 Wentao Sheng
Wentao Sheng Jianlan Deng2
Jianlan Deng2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 27 March 2023
Sec. Plant Biotechnology
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1140043
This article is part of the Research Topic Advances in Conservation, Characterization, and Use of Asparagus Genetic Resources View all 6 articles
Garden asparagus (Asparagus officinalis L.) is a horticultural crop with high nutritional and medical value, considered an ideal plant for sex determination research among many dioecious plants, whose genomic information can support genetic analysis and breeding programs. In this research, the entire mitochondrial genome of A. officinalis was sequenced, annotated and assembled using a mixed Illumina and PacBio data. The garden asparagus circular mitochondrial genome measures 492,062 bp with a GC value of 45.9%. Thirty-six protein-coding genes, 17 tRNA and 6 rRNA genes were annotated, among which 8 protein-coding genes contained 16 introns. In addition, 254 SSRs with 10 complete tandem repeats and 293 non-tandem repeats were identified. It was found that the codons of edited sites located in the amino acids showed a leucine-formation trend, and RNA editing sites mainly caused the mutual transformation of amino acids with the same properties. Furthermore, 72 sequence fragments accounting for 20,240 bp, presentating 4.11% of the whole mitochondrial genome, were observed to migrate from chloroplast to mitochondrial genome of A. officinalis. The phylogenetic analysis showed that the closest genetic relationship between A. officinalis with onion (Allium cepa) inside the Liliaceae family. Our results demonstrated that high percentage of protein-coding genes had evolutionary conservative properties, with Ka/Ks values less than 1. Therefore, this study provides a high-quality garden asparagus mitochondrial genome, useful to promote better understanding of gene exchange between organelle genomes.
The genus Asparagus includes several important plant resources used in traditional Chinese medicine. Garden asparagus (Asparagus officinalis L.) is the most traditionally significant and best-known plant in Asparagaceae owing to its economical, nutritional, and medical value (Norup et al., 2015; Zhang et al., 2022; García et al., 2023). Over the past 20 years, the acreage of this plant has expanded significantly worldwide, especially in China (Nothnagel et al., 2022). At present, the main commercial asparagus varieties are diploid varieties that originate from the original “Violet Dutch.” (Knaflewski, 1996). It has been demonstrated that asparagus germplasm resources have a narrow genetic base (Nothnagel et al., 2012; Amian et al., 2018; Mousavizadeh et al., 2021; Ranjbar et al., 2022). Therefore, expanding the genetic diversity is a prominent goal in asparagus breeding. Interspecies hybridization is an important method for enriching genetic diversity and cultivating new cultivars (Takeuchi et al., 2018; Moreno-Pinel et al., 2021; García et al., 2022; Nothnagel et al., 2022). However, at present, the genetic relationship among many plants in the genus Asparagus remains unknown, which has hindered advances in the genetics and breeding of A. officinalis (Kubota et al., 2012; Mousavizadeh et al., 2021; García et al., 2022). Genetic sequence information is useful for clarifying the phylogenetic relationships within this genus (Kubota et al., 2012; Castro et al., 2013; Mousavizadeh et al., 2021). Although molecular markers and genomic information derived from the nuclear and chloroplast genome, which can be used to analyze the phylogeny among asparagus resources, have been examined, the taxonomic relationships and evolutionary history of this genus remains poorly understood (Castro et al., 2013; Norup et al., 2015; Wong et al., 2022). Notably, research on the phylogenetic analysis of the mitochondrial genome of garden asparagus is lacking.
As important organelles, plant mitochondria are related to the energy metabolism of cells, which affects plant growth, development, reproduction, and immunity (Bergthorsson et al., 2005; Cheng et al., 2021; Sibbald et al., 2021). Margulis’s endosymbiosis theory states that mitochondria originate from archaea, which live in nucleated cells and integrate into their organelles (Knoop, 2004; Bock, 2017; Christensen, 2021). For most higher plants, the nuclear genome is parentally inherited, whereas chloroplast and mitochondrial genomes are inherited maternally (Bergthorsson et al., 2005; Fields et al., 2022). This genetic approach eliminates the influence of paternal genes, reduces the difficulty of genetic research, and is conducive to research on genetic mechanisms (Kozik et al., 2019; Small et al., 2020). In many higher plants, mitochondrial DNA (mtDNA) is regarded as a closed-circular double-stranded structure (Wu et al., 2022). In general, mtDNA usually consists of 32-67 genes with variable lengths of coding and non-coding regions (Møller et al., 2021). Compared with nuclear and chloroplast genomes, the plant mitochondrial genome is characterized by a larger genome, rearrangement, rich repeat sequence recombination, and frequent gene acquisition (via deletion, transfer, and duplication) (Bock, 2017; Wu et al., 2022). Furthermore, mtDNA contains comprehensive information, which is valuable for phylogenetic analysis. Therefore, mtDNA can be considered a useful resource for genetic research (Rice et al., 2013; Xue et al., 2022). At present, 10,123 complete chloroplast and 585 plant mitochondrial genomes are available on NCBI (https://www.ncbi.nlm.nih.gov/genome/browse#!/overview/) (as of February 10, 2023), which provides a basis for the study of mitochondrial genomics.
A. officinalis is a model horticultural plant that is used for several studies, including those on sex-determining mechanisms, chromosome evolution, and gender diversity (Harkess et al., 2017 & Moreno et al., 2018; Harkess et al., 2020; You et al., 2022). To date, the complete nuclear and chloroplast genomes have been reported in A. officinalis and registered in the NCBI database (https://ncbi.nlm.nih.gov/) (Harkess et al., 2017; Sheng et al., 2017). However, the only registered mitochondrial genome of garden asparagus currently appears in the form of a briefing, and a detailed mitochondrial genome structural analysis has yet to be performed in A. officinalis (Sheng, 2020). In this study, we aimed to: (1) comprehensively analyze the mitochondrial genome structure and sequence variations of garden asparagus; (2) assess the gene transfer between chloroplast and mitochondria of garden asparagus; (3) explore the phylogenetic relationships and perform comparative genomic analysis of garden asparagus.
The plant material of the cultivar ‘Atlas’ in A. officinalis was first obtained from the greenhouse of Nanchang Normal University and was discriminated by Professor Guangyu Chen (Jiangxi Academy of Agricultural Sciences, Nanchang, China, http://www.jxaas.cn/index.html). The sample was deposited in the Botanical Specimen Museum (http://swx.ncnu.edu.cn), and its voucher specimen number was NCNU-B-1021. Then, young adventitious roots were sampled, and whole genomic DNA was extracted from the harvested roots using the modified CTAB method (Arseneau et al., 2017). Subsequently, Nanodrop, 1% agarose gel electrophoresis and Qubit3.0 were used for DNA purity, concentration, and integrity inspection, respectively. Qualified DNA samples were sent to BIOZERON Gene Technology (http://www.Biozeron.com/) for Illumina and PacBio sequencing.
We assembled PacBio’s long reads into contigs using the Hierarchical Genome Assembly Process workflow in SMRT Analysis software suite 2.3 (Pacific Biosciences Inc., CA, United States) (Xie et al., 2020). The Basic Local Alignment Search Tool (BLAST) was used to identify mitochondrial contigs in every draft assembly with reference to Oryza sativa (CP018169.1) and Allium cepa (NC_030100.1) (Chen et al., 2015). Then, a self-looping contig was chosen as the candidate mitochondrial genome, relying on the number of matched hits. Illumina short-read and PacBio long-read data were utilized for correcting hybrid error with racon (v1.4.20), pilon (v 1.23), (Walker et al., 2014; Vaser et al., 2016) and minimap2/miniasm (Li, 2016; Li, 2018). All raw reads (containing short-read and long-read data) were mapped into the assembled mitochondrial genome structure using Burrows-Wheeler Aligner and SAMtools (v0.1.19) (Li and Durbin, 2009; Li et al., 2009). This indicated that the correctness of this genome assembly with the Illumina short-read and PacBio long-read data was based on a near-even coverage. The mean depth of the assembled mitochondrial genome was 197× (long reads), indicating a high copy number in the garden asparagus.
The mitochondrial genome of A. officinalis was analyzed using the Geseq annotation website and was manually annotated (https://chlorobox.mpimp-golm.mpg.de/geseq.html).TRNAscan-SE software was used to predict the tRNA gene to determine its location using the default parameters (Schattner et al., 2005). The mitochondrial genome structure map was drawn using the organelle genome drawing software OGDRAW (http://ogdraw.mpimp-golm.mpg.de/cgi-bin/ogdraw.pl). The sequences most likely to be encoded by proteins were analyzed using the gene prediction software ORF Finder (https://www.ncbi.nlm.nih.gov/orffmder/).
The SSR repeats of the mitochondrial genome in A. officinalis were analyzed using MISA software (http://pgrc.ipkgatersleben.de/misa/) (Thiel et al., 2003). The length of tandem repeats and the minimum number of repeats were set as follows: repeat motifs (1, 2, 3, 4, 5, and 6 bases) with repeat numbers (8, 5, 4, 3, 3, and 3). The minimum distance between two SSRs was set to 100 bp. Tandem repeat detection was performed using tandem repeat finder software using the default parameter settings (Benson, 1999). To screen the type, length, and position of the scattered repeat sequences in A. officinalis, the REPuter software was used for prediction. The parameters were set as a minimum repeat sequence length of 30 bp, and the similarity between the repeat sequences was >90% (Kurtz et al., 2001).
The potential RNA editing sites of A. officinalis protein-coding genes were predicted using PREP-Mt software (http://prep.unl.edu/) with a cut-off value of 0.2 (Mower, 2009). We also calculated the values of synonymous (Ks) and non-synonymous (Ka) substitution rates among the protein-coding genes in the A. officinalis mitochondrial genome with six representative species (Arabidopsis thaliana, NC_037304, Nicotiana tabacum, NC_006581.1, Triticum aestivum, NC_036024.1, Ginkgo biloba, NC_027976.1, Funaria hygrometrica, KC784959.1, and Chlorella sorokiniana, NC_024626.1). MEGAX was used for sequence extraction and alignment (Kumar et al., 2018), and the Ka/Ks value was determined using DNAsP v.6.12.03 (http://www.ub.edu/dnasp/) (Rozas et al., 2017).
It is common for DNA to migrate in plants, and its frequency varies among species. DNA migration often occurs during fertilization, gametogenesis, and autophagy (Sloan and Wu, 2014). We downloaded the chloroplast genome of A. officinalis (NC_034777.1) from the NCBI database (https://www.ncbi.nlm.nih.gov/nuccore/NC_034777.1). In this study, the mitochondrial genome contigs were compared with the whole chloroplast genome sequence of A. officinalis using Blastn to determine the migration gene sequence.
