 Shengli Yao
Shengli Yao Meili Xie
Meili Xie Ming Hu2
Ming Hu2 XiaoBo Cui
XiaoBo Cui Haoming Wu
Haoming Wu Chaobo Tong
Chaobo Tong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 20 March 2023
Sec. Plant Breeding
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1118339
This article is part of the Research TopicGenetics, Breeding and Engineering to Enhance Oil Quality and YieldView all 11 articles
Ubiquitin-conjugating enzyme (UBC) is a critical part of the ubiquitin–proteasome pathway and plays crucial roles in growth, development and abiotic stress response in plants. Although UBC genes have been detected in several plant species, characterization of this gene family at the whole-genome level has not been conducted in Brassica napus. In the present study, 200 putative BnUBCs were identified in B. napus, which were clustered into 18 subgroups based on phylogenetic analysis. BnUBCs within each subgroup showed relatively conserved gene architectures and motifs. Moreover, the gene expression patterns in various tissues as well as the identification of cis-acting regulatory elements in BnUBC promoters suggested further investigation of their potential functions in plant growth and development. Furthermore, three BnUBCs were predicted as candidate genes for regulating agronomic traits related to oil content and yield through association mapping. In conclusion, this study provided a wealth of information on the UBC family in B. napus and revealed their effects on oil content and yield, which will aid future functional research and genetic breeding of B. napus.
Ubiquitination is a crucial regulatory process for the selective protein degradation mechanism which regulates cell physiology through the ubiquitin-26S proteasome pathway in plants (Pickart, 2001; Smalle and Vierstra, 2004; Vierstra, 2009; Ye and Rape, 2009). Ubiquitination regulates a wide range of various plant growth and developmental processes including photomorphogenesis, protein translocation within cells, flower development, cell cycle control, both abiotic and biotic stress responses, phytohormone, regulation of proteome homeostasis as well as light signaling (Welchman et al., 2005; Dreher and Callis, 2007; Vierstra, 2009; Sadanandom et al., 2012; Doroodian and Hua, 2021; Yu and Hua, 2022). In addition, the ubiquitin-26S proteasome system (UPS) plays critical role in plant adaption (Hua and Yu, 2019). Protein ubiquitination involves the covalent attachment of a 76-amino acid (aa) sequence, through one of the encompassing seven lysine residues (Lys-6, Lys-11, Lys-27, Lys-29, Lys-33, Lys-48, Lys-63), to substrate protein (Welchman et al., 2005; Kim et al., 2013). Moreover, the fate of the ubiquitinated substrate is determined by the type of ubiquitination as well as the choice of the Lys residue for the modification, resulting in different linkages with various functions (Fang and Weissman, 2004; Sun and Chen, 2004).
The protein ubiquitin-proteasome system (UPS) is a multistep reaction mediated by three enzymes, E1 (ubiquitin-activating enzyme, UBA), E2 (ubiquitin-conjugating enzyme, UBC) and E3 (Ubiquitin-ligase enzyme) (Glickman and Ciechanover, 2002). Ubiquitin is initially activated by E1 through the ATP-dependent reaction, which results in the formation of a thioester-linked ubiquitin (Ramadan et al., 2015). Then, E1 transfers the thioester-linked ubiquitin to the cysteine (Cys) residue (active site) of the UBC domain through the passing of E2 via transesterification (Hershko et al., 1983). Subsequently, E2 transfers the ubiquitin moiety to the substrate protein with the help of E3 directly via a second ans-thiolation reaction, which mediates the formation of polyubiquitin chains on target proteins and determines the specificity of the substrate in the ubiquitination system (Bae and Kim, 2013; Bae and Kim, 2014). Finally, the 26S proteasome degrades the target protein. The family of UBCs (E2s) is central to this enzymatic cascade, which offers a platform for the attachment of ubiquitin to the target proteins (Hershko et al., 1983; Burroughs et al., 2008).
Most ubiquitin-like conjugating enzymes (UBLs) and E2s comprise a conserved catalytic core consisting of approximately 140-150 aa known as the UBC domain harboring the active site cysteine residue required for enzyme-ubiquitin (Criqui et al., 2002; Michelle et al., 2009; Schumacher et al., 2013). Moreover, several studies indicated that the UBC domain contributes to the mediation of the interaction between E2 and E3 (Huang et al., 1999; Christensen et al., 2007; Poyurovsky et al., 2007). The UBC domains in various members of the E2 family is highly conserved, both in terms of amino acid sequence and three-dimensional structure, which possessed four α-helices, an anti-parallel β-sheet formed by four strands, and a short 310–helix (Lin et al., 2002; Ozkan et al., 2005; Wenzel et al., 2011). Among the multiple binding sites, one highly conserved active-site (Cys residue) is located at a shallow groove formed by a short loop connecting α-helix 2 with α-helix 3 as well as a proximal long loop (Ye and Rape, 2009; van Wijk and Timmers, 2010). The formation of E2-ubiquitin thioester requires E2s to interact with ubiquitin as well as with E1 (or its cognates) which results in considerable evolutionary constraints on E2 structure and the formation of the conserved active site (Silver et al., 1992). Aside from the core E2 domain, some detected E2 enzymes comprise diverse N- and C-terminal extensions that are thought to contribute to the intracellular localization of the enzyme and its substrate specificity (Arrigoni et al., 2012; Hodson et al., 2014). Furthermore, UBC enzymes are divided into four classes, based on the N- and C-terminal extensions as well as the UBC domain. Class I UBCs contain only the UBC domain with a region of approximately 150 conserved residues, whereas Class II UBCs comprise the N-terminal extension and the UBC domain; Class III E2s harbor the C-terminal extension as well as the UBC domain; and Class IV UBCs possess both the N- and C-terminal extensions and the UBC domain (Papaleo et al., 2012; Schumacher et al., 2013).
The genes encoding UBCs usually form a multigene family and the number of UBCs in the multigene family is greater in the higher eukaryotes than in the lower eukaryotes, due to the expansion during the process of evolution (Jue et al., 2015). Moreover, UBLs, including the SUMO-conjugating enzymes, ubiquitin (RUB) conjugating enzymes and ubiquitin-conjugating enzyme variants (UEVs) also belong to the E2 category. Among the 48 UBC domain-containing proteins identified in Arabidopsis (Kraft et al., 2005), except 37 potential E2s, three are thioester-linked UBLs, one is a SUMO-conjugating enzyme [AtSCE1, At3g57870), and two are RUB-conjugating related enzymes (At4g36800 (RCE1) and At2g18600 (RCE2)]. These UBL-specific enzymes perform the same function as E2s, but they do not belong to the UBC family. In addition, eight UBC proteins are termed as ubiquitin-conjugating enzyme variants (UEVs), since they lack the active-site Cys residue, which is not active on their own (Callis, 2014). Based on sequence homology, the 48 Arabidopsis UBCs were classified into 16 groups (Kraft et al., 2005). Plant UBC proteins, as previous studies reported, play a crucial role in regulating plant development, growth and stress response (Dreher and Callis, 2007; Sadanandom et al., 2012; E et al., 2015; Millyard et al., 2016; Jue et al., 2018; Kravic et al., 2020; Liu et al., 2020). For example, functional analyses have revealed that AtUBC1 and AtUBC2 are involved in leaf development, tolerance response to UV stress, as well as activation of the floral repressor gene (Xu et al., 2009). AtUBC13 is involved in the plant response to DNA damage, iron deficiency and epidermal cell differentiation (Welchman et al., 2005; Wen et al., 2006; Wen et al., 2008; Li and Schmidt, 2010; Sadanandom et al., 2012). AtUBC19 and AtUBC20, which belong to the E2-C gene family, potentially contribute to protein ubiquitination reactions and play key functional roles in the cell cycle in differentiating or differentiated cells (Criqui et al., 2002). However, AtUBC21 (AtPEX4) specializes in protein for ubiquitination in peroxisome maintenance (Zolman et al., 2005). AtUBC22 has a function in female gametophyte development and potentially in Lys11-linked ubiquitination (Wang et al., 2016). AtUBC24 regulates the uptake, allocation as well as remobilization of inorganic phosphate (Dong et al., 1998; Lin et al., 2009), and is also the target gene of miR399s (Aung et al., 2006). AtUBC32 is a part of the endoplasmic reticulum-associated protein degradation (ERAD) complex, which is involved in salt stress tolerance mediated by brassinosteroid (BR) (Cui et al., 2012) and in plant growth promotion in Arabidopsis. Moreover, the viral pathogen Zymoseptoria tritici induced silence of Triticum aestivum ubiquitin-conjugating enzyme 4 (TaU4) gene, resulting in postponed progression of disease symptoms, and limited reproduction as well as the growth of Septoria in wheat leaves (Millyard et al., 2016). In Vitis vinifera, the UBC family is involved in the berry ripening process and cold and heat stress responses (Gao et al., 2017). In conclusion, UBC genes in plants play various important roles related to stress response and plant growth.
Allotetraploid Brassica napus (AnAnCnCn, 2n = 38) is a globally important oilseed crop, which is derived from the hybridization between Brassica rapa (ArAr, 2n=20) and Brassica oleracea (CoCo, 2n=18) followed by chromosome doubling approximately 7500 years ago (Rana et al., 2004; Lu et al., 2011; Chalhoub et al., 2014; Sun et al., 2017; Lu et al., 2019; Wu et al., 2019). During evolution, B. napus and other Brassica species underwent multiple rounds of whole-genome duplication events (Chalhoub et al., 2014; Liu et al., 2014), and thus serve as an ideal polyploid model for studying polyploid genome evolution including gene family expansion and gene sequence and function divergence (Liu et al., 2014; Cheng et al., 2018). High-quality whole genome sequences of B. rapa (Zhang et al., 2018), B. oleracea (Belser et al., 2018) and B. napus (Chalhoub et al., 2014) have been published, which provide an opportunity to systematically investigate a specific gene family in B. napus.
So far, several studies have been performed to detect and analyze the UBC proteins on a genome-wide level in different plant species including 53 UBCs in sorghum (Jia et al., 2019), 57 in potato (Liu et al., 2019), 40 in longan (Jue et al., 2018), 39 in rice (E et al., 2015), 75 in maize (Jue et al., 2015), 34 in papaya (Jue et al., 2017), 59 in tomato (Sharma and Bhatt, 2017) and 74 in banana (Dong et al., 2016). However, systematic investigation of the UBCs in B. napus has been lacking. In this research, 200 BnUBCs were detected in B. napus, and their phylogenetic relationship, gene structure, conserved motif, cis-acting regulatory element, duplication pattern and target genes of miRNA were also systematically analyzed. The evolutionary history of the UBC family was explored through synteny analysis between B. napus, B. rapa, B. oleracea, and Arabidopsis. Additionally, the RNA-seq data of various tissues of B. napus were collected from publicly available databases for exploring the expression patterns of BnUBC genes. Furthermore, the genetic variations of UBC genes in germplasm from a global core collection of B. napus (Tang, 2019) were also investigated. The analysis of association mapping uncovered that some UBC genes were significantly associated with agronomic traits related to oil content and yield in B. napus. The results of this study will help us better understand the BnUBCs and lay the groundwork for future research on gene function and genetic breeding.
Genome sequence and annotation data of B. napus cv. ‘Darmor-bzh’ (v_5.0) were obtained from the Genoscope database (http://www.genoscope.cns.fr/brassicanapus/) (Chalhoub et al., 2014). B. rapa ‘Chiifu’ (v3.0) datasets were obtained from the database (https://bigd.big.ac.cn/gwh/Assembly/134/show) (Zhang et al., 2018) and B. oleracea ‘HDEM’ (broccoli) datasets were acquired from Bolbase database (http://www.ocri-genomics.org/bolbase/index.html) (Belser et al., 2018). The ubiquitin-conjugating enzyme (UQ_con) domain (PF00179) annotation file obtained from the Pfam database (https://www.ebi.ac.uk/interpro/) was applied as queries to perform HMM search against protein sequences of annotated genes using HMMER version 3.1 (http://hmmer.org/) (Eddy, 2011) with E-value cutoff of 1e-5 (Finn et al., 2011). The amino acid sequences of UBCs (predicted above) were utilized as queries to carry out BLASTP searches against the full-length protein sequences of 48 AtUBCs from The Arabidopsis Information Resource (TAIR) database (http://www.arabidopsis.org/) and 39 OsUBCs from Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/downloads_gad.shtml) with E-value < 1e-5. The putative BnUBCs with hits of both AtUBCs and OsUBCs were deployed for further confirming the existence of the UQ_con domain using the Pfam (https://www.ebi.ac.uk/interpro/), SMART (http://smart.embl-heidelberg.de/) and CDD (https://www.ncbi.nlm.nih.gov/Structure/cdd) databases. The molecular weight (MW), isoelectric point (PI), instability index, aliphatic index and grand average of hydropathicity (GRAVY) of UBC proteins, were calculated using the ProtParam tool ExPASy (https://web.expasy.org/protparam/). The subcellular location of BnUBCs was predicted using Plant-mPLoc (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/#) (Chou and Shen, 2010).
The chromosomal location, coding sequence (CDS) and amino acid sequences were determined based on the genome annotation of B. napus in the Genoscope database (http://www.genoscope.cns.fr/brassicanapus/). The chromosomal distribution of BnUBCs was graphically represented using the RIdeogram package of the R software (https://github.com/TickingClock1992/RIdeogram) (Hao et al., 2020). Multiple alignments of the BnUBC protein sequences were performed using CLUSTAL v2.1, with default parameters. A schematic diagram of BnUBCs gene structure was conducted by Gene Structure Display Server 2.0 (http://gsds.cbi.pku.edu.cn/) (Hu et al., 2015). Conserved motifs in the BnUBC proteins were investigated using the online MEME server (http://meme-suite.org/tools/meme) with the following parameters settings: the maximum number of motifs, 10; minimum width of motifs, 6; maximum width of motifs, 100 aa; and E-value < 1e-10 (Bailey et al., 2009). Moreover, the 2-kb upstream sequence of the BnUBCs gene sequences was extracted and submitted to PlantCARE (Lescot et al., 2002) for the detection of the cis-elements.
