 Feng Liu
Feng Liu Nansheng Chen
Nansheng Chen Hongshu Wang
Hongshu Wang Jiamin Li
Jiamin Li Jing Wang
Jing Wang Fan Qu
Fan Qu- 1CAS Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences (IOCAS), Qingdao, Shandong, China
- 2Marine Ecology and Environmental Science Laboratory, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao, Shandong, China
- 3Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, Shandong, China
- 4Harbin University of Science and Technology, Weihai, Shandong, China
To understand the evolutionary driving forces of chloroplast (or plastid) genomes (plastomes) in the green macroalgal genus Ulva (Ulvophyceae, Chlorophyta), in this study, we sequenced and constructed seven complete chloroplast genomes from five Ulva species, and conducted comparative genomic analysis of Ulva plastomes in Ulvophyceae. Ulva plastome evolution reflects the strong selection pressure driving the compactness of genome organization and the decrease of overall GC composition. The overall plastome sequences including canonical genes, introns, derived foreign sequences and non-coding regions show a synergetic decrease in GC content at varying degrees. Fast degeneration of plastome sequences including non-core genes (minD and trnR3), derived foreign sequences, and noncoding spacer regions was accompanied by the marked decrease of their GC composition. Plastome introns preferentially resided in conserved housekeeping genes with high GC content and long length, as might be related to high GC content of target site sequences recognized by intron-encoded proteins (IEPs), and to more target sites contained by long GC-rich genes. Many foreign DNA sequences integrated into different intergenic regions contain some homologous specific orfs with high similarity, indicating that they could have been derived from the same origin. The invasion of foreign sequences seems to be an important driving force for plastome rearrangement in these IR-lacking Ulva cpDNAs. Gene partitioning pattern has changed and distribution range of gene clusters has expanded after the loss of IR, indicating that genome rearrangement was more extensive and more frequent in Ulva plastomes, which was markedly different from that in IR-containing ulvophycean plastomes. These new insights greatly enhance our understanding of plastome evolution in ecologically important Ulva seaweeds.
Introduction
The green algal class Ulvophyceae harbors at least 13 orders and more than 2700 species thus far, and ranks second in the number of species among Chlorophyta only next to the class Chlorophyceae (Guiry and Guiry, 2023). Species in the Ulvophyceae show great diversification of cytological types and morphological complexity, which varied from small unicellular species (e.g. Scotinosphaerales), to large multicellular thalli composed of uninucleate cells (e.g. Ulvales) or multinucleate cells (e.g. Cladophorales), to the gigantic single-celled coenocytic thalli (e.g. Bryopsidales and Dasycladales) (Cocquyt et al., 2010; Leliaert et al., 2012; Gulbrandsen et al., 2021). Meanwhile, their chloroplast or plastid genomes (plastomes, or cpDNAs, or ptDNAs) display miraculous variations in genome architecture, genome size, GC content, gene density, intron content and gene order (Lang and Nedelcu, 2012; Smith, 2017; Turmel and Lemieux, 2018), ranging from the circular 195.9-kb plastome with two inverted repeats (IRs) in Pseudendoclonium akinetum (Ulotrichales) (Pombert et al., 2005), which is the first ulvophycean cpDNA sequenced, to the 34 multiple hairpin cpDNA chromosomes in Boodlea composita (Cladophorales) with high GC content (average 57%) (Del Cortona et al., 2017), from the compact 74.5-kb IR-lacking plastome in Callipsygma wilsonis (Bryopsidales) (Cremen et al., 2018) to the approximately 2000-kb plastome in Acetabularia acetabulum (Dasycladales) with a high noncoding content (more than 85%) (de Vries et al., 2013).
The green macroalgal genus Ulva Linnaeus 1753 (Ulvophyceae, Chlorophyta) is the species-richest genus in the Ulvales. As more Ulva species have been accurately identified recently, 102 species names have been flagged as accepted taxonomically up to now (Guiry and Guiry, 2023). Globally, many Ulva seaweeds (e.g. Ulva prolifera, Ulva compressa, and Ulva meridionalis) are notorious for their rapid vegetative growth in eutrophic waters, leading to green tides formed by the accumulation of excess biomass (Wang et al., 2019; Liu et al., 2022b). Ulva simple morphologies show high similarity at the interspecific level, meanwhile cytological and morphological features could vary greatly at the intraspecific level, thus accurate identification of Ulva species has been challenging (e.g. Blomster et al., 2002; Hayden and Waaland, 2002). The use of molecular markers (e.g., ITS, rbcL, and tufA) for species identification has become the mainstream method to ensure the accuracy and credibility of identification results (e.g. Hofmann et al., 2010; Hughey et al., 2019; Steinhagen et al., 2019). However, due to the limited differentiation signals of these marker sequences, their resolution is inadequate for identifying closely related Ulva species. Organelle genomes (cpDNAs and mtDNAs) as super molecular markers have been proved to be powerful to understand the evolution and molecular species concepts in the genus Ulva, and are potential resources for developing specific high-resolution molecular markers (Mitsuhashi et al., 2020). Recently, phylogenomic analysis based on organelle genome data clearly depicted the evolutionary nature of double crown radiation in the phylogeny and speciation of Ulva species (Liu and Melton, 2021; Liu et al., 2022a; Liu et al., 2022b).
The data of Ulva plastomes have accumulated rapidly recently based on efficient high-throughput sequencing technology (Melton et al., 2015; Fort et al., 2021; Hughey et al., 2021), which makes it possible for more accurately understanding of the evolution trend of Ulva plastomes on a more detailed and specific sampling scale. A total of 33 plastomes from 17 Ulva species have been documented in the GenBank database thus far. The sequenced Ulva plastomes show many unique features when compared with the counterparts in other ulvophycean lineages. These Ulva plastomes belong to the compact circular IR-lacking plastomes with the smaller size (86.73 - 119.87 kb) and the lowest GC content (23.89 - 26.25%) within the Ulvophyceae (Table 1). Variations in the Ulva plastome size at interspecific and intraspecific level were mainly caused by differences in content of group I/II introns, integration of foreign DNA fragments and content of intergenic regions (Liu and Melton, 2021). The Ulva plastomes show high conservation in repertoire of canonical genes, and share the same set of 100 core genes including 71 protein-coding genes (PCGs), three ribosomal RNA (rRNA) genes and 26 transfer RNA (tRNA) genes (Wang et al., 2021). The organelle division inhibitor factor gene, minD, was observed to be present only in the plastome of Ulva aragoënsis (Liu and Melton, 2021), which used to be regarded as Ulva flexuosa (Cai et al., 2017). Only one group II (derived) intron (intron infA-62) were shared by all sequenced Ulva plastomes, and all other introns displayed highly variable and sporadic distribution pattern. Ulva plastome architectures were dynamic and plastic not conserved at the intrageneric level due to frequent genome rearrangements (Mitsuhashi et al., 2020; Liu and Melton, 2021).
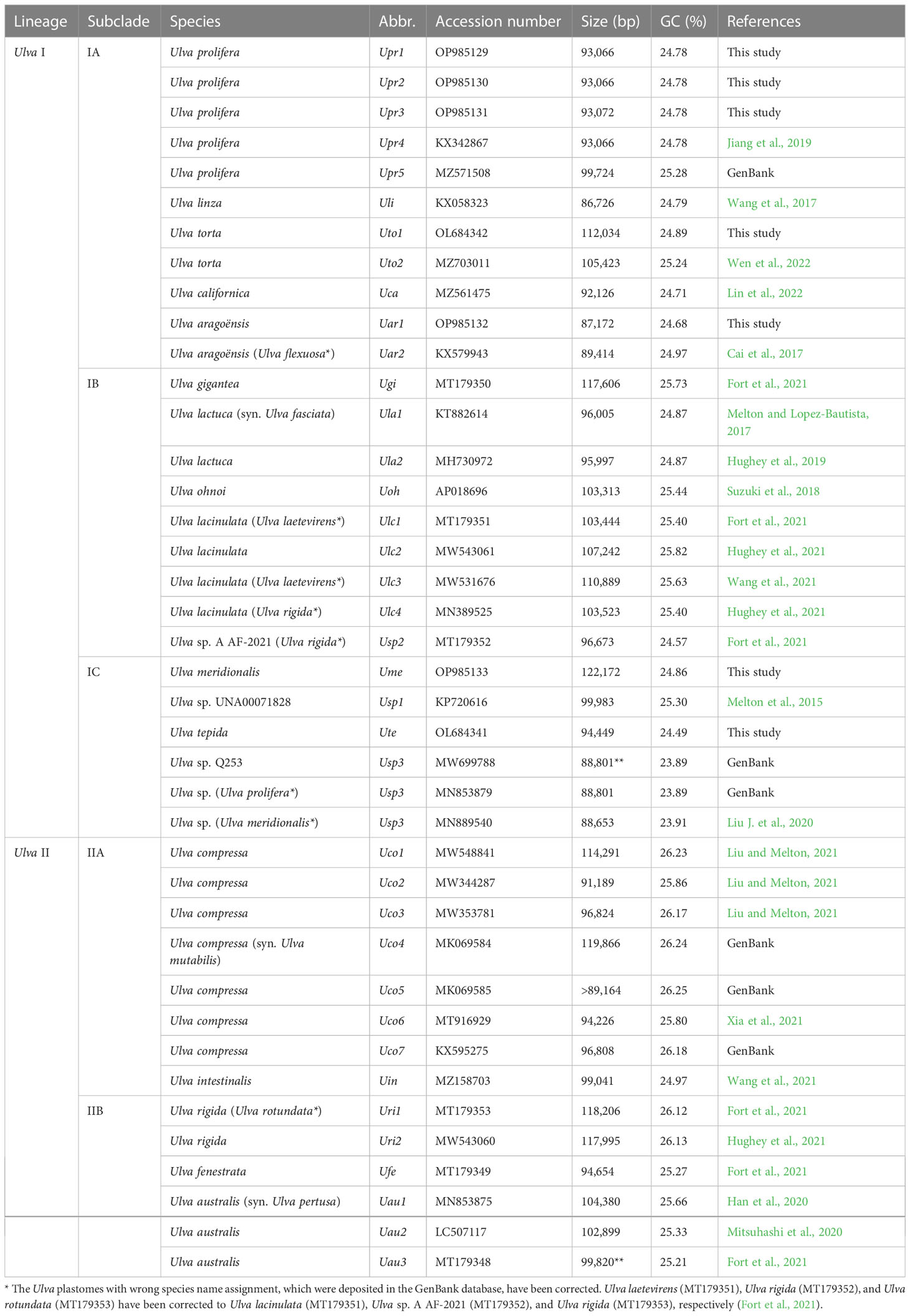
Table 1 The sequenced 40 plastomes of Ulva species for comparative analysis.
In this study, we sequenced and constructed seven complete plastomes from five Ulva species including Ulva prolifera O.F.Müller, Ulva aragoënsis (Bliding) Maggs, Ulva torta (Mertens) Trevisan, Ulva tepida Y. Masakiyo & S. Shimada, and Ulva meridionalis R. Horimoto & S. Shimada, and conducted comparative plastomic analysis to understand the evolutionary driving forces in ecologically important Ulva seaweeds.
Materials and methods
Sample collection and DNA extraction
Three free-floating algal samples of Ulva prolifera O.F.Müller (LF001, LF002 and LF003) were collected on 2 Jul. 2021 at the First (N36°05’53’’, E120°34’17’’), Second (N36°04′96″, E120°34′83″) and Third (N36°05′03″, E120°36′82″) bathing beaches along the coast of Qingdao, Shandong, China, respectively. The sessile samples of Ulva aragoënsis (Bliding) Maggs (LF005) and Ulva tepida Y.Masakiyo & S.Shimada (LF006) were collected on 11 Aug. 2021 at the Trestle Bridge (N36°06′09″, E120°31′08″) and the First bathing beach (N36°05′34″, E120°33′81″) along the coast of Qingdao, Shandong, China, respectively. The free-floating algal thalli of Ulva torta (Mertens) Trevisan (LF007) and Ulva meridionalis R.Horimoto & S.Shimada (LF010) were sampled on 4 Aug. 2021 in the Sakura Lake (37°07’30’’-56’’N, 122°27’03’’-50’’E), Rongcheng, Shandong, China. These Ulva samples were stored in coolers (5-8°C) after collection and transported back to the laboratory within 48 hours. Algal thallus for each individual Ulva thallus was cultured in a 9-cm diameter Petri dish containing 25-mL L1 medium with 0.5‰ GeO2, 50 µg/mL Dipterex (Fengcheng Animal Medicine Co., Ltd, China) and a suite of antibiotics (per mL: 50 µg streptomycin, 66.6 µg gentamycin, 20 µg ciprofloxacin, 2.2 µg chloramphenicol, and 100 µg ampicillin) (Shibl et al., 2020). The culture was maintained at 18°C, 100 - 120 μmol photons m-2 s-1 in the photoperiod of 12 h light: 12 h darkness in a GXZ-380C temperature-controlled incubator (Ningbo Jiangnan, China). After at least one week of culture, fresh algal tissue from each Ulva thallus was used for DNA extraction using a Plant Genome DNA Kit (DP305, Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. Species identification was conducted based on phylogenetic analyses of two common DNA marker datasets, including the nuclear ITS region including the 5.8S rDNA gene, and the chloroplast rbcL gene (Hayden and Waaland, 2002; Liu F. et al., 2020).
