 Shumpei Hashimoto1
Shumpei Hashimoto1 Satoshi Okada1
Satoshi Okada1 Satoko Araki-Nakamura1Kozue Ohmae-Shinohara1Kotaro Miura2Hideo Kawaguchi3
Satoko Araki-Nakamura1Kozue Ohmae-Shinohara1Kotaro Miura2Hideo Kawaguchi3 Chiaki Ogino4Shigemitsu Kasuga5
Chiaki Ogino4Shigemitsu Kasuga5 Takashi Sazuka1*
Takashi Sazuka1*- 1Bioscience and Biotechnology Center, Nagoya University, Nagoya, Japan
- 2Faculty of Bioscience and Biotechnology, Fukui Prefectural University, Fukui, Japan
- 3Engineering Biology Research Center, Kobe University, Kobe, Japan
- 4Department of Chemical Science and Engineering, Graduate School of Engineering, Kobe University, Kobe, Japan
- 5Faculty of Agriculture, Education and Research Center of Alpine Field Science, Shinshu University, Minamiminowa, Japan
Sorghum is the fifth most important cereal crop. Here we performed molecular genetic analyses of the ‘SUGARY FETERITA’ (SUF) variety, which shows typical sugary endosperm traits (e.g., wrinkled seeds, accumulation of soluble sugars, and distorted starch). Positional mapping indicated that the corresponding gene was located on the long arm of chromosome 7. Within the candidate region of 3.4 Mb, a sorghum ortholog for maize Su1 (SbSu) encoding a starch debranching enzyme ISA1 was found. Sequencing analysis of SbSu in SUF uncovered nonsynonymous single nucleotide polymorphisms (SNPs) in the coding region, containing substitutions of highly conserved amino acids. Complementation of the rice sugary-1 (osisa1) mutant line with the SbSu gene recovered the sugary endosperm phenotype. Additionally, analyzing mutants obtained from an EMS-induced mutant panel revealed novel alleles with phenotypes showing less severe wrinkles and higher Brix scores. These results suggested that SbSu was the corresponding gene for the sugary endosperm. Expression profiles of starch synthesis genes during the grain-filling stage demonstrated that a loss-of-function of SbSu affects the expression of most starch synthesis genes and revealed the fine-tuned gene regulation in the starch synthetic pathway in sorghum. Haplotype analysis using 187 diverse accessions from a sorghum panel revealed the haplotype of SUF showing severe phenotype had not been used among the landraces and modern varieties. Thus, weak alleles (showing sweet and less severe wrinkles), such as in the abovementioned EMS-induced mutants, are more valuable for grain sorghum breeding. Our study suggests that more moderate alleles (e.g. produced by genome editing) should be beneficial for improving grain sorghum.
1 Introduction
Sorghum (Sorghum bicolor L. Moench) is the fifth most cultivated cereal crop after wheat, maize, rice, and barley (FAOSTAT, 2019). It is an important cereal food consumed in semi-arid regions (e.g., Africa and Asia) due to its higher drought tolerance compared to other crops, highlighting its importance for global and regional food security. Sorghum grains are rich in phenolic compounds, micronutrients, dietary fiber, and minerals (Awika and Rooney, 2004; Moraes et al., 2015; Girard and Awika, 2018; Shen et al., 2018). However, it contains inadequate levels of some essential amino acids, particularly lysine, threonine, and tryptophan (Salunkhe et al., 1977). Sorghum grains are also a safe material and are suitable for people with celiac disease, a serious condition affecting millions of individuals (Ciacci et al., 2007; de Mesa-Stonestreet et al., 2010).
Starch is the main storage form of carbohydrates in plants and its synthesis is an important process that determines yield in cereal grains. The pathway of starch synthesis involves the conversion of sucrose to ADP-glucose, which is then converted into the two types of polysaccharides, namely amylose and amylopectin. Amylopectin synthesis is highly regulated by a group of enzymes comprising ADP-glucose pyrophosphorylases (AGPases), starch synthases (SSs), starch branching enzymes (SBEs), and debranching enzymes (DBEs) (Smith et al., 1997; Myers et al., 2000; Nakamura, 2002; Ball and Morell, 2003; Pfister and Zeeman, 2016). Of these enzymes, DBEs trim the shape of amylopectin clusters by hydrolyzing α-1,6-glucosidic linkages in α-polyglucans. They are considered to play an essential role in amylopectin synthesis (Ball et al., 1996). Two types of DBEs have been identified in higher plants and green algae, namely isoamylases (ISA1, ISA2, and ISA3) and pullulanases (PUL) (Nakamura, 2015).
