 Zhongjie Lu
Zhongjie Lu Zhengyu Yang2
Zhengyu Yang2 Qihui Gui
Qihui Gui Rui Dong
Rui Dong- 1Department of Grassland Science, College of Animal Science, Guizhou University, Guiyang, China
- 2Department of Vehicle Engineering, Guizhou Technological College of Machinery and Electricity, Duyun, China
Numerous studies have shown that plant microRNAs (miRNAs) play key roles in plant growth and development, as well as in response to biotic and abiotic stresses; however, the role of miRNA in legumes under aluminum (Al) stress have rarely been reported. Therefore, here, we aimed to investigate the role of miRNAs in and their mechanism of Al tolerance in legumes. To this end, we sequenced a 12-strand-specific library of Medicago truncatula under Al stress. A total of 195.80 M clean reads were obtained, and 876 miRNAs were identified, of which, 673 were known miRNAs and 203 were unknown. A total of 55 miRNAs and their corresponding 2,502 target genes were differentially expressed at various time points during Al stress. Further analysis revealed that mtr-miR156g-3p was the only miRNA that was significantly upregulated at all time points under Al stress and could directly regulate the expression of genes associated with root cell growth. Three miRNAs, novel_miR_135, novel_miR_182, and novel_miR_36, simultaneously regulated the expression of four Al-tolerant transcription factors, GRAS, MYB, WRKY, and bHLH, at an early stage of Al stress, indicating a response to Al stress. In addition, legume-specific miR2119 and miR5213 were involved in the tolerance mechanism to Al stress by regulating F-box proteins that have protective effects against stress. Our results contribute to an improved understanding of the role of miRNAs in Al stress in legumes and provide a basis for studying the molecular mechanisms of Al stress regulation.
1 Introduction
Aluminum (Al) toxicity is one of the most important factors limiting crop growth and production in acidic soils. Approximately 40% of global arable land is acidic, which is one of the main factors contributing to food shortages in regions such as those in Africa (Daspute et al., 2017). Al3+ is present in naturally acidic soils and is also the most harmful form of Al to plants, with micromolar concentrations of Al3+ inhibiting root elongation in a short period of time (Gui et al., 2022). Chandran et al. (2008) stained the root tips of Medicago truncatula seedlings grown at 10-μm Al concentration with hematoxylin and showed that the root tips treated with Al for the longest time stained the darkest, indicating that they were the most severely damaged. Li et al. (2020) found that Al stress significantly enhanced the expression of MsPG1 in the plasma membrane of Medicago sativa root apical epidermal cells, reduced the accumulation of Al in the cell wall, and improved the Al tolerance of M. sativa. However, some plants grown for a long time in acidic soils can chelate Al3+ in the vicinity of the root zone with organic acids such as malic, citric, and oxalic acids secreted by the root system, forming Al-organic acid complexes, and thus reducing the transport of Al to the interior of the root cell and enhancing Al tolerance (Chauhan et al., 2021).
MicroRNAs are a subset of the major non-coding RNAs in higher plants. They are typically 19–24 nucleotides long, and they are thought to post-transcriptionally regulate the cleavage of target mRNAs or repress their translation (Bartel, 2004; Kumar et al., 2017; Xu et al., 2019). miRNAs play key roles in gene expression, stress responses, growth and development, and other regulatory mechanisms in plants and animals (Zhang et al., 2015; Pegler et al., 2019). Liu et al. (2014) studied 22 conserved miRNA families in Zea mays and found that 72 genes targeted by 62 differentially expressed miRNAs may regulate maize ear development. Wang et al. (2011) identified two miRNAs, cbr-mir-241 and ath-miR854a, using microarray technology, that can directly regulate Glycine max resistance to blast rot through their targets (enzymes). Hoang et al. (2020) found that miRNAs play an important role in various processes of symbiotic nitrogen fixation with rhizobia in four legumes: Lotus japonicus, M. truncatula, G. max, and Phaseolus vulgaris.
Medicago truncatula is an annual legume that has been used as a model plant for legumes as it is a close relative of M. sativa and has the advantage of having a small genome, high similarity, and being a diploid plant (Chandran et al., 2008). In recent years, although researchers have conducted numerous studies on Al stress in plants, miRNA studies related to Al stress in legumes have rarely been reported. Therefore, we aimed to investigate the role of miRNAs in and their mechanism of Al tolerance in M. truncatula. High-throughput sequencing of root tip tissues under Al stress was performed to identify miRNAs associated with the Al stress response. The findings of this study provide new insights into the potential functions of miRNAs in the Al response mechanism.
2 Materials and methods
2.1 Plant materials and processing
Medicago truncatula A17 seeds were surface-disinfected with 1% NaClO solution for 10 min and rinsed five times in distilled water to remove residual disinfectant solution, then incubated in an artificial climate chamber at 25°C for 3 days protected from light, and then transferred to Hoagland’s medium (pH 5.8) for 7 days (25°C, light/dark cycle of 16/8 h). The composition of Hoagland’s culture solution is consistent with the experiments of Stephan and Procházka (1989).The nutrient solution was changed every 2 days during incubation. Seedlings with similar growth levels were divided into four groups, three of which were incubated in 10 μM AlCl3 and 0.5 mM CaCl2 (pH 4.5) solutions for 4, 24, and 48 h, respectively, and were recorded as T4, T24, and T48, respectively. The control group (T0) was incubated in a 0.5 mM CaCl2 (pH 4.5) solution for 48 h. To treat the four groups of seedlings, two groups of seedlings, T0 and T48, were incubated simultaneously. After 24 h and 44 h of incubation of the above two groups of seedlings, respectively, the seedlings of T24 and T4 groups were incubated, and the four groups of seedlings were harvested simultaneously at 48 h to reduce the circadian effect. Three replicates were set up for each group, with 60 seedlings per replicate. After treatment, root tips of ~ 1.5 cm from each treated plant were collected, immediately frozen in liquid nitrogen, and stored at –80 °C.
2.2 Physiological indicators and fluorescein diacetate staining test
The activities of three enzymes, superoxide dismutase (SOD, SOD-BC0170), catalase (CAT, CAT-BC0200), and peroxidase (POD, POD-BC0090), were measured in four sets of samples using kits provided by Beijing Solarbio Science and Technology Co., Ltd. (Beijing, China). The specific operational steps of the test are presented in the instruction manual.
An image scanner (Perfection V800 photo, Epson, Suwa, Japan) with a WinRhizo root analysis system (WinRhizo Tron Pro 2009, Regent Instruments Inc., Quebec, Canada) was used to measure the root length and root surface area of four sets of samples (Pang et al., 2018). When treating root length and root surface area, we included a control group of plant material grown under Al-free conditions in a 0.5 mM CaCl2 solution (pH 4.5) for the same duration as that of the corresponding Al treatment. The root vigor assay was performed using the naphthylamine method (BC5295, Beijing Solarbio Science and Technology Co., Ltd., Beijing, China). Sample root tips were treated in 2 g/mL FDA solution protected from light for 10 min according to the method of Ishikawa and Wagatsum (1998). The treated root tips were repeatedly rinsed at least five times with deionized water, followed by observation and photography using a fluorescence microscope (Axiolab5, ZEISS, Germany). Each sample was repeated three times. The aforementioned experimental operations were performed under dark conditions.
2.3 RNA extraction, library preparation of sRNA, and sequencing
RNA samples were extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and tested for concentration, purity, and integrity (Liu et al., 2016). The resulting 3′ sRNA and 5′ sRNA were ligated for splicing. The first strand was synthesized by reverse transcription. Finally, polymerase chain reaction (PCR) amplification and size selection were performed. The target fragments were screened by PAGE, and the cut gels were recovered as fragments to obtain sRNA libraries. Finally, the PCR products were purified (AMPure XP system) and the library quality was assessed.
The resulting libraries were sequenced on the Illumina NovaSeq 6,000 platform from Biomarker Co., Ltd. (BMKcloud, Beijing, China). Clean data were obtained by removing reads containing adapters, poly-N, and low-quality reads from the raw data (Zhao et al., 2020). Using Bowtie tools, clean reads were aligned against Silva and other databases to screen for other sRNAs, such as ribosomal RNA (rRNA), which were then eliminated. Finally, the unannotated reads were sequenced against the reference genome Medicago_truncatula.Mt4.0v2 using Bowtie2 (v1.0.0) software to obtain information on the position of the reads in the reference genome (Pertea et al., 2015).
