analysis
We excluded samples of Lobelia alsinoides and L. fangiana without the complete plastomes as well as repeated species of L. zeylanica_S11412. The length of the total plastome, the coding region, the non-coding region, and the GC content were calculated using Geneious 8.0.2. To observe the potential existence of IR contraction or expansion among all Asian lobelia plastomes, we used L. chinensis as a reference. We detected the IR/LSC and IR/SSC borders using the online software IRScope (Amiryousefifi et al., 2018). The percentage of sequence identity was plotted using the mVista program in shuffle-LAGAN mode (Brudno et al., 2003; Frazer et al., 2004). The IR region from each plastome was removed to check for repeats. REPuter tool (Kurtz et al., 2001) was used to investigate the dispersed repeats in each plastome, with a minimum repeat size of 20bp and a Hamming distance of 3. Repeats were categorized into three classes based on length (20-50 bp, 50-100 bp, and more than 100 bp), then visualized in R v.3.5.1 (R Core Team, 2020).
2.5 Identification of DNA markers
We examined SSRs throughout the genomes of 18 Asian lobelias using the Perl script MISA (Thiel et al., 2003). The minimum thresholds were 12, 6, 5, 5, 5, and 5 for the corresponding mono-, di-, tri-, tetra-, penta-, and hexanucleotide repeats, respectively. Because there were many structural rearrangements among the studied Lobelioideae plastomes, we compared nucleotide sequence divergences across the 74 plastid coding genes only to identify the plastid genome hotspots. Newly assembled and publicly available plastomes from Lobelioideae, including one with an inverted repeat region, were aligned using the MAFFT multiple sequence alignment program (Katoh and Standley, 2014). Nucleotide diversity (pi) throughout the plastid protein-coding regions was detected using the DNASP software package V.6.11.01 (Librado and Rozas, 2009). The window size was 600 bp, and the stepping size was 200 bp.
2.6 Phylogenetic informativeness for protein-coding genes
The PhyDesign online application was implemented with the standard settings (López-Giráldez and Townsend, 2011) to profile the phylogenetic informativeness of the 74 plastid coding sequences. We used the maximum likelihood reconstruction of concatenated coding genes. The ultrametric tree given in relative time was converted using the dnamlk program from Phylip v.3.2 (Felsenstein, 1989). Our input files in PhyDesign were the aligned 74 genes and the relative-time ultrametric tree.
2.7 Genes and sites under the positive selection
We excluded the ndh genes, rpl23, and infA from this analysis because some species had lost these sequences. To determine the PCG sequences affected by the species selection of Lobelioideae, we implemented the MUSCLE algorithm in MEGA 7.0 (Kumar et al., 2016) to align each gene. The ML phylogeny deduced from RAxML was employed as the constraint topology. First, we implemented the site-specific evolutionary selection, including M0, M1a, M2a, M3, M7, and M8 in PAML3.15 software (Yang, 2007). This model assumes that the ω ratio varies among sites but that all lineages share a single ω ratio. The following comparisons were performed: M0 (one-ratio)/M3 (discrete), M1a (nearly neutral)/M2a (positive selection), and M7 (β)/M8 (β and ω). The likelihood ratio test (LRT) was used to assess the strength of the selection. P values from chi-squared tests (x2) (<0.05) were regarded as significant.
To detect the influence of positive selection on the sites of the foreground branch, we constructed the branch-site model of the CodeML program in EasycodeML v.1.31 (Gao et al., 2019). This model allowed the ω ratio to vary among foreground lineages and within phylogenetic sites. The five sections were treated separately as foreground branches. We could also individually detect regions of positive selection in the above lineages using 63 PCGs. In cases where p-values were reported as significant (<0.05), we calculated posterior probabilities of positively selected sites on these lineages (Dhar et al., 2020) using the Bayes empirical Bayes (BEB) approach.
3 Results
3.1 Plastomes characteristics
Clean reads of the newly assembled genomes ranged in number from 12,024,606 to 20,781,623. Fourteen plastomes were complete, not including L. alsinoides, L. zeylanica_S11412, and L. fangiana since they could not be determined as circular, despite all the protein-coding genes being identified. All complete plastomes exhibited a typical quadripartite architecture comprising an LSC region, an SSC region, and two IRs. Their lengths ranged from 162,617 bp in L. heyneana to 174,130 bp in L. zeylanica (Table 1). Eleven protein-coding genes and seven tRNA genes were interrupted by introns, three of which contained two introns. Three trans-spliced exons were predicted from the rps12 gene. Plastomes shared the same gene order except for L. zeylanica and L. heyneana, which showed gene order changes in the LSC region (Figure 1). Species of L. sect. Hypsela, L. sect. Speirema, and L. sect. Rhynchopetalum had three typical ORFs (>400 bp) occurring in rps16-trnQ(UUG), ndhC-psaI, and rpoB-trnC(GCA) in the LSC regions (Table S3). However, L. zeylanica and L. heyneana did not exhibit these ORFs in the same regions. The largest number of dispersed repeats was observed in L. zeylanica and Pratia angulata, followed by L. melliana. Most accessions contained fewer than 300 dispersed repeats (Appendix S1).
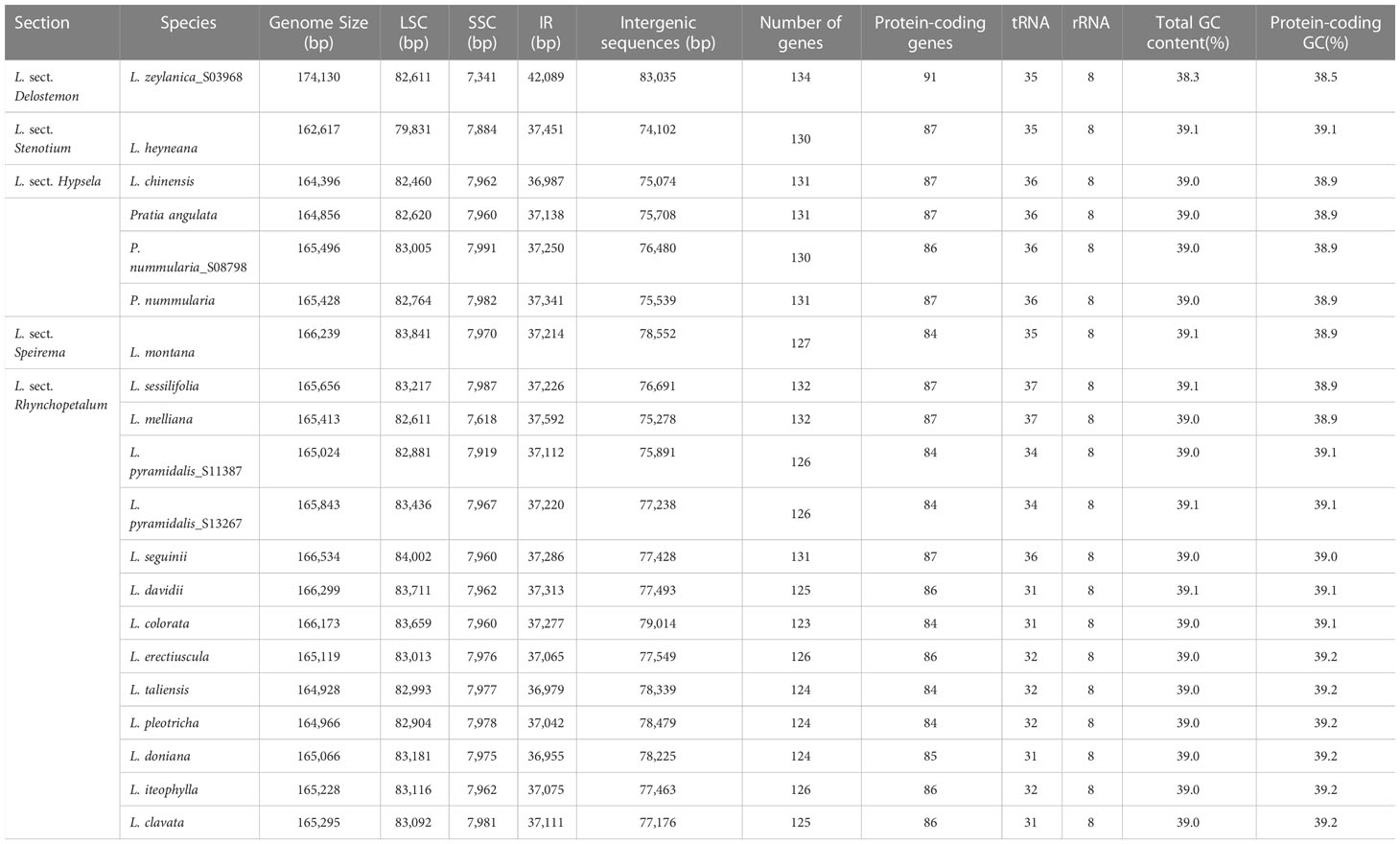
Table 1 Assembly information of twenty plastomes of East Asian lobelias.