A mutation in the Isa1 gene causes a dramatic change in the structure of amylopectin, whereby it becomes a randomly and more highly branched structure of α-1,6-/α-1-4-polyglucans referred to as phytoglycogen (James et al., 1995; Mouille et al., 1996; Zeeman et al., 1998; Kubo et al., 1999; Burton et al., 2002). These mutants have reduced starch content and accumulate more soluble sugars, resulting in a “sugary” endosperm phenotype. In maize, sugary endosperm has been widely used in sweet corn (Zea mays L.) breeding, which has a farm gate value of $1.4 billion per year in the United States (USDA-NASS, 2019). Sweet corn results from mutations in genes involved in the starch synthesis pathway, which modify the carbohydrate composition by increasing sugar content in the endosperm while reducing starch content. Several genetic loci have been shown to increase sugar content in the endosperm when mutated, including sugary1 (su1), shrunken2 (sh2), and sugary enhancer1 (se1) (Creech, 1965; Tsai and Nelson, 1966; Tracy, 1997; Rahman et al., 1998). Traditional sweet corn hybrids increased sugar content through mutations in the su1 gene, an SBE which encodes ISA1. Compared to normal field corn, su1 endosperms accumulate more sugar and phytoglycogen at the harvesting stage. When this enzyme is defective, the endosperm of mature kernels looks wrinkled and translucent.
Studies using isa1 mutants and its transgenic lines in rice have revealed its central role in starch synthesis (Nakamura et al., 1997; Kubo et al., 1999; Kubo et al., 2005; Fujita et al., 2012; Shufen et al., 2019). Kawagoe et al. (2005) used a rice isa1 mutant transformed with an amyloplast marker to track amyloplasts and starch granule formation and found that ISA1 is crucial during the early stages of starch granule formation (Kawagoe et al., 2005). Moreover, analysis of overexpression and mutant lines of ISAs revealed that ISA1 can exist as a homo-oligomer and can form a hetero-oligomer with ISA2, which is important for the synthesis of starch (Utsumi et al., 2011). As such, rice has played a central role as a model for functional studies of ISA1 in the starch synthesis pathway. However, these mutations in ISA1 have not been exploited in rice breeding because of the severe wrinkled phenotype, compared to maize, for which weak mutants were obtained.
Literature on the history of sugary sorghum may go back over half a century (Karper and Quinby, 1963). There, it was reported that sugary seeds contain at least twice as much total sugar as normal seeds. Moreover, the people of India have undoubtedly known of the peculiar sweetness of sugary endosperms for a long time. This sugary phenotype was assumed to be caused by a single recessive gene, and the corresponding gene was designated Su (Karper and Quinby, 1963). However, an analysis of the corresponding gene has not been conducted and the molecular mechanism of the sugary trait remains unknown. In this study, we studied a variety of sugary sorghum, namely ‘SUGARY FETERITA’, which shows the typical sugary endosperm phenotype. Molecular genetic studies revealed that the corresponding gene, Su, encodes sorghum ISA1 (SbISA1).
2 Materials and methods
2.1 Plant materials
The sorghum (Sorghum bicolor L. Moench) accessions ‘SUGARY FETERITA’ (SUF, JP No. 44623) and ‘FETERITA FAYOUMI D.S.21’ (FET, JP No. 246696) were obtained from Genebank Project, NARO (National Agriculture and Food Research Organization) and used for the phenotypic and genetic characterization of the sugary endosperm and gene expression. ‘Nakei MS-3B’ (MS3B) and SUF were used for segregation analysis and rough mapping. For complementation analysis using rice, the sugary-1 mutant SG0807 was obtained from Oryzabase (https://shigen.nig.ac.jp/rice/oryzabase/). Sorghum ethyl methanesulfonate (EMS) mutants (PI 678144, PI 678119, and PI 678151) were obtained from the Functional Gene Discovery Platform for Sorghum (https://www.purdue.edu/sorghumgenomics). For haplotype analysis, the Nagoya University Panel (NUP; 187 sorghum accessions) was obtained from the sorghum diversity research set of NARO (National Agriculture and Food Research Organization) (Shehzad et al., 2009), the US historic sweet sorghum collection (Wang et al., 2009), and the available lines in our lab, were used for this study (Supplementary Table S2).
