 Wenhao Yue
Wenhao Yue Kangfeng Cai
Kangfeng Cai Xue Xia
Xue Xia Lei Liu1,2
Lei Liu1,2 Junmei Wang
Junmei Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 13 February 2023
Sec. Plant Biophysics and Modeling
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1137434
SWEET (Sugars Will Eventually be Exported Transporter) proteins, an essential class of sugar transporters, are involved in vital biological processes of plant growth and development. To date, systematical analysis of SWEET family in barley (Hordeum vulgare) has not been reported. In this study, we genome-wide identified 23 HvSWEET genes in barley, which were further clustered into four clades by phylogenetic tree. The members belonging to the same clade showed relatively similar gene structures and conserved protein motifs. Synteny analysis confirmed the tandem and segmental duplications among HvSWEET genes during evolution. Expression profile analysis demonstrated that the patterns of HvSWEET genes varied and the gene neofunctionalization occurred after duplications. Yeast complementary assay and subcellular localization in tobacco leaves suggested that HvSWEET1a and HvSWEET4, highly expressed in seed aleurone and scutellum during germination, respectively, functioned as plasma membrane hexose sugar transporters. Furthermore, genetic variation detection indicated that HvSWEET1a was under artificial selection pressure during barley domestication and improvement. The obtained results facilitate our comprehensive understanding and further functional investigations of barley HvSWEET gene family, and also provide a potential candidate gene for de novo domestication breeding of barley.
All terrestrial life depends on the photosynthesis, that plants produce carbohydrates (e.g. sugars, starch) in source tissues (mesophyll cells) and transport the carbon assimilates long-distance via the phloem sieve element-companion cell complex to sustain the growth and development of sink tissues, such as roots, flowers, fruits and seeds (Ruan, 2014). In most plants, sucrose is the principal carbohydrates transported via either symplastic or apoplastic pathway, in which sugar transporters play important roles (Chen et al., 2015a). SWEET transporters were firstly characterized in last decades (Chen et al., 2010), mostly mediating sugar efflux following the concentration gradient (Baker et al., 2012) and involving in essential biological processes, for example sucrose export from mesophyll cells to apoplast for phloem loading (Chen et al., 2012; Bezrutczyk et al., 2018), sugars transfer from seed coat and endosperm to embryo during the seed filling stage (Chen et al., 2015b; Wang et al., 2019), sucrose export from nectary parenchyma to extracellular space to recruit pollinating insects (Lin et al., 2014), etc. However, pathovar-specific effectors can hijack the sugar efflux system and target the promoters of specific SWEET genes to induce gene expressions, increasing sugar content in the invasion sites to fuel their growth (Antony et al., 2010; Chen et al., 2010). The conserved domain in SWEET is MtN3/Saliva, which consists of two units of three transmembrane helices (TMHs) separated by a less conserved TMH in plants (Han et al., 2017). The Arabidopsis SWEET genes are mainly clustered into four clades, with clade I, II and IV preferentially transporting monosaccharides and clade III disaccharides, respectively (Eom et al., 2015). The SWEET proteins exhibit substrate recognition and selectivity based on the size of the substrate-binding pocket and function in oligomerization manner (Han et al., 2017). Given the importance of SWEET genes in sugar allocations, many SWEET genes underwent artificial selection during crop domestication. Maize and rice SWEET4, which mediate the hexose transport across the basal endosperm transfer layer, the entry point of nutrients into the seed, were strongly selected during domestication to sustain the development of the large starch-storing endosperm of cereal grains (Sosso et al., 2015). The artificial selection of soybean GmSWEET10a drove the initial domestication of multiple seed traits, such as seed size, oil content and protein content (Wang et al., 2020).
With the rapid development of sequencing technology, more and more reference genomes were available, leading to genome-wide identification of SWEET genes in various species, such as Arabidopsis (Arabidopsis thaliana) (Chen et al., 2010), rice (Oryza sativa) (Yuan and Wang, 2013), soybean (Glycine max) (Patil et al., 2015), sorghum (Sorghum bicolor) (Mizuno et al., 2016), wheat (Triticum aestivum) (Qin et al., 2020). The barley genome with size around 5G is characterized by high content of repetitive elements and large pericentromeric regions that are virtually devoid of meiotic recombination, and the first chromosome level reference genome publication (Mascher et al., 2017) makes it possible to genome-wide analyze the HvSWEET gene family. Although barley HvSWEET family members were identified in previously study (Mascher et al., 2017; Qin et al., 2020), they were not systematically investigated. In this study, we comprehensively analyzed phylogenetic relationships, gene structures and conserved protein motifs, syntenic relationships, expression patterns of HvSWEET family genes, and found that the plasma membrane (PM) localized hexose transporter HvSWEET1a, highly expressed in aleurone tissue during seed germination, underwent artificial selection during barley domestication and improvement.
Barley SWEET candidate genes were identified by combinations of conserved protein domain and BLAST searching methods. The barley reference genome and protein sequences (Mascher et al., 2017) were downloaded from Phytozome (https://phytozome-next.jgi.doe.gov/). A total of 17 AtSWEET proteins and 21 OsSWEET proteins identified previously (Chen et al., 2010) were obtained from corresponding Arabidopsis (Cheng et al., 2017) and rice (Ouyang et al., 2007) files in Phytozome. The Hidden Markov Model (HMM) of the MtN3_slv domain (PF03083), downloaded from Pfam 35.0 (http://pfam-legacy.xfam.org/), was used to identify putative barley SWEET proteins using HMMER (http://hmmer.org/) with the “trusted cutoff and E-value < 0.01” as the threshold. The 17 Arabidopsis SWEET proteins were used as queries to carry out a BLASTP search in the barley protein sequences with the E-value < 1e-5 as the threshold using DIAMOND (Buchfink et al., 2021) (Version 2.0.11). The barley SWEET proteins, identified by both HMM and BLASTP methods, were manual checked and named in accordance with previously study (Mascher et al., 2017). The R package Peptides (Osório et al., 2015) (Version 2.4.4) was used to calculate the physical and chemical parameters of HvSWEET proteins, including protein length, molecular weight and theoretical pI. The Multiple sequence alignment of HvSWEET proteins was performed using MUSCLE program implemented in MEGA7 (Kumar et al., 2016) with the default parameters, and the phylogenetic tree was constructed by MEGA7 with the bootstrap of 1000 replications using neighbor-joining (NJ) method. The phylogenetic tree of SWEET family proteins of Arabidopsis, rice and barley was constructed using the same method without OsSWEET7d/LOC_Os09g08490, for short protein length (63 aa). The MEME software (Bailey et al., 2009) (Version 5.0.5) was used to investigate the conserved motifs of HvSWEET proteins with the parameters ‘-mod anr –nmotifs 10 –minw 6 –maxw 200’. The amino acid sequences of 10 conserved protein motifs were plotted using R package ggseqlogo (Wagih, 2017). The phylogenetic tree, conserved protein motifs and gene structures of HvSWEET genes were shown with iTOL (https://itol.embl.de/).
The reference primary protein files of Arabidopsis (Cheng et al., 2017), rice (Ouyang et al., 2007), maize (Jiao et al., 2017), sorghum (McCormick et al., 2018), barley (Mascher et al., 2017), and wheat (Zhu et al., 2021) were downloaded from Phytozome, and used to perform genome-wide syntenic analysis within barley or between barley and other plant species. Proteins of plant species were subject to homologous searching by DIAMOND BLASTP with the paramenters ‘–evalue 1e-10 –max-target-seqs 5’. The MCScanX (Wang et al., 2012) was used to deal with the BLASTP results to identify the gene duplication events and the synteny results were visualized by Circos (Krzywinski et al., 2009) (Version 0.69-8) or python version JCVI (https://github.com/tanghaibao/jcvi) of MCScan. The OrthoFinder (Emms and Kelly, 2019) was used to make gene family clustering. The orthogroups, containing three genes in wheat and only one gene in other plant species, were identified and the genes, with two wheat genes randomly discarded, were derived to assemble super genes to construct maximum-likelihood (ML) species phylogenetic tree using IQ-Tree (Nguyen et al., 2015) (Version 1.6.12) with the parameters ‘-m MFP -bb 1000’.
