
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 15 March 2023
Sec. Plant Breeding
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1127760
 Ziyin Li1,2,3†
Ziyin Li1,2,3† Yuzhen Shi3†
Yuzhen Shi3† Xianghui Xiao1,3
Xianghui Xiao1,3 Jikun Song1,3
Jikun Song1,3 Pengtao Li2,3
Pengtao Li2,3 Juwu Gong1,3
Juwu Gong1,3 Haibo Zhang2
Haibo Zhang2 Wankui Gong3Aiying Liu3
Wankui Gong3Aiying Liu3 Renhai Peng2
Renhai Peng2 Haihong Shang3Qun Ge3Junwen Li3Jingtao Pan3
Haihong Shang3Qun Ge3Junwen Li3Jingtao Pan3 Quanjia Chen1*
Quanjia Chen1* Quanwei Lu2,3*
Quanwei Lu2,3* Youlu Yuan1,3*
Youlu Yuan1,3*Cotton is an important fiber crop. The cotton fiber is an extremely long trichome that develops from the epidermis of an ovule. The trichome is a general and multi-function plant organ, and trichome birefringence-like (TBL) genes are related to trichome development. At the genome-wide scale, we identified TBLs in four cotton species, comprising two cultivated tetraploids (Gossypium hirsutum and G. barbadense) and two ancestral diploids (G. arboreum and G. raimondii). Phylogenetic analysis showed that the TBL genes clustered into six groups. We focused on GH_D02G1759 in group IV because it was located in a lint percentage-related quantitative trait locus. In addition, we used transcriptome profiling to characterize the role of TBLs in group IV in fiber development. The overexpression of GH_D02G1759 in Arabidopsis thaliana resulted in more trichomes on the stems, thereby confirming its function in fiber development. Moreover, the potential interaction network was constructed based on the co-expression network, and it was found that GH_D02G1759 may interact with several genes to regulate fiber development. These findings expand our knowledge of TBL family members and provide new insights for cotton molecular breeding.
Cotton is a primary source of natural textiles (Qin et al., 2015), and cotton fiber plays an important role in modern economic activities (Zhang et al., 2008). The trichomes on the cotton seed coat, generally referred to as fibers, are the main harvest product of cotton and a valuable resource for the textile industry (Qin and Zhu, 2011). Fiber has four continuous but overlapping developmental stages: initiation, elongation, secondary cell wall deposition, and maturation (Haigler et al., 2012; Wang et al., 2019). Fiber yield is mainly determined by the number of polarly-developed epidermal cells on the ovule during fiber initiation, with each elongated cell becoming a single fiber in the future. For mature cotton bolls, the percentage of fiber is calculated as the lint percentage (LP). As an important trait of fiber yield, LP has largely been the primary focus of many studies (Su et al., 2016; Wu et al., 2016; Song et al., 2019).
With the development of cotton genomics, many genes influencing fiber yield have been identified. The collection of fiber-yield-related genes has revealed a complex regulatory landscape involving genes from various pathways (Fang et al., 2017; Du et al., 2018; Ma et al., 2018; Huang et al., 2020; He et al., 2021; Wang et al., 2022). Given that the trichome in Arabidopsis thaliana has a similar development pattern to fiber in cotton, genes stimulating trichome development in A. thaliana are considered crucial factors in cotton fiber initiation (Wang et al., 2019). However, as genes contributing to trichome development, the role of trichome birefringence-like (TBL) genes in fiber initiation has rarely been studied.
TBL is a gene family that functions in trichome development. In A. thaliana, TBL3 is transcriptionally coordinated with CESA genes, and the knockout of TBL3 reduces crystalline secondary wall cellulose in both the trichome and stem (Bischoff et al., 2010). The loss of TBL34 and TBL35 will limit the number of xylem vessels, resulting in extremely slow plant growth (Yuan et al., 2016a). In addition, there have been some studies of tbl mutants in A. thaliana, which have revealed that the deletion of the tbl gene leads to plant dwarfism, weak stems, and stunted growth (Bischoff et al., 2010; Xiong et al., 2013; Schultink et al., 2015). In cotton, TBL34 was reported to improve verticillium wilt resistance (Zhao et al., 2021b). Zhang (2020) found that GhTBL38 affects cell wall acetylation. Improving our understanding of the role of TBL genes in cotton fiber initiation could provide an important gene resource for molecular breeding.
In this study, we identified several TBL genes in four cotton species, namely G. arboretum, G. raimondii, G. hirsutum, and G. barbadense. After comprehensive characterization of the TBLs and quantitative trait locus (QTL) mapping interval (QTL2) of LP by our research group (Zhao et al., 2021a), we identified GH_D02G1759 (GhTBL82) as a candidate gene contributing to LP. Its role has been validated by overexpression in A. thaliana. This genome-wide investigation of TBLs will provide a theoretical basis for cotton molecular breeding.
The protein sequences of AtTBLs were obtained from http://www.arabidopsis.org. Based on these protein sequences from the Arabidopsis genome, we identified TBL members among the protein sequences from four Gossypium genomes, namely, G. arboreum (A2), G. raimondii (D5), G. hirsutum (AD1), and G. barbadense (AD2), by sequence alignment with an e-value of 1e−10 (Paterson et al., 2012; Du et al., 2018; Hu et al., 2019). The protein sequences of the TBLs selected by alignment were submitted to the pfam database (http://pfam.xfam.org/) and SMART database (http://smart.embl-heidelberg.de/) for further confirmation (Finn et al., 2014; Letunic et al., 2015).
The TBL members in the four Gossypium species were aligned to each other by multiple sequence alignment using clusterW (http://www.ebi.ac.uk/Tools/msa/clustalw2) (Larkin et al., 2007). Based on the multiple sequence alignments, phylogenetic trees of the TBL members were constructed in MEGA (v.7.0) using the neighbor-joining (NJ) method (Tamura et al., 2013). Branch support was tested based on 1000 bootstrap replicates.
For the gene structure characterization of the TBLs, the gene feature files of the four Gossypium members were downloaded from the CottonGen database (https://www.cottongen.org). The conserved motifs in TBLs were identified by MEME (http://meme-suite.org/) (Bailey et al., 2009). For the protein domain analysis, the NCBI database was used to detect the protein domains in the TBLs. TBtools (v.1.100) software was used for visualization (Chen et al., 2020). For molecular property characterization, the ProtParam tool (https://web.expasy.org/) was used (Gasteiger et al., 2003).
The locations of the TBLs in G. raimondii, G. arboreum, G. barbadense, and G. hirsutum were displayed on the corresponding chromosomes by MapChart (v.2.2) (Voorrips, 2002). Gene duplication and syntenic regions were identified by MCScanX, and the results were visualized by TBtools (v.1.100) (Wang et al., 2012; Chen et al., 2020).
The transcription landscape of the TBLs was characterized based on previously published transcriptome data (SRA; accession number SRP084203) (Lu et al., 2017). This transcriptome dataset consisted of 12 samples, including two materials (CCRI45 and MBI7747) from six different periods, 5 days post anthesis (DPA), 7 DPA, 10 DPA, 15 DPA, 20 DPA, and 25 DPA. Genes were selected by transcription abundance based on the condition that log2(FPKM+1)≥1. The transcription patterns of the candidate genes were visualized by the seaborn (v.0.9.0) package (installed through anaconda).
The selected genes by condition log2(FPKM+1)≥1 were retained for the WGCNA using R (v.4.2.1). The soft threshold was selected based on the R-square (≥0.85) and mean connectivity (≤500). With the selected soft threshold, the co-expression network was constructed and classified into several modules. The median transcription abundance of the genes in classified modules was linked to the phenotype by Pearson’s correlation analysis.