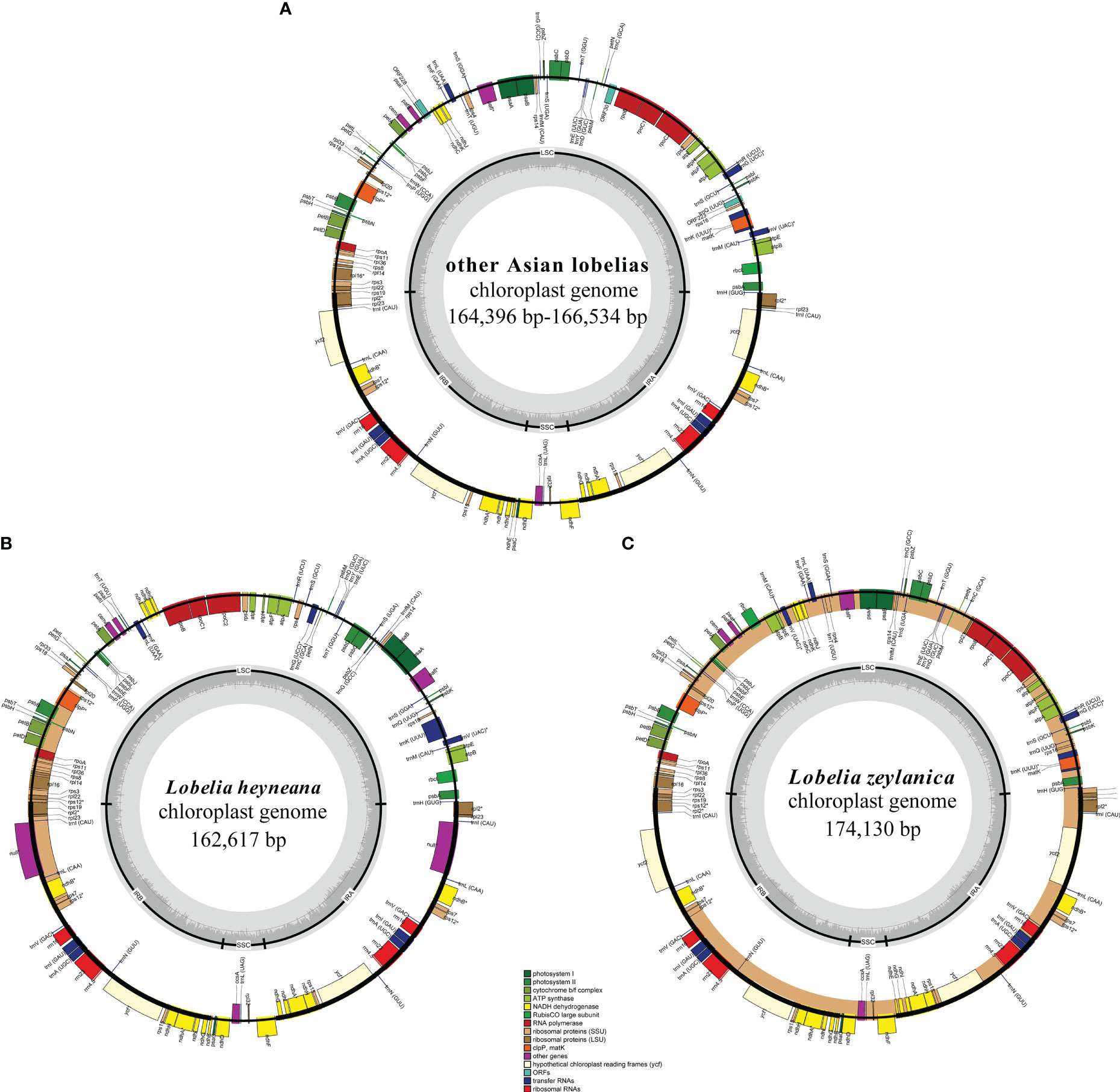
Figure 1 Plastid genomes of other East Asian lobelias (A), Lobelia heyneana (B), and Lobelia zeylanica (C). Genes inside the circle are transcribed clockwise, and genes outside the circle are transcribed counter-clockwise. The dark-gray inner circle corresponds to the GC content, and the light-gray represents the AT content.
The pattern of IR regions was not highly consistent among the plastomes of Asian lobelias with a length of 36,955-42,089 bp. The intergenic region between rpl16 and rps3 was inserted at the IRb/LSC junction in the L. zeylanica plastome. This junction was located within the rpl2 gene of L. heyneana, L. doniana, L. taliensis, and L. pleotricha plastomes. The IRb/LSC junctions in all other species were nested within the rps19 gene. The junctions of the IRa/SSC regions in most accessions were situated in the ndhE gene, although it laid between ndhF and ndhE in L. zeylanica plastome. The rpl2 and rps19 genes were found at the IRb/LSC junction of Pratia nummularia_S087983 and P. nummularia_MF061203, respectively (Figure 2).

Figure 2 Comparison of IR-SC border positions across plastomes of twenty East Asian lobelias. Genes are denoted by colored boxes. The gaps between the genes and the boundaries are indicated by the base lengths (bp).
To explore the sequence divergence of the plastome regions, we implemented the mVISTA program to plot the percentages of sequence identities, with L. chinensis as a reference. In addition to the plastomes of L. zeylanica and L. heyneana, the high similarity was found among other accessions, revealing that these plastome sequences were conserved. The IRs were more conserved than the LSC and SSC regions (Figure 3). Divergences were observed in both coding regions and non-coding regions. Coding regions, such as ycf1, ycf2, rpoC2, ccsA, ndhD, and ndhF, were frequently non-conserved. Seven non-coding regions showed substantial divergence in all intergenic spacers: rps16-trnQ(UUG), rpoB-trnC(GCA), ndhC-psaI, petA-psbJ, psbE-petL, trnL(UAG)-rpl32, rpl32-ndhF (Figure 3). The L. heyneana plastome possessed the ~25, ~1.5, and ~0.8 kb inversions, and the most significant inversion was located between ropB and psaI. L. zeylanica had a ~6 kb inversion between the psbA and matK.
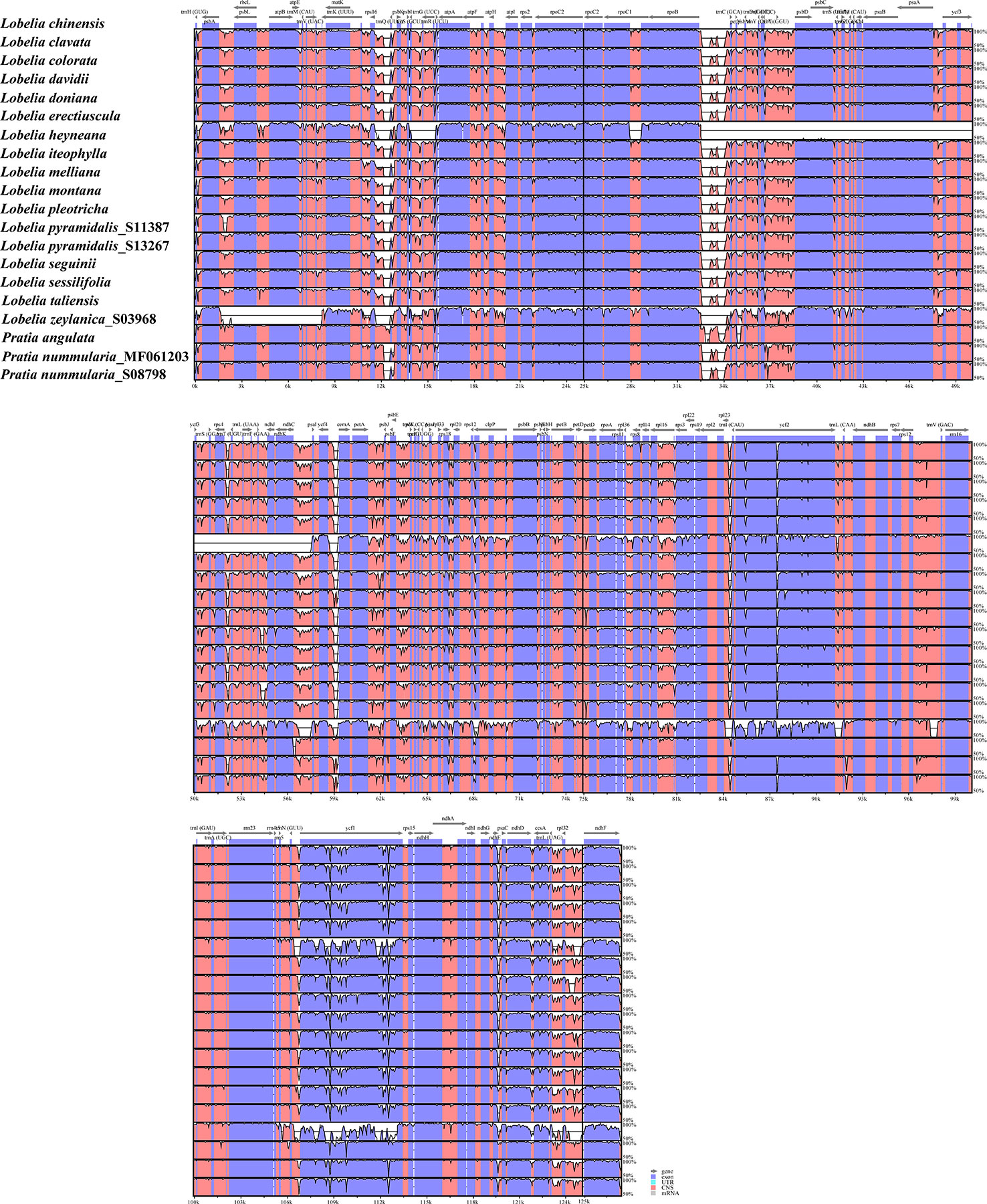
Figure 3 Visualization of the alignments of twenty plastomes representing all sections of East Asian lobelias using mVISTA, with Lobelia chinensis as the reference sequence. The gray arrows above the alignment indicate genes. Different colors represent different regions (coding and non-coding). The horizontal axis indicates the coordinates within the plastid genome. The vertical scale represents the percentage of identity, ranging from 50% to 100%.