To investigate the HvSWEET gene expression patterns, the RNA-seq raw sequencing data of 16 different barley developmental tissues (leaf, root, inflorescence, etc) (Mascher et al., 2017) and seed tissues during germination (embryo, aleurone, scutellum and grain) (Betts et al., 2017) were downloaded from NCBI, and filtered using fastp (Chen et al., 2018) (Version 0.12.4) with the default parameters. The high-quality cleaned reads were aligned to the barley reference genome (Mascher et al., 2017) with HISAT2 (Kim et al., 2019). Following alignments, raw counts for each gene were derived using featureCounts implemented in R package Rsubread (Liao et al., 2019), and normalized into the number of transcripts per kilobase of exon sequence in a gene per million mapped reads (TPM) with TMM method (Robinson and Oshlack, 2010). Heatmaps of HvSWEET gene expression profiles were generated using the R package ComplexHeatmap (Gu et al., 2016) (Version 2.10.0) based on the log2 (TPM +1) transformation or expression proportions of HvSWEET genes in that of the whole gene family calculated by TPM values.
The exome SNP data of 360 barley accessions (20 wild accessions, 166 landraces and 174 cultivars) with clearly known breeding history from previously published study (Bustos-Korts et al., 2019) were derived and used to analyze the genetic diversity of HvSWEET genes. The SNPs with missing data >10% or minor allele frequency (MAF) < 5% were filtered using VCFtools (Danecek et al., 2011) (Version 0.1.17), and then the missing data were imputated using the package beagle (Browning et al., 2018) (Version 5.4) with the default parameters. The SNPs in HvSWEET genes were extracted to perform haplotype analysis and annotated according to the barley genome (Mascher et al., 2017) using the package ANNOVAR (Wang et al., 2010) (Version 2019-10-24). The pairwise linkage disequilibrium (R2) of SNPs in HvSWEET genes were calculated using PLINK (Purcell et al., 2007) (Version 1.90) and displayed using R package LDheatmap (Shin et al., 2006) (Version 1.0-6). The network 10 (https://www.fluxus-engineering.com/) was used to construct median-joining network of different haplotypes (Bandelt et al., 1999).
Primers were designed according to open reading frames of HvSWEET1a, HvSWEET4 and AtSWEET1 genes. The corresponding sequences were amplified from cDNA of barley (Golden Promise) or Arabidopsis, and cloned into pDONR201 vector (Invitrogen). The clones were selected by PCR and sequenced to confirmation. For subcellular localization of HvSWEET1a and HvSWEET4 in tobacco leaves, the corrected pDONR201 vectors were recombinant with destination vectors pH7WGF2.0 to obtain GFP-HvSWEET1a and GFP-HvSWEET4 constructs using Gateway system. For complementation of yeast mutant EYB.VW4000 (Wieczorke et al., 1999), lacking 18 hexose transporters, and subcellular localization of SWEET proteins in yeast cells, the seamless cloning was used to introduce SWEET gene and GFP gene amplified from pFA6a-GFP(S65T)-His3MIX6 vector, into yeast expression vector pYEPlac195 vector, which was inserted by ADH1 promoter amplified from pADGT7 vector in advance, to obtain pADH1-SWEET-GFP(S65T) constructs. The primer information is listed in Supplementary Table 1.
The subcellular localization vectors were transiently expressed in tobacco (Nicotiana benthamiana) leaves by Agrobacterium-mediated infiltration. AtPIP2A-meCherry was used as plasma membrane (PM) marker. The Agrobaterium strain C58C1 harboring p19 was used to prevent the onset of PTGS (post-transcriptional gene silencing) in the infiltrated leaves. Infiltrated tobacco plants were grown for another 3 days for GFP and mCherry imaging using a Zeiss LSM710NLO confocal laser-scanning microscope. Excitation/emission wavelength were 488 nm for GFP, and 561 nm for mCherry.
The yeast complementation vectors or empty vectors were transformed into the hexose-uptake deficient yeast mutant EYB4000 (Wieczorke et al., 1999). Then, transformed yeasts were screened in synthetic dropout (SD)-Ura media, supplemented with 2% maltose. For complementation growth assays, yeasts were grown overnight in liquid SD media to an optical density at 600 nm (OD600) of ~0.6, then OD600 was adjusted to ~0.3 with water. Five-microliter aliquots of serial dilutions were plated on SD media containing 2% maltose (as control) or 2% other hexoses. Growth photographs were taken after incubation at 30 °C for three days.
For subcellular localization of SWEET proteins in yeast cells, transformed yeasts cultured in SD media supplemented with 2% maltose were collected, washed three times with water, and then applied on microscope slide. Fluorescence signals were detected using a Zeiss LSM710NLO confocal laser-scanning microscope. Excitation/emission wavelength were 488 nm for GFP signal.
A total of 23 SWEET genes were identified in barley, consistent with previously studies (Mascher et al., 2017; Qin et al., 2020), and were named accordingly (Supplementary Table 2). We analyzed the characteristics of HvSWEET genes, including exon number, protein length, the number of MtN3/saliva domain, molecular weight (MW) and isoelectric point (pI), and found that most proteins contained two MtN3/saliva domains and four contained only one domain (HvSWEET7b/c and HvSWEET15b/c), the protein length of HvSWEET ranged from 91 amino acid (aa; HvSWEET7b/c) to 353 aa (HvSWEET13a), the MW ranged from 10.19 kDa (HvSWEET7b/c) to 38.74 kDa (HvSWEET13a), and the pI ranged from 6.51 (HvSWEET12) to 11.3 (HvSWEET15c) (Supplementary Table 2).
To investigate the evolutionary relationship between HvSWEET and SWEET proteins from other species, including Arabidopsis and rice, we constructed a neighbor-joining (NJ) phylogenetic tree and found that HvSWEET proteins could be clustered into four clades, with clade I containing five members (HvSWEET1a/b, HvSWEET2a/b and HvSWEET3), clade II containing seven members (HvSWEET4, HvSWEET5, HvSWEET6a/b and HvSWEET7a/b/c), clade III containing 10 members (HvSWEET11a/b, HvSWEET12, HvSWEET13a/b, HvSWEET14a/b, HvSWEET15a/b/c) and clade IV containing one member (HvSWEET16), respectively (Figure 1). A small extension of HvSWEET11, HvSWEET13, HvSWEET14 and HvSWEET15 were observed, with each containing two or three members in barley compared with only one orthologue in Arabidopsis and rice, consistent with previous study (Mascher et al., 2017).
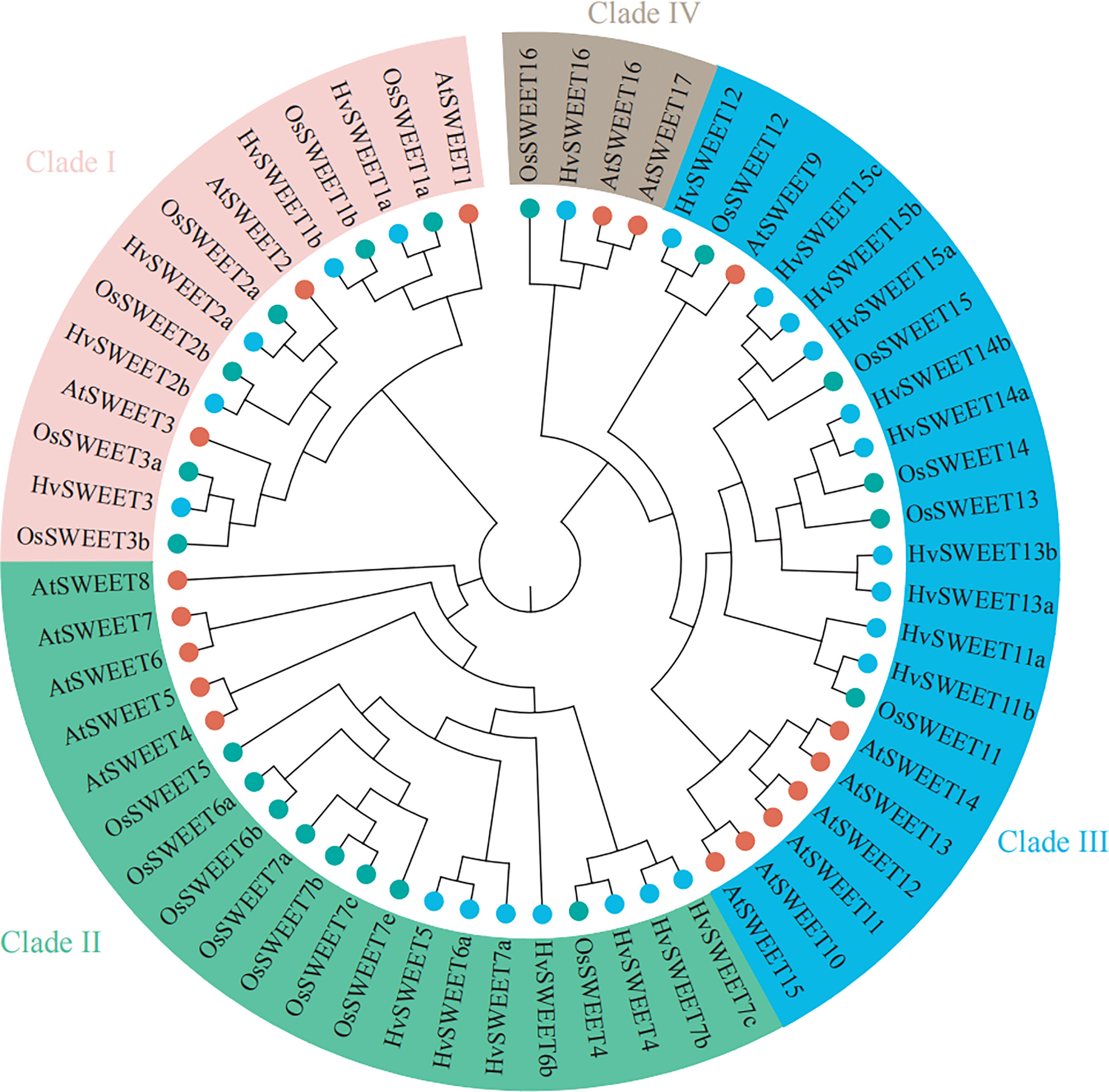
Figure 1 Phylogenetic relationship of SWEET family proteins of Arabidopsis, rice and barley. The phylogenetic tree is constructed using the neighbor-joining (NJ) method with 1000 bootstrap replications. Proteins of Arabidopsis, rice and barley are indicated with red, green and blue circles, respectively.