To better understand the evolutionary relationships among the UBC of B. rapa, B. oleracea, B. napus and A. thaliana, a phylogenetic analysis was performed. A phylogenetic tree was constructed applying MEGA 5.2 software (Tamura et al., 2011), based on the neighbor-joining (NJ) method and 1,000 bootstrap replications. The online software Interactive Tree Of Life (iTOL, http://itol.embl.de/) (Letunic and Bork, 2016) was used to decorate this phylogenetic tree. Genes orthologous between B. napus and its ancestors (B. rapa, B. oleracea, and A. thaliana) were identified by BLASTn searches of their CDSs based on two criteria: coverage of sequence length > 80%, and identity of aligned regions > 80% (Kong et al., 2013). In addition, DupGen_finder (https://github.com/qiao-xin/DupGen_finder) (Qiao et al., 2019) was employed to identify the modes of gene duplication of UBC paralogous genes in A. thaliana, B. napus, B. rapa and B. oleracea. The syntenic relationship among these paralogs was presented using the fmsb package of the R software.
The KaKs calculator (Wang et al., 2010) was applied to estimate the divergence between pairwise nonsynonymous substitution rates (Ka) and synonymous substitution rates (Ks). The evolutionary constraint (Ka/Ks) of UBC orthologous genes between B. napus and the other three species (A. thaliana, B. rapa and B. oleracea) was calculated according to their CDSs. In addition, to minimize errors, only the gene pairs with Ks < 1 were remained for further analysis (Guo et al., 2017; Wang T, et al., 2020).
The psRNATarget Server (Dai et al., 2018) was used to predict the miRNAs potentially targeting BnUBCs. The gene sequences of BnUBCs were submitted as candidates to search against the available sequences of reference B. napus miRNA with default arguments. A network of interactions between the predicted miRNAs and their target UBC genes in B. napus was visualized using the Cytoscape software (Shannon et al., 2003).
The publicly available RNA-seq datasets of five different B. napus tissues (callus, root, leaf, bud and silique) were collected from the NCBI SRA database (accession no. SRP136038) (Yao et al., 2020). The expression levels of BnUBCs were quantified based on their fragments per kilobase of exon per million reads mapped (FPKM) values using Cufflinks with default parameters (Trapnell et al., 2012). Furthermore, the expression levels of BnUBC genes were used to construct clustered heatmaps with TBtools (Chen et al., 2020). Then, GO enrichment analysis of BnUBC genes was performed using the clusterProfiler package of the R software.
The agronomic traits of 324 worldwide accessions comprising a natural population of B. napus were applied to detect the natural sequence variations of BnUBCs (Tang, 2019; Xie et al., 2022). Single nucleotide polymorphisms (SNPs) in BnUBCs were annotated using the SnpEff software (Cingolani et al., 2012). Moreover, agricultural traits involving the primary flowering time (PFT), full flowering time (FFT1), final flowering time (FFT2), plant height (PH), thousand seed weight (TSW), main inflorescence silique density (MISD), main inflorescence silique number (MISN), oil content (OC), protein content (PC) and main inflorescence length (MIL) were used to conduct associated mapping (Tang, 2019). The EMMAX software was applied to perform family-based association mapping analysis with a mixed linear model (Kang et al., 2010). Then, the visualization of haplotype blocks and linkage disequilibrium was conducted using the LDBlockShow software (Dong et al., 2021b). The interaction networks of B. napus proteins were obtained from the STRING database (Damian et al., 2020).
To identify the members in the UBC family, the annotation file of UBC domain obtained from the Pfam database (http://pfam.xfam.org/) was applied as a query for searching against the protein dataset of B. napus. The peptides of assumed BnUBCs showing hits of both AtUBCs and OsUBCs were further employed to confirm the presence of the UBC domains by searching in the Pfam, SMART and CDD databases. In total, 200 BnUBCs were identified in B. napus (Supplementary Table 1). The full-length transcript sequences of BnUBCs ranged from 168 bp (BnaA08g09170D) to 5,656 bp (BnaC07g05960D) with the deduced amino acid sequences varying from 55 aa (BnaA08g09170D) to 1,649 aa (BnaA07g03330D) (Supplementary Table 1). The predicted MW of the 200 deduced BnUBC proteins ranged from 6.10 kDa (BnaA08g09170D) to 184.41 kDa (BnaA07g03330D), and their GRAVY and pIs ranged from -0.904 (BnaC04g10080D) to 0.112 (BnaA08g13930D) and from 4.19 (BnaA04g11090D) to 9.57 (BnaA09g09660D), respectively (Supplementary Table 1). As previously reported in other plant species (Die et al., 2018; Zhou et al., 2018), a wide range of pIs suggested that BnUBCs function in different subcellular environments. Furthermore, the instability index of 38 of the 200 BnUBC proteins was less than 39, and these proteins were classified as stable. In addition, the prediction of their subcellular localization indicated that 172 BnUBCs were located in the nucleus, 27 in the cytoplasm and the other one in the cell membrane. In addition, to explore the evolutionary relationships among members of this family across Brassicaceae crops, 93 BraUBCs and 95 BolUBCs were identified in the reference genomes of B. rapa and B. oleracea respectively, following the same identification pipeline (Supplementary Table 2). Details of these BraUBCs and BolUBCs were listed in Supplementary Table 2.
The chromosomal positions of detected BnUBCs were drafted to corresponding chromosomes with the RIdeogram package of R software. The 200 BnUBCs were unevenly distributed across all 19 chromosomes with 97 and 103 genes located in the An and Cn subgenomes, respectively (Figure 1). The An subgenome carried, on average 9.7 BnUBCs on 10 chromosomes; A10 had the lowest number of BnUBCs (6), while A03 and A05 harbored the highest of BnUBCs (11 each). The average number of BnUBCs in the Cn subgenome was 11.4, with the lowest number (5) on C09 and the highest number on C03. Hence, no biased tendency was observed between the two subgenomes. Furthermore, on each chromosome, BnUBCs were unevenly distributed, especially, since the genes on A06 and A08 were located at the ends.
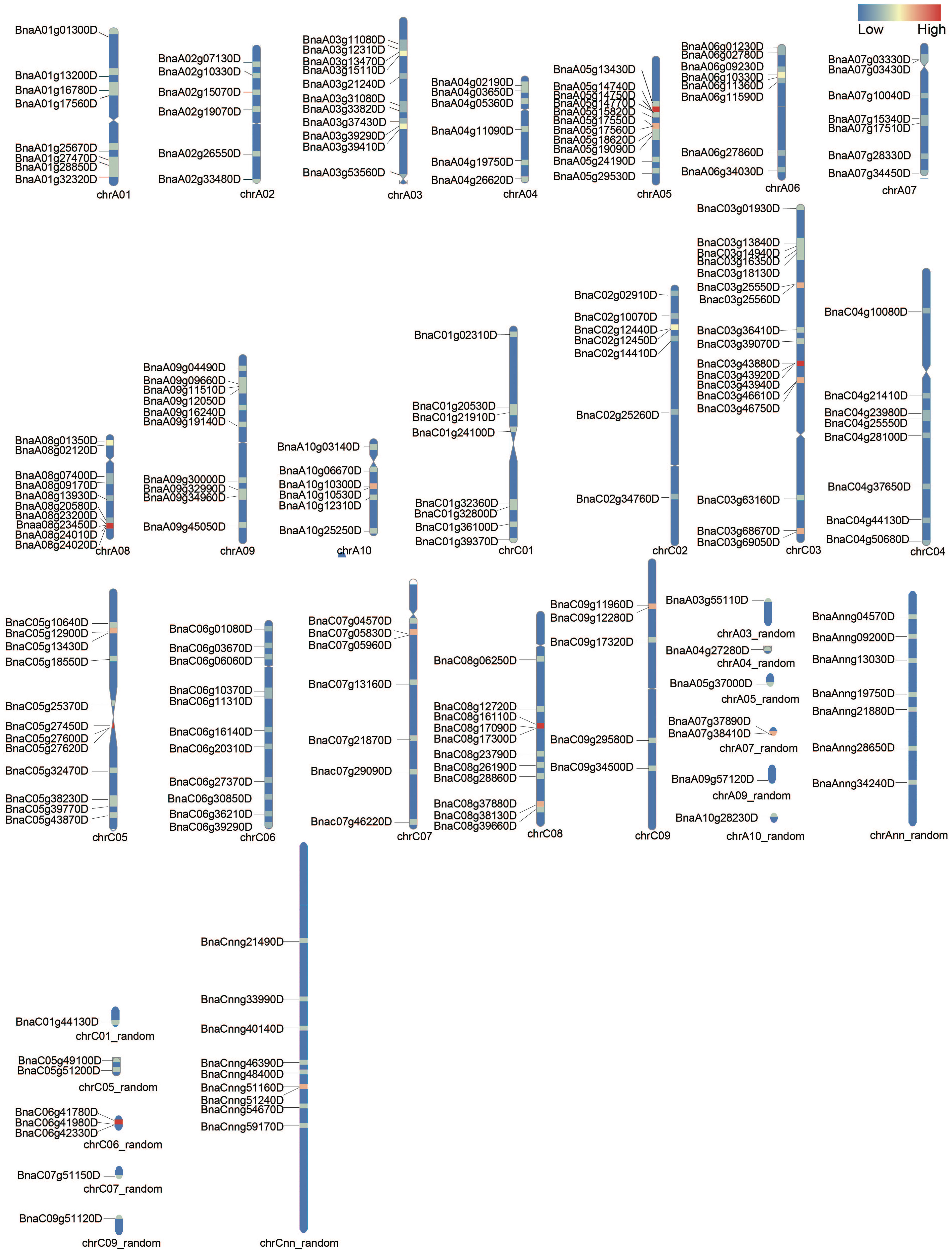
Figure 1 Chromosomal distribution of BnUBCs. The BnUBCs’ genome locations are plotted based on the location of genes, length of chromosomes, and positions of centromeres. The chromosome name is marked at the bottom of each bar. Heatmap represents the gene density (number of genes per Mb) of each chromosome.
To explore the evolutionary relationships among the UBC genes in A. thaliana, B. rapa, B. oleracea, and B. napus, a tree of phylogenetic was constructed based on the multiple sequence alignments of 48 AtUBCs, 93 BraUBCs, 95 BolUBCs, and 200 BnUBCs. All 200 BnUBCs could be categorized into 18 subfamilies and the number of groups was consistent with a previous report (Jia et al., 2019) (Figure 2). Among them, Group XVIII possessed the highest number (37) of the BnUBCss. Furthermore, group III (two BnUBCs) and group VIII (ten BnUBCs) members were involved in RUB and SUMO conjugation pathways, respectively, and other three subgroups (XVI, XV, VI) contained the members lacking the Cys active-site (UEVs).
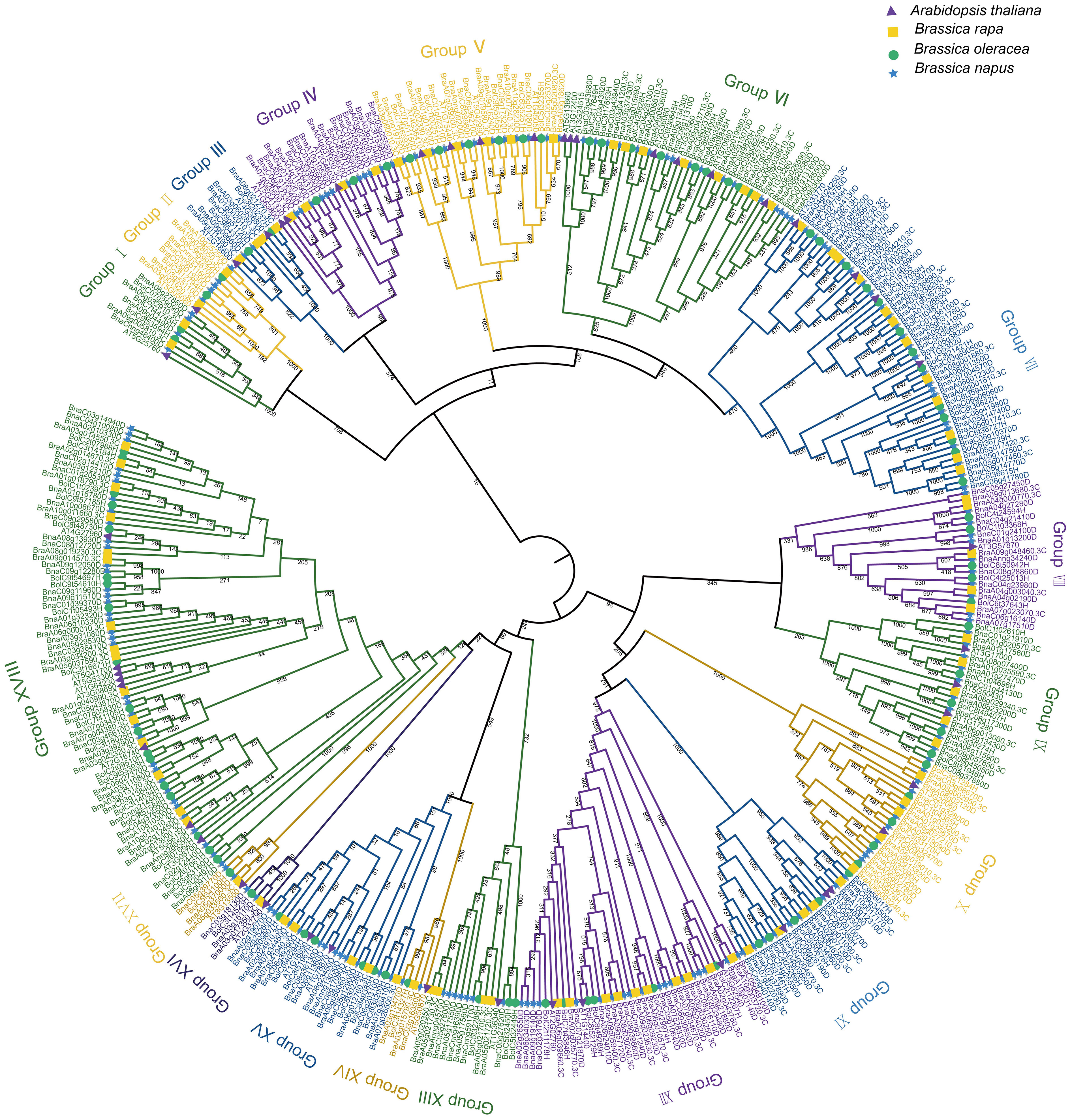
Figure 2 The phylogenetic relationship of UBC genes among A. thaliana, B. rapa, B. oleracea and B. napus. The construction of a phylogenetic tree using the UBC gene family among A. thaliana, B. rapa, B. oleracea, and B. napus. MEGA7.0 generated the phylogenetic tree topology using the neighbor-joining method with 1,000 bootstrap replicates. The members belonged to the same subgroup according to the node and branch they located and the characteristic of the tree. The different subgrouops were depicted in various colors.