The KEGG enrichment analysis for genes in the candidate modules was performed on cottonFGD (https://cottonfgd.net/analyze/) with a q-value threshold of 1e-5 (Zhu et al., 2017). The results of the KEGG analysis were displayed by the matplotlib package in python.
For the construction of the potential interaction network, we calculated Pearson’s correlation coefficient as an interaction weight between the target gene and candidate genes. The interaction network construction result was inputted into Cytoscape (v.3.7.1) for visualization.
For qRT-PCR of the candidate genes, CCRI45 and MBI7747, which were planted in a field in Henan Province, Anyang City, were selected as plant materials. The fibers of two plant materials at 5 DPA, 7 DPA, 10 DPA, 15 DPA, 20 DPA, and 25 DPA were collected for qRT-PCR. Each sample had three biological replicates, and the samples were immediately placed into liquid nitrogen and stored in a −80°C environment. Total RNA was extracted using an RNAprep Pure Plant Kit (Tiangen, Beijing, China). The ChamQ Universal SYBR qPCR Master Mix Kit (Vazyme) was used to perform qRT-PCR with Gh_D03G0370 (GhActin3) and AT3G18780 (AtActin2) as internal controls.
The 1,782 bp complete coding sequence (CDS) of GH_D02G1759 was amplified using the primers 35S::GH_D02G1759-F and 35S:: GH_D02G1759-R (Table S1). Then, the CDS of GH_D02G1759 was connected to the pCAMBIA3301 vector, which was digested by NcoI and BstE II. The re-constructed vector was transferred into Agrobacterium (GV3101), and the recombinant plasmid GV3101 was used to infect A. thaliana (Col-0). After genetic transformation and selfing for three generations, T3 lines were obtained for further analysis.
To investigate the subcellular localization of the GH_D02G1759 protein, the full-length coding region of GH_D02G1759 was inserted into the pBI121-EGFP plasmid to generate Pro35S::GH_D02G1759-EGFP constructs and introduced into GV3101, which were transformed into tobacco leaves. The GFP fluorescence in leaf epidermal cells was observed using a laser-scanning confocal microscope (TCS SP8, Leica, Germany).
To identify all TBL members in G. hirsutum, G. barbadense, G. arboreum, and G. raimondii, we first collected the protein sequences of AtTBLs in Arabidopsis and aligned them to all protein sequences in the four cotton species. The protein sequences that were similar to AtTBLs were selected as candidate TBL members, and these were further inputted into the NCBI database for RING_Ubox confirmation. Finally, we identified 73, 73, 143, and 146 TBL genes in G. arboreum, G. raimondii, G. hirsutum, and G. barbadense, respectively. After the identification of TBL members, we evaluated the molecular properties of these TBLs and found that great divergence existed in aspects of protein length (from 87 to 1423 aa) and molecular mass (from 9913.21 to 160422.21 Da) (Table S2).
Through investigating the evolutionary trajectories of TBL members in cotton, we built a phylogenetic tree using TBLs from four cotton species and A. thaliana (Figure 1). From the result of the phylogenetic tree, we noticed that the TBL members could be divided into six groups consistent with a previous report on Arabidopsis (Bischoff et al., 2010). Among the six groups, groups II, V, and VI had the most members, containing 118, 110, and 112 members, respectively. By contrast, group III had only 12 members. Based on the QTL mapping interval of LP in our research group, the candidate gene GH_D02G1759 was identified (Zhao et al., 2021a). The homologous gene of AT1G60790, GH_D02G1759, was assigned as a member in group IV, which contained 54 members. Therefore, we focused on the TBLs in group IV. Table 1 lists the specific information of the TBL genes in group IV from G. hirsutum, such as gene ID, chromosomal location, protein size (aa), and molecular weight (Da).
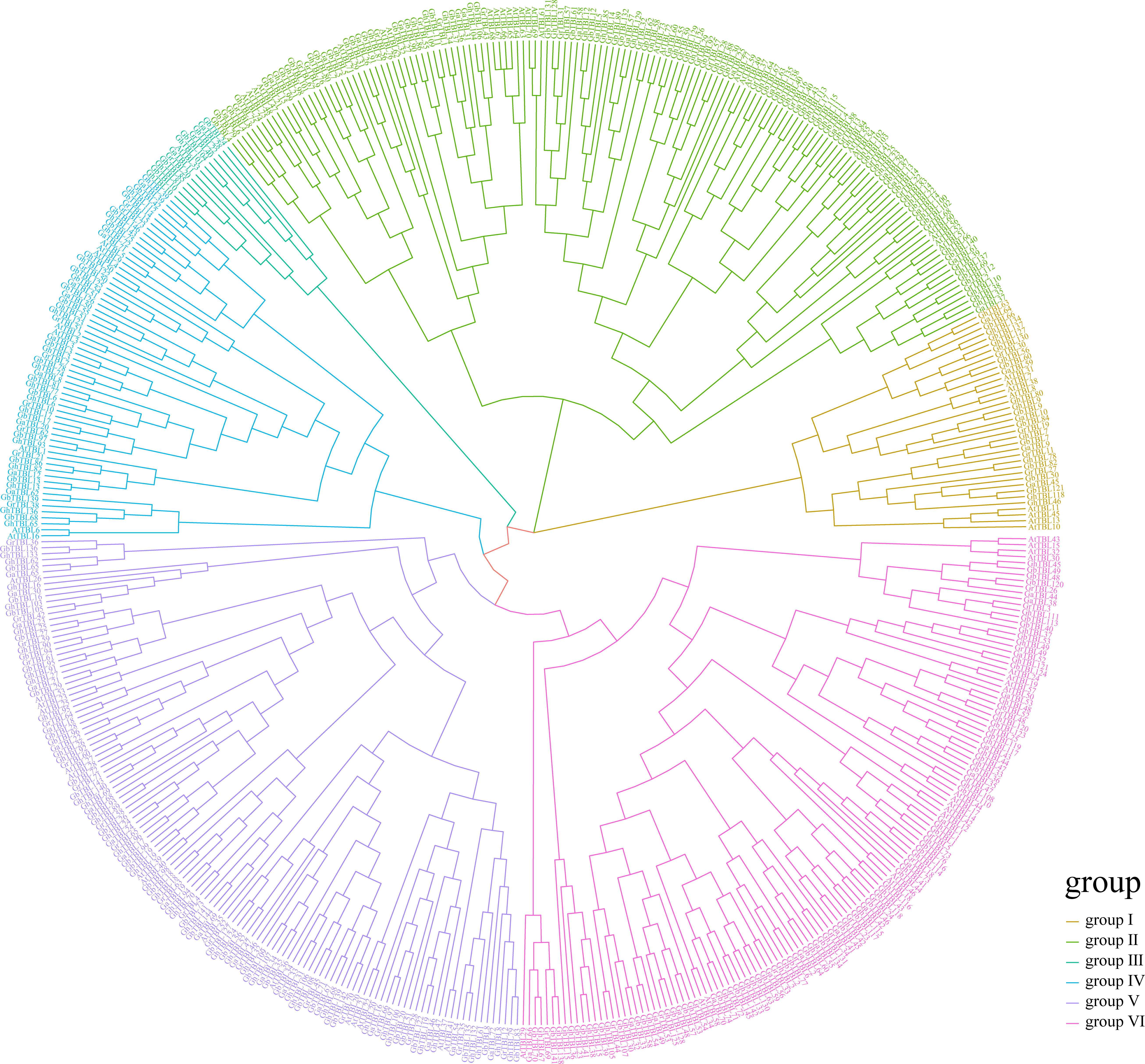
Figure 1 The phylogenetic analysis of TBL members across Gossypium and Arabidopsis.