Given that segmental and tandem duplications play important roles in gene family expansion during evolution, we detected these duplication events, involving HvSWEET genes, in barley. Two segmental duplications (HvSWEET1a/b, HvSWEET11a/b) and two tandem duplications (HvSWEET6a/b, HvSWEET15b/c) were observed (Figure 2A). Additionally, HvSWEET genes were observed uneven distributed over barley chromosomes, with chromosome 6 containing five members, chromosome 1 and 7 each containing four members, chromosome 3 and 4 each containing three members, chromosome 2 containing two members, and chromosome 5 and chromosome unscaffold each containing one member, respectively (Figure 2A).
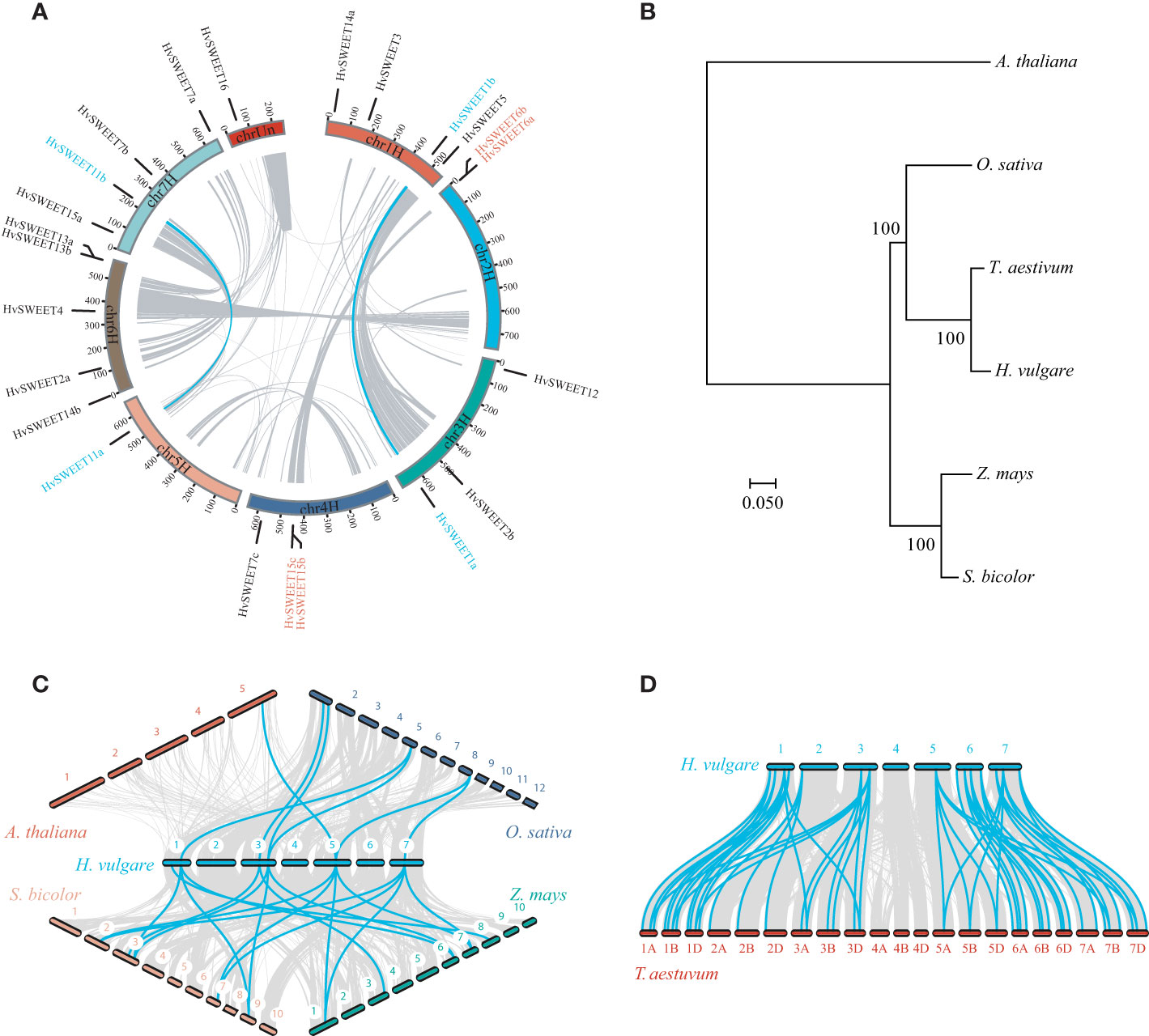
Figure 2 Snyteny analysis of SWEET family genes. (A) Snyteny analysis of HvSWEET genes within barley genome. The gray and blue lines in the inner circle denote syntenic blocks and duplications of HvSWEET gene pairs, respectively. The chromosome locations of HvSWEET genes are indicated with black lines linked with gene names, and segmental and tandem duplicated HvSWEET genes are texted in the blue and red colors, respectively. (B) Phylogenetic species tree of barley, Arabidopsis, rice, sorghum, maize and wheat using maximum-likelihood method with 1000 bootstrap replications. (C) and (D) Synteny analysis of SWEET family genes between barley and other plant species. The gray and blue lines represent snytenic blocks and homologous SWEET gene pairs, respectively.
To explore the evolution relationships of SWEET family genes between barley and other plant species, we performed synteny analysis between barley and Arabidopsis or Poaceae plant species, including rice, maize, sorghum and wheat. While only one pair of orthologous SWEET genes was observed between barley and Arabidopsis, which might be caused by evolutionally far genetic relationship between dicots and monocots (Figure 2B), there were six, eight, nine and 51 pairs of orthologous genes identified between barley and rice, maize, sorghum and wheat, respectively (Figures 2C, D). The closer evolution relationship and high ploidy levels of wheat might lead to more syntenic orthologous SWEET gene pairs between barley and wheat (Figure 2B).
To gain more insights into the characteristics of HvSWEET genes, the gene structural diversity was examined, and 11 members were found to have five exons, six members six exons, five members four exons and one member three exons (Figure 3C). The exon lengths were similar, whereas the intron lengths varied, with HvSWEET4, HvSWEET15c and HvSWEET16 containing very long introns (Figure 3C). Furthermore, we found clade I members contain six exons except HvSWEET2b containing five exons, clade II members contain five exons except HvSWEET7b and HvSWEET7c containing four exons, clade IV member contains six exons, and clade III members contain three to six exons (Figures 3A–C), suggesting that the gene structures of clade I and clade II are relatively conserved.