2.4 Identification of miRNAs and prediction of new miRNAs
The reads that were aligned to the reference genome were then compared to the mature sequences of known miRNAs in the miRBase (v22) database and their upstream 2nt and downstream 5nt ranges, and the identified reads were considered known miRNAs. As miRNA precursors have a signature hairpin structure, the formation of the mature body is achieved by shearing of the Dicer/DCL enzyme. Based on these features, the miRDeep2 software package (Friedlander et al., 2012) was used, and the final prediction of new miRNAs was achieved by scoring using a Bayesian model (Zhang, 2015).
2.5 Analysis of differentially expressed miRNAs
Differential expression analysis of the two conditions/groups was performed using the DESeq2 R package (1.10.1) (Love et al., 2014). The resulting P-values were adjusted using Benjamini and Hochberg’s approach to control for the false discovery rate. miRNAs with |log2(FC)| ≥ 0.58; P-value ≤ 0.05, found by DESeq2, were assigned as differentially expressed. Fold Change (FC) indicates the ratio of expression between two samples (groups).
2.6 Prediction, annotation, and functional analysis of DE miRNAs target genes
Target gene prediction was performed using TargetFinder (v1.6) software based on the gene sequence information of known miRNAs and newly predicted miRNAs in the corresponding species (Allen et al., 2005). The predicted target gene sequences were compared with the NCBI non-redundant protein sequence (Nr), SwissProt Protein databases, Gene Ontology database (GO), Clusters of Orthologous Groups of proteins (COG), Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Protein homology (KOG), Evolutionary genealogy of genes (eggNOG), and protein family (Pfam) (E-value < 10-5) (Yu et al., 2022) databases using BLAST (v2.2.26) software to obtain the annotation information of the target genes. GO enrichment and KEGG enrichment of miRNA target genes were analyzed using Cluster Profiler (v3.10.1) software and KOBAS software (Mao et al., 2005), respectively.
2.7 Quantitative real-time polymerase chain reaction analysis
The extracted total RNA, combined with reverse random primers, was used for reverse transcription of miRNA. The qRT-PCR was performed using the MyiQ Single Color Real-time PCR system (Bio-Rad, Hercules, CA, USA) (95°C for 3 min and 45 cycles of 95°C for 5 s and 60°C for 30 s). Primers were designed using Primer Premier (v6.0) software and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China) (Supplementary Table S1). The 2-ΔΔCt method was used to the calculate relative gene expression levels.
3 Results
3.1 Physiological characteristics of roots under Al stress
The root length, root surface area, root activity, and plant growth of M. truncatula plants after Al treatment are shown in Figures 1A–C. With increasing duration of Al stress, the root length of plants in the Al-treated group differed significantly from that of the control at 48 h (Figure 1A), and their root length decreased by 5.0% compared with the control. The plant heel surface area of the Al-treated group was significantly different from that of the control group at both 24 and 48 h (Figure 1B), with reductions of 3.9% and 4.5%, respectively. Root activity showed an increasing trend and then decreasing, and was significantly lower at 48 h than at 24 h (Figure 1C). As shown in Figure 1D (I–IV), the root morphology of the Al-treated and control plants also showed significant differences as the stress time increased.
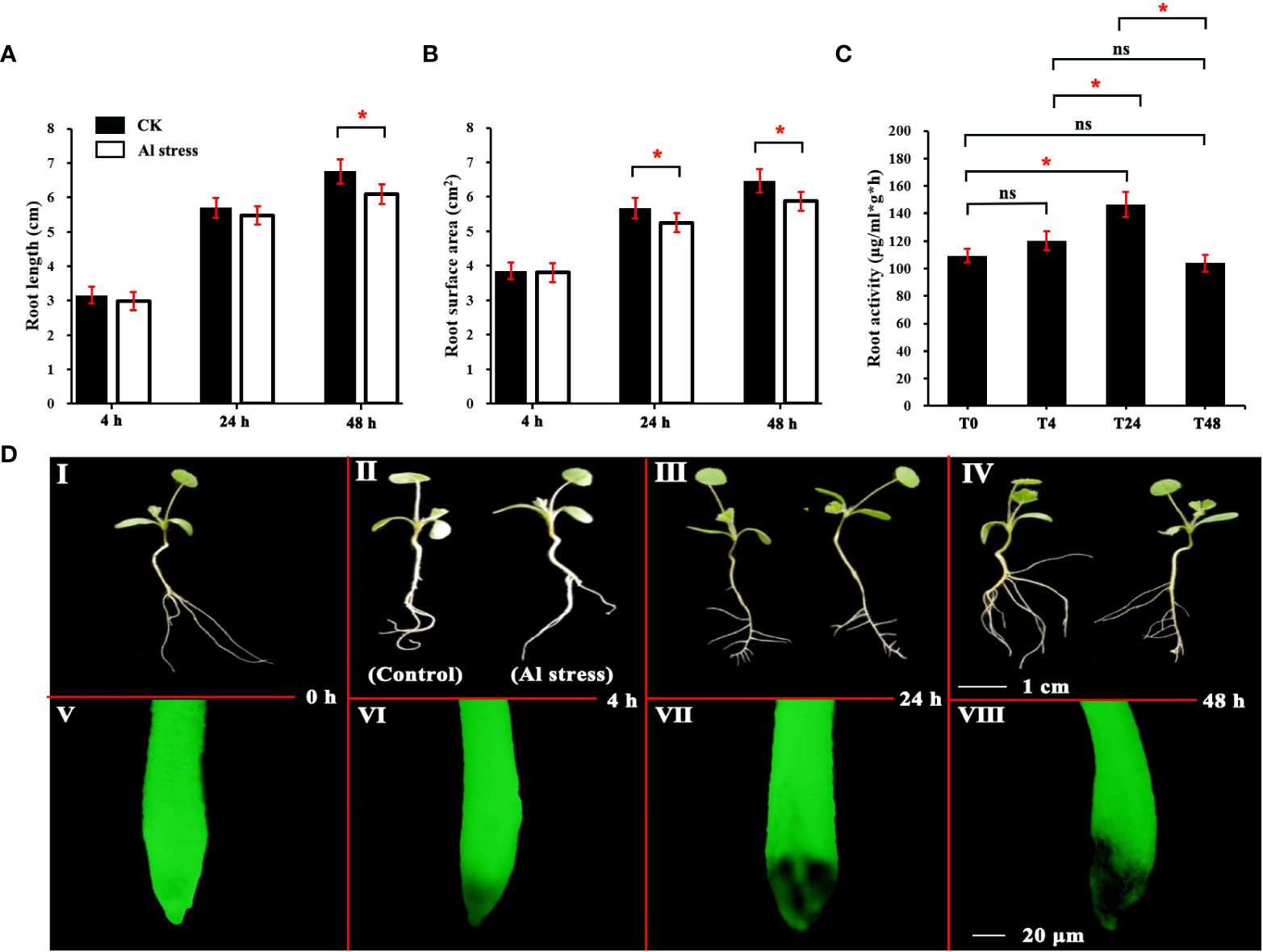
Figure 1 Analysis of root physiological characteristics of M. truncatula under aluminum stress. (A) Analysis of total root length of M. truncatula. (B) Analysis of root surface area of M. Truncatula. (C) Analysis of root activity of M. Truncatula. (D) I–IV is the growth comparison of plants under Al stress and the blank control at the same time point. Plant material grown under normal conditions and treated for the same amount of time as the corresponding aluminum treatment group were used as a control, scale = 1 cm; V–VIII is the comparison of root fluorescence of plants under Al stress and the blank control after FDA staining, scale = 20 μm. * indicates that there is a significant difference at the level of 0.05 according to Duncan’s multiple range test, while ns indicates that the difference is not significant.
The FDA fluorescent staining method only stains live cells. Therefore, the FDA staining of the root system at each treatment time point revealed that damage to the plant root tip tissues had already begun after 4 h of Al treatment, and the damage to the root-tip tissues gradually increased with the Al treatment time (Figure 1D V–VIII).
To investigate whether the redox system was activated in M. truncatula subjected to Al stress, the enzymatic activities of SOD, CAT, and POD were examined in this study. The SOD activity reached its maximum at 4 h, after which, it started to decline, and decreased significantly at 48 h (Figure 2A). CAT and POD showed the same trend, with both significantly increasing at 4 h versus 24 h and significantly decreasing at 48 h (Figures 2B, C). These results indicated that the redox system in M. truncatula was rapidly activated under Al stress.