DNA sequencing and plastome assembly
The quality and concentration of total genomic DNA extracted were checked using a NanoPhotometer spectrophotometer (Implen, CA, USA), and a Qubit 2.0 Flurometer (Life Technologies, CA, USA), respectively. Qualified DNA samples were fragmented into 350 bp by Covaris S220 ultrasonic crater for library construction. The libraries were sequenced on an Illumina NovaSeq platform (Illumina, USA) using paired-end sequencing, yielding about 10 Gb sequencing raw data of paired-end reads with 150 bp in length for each Ulva sample. Clean data were harvested by trimming sequencing adapters and removing short or low-quality reads from the raw data. Complete Ulva plastomes were constructed by the GetOrganelle v1.7.1 (Jin et al., 2020). The plastome of U. compressa (MW353781) was used as the reference genome for assembly. Plastome assemblies were re-examined by aligning reads against the assembled plastome sequence using the MEM algorithm of BWA v0.7.17 (Li and Durbin, 2010). VarScan v2.3.9 (Koboldt et al., 2009) and IGV v2.8.12 (Robinson et al., 2011) were employed to examine mutation sites and to verify assembly results, respectively.
Annotation of Ulva plastomes
Protein-coding genes (PCGs) were annotated by using the Open Reading Frame Finder at the National Center for Biotechnology Information (NCBI) website (https://www.ncbi.nlm.nih.gov/orffinder/), and by aligning homologous PCGs from Ulva cpDNAs deposited in the GenBank database with the newly sequenced Ulva plastomes. Transfer RNA genes (tRNAs) were searched for by reconstructing their cloverleaf structures using the tRNAscan-SE 2.0 software with default parameters (Chan et al., 2021). Ribosomal RNA genes (rRNAs) were identified by using the RNAweasel (https://megasun.bch.umontreal.ca/apps/rnaweasel/), and by aligning homologous rRNAs. The free-standing and intronic open reading frames (orfs) were found by using the Open Reading Frame Finder at the NCBI website. Intron insertion-sites were determined manually by aligning the intron-containing homologous genes, and corresponding genes in the U. compressa (MW353781) plastome were used as a reference (Liu and Melton, 2021). Intron name was defined as host gene plus insertion site. The class and core structure of all these introns were determined by using the RNAweasel and Mfold (Zuker, 2003). The core domains of intron-encoded proteins (IEPs) and free-standing specific ORFs were determined by significant Pfam-A matches (Bateman et al., 2000). To ensure the accuracy of comparative analysis, we have re-annotated the plastomes of Ulva species and Blidingia minima (MK408749 and MT948112) deposited in the GenBank database with the same method as above. All annotation results (including genes and introns) were manually verified. In some Ulva plastomes (e.g. MZ561475, MK069585, MT916929, and KX342867), incorrect annotations and abnormal sequence errors have been corrected in our subsequent comparative analysis.
Plastome comparison and phylogenetic analysis
Base composition of Ulva plastomes and other DNA sequences was determined by using MEGA 7.0 (Kumar et al., 2016). Tandem repeats were analyzed by using Tandem Repeats Finder with parameter settings of two for matches and seven for mismatches and indels (Benson, 1999). Differences and identity values of DNA sequences were calculated by use of BioEdit v7.1.9 (Hall, 1999). Synteny analysis of Ulva plastomes was executed by using Mauve v2.3.1 software with default parameters (Darling et al., 2010). A new class of specific orfs named Ucp-orf was found in Ulva plastomes. The thorough search for Ucp-orf-like sequences in Ulva plastomes was conducted against the NCBI nucleotide database. A total of 29 full-length Ucp-orfs were detected in 17 of 40 Ulva cpDNAs. Multiple sequence alignments of Ucp-ORFs were conducted by using ClustalX 1.83 with the default settings (Thompson et al., 1997). The structural domain or motif in Ucp-ORFs was searched on the HMMER website (https://www.ebi.ac.uk/Tools/hmmer/search/phmmer) and using InterProScan tool (Paysan-Lafosse et al., 2023). The phylogenetic relationships were inferred with the Maximum Likelihood (ML) method based on the JTT matrix-based model (Jones et al., 1992) by using MEGA 7.0 (Kumar et al., 2016). There was a total of 113 positions in the final dataset of Ucp-ORFs. Phylogenomic trees were constructed based on two plastome datasets including nucleotide (nt) sequences of 100 common genes and amino acid (aa) sequences of 71 common PCGs from 40 Ulva. The nt sequences of 100 genes and the aa sequences of 71 PCGs were individually aligned, checked and concatenated using ClustalX 1.83 with default settings (Thompson et al., 1997). Maximum-likelihood trees were constructed by IQ-TREE (Trifinopoulos et al., 2016) using default parameters with 1000 ultrafast bootstrap analysis (Minh et al., 2013). The substitution model conducted by IQ-TREE was GTR+G for nt dataset and cpREV+F+I+G4 for aa dataset, respectively. Blidingia minima (MK408749 and MT948112) was used as the outgroup.
Results and discussion
Ulva plastomes show a clear evolutionary trend of becoming smaller and more compact
These seven newly obtained plastomes from five Ulva species were successfully assembled as circular-mapping molecules, with sizes ranging from 87.2 kb in U. aragoënsis (Uar1) to 122.2 kb in U. meridionalis (Ume) (Table 1). The 122.2-kb cpDNA of Ume is the largest Ulva plastome sequenced thus far, and is 1.4 times the smallest one which is the 86.7-kb cpDNA of U. linza (Uli) (Wang et al., 2017). To clarify the unique evolutionary trend of Ulva plastomes, we built the Ulva plastome dataset composed of newly sequenced plastomes and the data deposited in the GenBank database (Table 1), with a total of 40 Ulva plastomes which represented 19 Ulva species from two independent Ulva evolutionary lineages (I and II) (Liu and Melton, 2021; Liu et al., 2022a), and compared Ulva plastomes with those in other lineages of Ulvophyceae (Supplementary Table 1).
On the whole, Ulva cpDNAs show a clear evolutionary trend of becoming smaller and more compact when compared with all circular complete counterparts in Ulvophyceae (Figure 1). The Ulva plastomes only encoded a total of 100 conserved canonical genes, including 71 protein-coding genes (PCGs), three rRNA genes and 26 tRNA genes, which are the least among the sequenced circular ulvophycean plastomes. The overall coding regions composed of these 100 canonical genes occupy approximately 71.2 - 72.5 kb in size. Only small repeat sequences were observed to be concentrated either in intergenic spacer regions or intronic regions, and some specific repeat sequences reside in several PCGs (e.g. rpoB, rpoC1, and rpoC2). Unlike those in Sykidiales (Pseudoneochloris marina), Ulotrichales, Oltmannsiellopsidales, Ignatiales and Trentepohliales (Pombert et al., 2005; Turmel and Lemieux, 2018; Kim et al., 2019; Fang et al., 2021), most of intergenic regions are relatively short and the rRNA operon-encoding inverted repeat (IR) has been completely lost in Ulva plastomes. The minimum size of overall non-coding intergenic regions was only approximately 11.6 kb, which was observed in U. californica (Uca) (Supplementary Table 2).
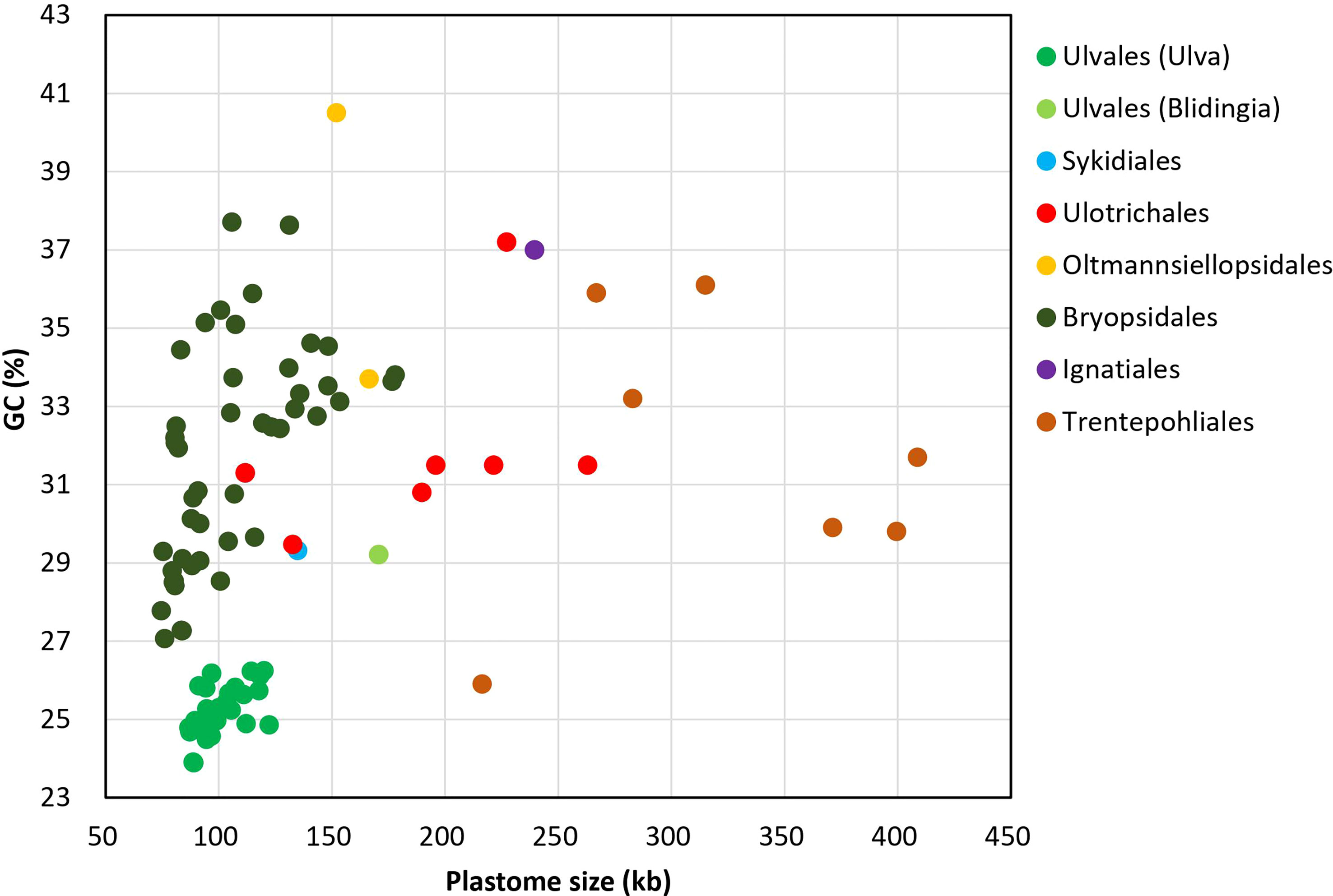
Figure 1 Comparison of GC composition and plastome size in different lineages of Ulvophyceae, including Ulva (39 plastomes. Uco5 was not included here) and Blidingia (2) in Ulvales, Sykidiales (1), Ulotrichales (8), Oltmannsiellopsidales (2), Bryopsidales (48), Ignatiales (2) and Trentepohliales (7) (Supplementary Table 1).
The ongoing gene loss or transfer can be clearly observed in Ulva plastomes. An intact organelle division inhibitor factor gene, minD (orf306), was observed to be present only in the U. aragoënsis (Uar) cpDNA (Liu and Melton, 2021). Our further comparative analysis shows that this gene or its residue exists in the trnL2-trnS2 intergenic region of cpDNAs in the U. aragoënsis-U. californica-U.torta (Uar-Uca-Uto) subclade, while it was completely lost in the other Ulva plastomes as well as the Blidingia cpDNAs (Gao et al., 2022). The minD was split into two parts (orf68 and orf209) in the Uca cpDNA, due to a 5-bp insertion mutation, whereas this gene has degenerated more seriously in the Uto cpDNAs, leaving only the residue orf44. The fracture and degeneration of minD, as well as the decreased GC content in its homologous sequences (Supplementary Table 2), indicated that there was no selective pressure to retain the integrity of this gene in Uca and Uto. Considering that homologues of minD are present in other lineages of core Ulvophyceae (Turmel and Lemieux, 2018), this gene is likely to have been transferred to the nuclear genome through horizontal transfer in Ulva species containing minD-lacking plastomes. However, we have not found this gene in the nuclear genome of U. mutabilis yet (De Clerck et al., 2018), but it cannot be determined that it does not exist in the nuclear genome, considering the incompleteness of the sequenced Ulva genome.