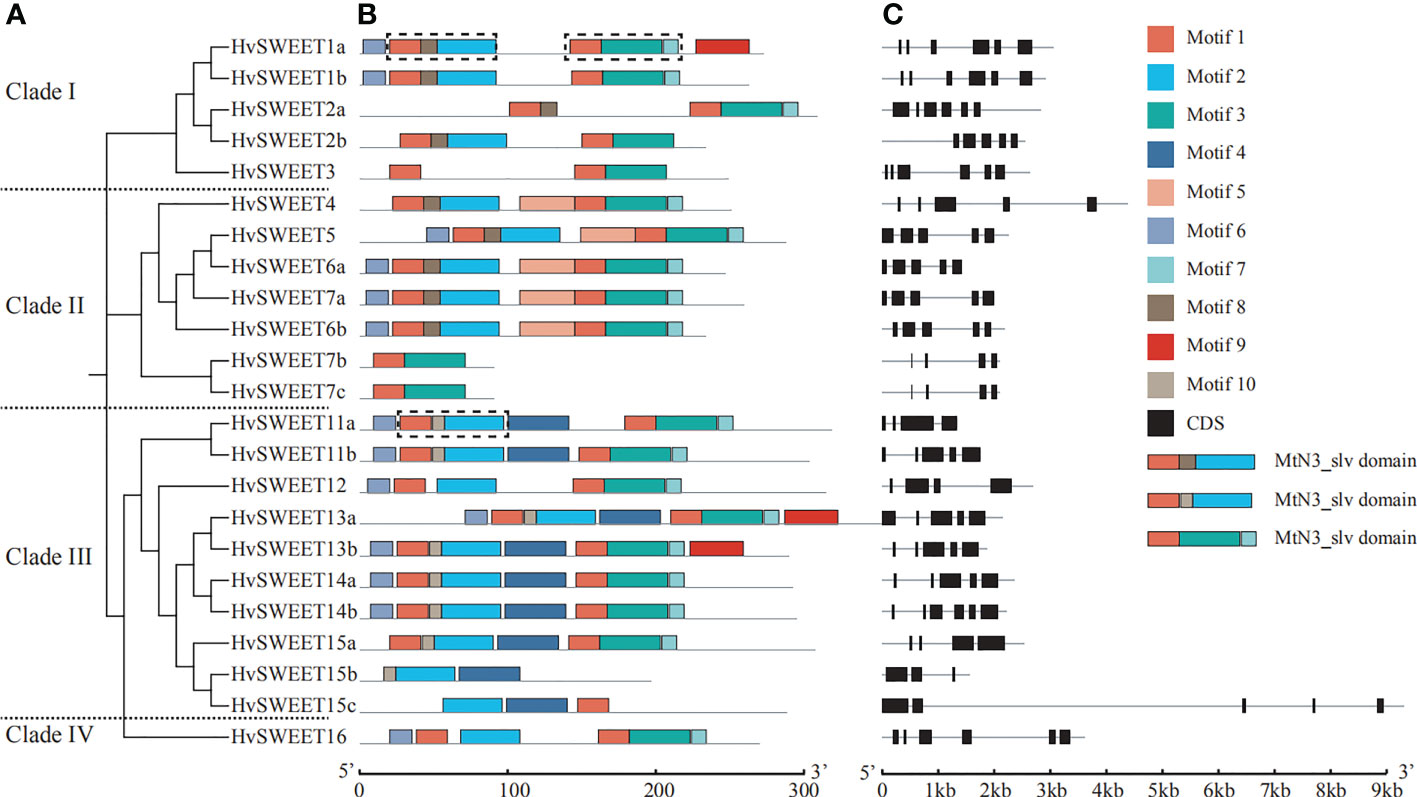
Figure 3 Phylogenetic relationship, conserved protein motifs and gene structures of HvSWEET genes. (A) Phylogenetic relationship analysis of HvSWEET genes using the neighbor-joining (NJ) method with 1000 bootstrap replications. (B) Conserved motif analysis of HvSWEET proteins. Different motifs are indicated with different colored boxes. The MtN3_slv domain is outlined using black dashed lines. (C) Gene structures of HvSWEET genes. The black boxes represent CDS regions.
To further investigate the gene structural diversity, the conserved motifs of all HvSWEET proteins were examined. In total, ten conserved protein motifs, with the range from eight to 41 aa (Supplementary Figure 1), were identified, among which the group containing motif 1, 8/10 and 2 and the group containing motif 1, 3 and 7 were annotated as MtN3_slv domain (Figure 3B). In addition, the result demonstrated that the motif 1, 2, 3, 6 and 7 exist in most HvSWEET members (96%, 83%, 91%, 61% and 74%, respectively), and motif 4 was identified only in the clade III. The HvSWEET protein in the same clade share relatively similar conserved motifs.
To comprehensively study the physiological functions of HvSWEET genes, the raw RNA-seq sequencing data of 16 different developmental tissues and seed tissues during germinating were derived from previously study (Betts et al., 2017; Mascher et al., 2017), and used to map against barley reference genome (Mascher et al., 2017). Different expression patterns were observed for HvSWEET genes in the detected tissues (Figure 4A). Five HvSWEET genes, including HvSWEET1a, HvSWEET2a/b, HvSWEET4 and HvSWEET15a, were expressed in almost all detected tissues, whereas four HvSWEET genes, including HvSWEET5, HvSWEET12 and HvSWEET15b/c, were nearly not detected (Figure 4A; Supplementary Tables 3, 4). Moreover, several HvSWEET genes were expressed during seed germination stages, such as HvSWEET7b/c, and several genes showed high expression levels in specific tissues, such as HvSWEET11b in developing caryopsis and inflorescence rachis, and HvSWEET13a/b in senescing leaf and epidermal strips (Figure 4A; Supplementary Tables 3, 4). Additionally, we examined the expression patterns of duplicated HvSWEET gene pairs, and found that some duplicated genes exhibited divergent expression patterns, such as SWEET1a/b and HvSWEET11a/b, suggesting they underwent neofunctionalization after duplications (Figure 4A; Supplementary Tables 3, 4).
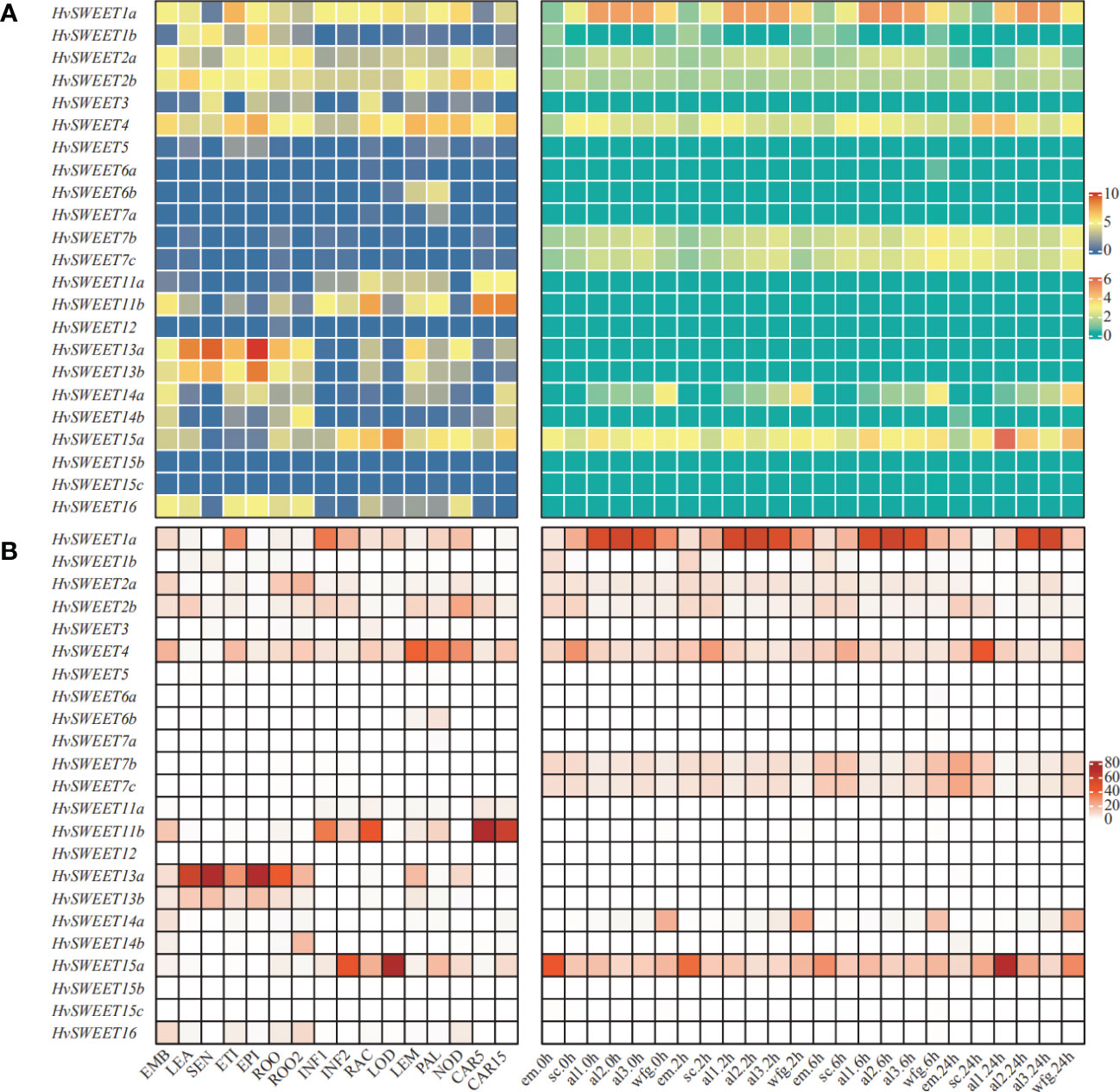
Figure 4 Expression profiles (A) and expression proportions of HvSWEET genes in that of the whole gene family (B) in the published different developmental tissues (Mascher et al., 2017) (left panel) and seed tissues during germination (Betts et al., 2017) (right panel). The TPM values of each gene, normalized by TMM method, are used for (B) and the transformation values by log2 (TPM+1) are used for (A). Left panel: EMB, 4 days after planting (dap) embryo; LEA, 17 dap leaf; SEN, 56 dap senescing leaf; ETI, 10 dap etiolated leaf grown in dark; EPI, 28 dap epidermal strips; ROO, 17 dap root; ROO2, 28 dap root; INF1, 30 dap inflorescences; INF2, 50 dap inflorescences; RAC, 35 dap inflorescence rachis; LOD, 42 dap lodicule; LEM, 42 dap lemma; PAL, 42 dap palea; NOD, the third stem internode (42 dap); CAR5, 5 days post-anthesis (dpa) caryopsis; CAR15, 15 dpa caryopsis. Right panel: em, embryo; sc, scutellum; al1, one third of aleurone proximal to the embryo; al2, the central third of aleurone; al3, one third of aleurone distal to embryo; wfg; whole fixed grain; 0h, 2h, 6h and 24h represent the corresponding time after seed germination.