To determine the differences between the mitochondrial genomes of A. officinalis and the six species (Arabidopsis thaliana, Nicotiana tabacum, Triticum aestivum, Ginkgo biloba, Funaria hygrometrica, and Chlorella sorokiniana), the mitochondrial genome size, GC content, coding genes, gene sequence insertions, and deletions were statistically analyzed and compared in this study. The plant species used for comparative analysis included algae, bryophytes, ferns, gymnosperms, and angiosperms in key positions of mitochondrial phylogenetic evolution (Ye et al., 2017). The mitochondrial genome information was obtained from the NCBI organelle genome resource library (https://www.ncbi.nlm.nih.gov/genome/browse#!/overview/).
We downloaded the mitogenomes of A. officinalis and other 41 taxa from the NCBI database (http://www.ncbi.nlm.nih.gov/genome/organelle/) (Supplementary Table S1). Among these species, not only do they have complete mitochondrial genome sequences, but they are also positioned clearly in plant taxonomy and are widely applied in molecular systematic research (Bi et al., 2016; Ye et al., 2017).
Phylogenetic tree re-constructing was carried out on 23 conserved protein-coding genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9, ccmB, ccmC, ccmFc, ccmFn, cox1, cox2, cox3, atp1, atp4, atp6, atp8, atp9, cob, and matR), which were extracted by Perl scripts. We aligned these selected gene sequences with the Muscle function plug-in in MEGAX (Kumar et al., 2018) and deleted gaps and missing data manually. Finally, we chose the AIC standard of jModelTest2.1.5 software (http://evomics.org/learning/phylogenetics/jmodeltest/) to select the best alternative model for the processed nucleotide sequence as GTR+F+R5 and constructed a phylogenetic tree using the maximum likelihood method of PhyML3.0, according to the best model (http://www.atgc-montpellier.fr/phyml/?tdsourcetag=s_pcqq_aiomsg). The degree of support of the maximum likelihood method node was obtained through 1000 replications. The gymnospermous plant Ginkgo biloba and the bryophyta plant of Marchantia paleacea were selected as the outgroup.
The total mitochondrial genome length from garden asparagus was 492,062 bp, containing 58 genes (NCBI accession no. NC_053642.1), which displayed a typical single circular structure with 45.9% GC content (Figure 1). The base compositions of A, T, G, and C were 23.89, 24.35, 26.65, and 25.11%, respectively. Seventeen transfer RNA (tRNA), six ribosomal RNA (rRNA), and 36 protein-coding genes were annotated (Table 1). Based on their functional characteristics, the protein-coding genes were categorized into nine types: NADH dehydrogenase genes (9), ATP synthase genes (5), cytochrome c biogenesis genes (4), cytochrome c oxidase genes (3), ubiquinol cytochrome c reductase genes (1), transport membrane protein genes (1), maturase genes (1), large ribosomal proteins (LRP) genes (2), and small ribosomal proteins (SRP) genes (10) (Table 1). The whole length of the protein-coding genes was 31,773 bp, accounting for 6.46% of the whole mitochondrial genome. The intergenic region length was 460,289 bp, with 46.08% GC content, which comprised 93.54% of the mitochondrial genome size. Three rRNA genes were identified in the genome, including rrn5 (117 bp), rrn18 (1,970 bp), and rrn26 (3,403 bp). Each rRNA gene contained two copies. In addition, 17 tRNA genes were identified in this genome, of which trnM-CAU had three copies and trnM-CAU was a chloroplast-derived gene.
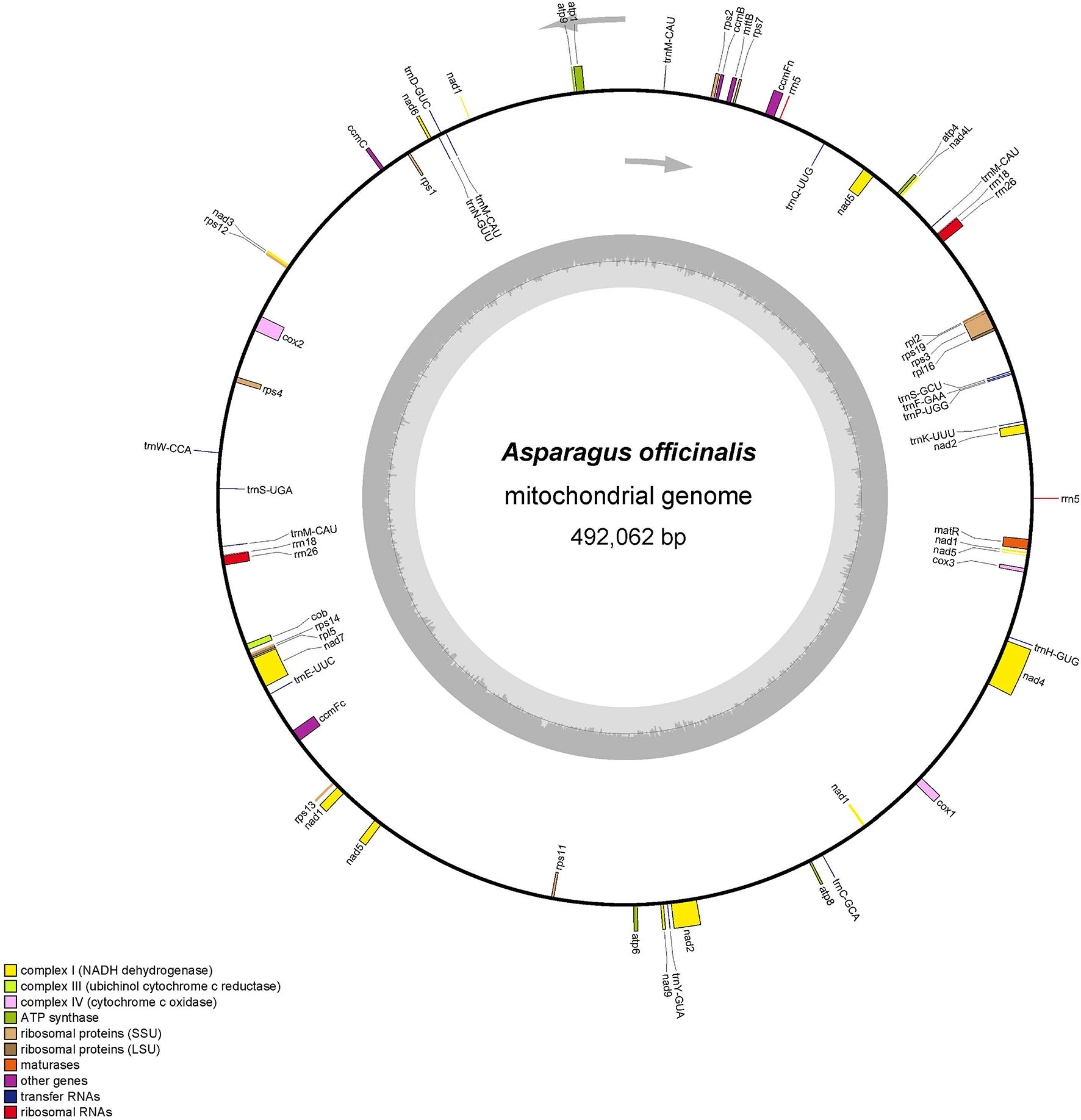
Figure 1 The mitochondrial genome organization structure of A. officinalis. The protein-coding region is marked with green, rRNA is marked with red, tRNA is marked with pink, and the repeat region is marked with brown in the circular mitochondrial genome structure. The copy starting point and direction of the L-chain replication are shown by a gray arrow.
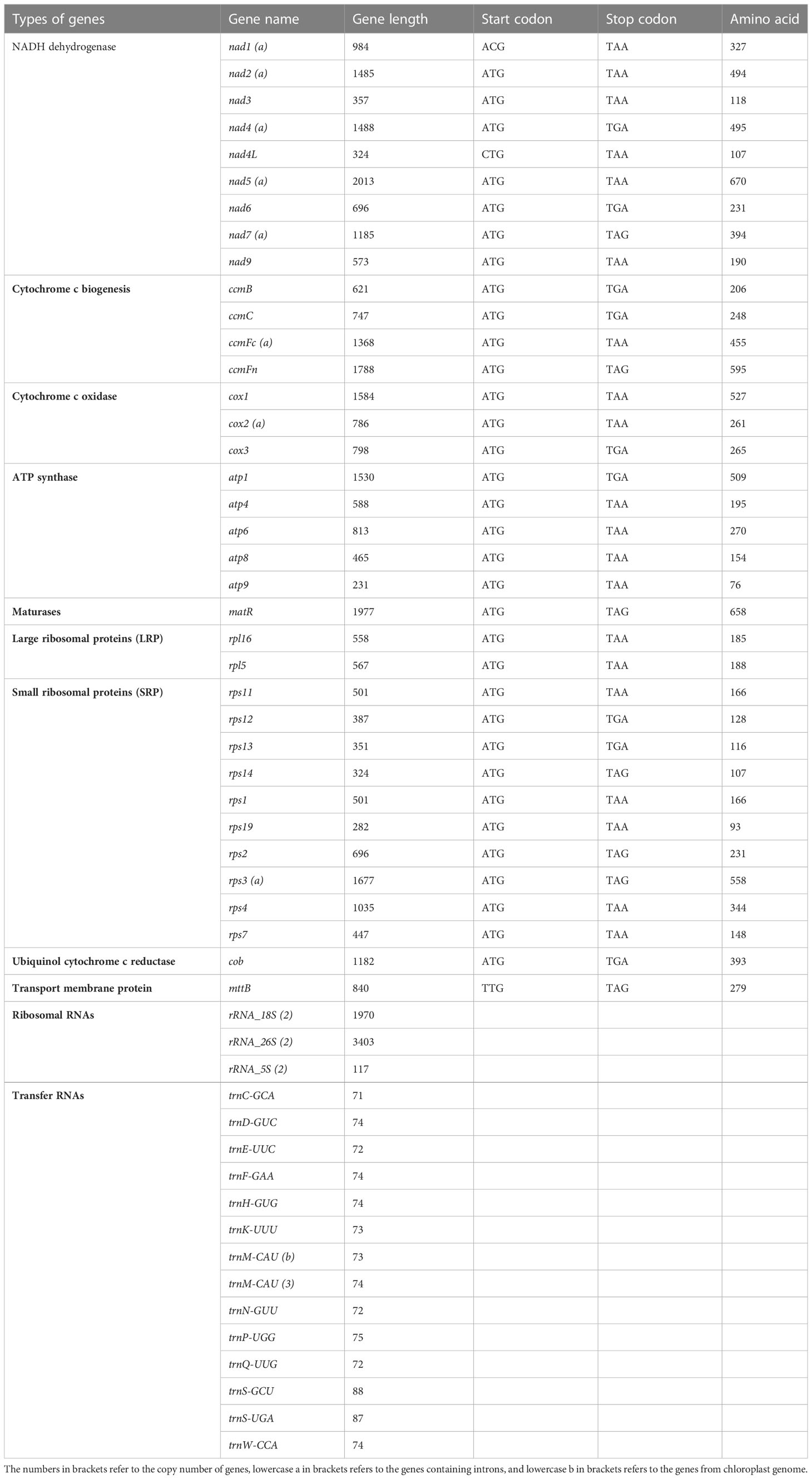
Table 1 Gene organization in the mitochondrial genome of A. officinalis.