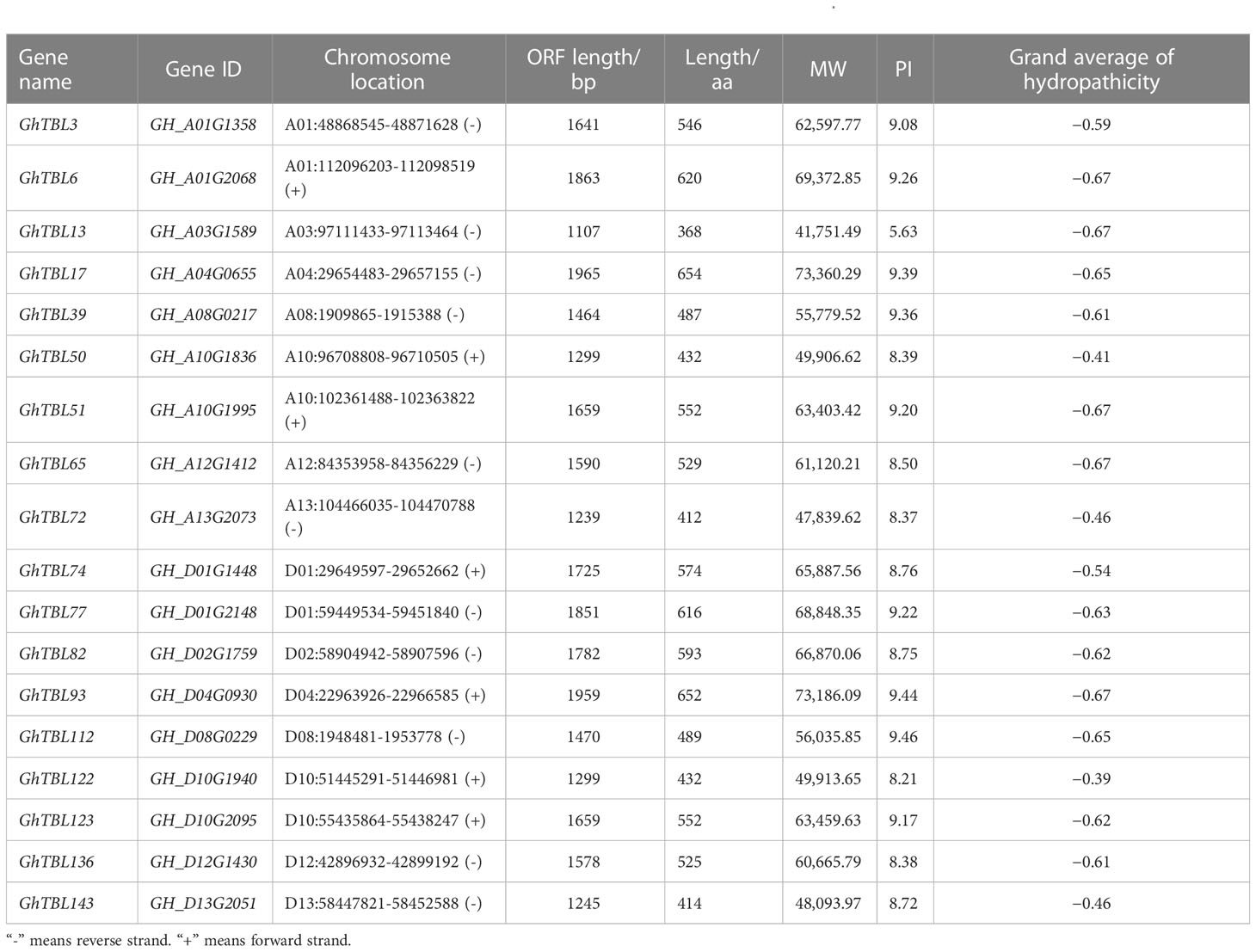
Table 1 Physico-chemical and biochemical features of the TBL genes in group IV from G. hirsutum.
The group IV members were distributed in different chromosomes in the four cotton species. In G. arboreum, nine GaTBLs were located on eight chromosomes (A01, A02, A03, A04, A08, A10, A12, and A13) (Figure 2A). In G. raimondii, nine GrTBLs were located on seven chromosomes (D01, D02, D04, D08, D10, D12, and D13) (Figure 2B). In G. hirsutum, 18 GhTBLs were mapped on 14 chromosomes, including seven chromosomes from the At sub-genome and Dt subgenome, respectively (Figure 2C). The number of chromosomes containing GbTBLs in G. barbadense was the same as that in G. hirsutum, and the number of GbTBLs was also 18 (Figure 2D).

Figure 2 The stance of the TBL genes in group IV on cotton chromosomes. Chromosome distribution in G. arboreum (A), G. raimondii (B), G. barbadense (C), and G. hirsutum (D).
The gene structure of group IV genes showed that most TBLs contained five exons, a few TBLs contained four exons, and only GbTBL139 had 16 exons (Figure 3A). Apart from gene structure, the motifs of the TBLs in group IV were detected by MEME, and motif detection showed that the TBLs had conserved motifs within gene regions (Figure 3B). Furthermore, we also investigated the protein domain distribution of the group IV TBL members (Figure 3C). All TBLs in group IV had PMR5N and PC-Esterase domains. GbTBL139 and GaTBL62 contained a DAP2 domain, and GhTBL13 and GbTBL13 contained a PLN02629 domain. From these results, we found that although most TBL members from group IV were conserved in cotton, a small amount of divergence remained in terms of gene structure and protein sequences.