Given the functional redundancy of HvSWEET genes, the relative expression level determines which member plays the main role in sugar transporting in the specific tissue within the gene family. We calculated proportion of each HvSWEET gene expression in that of the whole family, and observed that the sugar transporting activities of the gene family in leaf, senescing leaf, epidermal strips and root were mainly dependent on HvSWEET13a, the inflorescence, rachis and lodicule mainly dependent on HvSWEET11b and HvSWEET15a, the lemma and palea mainly dependent on HvSWEET4, the developing caryopsis mainly dependent on HvSWEET11b, and the germinating seed aleurone, embryo and scutellum mainly dependent on HvSWEET1a, HvSWEET15a and HvSWEET4, respectively (Figure 4B). These results demonstrate HvSWEET genes function redundantly and divergently to mediate sugar transport in barley developmental growth.
Barley was selected by early humans in the Fertile Crescent ground 10,000-12,000 years ago and is primarily used for animal feed, and malting and brewing, with a small percentage devoted to human food (Stein and Muehlbauer, 2018). The uniform and fast germination is a typical domestication syndrome trait, which ensure even maturity and enable crop management (Fuller and Allaby, 2009). Moreover, high malting quality is of importance for barley and also requires uniform and fast seed germination, during which the malting grains develop the enzymes required for modifying starches into various types of sugar to fuel the embryonic axis growth (Ma et al., 2017). Given overexpression of the sugar carrier AtSWEET16 resulted in improved seed germination rates (Klemens et al., 2013), we thought HvSWEET1a and HvSWEET4, relatively high expressed in aleurone and scutellum during seed germination, respectively, might be related with seed germination and could undergo artificial selection during barley domestication (wild accessions versus landraces) and improvement (landraces versus cultivars) (Figure 4B). To verify our hypothesis, we detected the genetic variations of HvSWEET1a using the exome SNP information of 360 accessions, consisting of 20 wild accessions, 166 landraces and 174 cultivars, from previously study (Bustos-Korts et al., 2019). In total, 20 SNPs, consisting of two SNPs in 5’ UTR, seven SNPs in introns and 11 SNPs in 3’ UTR, were detected in HvSWEET1a genetic region and classified the population into 27 haplotypes, each represented by one to more than one hundred accessions (Figure 5A). Median-joining network analysis sorted the 27 haplotypes into three main groups, namely H_I group (including H_1-1 to H_I-8), H_II group (including H_II-1 to H_II-16) and H_III group (including H_III-1 to H_III-3) (Figure 5B). Allele-frequency analysis demonstrated that the proportion of H_I significantly increased in landraces compared with that of wild accessions, and further increased in cultivars, indicating strong artificial selection of HvSWEET1a during barley domestication and improvement (Figure 5C). In addition, two groups of SNPs in the 3’ UTR region of HvSWEET1a gene showed strong linkage disequilibrium (LD), which might be the artificial selected regions (Figure 5A).
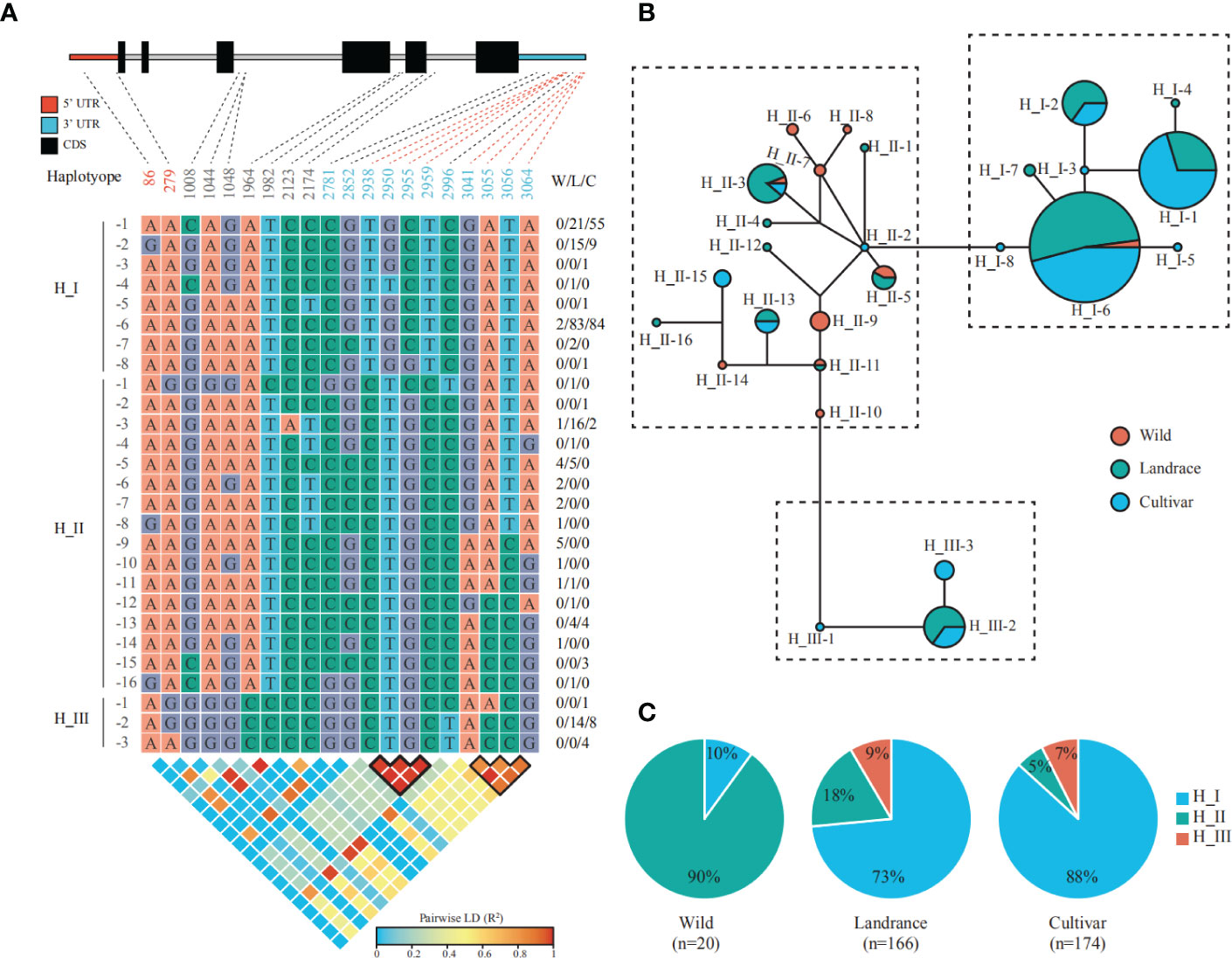
Figure 5 Artificial selection of HvSWEET1a gene during barley domestication and improvement. (A) Genetic variations detected in the genomic region of HvSWEET1a in the published exome re-sequencing data of 360 accessions (Bustos-Korts et al., 2019). The SNP positions are indicated relative to the gene start site of HvSWEET1a, 634,920,942 on chromosome 3, and displayed below gene structure, linked with dashed lines. The red dashed lines and black outlines denote the SNPs in strong LD regions in top and bottom panel, respectively. The W/L/C indicates the number of wild accessions, landraces and cultivars, respectively. The pairwise linkage disequilibria (LD) between SNPs are measured using R2. (B) Median-joining network representing the relatedness of 27 HvSWEET1a haplotypes, each represented by a circle with size proportional to the corresponding haplotype population size. Wild accessions, landraces and cultivars are indicated by red, green and blue colors, respectively. (C) Allele frequency distribution of three main haplotypes. H_I, blue; H_II, green; H_III, red. The accession number (n) is shown below the corresponding population.