Concerning the evolution of multigene families, the variety of gene structures provided an important resource (Ischoff et al., 2002; Yunpeng et al., 2016; Pellicer et al., 2018). For exploring the multiplicity of the gene structure of BnUBCs among the different groups, the intron-exon structure of BnUBCs was compared based on the phylogenetic relationships of these genes (Figure 3A, Supplementary Table 3). The results indicated that the gene architecture of BnUBCs was relatively complex (Figure 3B). Only five genes (BnaA10g10530D, BnaA05g17560D, BnaC04g10080D, BnaC07g46220D and BnaA08g09170D) contained a single intron, while the others harbored multiple introns. BnaA07g03330D contained the most introns and exons. Besides, 33 of 200 BnUBCs lacked untranslated regions (UTR) at both ends or one end because of inaccurate annotation. The number of introns among the BnUBCs ranged from 1 to 26, and the length of introns ranged from 14 to 6,632 bp. Moreover, exon numbers inBnUBCs also varied widely across distinct subfamilies, ranging from 2 to 26. However, the majority of the members within the same subfamily tended to show similar intron-exon distribution patterns. For example, members of Group X generally possessed five exons, whereas those of Group XV, harbored eight exons. Therefore, the organization of introns and exons presented valuable evidence for the phylogenetic relationship among members of this gene family.
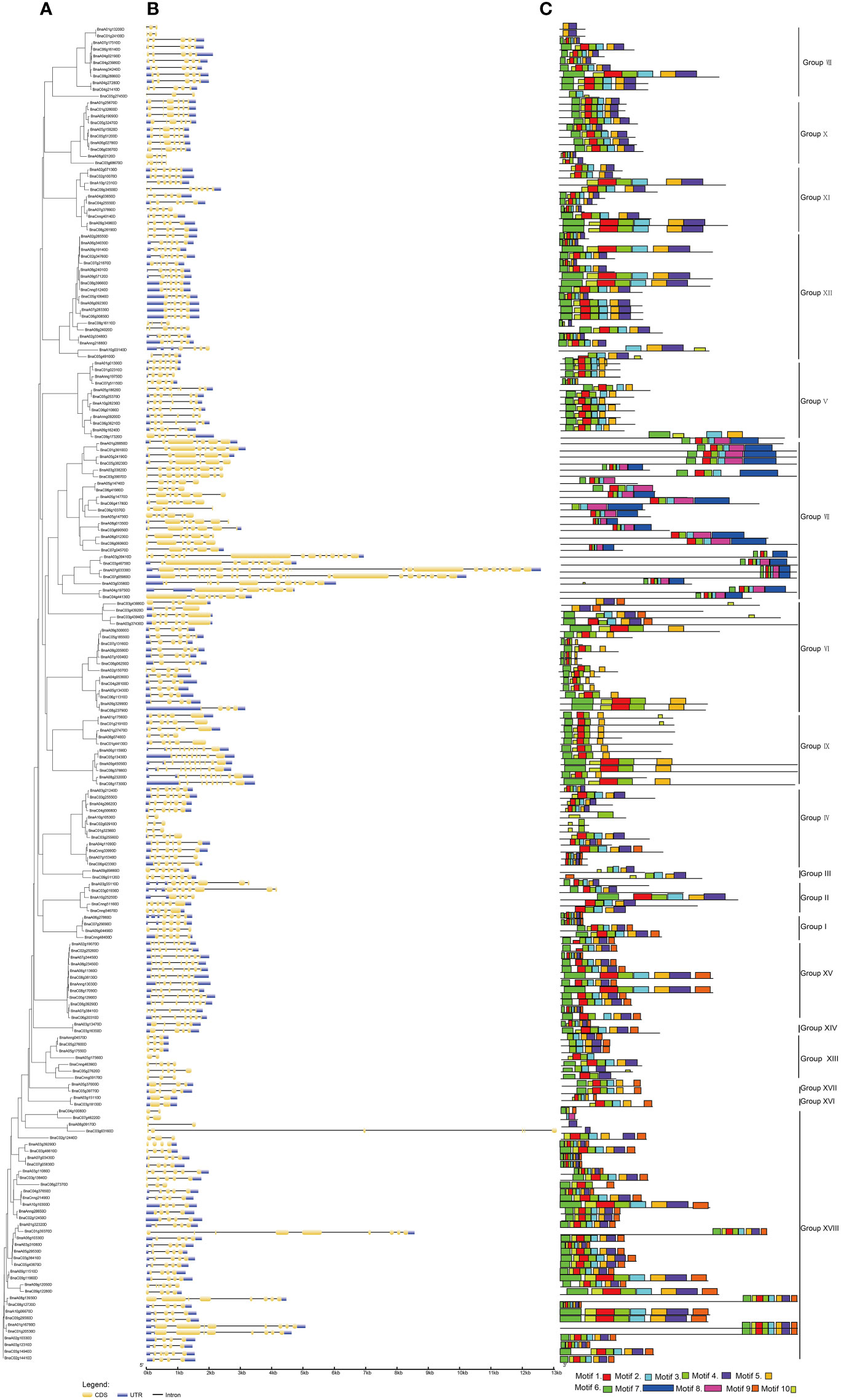
Figure 3 Gene structure and motif analysis of B. napus UBC genes according to their phylogenetic relationships. (A) Phylogenetic analysis. (B) Gene structures of BnUBCs. Yellow and blue-filled boxes represented CDS and UTR, respectively. Black lines indicated introns. (C) Conserved motifs in BnUBC proteins were detected by MEME analysis.
Furthermore, a total of 10 conserved motifs were recognized in 200 BnUBC genes (Figure 3C). Conserved motifs ranged in length from 11 to 74 aa, and among 10 motifs motif 3 as well as motif 1 were the most abundant. Moreover, the motif 1-4 were significantly matched to the UQ_con domain (Pfam: PF00179). Furthermore, a motif sequence search in the CDD database revealed that motifs 5 and 6 belonged to UBCc family. In addition, motifs 7-8 and motif 10 were matched to UBQ-conjugating_enzyme/RWD based on the search in the InterPro database. Motif composition varied across distinct subfamilies, whereas the conserved character of motif distributions in the same subcategory was similar, which emphasized their phylogenetic relationship. For example, all Group IX members possessed motif 1, motif 4-6 and motif 10, whereas in Group X, members generally contained motifs 1-6 and motif 10. In addition, three structural properties were detected and refined in BnUBC proteins (Supplementary Figure 1).
The cis-acting elements in the promoter region of a gene can modulate the initiation and efficiency of gene expression through the binding of specific transcription factors, associated with plant development, growth and stress response (Ibraheem et al., 2010). To better understand the potential functions and transcriptional regulation of BnUBCs, we isolated the sequences 2-kb upstream of the BnUBC CDSs from the B. napus genome sequence to search for cis-elements (Supplementary Table 4) (Chalhoub et al., 2014). A total of 115 functional cis-elements were detected in the promoter regions of BnUBCs (Supplementary Figure 2, Supplementary Table 5). Among these cis-elements, many were light-responsive and hormone-related related elements. Besides, majority of BnUBCs (197/200, 98.50%) contained both CAAT-box and TATA-box elements, which usually existed in eukaryon. Moreover, the MYB element, which participates in the regulation of phenylpropanoid secondary metabolism in plants was identified in 192 out of 200 BnUBCs. The MYC element was identified in 183 of the 200, (91.50%) BnUBCs, which were involved in the regulation of plant growth and development as well as resistance to environmental stress and secondary metabolite synthesis. Furthermore, many BnUBCs contained light-responsive elements such as Box 4 element (part of a conserved DNA module involved in the light response), G-box element (cis-acting regulatory element involved in the light response), GT1-motif (light responsive element), and TCT-motif (part of a light responsive element) (Supplementary Figure 2, Supplementary Table 5). Additionally, the stress-related elements, including the LTR element and TC-rich repeats were also detected in BnUBCs, suggesting that the BnUBCs played an important role in plant development and growth as well as in response to biotic and abiotic stresses.
Gene family expansion occurred mainly via the duplication of genes through whole-genome duplication (WGD), tandem duplication as well as segmental duplication events (Bodt et al., 2005; Mun et al., 2009). Moreover, the duplication of ubiquitin members through WGD events plays important role in plants (Hua et al., 2018). To further explore the evolutionary history of BnUBC gene family expansion we performed a syntenic comparison of genome sequences between A. thaliana, B. napus, B. rapa and B. oleracea. A. thaliana is the ancestor of Brassica species, whose structural genes have been identified and functionally annotated, therefore it is served as a prominent model system for the investigation of the evolutionary history of Brassica. homologous genes from inter-species comparison and paralogous genes from intra-species comparison were identified to trace the duplicated gene pairs of BnUBCs. We identified 268, 104, 86 and 29 paralogous UBC gene pairs of within B. napus, B. rapa, B. oleracea and A. thaliana, respectively (Figure 4B). Out of 286 paralogous UBC gene pairs in B. napus, 50 and 44 pairs were detected in the An subgenome, and Cn subgenome, respectively, while the remaining 174 pairs were identified across the An and Cn subgenomes. Moreover, we classified these paralogous gene pairs into five types: dispersed, proximal, tandem, WGD and transposed (Table 1, Supplementary Table 6). In B. napus, 195 genes, accounting for 97.5% of all BnUBCs, were derived from gene duplication, of which WGD (64.5%) and transposed (17.4%) gene duplication events were primarily responsible for gene family expansion (Table 1, Supplementary Table 6).
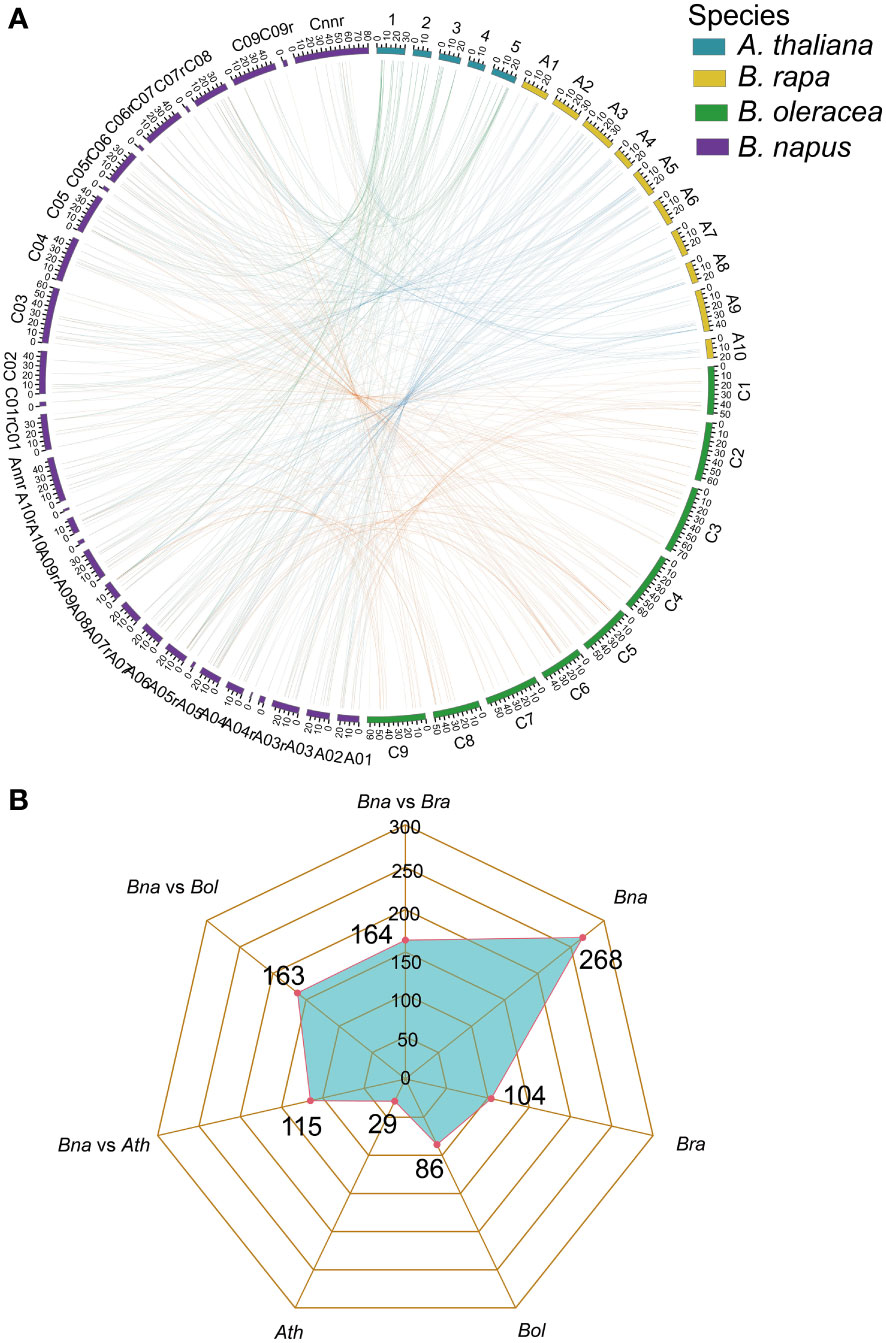
Figure 4 Genome-wide synteny analysis of UBC genes among A. thaliana, B. napus, B. rapa, and B. oleracea (A) Collinearity among the UBC genes in A. thaliana, B. rapa, B. oleracea, and B. napus. The chromosomes of A. thaliana, B. rapa, B. oleracea and B. napus were indicated with different colors corresponding to the color legend at the top right. Green lines represented the collinearity between A. thaliana and B. napus. The blue lines represented the collinearity between B. rapa and B. napus. The orange lines represented the collinearity between B. oleracea and B. napus. The visualization of the collinear correlations was performed by CIRCOS software. (B) Radar charts showed the number of orthologous and paralogous gene pairs of UBCs across four plant species.
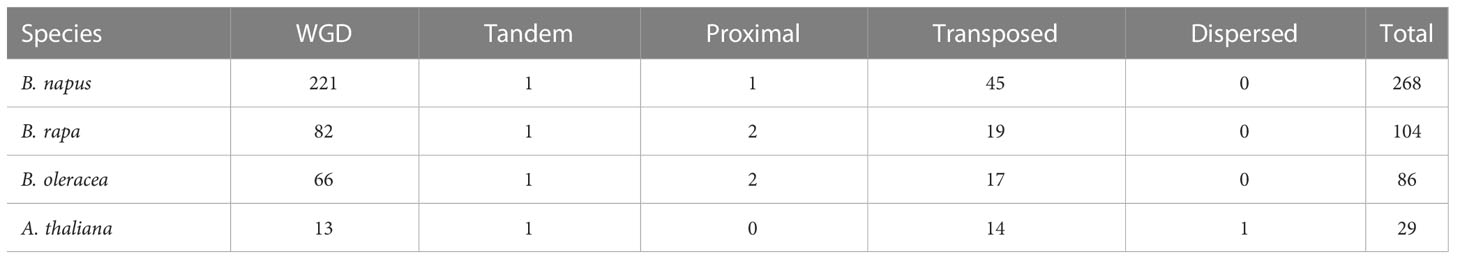
Table 1 The identification of duplicated type for UBC genes in B. napus and other three Brassicaceae species.