One specific trnR3(ccu) gene is present in plastomes of some Ulva species including U. prolifera (Upr1-5), U. gigantea (Ugi), U. rigida (Uri1-2), U. torta (Uto1), U. meridionalis (Ume), U. lacinulata (Ulc1-4) and Ulva sp. (Usp2) (Figure 2). This gene is conservatively located in the downstream region adjacent to psbC (Liu and Melton, 2021), but it is situated between psbA and trnT in the Uto1 cpDNA. Comparative analysis shows that this gene is highly similar with trnR2(ucu), indicating that it originated from the duplication of trnR2(ucu), and then its anti-codon mutated from UCU to CCU. More mutation sites were detected in trnR3(ccu) when compared with trnR2(ucu), and the latter maintains a highly conserved sequence in Ulva cpDNAs. A 5-bp (ACAAG) duplication mutation was detected in the dihydrouridine (DHU) stem-loop structure of trnR3(ccu) only in Upr5. The rates of sequence evolution for trnR3(ccu) appear to be dramatically higher than for trnR2(ucu), and the GC contents of trnR3(ccu) tend to decrease in varying degrees (Figure 2), indicating that trnR3(ccu) is subject to different selection pressures when compared with trnR2(ucu). We found that the 26 core tRNA genes are sufficient to meet all the requirements of protein synthesis in Ulva chloroplast genomes (Figure 3). Redundant trnR3(ccu) can be completely replaced by trnR2(ucu) in function, which should be the reason why it underwent significantly accelerated sequence evolution or a complete loss that had occurred.
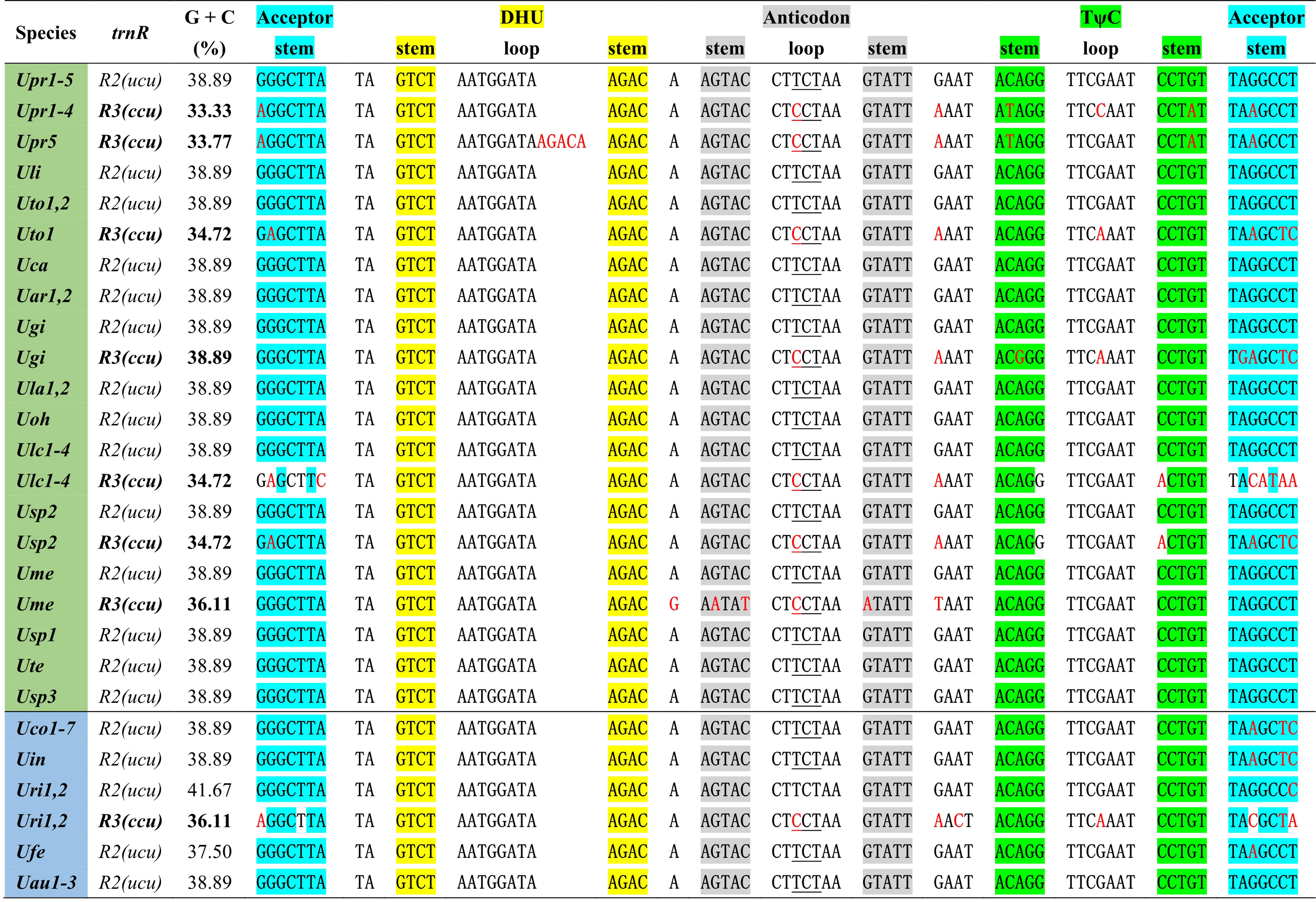
Figure 2 The aligned sequences of trnR2(ucu) and trnR3(ccu) in Ulva plastomes. Shaded nucleotides indicated that bases could be paired. Red letters in the sequence represent base mutations.
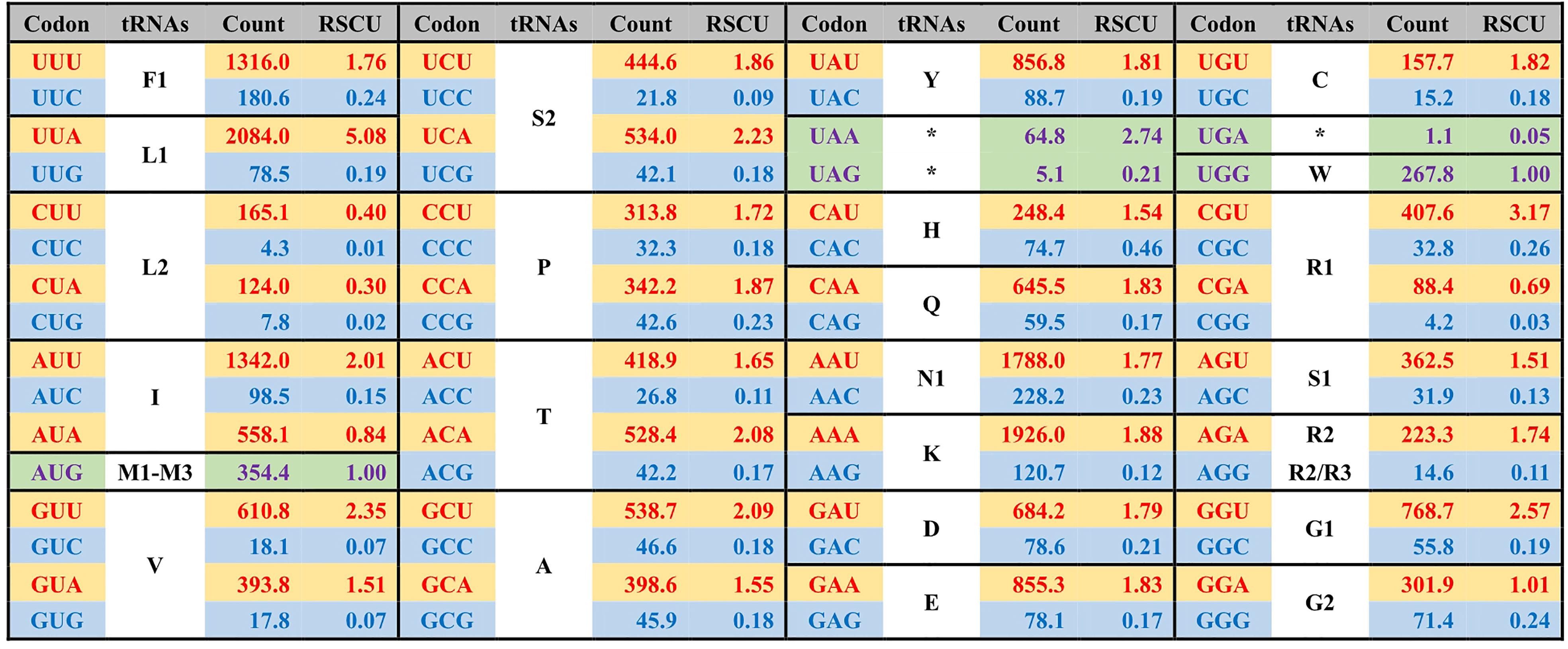
Figure 3 The average codon frequency and relative synonymous codon usage (RSCU) among the 71 core PCGs shared by Ulva plastomes. All frequencies are averages over 39 complete Ulva cpDNAs, and Uco5 is not included because some of its PCGs are incomplete (e.g. rpoA, rpoB, rpoC1 and rpoC2). The serial number of chloroplast tRNA genes is consistent with that previously reported (Liu and Melton, 2021).
In addition, these IR-lacking Ulva plastomes could sporadically incorporate foreign sequences in some specific intergenic regions, and accept group I/II introns in some housekeeping genes, which obviously increased their size. The current size of Ulva plastomes was the result of dynamic changes caused by several of the above factors. Especially, marked intraspecific differences in Ulva plastome sizes are common, involving gain or loss of introns, integration of foreign fragments and abundance of repetitive sequences (Liu and Melton, 2021).
Strong selection against GC in Ulva plastomes
The GC composition of ulvophycean plastomes varies significantly among different lineages (Turmel et al., 2017; Zhu et al., 2019; Fang et al., 2021), but particularly remarkable is that the GC content of Ulva cpDNAs was the lowest among the Ulvophyceae so far, ranging from 23.89 to 26.25% (Figure 1), indicating the strong selection against GC to shape the nucleotide composition in Ulva plastomes. Due to that GC content showed greatly heterogeneity in distinct regions of Ulva plastomes, we analyzed the differences in GC content of overall core-gene coding regions, intronic regions, foreign sequence regions, and non-coding intergenic spacer regions among these 40 Ulva cpDNAs.
The GC content is 26.53 - 27.71% in overall coding regions composed of 100 canonical genes (Supplementary Table 2). The GC composition of core-gene coding region as well as its total size showed very similar values at intraspecific level or among closely related species, but there are significant differences in the GC content and size of core-gene coding regions among different Ulva lineages (Table 1; Supplementary Table 2). The 26 tRNA regions have the highest GC content (51.32 - 51.88%), followed by the 3 rRNA regions (44.61 - 45.20%) and then the 71 PCG regions (24.50 - 25.82%) (Supplementary Table 2).
The GC composition is relatively stable in chloroplast rRNAs and tRNAs, but their variations in PCGs fluctuate greatly. Some PCGs related to photosynthesis have much higher GC content, e.g. psbA (40.96 ± 0.17%), psbB (38.60 ± 0.41%), psbC (38.20 ± 0.22%), psbD (38.76 ± 0.35%), and psaC (38.02 ± 0.91%), while some PCGs with large molecular weight, which are mainly involved in transcription and proteolysis, show very low GC content, e.g. ftsH (14.81 ± 0.68%), rpoA (16.96 ± 0.45%), rpoB (19.25 ± 0.65%), rpoC1 (16.24 ± 0.69%), and rpoC2 (14.76 ± 0.95%) (Figure 4). On the whole, codon usage pattern in Ulva chloroplast PCGs showed a much stronger preference for codons with A or T at the third position (Figure 3). The difference in GC content between chloroplast PCGs is mainly determined by their different amino acid composition and the different usage frequency of synonymous codons. The long PCGs with low GC content employ a large number of codons composed of only A and T. For example, the seven most frequently used codons in rpoC2 are AAU(N), AAA(K), UUA(L), UUU(F), UAU(Y), AUU(I), and AUA(I). However, GC-biased PCGs tend to prefer some codons with C at the third position. For example, UUC(F), AAC(N), AUC(I), UAC(Y) and CAC(H) were used more frequently than their synonymous codons in psbA.