Similarly, the genetic variations of HvSWEET4 were also examined and nine SNPs were detected, which sorted the population into 12 haplotypes (Supplementary Figure 2). Median-joining network sorted the 12 haplotypes into three main groups, namely H_I group (including H_I-1), group H_II (including H_II-1 to H_II-8) and group H_III (including H_III-1 to H_III-3) (Supplementary Figure 2). However, no significant allele-frequency variations of three main groups were observed among wild accessions, landraces and cultivars (Supplementary Figure 2).
To validate the sugar transporter function of HvSWEET1a and HvSWEET4 in yeast, the N-terminal HvSWEET1a or HvSWEET4 in fusion with GFP were transformed into hexose-uptake deficient yeast EYB.VW4000 (Wieczorke et al., 1999), which lacks 18 hexose transporters and could not grow on media with hexose as the only carbon source, with AtSWEET1 as the positive control. The results revealed that the plasma membrane (PM) localized HvSWEET1a and HvSWEET4 could transport glucose, galactose and fructose in yeast cells, and HvSWEET1a showed higher transport activity than HvSWEET4 (Figures 6A, B). To study the subcellular localization of HvSWEET1a and HvSWEET4 in plant cells, GFP fused with HvSWEET1a or HvSWEET4 was co-expressed with Arabidopsis PM intrinsic protein 2A fused to mCherry (AtPIP2A-mcherry) (Prak et al., 2008) in tobacco leaves. GFP fluorescence signals of HvSWEET1a or HvSWEET4 coincided to mCherry signals of PM maker in tobacco leaves (Supplementary Figure 3). These results demonstrate that HvSWEET1a and HvSWEET4 function as PM hexose transporters.

Figure 6 HvSWEET1a and HvSWEET4 have transport activity of hexoses. (A) Complementation of hexose uptake-deficiency of the yeast mutant EYB.VW 4000. Glucose, Galactose, Fructose and Maltose denote the corresponding carbon source used in the media. Photographs were taken 3 days after incubation. Experiment was repeated three times with similar results. AtSWEET1, positive control; Empty vector, negative control. (B) Plasma membrane subcellular localizations of HvSWEET1a, HvSWEET4 and AtSWEET1 in yeast cells. Bars = 5 μm.
Sucrose is produced in photosynthetically active tissues of the leaf and stem and actively loaded into phloem, and then translocated to various tissues and cells that depend on external sugar supply. SWEET transporters play vital roles in the sugar transport process (Eom et al., 2015), especially in the angiosperm grain filling, where only apoplastic pathway exists between maternal and filial tissues including endosperm and embryo (Yang et al., 2018). Genome-wide identification and analysis is an essential method to study the specific functions of the gene family. However, high content of repetitive elements and large pericentromeric regions lead to relatively late publication of chromosome-level barley reference genome (Mascher et al., 2017), hindering the gene family identification compared with other plant species (Chen et al., 2010; Patil et al., 2015; Xie et al., 2019; Zhang et al., 2019; Qin et al., 2020). The barley SWEET family members were identified in previously study (Mascher et al., 2017; Qin et al., 2020), but they were not systematically investigated. In this study, we comprehensively analyzed the barley SWEET family. We carried out genome-wide identification of HvSWEET genes and found 23 members, consistent with previously study (Mascher et al., 2017; Qin et al., 2020). The barley HvSWEET members were clustered into four clades, with a litter family member expansions (HvSWEET11, HvSWEET13, HvSWEET14 and HvSWEET15) compared with Arabidopsis and rice (Figure 1). The HvSWEET genes in the same clade shared relatively similar gene structures and conserved protein motifs (Figure 3). Moreover, two tandem and segmental duplicated HvSWEET gene pairs were observed in the barley genome, and the phylogenetic species relationship and polyploidization might account for the differences of SWEET gene pairs identified between barley and other plant species (Figure 2). HvSWEET genes showed different expression patterns (Figure 4A), indicative of their divergent functions in various biological processes. Interestingly, different expression patterns of duplicated HvSWEET genes were observed, demonstrating that gene neofunctionalization occurred after duplications (Figure 4A). Considering functional redundancy, we also examined the proportion of single HvSWEET gene in that of the whole family to identify the main functional member in specific tissue and observed the relatively high expression of HvSWEET1a and HvSWEET4 in seed aleurone and scutellum during germination, respectively (Figure 4B). Given seed germination is a typical domesticated trait and SWEET genes is related with seed germination rates (Klemens et al., 2013), we performed haplotype analysis and observed the artificial selection of HvSWEET1a during domestication and improvement (Figures 5, 6). Considering the SNP positions and LD values, the 3’ UTR of HvSWEET1a might be the region under selection (Figure 5A). Finally, the yeast complementary experiment and subcellular localization demonstrated that HvSWEET1a is a PM hexose transporter (Figure 6; Supplementary Figure 3). Although it is plausible to infer that HvSWEET1a, highly expressed in aleurone tissue during seed germination, was artificial selected due to facilitate seed germination, we could not exclude the possibilities that HvSWEET1a underwent selection for other important traits, considering the tissue expression profile of HvSWEET1a (Figure 4), and further experiments are needed to investigate the physiological function and the domestication mechanism of HvSWEET1a.
The reference genome assembly quality plays an important role in gene family identification and the recent development of fast and accurate long-read sequencing by circular consensus sequencing (CCS) facilitated the barley reference genome assembly version 3 (Mascher et al., 2021). To compare our genome-wild identification result of HvSWEET gene family, which was based on barley reference genome version 1 (Mascher et al., 2017), with that based on the version 3 (Mascher et al., 2021), we performed the same methods to identify HvSWEET family genes in the version 3. Surprisingly, 26 HvSWEET members were observed in version 3 with three more members (Supplementary Table 5). The phylogenetic analysis also classified the 26 members into four clades (Supplementary Figure 4). To investigate whether differences of HvSWEET genes exist between version 1 and version 3, we compared the protein similarity, exon numbers and MtN3/Saliva domain numbers of the two version members, and found that almost all the HvSWEET genes (version 1) share the same exon numbers and MtN3/Saliva domain numbers with corresponding ones in version 3 (Supplementary Table 5), and the majority (18/23) of HvSWEET genes (version 1) shared 100% similarity with those in version 3, four members more than 90% and one member more than 80% (Supplementary Table 5). Interestingly, HvSWEET16, which is located in chromosome unscaffold in version 1, is relocated to chromosome 5 in version 3 (Supplementary Table 5). The scaffold N50 length of barley reference genome version 3 is more than 60 times of that version 1 (Mascher et al., 2017; Mascher et al., 2021), and the better assembly quality and gene annotations account for more HvSWEET family members identified and chromosome location of HvSWEET16 in version 3. In the near future, more and more omics data based on the barley reference genome version 3 will further facilitate our understanding of HvSWEET gene family, especially for the newly identified HvSWEET members.