For the annotated protein-coding genes, the ATG base combination was used as the initiation codon, except nad1 (ACG), nad4L (CTG), and mttB (TTG). Furthermore, three types of termination codons (TAA, TGA, and TAG) were detected, with utilization frequencies of 55.6%, 25%, and 19.4%, respectively. And aundant intronic variation were shown in terrestrial plants. It was found that the number of introns in the mitochondrial genome is usually 1–4 (Li et al., 2021). The rps3, ccmFc, and nad1 genes were annotated with one intron, cox2 and nad5 were composed of two introns, and nad4, nad2, and nad7 had three introns each. Moreover, the length of the intron ranged from 861 bp (nad7) to 3,339 bp (nad4) (Table 2).
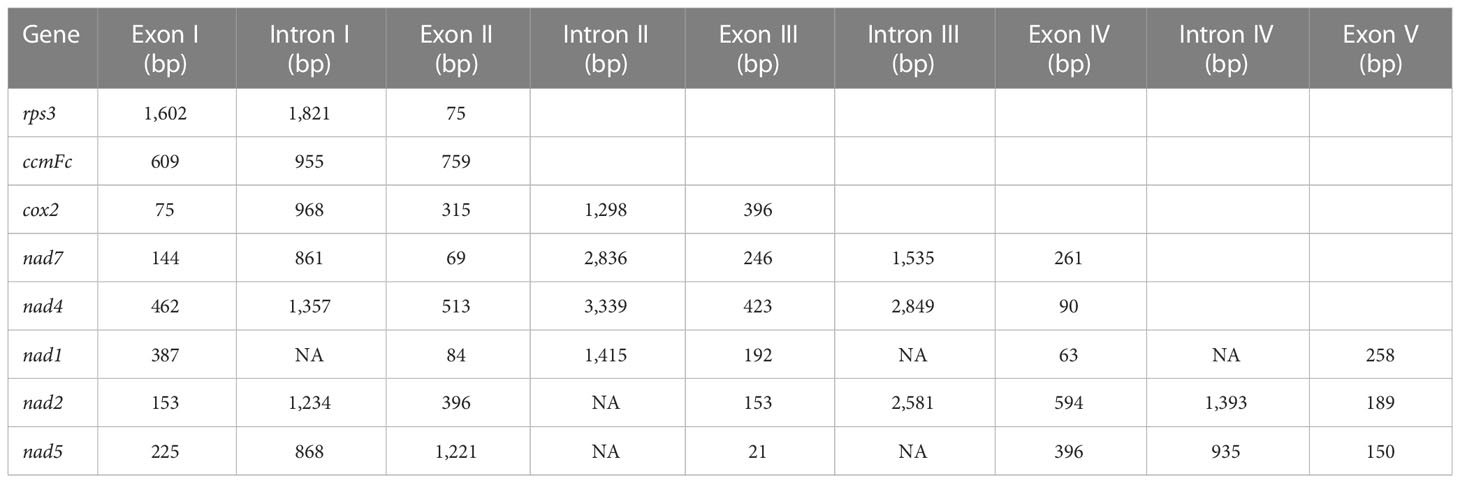
Table 2 The length of exon and intron in the A. officinalis mitochondrial genome.
Based on the sequence length, repeat sequences can be divided into short, long, and tandem repeats (Alverson et al., 2011; Ye et al., 2017). Microsatellites or simple sequence repeats (SSRs) have a sequence repeat unit length of 1–6 base pairs. Microsatellites are characterized by abundant polymorphism, dominant inheritance, high occurrence frequency, uniform genome coverage, and speediness and simplicity in PCR detection (Liu et al., 2014). SSRs in the mitochondrial genome of A. officinalis were analyzed using tandem repeat analysis software (Benson, 1999). The result showed that 254 SSRs were detected in the A. officinalis mtDNA genome, of which the number of nucleotide repeats was monomer forms (206 SSRs), dimer forms (24), trimer forms (20), tetramer forms (28), and pentamer forms (3), respectively (Supplementary Data Sheet 1). The monomer SSR form had the highest proportion of repeat types, accounting for 81.1% of total SSRs. Among the monomer SSR, adenine (A) repeat type had the highest proportion (87.9%), AG repeat had the highest proportion (75%) of dinucleotide repeat types, and there was no sequence length with repeat numbers greater than 20 bp for all repeat types.
Tandem repeats, also known as satellite DNA, are characterized by repeat lengths of 1–200 bases, with varying numbers of repeats (Liu et al., 2014). They are mainly distributed in eukaryotic genome sequences. Ten perfect tandem repeats were identified with lengths ranging from 31 to 97 bp in the A. officinalis mitochondrial genome (Table 3). The majority of the ten tandem repeats were located in the intergenic region, but only one tandem repeat with a length of 48 bp was distributed within the rpl16 gene.
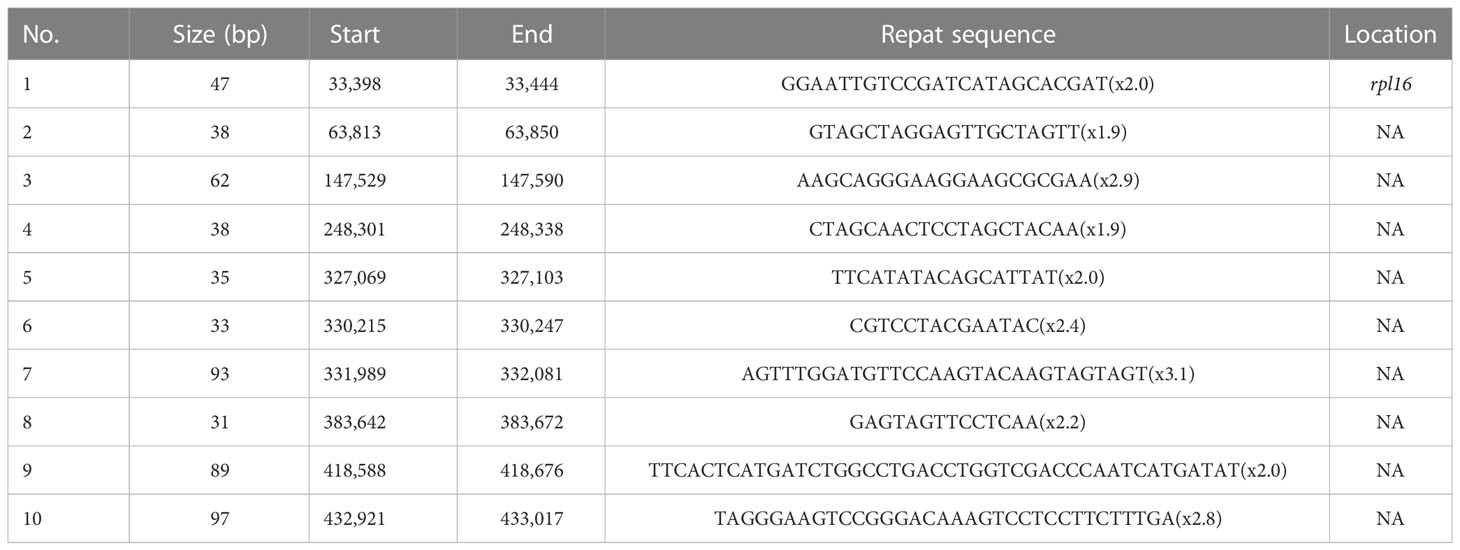
Table 3 Characteristics distribution of perfect tandem repeats in A. officinalis mitochondrial genome.
We also utilized Reputers software (http://bibiserv.techfak.uni-bielefeld.de/reputer/) to detect non-tandem repeat variations (Kurtz et al., 2001). For the long repeat sequence analysis, the parameters were set as minimum repeat size = 30 bp and edit distance = 3, to discriminate the following four repeat types: forward, reverse, complex, and palindromic repeat. It was found that 293 repeats had a length equal to or longer than 30 bp, of which 141 repeats were in the same direction, two were in the opposite direction, and 150 were in the palindrome structure (Supplementary Data Sheet 2). The longest forward repeat was 5,875 bp and the longest palindromic repeat was 12,348 bp. The length distribution of the entire repeat sequence is shown in Figure 2. The 30–34 bp segment was the most common, including 104 repeats. In contrast, the 55–59 bp segment was the least frequent type with only one repeat number.
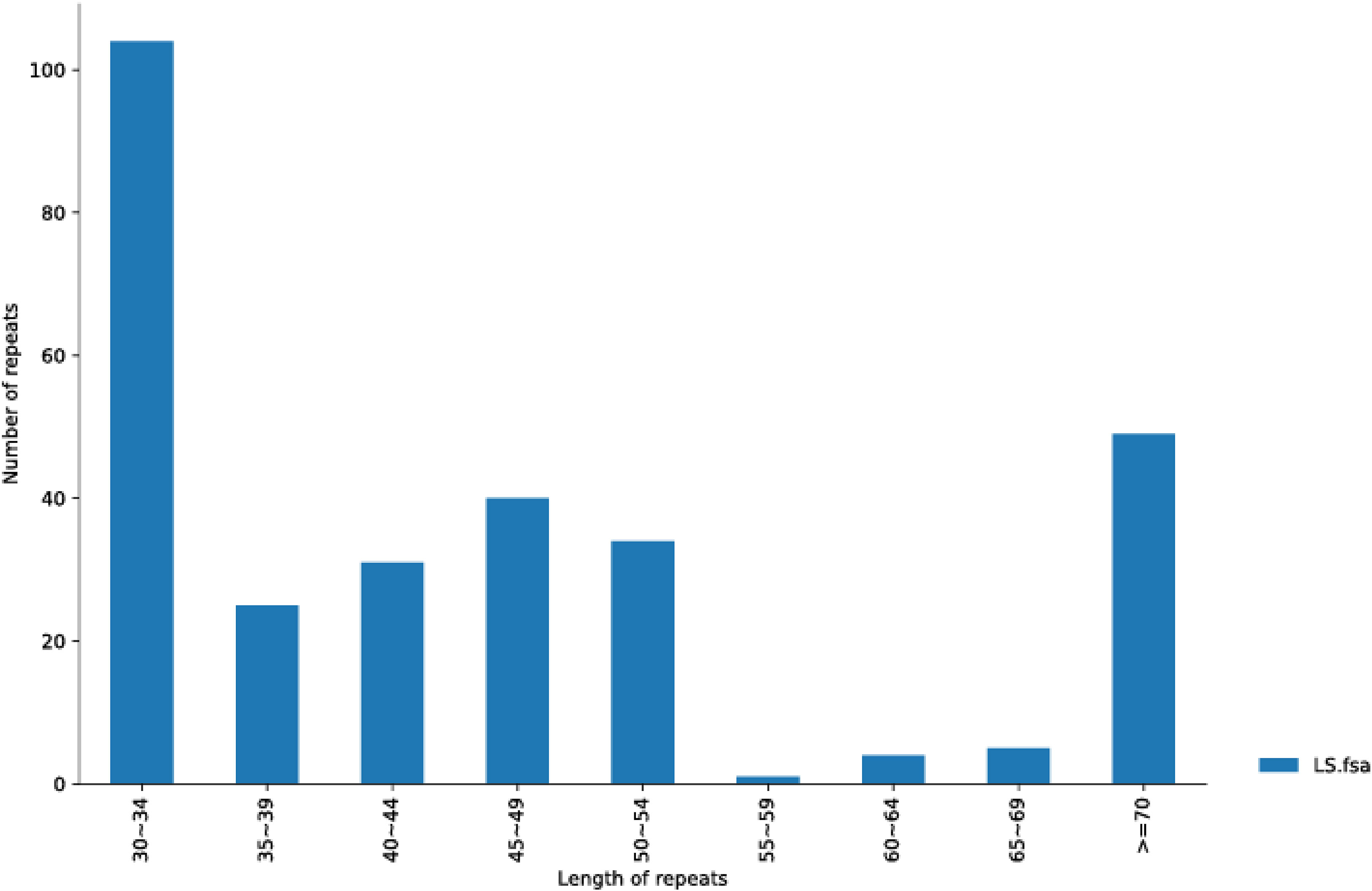
Figure 2 The repeat length distribution is shown in A. officinalis mitochondrial genome. The range size of the repetition length is displayed in the abscissa, and the number of repetitions is shown in the ordinate.