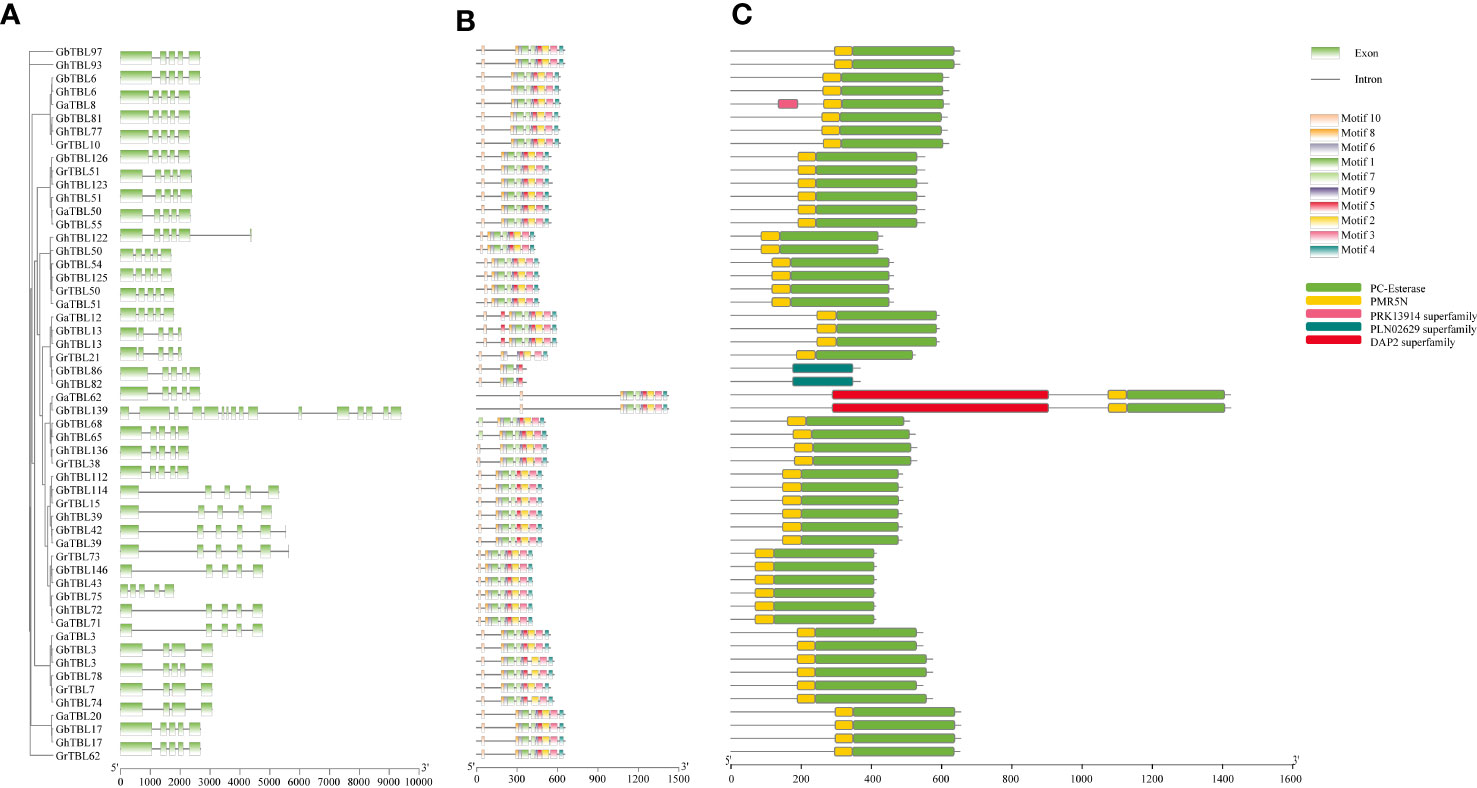
Figure 3 Gene structure and protein domain analysis of the TBL genes in group IV. The exon-intron structure (A) and motifs (B) of the TBL genes. (C) TBL protein domain prediction.
Gene duplication events are common during plant evolution. As we found a sequence divergence in group IV members, we inferred that the TBL members in group IV may have experienced gene duplication events during tetraploidy. The inter-specific collinearity of the 54 TBLs among G. arboreum, G. hirsutum, G. barbadense, and G. raimondii was evaluated to detect gene duplication events (Figures 4A, B). We collected TBLs from G. arboreum, G. hirustum (At), and G. barbadense (At) to perform collinearity analysis on A genomes, while the same analysis was also performed on TBLs from G. raimondii and D sub-genomes of G. hirustum and G. barbadense to investigate collinearity within D genomes. We found 7 and 8 TBLs with collinearity within A sub-genomes and D sub-genomes, respectively (Figures 4A, B). The intra-specific collinearity of 54 TBLs among G. hirsutum and G. barbadense which includes the collinearity between A and D sub-genomes was also evaluated (Figures 4C, D). Both G. hirsutum and G. barbadense contained 18 TBLs. Most TBLs have good collinearity in the At-genome or Dt-genome of 2 tetraploids (Figures 4C, D).
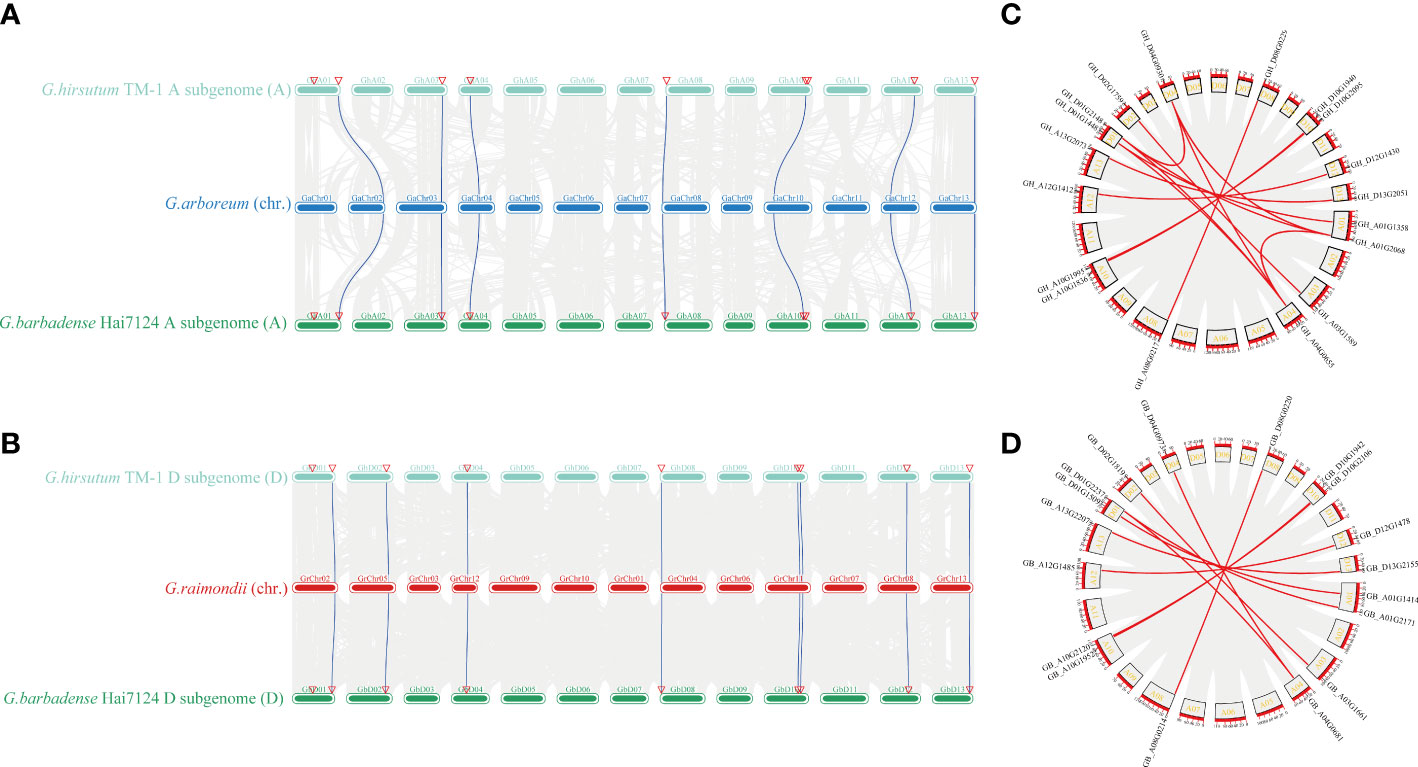
Figure 4 Analysis of synteny among multiple Gossypium genomes regarding the TBL genes in group IV. (A) Synteny analysis among G. arboreum, G. hirsutum (At subgenome), and G. barbadense (At subgenome). (B) Synteny analysis among G. raimondii, G. hirsutum (Dt subgenome), and G. barbadense (Dt subgenome). (C) Synteny analysis among G. hirsutum (At subgenome) and G. hirsutum (Dt subgenome). (D) Synteny analysis among G. barbadense (At subgenome) and G. barbadense (Dt subgenome).