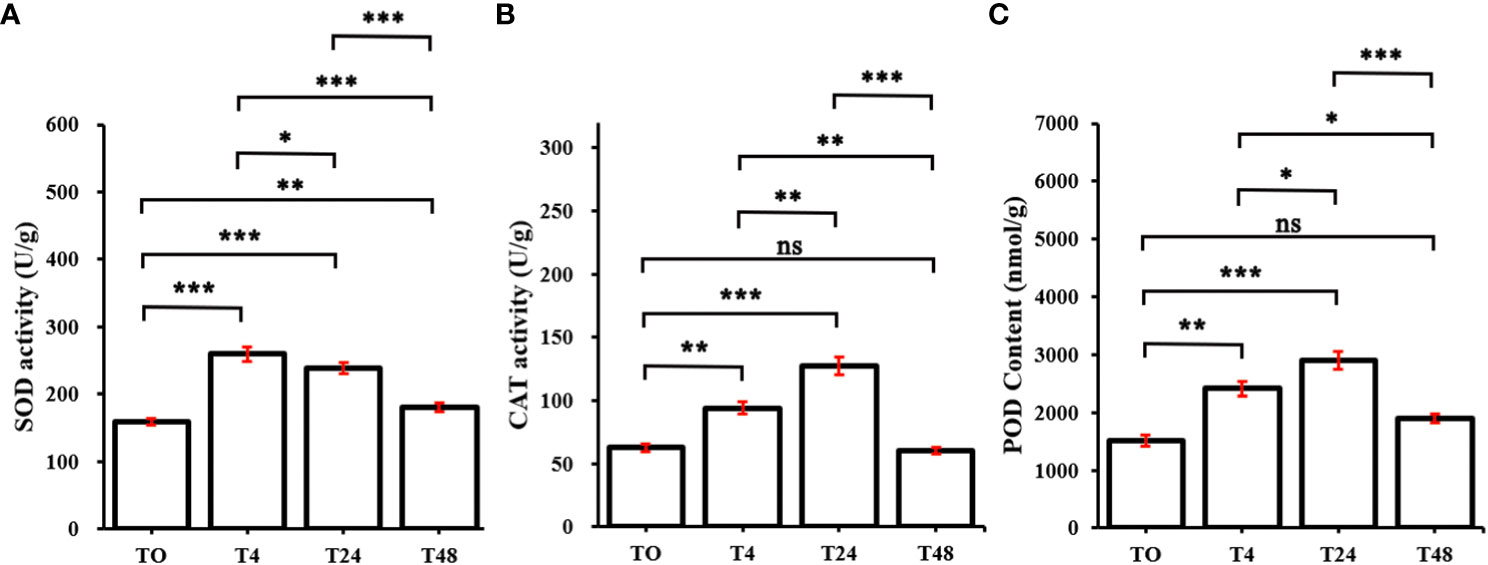
Figure 2 Changes of oxidoreductase in M. truncatula under continuous aluminum stress. (A) superoxide dismutase (SOD) activity. (B) Catalase (CAT) activity. (C) peroxidase activity (POD). The plotting data is the average and standard deviation of three repetitions. T0~T48 represent M. truncatula plant samples treated with aluminum for 0h, 4h, 24h, and 48h, respectively. According to Duncan’s multiple range test, the asterisk represents the significant difference between aluminum treated samples and the control (*, P < 0.05; **, P < 0.01; ***, P < 0.001), ns indicates that the difference is not significant.
3.2 MicroRNA sequence analysis and mapping
In this study, 12 M. truncatula samples were sequenced for small RNA and a total of 296.01 M raw reads were obtained. After removing contaminants and reads of length < 18 and > 30 nucleotides, 195.80 M clean reads were obtained. There were not less than 11.96 M clean reads for each sample, the average GC content and base number were 50.99% and 1258.05 Mb, respectively, and the average Q30 was 86.82% (Supplementary Table S2). The average mapped reads, uniquely mapped reads, and multiple-mapped reads constituted 61.46%, 40.42%, and 21.05% of all libraries, respectively, and the comparison efficiency of the reads in each sample against the reference genome ranged from 51.76–68.47% (Supplementary Table S3). The length distribution of sRNAs was similar among the 12 libraries, with the highest abundance of 21-nucleotide sRNAs, followed by 24-nucleotide sRNAs (Supplementary Table S4), which is consistent with the previous findings of Bao et al. (2019).
3.3 Identification of known and novel miRNAs, predictive analysis of target genes, and functional annotation
A total of 876 miRNAs were identified in this study, of which, 673 were known and 203 were unknown. There were 556 known miRNAs with family affiliation and 95 unknown miRNAs with family affiliation (Supplementary Table S5). Subsequently, miRNA target genes were predicted using TargetFinder software; 664 of the known miRNAs predicted 10,845 target genes, and 194 of the unknown miRNAs predicted 3,610 target genes (Supplementary Table S6). According to our functional annotation results, a total of 13,057 out of 13,148 target genes were annotated, with the number ranging from 3,852 (29.50%, KEGG) to 13,055 (99.98%, Nr), among which, the most genes were annotated in Nr and eggNOG with 13,055 and 10,935, respectively (Supplementary Table S7).
3.4 MicroRNA differential expression analysis, predictive analysis, and functional annotation of target genes
In total, 55 differentially expressed miRNAs (Supplementary Table S8) were identified in this study. Comparison of the T0 vs. T4 treatment groups showed the highest number of differentially expressed miRNAs, 48, of which, 31 were upregulated and 17 were downregulated. Five miRNAs were differentially expressed in the comparison between the T0 vs. T24 treatment groups, with all of them exhibiting upregulation. Ten miRNAs were differentially expressed in the comparison between the T0 vs. T48 treatment groups, of which, five were upregulated and five were downregulated. A total of 2,502 differential target genes were predicted for the 55 differentially expressed miRNAs; the maximum number of target genes of differentially expressed miRNAs was 2,088 in the T0 vs. T4 treatment group and the minimum was 101 in the T0 vs. T24 treatment group. Subsequently, the target genes of the differentially expressed miRNAs were annotated using eight databases, including Nr, KOG, COG, Pfam, Swiss-Prot, eggNOG, GO, and KEGG, and all 2,502 target genes were annotated. The target genes of all three treatment groups were the most abundant annotated genes in the NR database, with 2088, 101, and 307, respectively (Supplementary Table S9).
3.5 Gene ontology function and KEGG pathway enrichment analysis
To determine whether miRNAs are functionally involved in the Al stress response and defense processes, we performed GO functional analysis of differential target genes of miRNAs. A total of 2,714 DEGs were enriched in the 145 GO treatments (Supplementary Table S10). Based on the functions of the enriched genes in each GO tree, a cluster analysis of 145 GO terms could be clustered into 30 major classes (Figure 3A). The three GO terms “Cellular metabolic process,” “Organic substance metabolic process,” and “Cellular component organization” had the highest number of DEGs, at 1,382, 378, and 333, respectively (Figure 3A).
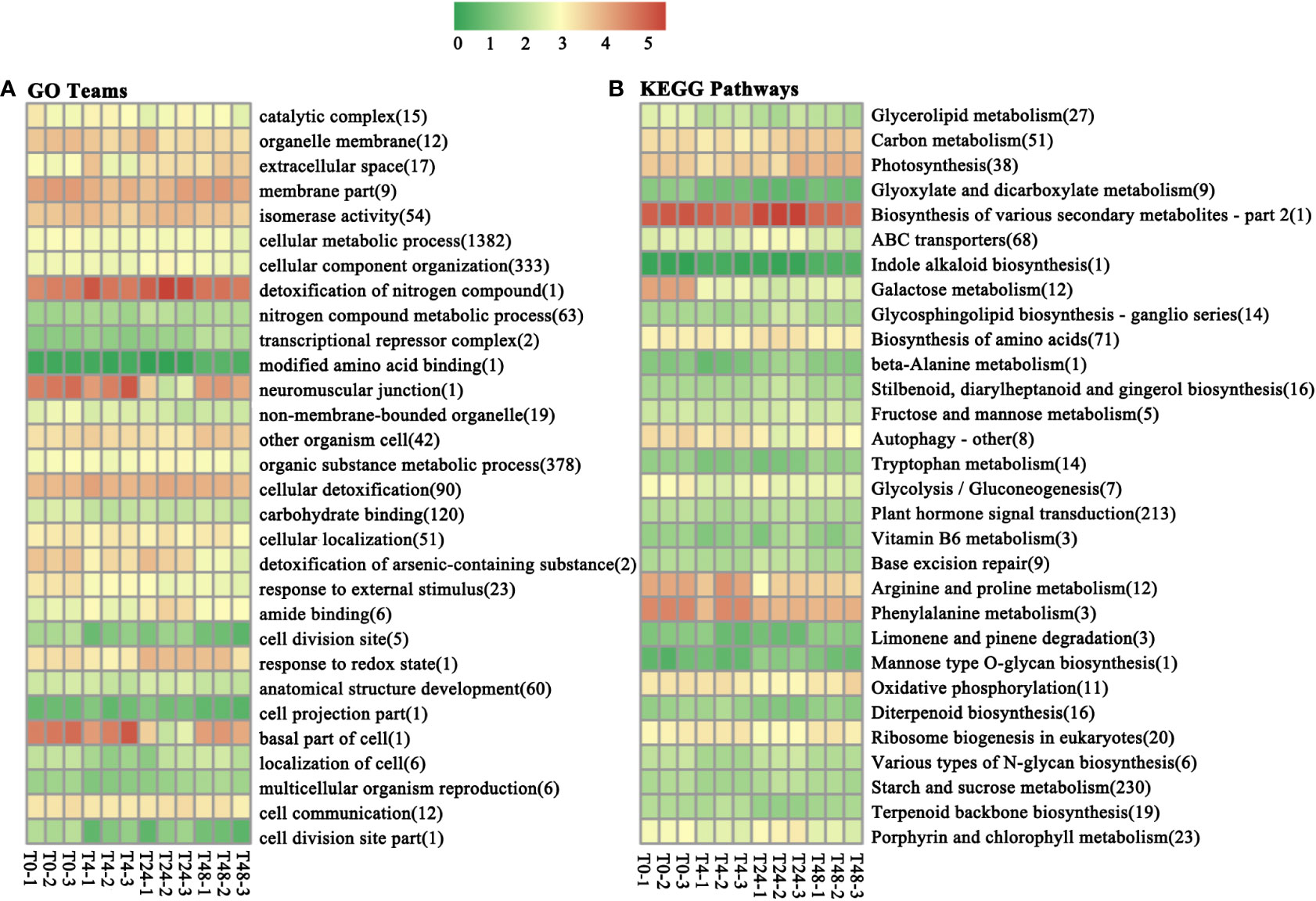
Figure 3 Under aluminum stress, 30 GO terms and KEGG pathways were enriched for the most DEGs. (A) GO teams. (B) KEGG pathways. The numbers in the abscissa represent the number of DEGs enriched for the GO term or KEGG pathway. T0 to T48 represent M. truncatula plant samples treated with aluminum for 0h, 4h, 24h, and 48h, respectively. The comparison is described as A vs B, which means the DEG was found in the A treatment relative to the B treatment.