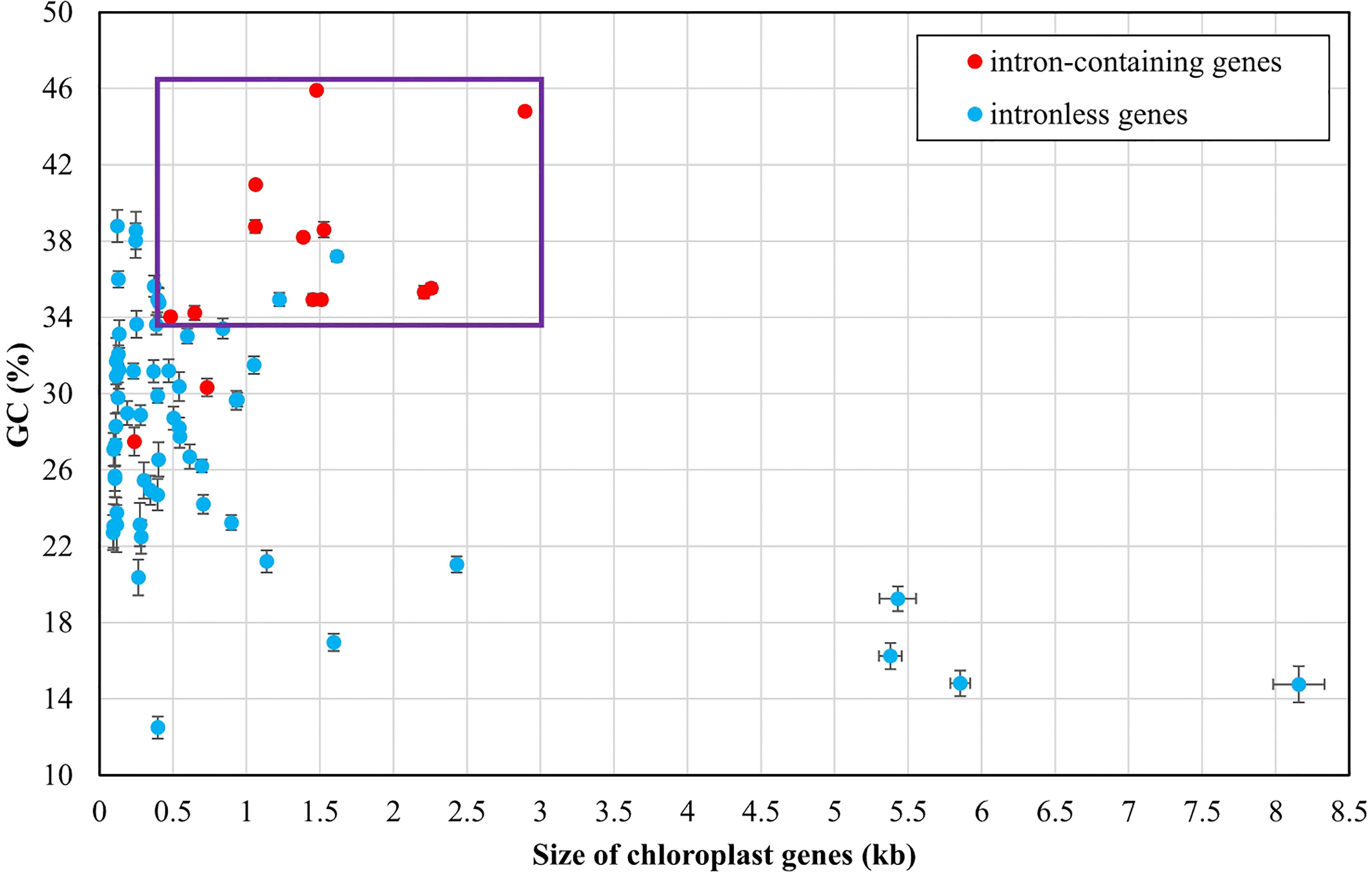
Figure 4 Comparison of GC composition and gene size among 71 PCGs and three rRNAs of Ulva plastomes. Error bars represent the standard deviation (SD). Purple box represents the distribution area of most intron-containing genes.
The overall intronic regions show great difference in GC composition among different Ulva cpDNAs, ranging from 22.59% in U. linza (Uli) to 34.72% in U. compressa (Uco2) (Supplementary Table 2), as mainly depends on type and content of introns which cpDNA harbors. The GC content of overall foreign sequence regions range from 22.79 to 35.83% in nearly all Ulva cpDNAs with the exception of Usp3 cpDNAs which show much lower values (18.69 - 18.87%) (Supplementary Table 2). The GC content in noncoding intergenic regions is obviously the lowest in the range from 8.38% in U. californica (Uca) to 19.25% in U. australis (Uau1) (Supplementary Table 2).
Low GC content in Ulva plastomes is mainly attributed to strong selection pressure driving A + T richness at a genomic level. This selection pressure seems to act on the overall plastome sequences in the microenvironment of Ulva chloroplasts, including coding regions, introns, foreign sequences and noncoding regions. The GC composition of all these regions has been markedly reduced when compared with the counterparts in other ulvophycean plastomes (Leliaert and Lopez-Bautista, 2015; Turmel et al., 2016). Plastomes in order Bryopsidales also show a trend of decreasing in size (74.5 - 177.8 kb) and GC content (27.07 - 37.71%) (Figure 1), and some species in Bryopsidales adopt the strategy of increasing the number of overlapping regions to make plastomes compact. The lowest GC content in Bryopsidales was 27.07%, which was observed in Boodleopsis plastome (MH591104). Its GC content in overall coding region (28.15%) is more than twice that in non-coding region (12.82%), and it was higher than that in coding regions of Ulva plastomes (26.53 - 27.71%).
Considering the high energy consumption and high nitrogen demand for GTP and CTP synthesis and the shortening of the sequence length in most PCGs (Mann and Chen, 2010), the advantage conferred by selection against GC observed in Ulva plastomes not in other ulvophycean plastomes seems to be more effective in saving the energy cost and serving photosynthesis and biomass synthesis in Ulva species, which is more conducive to supporting rapid and abundant growth of Ulva species. Meanwhile, the GC content (32.17 - 38.84%) in mitogenomes of Ulva species (Liu et al., 2022a; Liu et al., 2022b) does not show a significant difference from those in Ulotrichales and Oltmannsiellopsidales (Turmel et al., 2016). This strong selection of A + T preference seen in plastomes does not appear in nuclear genomes of Ulva species (e.g. 57.2% in U. mutabilis and 57.3% in U. compressa) (De Clerck et al., 2018; Osorio et al., 2022).
Distribution and diversity of Ulva plastome introns
These newly sequenced Ulva plastomes harbor different intron contents ranging from two in U. aragoënsis (Uar1) to 16 in U. meridionalis (Ume), occupying 3.5 - 17.1% of cpDNAs, which are in the range of the reported Ulva plastomes (Liu and Melton, 2021). To further understand the evolutionary trend of Ulva chloroplast introns in distribution and diversity, we systematically excavated and compared introns at intrageneric level. A total of 34 intron families were found among these 40 known Ulva chloroplast genomes. Among them, 33 intron families were found by RNAweasel (Lang et al., 2007), and only one (intron rns-476) was detected by alignment of homologous gene sequences (Table 2).
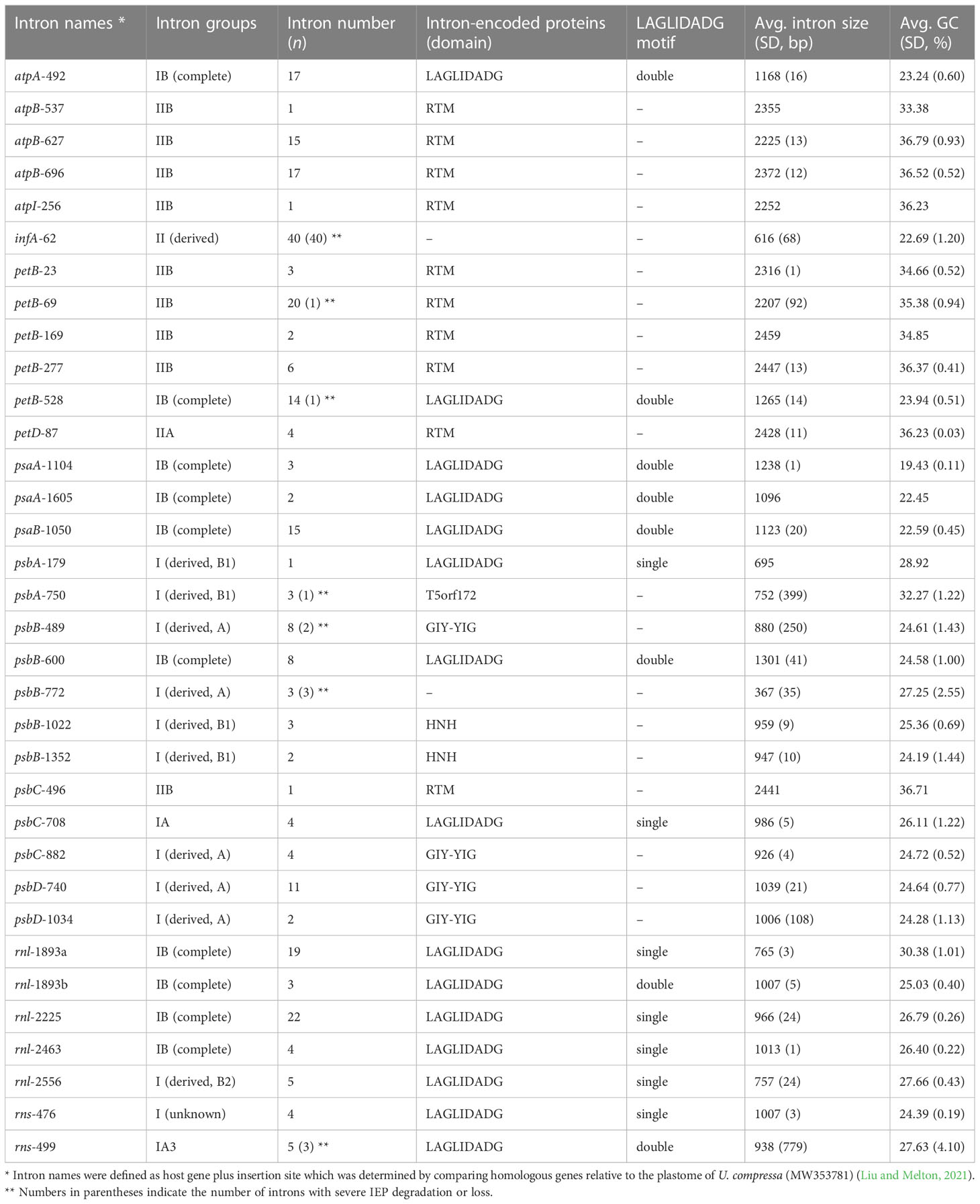
Table 2 General features of Ulva chloroplast introns detected among the 40 Ulva plastomes.
The Ulva chloroplast introns were detected at 33 insertion sites of 14 host genes including atpA (1 site), atpB (3), atpI (1), infA (1), petB (5), petD (1), psaA (2), psaB (1), psbA (2), psbB (5), psbC (3), psbD (2), rnl (4) and rns (2) (Table 2). Obviously, intron densities varied widely among chloroplast genes at interspecific and intraspecific levels (Supplementary Figure 1). These introns were mainly distributed in two rRNA genes (rnl, and rns) and some more conserved PCGs involving photosystem I and II, electron transport and ATP synthesis, indicating that different functional groups of genes have different propensities for intron insertion. Further comparative analysis showed that introns preferentially resided in conserved housekeeping genes with high GC content (usually more than 34%) and long size (usually more than 0.5 kb) (Figure 4). We speculate that this may be related to higher GC content in target site sequences required for IEP recognition in host genes and more target sites contained by long GC-rich genes. However, some PCGs (e.g. rbcL, and tufA) with the above similar characteristics have high expression in chloroplasts and tend to resist intron invasion to economically and effectively ensure unnecessary consumption and time cost in transcription and processing (Jeffares et al., 2006).
A total of 23 intron families were observed to belong to group I introns, and the remaining 11 were group II introns. Eight intron families including intron atpB-537, psaA-1605, psbA-179, psbB-1022, psbB-1352, psbC-708, psbD-1034 and rnl-1893b were found for the first time in Chlorophyta. The size and GC content of different intron families fluctuate markedly in Ulva plastomes (Table 2), which are largely determined by type of introns and degeneration degree of intron-encoded proteins (IEPs) (Figure 5A). Except for the degenerated group II (derived) intron infA-62 (Liu and Melton, 2021), the size of all other group II introns is significantly longer than that of group I introns, due to the different size of their IEP genes (Table 2). The GC content is positively correlated with group II intron size with the excellent coefficient of determination (R2 = 0.9809) (Figure 5B). Group IIA/IIB introns have the high GC content ranging from 33.38% in intron atp-537 to 36.79 ± 0.93% in intron atpB-627, while the GC content of group II (derived) intron infA-62 is only 22.69 ± 1.20% mainly due to the loss of the entire IEP (Table 2). Contrary to the group II introns, there is a weak negative correlation between GC content and size in group I intron (Figure 5B). The GC content in group I introns shows complex changes, which is mainly due to the diversity of types (related to secondary structure) and IEPs.