3.2 Molecular markers
The plastomes showed similar SSRs counts (Figure 4). However, L. zeylanica, L. heyneana, and L. sessilifolia had higher SSRs than the other studied accessions. The number of simple sequence repeats ranged from 27 (P. angulata) to 51 (L. zeylanica) (Figure 4A). A frequency of 60% was calculated for the mononucleotide repeat patterns. Dinucleotide and trinucleotide repeats had a frequency of more than 10% (Figure 4B). The frequencies of compound, tetranucleotide, pentanucleotide, and hexanucleotide repeats were less than 10%. For sliding window analyses (Figure 5), we computed a mean value of 0.03853 (pi) for the Campanulaceae species. We also observed some highly divergent regions, namely ccsA, clpP, matK, rpl22, rpoA, rpoC2, rps3, rps11, and rps18, in the SSC and LSC regions.
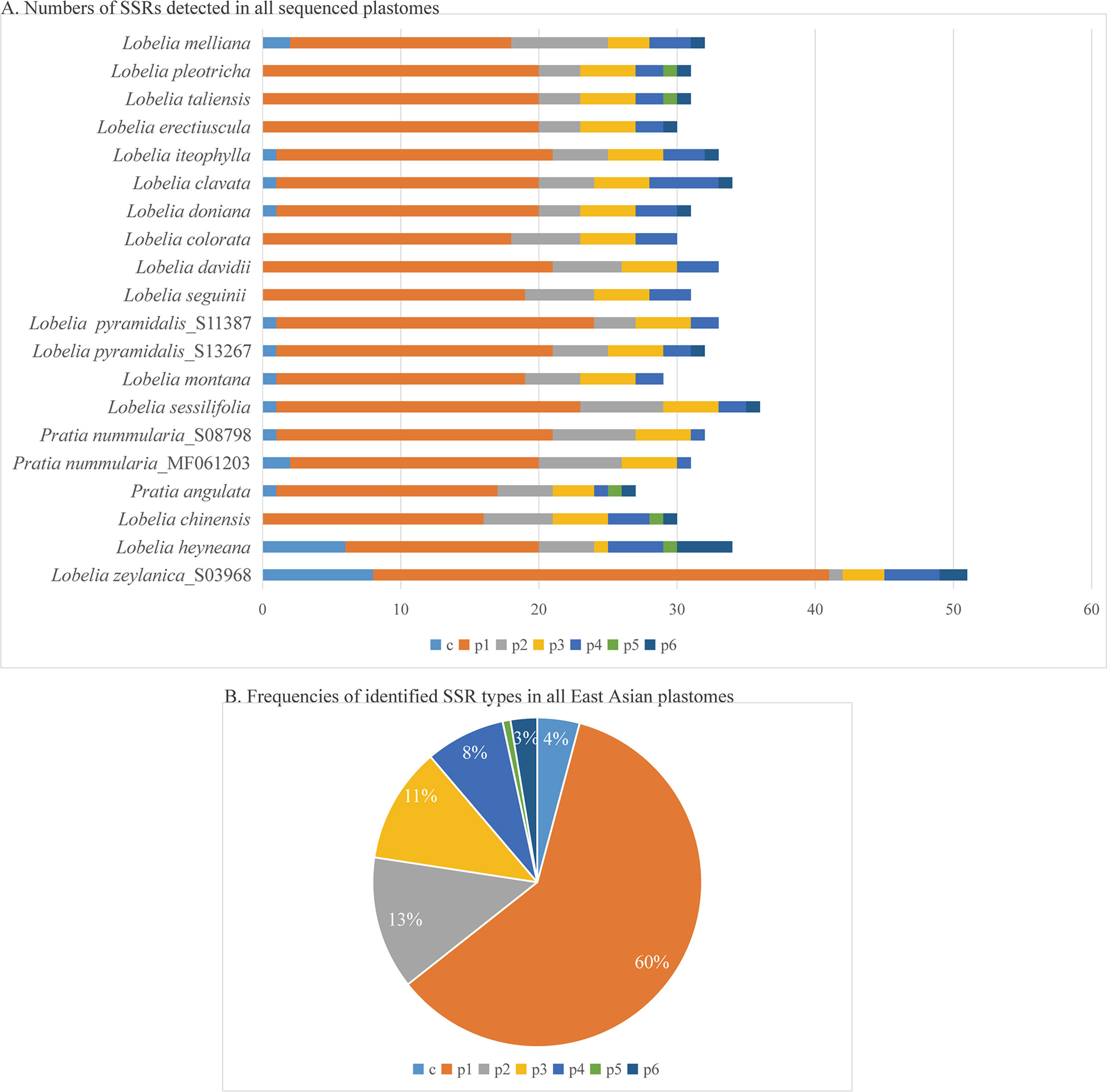
Figure 4 Comparison of simple sequence repeats (SSRs) among Asian plastomes. (A) Numbers of SSRs detected in all sequenced plastomes. (B) Frequencies of identified SSR types in all plastomes of East Asian species. p1, mononucleotides (mono-); p2, dinucleotides (di-); p3, trinucleotides (tri-); p4, tetranucleotides (tetra-); p5, pentanucleotides (penta-); p6, hexanucleotides (hex-); c, compound.
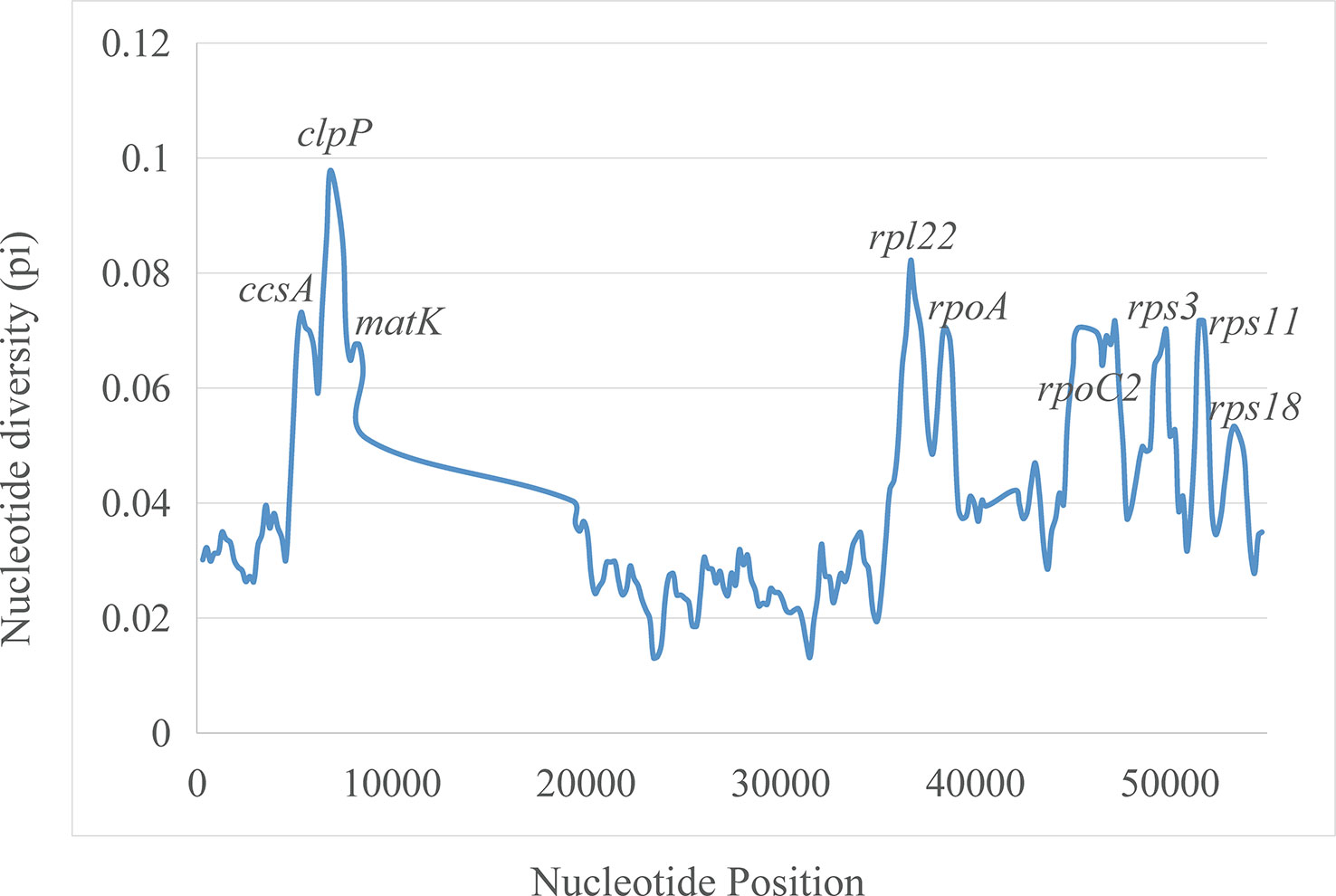
Figure 5 Sliding window analysis of nucleotide diversity (pi) along the twenty aligned plastomes of East Asian lobelias with one IR copy included. Genes underlying peaks of nucleotide diversity are labeled.
3.3 Phylogenetic relationships
The obtained alignment of the 74 plastid PCGs from the 84-species data set comprised 55,000 bp with 14,492 parsimony-informative sites. Partition Finder detected 22 subsets, and the best-fit partitioning schemes are described in Table S4. ML and BI nucleic acid analyses using partitioning schemes yielded identical tree topologies based on the 74 aligned protein-coding genes shared by all sampled species (Figure 6). Plastid phylogenomics has clarified the relationships between the major lineages of Lobelioideae. Bootstrap values (less than 100%) and posterior probabilities (PP<1.0) are labeled in the maximum likelihood tree.