KEGG pathway enrichment was performed to explore the function of genes differentially expressed under Al stress as well as metabolic pathways involved in the response. A total of 912 DEGs were enriched in 111 pathways (Supplementary Table S9). The 111 enriched pathways were clustered into 30 broad categories based on the function of the enriched genes in each pathway (Figure 3B). The highest number of DEGs was found in three enrichment pathways: “starch and sucrose metabolism,” “plant hormone signal transduction,” and “biosynthesis of amino acids,” at 230, 213, and 71, respectively.
3.6 qRT-PCR verification of DE miRNA and differential genes
Ten DE miRNAs were randomly selected for qRT-PCR validation to verify the reliability of the transcriptome data. The results showed that these miRNAs had similar expression levels and trends as observed in the miRNA-seq results (Figure 4) and the qRT-PCR results also demonstrated the reliability of the transcriptome data.
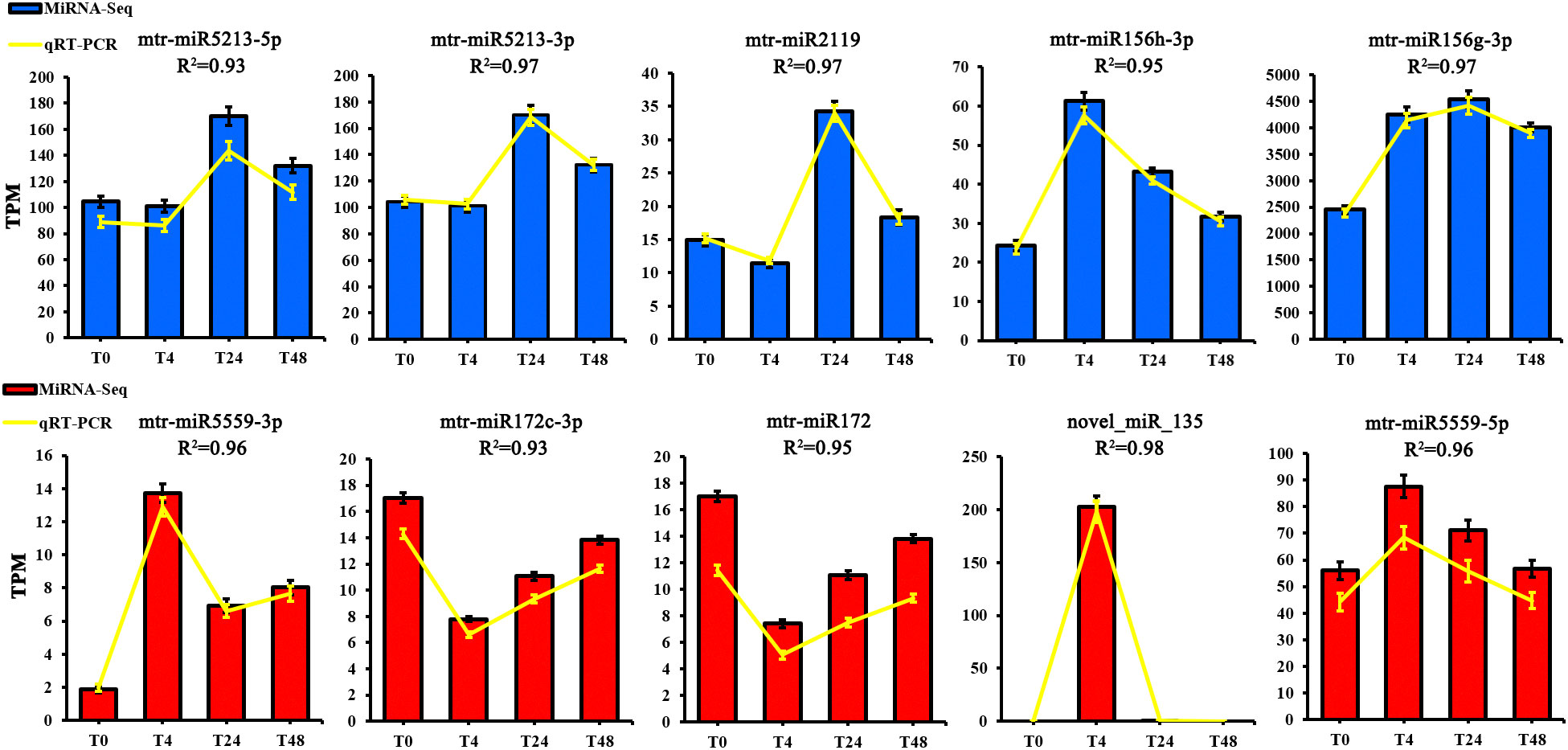
Figure 4 The expression patterns of ten selected miRNAs identified by RNA-Seq were verified by qRT-PCR. The Y-axis to the left of each histogram represents the expression level of RNA-seq (TPM). T0 to T48 represents M. truncatula plant samples treated with aluminum for 0h, 4h, 24h, and 48h, respectively.
4 Discussion
Al stress contributes majorly to altered metabolic activity, root damage, cell wall damage, and cytoplasmic lysis, resulting in reduced photosynthetic efficiency, impaired water and nutrient uptake, and increased respiratory consumption and toxin accumulation; these effects limit crop growth (Xu et al., 2021), ultimately leading to plant death and yield loss (Osman et al., 2021; Abdel Latef et al., 2021). MicroRNAs widely regulate various physiological processes, such as development, signal transduction, and stress response in plants (Chen, 2012; Sunkar et al., 2012), and their mechanisms in plant stress tolerance have been demonstrated in G.max (Ning et al., 2019), Z. mays (Fu et al., 2017; Shan et al., 2020), and Arabidopsis thaliana (Shukla et al., 2018).
In this study, a total of 12 sRNA samples of M. truncatula under Al stress for 4 h, 24 h, 48 h and a control group were subjected to whole transcriptome sequencing, and a total of 876 miRNAs were obtained, among which, 203 were unknown miRNAs. Cao et al. (2018) similarly identified 876 miRNAs under salt/alkali stress in M. truncatula, supporting the reliability of the data in this study.