2.2 Rough mapping of the sugary locus in SUF
For rough mapping, seeds exhibiting a wrinkled trait in the F2 progeny, resulting from a cross between MS3B and SUF, were used. Genomic DNA was isolated from ca. 2-week-old leaves from 96 individuals using the cetyltrimethylammonium bromide (CTAB) extraction method (Murray and Thompson, 1980) with some modifications. Briefly, leaf samples were ground using a MultiBead Shocker (Yasui Kikai, Osaka, Japan) in 2 × CTAB extraction buffer (100 mM Tris-HCl [pH 8.0], 50 mM EDTA, 1.4 M NaCl, 2% [w/v] CTAB, 1% [w/v] PVP). After incubating at 60°C for 30 min, an equal volume of chloroform was added. After centrifugation at 15,000 rpm for 5 min, the supernatant was recovered, and an equal volume of isopropyl alcohol was added. The sample was recovered by centrifugation at 15,000 rpm for 5 min and the pellet was washed with 70% (v/v) ethanol. The DNA pellet was dried and dissolved in 1 × Tris/EDTA (TE) solution (10 mM Tris-HCl [pH 8.0], 1 mM EDTA [pH 8.0]). The purified DNA samples were then used for genotyping using a polymerase chain reaction (PCR). Simple sequence repeat (SSR) markers, as reported by Yonemaru et al. (Yonemaru et al., 2009), were screened and 178 markers showing polymorphisms between MS3B and SUF were selected (Supplementary Table S3). DNA segments were amplified by PCR using the following program: 95°C for 1 min; 30 cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec; 72°C for 7 min. The PCR products were analyzed by agarose gel electrophoresis.
2.3 Gene expression analysis
‘SUGARY FETERITA’ and ‘FETERITA FAYOUMI D.S.21’ were grown in the greenhouse and immature seeds were sampled 0, 7, 14, and 21 days after flowering (DAF). These samples were immediately frozen in liquid nitrogen and crushed with a mortar and pestle until a powder was obtained. Next, RNA extraction buffer (50 mM Tris-HCl [pH 9.0], 20 mM EDTA [pH 8.0], 200 mM NaCl, 1% (w/v) Sodium N-lauroyl sarcosinate, and 5 mM DTT) was added to each sample and centrifuged at 14,000 rpm for 5 min at 4 °C. The supernatant was used in the subsequent steps. Total RNA was isolated from each sample using the TRIzol® reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Next, 500 ng of total RNA was used to synthesize first-strand cDNA using an Omniscript reverse transcription (RT) kit (Qiagen, Hilden, Japan), according to the manufacturer’s instructions. Quantitative RT-PCR (qRT-PCR) was conducted using the KOD SYBR® qPCR Mix (TOYOBO, Osaka, Japan) and a real-time thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA). The ubiquitin (UBQ) gene served as the internal reference for the qRT-PCR analyses. Primers used in the analysis were listed in Supplementary Table S3.
2.4 Phenotypic analysis of grains
We examined the morphological traits of sorghum kernels using a stereo microscope. Seeds were cut with a razor blade and cross-sections were observed using the same microscope. For histological analysis, seed samples were fixed in FAA fixative (formaldehyde and acetic acid in 50% [v/v] ethanol) under a vacuum and dehydrated in a graded ethanol series. Samples were embedded in Technovit 7100 (Kulzer, Hanau, Germany) and sectioned at a thickness of 1 μm with a manually operated rotary microtome (Leica RM2125RT). Sections were stained with iodine solution (1% [w/v] KI, 0.1% [w/v] I2) and observed under a light microscope (Olympus BX5). Photographs were taken using a digital camera (Olympus DP74) connected to the microscope. Brix values of the seeds were obtained as follows; first, 0.2 g of seeds were roughly crushed with a hammer and placed in a tube with beads. They were then crushed in a grinding apparatus (Bio-Medical Science Co., Ltd, BMS-A20TP) for at least 20 min at 1,100 rpm. To the powdered sample, 200 µL of water was added and mixed in the grinding device for a further 5 min at 1,100 rpm. Finally, the sample was centrifuged at 15,000 rpm for 5 min and the supernatant was measured with a refractometer (AS ONE, APAL-1).