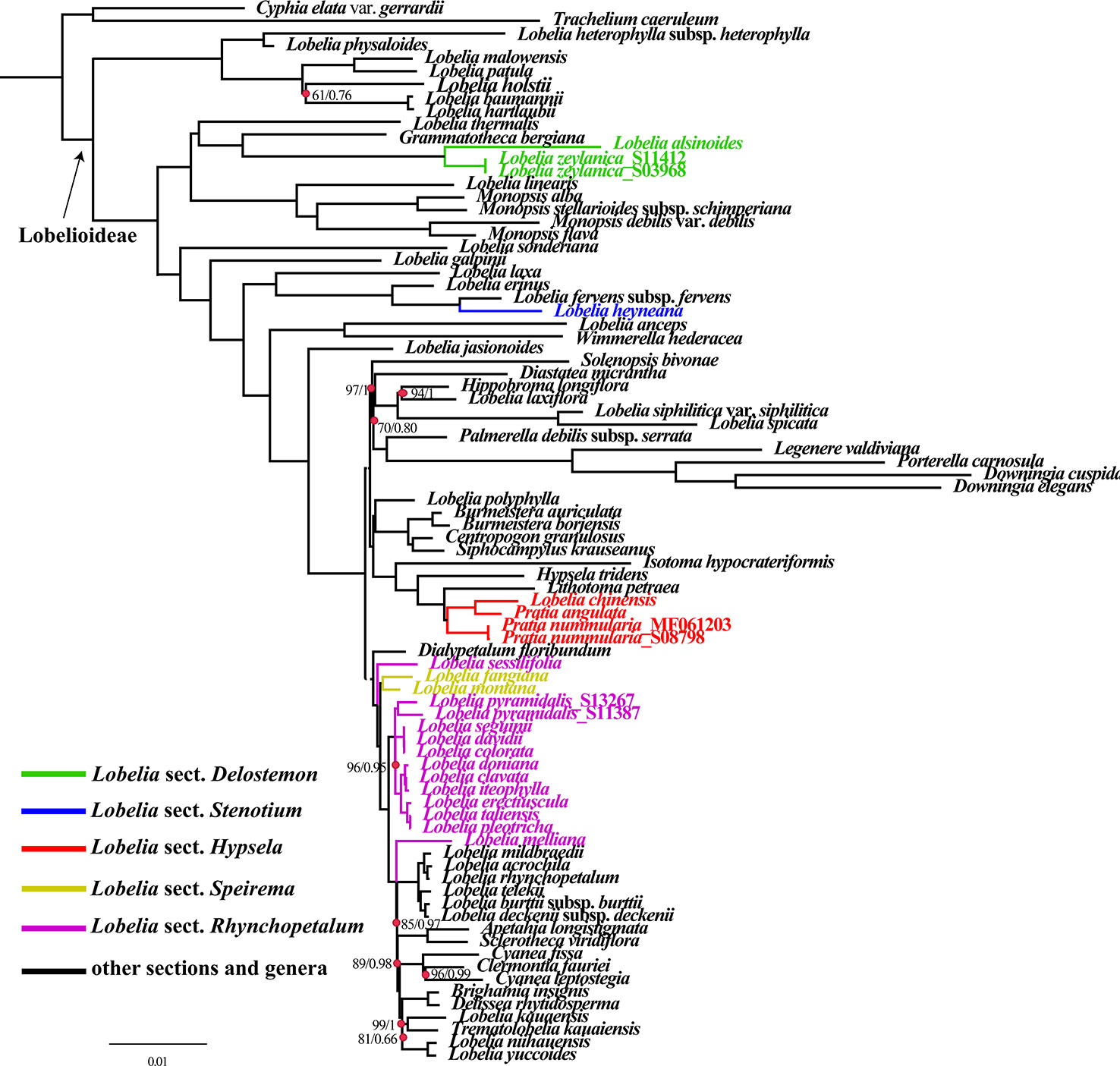
Figure 6 Phylogenetic relationships of all Lobelioideae species using the 74 protein-coding genes, based on the analyses of Maximum likelihood (ML) and Bayesian inference (BI).
Phylogenetic analyses revealed that the genus Lobelia was polyphyletic. Lobelia sect. Delostemon was polyphyletic because this section consisted of not only the East Asian species, namely L. zeylanica and L. alsinoides (Figure 6) but also L. baumannii and L. holstiie. L. sect. Stenotium, including L. galpinii, L. anceps, and the Asian L. heyneana, were polyphyletic. L. sect. Rhychopetalum included the monophyletic L. sect. Speirema (L. fangiana and L. montana), indicating that this section is not monophyletic. Most of the included sections were polyphyletic. L. sect. Hypsela was found to be monophyletic based on the taxa selected in our phylogenetic analyses. East Asia lobelias were scattered throughout the phylogenetic tree of Lobelioideae. L. zeylanica_S11412, L. zeylanica_S03968, and L. alsinoides were nested in a sister clade to Grammatotheca bergiana. L. heyneana emerged as a sister species to L. fervens subsp. fervens. Pratia. nummularia, P. angulata, and L. chinensis formed a sister clade with Lithotoma petraea. L. sessilifolia (collected from the Russian Federation, based on a previous study) showed a sister relationship with a lineage of many other East Asian lobelias and giant lobelias. L. fangiana and L. montana clustered as sister clades for most Asian taxa. L. pyramidalis_S13267 and L. pyramidalis_S11387 were placed as sisters to a clade that included the most of Asian lobelias with strong support (BP=96, PP=0.95). L. melliana was a sister to plants from East Africa, Hawaii, and the South Pacific, an evolutionary relationship that was moderately supported (BP=89, PP=0.98) (Figure 6).
3.4 Phylogenetic informativeness
PhyDesig was used to evaluate the profiles of the per-site and net phylogenetic informativeness (PI) values of the 74 genes (Figure 7). The maximum net PI value was observed for rpoC2, followed by rpoB, ndhF, and matK. PCGs with high net phylogenetic informativeness values had longer lengths, suggesting that gene length might contribute significantly to phylogenetic information. rpoC2 also performed well in the analysis of per-site PI, followed by rpoB and matK (Table S5). Genes with less net PI were relatively conservative and tended to be primarily related to the subunits of the ribosome or ATP synthase or shorter in length.
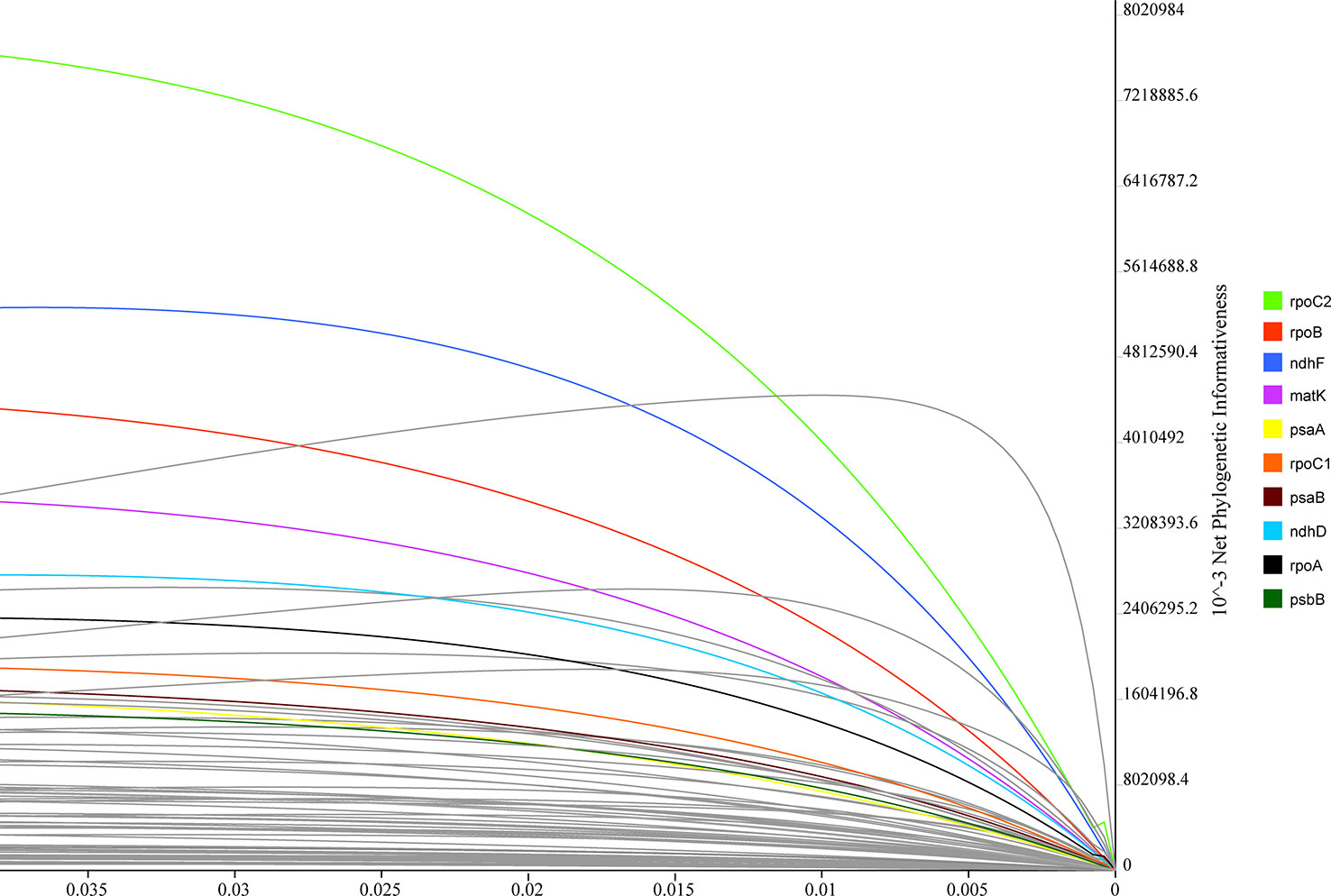
Figure 7 Phylogenetic net informativeness profiles of 74 protein-coding genes of Lobelioideae estimated using PhyDesign. Ten genes with the greatest informativeness are color-coded and indicated on the right. X- and Y-axes represent relative-time and net phylogenetic informativeness, respectively.