Although the sequence structure was thoroughly investigated, their potential roles in fiber development remained unclear. We collected published transcriptome data containing 12 samples (CCRI45 and MBI7747 from six periods) (Lu et al., 2017). CCRI45 has a high LP, while the LP of MBI7747 is relatively low. Therefore, characterizing the transcription landscape of GhTBLs from group IV is essential for illustrating their roles in fiber development. We filtered genes whose maximum FPKM values among 12 samples were smaller than 1, and 43,808 genes were retained. We found that some GhTBLs had material-specific transcription patterns, such as GH_D01G2148 and GH_D02G1759, while others had stage-specific transcription patterns, such as GH_D01G1448 and GH_A01G2068 (Figure S1A). For verification of the RNA-seq results, qRT-PCR analysis was performed to quantify the differential expression of the transcripts. The overall expression levels of the six genes were consistent with the RNA-seq data, confirming that the RNA-seq data were reliable and conducive to the identification of candidate genes during fiber development (Figure S1B). Divergent transcription patterns of GhTBLs in group IV indicated that the GhTBLs in group IV may play multiple roles during fiber development.
For further investigation of the potential association between GhTBLs from group IV and fiber development, we constructed a WGCNA network based on all of the retained genes (Figure 5A). To ensure the construction of a scale-free network, the soft threshold was selected based on both the R-square (≥ 0.85) and mean connectivity (≤ 200) (Figure 5B). After network construction, all of the retained genes were divided into 18 modules, which had various transcription patterns, implying complicated processes during fiber development. To dissect the associations between gene modules and fiber development, we associated the transcript abundance with the sample phenotype by Pearson’s correlation analysis. An absolute value of Pearson’s correlation coefficient larger than 0.3 and a P-value smaller than 0.05 were set as the threshold. We noticed that the blue and turquoise modules were linked to 5 DPA (R2 = 0.7 and 0.63, respectively) (Figure 5C). The red module was negatively related to 7 DPA (R2 = −0.61). The purple and magenta modules were related to 10 DPA, with R2 values of 0.65 and 0.73. Four modules were associated with 15 DPA, and apart from the brown module, another three modules, namely green, pink, and green-yellow, were positively related to 15 DPA (R2 = 0.72, R2 = 0.78, and R2 = 0.85, respectively). No modules were found to be associated with 20 DPA. Salmon and yellow were positively related to 25 DPA, while the black module was negatively associated with 25 DPA. The above modules were involved in fiber development, and other modules were found to be related to LP. The cyan module had a close association with the high LP phenotype (R2 = 0.97), while the midnight blue and grey60 modules were negatively correlated with high LP (Figure 5C).
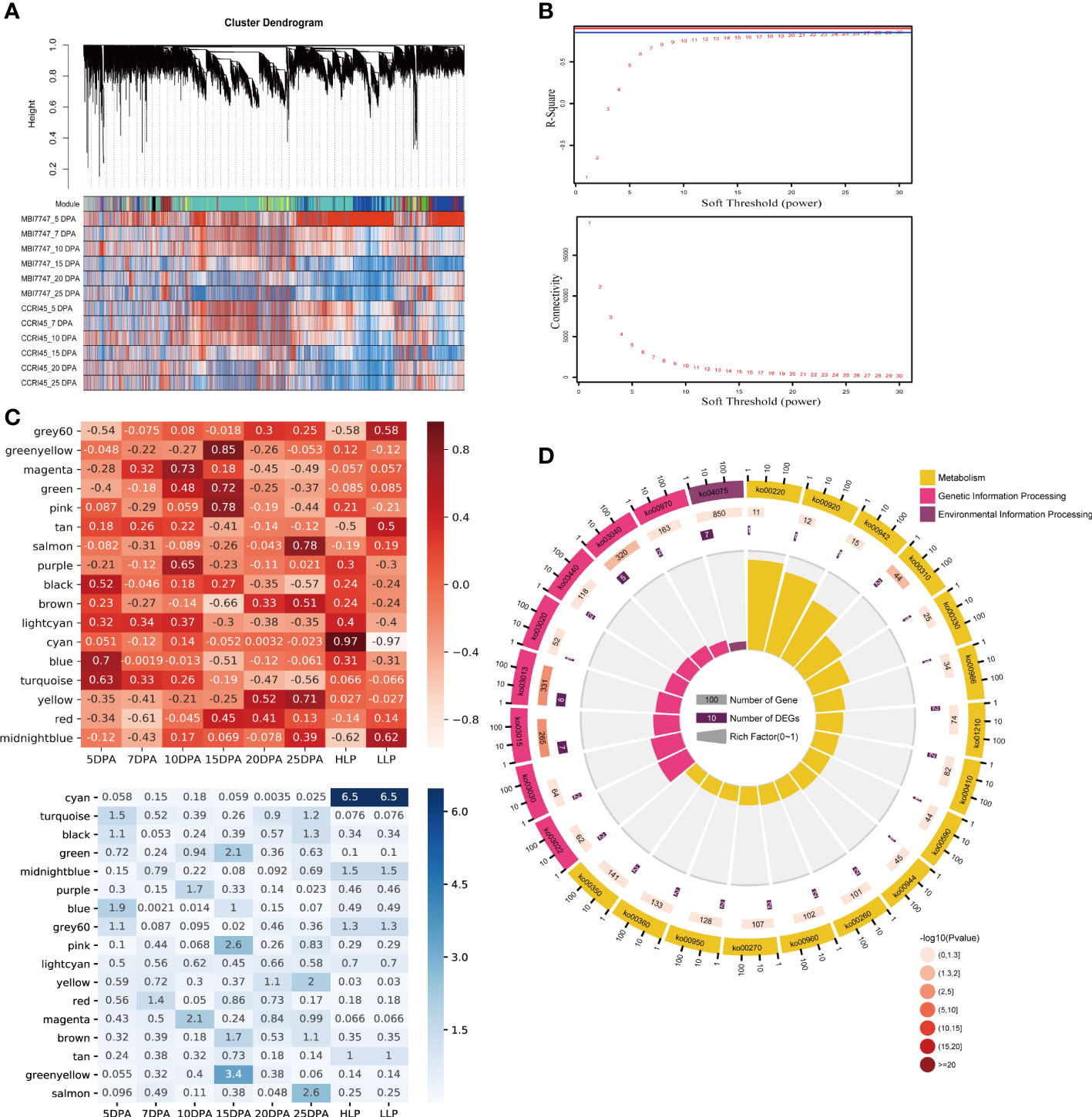
Figure 5 WGCNA for the transcriptome data in the study. (A) Results of the gene cluster analysis in WGCNA. (B) Mean connectivity and R-square of the WGCNA. With the increasing soft threshold, R-square rises and mean connectivity decreases. (C) Trait-module association results of the WGCNA. The upper heatmap is Pearson’s correlation between the modules and phenotype, and the lower heatmap is the −log10(p) value of Pearson’s significance test. (D) The KEGG pathway enrichment analysis of the blue module.
Among the retained 14 GhTBLs in group IV, we found that these 14 genes were from four modules, namely the blue, turquoise, red, and yellow modules, which were involved in fiber development at different stages. Although both the blue and turquoise modules contributed to the fiber initiation stage (5 DPA), their functions differed. Genes in the blue module were mainly involved in 18 pathway categories, including 51 KEGG pathways, among which the most abundant genes were enriched in “Global and overview maps” metabolic pathways (Figure 5D and Table S3). Genes in the turquoise module had pathways related to metabolism (Figure S2A). Interestingly, pathways related to fatty acid biosynthesis were enriched in the turquoise modules. As fatty acids are essential for fiber elongation, we inferred that the genes in the turquoise module played an important role in fiber development. The red module was enriched in various signaling pathways, implying that this module may regulate fiber elongation by influencing multiple signaling pathways (Figure S2B). Regarding the yellow module, which is a module related to secondary cell wall thickening, we found that fatty acid degradation (ko00071) was significantly enriched. All of these results showed that the GhTBLs in the fiber-related modules had various functions in fiber development (Figure S2C).