4.1 Effect of mtr-miR156g-3p on the root system of M. truncatula under Al stress
miR156 is one of the most abundantly expressed and highly evolutionarily conserved miRNAs in plants (Niu et al., 2015), and the physiological processes involved in the regulation of this family of miRNAs under abiotic stress have been demonstrated in A. thaliana (Guan et al., 2014; Niu et al., 2015), Panax notoginseng (Zheng et al., 2017), Phalaenopsis (Zhao et al., 2019), and other plants. miR156 is highly accumulated mainly in the seedling stage of plants, and its expression level decreases during the plant growth period, which is important for plant seedling development and for improving crop productivity and stress resistance (Wang et al., 2009; Wu et al., 2009; Jerome Jeyakumar et al., 2020). In the present study, mtr-miR156g-3p was the only DE miRNA involved in the response at 4, 24, and 48 h of Al stress, and it was upregulated compared to the control. Further annotation analysis of the target genes of mtr-miR156g-3p showed that six target genes, Medtr6g004260, Medtr4g056140, Medtr7g007440, Medtr5g077840, Medtr3g078260, and Medtr2g090610, regulate the formation of the root tip cell membrane. In addition, the expression levels of the above six target genes were significantly upregulated at 4 and 24 h of Al stress (P < 0.05) (Figure 5; Supplementary Table S10). Therefore, these findings suggest that mtr-miR156g-3p is activated in M. truncatula under Al stress and further overexpresses target genes with the ability to promote cell membrane formation in root tip cells to counteract the damage caused by stress. Niu et al. (2015) found that miR156 could enhance A. thaliana lateral root development by regulating related binding protein genes, which is consistent with the findings of the present study. Of note, in previous studies, mtr-miR156g-3p under abiotic stress mediated the negative expression of the SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) gene before regulating the expression of genes controlling root cell growth (Zheng et al., 2019; Rao et al., 2021). In contrast, there was no differential expression of genes associated with SPL in the present study, which may indicate that mtr-miR156g-3p in M. truncatula root tip tissue can directly regulate root cell growth genes. Zhao et al. (2019) found that mtr-miR156g-3p directly regulates anthocyanin formation in Phalaenopsis plants, and no expression of SPL-related genes was detected.
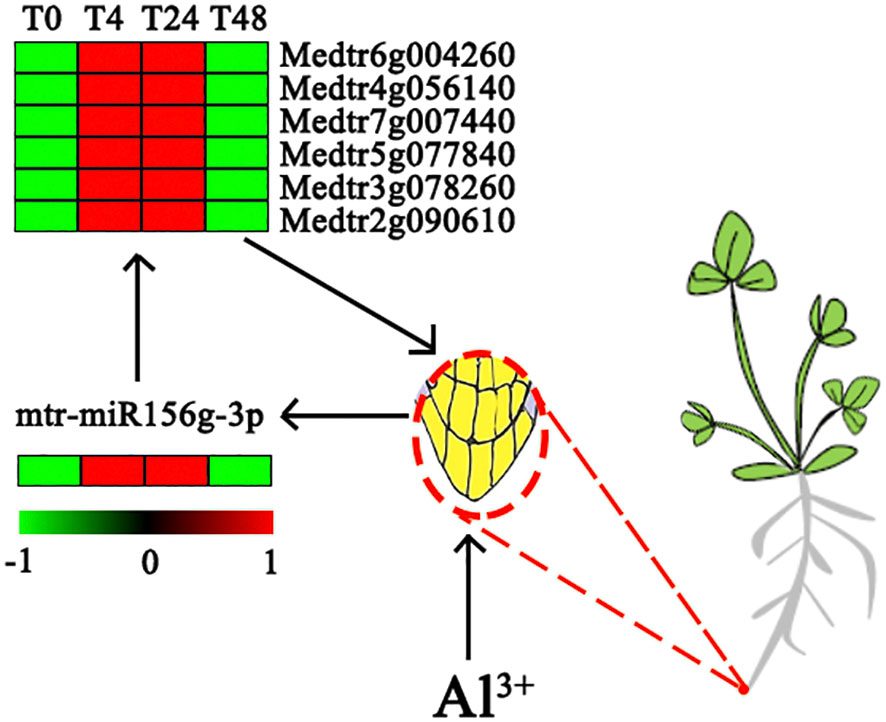
Figure 5 Analysis of expression patterns of miRNA and target genes affecting the root system of M. truncatula under aluminum stress.
4.2 Regulation of transcription factors by miRNAs and target genes in the mechanism of Al resistance
Transcription factors play an important role in Al stress-tolerance mechanisms (Lin et al., 2021; Shu et al., 2022). The GRAS TF family is involved in a variety of biological processes such as biotic and abiotic plant stress, rootstock development, and meristem tissue formation (Wang et al., 2018). In the present study, seven target genes of four DE miRNAs were involved in the regulated expression of GRAS TFs, and all seven target genes were significantly upregulated at 4 h of Al stress, but not at 24 h and 48 h (Table 1A; Supplementary Table S10). The GRAS TF family consists of three members: GAI (GIBBERELLIC ACID INSENSITIVE), RGA (REPRESSOR OF GA1-3 MUTANT), and SCR (SCARECROW) (Fan et al., 2021), which are mainly expressed in roots and vascular cells with SHR (SHORT ROOT) and can enhance plant stress resistance (Cui et al., 2014). The MYB TF family members play an important role in plant cell wall formation, growth, and development, and are one of the major components of stress response mechanisms in plants (Sun et al., 2014). In this study, 14 target genes of 10 DE miRNAs regulated the expression of MYB TFs at 4 h of Al stress treatment, and all the 14 target genes were significantly upregulated. In contrast, no DE miRNAs regulating MYB TFs or their corresponding target genes were activated at 24 h of Al stress treatment. At 48 h, a total of two target genes of one DE miRNA were involved in the regulation of MYB TF expression, both of which were significantly downregulated (Table 1A; Supplementary Table S10). The MYB TFs are one of the largest protein families in plants (Ambawat et al., 2013), and most MYB proteins belong to the R2R3-MYB subfamily, which positively regulates salt tolerance in plants by mediating the expression of abscisic acid (ABA) and regulating cuticle formation (Wang et al., 2021). WRKY is a plant-specific zinc finger TF that is involved in various physiological processes in plants (Wu et al., 2014; Wang et al., 2022). In this study, a total of 13 target genes of 7 DE miRNAs regulated the expression of WRKY TFs at 4 h of Al treatment, and all 13 target genes were significantly upregulated. At 24 h, no DE miRNAs that regulated WRKY TFs or target genes were activated. At 48 h, one target gene of one DE miRNA was involved in the expression regulation of WRKY TFs, and this target gene was significantly downregulated (Table 1A; Supplementary Table S10). The WRKY TFs are not only involved in the regulation of various biological functions by themselves but also interact with other TFs to form a signaling network that regulates different biological processes and are an important component of the plant stress tolerance system (Wani et al., 2021). The bHLH class of TFs is the second largest class of TFs in plants and is involved in biological processes, such as light signaling, hormone and other signal transduction, root hair development, and stress tolerance mechanisms (Nakata et al., 2013). In the present study, a total of 10 target genes of 4 DE miRNAs were involved in the regulated expression of bHLH TFs, and all 10 target genes were significantly upregulated at 4 h of Al stress, whereas they were not expressed at 24 h and 48 h of Al stress (Table 1B; Supplementary Table S10). In the present study, the expression pattern of bHLH TFs was consistent with that of GRAS TFs, and both TFs play an important role in the growth and development of plant roots under stress (Nakata et al., 2013; Wang et al., 2018). Therefore, the present study suggests that related miRNAs promote the development of plant roots by regulating the expression of bHLH TFs and GRAS TFs at the early stage of Al stress in M. truncatula, thus enhancing the Al resistance of M. truncatula.