Crop domestication is one of the most significant innovations in the history of humankind, which has enabled humans to survive, multiply, and ultimately give birth to the development of civilization (Doebley et al., 2006). However, the genetic diversities of modern crops significantly decreased compared with their progenitors for long term domestication and improvement (Huang et al., 2012; Hufford et al., 2012; Zhou et al., 2015; Milner et al., 2019), making them lack diversities in important genes, such as stress-resistance genes, and vulnerable to the rapid climate changes. More importantly, it is very difficult to develop super varieties based on the modern germplasms to meet the globally increasing food demand in face of extreme environmental challenges. Rapid de novo domestication of wild species is an alternative breeding strategy and has been successful in a few plant species by introducing desired mutations in specific genes, which were under selection pressure during domestication, in wild background with the powerful genome editing technology CRISPR/Cas9, and the elite lines with domestication traits and stress-resistance were obtained in a few years (Li et al., 2018; Zsogon et al., 2018; Yu et al., 2021). The pathogen, Xanthomonas oryzae pv. oryzae (Xoo), secretes transcription-activator-like effectors (TALes) to recognize effector-binding elements (EBEs) and induce, at minimum, one of OsSWEET11, OsSWEET13 and OsSWEET14 to increase sugar content in the invasion sites, and simultaneous introduction of mutations in EBE regions of all three OsSWEET promoters using CRISPR/Cas9 in rice line Kitaake and elite mega varieties IR64 (Mackill and Khush, 2018) and Ciherang-Sub1 (Toledo et al., 2015) confers them robust and broad-spectrum resistance to rice blight (Eom et al., 2019; Oliva et al., 2019). The promoter of maize ZmSWEET4c was strongly selected during domestication and the higher gene expression in maize than maize ancestor teosinte leads to larger grains (Sosso et al., 2015). The artificial selection of a 9-base pair deletion in the promoter of soybean GmSWEET10a during domestication and improvement upregulates the gene expression and results in higher oil content (Miao et al., 2020; Wang et al., 2020). Priority of De novo domestication is functional investigations of artificial selected genes, and our study provides a potential candidate gene for de novo domestication breeding of barley.
In this study, a total of 23 barley HvSWEET genes were identified, with two tandem and two duplicated, and divided into four clades. Most genes belonging to the same clade showed similar gene structures and conserved motifs. Expression patterns of HvSWEET genes varied, and HvSWEET1a and HvSWEET4, highly expressed in seed aleurone and scutellum during germination, respectively, showed PM subcellular localization and hexose transport activity. Moreover, haplotype analysis revealed the artificial selection of HvSWEET1a during barley domestication and improvement. The obtained results will be helpful for comprehensive understanding and further functional investigations of barley HvSWEET family, and also provide a potential candidate gene for de novo domestication breeding of barley.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
JW, WY, and KC conceived and designed the research. WY and KC performed data analysis. XX and LL carried out the experiment. WY and JW wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (32101642), Natural Science Foundation of Zhejiang Province (LQ21C130006), Major Program on Agricultural New Variety Breeding (2021c02064-3-2) and China Agriculture Research System of MOF and MARA (CARS-05-01A-06).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1137434/full#supplementary-material
Antony, G., Zhou, J., Huang, S., Li, T., Liu, B., White, F., et al. (2010). Rice xa13 recessive resistance to bacterial blight is defeated by induction of the disease susceptibility gene os-11N3. Plant Cell 22, 3864–3876. doi: 10.1105/tpc.110.078964
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Baker, R. F., Leach, K. A., Braun, D. M. (2012). SWEET as sugar: new sucrose effluxers in plants. Mol. Plant 5, 766–768. doi: 10.1093/mp/SSS054
Bandelt, H. J., Forster, P., Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Betts, N. S., Berkowitz, O., Liu, R., Collins, H. M., Skadhauge, B., Dockter, C., et al. (2017). Isolation of tissues and preservation of RNA from intact, germinated barley grain. Plant J. 91, 754–765. doi: 10.1111/tpj.13600
Bezrutczyk, M., Hartwig, T., Horschman, M., Char, S. N., Yang, J., Yang, B., et al. (2018). Impaired phloem loading in zmsweet13a,b,c sucrose transporter triple knock-out mutants in zea mays. New Phytol. 218, 594–603. doi: 10.1111/nph.15021
Browning, B. L., Zhou, Y., Browning, S. R. (2018). A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348. doi: 10.1016/j.ajhg.2018.07.015
Buchfink, B., Reuter, K., Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Bustos-Korts, D., Dawson, I. K., Russell, J., Tondelli, A., Guerra, D., Ferrandi, C., et al. (2019). Exome sequences and multi-environment field trials elucidate the genetic basis of adaptation in barley. Plant J. 99, 1172–1191. doi: 10.1111/tpj.14414
Chen, L. Q., Cheung, L. S., Feng, L., Tanner, W., Frommer, W. B. (2015a). Transport of sugars. Annu. Rev. Biochem. 84, 865–894. doi: 10.1146/annurev-biochem-060614-033904
Cheng, C. Y., Krishnakumar, V., Chan, A. P., Thibaud-Nissen, F., Schobel, S., Town, C. D. (2017). Araport11: a complete reannotation of the arabidopsis thaliana reference genome. Plant J. 89, 789–804. doi: 10.1111/tpj.13415
Chen, L. Q., Hou, B. H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X. Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nat. 468, 527–532. doi: 10.1038/nature09606
Chen, L. Q., Lin, I. W., Qu, X. Q., Sosso, D., McFarlane, H. E., Londono, A., et al. (2015b). A cascade of sequentially expressed sucrose transporters in the seed coat and endosperm provides nutrition for the arabidopsis embryo. Plant Cell 27, 607–619. doi: 10.1105/tpc.114.134585
Chen, L. Q., Qu, X. Q., Hou, B. H., Sosso, D., Osorio, S., Fernie, A. R., et al. (2012). Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335, 207–211. doi: 10.1126/science.1213351
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Doebley, J. F., Gaut, B. S., Smith, B. D. (2006). The molecular genetics of crop domestication. Cell 127, 1309–1321. doi: 10.1016/j.cell.2006.12.006
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Eom, J. S., Chen, L. Q., Sosso, D., Julius, B. T., Lin, I. W., Qu, X. Q., et al. (2015). SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 25, 53–62. doi: 10.1016/j.pbi.2015.04.005
Eom, J. S., Luo, D., Atienza-Grande, G., Yang, J., Ji, C., Thi Luu, V., et al. (2019). Diagnostic kit for rice blight resistance. Nat. Biotechnol. 37, 1372–1379. doi: 10.1038/s41587-019-0268-y
Fuller, D. Q., Allaby, R. (2009). Seed dispersal and crop domestication: shattering, germination and seasonality in evolution under cultivation. Annu. Plant Rev. 38, 238–295. doi: 10.1002/9781444314557.ch7
Gu, Z., Eils, R., Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. doi: 10.1093/bioinformatics/btw313
Han, L., Zhu, Y., Liu, M., Zhou, Y., Lu, G., Lan, L., et al. (2017). Molecular mechanism of substrate recognition and transport by the AtSWEET13 sugar transporter. Proc. Natl. Acad. Sci. 114, 10089–10094. doi: 10.1073/pnas.1709241114
Huang, X., Kurata, N., Wei, X., Wang, Z. X., Wang, A., Zhao, Q., et al. (2012). A map of rice genome variation reveals the origin of cultivated rice. Nat. 490, 497–501. doi: 10.1038/nature11532
Hufford, M. B., Xu, X., van Heerwaarden, J., Pyhajarvi, T., Chia, J. M., Cartwright, R. A., et al. (2012). Comparative population genomics of maize domestication and improvement. Nat. Genet. 44, 808–811. doi: 10.1038/ng.2309
Jiao, Y., Peluso, P., Shi, J., Liang, T., Stitzer, M. C., Wang, B., et al. (2017). Improved maize reference genome with single-molecule technologies. Nat. 546, 524–527. doi: 10.1038/nature22971