Furthermore, synteny comparison between B. napus and the other three species revealed that 180 BnUBCs were collinear to the orthologous genomic regions in one or more of the other three species, 164 orthologous gene pairs were identified between B. napus and B. rapa, 163 between B. napus and B. oleracea as well as 115 between B. napus and A. thaliana (Figures 4A, B, Supplementary Table 7). It was obvious that majority of the BnUBCs were inherited from their progenitors. In addition, Brassica species experienced an extra whole-genome triplication (WGT) event after divergence from A. thaliana (Feng et al., 2014). Therefore, a single A. thaliana gene was inherited as three copies in B. oleracea as well as B. rapa and as six copies in B. napus, if no gene loss occurred after the WGT event. We found that only 25.9% of the AtUBCs corresponded to six copies in B. napus, suggesting that a substantial gene loss occurred during the process of polyploidization. To estimate the selection pressure on BnUBCs, the non-synonymous to synonymous substitution ratios (Ka/Ks) of BnUBC orthologous genes to BraUBCs, BolUBCs, and AtUBCs were calculated. The Ka/Ks less than one represents purifying selection, whereas Ka/Ks equal one indicates neutral evolution and Ka/Ks more than one represents positive selection (Nekrutenko, 2002). Thus, the Ka/Ks ratio can predict the pressure of selection on each duplicated pair throughout evolution as well as their divergence time. The Ka/Ks ratios of all the BnUBCs paralogous gene pairs, except two duplicated gene pairs (BnaA07g03430D/BnaC07g05830D, BnaC02g02910D/BnaC03g25550D) were less than one, suggesting that these genes were subjected to strong purifying selection (Supplementary Table 8). Moreover, the duplicated gene pair BnaA07g03430D/BnaC07g05830D had Ka/Ks ratio considerably larger than 1, suggesting these genes were subjected to strong evolutionary pressure under positive selection. Furthermore, the Ka and Ks values of the identified orthologous gene pairs between A. thaliana and B. napus were calculated to estimate the selection pressure on orthologous gene pairs and the divergence time of the two species. The Ka/Ks ratios of all the orthologous gene pairs ranged from 0.001 to 0.25 with an average of 0.074, which indicated these genes were under purifying selection (Supplementary Table 9). The orthologous gene pairs among A. thaliana and B. napus diverged around 16 million years ago (MYA) based on the mutational rate estimate, R = 1.5 × 10-8 synonymous substitutions per site per year (Koch et al., 1999; Koch et al., 2001). This result was consistent with the previously estimated divergence time (14–24 MYA) between the A. thaliana and B. napus lineages (Koch et al., 2000; Cheung et al., 2009). In addition, the Ks values of the orthologous genes between B. napus and its progenitors were also calculated (Supplementary Table 9). The Ka/Ks ratio of the orthologous gene pairs between B. napus and B. oleracea was significantly higher than that between B. napus and B. rapa, showing that genes in the Cn subgenome underwent relatively weaker selection pressure in B. napus during the process of evolution (Figure 5). In addition, the Ka/Ks values of the orthologous gene pairs between B. napus and Arabidopsis were significantly lower than those of the orthologous gene pairs identified between B. napus and B. oleracea and between B. napus and B. rapa, indicating that orthologous gene pairs between B. napus and A. thaliana underwent intense purifying selection. Moreover, the sequences of their proteins might maintain more consistent characteristics during evolution (Figure 5).
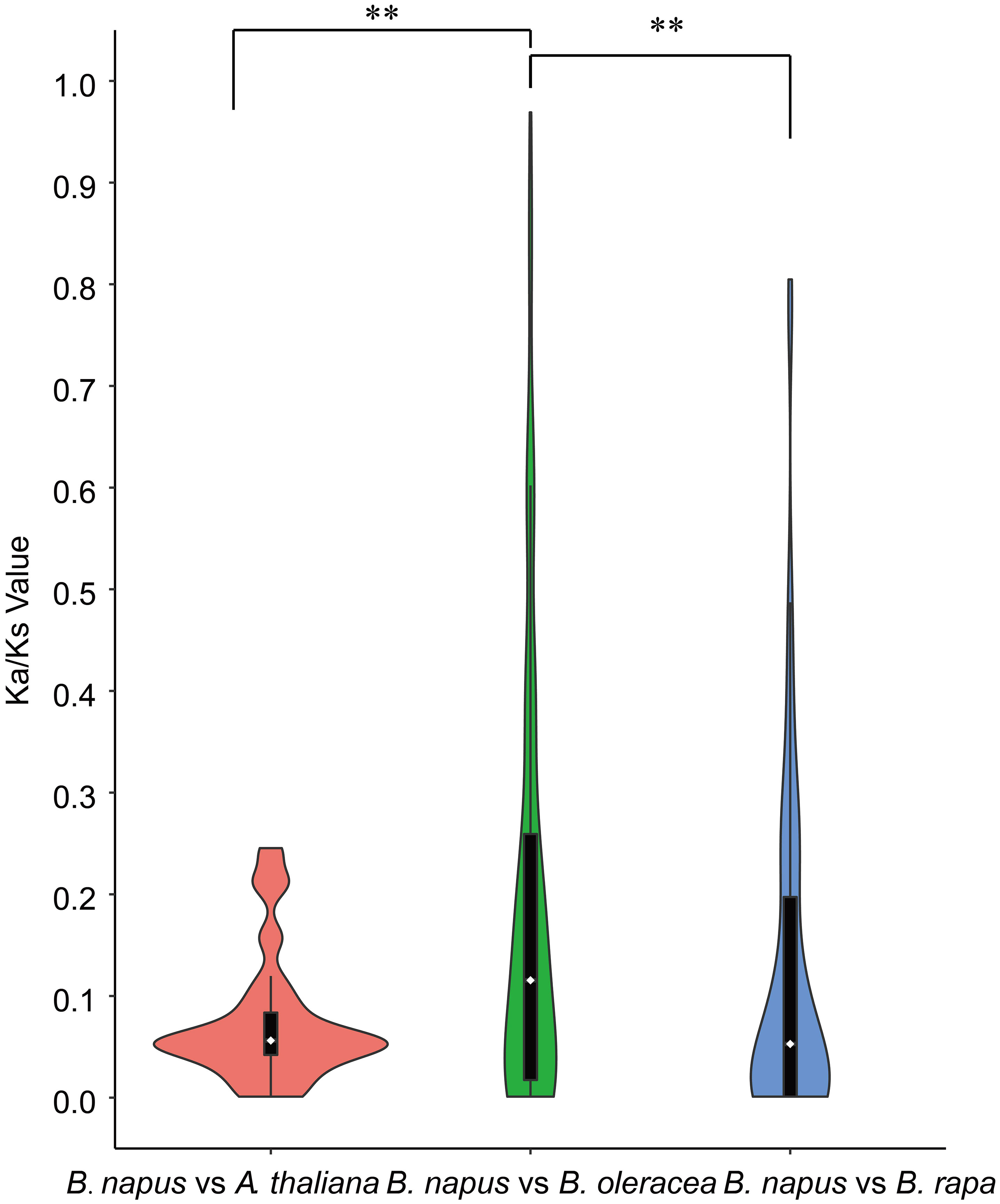
Figure 5 Violinplot of the pairwise Ka/Ks ratios of syntenic orthologous gene pairs. Only orthologous gene pairs with Ks <1 were considered. Wilcoxon Rank-Sum Tests were performed between different types of orthologous gene groups (**P < 0.01). Dotted lines within the violin plots represented the first, second, and third quartiles.
Many miRNAs were involved in regulating the function of their targeted genes in plants (Jones-Rhoades and Bartel, 2004; Yu et al., 2017; Song et al., 2019). To explore the miRNAs regulating BnUBCs, 106 BnUBC targeting genes of 71 putative miRNAs were identified, and their relationship network was constructed by Cytoscape software (Figure 6, Supplementary Table 10). It was found that BnaC06g03670D was targeted by the B. napus miRNA169 family based on an interaction network. A recent study showedthat miR169 regulated the function of the anaphase-promoting complex/cyclosome (APC/C), an essential ubiquitin-protein ligase, by targeting DUO POLLEN1 (DUO1). DUO1 upregulated the expression level of APC/C, which stimulates the production of miR159 (Zheng et al., 2011). Moreover, members of the miRNA156 family were found to regulate most of the BnUBCs. In a recent study, overexpression of miR156 inhibited gibberellic acid (GA)-induced and ubiquitination-mediated degradation of DELLA (Jerome Jeyakumar et al., 2020). In addition, the remaining BnUBCs were presumably targeted by members of the other 26 miRNA families (Figure 6).

Figure 6 Schematic representation of the interaction network between the miRNAs and their putative BnUBC targets.
Several studies reported that members of the UBC family mediated crosstalk among diverse signaling pathways for various abiotic stress responses (Gao et al., 2017; Jue et al., 2018; Jia et al., 2019). Besides, UBCs were predicted to play an important role in protein and ion binding (Liu et al., 2019). To predict the putative functions of BnUBCs, GO enrichment analysis was performed. The GO terms were divided into three categories: biological process (BP), molecular function (MF), and cellular component (CC) (Supplementary Table 11). The majority of enriched GO terms, such as postreplication repair, belonged to the BP category (Supplementary Figure 3). The remaining GO terms were broadly associated with vegetative growth (negative regulation of flower development, root epidermal cell differentiation), response to stress (response to iron ion, cellular response to water deprivation and response to gibberellin), cell growth and protein process. The CC category included UBC13−MMS2 complex and the perinuclear region of the cytoplasm. Moreover, various MF were detected in this analysis, which included acid-amino acid ligase activity, SUMO transferase activity, ubiquitin protein ligase binding and endopeptidase activity. The results of GO enrichment analysis suggested that BnUBCs play crucial roles in plant development, growth, cell protein quality regulation and response to stress, consistent with the findings of previous studies (Dreher and Callis, 2007; Sadanandom et al., 2012; Jue et al., 2018).
Moreover, the expression profiles of BnUBC genes were examined across five major tissues (callus, leaf, root, bud and silique) based on the previously published RNA-seq datasets of B. napus (Yao et al., 2020). The expression levels of BnUBCs were estimated with FPKM and displayed as a heatmap (Figure 7A). A total of 38 BnUBCs showed relatively weak expression (FPKM < 1) and 24 BnUBCs were silience in any of the five tissues. The remaining BnUBCs showed high expression levels (FPKM ≥ 1), the majority of which expressed at a specific organ or differentially expressed in various tissues (Figure 7B). BnaC06g16140D (ortholog of AT3G57870) presented the highest expression in all tissues, suggesting its critical roles in plant growth. Genes showing low expression levels (FPKM <1) across all five tissues could be regarded as pseudogenes (Figure 7, Supplementary Table 12). Notably, in our study, most of BnUBCs exhibited relatively lower expression levels in the leaf than in other tissues (Supplementary Table 12).
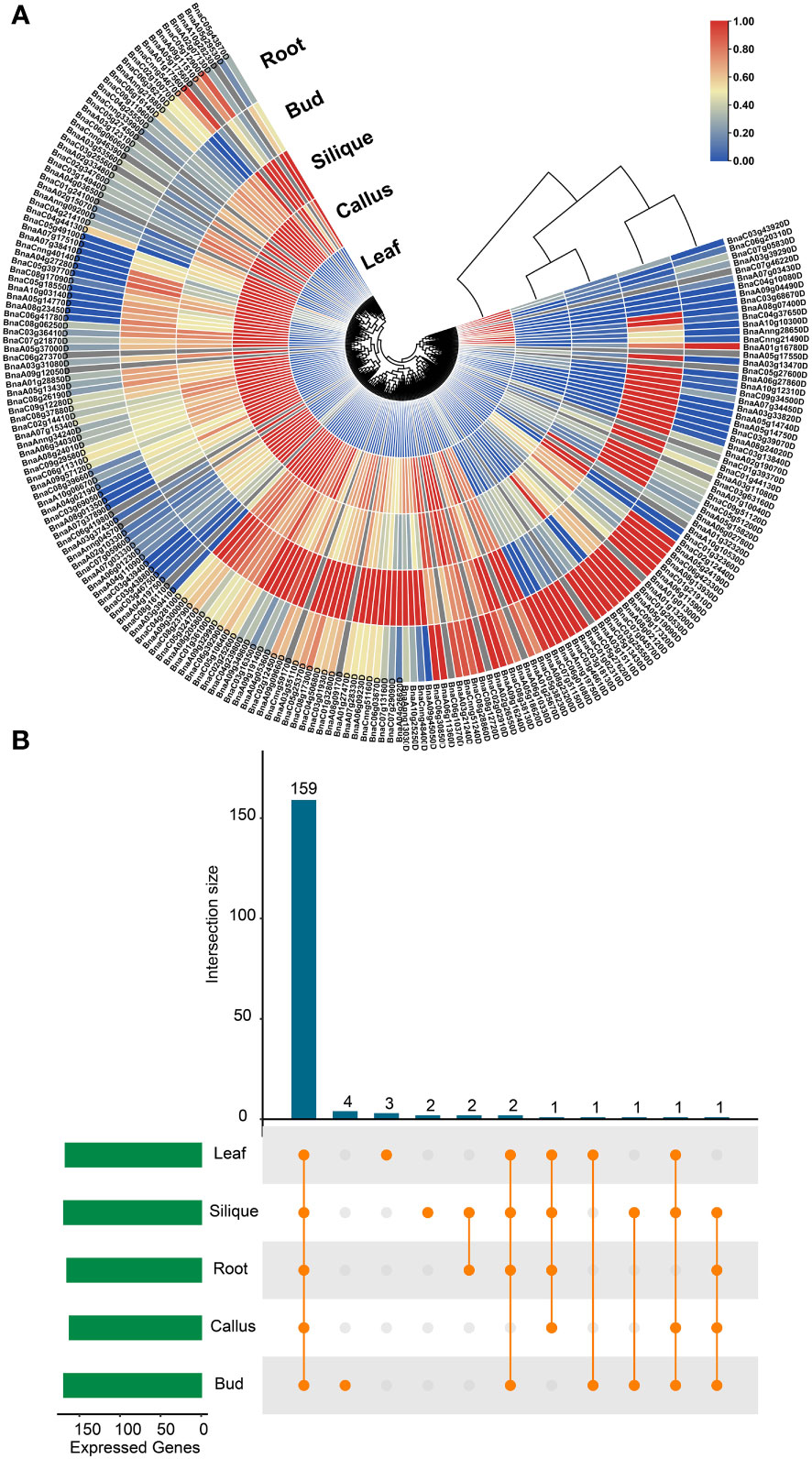
Figure 7 Expression profiles of BnUBCs across various tissues. (A) Expression data were represented by values of FPKM, and different colors showed different expression levels. (B) Number of BnUBCs expressed across various tissues.