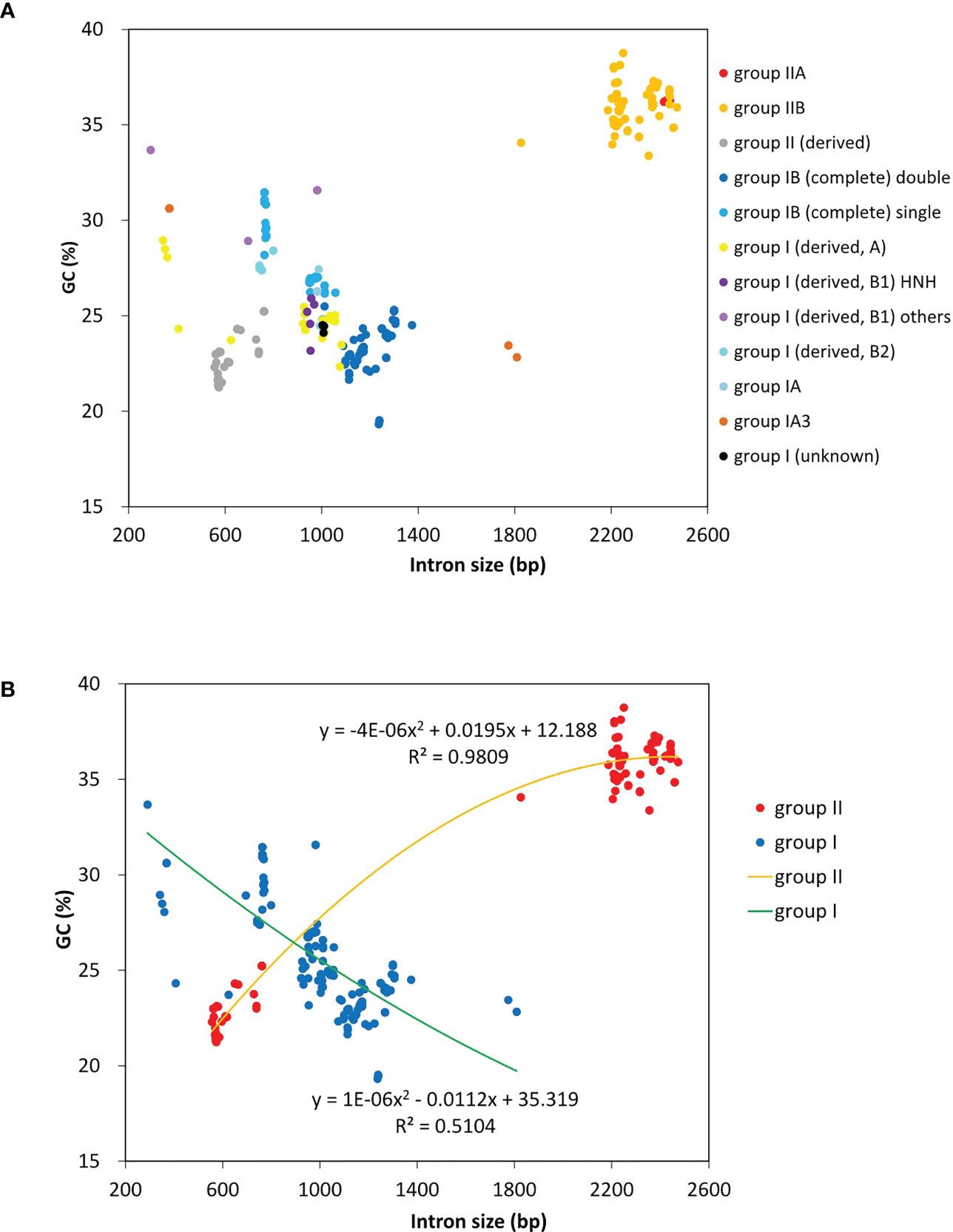
Figure 5 The GC composition and size of introns detected among 40 Ulva plastomes. (A) Comparison of GC content and size among different types of introns. (B) Comparison of GC content and size between group I and group II introns.
Group IIA/IIB introns harbor a reverse transcriptase/maturase (RTM) gene in Ulva plastomes. The vast majority (93.8%) of group I introns encode an intact IEP which is the member of the LAGLIDADG or GIY-YIG or HNH homing endonuclease (LHE or GHE or NHE) families. However, the IEPs from intron psbA-750 do not exhibit significant sequence similarity to common homing endonuclease families (e.g. LHE, GHE, and NHE), but contain a conserved T5orf172 domain which occurs in a stand-alone protein form in phage, virus and bacteria and is also found in DNA-binding regulatory proteins of bacterial and eukaryotic DNA viruses (Iyer et al., 2002). All chloroplast GHEs were encoded by group I (derived, A) introns, while the IEPs encoded by group I (derived, B1) introns showed diverse protein types including NHE, LHE, and T5orf172 domain-containing homing endonuclease (THE). All of chloroplast group IB introns and other group I introns encoded an LHE with one or two LAGLIDADG motifs.
Almost all introns displayed sporadic distribution pattern in Ulva plastomes, due to their nature of homing and mobility. Only the chloroplast intron infA-62 is an exception. This intron is shared by all Ulva plastomes, but absent in Blidingia cpDNAs, indicating that it might be acquired after its divergence from Blidingia. This intron has completely lost the ability to move and has been trapped in infA, because of its severe degeneration and the loss of IEP. This intron co-evolved with infA and showed a faster evolution rate than the host gene. Introns from the same insertion site were previously observed to be homologous among organelle genomes in Ulva (Liu and Melton, 2021; Liu et al., 2022a). However, two different intron families, intron rnl-1893a and rnl-1893b, were found to be present in the same insertion site. Although both of them belong to group IB intron and share similar secondary structure of ribozyme components, but their primary sequences and IEPs are markedly different. The IEPs in intron rnl-1893a were LHEs with only one LAGLIDADG motif, while those in intron rnl-1893b contained two LAGLIDADG motifs (Table 2). These facts indicated that these two different LHEs should recognize the same target site in rnl, although they can be a homodimer and a monomer (Haugen et al., 2005), respectively.
Novel insights into integration of foreign sequences and rearrangement of Ulva plastomes
Comparison of plastome intergenic regions shows that Ulva cpDNAs experienced frequent insertion of foreign DNA sequences which usually harbor some specific open reading frames (orfs), as were important indicators for insight into their source. The largest U. meridionalis (Ume) cpDNA contained 14.6-kb foreign sequence which encoded 15 specific orfs (Supplementary Figure 2), accounting for 11.9% of plastome. To elucidate the origin of exogenous sequences and their relationships, we systematically compared the sequence characteristics of large intergenic regions and the distribution of free-standing orfs among these 40 Ulva plastomes. A total of 154 specific free-standing orfs as well as many homologous residue DNA sequences of some specific orfs, which have no similarity to chloroplast canonical genes, were detected in intergenic regions of these 40 Ulva cpDNAs. These specific orfs were not randomly distributed but mainly located in some specific intergenic regions (e.g. psbA-psbB, trnT-psbA, psbB-psbD, psbC-psbB, trnS2-psbC, trnM3-psbD, psbC-trnM3, trnL2-psbD, trnL2-trnM3, trnM1-trnE, and trnW-psaJ) (Liu and Melton, 2021), indicating that these intergenic regions should be hot spots for the invasion of foreign sequences.
Some foreign DNA sequences integrated into different intergenic regions of Ulva plastomes harbor homologous orfs with significant high similarity (Supplementary Table 3). These facts indicated that these foreign sequences could have been derived from the same origin. It is very similar to the finding in Ulva mitogenomes where the derived foreign sequences mainly originated from mitochondrial plasmid DNA (Liu et al., 2022a; Liu et al., 2022b). Among these specific chloroplast homologous orfs, three classes of orfs show high similarities to the full length or partial sequences of putative bacterial tyrosine-type recombinase/integrase (tri), NAD-dependent DNA ligase (lig), and phage/plasmid DNA primase (Supplementary Table 3), respectively, based on tblastn search, which were also detected in cpDNAs of some siphonous green algae (Bryopsidales) (Leliaert and Lopez-Bautista, 2015; Cremen et al., 2018). It is worth noting that a new class of specific free-standing orfs was found only in Ulva plastomes, which we named Ucp-orf. A total of 29 full-length Ucp-orfs were detected in 17 of 40 Ulva plastomes, which belonged to different Ulva lineages (Figure 6), and none of such orf was found in other ulvophyceaen cpDNAs sequenced thus far. Interestingly, all of Ucp-orfs reside in several intergenic regions where genome rearrangement occurred. In the Ume cpDNA, there are five Ucp-orfs in three intergenic regions (i.e. psbA-psbB, psbC-trnS2, and trnL2-trnM3). One conserved domain was shared by all of these Ucp-orfs (Figure 7), but no information on its function can be obtained based on blastp search. In addition, the remaining orfs contain some recognizable protein domains acting on DNA or RNA, but their functions are still unknown (Supplementary Table 3).
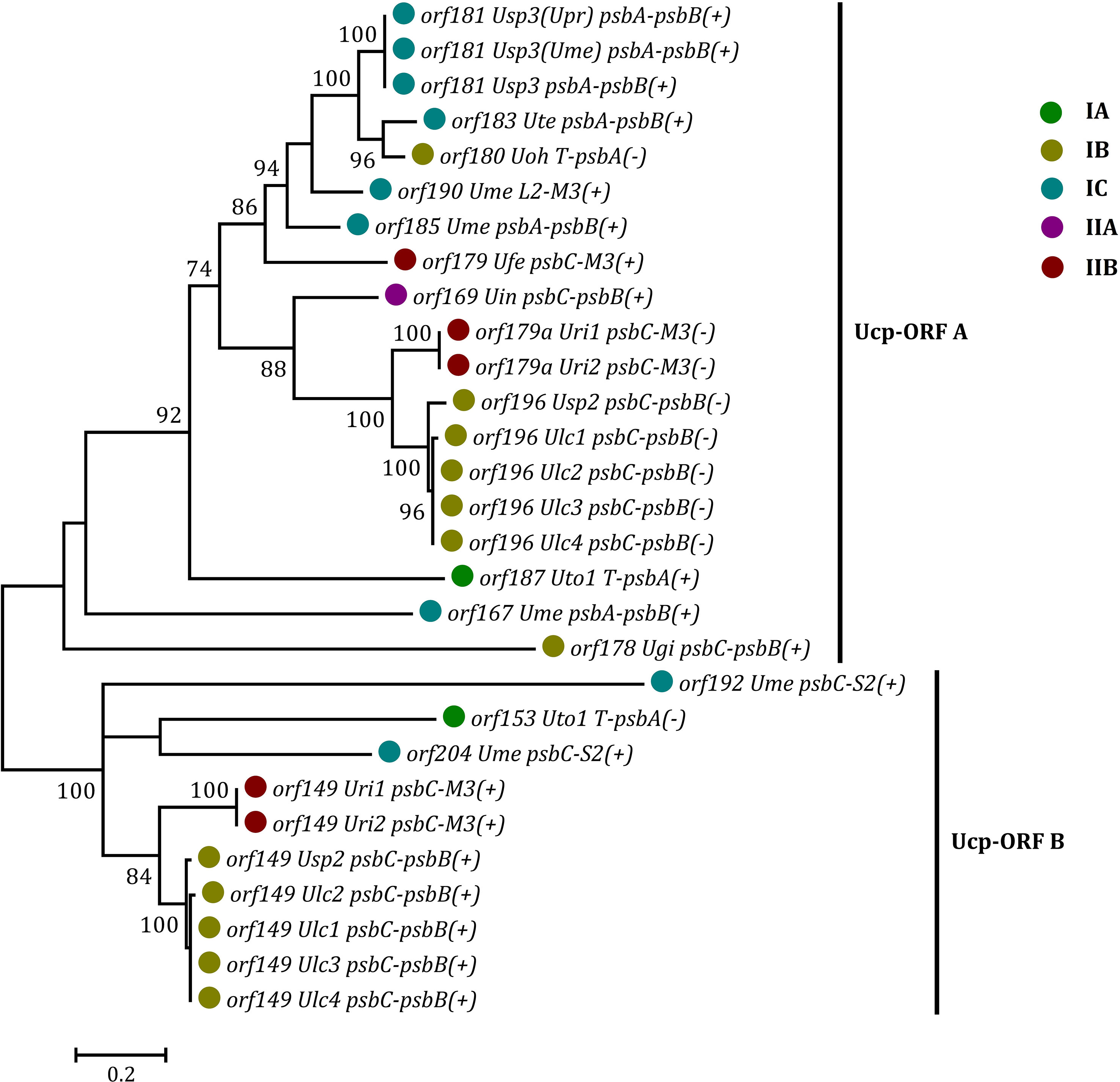
Figure 6 Phylogenetic analysis of the 29 full-length free-standing Ucp-ORFs found in Ulva plastomes. The bootstrap support values greater than 70% were displayed at branches. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees. Different colored circles represent different Ulva lineages.
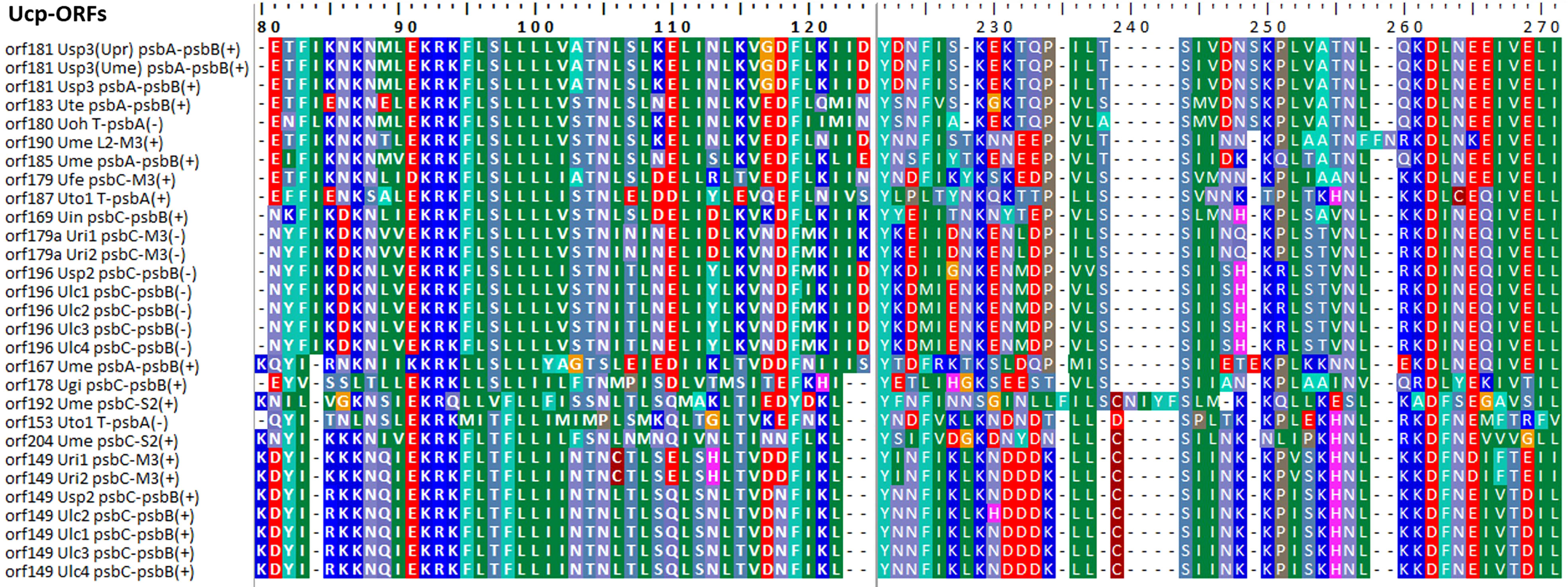
Figure 7 The conserved domain shared by the 29 Ucp-ORFs detected in Ulva plastomes.