In this study, the RNA editing site of each coding gene was analyzed using the prep software (http://prep.unl.edu/), and the change in the C-U editing site was predicted by homologous protein comparison (Bergthorsson et al., 2005; Mower, 2009). The results showed that rps7 contained the least predicted editing sites (1), whereas nad4 had the most predicted editing sites (53) among these protein-coding genes (Figure 3).
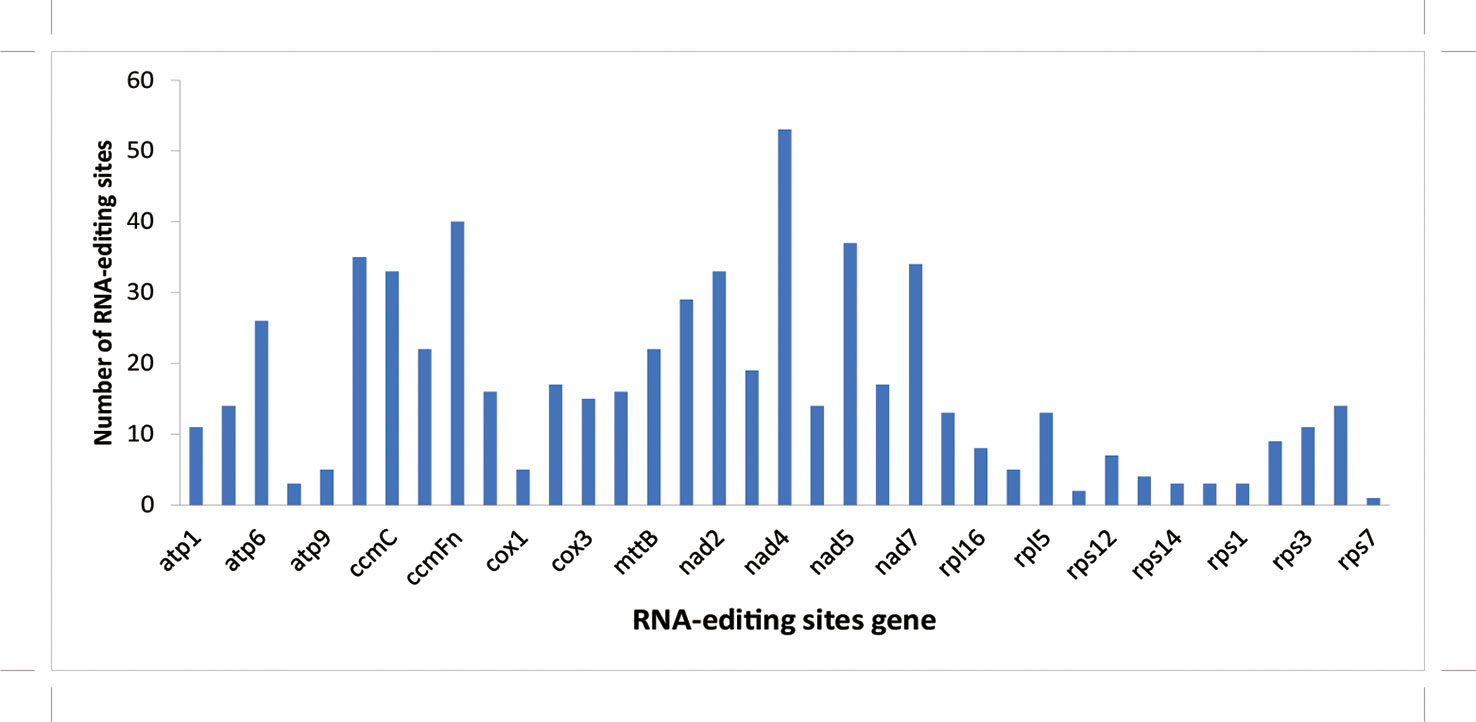
Figure 3 Prediction of RNA editing sites in protein-coding genes of mitochondrial genome in A. officinalis. The horizontal axis refers to the type of gene and the vertical axis refers to the number of editing sites.
For all identified editing sites, 215 sites were checked in the first encoding position, accounting for 35.19%, while 595 sites were marked in the second encoding position, accounting for 64.81%. Furthermore, an interesting phenomenon occurred in the predicted sites, where two base sites (CCT to TTT, CCC to TTC) changed at the same time. Meanwhile, 32.68% (200) of the hydrophobic amino acid sites remained unchanged, 11.93% (73) of the hydrophilic amino acid sites did not change, 9.15% (56) of the total sites changed from hydrophobic to hydrophilic amino acids, 45.42% (278) of hydrophilic amino acids had turned into hydrophobic amino acids, and 0.82% (5) of amino acid coding sites resulted in stop codons, located in the atp9 and ccmFc gene (CGA-TGA), rps11 and atp6 gene (CAA-TAA), and rpl16 gene (CAG-TAG), respectively. In this study, the amino acids in the codons of the edited sites were found to exhibit a leucine formation trend, which was supported by the fact that 510 amino acid editing sites (83.4%) were predicted to be converted into leucine (Table 4).
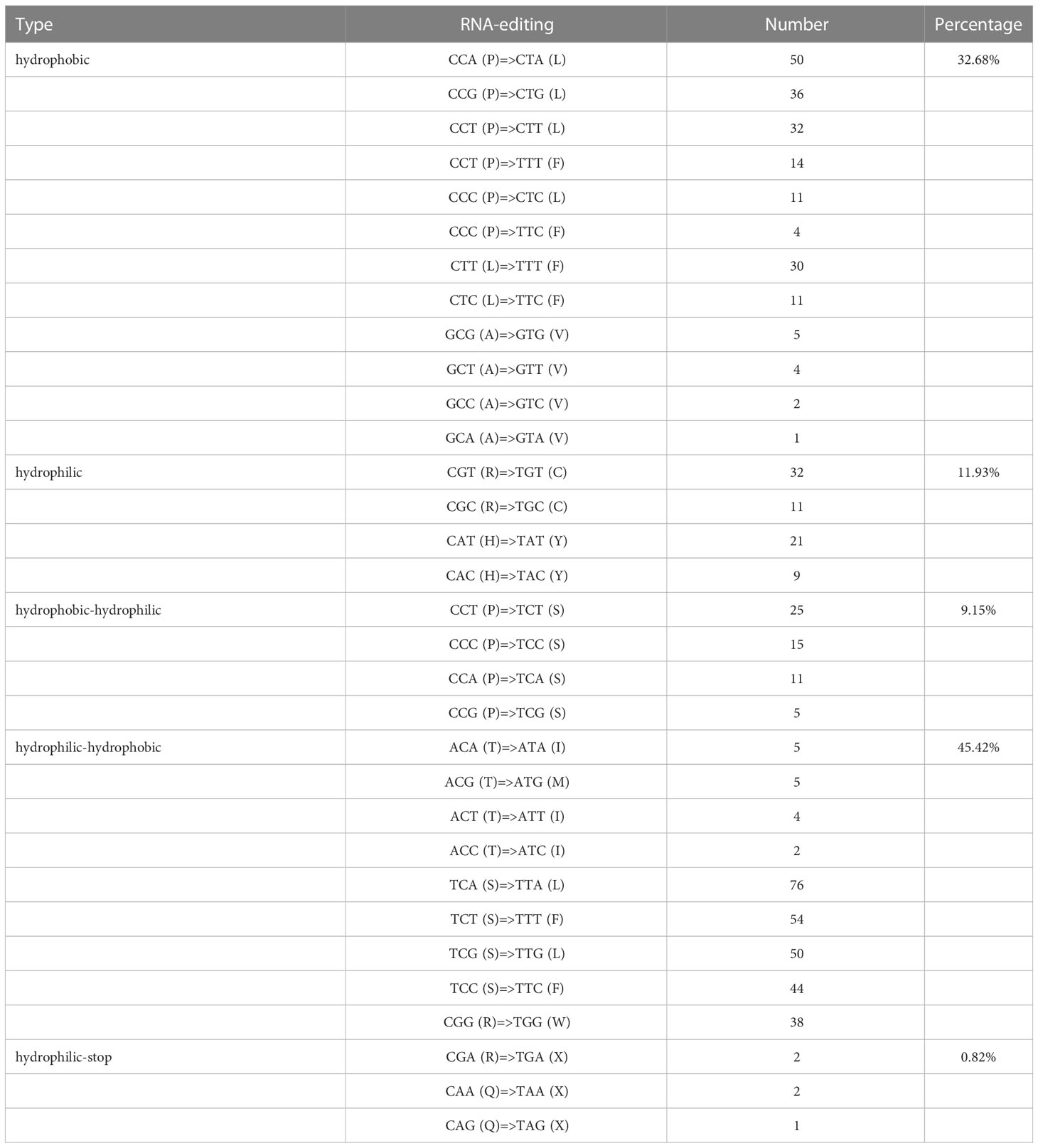
Table 4 Characteristics of hydrophilic and hydrophobic changes in RNA editing sites.