3.5 Positive selection
We used the results of BEB to determine sites under highly positive selection (P>99%,**). Ten slightly positive sites (P>95%) were also detected. Sites under positive selection within twenty-four genes were predicted (Table S6). The clpP and rps7 genes contained the most sites under positive selection, at more than 20 sites, followed by cemA (2, 2), matK (8, 3), rbcL (9, 6), rpl2 (0, 3), rpl16 (5, 5), rpoA (3, 3), rpoB (0, 8), rpoC1 (0, 1), rps2 (2, 3), rps3 (2, 3), rps4 (2, 3), rps8 (5, 7), rps11 (7, 7), rps14 (4, 4), rps15(1, 2), and rps18 (2, 2). The LRT results further supported the positively selected codon sites identified (Table S7). A branch-site model that does not permit positive selection (Model Anull) was compared with a model in which positive selection on the specified branches was allowed (Model A). For L. sect. Stenotium and L. sect. Delostemon, we used this model to detect 11 positive selections in five PCGs: atpF (two sites), clpP (three sites), matK (two sites), psbK (one site), and rbcL (three sites) (Table 2).
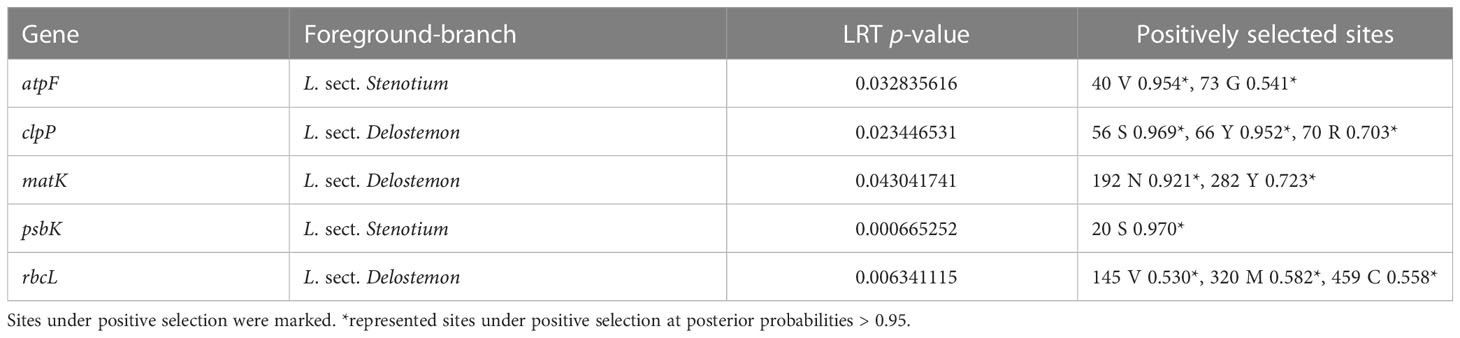
Table 2 Genes and sites under positive selection in the plastomes of East Asian lobelias.
4 Discussion
4.1 Plastome features
In the present study, the plastomes of all East Asian lobelias were found to have many dispersed and longer repeats and exhibited structural variations. Repeats have been reported to be associated with architectural variations in the plastid genome (Huang et al., 2014; Knox, 2014; Choi et al., 2019). Large ORF was not observed in the LSC regions of L. zeylanica and L. heyneana. However, the other 18 accessions with large LSC regions contained three typical ORFs, suggesting that ORFs existing in the LSC regions might be relevant to the expansion of the LSC region. These ORFs have been found in some plastomes of Campanulaceae (Knox, 2014), indicating that such a genome structure may reflect their shared evolutionary history.
We demonstrated that the IRb/LSC border occurs at different positions within rps19 or rpl2. In contrast, the IRa/LSC borders were located in the regions of rpl2-trnH-GUG for most Asian lobelias. Angiosperms shared this common IRa/LSC boundary, except for monocots whose trnH gene is located in the regions of rpl2-rps19 (Wang et al., 2008; Givnish et al., 2018). The L. zeylanica plastid genome had a shorter LSC region (82,611 bp) but a larger IR region (42,089 bp) than the other Asian lobelias studied, which might be associated with the expansion of the junctions between the LSC and IR regions (Bendich, 2004; Li et al., 2020). L. taliensis, L. doniana, and P. nummularia_S08798 exhibited contractions of the IR region that had not yet been identified. A possible interpretation is that it is an illegitimate recombination event (Blazier et al., 2011; Downie and Jansen, 2015). P. nummularia_S08798 and P. nummularia_MF061203 showed different LSC/IRb junctions, which require further research in the future. Large-scale IR expansion/contraction has been illustrated to be a vital contributor to the plastome size variation (Ruhlman and Jansen, 2014; Shen et al., 2020). IR expansion/contraction is not uncommon in angiosperm plastid genomes and is known to occur in some groups, such as Pelargonium (Weng et al., 2017), Passiflora (Rabah et al., 2019), and Euphorbia (Wei et al., 2021).
We analyzed 20 complete plastomes of East Asian lobelias. The gene content of the plastomes was similar to that of other angiosperms. Despite the shared gene structure among the plastomes of L. sect. Hypsela, L. sect. Speirema, L. sect. Rhynchopetalum, IR/LSC boundaries, genome size, and the number of repeats were variable, indicating the non-conservative nature of plastomes evolution within these sections. L. zeylanica and L. heyneana had structural rearrangements (including large inversions) in the LSC regions that were not identified in other accessions. Therefore, this could be attributed to the different evolutionary histories of the L. sect. Delostemon and L. sect. Stenotium. Based on a comparison of plastome architectures from diverse land plant species, IR boundary shifts and inversions are the main processes of structural rearrangements, with frequency variations among lineages (Mower and Vickrey, 2018; Mower et al., 2019). Inversions have been identified in scattered angiosperm lineages; for example, the ~36 kb inversion detected in most core Genistoids (Martin et al., 2014), the ~125 kb inversion identified across the family Campanulaceae (Knox, 2014), and the ~49 kb inversion found in Circaeasteraceae (Sun et al., 2017). Large inversions are predicted to have significant value in constructing phylogenies because of their rarity, easily identified homologous features, and the deduced state polarization (Jansen et al., 2008; Shrestha et al., 2019).
4.2 Molecular markers
East Asian lobelias are traditional medicinal plant species because of their bioactivity and many chemical constituents (Anand et al., 2009; Han et al., 2013; Zheng et al., 2021). However, the scarcity of genomic resources for East Asian lobelias has hampered plant genetics, ecology, taxonomy, and resource utilization. The plastomes assembled in the present study provide new insights into the evolutionary history of plastids and offer valuable plant genetic resources which will be helpful in future genetic and taxonomic research of lobelia species in East Asia (Lee et al., 2019). Mononucleotide repeat and microsatellites were the most numerous markers and have also been described in other lineages such as Cyatheaceae (Zhu et al., 2021) and Campanuloideae (Li et al., 2020).
Our results indicated that the plastomes of Lobelioideae exhibited high levels of divergence in eight PCGs, which could function as useful genetic information beneficial for species authentication and phylogenetic assessment. The protein-coding region of clpP (pi close to 0.1) exhibited higher nucleotide diversity in all plastomes. Several studies (Kuo et al., 2011; Dugas et al., 2015; Peterson et al., 2022) have revealed different plastid genes such as matK, ccsA, and clpP used in the phylogenetic reconstructions at various taxonomic levels. For the herbal medicinal genus Lobelia, the highly variable regions acquired from this study may be promising molecular barcodes for authenticating herbarium specimens and medicinal quality assurance.
4.3 Phylogenetic relationships of East Asian lobelias
Twenty-nine genera constitute the Lobelioideae (Lammers, 2007a; Lammers, 2007b). Phylogenetic relationships within the broad Lobelioideae remain unclear, mainly because of the many segregates detached from Lobelia. This large genus has been found to be polyphyletic using molecular data (Antonelli, 2008; Knox, 2014; Knox and Li, 2017), which was supported by the synthesis of diverse phenotypic data (Lammers, 2011). Lobelia is one of the most complex genera across Lobelioideae and shows morphological diversity and a sub-cosmopolitan distribution (Lammers, 2011). None of the previous phylogenetic studies have sampled all five sections of the East Asian lobelias. Therefore, in this study, 17 new plastomes from all sections were sequenced and combined with previously published data to reconstruct the phylogeny. Most nodes were supported with higher BP and PP values, indicating that the relationships were robust. This phylogenomic framework confirmed the phylogenetic position of the East Asian lobelias within the Lobelioideae subfamily. The sectional partition of these species, shown in the phylogenetic tree, coincided with the classification of Lammers (2011). Our results indicated that L. sect. Delostemon was the earliest diverging group and L. sect. Stenotium was the transitional group, whereas L. sect. Rhynchopetalum was the derived group and comprised giant lobelias.