Furthermore, the expression divergence between the duplicated genes in B. napus investigated. For example, BnaA06g10330D was silence across all tissues, whereas its paralog BnaA03g31080D presented a high expression level in various tissues. Similarly, BnaA04g02190D expressed in all tissues, however, its paralogs BnaA01g13200D and BnaC01g24100D didn’t express in any tissue. All these results could provide a clue for the functional divergence between duplicated gene pairs in BnUBCs.
SNPs in a natural population with 324 B. napus accessions collected from worldwide countries were identified (Tang, 2019) (Supplementary Table 13) for investigating the genetic variants of BnUBCs in germplasm. Each BnUBC gene contained 34 SNPs on average, which was close to the 36 SNPs in each gene across the entire genome. Taking genome size into account, the number of SNP in each kilobase (kb) among the BnUBCs was calculated (17 SNPs/kb), whereas 13 SNPs/kb for the genes across entire genome. BnUBCs in the An subgenome contained slightly higher SNP density (20 SNPs/kb) than that in the Cn subgenome (14 SNPs/kb). Moreover, variations in SNP number between some paralogous gene pairs of BnUBCs were observed. For example, BnaA03g11080D possessed 65 SNPs, however, its paralog BnaC02g12440D contained no SNP. Moreover, the paralogs BnaA07g03330D/BnaC03g46750D, contained 235 and 33 SNPs, respectively. Based on the paired t-test, no significant difference was detected in SNP density between the BnUBC paralogs. Additionally, SNP annotation presented that 1,671 SNPs existed in the exon regions of BnUBCs and 600 SNPs lead to missense mutations.
Ubiquitination is a well-characterized post-translational that regulates plant growth and developmental processes contributing to diverse phenotypes, and affect agronomic traits (Dreher and Callis, 2007; Vierstra, 2009; Sadanandom et al., 2012; Linden and Callis, 2020; Yu and Hua, 2022). Association mapping analysis was performed to explore the impact of BnUBCs on agronomic phenotype. In total, 96 SNPs in 33 BnUBC genes (Supplementary Table 13) were significantly associated with the investigated agronomic traits (p < 0.001). Moreover, BnaC02g25260D was significantly associated with plant height (Figure 8A, B). Furthermore, the two genotypes of accessions were divided according to the most significantly associated SNP of BnaC02g25260D, and the plant height between the two genotypes was statistically significant according to the t-test (p < 2.7 e–4) (Figure 8C). The protein interaction network of BnaC02g25260D was obtained from STRING (Damian et al., 2020) (Figure 8D) and the interacted genes were enriched in the regulation of embryonic development (GO:0045995), positive regulation of cell size (GO:0045793), cellulose biosynthetic process (GO:0030244), circadian rhythm (GO:0007623), positive regulation of flower development (GO:0009911), detection of visible light (GO:0009584), leaf development (GO:0048366), regulation of hormone levels (GO:0010817) and chlorophyll biosynthetic process (GO:0015995) (Figure 8E). Overall, the above categories were related to plant development and growth, the regulation of hormones and oil content, which affected the plant height and the cell size in seed formation (Wang X, et al., 2020; Dong et al., 2021a). Furthermore, SNPs distributed in BnaAnng34240D were significantly associated with flowering time and silique density (Supplementary Figure 4A-D, G-H). The protein interaction networks of BnaAnng34240D were also obtained from STRING (Supplementary Figure 5A), and its interacted proteins enriched in the protein modification process, regulation of transcription and so on (Supplementary Figure 5B). Besides, BnaA07g28330D was significantly associated with TSW, which affect the crop yield (Supplementary Figure 4E, F). Furthermore, we found that BnaA07g28330D exhibited a higher expression level in the silique of high oil content accession than in that in the silique of low oil content accession (https://bnaomics.ocri-genomics.net/tools/exp-view/index.php) (Tang, 2019).
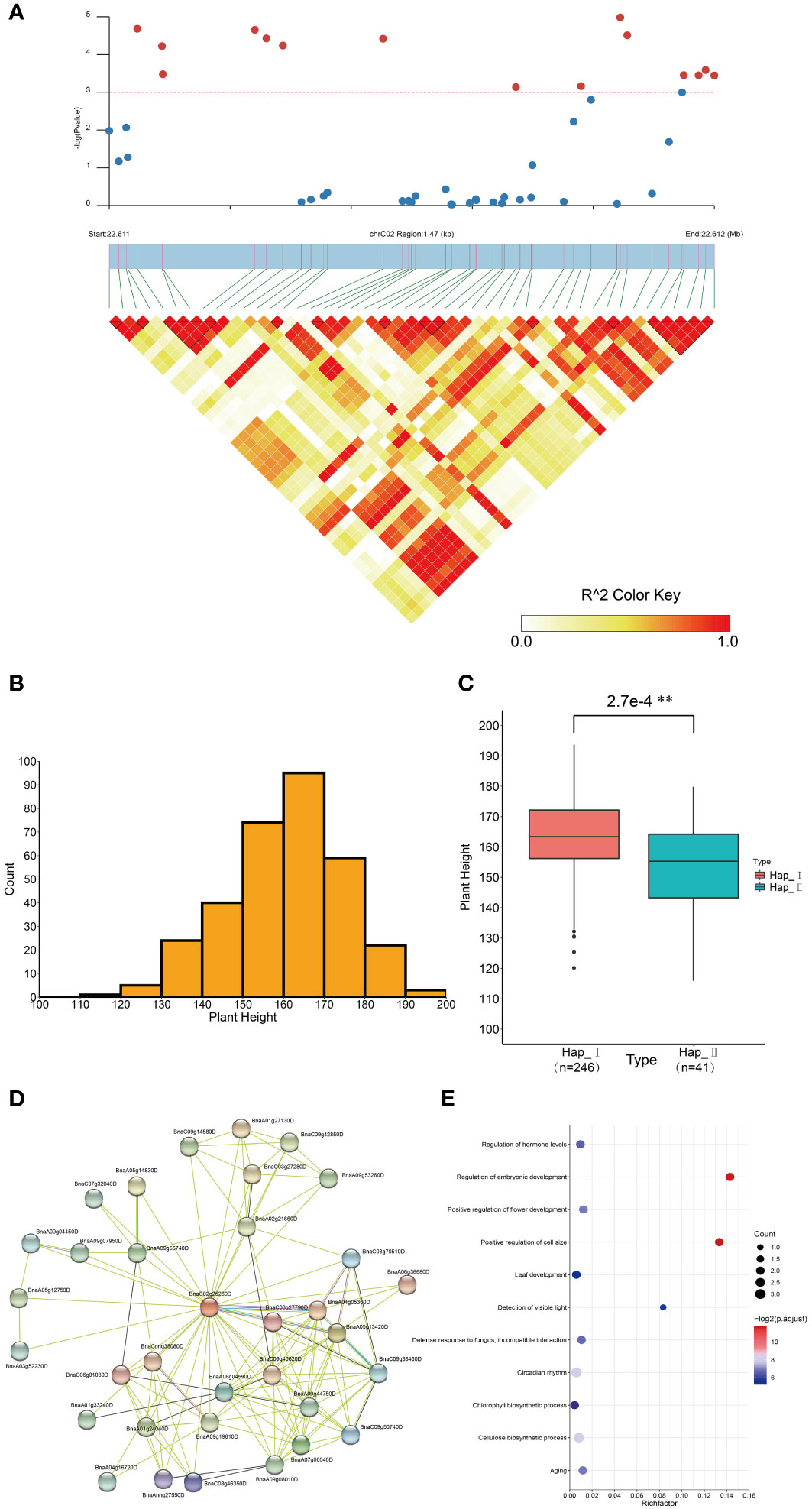
Figure 8 Association mapping analysis of BnaC02g25260D in B. napus germplasm with 324 core collections. (A) Significant association of BnaC02g25260D with tplant height. (B) Distribution of the plant height of 324 accessions. (C) Box plot showed the comparison of plant height between two haplotypes divided based on the most significantly associated SNP in BnaC04g00810D. (D) Protein-protein interaction network of BnaC02g25260D. (E) GO enrichment analysis of BnaC02g25260D interacting proteins. **p < 0.01.
Ubiquitin-conjugating enzymes transfer ubiquitin from ubiquitin-activating enzymes to ubiquitin ligases, which is a key step in protein ubiquitination (Bae and Kim, 2013). In plants, UBC proteins are involved in multiple crucial processes, including growth, development and abiotic stress response (Welchman et al., 2005; Dreher and Callis, 2007; Sadanandom et al., 2012). However, only a few members of the UBC family have been characterized in plants such as Arabidopsis (Kraft et al., 2005), rice (E et al., 2015), maize (Jue et al., 2015) and tomato (Sharma and Bhatt, 2017) on the genome-wide level. B. napus is a valuable oil crop and an ideal polyploid model for studying the evolution, domestication and genetics of polyploids. However, a systematical investigation of the BnUBC family s has not been conducted to date. In this study, the availability of the B. napus whole-genome sequence provides opportunities for characterizing the BnUBC family and revealing its genetic effects on agronomic traits.
In this study, 200 UBC genes including two RUB genes, ten SUMO and 31 UEV genes, encoding UBC domain-containing ubiquitin-conjugating enzymes were detected in B. napus. The number of UBC gene in A. thaliana, maize, tomato, rice and B. napus (48, 75, 59, 39 and 200, respectively) are not correlated with the genome size (~125, ~466, ~2300, ~900, and ~825 Mb, respectively). Increasing evidence suggest that segmental duplications may be the main factor responsible for the expansion of gene families in plants (Wang et al., 2015; Liu et al., 2017; Wang et al., 2017). The loss and gain of genes during the process of polyploidization determine the genetic variability, which ultimately affects the function of the protein (Albalat and Cañestro, 2016). Analysis of gene structures and motifs of BnUBCs revealed that variation in the number of exons and introns as well as motif constitution may lead to their functional diversity.
Cis-acting elements in promoter regions regulate the transcription of genes that participate in various physiological processes (responses to hormone stress, plant growth and development) and that form the basic functional links among the complex regulatory networks (Nuruzzaman et al., 2014). Abundant cis-acting elements which were light-responsive and related to the hormone, plant growth and development were detected in the promoter regions of BnUBCs. Two cis-acting elements (TATA-box and CAAT-box) were presented in most BnUBCs (Supplementary Figure 2). Some cis-acting elements widely distributed in the eukaryotes, such as TATA-box and CAAT-box were also found in BnUBCs. These cis-acting elements constitute the RNA transcription factors binding sites (Laloum et al., 2013) and regulate the gene transcription process in conjunction with its binding factor. Moreover, other cis-acting elements such as MYB, MYC, ARE and ABRE, which are related to plant growth and development, were also found in the majority of BnUBCs. For example, MYB transcription factors function in plant breeding and response to stresses (Li et al., 2019) and MYC proteins function as transcriptional activators in abscisic acid (ABA) signaling (Abe et al., 2003). Besides, the abscisic-responsive element (ARE) cis-acting element is necessary for the anaerobic induction and the ABA-responsive element (ABRE) cis-acting element functioned in the response to ABA stress (Narusaka et al., 2003). Furthermore, many cis-acting elements involved in response to light were found in the BnUBC promoter regions, suggesting their potential role in regulating the pathways associated with light responsiveness. Overall, the cis-acting elements detected in BnUBC promoters suggested that they played crucial roles in plant growth, development and responses to abiotic stress and light.
Phylogenetic analysis divided BnUBC proteins into 18 groups, which is similar to the previous studies (Kraft et al., 2005; Dong et al., 2016; Sharma and Bhatt, 2017). In addition, the UBC domains showed uniform distribution in all the UBCs and gene expansion caused the variation of this core element during the evolution. Group XVIII possessed the largest number of BnUBCs with the most variation in gene structure and motifs, indicating that members in this group evolved more diverse functions than the other groups. Moreover, the phylogenetic classification of BnUBCs was further supported by the analyses of gene architecture and conserved motifs. Additionally, most of the members in the same subgroup shared similar gene structures and conserved motifs, but showed obvious different in cis-acting regulatory elements (Supplementary Figure 2), indicating their potential function diversity (Zou et al., 2011; Oudelaar and Higgs, 2021). Furthermore, the phylogenetic relationships of AtUBCs and BnUBCs suggested that gene duplication was likely a major factor in the diversification of BnUBC genes during evolution.
Gene duplication is considered a major mechanism leading to gene family expansion and functional diversification (Lynch and Conery, 2000; Prince and Pickett, 2002; Bianconi et al., 2018). In plants, three types of genome duplication have been reported, including WGD, tandem duplication and chromosomal segmental duplication (Ramsey and Schemske, 1998). According to many previous studies, WGD and segmental duplication are important factors for genome duplication and gene expansion (Ma et al., 2017; Wu et al., 2018; Zhu et al., 2020). After divergence from the ancestor of Arabidopsis lineage, Brassica species underwent WGT of approximately 13 MYA. Allotetraploid B. napus was formed by the interspecific hybridization between B. rapa and B. oleracea (Allender and King, 2010; Chalhoub et al., 2014). Thus, the genome size of B. napus was expanded and a single gene in A. thaliana corresponded to six copies in B. napus (Lysak et al., 2005). According to collinearity analysis, WGD showed a large contribution to genome expansion in Brassica species, followed by transposed duplication (Table 1). In this study, the number of BnUBCs (200) was approximately 4-fold higher than that of AtUBCs, while the number of BraUBC and BolUBC genes was <3-fold higher than that of AtUBCs, indicating gene loss in Brassica species during evolution (Albalat and Cañestro, 2016). Moreover, the numbers of BnUBCs in the An and Cn subgenomes were 97 and 103, respectively, which was almost consistent with that in the genomes of its progenitors B. rapa and B. oleracea genomes. These results demonstrate that most gene loss of UBCs occurred after whole genome triplication in Brassica ancestors and BnUBCs were mainly inherited from their progenitors (Chalhoub et al., 2014). Notably, several orthologs of AtUBCs (AT3G57870, AT1G14400, AT5G56150, AT1G23260, AT1G17280, AT2G36060 and AT3G08690) maintained six copies in B. napus and were not loss after WGD, implying their crucial roles in plant growth and development. In B. napus, the Ka/Ks ratios between UBC paralogs were obviously less than one, indicating that these genes were undergone purifying selection during evolution.