GH_D02G1759, as the target gene, was from the blue module. The transcription patterns of GH_D02G1759 and the blue module were similar, having high transcription abundance in fiber initiation. To address the temporal restriction of the present transcriptomic assay (http://cotton.zju.edu.cn/2.search_gene_locus.php), we checked the transcription abundance of GH_D02G1759 from −3 DPA to 3 DPA in a previous assay (Figure S3). The results showed that GH_D02G1759 was expressed in fiber initiation. We assessed the location of GH_D02G1759 in vivo by subcellular localization on tobacco leaves and found that GH_D02G1759 was located on the membrane (Figure 6A). Furthermore, we overexpressed GH_D02G1759 in A. thaliana to explore its role in trichome development. Compared with the wild type, the overexpressing plants had more trichomes on the stem surface, implying its potential role in cellular elongation. We inferred that GH_D02G1759 could enhance the LP by stimulating the elongation of ovule epidermal cells (Figure 6B).
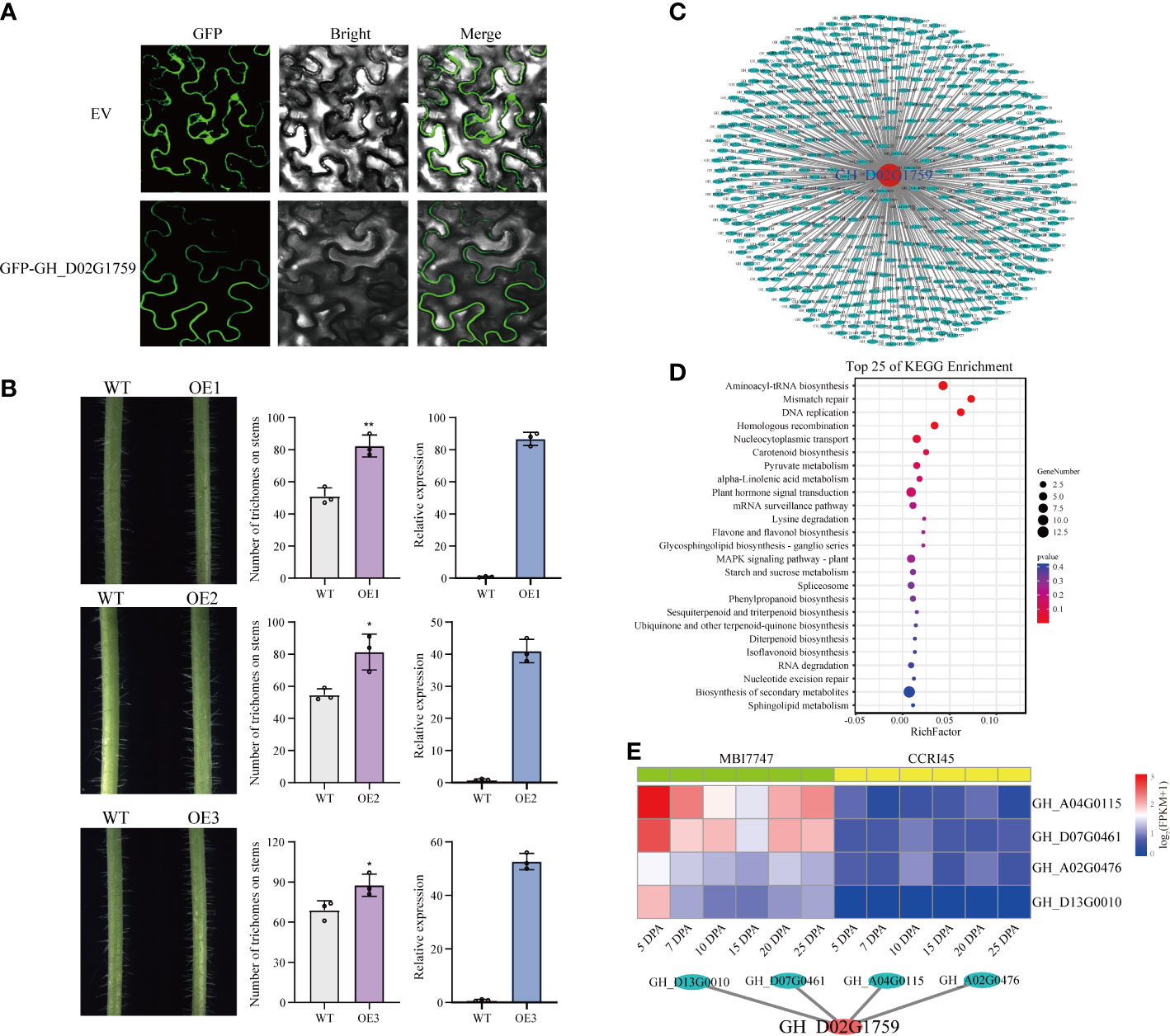
Figure 6 Functional investigation of the candidate gene GH_D02G1759. (A) Subcellular localization of the GH_D02G1759 protein in the epidermal cells of the tobacco leaf. The names of the constructs are shown on the left. The scale bar is 20 μm. (B) Overexpression of GH_D02G1759 in A. thaliana. The left images are epidermal trichomes on the stems, the middle images are the statistics on the number of trichomes (t-test), and the right images are the relative expression of GH_D02G1759 in three overexpressed lines. (C) The entire network of GH_D02G1759 constructed based on genome-wide transcriptome data. (D) The KEGG pathway enrichment analysis of the 491 genes. (E) The interaction network of GH_D02G1759 constructed based on cyan module members. The heatmap indicates the transcriptional abundance of genes among samples.
Although the function of GH_D02G1759 has been validated by overexpression, its interaction network is still unclear. The results of the WGCNA provided us with a platform to construct a potential interaction network of GH_D02G1759. GH_D02G1759 belongs to the blue module, and we constructed the interaction network of GH_D02G1759 and genes from the blue modules (Figure 6C). A total of 491 genes from the blue module with a 0.95 Pearson’s correlation with GH_D02G1759 were regarded as candidate-interacted genes (Figure 6C and Table S4). To detect the potential role of the interaction network of GH_D02G1759, we performed KEGG analysis on these 491 genes (Figure 6D). We found that both basic pathways (mismatch repair, DNA replication, and homologous recombination) and specific pathways (plant hormone signal transduction and starch and sucrose metabolism) were enriched in this network. Interestingly, starch is the source of fiber component synthesis, and GH_D02G1759 may interact with genes in starch metabolism to enhance fiber development. Moreover, GH_D02G1759 could be also regulated by genes in signal transduction pathways. There were also some secondary metabolism-related pathways in the interaction network, such as sphingolipid metabolism and flavone and flavonol biosynthesis, indicating that GH_D02G1759 may also participate in resistance to abiotic stress (Table S5). As a module with a positive correlation with high LP, there were only 25 genes in the cyan module. We investigated the relationship between GH_D02G1759 and cyan members and found that four genes, namely GH_A02G0476 (GhSPL7), GH_A04G0115 (GhMOCS3), GH_D07G0461 (GhOMA1), and GH_D13G0010 (At5g05130) were correlated to GH_D02G1759. SPL7 was proved to activate miRNAs in response to several biological processes, implying that GH_D02G1759 may influence cellular elongation via a complicated mechanism (Figure 6E).