We showed for the first time that L. heyneana is a sister species to various African species. L. zeylanica and L. alsinoides are closely related, which is consistent with their morphological classification (Hong et al., 2011). L. zeylanica and L. alsinoides were sisters to Grammatotheca, which is endemic in South Africa. L. sect. Hypsela and Australasian species were in sister clades, in agreement with the results of previously published studies (Chen et al., 2016; Knox and Li, 2017). The close relationships among L. seguinii, L. colorata and L. davidii, were strongly supported, revealing the discordance between our study and the observations of an earlier study which positioned L. sequinii close to L. melliana (Chen et al., 2016). This inconsistency likely resulted from the sparse taxon samples and the few phylogenetic markers used. L. pyramidalis differs from L. seguinii only in corolla color and capsule shape (white or rose corolla and subglobose capsule versus purple-red or purple-blue corolla and ellipsoid capsule) (Hong et al., 2011). Although L. seguinii and L. pyramidalis are morphologically similar, they are not closely related. L. doniana was once considered a variant of L. seguinii by Wimmer (Pfl. R. Heft 107: 651. 1957) as their appearances are similar, albeit with subtle morphological differences. These are now considered separate species (Lammers, 2011), as confirmed by our phylogenetic analysis which did not find a close relationship. Although some pairwise species are morphologically similar, they may not be each other most relative lineages if two closely related taxa are present in divergent surroundings or if the lineages are undergoing adaptive evolution (Losos, 2011). The results of our study did not alter the ancestry of giant lobelias established by previous sampling studies (Chen et al., 2016; Knox and Li, 2017). The newly sequenced East Asian samples added in the present study will limit long-branch attraction in phylogenetic analysis, which should improve resolution and support at the base of the Oceania clades.
4.4 Phylogenetic informativeness of plastid genes
Although some advances in the analyses of phylogenetic reconstructions within Asian lobelias have been made using plastid markers, few studies have detected the sequence utility of plastomes for phylogenetic inferences in this group (Knox and Li, 2017). The severe bottleneck in the molecular phylogeny of recent diversification can be attributed to the insufficient resolution generated by the lack of phylogenetic signals (Chen et al., 2016). Therefore, identifying the plastid markers which show the highest PI values will help consolidate efforts to resolve complex phylogenies at the species level and identify plant DNA barcodes. matK and ndhF genes have been extensively used in the phylogenetic analyses of Lobelioideae (Koopman and Ayers, 2005; Antonelli, 2008). However, two longer genes (rpoC2, rpoB) with higher PI values have not yet been used in the phylogenetic reconstruction of Lobelioideae. It has been reported that the rpoC2 gene performed well in reconstructing angiosperm phylogenies (Walker et al., 2019). Therefore, the utility of the rpoC2 gene as a valuable plastid locus may be helpful in the phylogenetic inferences of Lobelioideae. The present study provides a standard procedure for genetic marker selection. Primer availability is crucial for a realistic assessment of the PI of markers in deep learning.
4.5 Signatures of positive selection on plastid genes and sites
Lobelia is almost cosmopolitan but is most diverse in the tropics and subtropics, with several taxa extending into temperate regions. Most East Asian lobelia species are found in high-altitude environments (Lammers, 2011). Thus these plants are continuously exposed to super ultraviolet radiation, a capricious climate, low temperatures, and high oxygen levels (Askew, 2002; Zhang et al., 2016). The site-specific model revealed 173 sites under highly positive selection in the 63 PCGs with posterior probabilities greater than 0.99. Positively selected genes have functions in both photosynthesis and the plastid genetic system, illustrating that the functional genes in the plastid genome significantly affect plant evolution (Hao et al., 2010; Jiang et al., 2018; Gao et al., 2022). In addition, positive selection is associated with shifts in function and the environment (Hu et al., 2014; Li et al., 2018). Thus, the sites under positive selection determined in this study might drive the changes in PCGs, allowing adaptation to diverse and harsh habitats (Li et al., 2018; Dhar et al., 2020; Peng et al., 2022).
We identified five genes (atpF, clpP, matK, psbK, and rbcL) that putatively underwent positive selection within the species of L. sect. Stenotium and L. sect. Delostemon. The atpF gene encodes a subunit of ATP synthase, which is essential for photosynthetic energy transduction (Walker, 2013). The accelerated evolutionary rate of atpF may enhance the efficiency of photosynthetic energy transduction systems. clpP, which encodes a proteolytic subunit of the Clp protease involved in importing proteins into plastids (Krause, 2012; Ravin et al., 2016), is presumed necessary for maintenance (Delannoy et al., 2011). A previous study has indicated that positive selection in the clpP gene is widespread in flowering plants (Erixon and Oxelman, 2008; Rockenbach et al., 2016; Williams et al., 2022). matK gene is suggested to be the plastid-encoded maturase of group II intron, and is therefore involved in the post-transcriptional processing of chloroplasts (Barthet and Hilu, 2007; Qu et al., 2018). In contrast to other groups of IIA ORFs, matK proteins lack domains that perform reverse transcriptase and endonuclease functions (Wicke et al., 2011). Positive selection sites in matK suggest positive selection fixes beneficial changes within East Asian lobelias. psbK, a subunit of photosystem II (Wu et al., 2020), was also positively selected. To our knowledge, the positive selection of psbK is uncommon in angiosperms. Positive selection may imply a unique ability of East Asian species to adapt to different light environments. rbcL is influenced by positive selection. This gene encodes the RuBisCO large subunit and plays a vital role in the adaptation of plants to environmental changes related to drought and low temperature. The positive selection of rbcL has also been described in some groups, such as Brassicaceae (Liu et al., 2012) and IIex (Aquifoliaceae) (Yao et al., 2019).
5 Conclusion
Early analyses used DNA fragmentation data to reconstruct the phylogenetic relationships of East Asian lobelias. Most current phylogenomic studies have suffered from the need to use more samples from the L. sect. Hypsela, L.sect. Rhynchopetalum, and L. sect. Speirema. Our phylogenomic framework inferred from complete plastomes illustrates the likely evolutionary relationships among East Asian lobelias based on all five representative sections. L. melliana was still found to be a sister species to the Oceanian and Afrotropic giant lobelias. Our study also demonstrates that the rarely used plastid marker in Lobelioideae phylogenetic reconstruction, rpoC2, shows considerable phylogenetic informativeness, which will advance the phylogenetic analyses of this subfamily. The IR/LSC boundaries, genome size, and the number of plastome repeats in L. sect. Hypsela, L. sect. Speirema, and L. sect. Rhynchopetalum were variable, indicating the non-conservative nature of the plastome structural evolution within these sections. In contrast, L. zeylanica and L. heyneana had structural rearrangements in their LSC regions, suggesting they might have undergone different evolutionary histories. Identifying and authenticating species with medicinal significance are essential for the quality control of medicinal plants. We found highly divergent plastomes sequences, which would benefit future taxonomic work and population genetics studies of East Asian lobelias. Furthermore, the signatures of positive selection identified in some of the 74 protein-coding genes detected here provide insights into the role of plastomes in adaptive evolution. However, a more intensive sampling of Lobelioideae is essential for understanding their diversity. Moreover, the availability of plant plastomes will help elucidate the mode of molecular evolution.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Genbank accession numbers: OQ148738-OQ148751.
Author contributions
C-JL collected samples, and wrote the manuscript; X-TX and H-XL collected samples and performed the analyses; R-NW performed the analyses; D-ZL designed the research and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Chinese Academy of Sciences, grant number XDB31000000. This work was also facilitated by the Shenyang Normal University Ph.D., Introduced Talents Scientific Research Project Startup Fund, grant number BS202211.
Acknowledgments
We appreciate the help from the Herbarium of Kunming Institute of Botany (KUN) for providing materials.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1144406/full#supplementary-material
Appendix S1 | Repeats among the East Asian lobelias.