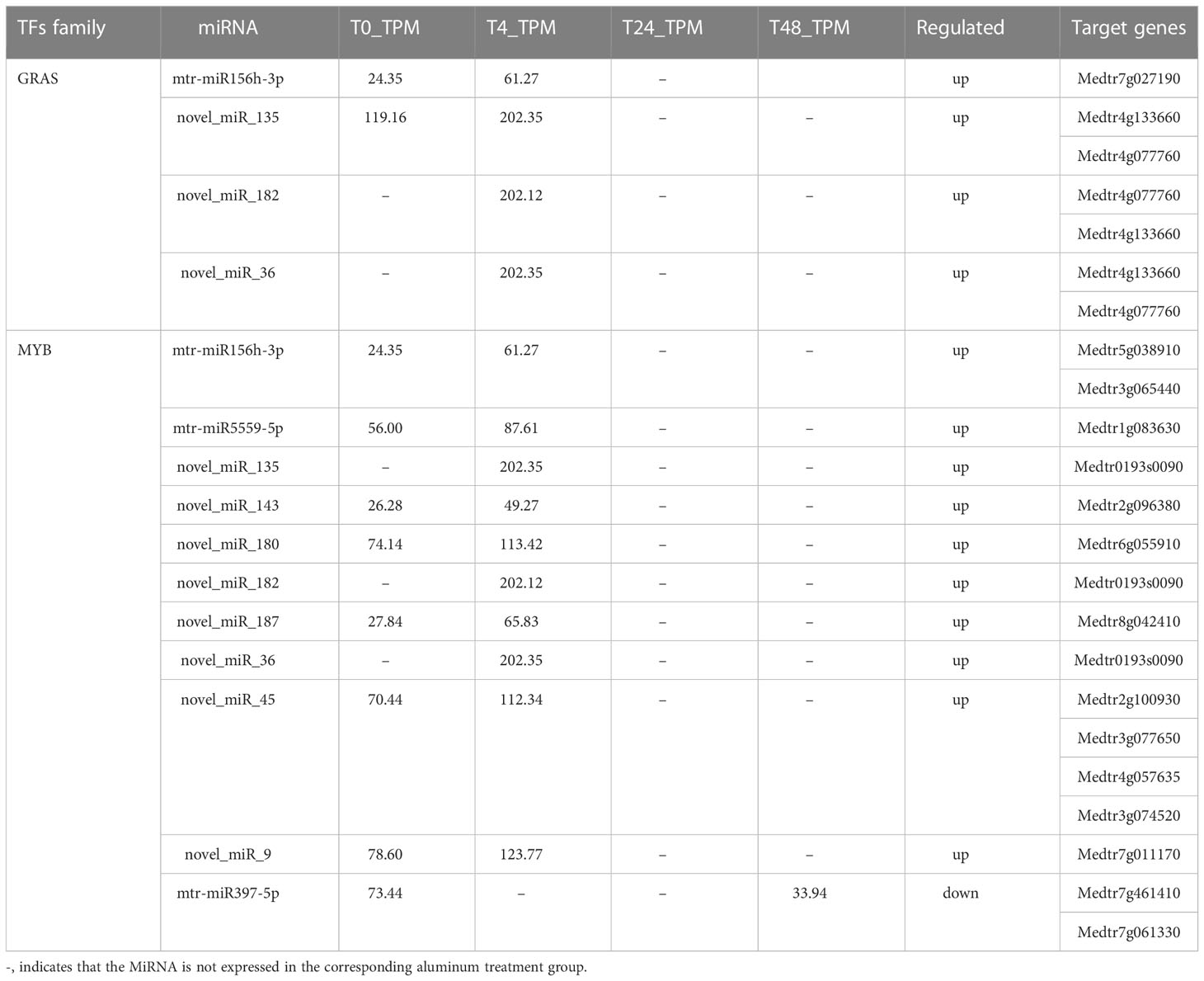
Table 1A Information About Special MiRNA.
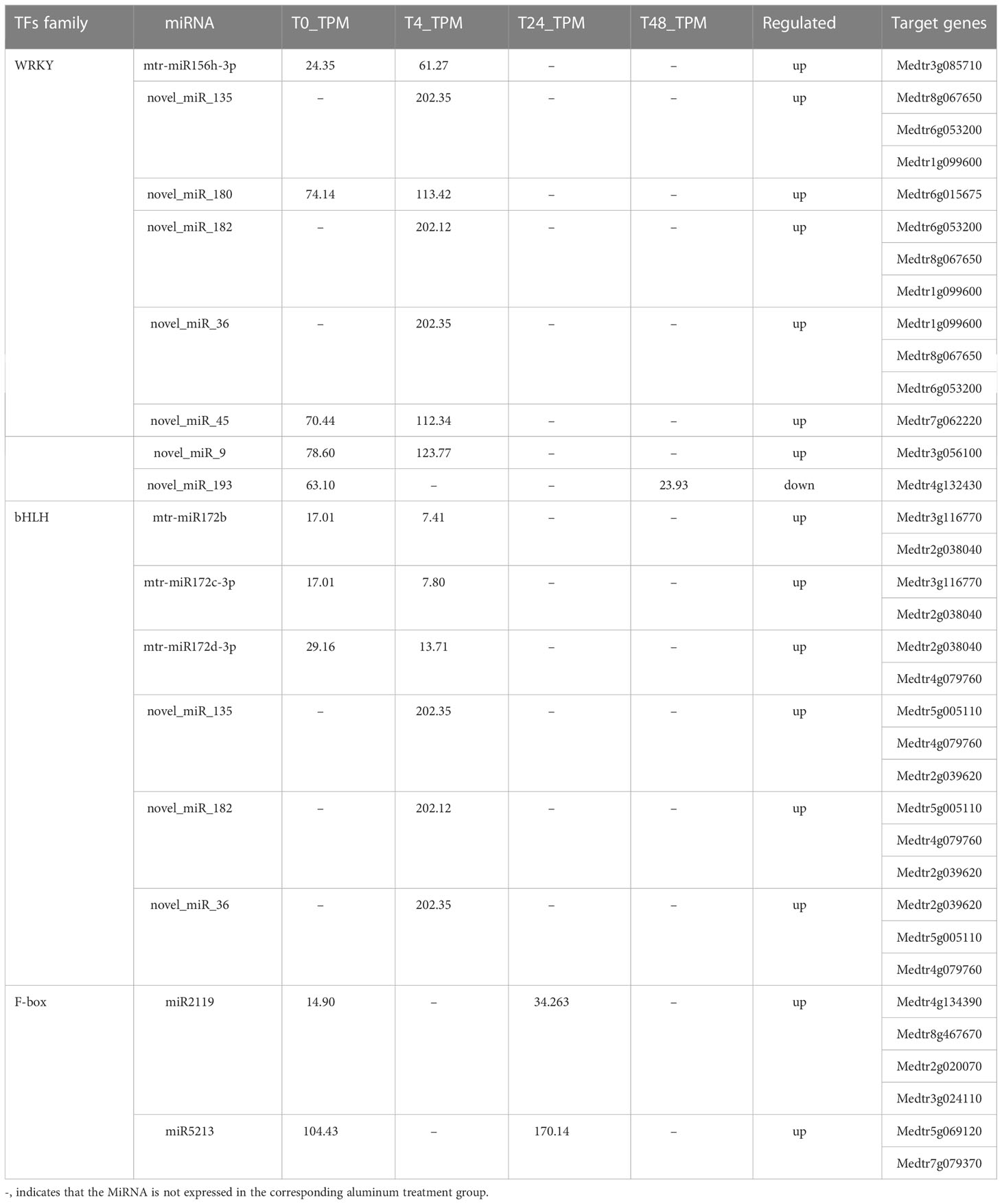
Table 1B Information About Special MiRNA.
MicroRNAs and target genes mediate TFs, such as GRAS, MYB, WRKY, and bHLH, which play an important role in the Al response mechanism of M. truncatula. However, all target genes regulating the above TFs were significantly upregulated at 4 h during the initial stage of Al stress. In contrast, no genes regulating TFs were activated at 24 h. Although some target genes of individual TFs were activated at 48 h, they were all downregulated. This study concluded that the above results could be attributed to the severe damage to the root tip tissue of M. truncatula with the increase in Al stress time, resulting in a large amount of cell inactivation. This reduces the ability of root tip cells to express Al tolerance genes and secrete secondary substances involved in the Al stress response. The above expression pattern of TFs was also consistent with the trend of the root physiological indicators in Figure 1. Notably, three miRNA target genes, novel_miR_135, novel_miR_182, and novel_miR_36, were involved in regulating the expression of the above four TFs, and they were significantly upregulated at the early stage of stress. Therefore, this study suggests that three miRNAs, novel_miR_135, novel_miR_182, and novel_miR_36, and their target genes may have potential roles in regulating Al responsive TFs.
4.3 Role of legume-specific miRNAs in the Al response mechanism of M. truncatula
miR2111, miR2119, and miR5213 are considered legume-specific miRNAs that regulate the expression of relevant defense genes when legumes are attacked by pathogens, thereby enhancing their own defense mechanisms (Kohli et al., 2014). This has been verified in plants such as Prunella vulgaris (Rosa et al., 2019) and Cicer arietinum (Kohli et al., 2014). In the present study, three target genes of miR2119 and one target gene of miR5213 were significantly upregulated at 24 h of Al stress, while their expression levels at other time points of Al stress were consistent with the treatment group Table 1A; Supplementary Table S10). Functional analysis of the four target genes revealed that they were involved in the regulation of an F-box protein containing a TIR structural domain (Supplementary Table S8). F-box proteins are receptors for plant growth hormones and are involved in plant defense responses (Dharmasiri et al., 2005; Navarro et al., 2006). Therefore, our findings suggest that legume-specific miR2119 and miR5213 may enhance Al tolerance in M. truncatula by regulating F-box protein overexpression via their target genes.