The majority of BnUBCs localized to the nucleus, and only a few localized to the cytoplasm (Supplementary Table 1). This is consistent with a previous study, which showed their existence in the nuclear region for their involvement in fruit ripening (Wang et al., 2014). Moreover, these findings were also consistent with the results of GO enrichment analysis (Supplementary Figure 3). GO enrichment analyses predicted that BnUBCs are mainly involved in ubiquitination, cell growth and response to stresses (Supplementary Figure 3, Supplementary Table 11). However, the functional analysis of BnUBCs had been lacking. Therefore, we analyzed the expression profiles of these BnUBCs in various tissues (bud, callus, root, silique, and leaf) to predict their possible functions. The RNA-seq data (Yao et al., 2020) showed that the expression levels of BnUBCs varied across various tissues, consistent with previous studies (Cui et al., 2012; Jeon et al., 2012; E et al., 2015; Jue et al., 2018). For instance, AtUBC1 and AtUBC2 expressed in root, leaf, flower and seedling (Xu et al., 2009) And their orthologs in B. napus showed relatively high expression in bud, root and silique. The average expression level of BnUBCs was low in the leaf and the least number of genes expressed in callus (Figure 7, Supplementary Table 12). Moreover, diverse expression patterns were found between several duplicated genes, suggesting their divergence through subfunctionalization, neofunctionalization or pseudogenization in B. napus polyploidization (Chaudhary et al., 2009). Moreover, the difference of cis-acting regulatory elements in the promoter regions between BnUBCs may contribute to their expression level and function divergences.
To reveal the genetic effects of BnUBCs on agronomic traits, SNPs of BnUBCs were detected in accessions comprising a B. napus natural population (Tang, 2019) (Supplementary Table 13). The average SNP density of BnUBCs (17 SNPs/kb) was slightly higher than that of genes in the entire genome (13 SNPs/kb), suggesting that a large number of polymorphisms accumulated in BnUBCs during evolution. The SNP density of BnUBCs in the An subgenome was higher than that in the Cn subgenome, which is consistent with some other gene families in B. napus (Zhu et al., 2020; Wahid et al., 2022; Xie et al., 2022). Classically, the difference in genetic variations between paralogs may lead to subfunctionalization, pseudogenization or neofunctionalization (Schiessl et al., 2017). Several duplicated BnUBC gene pairs, such as BnaA03g11080D/BnaC02g12440D and BnaA07g03330D/BnaC03g46750D, showed significant different in the number of SNPs. Differences were also observed in the expression levels of these duplicated genes, implying their functional divergence. Furthermore, association mapping analysis of BnUBCs, based on SNPs, revealed their genetic effects on agronomic traits. For example, BnaC02g25260D was significantly associated with plant height (Figure 8). According to the results of GO enrichment analysis, BnaC02g25260D -interacting protein were likely involved in the regulation of plant cell growth, hormone levels and chlorophyll biosynthetic process, and could eventually influence plant height (Wang X, et al., 2020; Dong et al., 2021a). In addition, BnaAnng34240D was significantly associated with agronomic traits such as flowering time and main inflorescence silique density (Supplementary Figure 4). Ubiquitination, as reported in previous studies, is one of the crucial mechanisms that control the photoperiodic regulation of floral organ development (Sadanandom et al., 2012; Piñeiro and Jarillo, 2013). Moreover, the interacting proteins of BnaAnng34240D were involved in protein modification, regulation of transcription and so on. AtSUMO1/2 and S AtSCE1a (ortholog of BnaAnng34240D) are implicated in ABA responses and the ubiquitin-like SUMO protease 1 (ULP1) gene AtESD4 is involved in flowering time regulation (Miura et al., 2007). In addition, BnaA07g28330D was significantly associated with TSW, an important trait affecting crop yield. The obviously different expression levels of BnaA07g28330D between high and low oil content accessions in silique, implying its effects on oil content. Thus, the results of this analysis in the present study provided a valuable resource including candidate UBC genes affecting agronomic traits in B. napus.
To date, the UBC family were reported in several plant species. In this study, the characteristics of the BnUBC were compared with those of the UBC family in other plant species to help broaden our understanding of the differences or similarities in E2s across species. When compared with other species, this family in B. napus was largest and contained most subgroups suggesting the complexity of the allotetraploid genome. Moreover, majority of the UBCs were derived from WGD in B. napus and Vitis vinifera (Gao et al., 2017), whereas by segmental duplication in rice (E et al., 2015), tomato (Sharma and Bhatt, 2017), maize (Jue et al., 2015) and banana (Dong et al., 2016). The physicochemical properties of BnUBCs were similar to those in potato (Liu et al., 2019), maize (Jue et al., 2015) and banana (Dong et al., 2016). Additionally, and the chromosomal locations of the BnUBCs predicted in this study were partially consistent with previous reports (Miura et al., 2011; Cui et al., 2012; Cheng et al., 2017).
Our study focused on the UBC family, which represents only a part of the UPS, which generally contains the sequential action of E1, E2, and E3 enzymes. To gain better insight into the regulatory functions of the UPS, we compared our results with the other family in UPS. WGD played a predominant role in the expansion of the ubiquitin family (Hua et al., 2018), which is consistent with BnUBCs. Different from the similarity of physicochemical properties within the UBC family across species, significant sequence diversities were discovered between Poaceae and Brassicaceae (Hua and Yu, 2019). Only two members of the E1 family in A. thaliana were detected with experimental verification (Hatfield et al., 1997). In addition, these two genes did not exhibit different expression patterns and the encoded E1 proteins showed no significant difference in enzymatic activities. However, the BnUBCs differentially expressed in various tissues and displayed various gene structures and motifs between different subgroups. With regard to the E3 family, it can be divided into three types according to their subunit composition and functional mechanism, including homologous to E6-AP carboxyl terminus (HECT), Really Interesting New Gene (RING) and U- box type and cullin-RING ligase (CRL) (Vierstra, 2009). Thus, the E3 family was more complicated than the UBC family based on the classification of its domains. The U-box type E3 family contains 79 members in maize (Li et al., 2022), 68 in sorghum (Fang et al., 2022) and 62 in tomato (Sharma and Taganna, 2020); this family is slightly larger in size than the E2 family. Furthermore, the E3 genes are involved in plant development, growth and abiotic stress response, which is consistent with the functions of E2 genes reported previously (Sharma and Taganna, 2020; Wang et al., 2021; Fang et al., 2022; Li et al., 2022; Lin et al., 2022).
In the present research, we conducted a comprehensive investigation of the UBC family in B. napus. A total of 200 BnUBCs were identified and classified into 18 groups. The gene structures and motifs were highly conserved among members of the same phylogenetic subgroup. Moreover, cis-acting elements found in the BnUBCs promoters and the expression patterns of BnUBCs in diverse tissues demonstrated that these genes play a crucial role in plant growth and development. In addition, synteny analysis of the UBCs between B. napus and three ancestors (A. thaliana, B. oleracea and B. rapa) revealed the expansion history of the BnUBC gene family. Furthermore, genetic variations identification and association mapping analyses of BnUBCs uncovered their potential genetic effects on agronomic traits related to oil content and yield in B. napus. Overall, this study provided useful information about BnUBCs and will facilitate functional studies as well as the genetic breeding of B. napus in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
SY, CT, and XY designed the research. SY, HW, XL and PH performed the experiments. SY, MX, MH and XC analyzed the data. SY, CT, and XY wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
The project was supported by the National Natural Science Foundation of China (nos 32201220, 31770250) and the Young Top-notch Talent Cultivation Program of Hubei Province for CB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1118339/full#supplementary-material
Abe, H., Urao, T., Ito, T., Seki, M., Shinozaki, K., Yamaguchi-Shinozaki, K. (2003). Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15 (1), 63–78. doi: 10.1105/tpc.006130
Albalat, R., Cañestro, C. (2016). Evolution by gene loss. Nat. Rev. Genet. 17 (7), 379–391. doi: 10.1038/nrg.2016.39
Allender, C. J., King, G. J. (2010). Origins of the amphiploid species Brassica napus l. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 10, 54. doi: 10.1186/1471-2229-10-54
Arrigoni, A., Grillo, B., Vitriolo, A., De Gioia, L., Papaleo, E. (2012). C-terminal acidic domain of ubiquitin-conjugating enzymes: a multi-functional conserved intrinsically disordered domain in family 3 of E2 enzymes. J. Struct. Biol. 178 (3), 245–259. doi: 10.1016/j.jsb.2012.04.003
Aung, K., Lin, S. I., Wu, C. C., Huang, Y. T., Su, C. L., Chiou, T. J. (2006). pho2, a phosphate overaccumulator, is caused by a nonsense mutation in a microRNA399 target gene. Plant Physiol. 141 (3), 1000–1011. doi: 10.1104/pp.106.078063
Bae, H., Kim, W. T. (2013). The n-terminal tetra-peptide (IPDE) short extension of the U-box motif in rice SPL11 E3 is essential for the interaction with E2 and ubiquitin-ligase activity. Biochem. Biophys. Res. Commun. 433 (2), 266–271. doi: 10.1016/j.bbrc.2013.03.005
Bae, H., Kim, W. T. (2014). Classification and interaction modes of 40 rice E2 ubiquitin-conjugating enzymes with 17 rice ARM-u-box E3 ubiquitin ligases. Biochem. Biophys. Res. Commun. 444 (4), 575–580. doi: 10.1016/j.bbrc.2014.01.098
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37 (Web Server issue), W202–W208. doi: 10.1093/nar/gkp335
Belser, C., Istace, B., Denis, E., Dubarry, M., Baurens, F. C., Falentin, C., et al. (2018). Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 4 (11), 879–887. doi: 10.1038/s41477-018-0289-4
Bianconi, M. E., Dunning, L. T., Moreno-Villena, J. J., Osborne, C. P., Christin, P. A. (2018). Gene duplication and dosage effects during the early emergence of C4 photosynthesis in the grass genus alloteropsis. J. Exp. Bot. 69 (8), 1967–1980. doi: 10.1093/jxb/ery029
Bodt, S. D., Maere, S., Peer, Y. (2005). Genome duplication and the origin of angiosperms. Trends Ecol. Evol. 20 (11), 591–597. doi: 10.1016/j.tree.2005.07.008
Burroughs, A. M., Jaffee, M., Iyer, L. M., Aravind, L. (2008). Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J. Struct. Biol. 162 (2), 205–218. doi: 10.1016/j.jsb.2007.12.006
Callis, J. (2014). The ubiquitination machinery of the ubiquitin system. Arabidopsis Book 12, e0174. doi: 10.1199/tab.0174
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Plant genetics. early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 345 (6199), 950–953. doi: 10.1126/science.1253435
Chaudhary, B., Flagel, L., Stupar, R. M., Udall, J. A., Verma, N., Springer, N. M., et al. (2009). Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (gossypium). Genetics 182 (2), 503–517. doi: 10.1534/genetics.109.102608
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, M. C., Kuo, W. C., Wang, Y. M., Chen, H. Y., Lin, T. P. (2017). UBC18 mediates ERF1 degradation under light-dark cycles. New Phytol. 213 (3), 1156–1167. doi: 10.1111/nph.14272
Cheng, F., Wu, J., Cai, X., Liang, J., Freeling, M., Wang, X. (2018). Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4 (5), 258–268. doi: 10.1038/s41477-018-0136-7
Cheung, F., Trick, M., Drou, N., Lim, Y. P., Park, J. Y., Kwon, S. J., et al. (2009). Comparative analysis between homoeologous genome segments of Brassica napus and its progenitor species reveals extensive sequence-level divergence. Plant Cell 21 (7), 1912–1928. doi: 10.1105/tpc.108.060376
Chou, K. C., Shen, H. B. (2010). Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PloS One 5 (6), e11335. doi: 10.1371/journal.pone.0011335
Christensen, D. E., Brzovic, P. S., Klevit, R. E. (2007). E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol. 14 (10), 941–948. doi: 10.1038/nsmb1295
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6 (2), 80–92. doi: 10.4161/fly.19695
Criqui, M. C., de Almeida Engler, J., Camasses, A., Capron, A., Parmentier, Y., Inzé, D., et al. (2002). Molecular characterization of plant ubiquitin-conjugating enzymes belonging to the UbcP4/E2-C/UBCx/UbcH10 gene family. Plant Physiol. 130 (3), 1230–1240. doi: 10.1104/pp.011353
Cui, F., Liu, L., Zhao, Q., Zhang, Z., Li, Q., Lin, B., et al. (2012). Arabidopsis ubiquitin conjugase UBC32 is an ERAD component that functions in brassinosteroid-mediated salt stress tolerance. Plant Cell 24 (1), 233–244. doi: 10.1105/tpc.111.093062
Dai, X., Zhuang, Z., Zhao, P. X. (2018). psRNATarget: a plant small RNA target analysis server, (2017 release). Nucleic Acids Res. 46 (W1), W49–W54. doi: 10.1093/nar/gky316
Damian, S., Gable, A. L., Nastou, K. C., David, L., Rebecca, K., Sampo, P., et al. (2020). The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49 (D1), D605-D612. doi: 10.1093/nar/gkaa1074
Die, J. V., Gil, J., Millan, T. (2018). Genome-wide identification of the auxin response factor gene family in cicer arietinum. BMC Genomics 19 (1). doi: 10.1186/s12864-018-4695-9
Dong, Z., Alam, M. K., Xie, M., Yang, L., Liu, S. (2021a). Mapping of a major QTL controlling plant height using a high-density genetic map and QTL-seq methods based on whole-genome resequencing in Brassica napus. G3-Genes Genomes Genet. 11 (7), jkab118. doi: 10.1093/g3journal/jkab118
Dong, S. S., He, W. M., Ji, J. J., Zhang, C., Yang, T. L. (2021b). LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Briefings Bioinf. 22 (4), bbaa227. doi: 10.1101/2020.06.14.151332
Dong, C., Hu, H., Jue, D., Zhao, Q., Chen, H., Xie, J., et al. (2016). The banana E2 gene family: Genomic identification, characterization, expression profiling analysis. Plant Sci. 245, 11–24. doi: 10.1016/j.plantsci.2016.01.003
Dong, B., Rengel, Z., Delhaize, E. (1998). Uptake and translocation of phosphate by pho2 mutant and wild-type seedlings of Arabidopsis thaliana. Planta 205 (2), 251–256. doi: 10.1007/s004250050318
Doroodian, P., Hua, Z. (2021). The ubiquitin switch in plant stress response. Plants (Basel) 10 (2). doi: 10.3390/plants10020246
Dreher, K., Callis, J. (2007). Ubiquitin, hormones and biotic stress in plants. Ann. Bot. 99 (5), 787–822. doi: 10.1093/aob/mcl255
E, Z., Zhang, Y., Li, T., Wang, L., Zhao, H. (2015). Characterization of the ubiquitin-conjugating enzyme gene family in rice and evaluation of expression profiles under abiotic stresses and hormone treatments. PloS One 10 (4), e0122621. doi: 10.1371/journal.pone.0122621
Eddy, S. R. (2011). Accelerated profile HMM searches. PloS Comput. Biol. 7 (10), e1002195. doi: 10.1371/journal.pcbi.1002195
Fang, Y., Du, Q., Yang, Q., Jiang, J., Hou, X., Yang, Z., et al. (2022). Identification, characterization, and expression profiling of the putative U-box E3 ubiquitin ligase gene family in Sorghum bicolor. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.942302
Fang, S., Weissman, A. M. (2004). A field guide to ubiquitylation. Cell Mol. Life Sci. 61 (13), 1546–1561. doi: 10.1007/s00018-004-4129-5
Feng, C., Jian, W., Wang, X. (2014). Genome triplication drove the diversification of brassica plants. Horticult Res. 1, 14024. doi: 10.1038/hortres.2014.24
Finn, R. D., Clements, J., Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39 (Web Server issue), W29–W37. doi: 10.1093/nar/gkr367
Gao, Y., Wang, Y., Xin, H., Li, S., Liang, Z. (2017). Involvement of ubiquitin-conjugating enzyme (E2 gene family) in ripening process and response to cold and heat stress of Vitis vinifera. Sci. Rep. 7 (1), 13290. doi: 10.1038/s41598-017-13513-x
Glickman, M. H., Ciechanover, A. (2002). The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82 (2), 373–428. doi: 10.1152/physrev.00027.2001
Guo, Y., Liu, J., Zhang, J., Liu, S., Du, J. (2017). Selective modes determine evolutionary rates, gene compactness and expression patterns in Brassica. Plant J. 91 (1), 34–44. doi: 10.1111/tpj.13541
Hao, Z., Lv, D., Ge, Y., Shi, J., Weijers, D., Yu, G., et al. (2020). RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 6, e251. doi: 10.7717/peerj-cs.251
Hatfield, P. M., Gosink, M. M., Carpenter, T. B., Vierstra, R. D. (1997). The ubiquitin-activating enzyme (E1) gene family in Arabidopsis thaliana. Plant J. 11 (2), 213–226. doi: 10.1046/j.1365-313x.1997.11020213.x
Hershko, A., Heller, H., Elias, S., Ciechanover, A. (1983). Components of ubiquitin-protein ligase system. resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258 (13), 8206–8214.