The plastome architecture is not as conserved as that of mitogenomes in Ulva species (Supplementary Figure 2), but has experienced several rearrangement events to varying degrees (Liu and Melton, 2021). It is worth noting that the intergenic regions where foreign sequences frequently invade match well with the regions where plastome rearrangement occurs. Six conserved gene blocks can be detected in Ulva cpDNAs by comparing the plastome structure, and chloroplast genome recombination frequently occurs in the regions on both sides of the psbD-psbC gene block, the upstream region of psbB and the downstream region of trnT (Figure 8). These regions are exactly the regions where foreign DNA sequences are inserted most frequently. It seems that the invasion of foreign fragments causes the instability of genome architecture and triggered inversion of some gene blocks in Ulva cpDNAs. The Ulva chloroplast genomes belong to IR-lacking cpDNAs, and the IR was supposed to play an important role in stabilizing the architecture of cpDNAs (Turmel and Lemieux, 2018). The invasion of foreign sequences, especially in the context of IR loss, seems to be an important driving force for Ulva genome rearrangement.

Figure 8 Comparison of plastome organization and gene order between Ulva lineage I and II. Thick lines with different colors on the left represented different gene blocks. Thick lines with different colors on the top represented different Ulva lineages. The arrows indicated the direction of gene transcription. Only canonical genes were shown as filled boxes in different colors representing different gene types.
These acquired orfs display a different evolutionary trend from the chloroplast canonical genes. The levels of sequence divergence among homologous specific orfs or foreign DNA sequences in Ulva cpDNAs greatly exceed those observed in chloroplast genes. Frequent insertion and deletion mutations lead to serious fracture and degeneration of these sequences, accompanied by reduced GC content (Supplementary Table 2, 3). These evidences show that their existence is not a necessary requirement of Ulva chloroplast genomes, and there is no selection pressure to maintain their existence. Differential GC content can be used as an indicator to distinguish the background genome and the non-self (or introduced) DNA sequence (Mann and Chen, 2010). Because of rapid evolution and high divergence of derived foreign sequences, their GC content decreased at varying levels, as depends on their evolution time and rate after their insertion into different Ulva plastomes. Their changes from heterogeneity to homogeneity caused by rapid evolution make it difficult to distinguish some foreign sequences that completely lose coding ability from non-coding intergenic regions. The fate of these integrated foreign sequences will most likely be accelerated evolution and eventually lose, which reminds us of the similar phenomena observed in the Ulva mitogenomes where the frequently inserted plasmid-derived sequences underwent multiple mutations and rapid degeneration (Liu et al., 2022a; Liu et al., 2022b).
Novel insights into plastome architecture and phylogenomic analysis
Due to the limited data at present, it is difficult to reconstruct the plastome structure of the common ancestor of Ulva species. Based on comparative analysis of architectures in sequenced Ulvales-Sykidiales-Ulotrichales cpDNAs to date, plastomes have completely lost the IR in Ulvales (e.g. Ulva species and B. minima) (Figures 8, 9), which is a remarkable difference from those in Sykidiales (e.g. P. marina) and Ulotrichales carrying identical or non-identical IR copies (Turmel et al., 2017; Kim et al., 2019). Gene order and gene distribution show some new characteristics in these IR-lacking plastomes. In Ulva plastomes, gene clusters show a staggered distribution pattern on two strands (Figure 8), while in B. minima plastomes (MT948112 and MK408749), gene clusters tend to accumulate on one main strand and only one gene cluster (rpl20-rps8-rps4-rps9-rpl12) and 13 tRNAs are transcribed on another strand (Figure 9). We found that gene partitioning pattern changed after the loss of IR and distribution range of gene clusters became larger, indicating that genome rearrangement was more extensive and more frequent in IR-lacking Ulvales plastomes. Many new gene clusters, e.g. rpl23-rpl2-rps19-rps3-rpl16-rpl14-rpl5-rps8-infA-rpl36-rps11-rpoA-petB-petD, psaC-ycf20-ccsA-trnR1-ycf1-psbA, rns-trnI-trnA-rnl-rrn5-psaA, trnV-rpoB-rpoC1-rpoC2, ycf3-psbI-psaI-psbN, and psbB-psbT-psbH, were observed to be shared only by plastomes of Ulva and Blidingia (Figure 9), but they did not appear in plastomes of P. marina and other ulvophycean species (e.g. Pombert et al., 2005; Pombert et al., 2006; Leliaert and Lopez-Bautista, 2015; Turmel et al., 2017; Fang et al., 2021), indicating they have been formed and maintained in their common ancestor after divergence from P. marina. These findings provide important clues for us to understand genome structure and gene order of early IR-lacking plastomes in Ulvales.
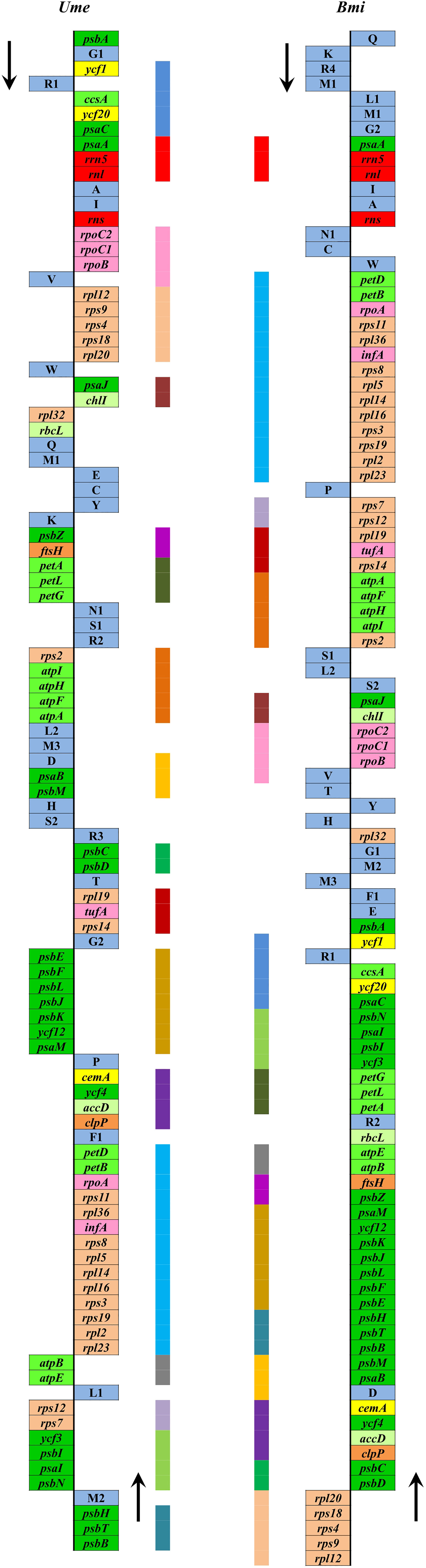
Figure 9 Comparison of plastome organization and gene order between Ulva meridionalis (OP985133) and Blidingia minima (MK408749 and MT948112). Thick lines with different colors between two plastomes represented different gene blocks. The arrows indicated the direction of gene transcription. Only canonical genes were shown as filled boxes in different colors representing different gene types.
Phylogenomic analyses of two Ulva plastome datasets (nt sequences of 100 canonical genes and aa sequences of 71 PCGs) showed that the common ancestor of Ulva species had a very early internal divergence and split into two major evolutionary lineages (Ulva I and II) (Liu and Melton, 2021). Ulva lineage I has evolved into at least three independent clades (IA, IB and IC), and Ulva lineage II into at least two clades (IIA and IIB) (Figures 10, 11). The inversion of psbD-psbC gene cluster is the most significant difference between lineage I and II in gene order, but the continuous inversion of this gene cluster makes the gene order in the Ulva intestinalis (Uin) plastome completely consistent with those in IA and IB clades (Figure 8). Ulva IA clade contains the U. linza-prolifera (LP) complex which harbor the rps19 gene with GTG start code and the Uar-Uca-Uto subclade which is the only minD-containing Ulva lineage. Plastomes in IA and IB clades shared the identical gene order, but were different from those in IC clade. In the IC plastomes, two gene clusters composed of 45 genes from psbB to trnT(ugu) and six genes (trnM3-trnD-psaB-psbM-trnH-trnS2) were inverted respectively, and then the latter had a secondary inversion in Usp1 and the psbD-psbC gene block was inverted in Ume. Like plastomes in the IC clade, the large gene cluster containing 45 genes was also inverted in the IIB clade but not in IIA (Figure 8; Supplementary Figure 3). Because of frequent inversion of gene clusters in plastomes, the gene order cannot well reflect their evolutionary relationship between Ulva species (Wang et al., 2021).
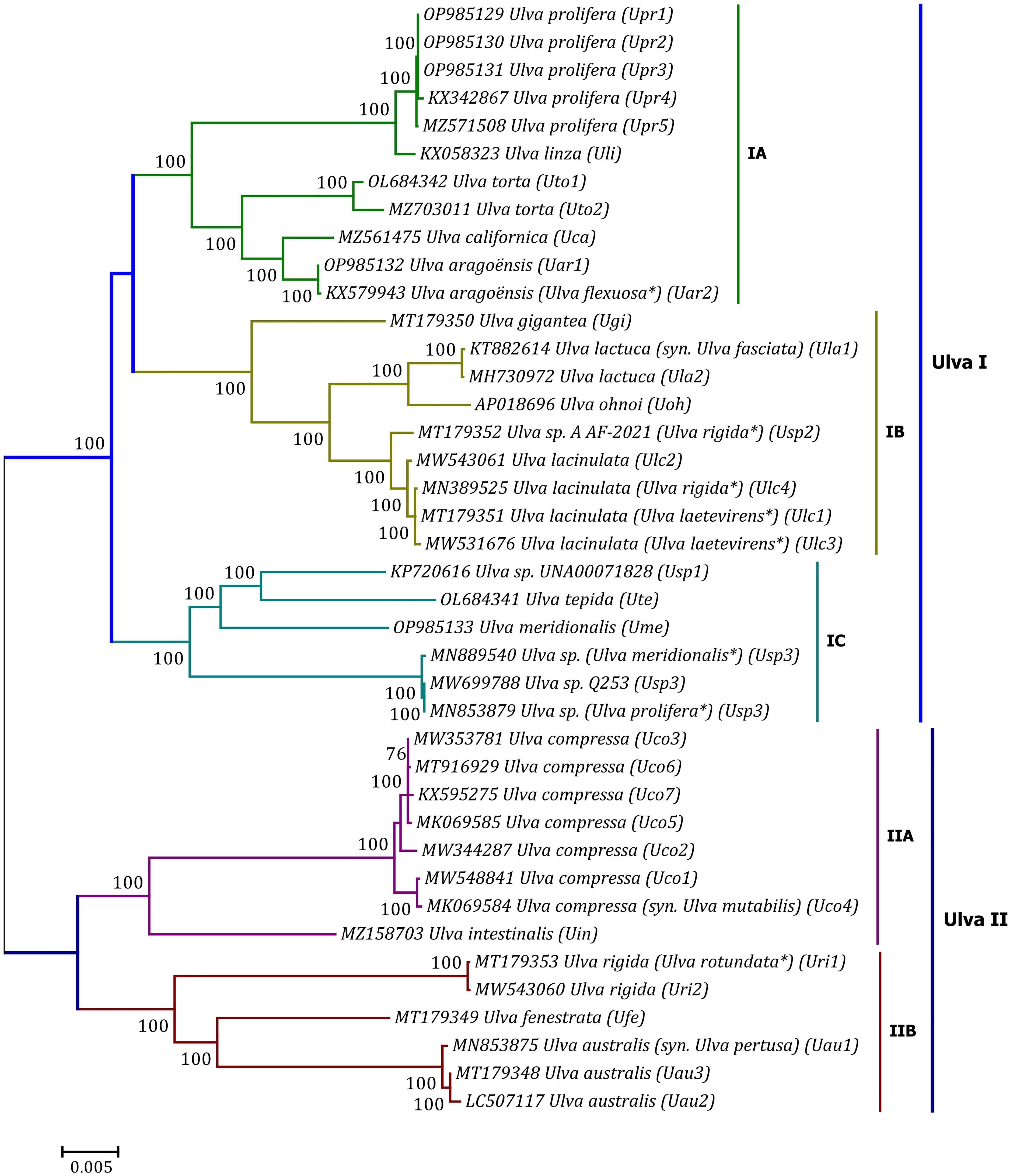
Figure 10 Unrooted phylogenomic tree based on Maximum Likelihood (ML) analysis of the nucleotide (nt) sequences of the 100 common genes in the 40 Ulva plastomes. The bootstrap support values greater than 70% were displayed at branches. Branch lengths are proportional to the amount of sequence change, which are indicated by the scale bar below the trees. The asterisk indicates that the species name has been corrected.