The plant mitochondrial genome is characterized by its sequence length size, genome structure and composition, gene content, and function (Kozik et al., 2019). In addition, fragment exchange between mitochondria and chloroplast is common. Approximately 5–10% of different species of the mitochondrial genome can find homologous sequences in its corresponding chloroplast genome (Sloan and Wu, 2014; Petersen et al., 2020). Homologous regions of the mitochondrial and chloroplast genomes were determined by Blastn (ncbi-blast-2.2.30+) comparison. As a result, 72 fragments of 20,240 bp were observed to migrate from the chloroplast to the mitochondrial genome of A. officinalis, accounting for 4.11% of the whole mitochondrial genome.
Moreover, four annotated gene fragments were transferred: including atp1 (460 and 89 bp segment), rrn26 (192, 192, 164, 164, 97, 97, 97, 97, 75, 69, and 69 bp segments), trnM-CAU (77 bp segment), and trnF-GAA (59 bp segment) (Supplementary Table S2). Among them, the transferred fragment in the atp1 gene accounted for 35.9% of the total length, while the rrn26 gene was 38.5%. However, nearly all trnM-CAU and trnF-GAA fragments were transferred. These data indicate that some rRNA genes, tRNA genes, and ATP synthesis-related genes are easy to transfer, presenting weaker sequence conservation during gene migration in A. officinalis.
The phylogenetic tree rebuilt using conserved mitochondrial protein-coding genes can be helpful for determining the molecular evolutionary relationships of green plants (Duminil and Besnard, 2021; Xue et al., 2022). To ascertain the taxonomic status of the mitochondrial genome in A. officinalis, phylogenetic analysis was carried out with 41 other species, including 26 eudicotyledons (rosids, 19 species; asterids, 7 species), 13 monocotyledon plants, one gymnosperm plant, and one bryophyta plant (designated as the out-group). A phylogenetic tree was reconstructed after sequence comparison based on 23 conserved protein-coding genes from the selected species.
The results showed that the phylogenetic trees strongly supported the partitioning of dicotyledons from monocotyledons and the separation of angiosperms from gymnosperms. In addition, taxa from 24 families (Leguminosae, Vitaceae, Rosaceae, Malvaceae, Caricaceae, Bataceae, Brassicaceae, Salicaceae, Cucurbitaceae, Apiaceae, Solanaceae, Amaranthaceae, Lamiaceae, Apocynaceae, Poaceae, Amaryllidaceae, Butomaceae, Arecaceae, Araceae, Hydrocharitaceae, Zosteraceae, Ginkgoaceae, and Marchantiaceae) were well clustered. The relationship in the phylogenetic tree was in line with the traditional taxonomic relationship of these species, indicating consistency between traditional classification and molecular classification at the family level. Phylogenetic analysis showed that A. officinalis had the closest genetic relationship with Allium cepa L.(Figure 4). To determine the phylogenetic relationship of A. officinalis in the genus Asparagus, it is necessary to obtain further mitochondrial genome information of smaller taxa.
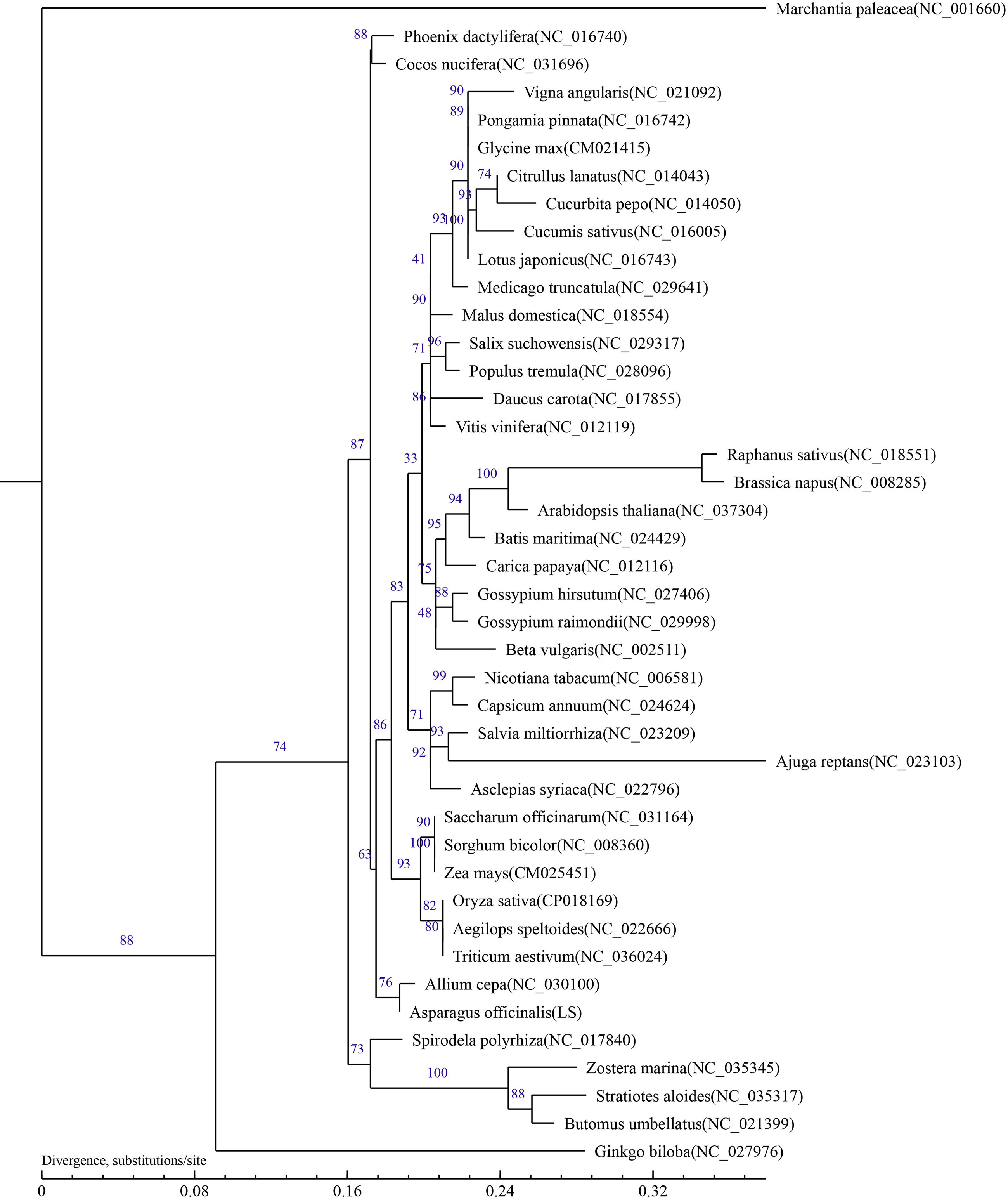
Figure 4 Phylogenetic tree inferred based on 23 conserved protein-coding genes of 42 typical plant mitochondrial genomes. Numbers on each node were bootstrap support values. Marchantia paleacea was used as the out-group.
The genome size and GC content in organelles are the two principal features of mitochondiral DNA genome research (Ye et al., 2017; Robles and Quesada, 2021). Herein, we compared A. officinalis with the six typical plants (Arabidopsis thaliana, Nicotiana tabacum, Triticum aestivum, Ginkgo biloba, Funaria hygrometrica, and Chlorella sorokiniana) in terms of their mitochondrial genome characteristics. For the lower plant types, the proportion of protein-coding genes in Chlorella sorokiniana and Funaria hygrometrica was 42.26% and 29.51%, respectively, indicating that the coding sequences of genes accounted for a high proportion in the genome (Table 5). Among the higher plant types, the ratio of protein-coding genes in A. officinalis and Arabidopsis thaliana was 6.45% and 8.53%, respectively, showing that the coding sequence of genes occupies a lower proportion in the mitochondrial genome. Related to this, the number of non-coding sequences was roughly the same as that of coding gene sequences in lower plants, but accounted for a high proportion in higher plants. These data demonstrate that with the evolution of species, the number of non-coding sequences continues to increase, resulting in a significant increase in the mitochondrial genome, which plays an important role in the composition and structure of the genome. Moreover, rRNA and tRNA genes showed the same downward trend as the species evolved. The presence of a large number of cis-splicing introns in the mitochondrial genome is also an important reason for the change in mitochondrial genome size.
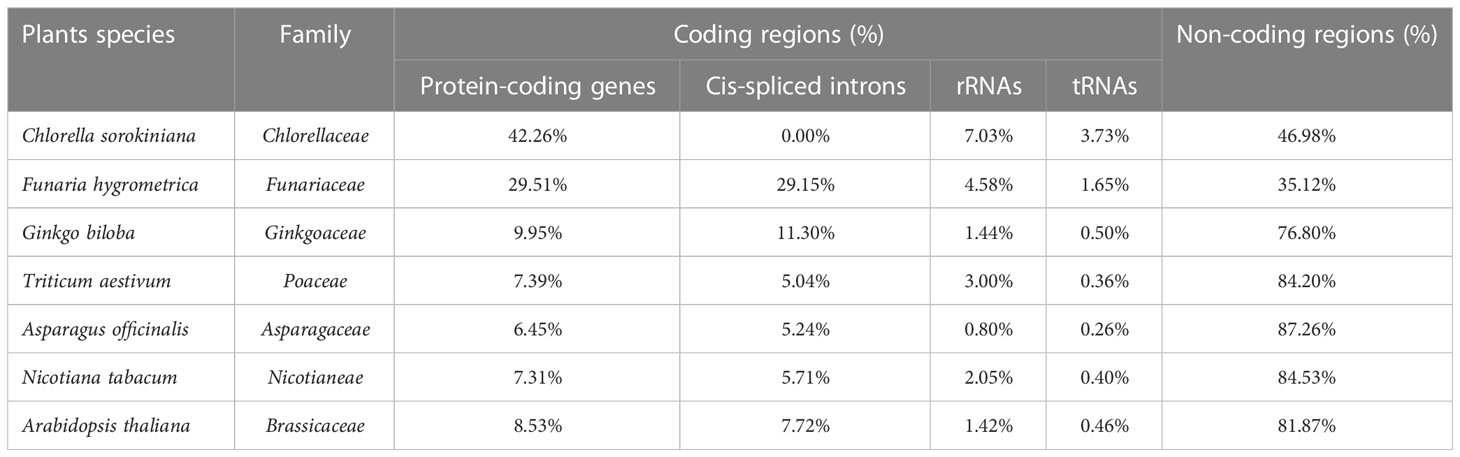
Table 5 Comparison of genome characteristics in seven plant mitochondrial genome.
The GC content of our observed plant mitochondrial genome varied from 29.11% in Chlorella sorokiniana to 50.36% in Ginkgo biloba. The results showed that the genome size of angiosperms was higher than that of algae and gymnosperms, and its size also fluctuated significantly; however, its GC content fluctuated slightly and was conservative in angiosperm plants (Supplementary Table S3). For the mitochondrial protein-coding genes, we found that the mitochondrial complex I, III, IV, and V genes and the cytochrome C synthesis gene were not lost with strong conservatism. However, the ribosomal synthetic protein, tRNA, complex II, and mttR and mattB genes were often missing in different species with high volatility and weak conservatism during plant evolution. Meanwhile, some genes, such as rps19, became pseudogenes in Triticum aestivum (Supplementary Table S3).