Hodson, C., Purkiss, A., Miles, J. A., Walden, H. (2014). Structure of the human FANCL RING-Ube2T complex reveals determinants of cognate E3-E2 selection. Structure 22 (2), 337–344. doi: 10.1016/j.str.2013.12.004
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31 (8), 1296–1297. doi: 10.1093/bioinformatics/btu817
Hua, Z., Doroodian, P., Vu, W. (2018). Contrasting duplication patterns reflect functional diversities of ubiquitin and ubiquitin-like protein modifiers in plants. Plant J. 95 (2), 296–311. doi: 10.1111/tpj.13951
Hua, Z., Yu, P. (2019). Diversifying evolution of the ubiquitin-26S proteasome system in brassicaceae and poaceae. Int. J. Mol. Sci. 20 (13). doi: 10.3390/ijms20133226
Huang, L., Kinnucan, E., Wang, G., Beaudenon, S., Howley, P. M., Huibregtse, J. M., et al. (1999). Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science 286 (5443), 1321–1326. doi: 10.1126/science.286.5443.1321
Ibraheem, O., Botha, C. E., Bradley, G. (2010). In silico analysis of cis-acting regulatory elements in 5' regulatory regions of sucrose transporter gene families in rice (Oryza sativa japonica) and Arabidopsis thaliana. Comput. Biol. Chem. 34 (5-6), 268–283. doi: 10.1016/j.compbiolchem.2010.09.003
Ischoff, E. B., Barale, J. C., Mercereaupuijalon, O. (2002). Three multigene families in plasmodium parasites: facts and questions. Int. J. Parasitol. 32 (11), 1323–1344. doi: 10.1016/s0020-7519(02)00111-x
Jeon, E. H., Pak, J. H., Kim, M. J., Kim, H. J., Shin, S. H., Lee, J. H., et al. (2012). Ectopic expression of ubiquitin-conjugating enzyme gene from wild rice, OgUBC1, confers resistance against UV-b radiation and botrytis infection in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 427 (2), 309–314. doi: 10.1016/j.bbrc.2012.09.048
Jerome Jeyakumar, J. M., Ali, A., Wang, W. M., Thiruvengadam, M. (2020). Characterizing the role of the miR156-SPL network in plant development and stress response. Plants (Basel) 9 (9). doi: 10.3390/plants9091206
Jia, L., Zhao, Q., Chen, S. (2019). Evolution and expression analysis of the sorghum ubiquitin-conjugating enzyme family. Funct. Plant Biol. 46 (3), 236–247. doi: 10.1071/FP18184
Jones-Rhoades, M. W., Bartel, D. P. (2004). Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 14 (6), 787–799. doi: 10.1016/j.molcel.2004.05.027
Jue, D., Sang, X., Liu, L., Shu, B., Wang, Y., Xie, J., et al. (2018). The ubiquitin-conjugating enzyme gene family in longan (Dimocarpus longan lour.): Genome-wide identification and gene expression during flower induction and abiotic stress responses. Molecules 23 (3). doi: 10.3390/molecules23030662
Jue, D., Sang, X., Lu, S., Dong, C., Zhao, Q., Chen, H., et al. (2015). Genome-wide identification, phylogenetic and expression analyses of the ubiquitin-conjugating enzyme gene family in maize. PloS One 10 (11), e0143488. doi: 10.1371/journal.pone.0143488
Jue, D., Sang, X., Shu, B., Liu, L., Wang, Y., Jia, Z., et al. (2017). Characterization and expression analysis of genes encoding ubiquitin conjugating domain-containing enzymes in carica papaya. PloS One 12 (2), e0171357. doi: 10.1371/journal.pone.0171357
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B., et al. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42 (4), 348–354. doi: 10.1038/ng.548
Kim, D. Y., Scalf, M., Smith, L. M., Vierstra, R. D. (2013). Advanced proteomic analyses yield a deep catalog of ubiquitylation targets in Arabidopsis. Plant Cell 25 (5), 1523–1540. doi: 10.1105/tpc.112.108613
Koch, M., Bishop, J., Mitchell-Olds, T. (1999). Molecular systematics and evolution of arabidopsis and Arabis. Plant Biol. (Stuttg) 1 (05), 529–537. doi: 10.1111/j.1438-8677.1999.tb00779.x
Koch, M. A., Haubold, B., Mitchell-Olds, T. (2000). Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 17 (10), 1483–1498. doi: 10.1093/oxfordjournals.molbev.a026248
Koch, M., Haubold, B., Mitchell-Olds, T. (2001). Molecular systematics of the brassicaceae: evidence from coding plastidic matK and nuclear chs sequences. Am. J. Bot. 88 (3), 534–544. doi: 10.2307/2657117
Kong, X., Lv, W., Jiang, S., Zhang, D., Cai, G., Pan, J., et al. (2013). Genome-wide identification and expression analysis of calcium-dependent protein kinase in maize. BMC Genomics 14 (433). doi: 10.1186/1471-2164-14-433
Kraft, E., Stone, S. L., Ma, L., Su, N., Gao, Y., Lau, O. S., et al. (2005). Genome analysis and functional characterization of the E2 and RING-type E3 ligase ubiquitination enzymes of arabidopsis. Plant Physiol. 139 (4), 1597–1611. doi: 10.1104/pp.105.067983
Kravic, B., Behrends, C., Meyer, H. (2020). Regulation of lysosome integrity and lysophagy by the ubiquitin-conjugating enzyme UBE2QL1. Autophagy 16 (1), 179–180. doi: 10.1080/15548627.2019.1687217
Laloum, T., De Mita, S., Gamas, P., Baudin, M., Niebel, A. (2013). CCAAT-box binding transcription factors in plants: Y so many? Trends Plant Sci. 18 (3), 157–166. doi: 10.1016/j.tplants.2012.07.004
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Peer, Y., et al. (2002). PlantCARE, a database of plant cis -acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30 (1), 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44 (W1), W242–W245. doi: 10.1093/nar/gkw290
Li, J., Han, G., Sun, C., Sui, N. (2019). Research advances of MYB transcription factors in plant stress resistance and breeding. Plant Signal Behav. 14 (8), 1613131. doi: 10.1080/15592324.2019.1613131
Li, W., Schmidt, W. (2010). A lysine-63-linked ubiquitin chain-forming conjugase, UBC13, promotes the developmental responses to iron deficiency in Arabidopsis roots. Plant J. 62 (2), 330–343. doi: 10.1111/j.1365-313X.2010.04150.x
Li, X., Zhu, L., Wu, Z., Chen, J., Wang, T., Zhang, X., et al. (2022). Classification and expression profile of the U-box E3 ubiquitin ligase enzyme gene family in maize (Zea mays l.). Plants (Basel) 11 (19). doi: 10.3390/plants11192459
Lin, Y., Hwang, W. C., Basavappa, R. (2002). Structural and functional analysis of the human mitotic-specific ubiquitin-conjugating enzyme, UbcH10. J. Biol. Chem. 277 (24), 21913–21921. doi: 10.1074/jbc.M109398200
Lin, W. Y., Lin, S. I., Chiou, T. J. (2009). Molecular regulators of phosphate homeostasis in plants. J. Exp. Bot. 60 (5), 1427–1438. doi: 10.1093/jxb/ern303
Lin, Z., Nie, H., Zhang, Y., Yin, Z., Yan, X. (2022). Genome-wide identification and analysis of HECT E3 ubiquitin ligase gene family in Ruditapes philippinarum and their involvement in the response to heat stress and vibrio anguillarum infection. Comp. Biochem. Physiol. Part D Genomics Proteomics 43, 101012. doi: 10.1016/j.cbd.2022.101012
Linden, K. J., Callis, J. (2020). The ubiquitin system affects agronomic plant traits. J. Biol. Chem. 295 (40), 13940–13955. doi: 10.1074/jbc.REV120.011303
Liu, S., Liu, Y., Yang, X., Tong, C., Edwards, D., Parkin, I. A., et al. (2014). The brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930. doi: 10.1038/ncomms4930
Liu, W., Tang, X., Zhu, X., Qi, X., Zhang, N., Si, H. (2019). Genome-wide identification and expression analysis of the E2 gene family in potato. Mol. Biol. Rep. 46 (1), 777–791. doi: 10.1007/s11033-018-4533-9
Liu, C., Xie, T., Chen, C., Luan, A., Long, J., Li, C., et al. (2017). Genome-wide organization and expression profiling of the R2R3-MYB transcription factor family in pineapple (Ananas comosus). BMC Genomics 18 (1), 503. doi: 10.1186/s12864-017-3896-y
Liu, F., Zhu, C., Gao, P., Zheng, S., Li, C. (2020). Ubiquitin-conjugating enzyme E2T regulates cell proliferation and migration in cholangiocarcinoma. Anticancer Drugs 31 (8), 836–846. doi: 10.1097/CAD.0000000000000955
Lu, C., Napier, J. A., Clemente, T. E., Cahoon, E. B. (2011). New frontiers in oilseed biotechnology: meeting the global demand for vegetable oils for food, feed, biofuel, and industrial applications. Curr. Opin. Biotechnol. 22 (2), 252–259. doi: 10.1016/j.copbio.2010.11.006
Lu, K., Wei, L., Li, X., Wang, Y., Wu, J., Liu, M., et al. (2019). Whole-genome resequencing reveals brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 10 (1), 1154. doi: 10.1038/s41467-019-09134-9
Lynch, M., Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290 (5494), 1151–1155. doi: 10.1126/science.290.5494.1151
Lysak, M. A., Koch, M. A., Pecinka, A., Schubert, I. (2005). Chromosome triplication found across the tribe brassiceae. Genome Res. 15 (4), 516–525. doi: 10.1101/gr.3531105
Ma, J. Q., Jian, H. J., Yang, B., Lu, K., Zhang, A. X., Liu, P., et al. (2017). Genome-wide analysis and expression profiling of the GRF gene family in oilseed rape (Brassica napus l.). Gene 620, 36–45. doi: 10.1016/j.gene.2017.03.030
Michelle, C., Vourc'h, P., Mignon, L., Andres, C. R. (2009). What was the set of ubiquitin and ubiquitin-like conjugating enzymes in the eukaryote common ancestor? J. Mol. Evol. 68 (6), 616–628. doi: 10.1007/s00239-009-9225-6
Millyard, L., Lee, J., Zhang, C., Yates, G., Sadanandom, A. (2016). The ubiquitin conjugating enzyme, TaU4 regulates wheat defence against the phytopathogen Zymoseptoria tritici. Sci. Rep. 6, 35683. doi: 10.1038/srep35683
Miura, K., Jin, J. B., Hasegawa, P. M. (2007). Sumoylation, a post-translational regulatory process in plants. Curr. Opin. Plant Biol. 10 (5), 495–502. doi: 10.1016/j.pbi.2007.07.002
Miura, K., Sato, A., Ohta, M., Furukawa, J. (2011). Increased tolerance to salt stress in the phosphate-accumulating Arabidopsis mutants siz1 and pho2. Planta 234 (6), 1191–1199. doi: 10.1007/s00425-011-1476-y
Mun, J. H., Kwon, S. J., Yang, T. J., Seol, Y. J., Jin, M. (2009). Genome-wide comparative analysis of the brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol. 10 (10), R111. doi: 10.1186/gb-2009-10-10-r111
Narusaka, Y., Nakashima, K., Shinwari, Z. K., Sakuma, Y., Furihata, T., Abe, H., et al. (2003). Interaction between two cis-acting elements, ABRE and DRE, in ABA-dependent expression of Arabidopsis rd29A gene in response to dehydration and high-salinity stresses. Plant J. 34 (2), 137–148. doi: 10.1046/j.1365-313x.2003.01708.x
Nekrutenko, A. (2002). The K A/K s ratio test for assessing the protein-coding potential of genomic regions: An empirical and simulation study. Genome Res. 12 (1), 198–202. doi: 10.1101/gr.200901
Nuruzzaman, M., Sharoni, A. M., Satoh, K., Kumar, A., Leung, H., Kikuchi, S. (2014). Comparative transcriptome profiles of the WRKY gene family under control, hormone-treated, and drought conditions in near-isogenic rice lines reveal differential, tissue specific gene activation. J. Plant Physiol. 171 (1), 2–13. doi: 10.1016/j.jplph.2013.09.010
Oudelaar, A. M., Higgs, D. R. (2021). The relationship between genome structure and function. Nat. Rev. Genet. 22 (3), 154–168. doi: 10.1038/s41576-020-00303-x
Ozkan, E., Yu, H., Deisenhofer, J. (2005). Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 102 (52), 18890–18895. doi: 10.1073/pnas.0509418102
Papaleo, E., Casiraghi, N., Arrigoni, A., Vanoni, M., Coccetti, P., De Gioia, L. (2012). Loop 7 of E2 enzymes: an ancestral conserved functional motif involved in the E2-mediated steps of the ubiquitination cascade. PloS One 7 (7), e40786. doi: 10.1371/journal.pone.0040786
Pellicer, J., Hidalgo, O., Dodsworth, S., Leitch, I. J. (2018). Genome size diversity and its impact on the evolution of land plants. Genes (Basel) 9 (2), 88. doi: 10.3390/genes9020088