2.5 Agrobacterium-mediated rice transformation
Genomic DNA fragments (8.6 kb) containing Sobic.007G204600 from MS3B and SUF were introduced into pCAMBIA1300 (Cambia) using the KpnI/EcoRI sites. Each construct was transformed into Agrobacterium tumefaciens EHA105 using an electroporator (Eppendorf) at 2,500 V and used to infect calli of sugary-1 mutant (SG0807), whose genetic background is Oryza sativa (L.) cv. ‘Kinmaze’ as described by Ozawa (Ozawa, 2012). Transformed calli and plants were selected by hygromycin resistance, with regenerants grown in pots in a greenhouse.
2.6 Haplotype analysis
All accessions were sequenced using an Illumina Hiseq X Ten sequencer (Illumina, San Diego, CA, USA) by pair-end sequencing to obtain SNPs as previously reported (Hashimoto et al., 2021). For SNP calling, clean reads were aligned with the BTx623 (v3.0.1) reference genome using Burrows-Wheeler Alignment (BWA, v0.7.17) (Li and Durbin, 2009) with default parameters. Variants were called independently and merged using the Genome Analysis Toolkit (GATK, v4.1.8, HaplotypeCaller) (McKenna et al., 2010). After the genomic region of SbSu (Sobic.007G204600) was extracted using VCFtools v0.1.16 (Danecek et al., 2011) from the resulting merged VCF file, haplotypes were categorized based on nonsynonymous SNPs associated with single amino acid substitutions in the coding region.
3 Results
3.1 Phenotypic characterization of ‘SUGARY FETERITA’
During the vegetative and heading stages, SUF showed no obvious differences from the wild type FETERITA (FETERITA FAYOUMI D.S.21; FET) (Supplementary Figure S1). However, at the hard dough stage, grains of SUF exhibited wrinkled and translucent phenotypes whereas FET had normal, smooth grains (Figures 1A–D). Grains stained with iodine solution revealed that the endosperm of FET stained well in dark blue (Figures 1E, G), while that of SUF stained less overall, with no staining in the central region (Figure 1F, H). The detailed observation of the cross-sections revealed that SUF had loosely packed and abnormal starch granules compared to the densely packed granules of FET (Figures 1I-L). The Brix value of SUF was 2 fold higher than FET (Figure 1M). A decrease of grain weight was also observed in SUF (Figure 1N). These results indicate that the starch components of the endosperm of SUF differ from FET, resulting in the wrinkled grain phenotype and the accumulation of soluble sugars.