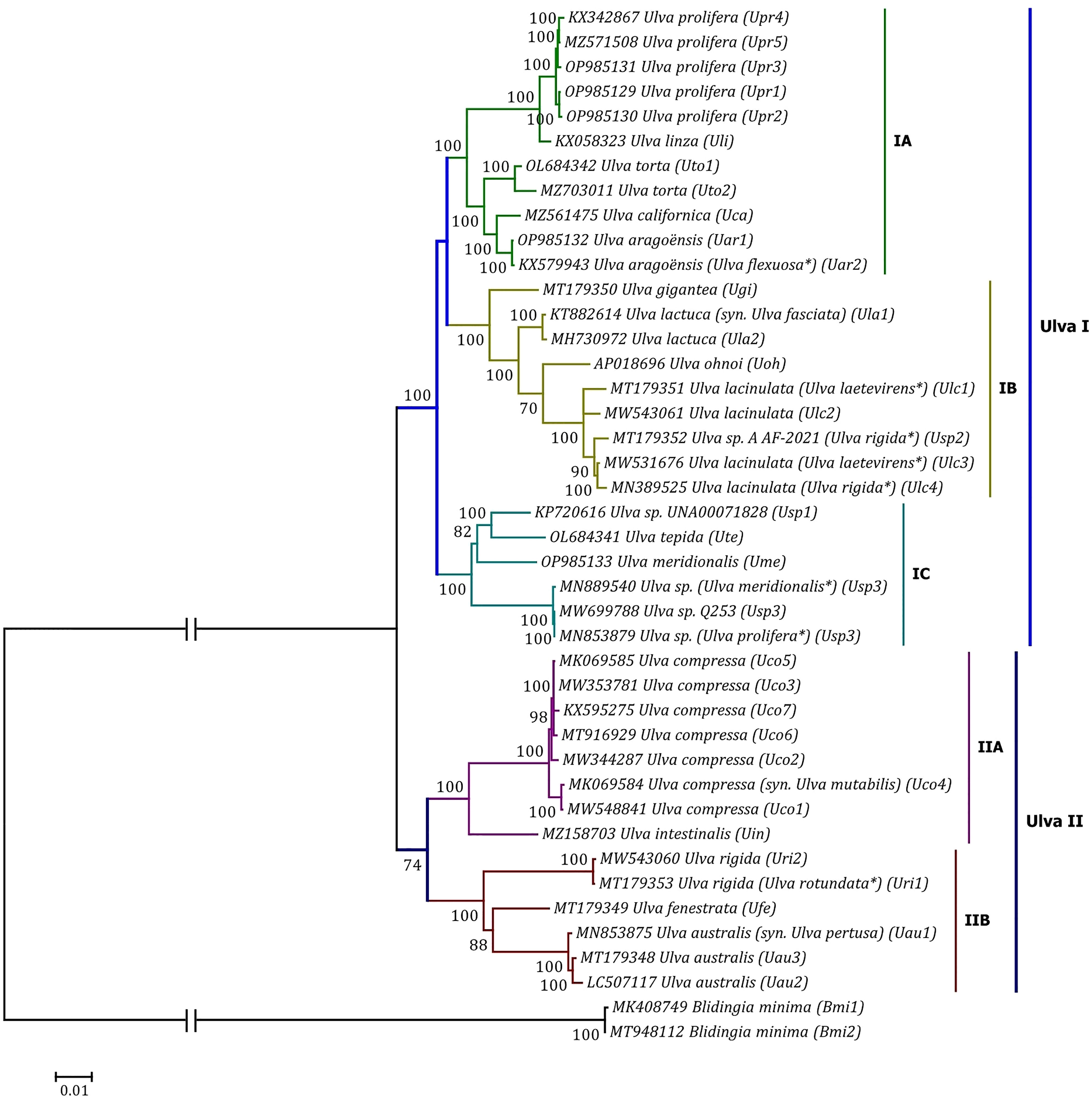
Figure 11 Phylogenomic tree based on Maximum Likelihood (ML) analysis of the amino acid (aa) sequences of the 71 common PCGs in the 40 Ulva plastomes. The bootstrap support values greater than 70% were displayed at branches. Branch lengths are proportional to the amount of sequence change, which are indicated by the scale bar below the trees. The tree was rooted with Blidingia minima as the outgroup. The asterisk indicates that the species name has been corrected.
Our results of phylogenomic analysis well supported taxonomic revisions of some species names at the genomic level (Figures 10, 11), e.g. U. mutabilis Föyn, U. pertusa Kjellman and U. fasciata Delile are taxonomic synonyms of U. compressa Linnaeus (Steinhagen et al., 2019), U. australis Areschoug (Couceiro et al., 2011) and U. lactuca Linnaeus (Hughey et al., 2019), respectively. It is worth emphasizing that our results show that eight of 40 Ulva plastomes were assigned wrong species names reported at first (Liu J. et al., 2020; Wang et al., 2020; Fort et al., 2021; Hughey et al., 2021). The fact that inaccurate species identification occurred frequently leads to incorrect Ulva species names matched by DNA sequences deposited in the GenBank database. Therefore, the way to fundamentally eliminate the mismatch between species names and sequences is to use molecular marker technology, especially the comparative analysis of organelle genomes, to more clearly reveal the genotype differences between individuals of Ulva species, especially the closely related species.
Conclusion
The accumulation of Ulva cpDNA data provides us with an opportunity to decipher the unique evolution of plastomes in these globally distributed green macroalgae. In this study, more new insights into plastome evolution of Ulva species have been gained. First, Ulva plastome evolution reflects the strong selection pressure driving the compactness of genome organization and the decrease of overall genomic GC content. The overall plastome sequences including canonical genes, introns, derived foreign sequences and non-coding regions show a synergetic decrease in GC content at the varying degree. Fast degeneration of plastome sequences including non-core genes (minD and trnR3), derived foreign sequences, and noncoding spacer regions was accompanied by the marked decrease of their GC composition. Second, introns preferentially resided in conserved housekeeping genes with high GC content and long length in Ulva plastomes. It might be related to high GC content in target site sequences required for IEP recognition and more target sites contained by long GC-rich genes. Third, many foreign DNA sequences integrated into different intergenic regions harbor some homologous specific orfs with high similarity, indicating that they could have been derived from the same origin. Fourth, the invasion of foreign sequences seems to be an important driving force for plastome rearrangement in these IR-lacking Ulva cpDNAs. It seems that the invasion of foreign fragments causes the instability of genome architecture and triggered inversion of some gene blocks in Ulva cpDNAs. Finally, gene partitioning pattern changed after the loss of IR, and genome rearrangement was more extensive and more frequent in IR-lacking Ulvales plastomes. Gene clusters show a staggered distribution pattern on two strands in Ulva plastomes. Our new findings have deepened our understanding of the evolutionary trend of the plastomes in ecologically important Ulva seaweeds.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank OL684341, OL684342, and OP985129-OP985133.
Author contributions
FL designed the study. FL, NC, HW, JL, JW and FQ performed the experiments. FL and HW performed the analysis. FL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 42276133/41876165), the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDA23050302/XDA23050403), the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (No. QYZDB-SSW-DQC023), the Science and Technology Basic Resources Investigation Program of China (No. 2018FY100200), the Major Scientific and Technological Innovation Project of Shandong Province (No. 2019JZZY020706), and Natural Science Foundation of Shandong Province for Young Scholars (No. ZR2022QD066).
Acknowledgments
We are thankful to reviewers for their valuable comments and all staffs of marine ecological environment genomics research group in Institute of Oceanology, Chinese Academy of Sciences.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1126175/full#supplementary-material
References
Bateman, A., Birney, E., Durbin, R., Eddy, S. R., Howe, K. L., Sonnhammer, E. L. (2000). The pfam protein families database. Nucleic Acids Res. 28, 263–266. doi: 10.1093/nar/28.1.263
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Blomster, J., Bäck, S., Fewer, D. P., Kiirikki, M., Lehvo, A., Maggs, C. A., et al. (2002). Novel morphology in Enteromorpha (Ulvophyceae) forming green tides. Am. J. Bot. 89, 1756–1763. doi: 10.3732/ajb.89.11.1756
Cai, C., Wang, L., Zhou, L., He, P., Jiao, B. (2017). Complete chloroplast genome of green tide algae Ulva flexuosa (Ulvophyceae, chlorophyta) with comparative analysis. PloS One 12 (9), e0184196. doi: 10.1371/journal.pone.0184196
Chan, P. P., Lin, B. Y., Mak, A. J., Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49 (16), 9077–9096. doi: 10.1101/614032
Cocquyt, E., Verbruggen, H., Leliaert, F., De Clerck, O. (2010). Evolution and cytological diversification of the green seaweeds (Ulvophyceae). Mol. Biol. Evol. 27 (9), 2052–2061. doi: 10.1093/molbev/msq091
Couceiro, L., Cremades, J., Barreiro, R. (2011). Evidence for multiple introductions of the pacific green alga Ulva australis areschoug (Ulvales, chlorophyta) to the Iberian peninsula. Bot. Mar. 54 (4), 391–402. doi: 10.1515/bot.2011.044
Cremen, M. C. M., Leliaert, F., Marcelino, V. R., Verbruggen, H. (2018). Large Diversity of non-standard genes and dynamic evolution of chloroplast genomes in siphonous green algae (Bryopsidales, chlorophyta). Genome Biol. Evol. 10 (4), 1048–1061. doi: 10.1093/gbe/evy063
Darling, A. E., Mau, B., Perna, N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PloS One 5 (6), e11147. doi: 10.1371/journal.pone.0011147
De Clerck, O., Kao, S. M., Bogaert, K. A., Blomme, J., Foflonker, F., Kwantes, M., et al. (2018). Insights into the evolution of multicellularity from the Sea lettuce genome. Curr. Biol. 28 (18), 2921–2933. doi: 10.1016/j.cub.2018.08.015
Del Cortona, A., Leliaert, F., Bogaert, K. A., Turmel, M., Boedeker, C., Janouškovec, J., et al. (2017). The plastid genome in cladophorales green algae is encoded by hairpin chromosomes. Curr. Biol. 27, 3771–3782. doi: 10.1016/j.cub.2017.11.004
de Vries, J., Habicht, J., Woehle, C., Huang, C., Christa, G., Wägele, H., et al. (2013). Is ftsH the key to plastid longevity in sacoglossan slugs? Genome Biol. Evol. 5 (12), 2540–2548. doi: 10.1093/gbe/evt205
Fang, J., Liu, B., Liu, G., Verbruggen, H., Zhu, H. (2021). Six newly sequenced chloroplast genomes from trentepohliales: the inflated genomes, alternative genetic code and dynamic evolution. Front. Plant Sci. 12, 780054. doi: 10.3389/fpls.2021.780054
Fort, A., McHale, M., Cascella, K., Potin, P., Usadel, B., Guiry, M. D., et al. (2021). Foliose Ulva species show considerable inter-specific genetic diversity, low intra-specific genetic variation, and the rare occurrence of inter-specific hybrids in the wild. J. Phycol. 57, 219–233. doi: 10.1111/jpy.13079
Gao, D., Sun, Z., Bi, G., Zhang, X. (2022). The complete plastome of Blidingia marginata and comparative analysis with the relative species in ulvales. Aquat. Bot. 183, 103568. doi: 10.1016/j.aquabot.2022.103568
Guiry, M. D., Guiry, G. M. (2023). AlgaeBase. world-wide electronic publication (Galway: National University of Ireland).