As an important economic crop in the textile industry, improving the fiber yield of cotton is crucial for crop modification (Song et al., 2019). Given that fiber develops from epidermal cells, the number of epidermal cells with extreme elongation determines fiber yield (Qin et al., 2015). Therefore, stimulating the cellular elongation of epidermal cells from an ovule is a reasonable way to increase fiber yield. With the development of genomics, many large population-scale studies have been implemented to detect the functional genes involved in fiber development (Ma et al., 2018; He et al., 2021). Although a large number of functional genes have been detected, their functional validations are still limited because of the low transgenic efficiency of cotton. In response, researchers have used A. thaliana as a substitute to obtaining higher transgenic efficiency, as the cell structure of the trichomes in A. thaliana is similar to that of the fiber in cotton (Wang et al., 2004; Yang and Ye, 2013).
TBL proteins, which contribute to cellulose formation in A. thaliana, have been characterized (Bischoff et al., 2010). Among the 46 TBL members in A. thaliana, the functions of only a few TBLs have been validated. TBL44 is related to resistance to powdery mildew fungi (Vogel et al., 2004), while TBL3 is essential for xylan acetylation (Xiong et al., 2013; Yuan et al., 2013; Yuan et al., 2016b). In rice, TBL1 and TBL2 affect the acetylation level and response to rice blight disease (Gao et al., 2017). In cotton, the overexpression of GhTBL34 can improve verticillium wilt resistance (Zhao et al., 2021b). Zhang (2020) found that the overexpression of GhTBL38 affected cell wall acetylation. Although TBLs have also been found to participate in other biological processes such as pathogen response, reports on their roles during fiber development remain limited (Zhao et al., 2021a). Altogether, these studies highlight that TBL genes have significant value in breeding and play an important regulatory role in controlling trichome development.
We identified 73, 73, 146, and 143 TBL genes in G. raimondii, G. arboretum, G. barbadense, and G. hirsutum, respectively. The chromosomal distribution, evolutionary relationship, and expression patterns of the TBL genes in group IV were analyzed. It has been found that all TBL genes in group IV have typical PMR5N and PC-Esterase domains, which have acyl esterase activity and are predicted to modify cell-surface biopolymers such as glycans and glycoproteins (Anantharaman and Aravind, 2010). Gene structure analysis revealed that almost all TBL genes in group IV have four exons, which indicates that this gene may be functionally conserved during evolution. According to the selection pressure analysis, the Ka/Ks ratio was less than 1, further supporting the evolutionary conservation of these genes. Using published transcriptome data, we found that TBLs in cotton could influence fiber development via multiple pathways such as fatty acid biosynthesis and various signal transductions. In this study, we overexpressed the selected gene in A. thaliana and observed changes in the trichomes to infer the function of the candidate genes in fiber development. With the combination of transcriptome profiling and the A. thaliana phenotype, we confirmed the role of GH_D02G1759 in fiber development. With the increase in the number of trichomes in the stems, we inferred that it could enhance the number of fibers on a single ovule by promoting cellular elongation. Moreover, we constructed the potential interaction network of GH_D02G1759 based on transcription abundance. Although GH_D02G1759 does not belong to the cyan module, which is linked to LP, it still had correlated transcription patterns with four members in the cyan module. One of these four genes, SPL7, activates miRNAs in response to several biological processes in which GH_D02G1759 may participate.
All of these characterizations showed the potential role of the TBLs in fiber development, and the candidate gene GH_D02G1759 detected in this study could be used as an important gene resource for improving the fiber yield of cotton.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
ZL analyzed and summarized all of the data, drew the figures, and wrote the manuscript. YS and XX participated in sample preparation. JS, PL, JG, HZ, WG, AL, RP, HS, QG, JL, and JP participated in data collection and analysis. QC, QL, and YY performed the experiments and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by the National Natural Science Foundation of China (32272188 and 32070560), the Natural Science Foundation of Henan Province (202300410549), China Agriculture Research System of MOF and MARA, Hainan Yazhou Bay Seed Lab (B21HJ0210), the National Agricultural Science and Technology Innovation Project for CAAS (CAAS-ASTIP-2016-ICR), the Xinjiang Production and Construction Corps Innovation Program for Science and Technology Talents (2020CB005), and the National Key R&D Program for Crop Breeding (2016YFD0100306).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MW declared a shared parent affiliation with the authors ZL, YS, XX, JS, PT, JG, WG, AL, HS, QG, JL, JP and YY to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1127760/full#supplementary-material
Anantharaman, V., Aravind, L. (2010). Novel eukaryotic enzymes modifying cell-surface biopolymers. Biol. Direct. 5, 1. doi: 10.1186/1745-6150-5-1
Bailey, T.L., Boden, M., Buske, F.A., Frith, M., Grant, C.E., Clementi, L., et al. (2009). MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 37, 202–208. doi: 10.1093/nar/gkp335
Bischoff, V., Nita, S., Neumetzler, L., Schindelasch, D., Urbain, A., Eshed, R., et al. (2010). Trichome birefringence and its homolog AT5G01360 encode plant-specific DUF231 proteins required for cellulose biosynthesis in arabidopsis. Plant Physiol. 153 (2), 590–602. doi: 10.1104/pp.110.153320
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Du, X., Huang, G., He, S., Yang, Z., Sun, G., Ma, X., et al. (2018). Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nat. Genet. 50 (6), 796–802. doi: 10.1038/s41588-018-0116-x
Fang, L., Wang, Q., Hu, Y., Jia, Y. H., Chen, J. D., Liu, B. L., et al. (2017). Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 49 (7), 1089–108+. doi: 10.1038/ng.3887
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42 (Database issue), D222–D230. doi: 10.1093/nar/gkt1223
Gao, Y., He, C., Zhang, D., Liu, X., Xu, Z., Tian, Y., et al. (2017). Two trichome birefringence-like proteins mediate xylan acetylation, which is essential for leaf blight resistance in rice. Plant Physiol. 173 (1), 470–481. doi: 10.1104/pp.16.01618
Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D., Bairoch, A. (2003). ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31 (13), 3784–3788. doi: 10.1093/nar/gkg563
Haigler, C. H., Betancur, L., Stiff, M. R., Tuttle, J. R. (2012). Cotton fiber: a powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 3. doi: 10.3389/fpls.2012.00104