Selection pressures containing the non-synonymous (Ka) and synonymous (Ks) nucleotide substitution models are useful parameters in molecular evolution research (Zhang et al., 2006; Shtolz and Mishmar, 2019). The Ka/Ks ratio can be used to justify the selective pressure on specific protein-coding genes in the process of evolution, among which Ka/Ks > 1 represents positive selection, Ka/Ks = 1 represents neutral selection, and Ka/Ks < 1 refers to negative selection (Li et al., 2009; Sloan et al., 2012).
The Ka/Ks value of the common protein-coding genes was computed in the A. officinalis mitochondrial genome and compared to six typical plant species (Arabidopsis thaliana, Nicotiana tabacum, Triticum aestivum, Ginkgo biloba, Funaria hygrometrica, and Chlorella sorokiniana) (Figure 5). In total, the mean Ka/Ks value of 18 identical protein-coding genes was 0.35 in the seven analyzed species. The data showed that the Ka/Ks ratio of A. officinalis nad2 and nad3 compared to Ginkgo biloba and Triticum aestivum were larger than 1, and its ratio of A. officinalis rps12 compared to Ginkgo biloba was higher than 1, indicating a positive selection effect on its evolution. However, the majority of the protein-coding genes with Ka/Ks values < 1 indicated a negative selection effect occurring in the genes of A. officinalis compared to the other six species. This indicates that most protein-coding genes in the mitochondrial genome of A. officinalis are highly conserved during the process of molecular evolution.
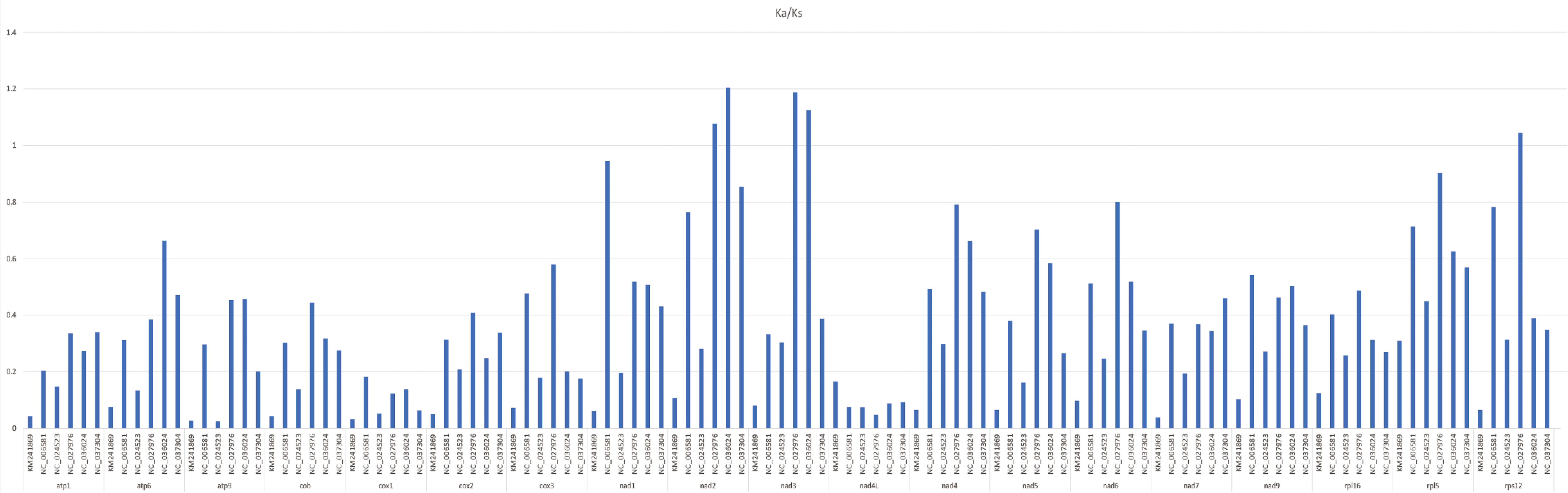
Figure 5 Ka/Ks of 18 similar protein-coding genes of A. officinalis versus six species. Deep gray and light gray boxes indicate Ka/Ks ratio of A. officinalis versus Chlorella sorokiniana, Nicotiana tabacum, Funaria hygrometrica, Ginkgo biloba, Triticum aestivum, Arabidopsis thaliana, respectively.
Generally, the plant mitochondrial genome is characterized by a complex composition, diverse structure, fluctuating non-coding sequences, and high recombination of repetitive sequences (Skippington et al., 2015; Small et al., 2020). Owing to these features, it is difficult to study plant mitochondria. In the current study, a strategy was proposed to obtain the plant mitochondrial genome based on the combined splicing of second- and third-generation whole-genome sequencing data. This principle benefits from the fact that the copy number of the plant organelle genome is much higher than that of its corresponding nuclear genome (Shtolz and Mishmar, 2019). Therefore, we applied a combination of Illumina and PacBio sequencing technologies to the study of plant mitochondria and obtained the mitochondrial genome sequence of A. officinalis, which is the first reported mitochondrial genome sequence in Asparagaceae and provides a reference for future mitochondrial genome research.
Herein, the mitochondrial genome of A. officinalis was assembled into a single circular molecule after genome sequencing, assembly, and gap-filling steps. Previous studies have found that the genome length of most plant mitochondria ranges from 200 to 700 kb, among which the smallest genome is Viscum scurruloideum (with only 66 kb) (Skippington et al., 2015) and the largest genome is Silene conica (11.3 Mb) (Richardson et al., 2013). At the same time, the content of genes also changed significantly, mainly in the range of 32 to 67 genes, with some genes, such as NADH dehydrogenase, losing or regaining their functions during evolution (Knoop, 2004). In this study, the mitochondrial genome of A. officinalis was assembled into a circular structure with a total length of 492,062 bp, encoding 58 genes, and the genome size was in the middle range. The gene types and numbers in A. officinalis mitochondria were consistent with the majority of reported circular mitochondrial genomes in most land plants (Alverson et al., 2010; Møller et al., 2021). Comparative genomic analysis revealed that the size of the genome contained in different plants was mainly caused by the spacer of the genes. A large number of unknown sequences and repetitive fragments are known to accumulate in the interval of genes. Furthermore, the intracellular transfer of genes can cause changes in the spacer (Choi et al., 2019; Fields et al., 2022). Accordingly, a large number of non-coding gene sequences were also found in the mitochondrial genome of A. officinalis, and abundant sequences from chloroplast genome transfers were observed.
Gene transfer between the chloroplast and mitochondrial genomes is an important feature of plant mitochondrial genome evolution (Notsu et al., 2002; Choi et al., 2019). Thus, tracking gene migration is essential to explore the evolution of the plant mitochondrial genome. This phenomenon is the main reason for the differences in the number of coding genes observed in different plant species (Sibbald et al., 2021). For example, the mitochondrial genome in Amborella trichopoda contains more than 250 coding genes, which may have been caused by horizontal gene transfer during evolution (Rice et al., 2013). The chloroplast genes in angiosperms are transferred to the mitochondrial genome (Sloan and Wu, 2014). Based on the data, it was found that 34 migration fragments were equal to or greater than l00 bp, of which the longest fragment was 2,013 bp. The transfer length of rrn26 was 1,388 bp, whereas in Suaeda gluaca, the gene length was only 1,369 bp, which may be the reason for the large rrn26 gene in A. officinalis.
The mechanism of male sterility in crops is related to RNA editing (Tang and Luo, 2018; Robles and Quesada, 2021). RNA editing is a means of modifying genetic information at the transcriptional level by inserting, knocking out, and replacing RNA base sequences. RNA editing in plants mainly occurs from cytosine (C) to uracil (U) (Malek et al., 1996; Tang and Luo, 2018). RNA editing regulates plant growth and development by correcting “wrong” sequences based on organelle transcripts. RNA editing usually occurs at the first or second base of the codon (Sloan and Wu, 2014; Christensen, 2021). By generating a new start codon, a new stop codon, or changing the sequence of amino acids, it promotes the formation of correct secondary protein structures, which plays important roles in gene regulation, ensuring the accurate expression of genetic information in cells (Small et al., 2020). In A. officinalis, the G/C codon usage is preferred. This preference may be due to the high binding energy of G/C codons, which is conducive to ensuring translation accuracy (Tang and Luo, 2018; Christensen, 2021). The tRNA genes encoded by the mitochondrial genome are highly conserved and can be divided into two categories: 10–12 tRNAs in mitochondria and 4–13 chloroplast-like tRNAs with high homology with chloroplast DNA in A. officinalis. RNA editing is ubiquitous in higher plant mitochondria and is an essential process for the production of functional proteins by mitochondria (Warren and Sloan, 2020). The start codon in the A. officinalis mitochondrial genome was different from the standard AUG start codon. Therefore, the discovery of RNA editing reasonably explains why mitochondria are inconsistent with the use of standard codons in nuclear heredity. Thus, in future research, sterile male line materials could be created according to the RNA editing sites of A. officinalis to promote genetic breeding.
Repetitive sequences play an important role in genome recombination, mediating frequent recombination and producing a large number of chimeric genes, leading to crop abortion (Wu et al., 2022). Differences in mitochondrial genome size in the same species are mainly caused by repetitive sequences, especially the noncoding sequences of gene spacers (Duminil and Besnard, 2021; Li et al., 2021). Recombination is the main mechanism underlying mitochondrial genome evolution (Tomal et al., 2019; Welchen et al., 2021). The difference between the mitochondrial genome size of dicotyledonous plants Silene conica (11,318,806 nt) and that of Silene latifolia (253,413 nt) in the Dianthus family were found to be approximately 44.7-fold (Sloan et al., 2012). The mitochondrial genome sizes of Cucurbitaceae melon (approximately 2.9 M) and watermelon (379,236 nt) differed by over seven-fold (Alverson et al., 2010). Altogether, 254 SSRs and 293 non-tandem repeats were identified in A. officinalis repeat types. Moreover, a large palindromic sequence (12,348 bp) was identified. Similar large fragments were detected in Gossypium raimondii (Bi et al., 2016), Salix hypochonsis (Ye et al., 2017), and Acer truncatum (Ma et al., 2022). This suggests that the mitochondrial genome is relatively complex, with the characteristics of structural polymorphism, composition heterogeneity, and gene sequence variability in A. officinalis.