5 Conclusions
In this study, a total of 195.80 M clean reads were obtained from 12 sRNA libraries, and 876 miRNAs were identified, along with their corresponding 14,455 target genes. A total of 55 miRNAs were differentially expressed during Al stress, with different miRNAs enhancing Al tolerance in M. truncatula through mechanisms such as the promotion of root tip cell formation, expression of Al-resistant TFs, and regulation of defense gene expression. These findings could provide useful genetic resources for subsequent biotechnological studies, such as miRNA silencing or gene overexpression, and may inform genetic improvement programs for the development of Al stress-tolerant genotypes in legume crops.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA908067.
Author contributions
RD and CC conceived the experiment. ZL, ZY, ZT, and QG carried it out. ZL and RD analyzed the data. RD and ZL wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (32060392) and Support by Guizhou Province Science and Technology Projects (Qian Ke He Zhi Cheng [2020]1Y074) and GZMARS- Forage Industry Technology System.
Conflict of interest
The authors declare that the research has been conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional materials have been uploaded separately at the time of submission.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1137764/full#supplementary-material
References
Abdel Latef, A. A., Omer, A. M., Badawy, A. A., Osman, M. S., Ragaey, M. M. (2021). Strategy of salt tolerance and interactive impact of azotobacter chroococcum and/or alcaligenes faecalis inoculation on canola (Brassica napus l.). plants grown in saline soil. Plants 10, 110. doi: 10.3390/plants10010110
Allen, E., Xie, Z., Gustafson, A. M., Carrington, J. C. (2005). microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121, 207–221. doi: 10.1016/j.cell.2005.04.004
Ambawat, S., Sharma, P., Yadav, N. R., Yadav, R. C. (2013). MYB transcription factor genes as regulators for plant responses: an overview. Physiol. Mol. Biol. Plants 19, 307–321. doi: 10.1007/s12298-013-0179-1
Bao, H., Chen, H., Chen, M., Xu, H., Huo, X., Xu, Q., et al. (2019). Transcriptome-wide identification and characterization of microRNAs responsive to phosphate starvation in populus tomentosa. Funct. Integr. Genomics 19, 953–972. doi: 10.1007/s10142-019-00692-1
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Cao, C., Long, R., Zhang, T., Kang, J., Wang, Z., Wang, P., et al. (2018). Genome-wide identification of microRNAs in response to salt/alkali stress in Medicago truncatula through high-throughput sequencing. Int. J. Mol. Sci. 19, 4076. doi: 10.3390/ijms19124076
Chandran, D., Sharopova, N., VandenBosch, K. A., Garvin, D. F., Samac, D. A. (2008). Physiological and molecular characterization of aluminum resistance in Medicago truncatula. BMC Plant Biol. 8, 89. doi: 10.1186/1471-2229-8-89
Chauhan, D. K., Yadav, V., Vaculík, M., Gassmann, W., Pike, S., Arif, N. (2021). Aluminum toxicity and aluminum stress-induced physiological tolerance responses in higher plants. Crit. Rev. Biotechnol. 41, 715–730. doi: 10.1080/07388551.2021.1874282
Chen, X. (2012). Small RNAs in development-insights from plants. Curr. Opin. Genet. Dev. 22, 361–367. doi: 10.1016/j.gde.2012.04.004
Cui, H., Kong, D., Liu, X., Hao, Y. (2014). SCARECROW, SCR-LIKE 23 and SHORT-ROOT control bundle sheath cell fate and function in Arabidopsis thaliana. Plant J. 78, 319–327. doi: 10.1111/tpj.12470
Daspute, A. A., Sadhukhan, A., Tokizawa, M., Kobayashi, Y., Panda, S. K., Koyama, H. (2017). Transcriptional regulation of aluminum-tolerance genes in higher plants: Clarifying the underlying molecular mechanisms. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01358
Dharmasiri, N., Dharmasiri, S., Estelle, M. (2005). The f-box protein TIR1 is an auxin receptor. Nature 435, 441–445. doi: 10.1038/nature03543
Fan, Y., Yan, J., Lai, D., Yang, H., Xue, G., He, A., et al. (2021). Genome-wide identification, expression analysis, and functional study of the GRAS transcription factor family and its response to abiotic stress in sorghum [Sorghum bicolor (L.) moench]. BMC Genomics 22 (1), 1–21. doi: 10.1186/s12864-021-07848-z
Friedlander, M. R., Mackowiak, S. D., Li, N., Chen, W., Rajewsky, N. (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi: 10.1093/nar/gkr688
Fu, R., Zhang, M., Zhao, Y., He, X., Ding, C., Wang, S., et al. (2017). Identification of salt tolerance-related microRNAs and their targets in maize (Zea mays l.) using high-throughput sequencing and degradome analysis. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00864
Guan, X., Pang, M., Nah, G., Shi, X., Ye, W., Stelly, D. M., et al. (2014). miR828 and miR858 regulate homoeologous MYB2 gene functions in arabidopsis trichome and cotton fibre development. Nat. Commun. 5, 3050. doi: 10.1038/ncomms4050
Gui, Q., Yang, Z., Chen, C., Yang, F., Wang, S., Dong, R. (2022). Identification and characterization of long noncoding RNAs involved in the aluminum stress response in Medicago truncatula via genome-wide analysis. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1017869
Hoang, N. T., Tóth, K., Stacey, G. (2020). The role of microRNAs in the legume– Rhizobium Nitrogen-fixing symbiosis. Journal of Experimental Botany 71, 1668–1680. doi: 10.1093/jxb/eraa018
Ishikawa, S., Wagatsum, T. (1998). Plasma membrane permeability of root-tip cells following temporary exposure to Al ions is a rapid measure of Al tolerance among plant species. Plant Cell Physiol. 39, 516–525. doi: 10.1093/oxfordjournals.pcp.a029399
Jerome Jeyakumar, J. M., Ali, A., Wang, W. M., Thiruvengadam, M. (2020). Characterizing the role of the miR156-SPL network in plant development and stress response. Plants 9, 1206. doi: 10.3390/plants9091206
Kohli, D., Joshi, G., Deokar, A. A., Bhardwaj, A. R., Agarwal, M., Katiyar-Agarwal, S., et al. (2014). Identification and characterization of wilt and salt stress-responsive MicroRNAs in chickpea through high-throughput sequencing. PloS One 9, e108851. doi: 10.1371/journal.pone.0108851
Kumar, V., Khare, T., Shriram, V., Wani, S. H. (2017). Plant small RNAs: the essential epigenetic regulators of gene expression for salt-stress responses and tolerance. Plant Cell Rep. 26, 1–5. doi: 10.1007/s00299-017-2210-4
Lin, Y., Liu, G., Xue, Y., Guo, X., Luo, J., Pan, Y., et al. (2021). Functional characterization of aluminum (Al)-responsive membrane-bound NAC transcription factors in soybean roots. Int. J. Mol. Sci. 22, 12854. doi: 10.3390/ijms222312854
Li, J., Su, L., Lv, A., Li, Y., Zhou, P., An, Y. (2020). MsPG1 alleviated aluminum-induced inhibition of root growth by decreasing aluminum accumulation and increasing porosity and extensibility of cell walls in alfalfa (Medicago sativa). Environ. Exp. Bot. 175, 104045. doi: 10.1016/j.envexpbot.2020.104045
Liu, H., Qin, C., Chen, Z., Zuo, T., Yang, X., Zhou, H., et al. (2014). Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics 15, 25. doi: 10.1186/1471-2164-15-25
Liu, W., Zhang, Z., Chen, S., Ma, L., Wang, H., Dong, R., et al. (2016). Global transcriptome profiling analysis reveals insight into saliva-responsive genes in alfalfa. Plant Cell Rep. 35, 561–571. doi: 10.1007/s00299-015-1903-9
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Mao, X., Cai, T., Olyarchuk, J. G., Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Nakata, M., Mitsuda, N., Herde, M., Koo, A., Moreno, J. E., Suzuki, K., et al. (2013). A bhlh-type transcription factor, aba-inducible bhlh-type transcription factor/ja-associated myc2-like1, acts as a repressor to negatively regulate jasmonate signaling in arabidopsis. Plant Cell 25, 1641–1656. doi: 10.1105/tpc.113.111112
Navarro, L., Dunoyer, P., Jay, F., Arnold, B., Dharmasiri, N., Estelle, M., et al. (2006). A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 21, 436–439. doi: 10.1073/pnas.0510928103
Ning, L. H., Du, W. K., Song, H. N., Shao, H. B., Qi, W. C., Sheteiwy, M. S. A., et al. (2019). Identification of responsive miRNAs involved in combination stresses of phosphate starvation and salt stress in soybean root. Environ. Exp. Bot. 167, 103823. doi: 10.1016/j.envexpbot.2019.103823
Niu, Y., Niu, Q. W., Ng, K. H., Chua, N. H. (2015). The role of miR156/SPLs modules in arabidopsis lateral root development. Plant J. 83, 673–685. doi: 10.1111/tpj.12919
Osman, M. S., Badawy, A. A., Osman, A. I., Abdel Latef, A. A. (2021). Ameliorative impact of an extract of the halophyte Arthrocnemum macrostachyum on growth and biochemical parameters of soybean under salinity stress. J. Plant Growth Regul. 40, 1245–1256. doi: 10.1007/s00344-020-10185-2
Pang, J. Y., Bansal, R., Zhao, H. X., Bohuon, E., Lambers, H., Ryan, M. H., et al. (2018). The carboxylate-releasing phosphorus-mobilizing strategy can be proxied by foliar manganese concentration in a large set of chickpea germplasm under low phosphorus supply. New Phytologist 219(2), 518–529. doi: 10.1111/nph.15200.