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Piñeiro, M., Jarillo, J. A. (2013). Ubiquitination in the control of photoperiodic flowering. Plant Sci. 198, 98–109. doi: 10.1016/j.plantsci.2012.10.005
Poyurovsky, M. V., Priest, C., Kentsis, A., Borden, K. L., Pan, Z. Q., Pavletich, N., et al. (2007). The Mdm2 RING domain c-terminus is required for supramolecular assembly and ubiquitin ligase activity. EMBO J. 26 (1), 90–101. doi: 10.1038/sj.emboj.7601465
Prince, V. E., Pickett, F. B. (2002). Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet. 3 (11), 827–837. doi: 10.1038/nrg928
Qiao, X., Li, Q., Yin, H., Qi, K., Li, L., Wang, R., et al. (2019). Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20 (1), 38. doi: 10.1186/s13059-019-1650-2
Ramadan, A., Nemoto, K., Seki, M., Shinozaki, K., Takeda, H., Takahashi, H., et al. (2015). Wheat germ-based protein libraries for the functional characterisation of the Arabidopsis E2 ubiquitin conjugating enzymes and the RING-type E3 ubiquitin ligase enzymes. BMC Plant Biol. 15, 275. doi: 10.1186/s12870-015-0660-9
Ramsey, J., Schemske, D. W. (1998). Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 29 (1), 467–501. doi: 10.1146/annurev.ecolsys.29.1.467
Rana, D., van den Boogaart, T., O'Neill, C. M., Hynes, L., Bent, E., Macpherson, L., et al. (2004). Conservation of the microstructure of genome segments in Brassica napus and its diploid relatives. Plant J. 40 (5), 725–733. doi: 10.1111/j.1365-313X.2004.02244.x
Sadanandom, A., Bailey, M., Ewan, R., Lee, J., Nelis, S. (2012). The ubiquitin-proteasome system: central modifier of plant signalling. New Phytol. 196 (1), 13–28. doi: 10.1111/j.1469-8137.2012.04266.x
Schiessl, S., Huettel, B., Kuehn, D., Reinhardt, R., Snowdon, R. (2017). Post-polyploidisation morphotype diversification associates with gene copy number variation. Sci. Rep. 7, 41845. doi: 10.1038/srep41845
Schumacher, F. R., Wilson, G., Day, C. L. (2013). The n-terminal extension of UBE2E ubiquitin-conjugating enzymes limits chain assembly. J. Mol. Biol. 425 (22), 4099–4111. doi: 10.1016/j.jmb.2013.06.039
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504. doi: 10.1101/gr.1239303
Sharma, B., Bhatt, T. K. (2017). Genome-wide identification and expression analysis of E2 ubiquitin-conjugating enzymes in tomato. Sci. Rep. 7 (1), 8613. doi: 10.1038/s41598-017-09121-4
Sharma, B., Taganna, J. (2020). Genome-wide analysis of the U-box E3 ubiquitin ligase enzyme gene family in tomato. Sci. Rep. 10 (1), 9581. doi: 10.1038/s41598-020-66553-1
Silver, E. T., Gwozd, T. J., Ptak, C., Goebl, M., Ellison, M. J. (1992). A chimeric ubiquitin conjugating enzyme that combines the cell cycle properties of CDC34 (UBC3) and the DNA repair properties of RAD6 (UBC2): implications for the structure, function and evolution of the E2s. EMBO J. 11 (8), 3091–3098. doi: 10.1002/j.1460-2075.1992.tb05381.x
Smalle, J., Vierstra, R. D. (2004). The ubiquitin 26S proteasome proteolytic pathway. Annu. Rev. Plant Biol. 55, 555–590. doi: 10.1146/annurev.arplant.55.031903.141801
Song, X., Li, Y., Cao, X., Qi, Y. (2019). MicroRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 70, 489–525. doi: 10.1146/annurev-arplant-050718-100334
Sun, L., Chen, Z. J. (2004). The novel functions of ubiquitination in signaling. Curr. Opin. Cell Biol. 16 (2), 119–126. doi: 10.1016/j.ceb.2004.02.005
Sun, F., Fan, G., Hu, Q., Zhou, Y., Guan, M., Tong, C., et al. (2017). The high-quality genome of Brassica napus cultivar 'ZS11' reveals the introgression history in semi-winter morphotype. Plant J. 92 (3), 452–468. doi: 10.1111/tpj.13669
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28 (10), 2731–2739. doi: 10.1093/molbev/msr121
Tang, M. (2019). Population genome variations and subgenome asymmetry in brassica napus l (Huazhong: Huazhong Agricultural University).
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat. Protoc. 7 (3), 562–578. doi: 10.1038/nprot.2012.016
van Wijk, S. J., Timmers, H. T. (2010). The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J. 24 (4), 981–993. doi: 10.1096/fj.09-136259
Vierstra, R. D. (2009). The ubiquitin-26S proteasome system at the nexus of plant biology. Nat. Rev. Mol. Cell Biol. 10 (6), 385–397. doi: 10.1038/nrm2688
Wahid, S., Xie, M., Sarfraz, S., Liu, J., Zhao, C., Bai, Z., et al. (2022). Genome-wide identification and analysis of Ariadne gene family reveal its genetic effects on agronomic traits of brassica napus. Int. J. Mol. Sci. 23 (11). doi: 10.3390/ijms23116265
Wang, S., Cao, L., Wang, H. (2016). Arabidopsis ubiquitin-conjugating enzyme UBC22 is required for female gametophyte development and likely involved in Lys11-linked ubiquitination. J. Exp. Bot. 67 (11), 3277–3288. doi: 10.1093/jxb/erw142
Wang, T., Hu, J., Ma, X., Li, C., Yang, Q., Feng, S., et al. (2020). Identification, evolution and expression analyses of whole genome-wide TLP gene family in Brassica napus. BMC Genomics 21 (1), 264. doi: 10.1186/s12864-020-6678-x
Wang, W., Jiang, W., Liu, J., Li, Y., Gai, J., Li, Y. (2017). Genome-wide characterization of the aldehyde dehydrogenase gene superfamily in soybean and its potential role in drought stress response. BMC Genomics 18 (1), 518. doi: 10.1186/s12864-017-3908-y
Wang, C., Song, B., Dai, Y., Zhang, S., Huang, X. (2021). Genome-wide identification and functional analysis of U-box E3 ubiquitin ligases gene family related to drought stress response in Chinese white pear (Pyrus bretschneideri). BMC Plant Biol. 21 (1), 235. doi: 10.1186/s12870-021-03024-3
Wang, Y., Wang, W., Cai, J., Zhang, Y., Qin, G., Tian, S. (2014). Tomato nuclear proteome reveals the involvement of specific E2 ubiquitin-conjugating enzymes in fruit ripening. Genome Biol. 15 (12), 548. doi: 10.1186/s13059-014-0548-2
Wang, Y., Wang, Q., Zhao, Y., Han, G., Zhu, S. (2015). Systematic analysis of maize class III peroxidase gene family reveals a conserved subfamily involved in abiotic stress response. Gene 566 (1), 95–108. doi: 10.1016/j.gene.2015.04.041
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., Yu, J. (2010). KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinf. 8 (1), 77–80. doi: 10.1016/s1672-0229(10)60008-3
Wang, X., Zheng, M., Liu, H., Zhang, L., Hua, W. (2020). Fine-mapping and transcriptome analysis of a candidate gene controlling plant height in Brassica napus l. Biotechnol. Biofuels 13 (42). doi: 10.1186/s13068-020-01687-y
Welchman, R. L., Gordon, C., Mayer, R. J. (2005). Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 6 (8), 599–609. doi: 10.1038/nrm1700
Wen, R., Newton, L., Li, G., Wang, H., Xiao, W. (2006). Arabidopsis thaliana UBC13: implication of error-free DNA damage tolerance and Lys63-linked polyubiquitylation in plants. Plant Mol. Biol. 61 (1-2), 241–253. doi: 10.1007/s11103-006-0007-x
Wen, R., Torres-Acosta, J. A., Pastushok, L., Lai, X., Pelzer, L., Wang, H., et al. (2008). Arabidopsis UEV1D promotes lysine-63-linked polyubiquitination and is involved in DNA damage response. Plant Cell 20 (1), 213–227. doi: 10.1105/tpc.107.051862
Wenzel, D. M., Stoll, K. E., Klevit, R. E. (2011). E2s: structurally economical and functionally replete. Biochem. J. 433 (1), 31–42. doi: 10.1042/bj20100985
Wu, Y., Ke, Y., Wen, J., Guo, P., Ran, F., Wang, M., et al. (2018). Evolution and expression analyses of the MADS-box gene family in Brassica napus. PloS One 13 (7), e0200762. doi: 10.1371/journal.pone.0200762
Wu, D., Liang, Z., Yan, T., Xu, Y., Xuan, L., Tang, J., et al. (2019). Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol. Plant 12 (1), 30–43. doi: 10.1016/j.molp.2018.11.007
Xie, M., Zuo, R., Bai, Z., Yang, L., Zhao, C., Gao, F., et al. (2022). Genome-wide characterization of Serine/Arginine-rich gene family and its genetic effects on agronomic traits of Brassica napus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.829668
Xu, L., Ménard, R., Berr, A., Fuchs, J., Cognat, V., Meyer, D., et al. (2009). The E2 ubiquitin-conjugating enzymes, AtUBC1 and AtUBC2, play redundant roles and are involved in activation of FLC expression and repression of flowering in Arabidopsis thaliana. Plant J. 57 (2), 279–288. doi: 10.1111/j.1365-313X.2008.03684.x
Yao, S., Liang, F., Gill, R. A., Huang, J., Cheng, X., Liu, Y., et al. (2020). A global survey of the transcriptome of allopolyploid Brassica napus based on single-molecule long-read isoform sequencing and illumina-based RNA sequencing data. Plant J. 103 (2), 843–857. doi: 10.1111/tpj.14754
Ye, Y., Rape, M. (2009). Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10 (11), 755–764. doi: 10.1038/nrm2780
Yu, P., Hua, Z. (2022). The ubiquitin-26S proteasome system and autophagy relay proteome homeostasis regulation during silique development. Plant J. 111 (5), 1324–1339. doi: 10.1111/tpj.15891
Yu, Y., Jia, T., Chen, X. (2017). The 'how' and 'where' of plant microRNAs. New Phytol. 216 (4), 1002–1017. doi: 10.1111/nph.14834
Yunpeng, C., Yahui, H., Dandan, M., Dahui, L., Qing, J., Yi, L., et al. (2016). Structural, evolutionary, and functional analysis of the class III peroxidase gene family in Chinese pear (Pyrus bretschneideri). Front. Plant Sci. 7, 1874. doi: 10.3389/fpls.2016.01874
Zhang, L., Cai, X., Wu, J., Liu, M., Grob, S., Cheng, F., et al. (2018). Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 5, 50. doi: 10.1038/s41438-018-0071-9
Zheng, B., Chen, X., McCormick, S. (2011). The anaphase-promoting complex is a dual integrator that regulates both MicroRNA-mediated transcriptional regulation of cyclin B1 and degradation of cyclin B1 during Arabidopsis male gametophyte development. Plant Cell 23 (3), 1033–1046. doi: 10.1105/tpc.111.083980
Zhou, X., Wu, X., Li, T., Jia, M., Liu, X., Zou, Y., et al. (2018). Identification, characterization, and expression analysis of auxin response factor (ARF) gene family in Brachypodium distachyon. Funct. Integr. Genomics 18 (6), 709–724. doi: 10.1007/s10142-018-0622-z
Zhu, W., Guo, Y., Chen, Y., Wu, D., Jiang, L. (2020). Genome-wide identification, phylogenetic and expression pattern analysis of GATA family genes in Brassica napus. BMC Plant Biol. 20 (1), 543. doi: 10.1186/s12870-020-02752-2
Zolman, B. K., Monroe-Augustus, M., Silva, I. D., Bartel, B. (2005). Identification and functional characterization of arabidopsis PEROXIN4 and the interacting protein PEROXIN22. Plant Cell 17 (12), 3422–3435. doi: 10.1105/tpc.105.035691
Keywords: Brassica napus, the UBC family, evolution, association mapping analysis, oil content and yield
Citation: Yao S, Xie M, Hu M, Cui X, Wu H, Li X, Hu P, Tong C and Yu X (2023) Genome-wide characterization of ubiquitin-conjugating enzyme gene family explores its genetic effects on the oil content and yield of Brassica napus. Front. Plant Sci. 14:1118339. doi: 10.3389/fpls.2023.1118339
Received: 07 December 2022; Accepted: 07 March 2023;
Published: 20 March 2023.
Edited by:
Liezhao Liu, Southwest University, ChinaReviewed by:
Maoteng Li, Huazhong University of Science and Technology, ChinaCopyright © 2023 Yao, Xie, Hu, Cui, Wu, Li, Hu, Tong and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaobo Tong, dG9uZ2NoYW9ib0AxMjYuY29t; Xiaoli Yu, eXhsbDI2OEAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.