Gulbrandsen, Ø.S., Andresen, I. J., Krabberød, A. K., Bråte, J., Shalchian-Tabrizi, K. (2021). Phylogenomic analysis restructures the ulvophyceae. J. Phycol. 57, 1223–1233. doi: 10.1111/jpy.13168
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98. doi: 10.1021/bk-1999-0734.ch008
Han, H., Yan, L., Song, W., Wang, Z., Zhang, X. (2020). Complete chloroplast genome of Ulva pertusa, one of the causal species of green macroalgal blooms in the coastal waters of qinhuangdao, China. Mitochondrial. DNA Part B. 5 (1), 1084–1086. doi: 10.1080/23802359.2020.1723448
Haugen, P., Simon, D. M., Bhattacharya, D. (2005). The natural history of group I introns. Trends Genet. 21, 111–119. doi: 10.1016/j.tig.2004.12.007
Hayden, H. S., Waaland, J. R. (2002). Phylogenetic systematics of the ulvaceae (Ulvales, ulvophyceae) using chloroplast and nuclear DNA sequences. J. Phycol. 38 (6), 1200–1212. doi: 10.1046/j.1529-8817.2002.01167.x
Hofmann, L. C., Nettleton, J. C., Neefus, C. D., Mathieson, A. C. (2010). Cryptic diversity of Ulva (Ulvales, chlorophyta) in the great bay estuarine system (Atlantic USA): introduced and indigenous distromatic species. Eur. J. Phycol. 45 (3), 230–239. doi: 10.1080/09670261003746201
Hughey, J. R., Gabrielson, P. W., Maggs, C. A., Mineur, F. (2021). Genomic analysis of the lectotype specimens of European Ulva rigida and Ulva lacinulata (Ulvaceae, chlorophyta) reveals the ongoing misapplication of names. Eur. J. Phycol. 57 (2), 143–153. doi: 10.1080/09670262.2021.1914862
Hughey, J. R., Maggs, C. A., Mineur, F., Jarvis, C., Miller, K. A., Shabaka, S. H., et al. (2019). Genetic analysis of the linnaean Ulva lactuca (Ulvales, chlorophyta) holotype and related type specimens reveals name misapplications, unexpected origins, and new synonymies. J. Phycol. 55, 503–508. doi: 10.1111/jpy.12860
Iyer, L. M., Koonin, E. V., Aravind, L. (2002). Extensive domain shuffling in transcription regulators of DNA viruses and implications for the origin of fungal APSES transcription factors. Genome Biol. 3 (3), research0012. doi: 10.1186/gb-2002-3-3-research0012
Jeffares, D. C., Mourier, T., Penny, D. (2006). The biology of intron gain and loss. Trends Genet. 22 (1), 16–22. doi: 10.1016/j.tig.2005.10.006
Jiang, T., Gu, K., Wang, L., Liu, Q., Shi, J., Liu, M., et al. (2019). Complete chloroplast genome of Ulva prolifera, the dominant species of green macroalgal blooms in yellow Sea, China. Mitochondrial. DNA Part B. 4 (1), 1930–1931. doi: 10.1080/23802359.2019.1610090
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., dePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Jones, D. T., Taylor, W. R., Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Kim, D., Lee, J. M., Choi, J. W., Yang, J. H. (2019). Flip-flop organization in the chloroplast genome of Capsosiphon fulvescens (Ulvophyceae, chlorophyta). J. Phycol. 55, 214–223. doi: 10.1111/jpy.12811
Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., McLellan, M. D., Mardis, E. R., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285. doi: 10.1093/bioinformatics/btp373
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lang, B. F., Laforest, M.-J., Burger, G. (2007). Mitochondrial introns: a critical view. Trends Genet. 23, 119–125. doi: 10.1016/j.tig.2007.01.006
Lang, B. F., Nedelcu, A. M. (2012). “Plastid genomes of algae,” in Advances in photosynthesis and respiration including bioenergy and related processes: genomics of chloroplasts and mitochondria. Eds. Bock, R., Knoop, V. (Dordrecht: Springer), 59–87.
Leliaert, F., Lopez-Bautista, J. M. (2015). The chloroplast genomes of Bryopsis plumosa and Tydemania expeditiones (Bryopsidales, chlorophyta): compact genomes and genes of bacterial origin. BMC Genomics 16, 204. doi: 10.1186/s12864-015-1418-3
Leliaert, F., Smith, D. R., Moreau, H., Herron, M. D., Verbruggen, H., Delwiche, C. F., et al. (2012). Phylogeny and molecular evolution of the green algae. Crit. Rev. Plant Sci. 31, 1–46. doi: 10.1080/07352689.2011.615705
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Lin, X., Liu, W., Wei, X., Jiang, P. (2022). Complete chloroplast genome of an invasive marine macroalga Ulva californica (Ulvophyceae, chlorophyta). Mitochondrial. DNA Part B. 7 (7), 1337–1339. doi: 10.1080/23802359.2022.2098854
Liu, F., Melton, J. T. (2021). Chloroplast genomes of the green-tide forming alga Ulva compressa: comparative chloroplast genomics in the genus Ulva (Ulvophyceae, chlorophyta). Front. Mar. Sci. 8, 668542. doi: 10.3389/fmars.2021.668542
Liu, F., Melton, J. T., Lopez-Bautista, J. M., Chen, N. (2020). Multiple intraspecific variations of mitochondrial genomes in the green-tide forming alga, Ulva compressa Linnaeus (Ulvophyceae, chlorophyta). Front. Mar. Sci. 7, 714. doi: 10.3389/fmars.2020.00714
Liu, F., Melton, J. T., Wang, H., Wang, J., Lopez-Bautista, J. M. (2022a). Understanding the evolution of mitochondrial genomes in the green macroalgal genus Ulva (Ulvophyceae, chlorophyta). Front. Mar. Sci. 9, 850710. doi: 10.3389/fmars.2022.850710
Liu, F., Wang, H., Song, W. (2022b). Tandem integration of circular plasmid contributes significantly to the expanded mitochondrial genomes of the green-tide forming alga Ulva meridionalis (Ulvophyceae, chlorophyta). Front. Plant Sci. 13, 937398. doi: 10.3389/fpls.2022.937398
Liu, J., Yang, X., Cui, J., Zhuang, M., Zhao, L., Li, J., et al. (2020). Complete chloroplast genome of Ulva meridionalis (Ulvales: ulvaceae): an extremely fast-growing green macroalgae. Mitochondrial. DNA Part B. 5 (2), 1390–1392. doi: 10.1080/23802359.2020.1735967
Mann, S., Chen, Y. P. (2010). Bacterial genomic G + c composition-eliciting environmental adaptation. Genomics 95 (1), 7–15. doi: 10.1016/j.ygeno.2009.09.002
Melton, J. T., Leliaert, F., Tronholm, A., Lopez-Bautista, J. M. (2015). The complete chloroplast and mitochondrial genomes of the green macroalga ulva sp. UNA00071828 (Ulvophyceae, chlorophyta). PloS One 10 (4), e0121020. doi: 10.1371/journal.pone.0121020
Melton, J. T., Lopez-Bautista, J. M. (2017). The chloroplast genome of the marine green macroalga Ulva fasciata delile (Ulvophyceae, chlorophyta). Mitochondrial. DNA Part A. 28 (1), 93–95. doi: 10.3109/19401736.2015.1110814
Minh, B. Q., Nguyen, M. A. T., von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30 (5), 1188–1195. doi: 10.1093/molbev/mst024
Mitsuhashi, C., Teramura, H., Shimada, H. (2020). Construction of genomic marker sets based on the chloroplast genome of a green alga, Ulva pertusa (syn. Ulva australis), leads to simple detection of Ulva species. Genes Genet. Systems. 95 (2), 55–63. doi: 10.1266/ggs.19-00054
Osorio, H., Tapia-Reyes, P., Espinoza, D., Laporte, D., González, A., Castro-Nallar, E., et al. (2022). The genome of the marine alga Ulva compressa (Chlorophyta) reveals protein-coding genes with similarity to plants and green microalgae, but also to animal, bacterial, and fungal genes. Int. J. Mol. Sci. 23, 7279. doi: 10.3390/ijms23137279
Paysan-Lafosse, T., Blum, M., Chuguransky, S., Grego, T., Pinto, B. L., Salazar, G. A., et al. (2023). InterPro in 2022. Nucleic Acids Res. 51 (D1), D418–D427. doi: 10.1093/nar/gkac993
Pombert, J.-F., Lemieux, C., Turmel, M. (2006). The complete chloroplast DNA sequence of the green alga Oltmannsiellopsis viridis reveals a distinctive quadripartite architecture in the chloroplast genome of early diverging ulvophytes. BMC Biol. 4, 3. doi: 10.1186/1741-7007-4-3
Pombert, J.-F., Otis, C., Lemieux, C., Turmel, M. (2005). The chloroplast genome sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) reveals unusual structural features and new insights into the branching order of chlorophyte lineages. Mol. Biol. Evol. 22 (9), 1903–1918. doi: 10.1093/molbev/msi182
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Shibl, A. A., Isaac, A., Ochsenkühn, M. A., Cárdenas, A., Fei, C., Behringer, G., et al. (2020). Diatom modulation of select bacteria through use of two unique secondary metabolites. P. Natl. Acad. Sci. U.S.A. 117, 27445–27455. doi: 10.1073/pnas.2012088117
Smith, D. R. (2017). Evolution: in chloroplast genomes, anything goes. Curr. Biol. 27 (24), R1305–R1307. doi: 10.1016/j.cub.2017.10.049
Steinhagen, S., Weinberger, F., Karez, R. (2019). Molecular analysis of Ulva compressa (Chlorophyta, ulvales) reveals its morphological plasticity, distribution and potential invasiveness on German north Sea and Baltic Sea coasts. Eur. J. Phycol. 54 (1), 102–114. doi: 10.1080/09670262.2018.1513167
Suzuki, S., Yamaguchi, H., Hiraoka, M., Kawachi, M. (2018). Mitochondrial and chloroplast genome sequences of Ulva ohnoi, a green-tide forming macroalga in the southern coastal regions of Japan. Mitochondrial. DNA Part B. 3 (2), 765–767. doi: 10.1080/23802359.2018.1483778
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., Higgins, D. G. (1997). The ClustalX windows interface flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A., Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44 (W1), W232–W235. doi: 10.1093/nar/gkw256
Turmel, M., Lemieux, C. (2018). Evolution of the plastid genome in green algae. Adv. Bot. Res. 85, 157–193. doi: 10.1016/bs.abr.2017.11.010
Turmel, M., Otis, C., Lemieux, C. (2016). Mitochondrion-to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns in Gloeotilopsis green algae (Ulotrichales, ulvophyceae). Genome Biol. Evol. 8 (9), 2789–2805. doi: 10.1093/gbe/evw190
Turmel, M., Otis, C., Lemieux, C. (2017). Divergent copies of the large inverted repeat in the chloroplast genomes of ulvophycean green algae. Sci. Rep. 7, 994. doi: 10.1038/s41598-017-01144-1
Wang, L., Cai, C., Zhou, L., He, P., Jiao, B. (2017). The complete chloroplast genome sequence of Ulva linza. conserv. Genet. Resour. 9, 463–466. doi: 10.1007/s12686-016-0682-0
Wang, Y., Liu, F., Liu, X., Shi, S., Bi, Y., Moejes, F. W. (2019). Comparative transcriptome analysis of four co-occurring Ulva species for understanding the dominance of Ulva prolifera in the yellow Sea green tides. J. Appl. Phycol. 31, 3303–3316. doi: 10.1007/s10811-019-01810-z
Wang, H., Liu, F., Wang, J., Chen, N. (2021). Phylogenomic analysis of the chloroplast genome of the green-tide forming macroalga Ulva intestinalis Linnaeus (Ulvophyceae, chlorophyta). Mitochondrial. DNA Part B. 6 (10), 3052–3054. doi: 10.1080/23802359.2021.1978889
Wang, J., Wang, Y., Wang, H., Liu, F., Chen, N. (2020). Comparative chloroplast genome analysis of new-green tide causative species in China: Ulva laetevirens. Oceanol. Et Linnol. Sinica. 52 (5), 1–13.
Wen, Q., Yang, W., Li, J., Liu, J., Zhao, S., Gao, S., et al. (2022). Characterization of complete chloroplast genome of Ulva torta (Mertens) trevisa. Mitochondrial. DNA Part B. 7 (6), 1041–1043. doi: 10.1080/23802359.2022.2081943
Xia, L., Qin, Y., Liu, J., Zhang, H., Wu, L., Gao, S., et al. (2021). Complete chloroplast genome of Ulva compressa (Ulvales: ulvaceae). Mitochondrial. DNA Part B. 6 (3), 720–722. doi: 10.1080/23802359.2020.1860696
Zhu, H., Hu, Y., Liu, F., Hu, Z., Liu, G. (2019). Characterization of the chloroplast genome of Trentepohlia odorata (Trentepohliales, chlorophyta), and discussion of its taxonomy. Int. J. Mol. Sci. 20, 1774. doi: 10.3390/ijms20071774
Keywords: chloroplast genome, Ulvophyceae, comparative genomics, GC content, group I/II intron, genome rearrangement
Citation: Liu F, Chen N, Wang H, Li J, Wang J and Qu F (2023) Novel insights into chloroplast genome evolution in the green macroalgal genus Ulva (Ulvophyceae, Chlorophyta). Front. Plant Sci. 14:1126175. doi: 10.3389/fpls.2023.1126175
Received: 17 December 2022; Accepted: 03 April 2023;
Published: 18 April 2023.
Edited by:
Enrico Vito Perrino, International Centre for Advanced Mediterranean Agronomic Studies, ItalyReviewed by:
Zoltan Fussy, Charles University, CzechiaEric Wade Linton, Central Michigan University, United States
Huan Zhu, Institute of Hydrobiology (CAS), China
Copyright © 2023 Liu, Chen, Wang, Li, Wang and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Liu, bGl1ZmVuZ0BxZGlvLmFjLmNu; cHJjbGl1ZmVuZ0BzaW5hLmNu