He, S., Sun, G., Geng, X., Gong, W., Dai, P., Jia, Y., et al. (2021). The genomic basis of geographic differentiation and fiber improvement in cultivated cotton. Nat. Genet. 53 (6), 916–924. doi: 10.1038/s41588-021-00844-9
Hu, Y., Chen, J., Fang, L., Zhang, Z., Ma, W., Niu, Y., et al. (2019). Gossypium barbadense and gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51 (4), 739–748. doi: 10.1038/s41588-019-0371-5
Huang, G., Wu, Z., Percy, R. G., Bai, M., Li, Y., Frelichowski, J. E., et al. (2020). Genome sequence of gossypium herbaceum and genome updates of gossypium arboreum and gossypium hirsutum provide insights into cotton a-genome evolution. Nat. Genet. 52 (5), 516–524. doi: 10.1038/s41588-020-0607-4
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23 (21), 2947–2948. doi: 10.1093/bioinformatics/btm404
Letunic, I., Doerks, T., Bork, P. (2015). SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43 (Database issue), D257–D260. doi: 10.1093/nar/gku949
Lu, Q., Shi, Y., Xiao, X., Li, P., Gong, J., Gong, W., et al. (2017). Transcriptome analysis suggests that chromosome introgression fragments from sea island cotton (Gossypium barbadense) increase fiber strength in upland cotton (Gossypium hirsutum). G3 (Bethesda) 7 (10), 3469–3479. doi: 10.1534/g3.117.300108
Ma, Z., He, S., Wang, X., Sun, J., Zhang, Y., Zhang, G., et al. (2018). Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 50 (6), 803–813. doi: 10.1038/s41588-018-0119-7
Paterson, A. H., Wendel, J. F., Gundlach, H., Guo, H., Jenkins, J., Jin, D., et al. (2012). Repeated polyploidization of gossypium genomes and the evolution of spinnable cotton fibres. Nature 492 (7429), 423–427. doi: 10.1038/nature11798
Qin, H., Chen, M., Yi, X., Bie, S., Zhang, C., Zhang, Y., et al. (2015). Identification of associated SSR markers for yield component and fiber quality traits based on frame map and upland cotton collections. PloS One 10 (1), e0118073. doi: 10.1371/journal.pone.0118073
Qin, Y. M., Zhu, Y. X. (2011). How cotton fibers elongate: a tale of linear cell-growth mode. Curr. Opin. Plant Biol. 14 (1), 106–111. doi: 10.1016/j.pbi.2010.09.010
Schultink, A., Naylor, D., Dama, M., Pauly, M. (2015). The role of the plant-specific ALTERED XYLOGLUCAN9 protein in arabidopsis cell wall polysaccharide O-acetylation. Plant Physiol. 167 (4), 1271–1283. doi: 10.1104/pp.114.256479
Song, C., Li, W., Pei, X., Liu, Y., Ren, Z., He, K., et al. (2019). Dissection of the genetic variation and candidate genes of lint percentage by a genome-wide association study in upland cotton. Theor. Appl. Genet. 132 (7), 1991–2002. doi: 10.1007/s00122-019-03333-0
Su, J., Fan, S., Li, L., Wei, H., Wang, C., Wang, H., et al. (2016). Detection of favorable QTL alleles and candidate genes for lint percentage by GWAS in chinese upland cotton. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.01576
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30 (12), 2725–2729. doi: 10.1093/molbev/mst197
Vogel, J. P., Raab, T. K., Somerville, C. R., Somerville, S. C. (2004). Mutations in PMR5 result in powdery mildew resistance and altered cell wall composition. Plant J. 40 (6), 968–978. doi: 10.1111/j.1365-313X.2004.02264.x
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93 (1), 77–78. doi: 10.1093/jhered/93.1.77
Wang, M., Li, J., Qi, Z., Long, Y., Pei, L., Huang, X., et al. (2022). Genomic innovation and regulatory rewiring during evolution of the cotton genus gossypium. Nat. Genet. 54 (12), 1959–1971. doi: 10.1038/s41588-022-01237-2
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi: 10.1093/nar/gkr1293
Wang, S., Wang, J. W., Yu, N., Li, C. H., Luo, B., Gou, J. Y., et al. (2004). Control of plant trichome development by a cotton fiber MYB gene. Plant Cell 16 (9), 2323–2334. doi: 10.1105/tpc.104.024844
Wang, Z., Yang, Z., Li, F. (2019). Updates on molecular mechanisms in the development of branched trichome in arabidopsis and nonbranched in cotton. Plant Biotechnol. J. 17 (9), 1706–1722. doi: 10.1111/pbi.13167
Wu, M., Zhang, L. Y., Li, X. H., Xie, X. B., Pei, W. F., Yu, J. W., et al. (2016). A comparative transcriptome analysis of two sets of backcross inbred lines differing in lint-yield derived from a gossypium hirsutum × gossypium barbadense population. Mol. Genet. Genomics 291 (4), 1749–1767. doi: 10.1007/s00438-016-1216-x
Xiong, G., Cheng, K., Pauly, M. (2013). Xylan O-acetylation impacts xylem development and enzymatic recalcitrance as indicated by the arabidopsis mutant tbl29. Mol. Plant 6 (4), 1373–1375. doi: 10.1093/mp/sst014
Yang, C., Ye, Z. (2013). Trichomes as models for studying plant cell differentiation. Cell Mol. Life Sci. 70 (11), 1937–1948. doi: 10.1007/s00018-012-1147-6
Yuan, Y., Teng, Q., Zhong, R., Ye, Z. H. (2013). The arabidopsis DUF231 domain-containing protein ESK1 mediates 2-o- and 3-o-acetylation of xylosyl residues in xylan. Plant Cell Physiol. 54 (7), 1186–1199. doi: 10.1093/pcp/pct070
Yuan, Y., Teng, Q., Zhong, R., Ye, Z. H. (2016a). Roles of arabidopsis TBL34 and TBL35 in xylan acetylation and plant growth. Plant Sci. 243, 120–130. doi: 10.1016/j.plantsci.2015.12.007
Yuan, Y., Teng, Q., Zhong, R., Ye, Z. H. (2016b). TBL3 and TBL31, two arabidopsis DUF231 domain proteins, are required for 3-O-Monoacetylation of xylan. Plant Cell Physiol. 57 (1), 35–45. doi: 10.1093/pcp/pcv172
Zhang, C. (2020). Genome-wide association analysis of fiber quality and yield and functional analysis of candidate genes in upland cotton (Northwest Agricultural and Forestry University). doi: 10.27409/d.cnki.gxbnu.2020.001462
Zhang, H. B., Li, Y., Wang, B., Chee, P. W. (2008). Recent advances in cotton genomics. Int. J. Plant Genomics 2008, 742304. doi: 10.1155/2008/742304
Zhao, Y., Feng, J. J., Xiao, X. H., Huang, J. J., Lu, Q. W., Qu, Y. F. (2021a). Identification of candidate genes associating with fiber lint percentage using BSA-seq. J. Plant Genet. Resour. 22 (6), 1723–1731. doi: 10.13430/j.cnki.jpgr.20210421002
Zhao, Y., Jing, H., Zhao, P., Chen, W., Li, X., Sang, X., et al. (2021b). GhTBL34 is associated with verticillium wilt resistance in cotton. Int. J. Mol. Sci. 22 (17), 9115. doi: 10.3390/ijms22179115
Keywords: cotton, TBL, expression pattern, lint percentage, WGCNA, GH_D02G1759
Citation: Li Z, Shi Y, Xiao X, Song J, Li P, Gong J, Zhang H, Gong W, Liu A, Peng R, Shang H, Ge Q, Li J, Pan J, Chen Q, Lu Q and Yuan Y (2023) Genome-wide characterization of trichome birefringence-like genes provides insights into fiber yield improvement. Front. Plant Sci. 14:1127760. doi: 10.3389/fpls.2023.1127760
Received: 20 December 2022; Accepted: 16 February 2023;
Published: 15 March 2023.
Edited by:
Linghe Zeng, United States Department of Agriculture (USDA), United StatesReviewed by:
Man Wu, Chinese Academy of Agricultural Sciences (CAAS), ChinaCopyright © 2023 Li, Shi, Xiao, Song, Li, Gong, Zhang, Gong, Liu, Peng, Shang, Ge, Li, Pan, Chen, Lu and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanjia Chen, Y2hxamlhQDEyNi5jb20=; Quanwei Lu, ZGF3ZWlhbnlhbmdAMTYzLmNvbQ==; Youlu Yuan, eXVhbnlvdWx1QGNhYXMuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.