References
Amiryousefifi, A., Hyvonen, J., Poczai, P. (2018). IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Anand, A., Roy, A., Bhargava, B., Raj, H., Barde, P. B., Vijayan, V.k. (2009). Early symptom-relief after valvulotomy in mitral stenosis indicates role of lobeline-sensitive intrapulmonary receptors. Respir. Physiol. Neurobiol. 169, 297–302. doi: 10.1016/j.resp.2009.09.007
Antonelli, A. (2008). Higher level phylogeny and evolutionary trends in campanulaceae subfam. lobelioideae: molecular signal overshadows morphology. Mol. Phylogenet. Evol. 46, 1–18. doi: 10.1016/j.ympev.2007.06.015
Antonelli, A., Verola, C. F., Parisod, C., Gustafsson, A. L. S. (2010). Climate cooling promoted the expansion and radiation of a threatened group of south American orchids (Epidendroideae: Laeliinae). Biol. J. Linn. Soc 100, 597–607. doi: 10.1111/j.1095-8312.2010.01438.x
Askew, E. W. (2002). Work at high altitude and oxidative stress: Antioxidant nutrients. Toxicology 180, 107–119. doi: 10.1016/S0300-483X(02)00385-2
Barthet, M. M., Hilu, K. W., Hilu, K. W. (2007). Expression of matK: functional and evolutionary implications[J]. Am. J. Bot. 94, 1402–1412. doi: 10.3732/ajb.94.8.1402
Bendich, A. J. (2004). Circular chloroplast chromosomes: The grand illusion. Plant Cell. 16, 166–1666. doi: 10.1105/tpc.160771
Blazier, J., Guisinger, M. M., Jansen, R. K. (2011). Recent loss of plastidencoded ndh genes within Erodium (Geraniaceae). Plant Mol. Biol. 76, 263–272. doi: 10.1007/s11103-011-9753-5
Braukmann, T. W. A., Broe, M. B., Stefanović, S., Freudenstein, J. C. (2017). On the brink: The highly reduced plastomes of nonphotosynthetic ericaceae. New Phytol. 216, 254–266. doi: 10.1111/nph.14681
Brudno, M., Do, C. B., Cooper, G. M., Kim, M. F., Davydov, E., Program, C. S., et al. (2003). LAGAN and multi-LAGAN: effiffifficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13, 721–731. doi: 10.1101/gr.926603
Chen, L. Y., Wang, Q. F., Renner, S. S. (2016). East Asian Lobelioideae and ancient divergence of a giant rosette lobelia in Himalayan Bhutan. Taxon 65, 293–304. doi: 10.12705/652.6
Choi, I. S., Jansen, R., Ruhlman, T. (2019). Lost and found: return of the inverted repeat in the legume clade defined by its absence. Genome Biol. Evol. 11, 1321–1333. doi: 10.1093/gbe/evz076
Crowl, A. A., Miles, N. W., Visger, C. J., Hansen, K., Ayers, T., Haberle, R., et al. (2016). A global perspective on campanulaceae: Biogeographic, genomic, and floral evolution. Am. J. Bot. 103, 233–245. doi: 10.3732/ajb.1500450
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Dhar, D., Dey, D., Basu, S., Fortunato, H. (2020). Understanding the adaptive evolution of mitochondrial genomes in intertidal chitons. BioRxivorg 3, 980664. doi: 10.1101/2020.03.06.980664
Delannoy, E., Fujii, S., Colas des Francs-Small, C., Brundrett, M., Small, I. (2011). Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 28, 2077–2086. doi: 10.1093/molbev/msr028
Downie, S. R., Jansen, R. K. (2015). A comparative analysis of whole plastid genomes from the apiales: expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 40, 336–351. doi: 10.1600/036364415X686620
Doyle, J. J. (1987). A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochemical Bull. 19, 11–15. doi: 10.1371/journal.pone.0077965
Dugas, D. V., Hernandez, D., Koenen, E. J. M., Schwarz, E., Straub, S., Hughes, C. E., et al. (2015). Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions and accelerated rate of evolution in clpP. Sci. Rep. 5, 1–13. doi: 10.1038/srep16958
Erixon, P., Oxelman, B. (2008). Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PloS One 3, e1386. doi: 10.1371/journal.pone.0001386
Felsenstein, J. (1989). PHYLIP–phylogeny inference package (version 3.2). Cladistics 5, 164–166. doi: 10.1126/science.246.4932.941
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, 273–279. doi: 10.1093/nar/gkh458
Gao, F., Chen, C., Arab, D. A., Du, Z., He, Y., Ho, S. Y. W. (2019). EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Gao, Y., Thiele, W., Saleh, O., Scossa, F., Arabi, F., Zhang, H., et al. (2022). Chloroplast translational regulation uncovers nonessential photosynthesis genes as key players in plant cold acclimation. Plant Cell. 34, 2056–2079. doi: 10.1093/plcell/koac056
Gitzendanner, M. A., Soltis, P. S., Wong, G. K. S., Ruhfel, B. R., Soltis, D. E. (2018). Plastid phylogenomic analysis of green plants: a billion years of evolutionary history. Am. J. Bot. 105, 291–301. doi: 10.1002/ajb2.1048
Givnish, T. J., Zuluaga, A., Spalink, D., Gomez, M. S., Lam, V. K. Y., Saarela, J. M., et al. (2018). Monocot plastid phylogenomics, timeline, net rates of species diversification, the power of multi-gene analyses, and a functional model for the origin of monocots. Am. J. Bot. 105, 1888–1910. doi: 10.1002/ajb2.1178
Han, S. R., Lv, X. Y., Wang, Y. M., Gong, H., Zhang, C., Tong, A. N., et al. (2013). A study on the effect of aqueous extract of Lobelia chinensis on colon precancerous lesions in rats. Afr J. Tradit Complement Altern. Med. 10, 422–425. doi: 10.4314/ajtcam.v10i6.2
Hao, D. C., Chen, S. L., Xiao, P. G. (2010). Molecular evolution and positive Darwinian selection of the chloroplast maturase matK. J. Plant Res. 123, 241–247. doi: 10.1007/s10265-009-0261-5
Heckenhauer, J., Paun, O., Chase, M. W., Ashton, P. S., Kamariah, A. S., Samuel, R. (2019). Molecular phylogenomics of the tribe shoreeae (Dipterocarpaceae) using whole plastid genomes. Ann. Bot. 23, 857–865. doi: 10.1093/aob/mcy220
Hernandez-Leon, S., Gernandt, D. S., Perez de la Rosa, J. A., Jardon-Barbolla, L. (2013). Phylogenetic relationships and species delimitation in Pinus section trifoliae inferrred from plastid DNA. PloS One 8 (7), e70501. doi: 10.1371/journal.pone.0070501
Hong, D. Y., Ge, S., Lammers, T. G., Klein, L. L. (2011). CampanulaceaeFlora of China Vol. 19. Ed. Turland, N. (St. Louis: Missouri Botanical Garden Press), 505–563. doi: 10.11646/phytotaxa.255.2.9
Hu, Q., Zhu, Y., Liu, Y., Wang, N., Chen, S. (2014). Cloning and characterization of wnt4a gene and evidence for positive selection in half-smooth tongue sole (Cynoglossus semilaevis). Sci. Rep. 4, 7167. doi: 10.1038/srep07167
Huang, H., Shi, C., Liu, Y., Mao, S. Y., Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14, 1–17. doi: 10.1186/1471-2148-14-151
Jansen, R. K., Wojciechowski, M. F., Sanniyasi, E., Lee, S. B., Daniell, H. (2008). Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 48, 1204–1217. doi: 10.1016/j.ympev.2008.06.013
Jiang, P., Shi, F. X., Li, M. R., Liu, B., Wen, J., Xiao, H. X., et al. (2018). Positive selection driving cytoplasmic genome evolution of the medicinally important ginseng plant genus Panax. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00359
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Kagame, S. P., Gichira, A. W., Chen, L. Y., Wang, Q. F. (2021). Systematics of lobelioideae (Campanulaceae): review, phylogenetic and biogeographic analyses. PhytoKeys 174, 13. doi: 10.3897/phytokeys.174.59555
Katoh, K., Standley, D. M. (2014). “MAFFT: iterative refinement and additional methods,” in Multiple sequence alignment methods (Totowa, NJ: Humana Press), 131–146. doi: 10.1007/978-1-62703-646-7_8
Kearse, M., Moir, R., Wilson, A., Stones, H. S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Knox, E. B. (2014). The dynamic history of plastid genomes in the campanulaceae sensu lato is unique among angiosperms. Proc. Natl. Acad. Sci. U.S.A. 111, 11097–11102. doi: 10.1073/pnas.1403363111
Knox, E. B., Li, C. (2017). The East Asian origin of the giant lobelias. Am. J. Bot. 104, 924–938. doi: 10.3732/ajb.1700025
Koopman, M. M., Ayers, T. J. (2005). Nectar spur evolution in the Mexican lobelias (Campanulaceae: Lobelioideae). Am. J. Bot. 92, 558–562. doi: 10.3732/ajb.92.3.558
Krause, K. (2012). Plastid genomes of parasitic plants: A trail of reductions and losses. In: Bullerwell CE ed. Organelle Genetics. Heidelberg: Springer. 79–103.
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kuo, L. Y., Li, F. W., Chiou, W. L., Wang, C. N. (2011). First insights into fern matK phylogeny. mol. Phylogenet. Evol. 59, 556–566. doi: 10.1016/j.ympev.2011.03.010
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lagomarsino, L. P., Antonelli, A., Muchhala, N., Timmermann, A., Mathews, S., Davis, C. C. (2014). Phylogeny, classification, and fruit evolution of the species-rich Neotropical bellflowers (Campanulaceae: Lobelioideae). Am. J. Bot. 101, 2097–2112. doi: 10.3732/ajb.1400339
Lammers, T. G. (2007a). World checklist and bibliography of campanulaceae (Kew, Richmond: Royal Botanic Gardens).
Lammers, T. G. (2007b). Revision of Lobelia sect. Galeatella (Campanulaceae: Lobelioideae). J. Bot. Res. Inst. 1, 789–810.
Lammers, T. G. (2011). Revision of the infrageneric classification of Lobelia l. (Campanulaceae: Lobelioideae). Ann. Mo. Bot. Gard. 98, 37–62. doi: 10.3417/2007150
Lanfear, R., Calcott, B., Ho, S. Y. W., Guindon, S. (2012). PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701. doi: 10.1093/molbev/mss020
Lee, S. R., Kim, K., Lee, B. Y., Lim, C. E. (2019). Complete chloroplast genomes of all six Hosta species occurring in Korea: molecular structures, comparative, and phylogenetic analyses. BMC Genomics 20, 833. doi: 10.1186/s12864-019-6215-y
Li, X. D., Jiang, G. F., Yan, L. Y., Li, R., Mu, Y., Deng, W. A. (2018). Positive selection drove the adaptation of mitochondrial genes to the demands of flight and high-altitude environments in grasshoppers. Front. Genet. 9, 605. doi: 10.3389/fgene.2018.00605
Li, C. J., Wang, R. N., Li, D. Z. (2020). Comparative analysis of plastid genomes within the Campanulaceae and phylogenetic implications. PloS One 15, e0233167. doi: 10.1371/journal.pone.0233167
Li, C., Lu, J., Li, Y., Zhu, J., Zhang, L. (2017). Comparative morphology of the leaf epidermis in Lobelia (Lobelioideae) from China. Microsc. Res. Tech. 80, 763–778. doi: 10.1002/jemt.22862
Librado, P., Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, L., Zhao, B., Zhang, Y., Wang, J. (2012). Adaptive evolution of the rbcL gene in brassicaceae. Biochem. Syst. Ecol. 44, 13–19. doi: 10.1016/j.bse.2012.04.007
Lohse, M., Drechsel, O., Kahlau, S., Ralph, B. (2013). Organellar genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, 575–581. doi: 10.1093/nar/gkt289
López-Giráldez, F., Townsend, J. P. (2011). PhyDesign: an online application for profiling phylogenetic informativeness. BMC Evol. Biol. 11, 152. doi: 10.1186/1471-2148-11-152
Losos, J. B. (2011). Lizards in an evolutionary tree: ecology and adaptive radiation of anoles: ecology and adaptive radiation of anoles (Vol. 10) (University of California Press), 187–220.