Pegler, J. L., Oultram, J. M. J., Grof, C. P. L., Eamens, A. L. (2019). Profiling the abiotic stress responsive microRNA landscape of Arabidopsis thaliana. Plants 8, 58. doi: 10.3390/plants8030058
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T., Mendell, J. T., Salzberg, S. L., et al. (2015). StringTie enables improved reconstruction of a transcriptome from RNAseq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.312
Rao, S., Li, Y., Chen, J. (2021). Combined analysis of MicroRNAs and target genes revealed miR156-SPLs and miR172-AP2 are involved in a delayed flowering phenomenon after chromosome doubling in black goji (Lycium ruthencium). Front. Genet. 12. doi: 10.3389/fgene.2021.706930
Rosa, C. D. L., Covarrubias, A. A., Reyes, J. L. (2019). A dicistronic precursor encoding miR398 and the legume-specific miR2119 coregulates CSD1 and ADH1 mRNAs in response to water deficit. Plant Cell Environ. 42, 133–144. doi: 10.1111/pce.13209
Shan, T., Fu, R., Xie, Y., Chen, Q., Wang, Y., Li, Z., et al. (2020). Regulatory mechanism of maize (Zea mays l.) miR164 in salt stress response. Russ J. Genet. 56, 835–842. doi: 10.1134/S1022795420070133
Shukla, P. S., Borza, T., Critchley, A. T., Hiltz, D., Norrie, J., Prithiviraj, B. (2018). Extract mitigates salinity stress in Arabidopsis thaliana By modulating the expression of miRNA involved in stress tolerance and nutrient acquisition. PloS One 13, e0206221. doi: 10.1371/journal.pone.020622
Shu, W., Zhou, Q., Xian, P., Cheng, Y., Lian, T., Ma, Q., et al. (2022). GmWRKY81 encoding a WRKY transcription factor enhances aluminum tolerance in soybean. Int. J. Mol. Sci. 23, 6518. doi: 10.3390/ijms23126518
Stephan, U. W., Procházka, Ž. (1989). Physiological disorders of the nicotianamine-auxotroph tomato mutant chloronerva at different levels of iron nutrition. i. growth characteristics and physiological abnormalities related to iron and nicotianamine supply. Acta Botanica Neerlandica 38, 147–153. doi: 10.1111/j.1438-8677.1989.tb02037.x
Sunkar, R., Li, Y. F., Jagadeeswaran, G. (2012). Functions of microRNAs in plant stress responses. Trends Plant Sci. 17, 196–203. doi: 10.1016/j.tplants.2012.01.010
Sun, P., Zhu, X., Huang, X., Liu, J. (2014). Overexpression of a stress-responsive MYB transcription factor of poncirus trifoliata confers enhanced dehydration tolerance and increases polyamine biosynthesis. Plant Physiol. Biochem. 78, 71–79. doi: 10.1016/j.plaphy.2014.02.022
Wang, J. W., Czech, B., Weigel, D. (2009). miR156-regulated SPL transcription factors define an endogenous flowering pathway in. Arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, C., Hao, X., Wang, Y., Maoz, I., Zhou, W., Zhou, Z., et al. (2022). Identification of WRKY transcription factors involved in regulating the biosynthesis of the anti-cancer drug camptothecin in ophiorrhiza pumila. Hortic. Res. 9, uhac099. doi: 10.1093/hr/uhac099
Wang, Y. X., Liu, Z. W., Wu, Z. J., Li, H., Wang, W. L., Cui, X., et al. (2018). Genome-wide identification and expression analysis of gras family transcription factors in tea plant (Camellia sinensis). Sci. Rep. 8, 3949. doi: 10.1038/s41598-018-22275-z
Wang, J., Liu, C., Zhang, L., Wang, J., Hu, G., Ding, J., et al. (2011). MicroRNAs involved in the pathogenesis of phytophthora root rot of soybean (Glycine max). Agricultural Sciences in China 10, 1159–1167. doi: 10.1016/S1671-2927(11)60106-5
Wang, X., Niu, Y., Zheng, Y. (2021). Multiple functions of MYB transcription factors in abiotic stress responses. Int. J. Mol. Sci. 122, 6125. doi: 10.3390/ijms22116125
Wani, S. H., Anand, S., Singh, B., Bohra, A., Joshi, R. (2021). And plant defense responses: latest discoveries and future prospects. Plant Cell Rep. 40, 1071–1085. doi: 10.1007/s00299-021-02691-8
Wu, G., Park, M. Y., Conway, S. R., Wang, J. W., Weigel, D., Poethig, R. S. (2009). The sequential action of miR156 and miR172 regulates developmental timing in arabidopsis. Cell 138, 750–759. doi: 10.1016/j.cell.2009.06.031
Wu, Y., Wei, W., Pang, X., Wang, X., Zhang, H., Dong, B. (2014). Comparative transcriptome profiling of a desert evergreen shrub, ammopiptanthus mongolicus, in response to drought and cold stresses. BMC Genomics 15, 671. doi: 10.1186/1471-2164-15-671
Xu, J., Hou, Q. M., Khare, T., Verma, S. K., Kumar, V. (2019). Exploring miRNAs for developing climate-resilient crops: a perspective review. Sci. Total Environ. 653, 91–104. doi: 10.1016/j.scitotenv.2018.10.340
Xu, T., Zhang, L., Yang, Z., Wei, Y., Dong, T. (2021). Identification and functional characterization of plant MiRNA under salt stress shed light on salinity resistance improvement through MiRNA manipulation in crops. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.665439
Yu, L., Huang, T., Qi, X., Yu, J., Wu, T., Luo, Z., et al. (2022). Genome-wide analysis of long non-coding RNAs involved in nodule senescence in Medicago truncatula. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.917840
Zhang, B. H. (2015). MicroRNA: a new target for improving plant tolerance to abiotic stress. Journal of Experimental Botany 66, 1749–1761. doi: 10.1093/jxb/erv013
Zhang, Z., Jiang, L., Wang, J., Gu, P., Chen, M. (2015). MTide: an integrated tool for the identification of miRNA-target interaction in plants. Bioinformatics 31, 290–291. doi: 10.1093/bioinformatics/btu633
Zhao, A., Cui, Z., Li, T., Pei, H., Sheng, Y., Li, X., et al. (2019). mRNA and miRNA expression analysis reveal the regulation for flower spot patterning in Phalaenopsis ‘Panda’. International Journal of Molecular Sciences. 20, 4250. doi: 10.3390/ijms20174250
Zhao, M., Wang, T., Sun, T., Yu, X., Tian, R., Zhang, W. H., et al. (2020). Identification of tissue-specific and cold-responsive lncRNAs in Medicago truncatula by high-throughput RNA sequencing. BMC Plant Biol. 20, 99. doi: 10.1186/s12870-020-2301-1
Zheng, Y., Chen, K., Xu, Z., Liao, P., Zhang, X., Liu, L., et al. (2017). Small RNA profiles from Panax notoginseng roots differing in sizes reveal correlation between miR156 abundances and root biomass levels. Sci. Rep. 7, 9418. doi: 10.1038/s41598-017-09670-8
Keywords: plant miRNA, Medicago truncatula, aluminum stress, transcript analysis, plant growth
Citation: Lu Z, Yang Z, Tian Z, Gui Q, Dong R and Chen C (2023) Genome-wide analysis and identification of microRNAs in Medicago truncatula under aluminum stress. Front. Plant Sci. 14:1137764. doi: 10.3389/fpls.2023.1137764
Received: 04 January 2023; Accepted: 09 January 2023;
Published: 27 January 2023.
Edited by:
Hui Song, Qingdao Agricultural University, ChinaReviewed by:
Ruochen Wang, Sichuan University, ChinaQingtao Wang, Hebei University of Engineering, China
Copyright © 2023 Lu, Yang, Tian, Gui, Dong and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Dong, cmRvbmdAZ3p1LmVkdS5jbg==; Chao Chen, Y2M2OTAwNzY3NDhAMTYzLmNvbQ==