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Klemens, P. A., Patzke, K., Deitmer, J., Spinner, L., Le Hir, R., Bellini, C., et al. (2013). Overexpression of the vacuolar sugar carrier AtSWEET16 modifies germination, growth, and stress tolerance in arabidopsis. Plant Physiol. 163, 1338–1352. doi: 10.1104/pp.113.224972
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Liao, Y., Smyth, G. K., Shi, W. (2019). The r package rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47. doi: 10.1093/nar/gkz114
Lin, I. W., Sosso, D., Chen, L. Q., Gase, K., Kim, S. G., Kessler, D., et al. (2014). Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nat. 508, 546–549. doi: 10.1038/nature13082
Li, T., Yang, X., Yu, Y., Si, X., Zhai, X., Zhang, H., et al. (2018). Domestication of wild tomato is accelerated by genome editing. Nat. Biotechnol. 36, 1160–1163. doi: 10.1038/nbt.4273
Ma, Z., Bykova, N. V., Igamberdiev, A. U. (2017). Cell signaling mechanisms and metabolic regulation of germination and dormancy in barley seeds. Crop J. 5, 459–477. doi: 10.1016/j.cj.2017.08.007
Mackill, D. J., Khush, G. S. (2018). IR64: a high-quality and high-yielding mega variety. Rice 11, 18. doi: 10.1186/s12284-018-0208-3
Mascher, M., Gundlach, H., Himmelbach, A., Beier, S., Twardziok, S. O., Wicker, T., et al. (2017). A chromosome conformation capture ordered sequence of the barley genome. Nat. 544, 427–433. doi: 10.1038/nature22043
Mascher, M., Wicker, T., Jenkins, J., Plott, C., Lux, T., Koh, C. S., et al. (2021). Long-read sequence assembly: a technical evaluation in barley. Plant Cell 33, 1888–1906. doi: 10.1093/plcell/koab077
McCormick, R. F., Truong, S. K., Sreedasyam, A., Jenkins, J., Shu, S., Sims, D., et al. (2018). The sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 93, 338–354. doi: 10.1111/tpj.13781
Miao, L., Yang, S., Zhang, K., He, J., Wu, C., Ren, Y., et al. (2020). Natural variation and selection in GmSWEET39 affect soybean seed oil content. New Phytol. 225, 1651–1666. doi: 10.1111/nph.16250
Milner, S. G., Jost, M., Taketa, S., Mazon, E. R., Himmelbach, A., Oppermann, M., et al. (2019). Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 51, 319–326. doi: 10.1038/s41588-018-0266-x
Mizuno, H., Kasuga, S., Kawahigashi, H. (2016). The sorghum SWEET gene family: stem sucrose accumulation as revealed through transcriptome profiling. Biotechnol. Biofuels 9, 127. doi: 10.1186/s13068-016-0546-6
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Oliva, R., Ji, C., Atienza-Grande, G., Huguet-Tapia, J. C., Perez-Quintero, A., Li, T., et al. (2019). Broad-spectrum resistance to bacterial blight in rice using genome editing. Nat. Biotechnol. 37, 1344–1350. doi: 10.1038/s41587-019-0267-z
Osório, D., Rondón-Villarreal, P., Torres, R. (2015). Peptides: A package for data mining of antimicrobial peptides. R. J. 7, 4–14. doi: 10.32614/rj-2015-001
Ouyang, S., Zhu, W., Hamilton, J., Lin, H., Campbell, M., Childs, K., et al. (2007). The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res. 35, D883–D887. doi: 10.1093/nar/gkl976
Patil, G., Valliyodan, B., Deshmukh, R., Prince, S., Nicander, B., Zhao, M., et al. (2015). Soybean (Glycine max) SWEET gene family: insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genom. 16, 520. doi: 10.1186/s12864-015-1730-y
Prak, S., Hem, S., Boudet, J., Viennois, G., Sommerer, N., Rossignol, M., et al. (2008). Multiple phosphorylations in the c-terminal tail of plant plasma membrane aquaporins: role in subcellular trafficking of AtPIP2;1 in response to salt stress. Mol. Cell. Proteomics 7, 1019–1030. doi: 10.1074/mcp.M700566-MCP200
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Qin, J. X., Jiang, Y. J., Lu, Y. Z., Zhao, P., Wu, B. J., Li, H. X., et al. (2020). Genome-wide identification and transcriptome profiling reveal great expansion of SWEET gene family and their wide-spread responses to abiotic stress in wheat (Triticum aestivum l.). J. Integr. Agric. 19, 1704–1720. doi: 10.1016/S2095-3119(19)62761-9
Robinson, M. D., Oshlack, A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, 1–9. doi: 10.1186/gb-2010-11-3-r25
Ruan, Y. L. (2014). Sucrose metabolism: gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 65, 33–67. doi: 10.1146/annurev-arplant-050213-040251
Shin, J. H., Blay, S., McNeney, B., Graham, J. (2006). LDheatmap: an r function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Software 16, 1–9. doi: 10.18637/jss.v016.c03
Sosso, D., Luo, D., Li, Q. B., Sasse, J., Yang, J., Gendrot, G., et al. (2015). Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 47, 1489–1493. doi: 10.1038/ng.3422
Toledo, A. M. U., Ignacio, J. C. I., Casal, C., Gonzaga, Z. J., Mendioro, M. S., Septiningsih, E. M. (2015). Development of improved ciherang-Sub1 having tolerance to anaerobic germination conditions. Plant Breed. Biotech. 3, 77–87. doi: 10.9787/PBB.2015.3.2.077
Wagih, O. (2017). Ggseqlogo: a versatile r package for drawing sequence logos. Bioinformatics 33, 3645–3647. doi: 10.1093/bioinformatics/btx469
Wang, K., Li, M., Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi: 10.1093/nar/gkq603
Wang, S., Liu, S., Wang, J., Yokosho, K., Zhou, B., Yu, Y. C., et al. (2020). Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 7, 1776–1786. doi: 10.1093/nsr/nwaa110
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. doi: 10.1093/nar/gkr1293
Wang, S., Yokosho, K., Guo, R., Whelan, J., Ruan, Y. L., Ma, J. F., et al. (2019). The soybean sugar transporter GmSWEET15 mediates sucrose export from endosperm to early embryo. Plant Physiol. 180, 2133–2141. doi: 10.1104/pp.19.00641
Wieczorke, R., Krampe, S., Weierstall, T., Freidel, K., Hollenberg, C. P., Boles, E. (1999). Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in saccharomyces cerevisiae. FEBS Lett. 464, 123–128. doi: 10.1016/S0014-5793(99)01698-1
Xie, H., Wang, D., Qin, Y., Ma, A., Fu, J., Qin, Y., et al. (2019). Genome-wide identification and expression analysis of SWEET gene family in litchi chinensis reveal the involvement of LcSWEET2a/3b in early seed development. BMC Plant Biol. 19, 499. doi: 10.1186/s12870-019-2120-4
Yang, J., Luo, D., Yang, B., Frommer, W. B., Eom, J. S. (2018). SWEET11 and 15 as key players in seed filling in rice. New Phytol. 218, 604–615. doi: 10.1111/nph.15004
Yuan, M., Wang, S. (2013). Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mol. Plant 6, 665–674. doi: 10.1093/mp/sst035
Yu, H., Lin, T., Meng, X., Du, H., Zhang, J., Liu, G., et al. (2021). A route to de novo domestication of wild allotetraploid rice. Cell 184, 1156–1170 e1114. doi: 10.1016/j.cell.2021.01.013
Zhang, W., Wang, S., Yu, F., Tang, J., Shan, X., Bao, K., et al. (2019). Genome-wide characterization and expression profiling of SWEET genes in cabbage (Brassica oleracea var. capitata l.) reveal their roles in chilling and clubroot disease responses. BMC Genom. 20, 93. doi: 10.1186/s12864-019-5454-2
Zhou, Z., Jiang, Y., Wang, Z., Gou, Z., Lyu, J., Li, W., et al. (2015). Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 33, 408–414. doi: 10.1038/nbt.3096
Zhu, T., Wang, L., Rimbert, H., Rodriguez, J. C., Deal, K. R., De Oliveira, R., et al. (2021). Optical maps refine the bread wheat triticum aestivum cv. Chinese spring genome assembly. Plant J. 107, 303–314. doi: 10.1111/tpj.15289
Keywords: barley, SWEET, expression profile, haplotype, domestication
Citation: Yue W, Cai K, Xia X, Liu L and Wang J (2023) Genome-wide identification, expression pattern and genetic variation analysis of SWEET gene family in barley reveal the artificial selection of HvSWEET1a during domestication and improvement. Front. Plant Sci. 14:1137434. doi: 10.3389/fpls.2023.1137434
Received: 04 January 2023; Accepted: 02 February 2023;
Published: 13 February 2023.
Edited by:
Zhong-Hua Chen, Western Sydney University, AustraliaReviewed by:
Gaofeng Zhou, Department of Primary Industries and Regional Development of Western Australia (DPIRD), AustraliaCopyright © 2023 Yue, Cai, Xia, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junmei Wang, MjF3YW5nam1Ac2luYS5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.