Plant mitochondrial genomes are characterized by the usual structural rearrangement, the loss or acquisition of a large number of genes, the usual intragenic transfer and exogenous DNA transfer, highly variable RNA editing processes, and extremely low nucleotide mutation rates (Duminil and Besnard, 2021; Xue et al., 2022). This provides unique information for the study of systematic molecular evolution (Christensen, 2021; Fields et al., 2022). The analysis of plant mitochondrial gene homology can reflect the relationships and evolutionary history of different species (Kozik et al., 2019). Herein, the phylogenetic relationship in A. officinalis was rebuilt using mitochondrial genome information based on representative species at the family level. The results indicated that a clear taxonomic relationship was reflected with the traditional classification of taxa. To classify the phylogenetic relationship of the whole species of the genus Asparagus, more species in different subgenera were sequenced to straighten their development relationships and perform more effective interspecific hybridization breeding.
The GC content in the mitochondrial genome was compared to that of some selective species and was found to be highly conserved in higher plants. Genome comparison and Ka/Ks analysis of protein-coding genes provide a good theoretical basis for exploring mitochondrial genome evolution. Generally, few coding genes in this study were affected by the positive selection effect, which is similar to existing research reports (Cheng et al., 2021; Ma et al., 2022). At the same time, this study also identified genes subject to neutral and negative selection pressure, most of which were affected by the negative selection effect. This analysis further indicated that protein-coding genes in the mitochondrial genome were conserved in green plants.
Previous studies have found that cytoplasmic male sterility is associated with changes in mitochondrial genes (Bi et al., 2016; Robles and Quesada, 2021). Therefore, the study of the mitochondrial genome may provide insights into the genetics and breeding of this plant. The expounding of gene composition, structure, and function in the mitochondrial genome is the basis of fertility, molecular-assisted breeding, and genetic evolution in plants (Shtolz and Mishmar, 2019; Ma et al., 2022). Given that garden asparagus is an important crop, its economic, edible, and nutritional health care value has been verified. As the most important plant of the family Asparagiaceae, garden asparagus is characterized by sex diversity, diverse propagation methods (e.g., ramet propagation, seed, and cutting propagation), a small chromosome genome (2n = 20), self-cross or hetero-cross pollination, and mature genetic transformation systems (Moreno et al., 2018; Moreno-Pinel et al., 2021; Encina and Regalado, 2022; García et al., 2023). Therefore, garden asparagus is an ideal material for many studies of dioecious plants. The garden asparagus industry has proposed it as a model plant for sex-determining mechanisms (Takeuchi et al., 2018; Harkess et al., 2020; Ahmad et al., 2022; You et al., 2022). Moreover, abundant variations were detected in garden asparagus mitochondria, including the accumulation of repetitive sequences, the acquisition of unknown sequences, variations in intron size, and variations in the number and size of genes. The mitochondrial gene composition and size, repeat sequences, and chloroplast-derived sequences provide an important basis for studying the origin and evolution of garden asparagus. Phylogenetic analysis using new genome sequences will provide insights into evolutionary pathways for the development of breeding programs in the genus Asparagus.
In this study, the mitochondrial genome sequence of garden asparagus was systematically investigated. After the assembling and annotating of its genome, we explored their basic composition, comparative analysis of coding gene differences, codon preference, repetitive sequences and SSRs, gene gain and loss, and sequence diversity analysis. Using the conserved protein-coding genes sequences, its phylogenetic status and evolutionary relationship of garden asparagus were discussed, which provided data support for the taxonomic study and the subsequent innovation and application of germplasm resources in garden asparagus.
The data presented in the study are deposited in the NCBI repository, accession number NC_053642.1. This data can be found here: https://www.ncbi.nlm.nih.gov/nuccore/NC_053642.1/.
The collection of specimen conformed to the requirement of International ethics, which did not cause damage to the local environment. The process and purpose of this experimental research were in line with the rules and regulations of our institute.
WS worked on genome assembly, performed the data analysis and wrote the original manuscript. QK designed the project and analyzed the data. WS, JD and CW contributed to the plant sample collection. JD, CW and QK wrote and improved the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of China (32060078), the Natural Science Foundation of Jiangxi (20171BAB214024, 20202BABL203044), the Special Program of Science and Technology Cooperation of Jiangxi Provincial Department of Science and Technology (20212BDH81022), the Science and Technology Program of Jiangxi Provincial Department of Education (GJJ202619), the “Biology and Medicine” discipline construction project of Nanchang Normal University (100/20149) and Jiangxi Province Key Laboratory of oil crops biology (YLKFKT202203).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1140043/full#supplementary-material
Supplementary Sheet 1 | SSR type and number distribution in mitochondrial genome of garden asparagus.
Supplementary Sheet 2 | Non-tandem repeat type and number distribution in mitochondrial genome of garden asparagus.
Supplementary Table 1 | Plant species used in phylogenetic studies.
Supplementary Table 2 | Fragments transferred from chloroplast to mitochondrial genome in garden asparagus.
Supplementary Table 3 | Organization of mitochondrial genomes in garden asparagus and other six plants.
Asparagus officinalis, A. officinalis; mitochondrial DNA, mtDNA; transfer RNA, tRNA; ribosomal RNA, rRNA; large ribosomal proteins, LRP; small ribosomal proteins, SRP; The Basic Local Alignment Search Tool, BLAST; simple sequence repeats, SSRs.
Ahmad, N., Tian, R. Z., Li, G. H., Zhao, C. Z., Fan, S. J., Sun, J., et al. (2022). Establishment of male-specific sequence-tagged site markers in Asparagus officinalis: An efficient tool for sex identification. Plant Breed. 141 (3), 471–481. doi: 10.1111/pbr.13016
Alverson, A. J., Wei, X. X., Rice, D. W., Stern, D. B., Barry, K., Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27 (6), 1436–1448. doi: 10.1093/molbev/msq029PMID: 20118192
Alverson, A. J., Zhuo, S., Rice, D. W., Sloan, D. B., Palmer, J. D. (2011). The mitochondrial genome of the legume vigna radiata and the analysis of recombination across short mitochondrial repeats. PloS One 6, e16404. doi: 10.1371/journal.pone.0016404
Amian, L., Rubio, J., Castro, P., Gil, J., Moreno, R. (2018). Introgression of wild relative asparagus spp. germplasm into the Spanish landrace ‘Morado de huétor’. Acta Hortic. 1223, 33–37. doi: 10.17660/ActaHortic.2018.1223.5
Arseneau, J. R., Steeves, R., Laflamme, M. (2017). Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 17, 686–693. doi: 10.1111/1755-0998.12616
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 (2), 573–580. doi: 10.1093/nar/27.2.573
Bergthorsson, U., Richardson, A. O., Young, G. J., Goertzen, L. R., Palmer, J. D. (2005). Massive horizontal transfer of mitochondrial genes from diverse land plant donors to the basal angiosperm amborella. Proc. Natl. Acad. Sci. 101 (51), 17747–17752. doi: 10.1073/pnas.0408336102
Bi, C. W., Paterson, A. H., Wang, X. L., Xu, Y. Q., Wu, D. Y., Qu, Y. S., et al. (2016). Analysis of the complete mitochondrial genome sequence of the diploid cotton gossypium raimondii by comparative genomics approaches. BioMed. Res. Int (3). doi: 10.1155/2016/5040598
Bock, R. (2017). Witnessing genome evolution: experimental reconstruction of endo-symbiotic and horizontal gene transfer. Annu. Rev. Genet. 51, 1–22. doi: 10.1146/annurev-genet-120215-035329
Castro, P., Gil, J., Cabrera, A., Moreno, R. (2013). Assessment of genetic diversity and phylogenetic relationships in asparagus species related to Asparagus officinalis. Genet. Resour Crop Evol. 60, 1275–1288. doi: 10.1007/s10722-012-9918-3
Chen, Y., Ye, W., Zhang, Y., Xu, Y. (2015). High speed BLASTN: An accelerated mega BLAST search tool. Nucleic Acids Res. 43, 7762–7768. doi: 10.1093/nar/gkv784
Cheng, Y., He, X. X., Priyadarshani, S. V. G. N., Wang, Y., Ye, L., Shi, C., et al. (2021). Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genomics 22, 167. doi: 10.1186/s12864-021-07490-9
Choi, I., Schwarz, E., Ruhlman, T., Khiyami, M., Sabir, J., Hajarah, N., et al. (2019). Fluctuations in fabaceae mitochondrial genome size and content are both ancient and recent. BMC Plant Biol. 19, 448. doi: 10.1186/s12870-019-2064-8
Christensen, A. C. (2021). Plant mitochondria are a riddle wrapped in a mystery inside an enigma. J. Mol. Evol. 89 (3), 151–156. doi: 10.1007/s00239-020-09980-y
Duminil, J., Besnard, G. (2021). Utility of the mitochondrial genome in plant taxonomic studies. Methods Mol. Biol. 2222, 107–118. doi: 10.1007/978-1-0716-0997-2_6
Encina, C. L., Regalado, J. J. (2022). Aspects of in vitro plant tissue culture and breeding of Asparagus: a review. Horticulturae 8, 439. doi: 10.3390/horticulturae8050439
Fields, P. D., Waneka, G., Naish, M., Schatz, M. C., Henderson, I. R., Sloan, D. B. (2022). Complete sequence of a 641-kb insertion of mitochondrial DNA in the Arabidopsis thaliana nuclear genome. Genome Biol. Evol. 14 (5), 5. doi: 10.1093/gbe/evac059
García, V., Castro, P., Die, J. V., Millán, T., Gil, J., Moreno, R. (2023). QTL analysis of morpho-agronomic traits in garden asparagus (Asparagus officinalis l.). Horticulturae 9, 41. doi: 10.3390/horticulturae9010041
García, V., Castro, P., Millán, T., Gil, J., Moreno, R. (2022). Genetic variability assessment of a diploid pre-breeding asparagus population developed using the tetraploid landrace ‘Morado de huétor’. Horticulturae 8, 859. doi: 10.3390/horticulturae8100859
Harkess, A., Huang, K., Hulst, R. V. D., Tissen, B., Leebens-Mack, J. (2020). Sex determination by two y-linked genes in garden Asparagus. Plant Cell. 32 (6), 1790–1796. doi: 10.1105/tpc.19.00859