Martin, G. E., Rousseau-Gueutin, M., Cordonnier, S., Lima, O., Michon-Coudouel, S., Naquin, D., et al. (2014). The first complete chloroplast genome of the genistoid legume Lupinus luteus: evidence for a novel major lineage-specifific rearrangement and new insights regarding plastome evolution in the legume family. Ann. Bot. 113, 1197–1210. doi: 10.1093/aob/mcu050
Mower, J. P., Ma, P. F., Grewe, F., Taylor, A., Michael, T. P., VanBuren, R., et al. (2019). Lycophyte plastid genomics: Extreme variation in GC, gene and intron content and multiple inversions between a direct and inverted orientation of the rRNA repeat. New Phytol. 222, 1061–1075. doi: 10.1111/nph.15650
Mower, J. P., Vickrey, T. L. (2018). “Structural diversity among plastid genomes of land plants,” in Advances in botanical research, vol. 85 . Eds. Chaw, S.-M., Jansen, R. K. (Cambridge, MA, USA: Academic Press), 263–292.
Peng, R., Xu, Y., Tian, S., Unver, T., Liu, Z., Zhou, Z., et al. (2022). Evolutionary divergence of duplicated genomes in newly described allotetraploid cottons. Proc. Natl. Acad. Sci. U.S.A. 119, e2208496119. doi: 10.1073/pnas.2208496119
Peterson, P. M., Romaschenko, K., Herrera, A. Y., Vorontsova, M. S. (2022). Phylogeny, classification, and biogeography of Afrotrichloris, Apochiton, Coelachyrum, Dinebra, Eleusine, Leptochloa, Schoenefeldia, and a new genus, Schoenefeldiella (Poaceae: Chloridoideae: Cynodonteae: Eleusininae). J. Syst. Evol. 60, 630–639. doi: 10.1111/jse.12803
Qu, Y., Legen, J., , J., Arndt, J., Henkel, S., Hoppe, G., Thieme, C., et al (2018). Ectopic transplastomic expression of a synthetic MatK gene leads to cotyledon-specific leaf variegation. Front. Plant Sci. 9, 1453. doi: 10.3389/fpls.2018.01453
Qu, X. J., Moore, M. J., Li, D. Z., Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 1–12. doi: 10.1186/s13007-019-0435-7
Rabah, S. O., Shrestha, B., Hajrah, N. H., Sabir, M. J., Alharby, H. F., Sabir, M. J., et al. (2019). Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Syst. Evol. 57, 1–14. doi: 10.1111/jse.12425
Rambaut, A., Suchard, M. A., Xie, D., Drummond, A. J. (2014) Tracer v1.6. Available at: http://beast.bio.ed.ac.uk/Tracer.
Rambaut, A. (2012) Fig. Tree. Tree Figure Drawing Tool, v. 1.4.0. Available at: http://tree.bio.ed.ac.uk/software/figtree/.
Ravin, N.V., Gruzdev, E.V., , Beletsky, A.V., Mazur, A.M., Prokhortchouk, E.B., Filyushin, M.A., et al. (2016). The loss of photosynthetic pathways in the plastid and nuclear genomes of the nonphotosynthetic mycoheterotrophic eudicot Monotropa hypopitys. BMC Plant Biol. 16, 153–161. doi: 10.1111/nph.14681
R Core Team (2020). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Rockenbach, K., Havird, J. C., Monroe, J. G., Triant, D. A., Taylor, D. R., Sloan, D. B. (2016). Positive selection in rapidly evolving plastid–nuclear enzyme complexes. Genetics 204, 1507–1522. doi: 10.1534/genetics.116.188268
Ronquist, F., Huelsenbeck, J., Teslenko, M. (2011) Draft MrBayes version 3.2 manual: tutorials and model summaries. Available at: http://brahms.biology.rochester.edu/software.Html.
Ruhlman, T. A., Jansen, R. K. (2014). “The plastid genomes of flowering plants,” in Chloroplast biotechnology (Totowa, NJ: Humana Press), 3–38.
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M., Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Shen, J., Zhang, X., Landis, J. B., Zhang, H., Deng, T., Sun, H., et al. (2020). Plastome evolution in Dolomiaea (Asteraceae, cardueae) using phylogenomic and comparative analyses. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.00376
Shrestha, B., Weng, M. L., Theriot, E. C., Gilbert, L. E., Ruhlman, T. A., Krosnick, S. E., et al. (2019). Highly accelerated rates of genomic rearrangements and nucleotide substitutions in plastid genomes of Passiflora subgenus Decaloba. Mol. Phylogenet. Evol. 138, 53–64. doi: 10.1016/j.ympev.2019.05.030
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Sun, Y., Moore, M. J., Lin, N., Adelalu, K. F., Meng, A., Jian, S., et al. (2017). Complete plastome sequencing of both living species of circaeasteraceae (Ranunculales) reveals unusual rearrangements and the loss of the ndh gene family. BMC Genom. 18, 592. doi: 10.1186/s12864-017-3956-3
Tamboli, A. M., Rub, R. A., Ghosh, P., Bodhankar, S. L. (2012). Antiepileptic activity of lobeline isolated from the leaf of Lobelia nicotianaefolia, and its effect on brain GABA level in mice. Asian Pac. J. Trop. Biomed. 2, 537–542. doi: 10.1016/S2221-1691(12)60092-6
Thawabteh, A., Juma, S., Bader, M., Karaman, D., Scrano, L., Bufo, S. A., et al. (2019). The biological activity of natural alkaloids against herbivores, cancerous cells and pathogens. Toxins 11, 656. doi: 10.3390/toxins11110656
Thiel, T., Michalek, W., Varshney, R. K., Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare l.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Thode, V. A., Lohmann, L. G., Sanmartín, I. (2020). Evaluating character partitioning and molecular models in plastid phylogenomics at low taxonomic levels: a case study using Amphilophium (Bignonieae, bignoniaceae). J. Syst. Evol. 58, 1071–1089. doi: 10.1111/jse.12579
Walker, J. E. (2013). The ATP synthase: The understood, the uncertain and the unknown. biochem. Soc Trans. 41, 1–16. doi: 10.1042/BST20110773
Walker, J. F., Stull, G. W., Walker-Hale, N., Vargas, O. M., Larson, D. A. (2019). Characterizing gene tree conflflict in plastome-inferred phylogenies. Peer J. 7, e7747. doi: 10.1101/512079
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8, 36. doi: 10.1186/1471-2148-8-36
Wei, N., Pérez-Escobar, O. A., Musili, P. M., Huang, W. C., Yang, J. B., Hu, A. Q., et al. (2021). Plastome evolution in the hyperdiverse genus euphorbia (Euphorbiaceae) using phylogenomic and comparative analyses: Large-scale expansion and contraction of the inverted repeat region. Front. Plant Sci. 12, 712064. doi: 10.3389/fpls.2021.712064
Weng, M. L., Ruhlman, T. A., Jansen, R. K. (2017). Expansion of inverted repeat does not decrease substitution rates in pelargonium plastid genomes. New Phytol. 214, 842–851. doi: 10.1111/nph.14375
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wicke, S., Schneeweiss, G. M., Müller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Williams, A. M., Carter, O. G., Forsythe, E. S., Mendoza, H. K., Sloan, D. B. (2022). Gene duplication and rate variation in the evolution of plastid ACCase and clp genes in angiosperms. Mol. Phylogenet. Evol. 168, 107395. doi: 10.1101/2021.09.13.460099
Wu, Z., Liao, R., Yang, T., Dong, X., Lan, D., Qin, R., et al. (2020). Analysis of six chloroplast genomes provides insight into the evolution of Chrysosplenium (Saxifragaceae). BMC Genom. 21, 1–14. doi: 10.5061/dryad.jdfn2z38m
Yang, Z. H. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yao, X., Tan, Y., Yang, J., Wang, Y., Corlett, R. T., Manen, J. F. (2019). Exceptionally high rates of positive selection on the rbcL gene in the genus Ilex (Aquifoliaceae). BMC Evol. Biol. 19, 1–13. doi: 10.1186/s12862-019-1521-1
Zhang, J., Tian, Y., Yan, L., Zhang, G., Wang, X., Zeng, Y., et al. (2016). Genome of plant maca (Lepidium meyenii) illuminates genomic basis for high-altitude adaptation in the central Andes. Mol. Plant 9, 1066–1077. doi: 10.1016/j.molp.2016.04.016
Zheng, Q., Wang, Y., Zhang, S. (2021). Beyond alkaloids: novel bioactive natural products from Lobelia species. Front. Pharmacol. 12. doi: 10.3389/fphar.2021.638210
Zhou, Q., Lin, C. W., Ng, W. L., Dai, J., Denda, T., Zhou, R., et al. (2019). Analyses of plastome sequences improve phylogenetic resolution and provide new insight into the evolutionary history of Asian Sonerileae/Dissochaeteae. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01477
Keywords: East Asian lobelias, plastomes, phylogenomic framework, highly divergent regions, positive selection, adaptive evolution
Citation: Li C-J, Xie X-T, Liu H-X, Wang R-N and Li D-Z (2023) Plastome evolution in the East Asian lobelias (Lobelioideae) using phylogenomic and comparative analyses. Front. Plant Sci. 14:1144406. doi: 10.3389/fpls.2023.1144406
Received: 14 January 2023; Accepted: 16 March 2023;
Published: 31 March 2023.
Edited by:
Gang Yao, South China Agricultural University, ChinaReviewed by:
Huasheng Peng, China Academy of Chinese Medical Sciences, ChinaHanghui Kong, South China Botanical Garden (CAS), China
Junwen Zhai, Fujian Agriculture and Forestry University, China
Copyright © 2023 Li, Xie, Liu, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: De-Zhu Li, ZHpsQG1haWwua2liLmFjLmNu