Harkess, A., Zhou, J. S., Xu, C. Y., Bowers, J. E., van der, R., Ayyampalayam, S., et al. (2017). The asparagus genome sheds light on the origin and evolution of a young y chromosome. Nat. Commun. 8 (1), 1279. doi: 10.1038/s41467-017-01064-8
Knaflewski, M. (1996). Genealogy of asparagus cultivars. Acta Hortic. 415, 87–91. doi: 10.17660/ActaHortic.1996.415.13
Knoop, V. (2004). The mitochondrial DNA of land plants: peculiarities in phylogenetic perspective. Curr. Genet. 46 (3), 123–139. doi: 10.1007/s00294-004-0522-8
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 15 (8), e1008373. doi: 10.1371/journal.pgen.1008373
Kubota, S., Konno, I., Kanno, A. (2012). Molecular phylogeny of the genus Asparagus (Asparagaceae) explains interspecific crossability between the garden asparagus (A. officinalis) and other asparagus species. Theor. Appl. Genet. 124, 345–354. doi: 10.1007/s00122-011-1709-2
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C. S., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Li, H. (2016). Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. doi: 10.1093/bioinformatics/btw152
Li, H. (2018). Minimap2:Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/BIOINFORMATICS/BTY191
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows- wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, X. L., Sun, M. D., Liu, S. J., Teng, Q., Li, S. H., Jiang, Y. S. (2021). Functions of PPR proteins in plant growth and development. Int. J. Mol. Sci. 22, 11274. doi: 10.3390/ijms222011274
Liu, Y. C., Liu, S., Liu, D. C., Wei, Y. X., Liu, C., Yang, Y. M., et al. (2014). Exploiting EST databases for the development and characterization of EST-SSR markers in blueberry (Vaccinium) and their cross- species transferability in vaccinium spp. Sci. Hortic. 176, 319–329. doi: 10.1016/j.scienta.2014.07.026
Møller, I. M., Rasmusson, A. G., Aken, O. V. (2021). Plant mitochondria-past, present and future. Plant J. 108, 912–959. doi: 10.1111/tpj.15495
Ma, Q. Y., Wang, Y. X., Li, S. S., Wen, J., Zhu, L., Yan, K. Y., et al. (2022). Assembly and comparative analysis of the frst complete mitochondrial genome of Acer truncatum bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 22, 29. doi: 10.1186/s12870-021-03416-5
Malek, O., Lättig, K., Hiesel, R., Brennicke, A., Knoop, V. (1996). RNA Editing in bryophytes and a molecular phylogeny of land plants. EMBO J. 15 (6), 1403–1411. doi: 10.1002/j.1460-2075.1996.tb00482.x
Moreno, R., Castro, P., Vrána, J., Kubaláková, M., Cápal, P., García, V., et al. (2018). Integration of genetic and cytogenetic maps and identification of sex chromosome in garden asparagus (Asparagus officinalis l.). Front. Plant Sci. 9. doi: 10.3389/fpls.2018.01068
Moreno-Pinel, R., Castro-López, P., Die-Ramón, J. V., Gil-Ligero, J. (2021). “Asparagus (Asparagus officinalis l.) breeding,” in Advances in plant breeding strategies: Vegetable crops. Eds. Al-Khayri, J. M., Jain, S. M., Johnson, D. V. (Cham: Springer). doi: 10.1007/978-3-030-66961-4_12
Mousavizadeh, S. J., Gil, J., Castro, P., Hassandokht, M. R., Moreno, R. (2021). Genetic diversity and phylogenetic analysis in Asian and European asparagus subgenus species. Genet. Resour Crop Evol. 68, 3115–3124. doi: 10.1007/s10722-021-01262-w
Mower, J. P. (2009). The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 37 (suppl_2), W253–W259. doi: 10.1093/nar/gkp337
Norup, M. F., Petersen, G., Burrows, S., Bouchenak-Khelladi, Y., Leebens-Mack, J., Pires, J. C., et al. (2015). Evolution of Asparagus l. (Asparagaceae): Out-of- south- Africa and multiple origins of sexual dimorphism. Mol. Phylogenet Evol. 92, 25–44. doi: 10.1016/j.ympev.2015.06.002
Nothnagel, T., König., J., Keilwagen, J., Graner, E.-M., Plieske, J., Budahn, H. (2022). Transfer of the dominant virus resistance gene AV-1pro from Asparagus prostratus to chromosome 2 of garden asparagus a. officinalis l. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.809069
Nothnagel, T., Kramer, R., Budahn, H., Schrader, O., Ulrich, D., Schreyer, L. (2012). Interspecific hybridization of asparagus for the enlargement of the genetic basis concerning resistance to biotic and abiotic stress. Acta Hortic. 960, 139–146. doi: 10.17660/actahortic.2012.960.19
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (Oryza sativa l.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Gen. Genomics 268 (4), 434–445. doi: 10.1007/s00438-002-0767-1
Petersen, G., Anderson, B., Braun, H. P., Meyer, E. H., Møller, I. M. (2020). Mitochondria in parasitic plants. Mitochondrion 52, 173–182. doi: 10.1016/j.mito.2020.03.008
Ranjbar, M. E., Ghahremani, Z., Mousavizadeh, S. J., Barzegar, T., Gil, J., Moreno, R. (2022). New hybrids between cultivated and wild species of asparagus (Asparagus spp.) and their validation by SSR markers. Eur. J. Hortic. Sci. 87 (4), 1–10. doi: 10.17660/eJHS.2022/044
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm amborella. Science. 342 (6165), 1468–1473. doi: 10.1126/science.1246275PMID: 24357311
Richardson, A. O., Rice, D. W., Young, G. J., Alverson, A. J., Palmer, J. D. (2013). The “fossilized” mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 11 (1), 1–17. doi: 10.1186/1741-7007-11-29
Robles, P., Quesada, V. (2021). Organelle genetics in plants. Int. J. Mol. Sci. 22, 2104. doi: 10.3390/ijms22042104
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34 (12), 3299–3302. doi: 10.1093/molbev/msx248
Schattner, P., Brooks, A. N., Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sheng, W. T. (2020). The complete mitochondrial genome of Asparagus officinalis. Mitochondrial DNA Part B Resour. 5 (3), 2627–2628. doi: 10.1080/23802359.2020.1780986
Sheng, W. T., Chai, X. W., Rao, Y. S., Du, S. G., Tu, X. T. (2017). The complete chloroplast genome sequence of asparagus (Asparagus officinalis l.) and its phylogenetic position within Asparagales. J. Plant Breed. Genet. 5 (3), 121–128.
Shtolz, N., Mishmar, D. (2019). The mitochondrial genome-on selective constraints and signatures at the organism, cell, and single mitochondrion levels. Front. Ecol. Evol. 7. doi: 10.3389/fevo.2019.00342
Sibbald, S. J., Lawton, M., Archibald, J. M. (2021). Mitochondrial genome evolution in pelagophyte algae. Genome Biol. Evol. 13 (3), 1–14. doi: 10.1093/gbe/evab018
Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. 112 (27), E3515–E3524. doi: 10.1073/pnas.1504491112
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10 (1), e1001241. doi: 10.1371/journal.pbio.1001241
Sloan, D. B., Wu, Z. Q. (2014). History of plastid DNA insertions reveals weak deletion and at mutation biases in angiosperm mitochondrial genomes. Genome Biol. Evol. 6 (12), 3210–3221. doi: 10.1093/gbe/evu253
Small, I. D., Schallenberg-Rudinger, M., Takenaka, M., Mireau, H., Ostersetzer- Biran, O. (2020). Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056. doi: 10.1111/tpj.14578
Takeuchi, M. Y., Kakizoe, E., Yoritomi, R., Iwato, M., Kanno, A., Ikeuchi, T., et al. (2018). Features in stem blight resistance confirmed in interspecific hybrids of Asparagus officinalis l. and Asparagus kiusianus. HORTICULT J. 87 (2), 200–205. doi: 10.2503/hortj.OKD-104
Tang, W., Luo, C. (2018). Molecular and functional diversity of RNA editing in plant mitochondria. Mol. Biotechnol. 60, 935–945. doi: 10.1007/s12033-018-0126-z
Thiel, T., Michalek, W., Varshney, R. K., Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare l.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Tomal, A., Kwasniak-Owczarek, M., Janska, H. (2019). An update on mitochondrial ribosome biology: The plant mitoribosome in the spotlight. Cells 8, 1562. doi: 10.3390/cells8121562
Vaser, R., Sovi´c, I., Nagarajan, N., Šiki´c, M. (2016). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Warren, J. M., Sloan, D. B. (2020). Interchangeable parts: The evolutionarily dynamic tRNA population in plant mitochondria. Mitochondrion 52, 144–156. doi: 10.1016/j.mito.2020.03.007
Welchen, E., Canal, M. V., Gras, D. E., Gonzalez, D. H. (2021). Cross-talk between mitochondrial function, growth, and stress signalling pathways in plants. J. Exp. Bot. 72 (11), 4102–4118. doi: 10.1093/jxb/eraa608
Wong, K. H., Kong, B. L. H., Siu, T. Y., Wu, H. Y., But, G. W. C., Shaw, P., et al. (2022). Complete chloroplast genomes of Asparagus aethiopicus l., a. densiflorus (Kunth) Jessop ‘Myers’, and A. cochinchinensis (Lour.) merr.: Comparative and phylogenetic analysis with congenerics. PloS One 17 (4), e0266376. doi: 10.1371/journal.pone.0266376
Wu, Z. Q., Liao, X. Z., Zhang, X. N., Tembrock, L. R., Broz, A. (2022). Genomic architectural variation of plant mitochondria-a review of multichromosomal structuring. J. Syst. Evol. 60 (1), 160–168. doi: 10.1111/jse.12655
Xie, H. Y., Yang, C. Y., Sun, Y. M., Igarashi, Y., Jin, T., Luo, F. (2020). PacBio long reads improve metagenomic assemblies, gene catalogs, and genome binning. Front. Genet. 11. doi: 10.3389/fgene.2020.516269
Xue, J. Y., Dong, S. S., Wang, M. Q., Song, T. Q., Zhou, G. C., Li, Z., et al. (2022). Mitochondrial genes from 18 angiosperms fill sampling gaps for phylogenomic inferences of the early diversification of flowering plants. J. Syst. Evol. 60, 773–788. doi: 10.1111/jse.12708
Ye, N., Wang, X. L., Li, J., Bi, C. W., Xu, Y. Q., Wu, D. Y., et al. (2017). Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant salix suchowensis. Peer J. 5, e3148. doi: 10.7717/peerj.3148
You, C., Wen, R. D., Zhang, Z. L., Cheng, G. Q., Zhang., Y. L., Li, N., et al. (2022). Development and applications of a collection of single copy genebased cytogenetic DNA markers in garden asparagus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1010664
Zhang, X. H., Han, C. Z., Liang, Y. Q., Yang, Y., Liu, Y., Cao, Y. P. (2022). Combined full length transcriptomic and metabolomic analysis reveals the regulatory mechanisms of adaptation to salt stress in asparagus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1050840
Keywords: asparagus officinalis, mitochondrial genome, PacBio and Illumina sequencing, phylogenetic analysis, asparagus
Citation: Sheng W, Deng J, Wang C and Kuang Q (2023) The garden asparagus (Asparagus officinalis L.) mitochondrial genome revealed rich sequence variation throughout whole sequencing data. Front. Plant Sci. 14:1140043. doi: 10.3389/fpls.2023.1140043
Received: 08 January 2023; Accepted: 08 March 2023;
Published: 27 March 2023.
Edited by:
Roberto Moreno, University of Cordoba, SpainReviewed by:
Francesco Sunseri, Mediterranea University of Reggio Calabria, ItalyCopyright © 2023 Sheng, Deng, Wang and Kuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Kuang, a3VhbmdxdWFuMjAyMkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.