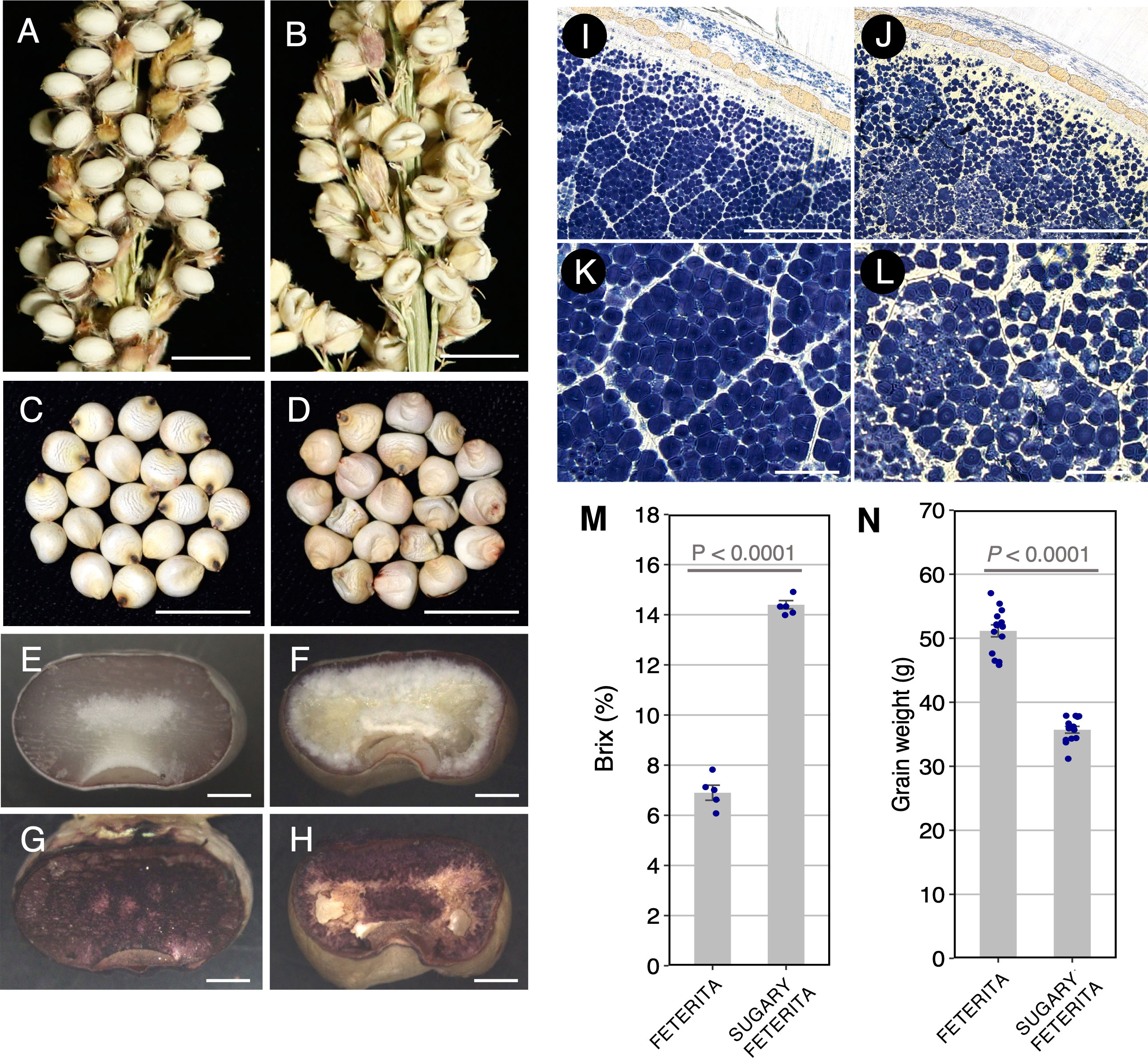
Figure 1 Seed phenotypes of the two varieties, ‘FETERITA’ (FET) and ‘SUGARY FETERITA’ (SUF). (A, D) Panicles of FET (A) and SUF (B). Kernels of FET (normal, C) and SUF (wrinkled, D). E–G, Transverse sections of FET and SUF stained with iodine solution (G, H). (K–N), Cross sections of FET (I, K) and SUF (J, L). Aberrant starch structures were observed in (L). (M, N) Brix values (M) and grain weights (N) of the seeds. Scale bars: 1 cm in (A–D), 1 mm in (E–H), 200 μm in (I, J), and 50 μm (K, L).
3.2 Positional mapping and identification of the corresponding gene
It is known that sugary mutants of maize and rice present wrinkled seeds, therefore, we used this phenotype as a morphological indicator of sugary endosperm in sorghum. For positional mapping, a Japanese sorghum variety, MS3B (a maintainer line with a normal phenotype), and SUF were crossed. The resulting MS3B x SUF (F1) seeds showed a normal phenotype (Supplementary Figure S2), suggesting that the sugary endosperm phenotype is recessive. We then analyzed the segregation pattern of the 1,627 F2 seeds and found 1,212 were wild-type seeds and 415 seeds with the wrinkled phenotype. The segregation ratio of normal to wrinkled seeds in the population fit to 3:1 via the chi-square test (Supplementary Table S1), suggesting that the seed wrinkling trait is a recessive, single-gene trait.
Positional mapping using 96 F2 wrinkled phenotype seeds revealed genomic regions on chromosome 7 (60.6–65.0 Mb) and 9 (55.1–59.2 Mb) with biased segregation ratios (Supplementary Figure S3). Although the bias on the latter region did not fit the segregation pattern described above (the recessive mutation model), the former biased for the SUF homozygotes fit it (Supplementary Table S1). In the region on chromosome 7, we searched candidate genes (i.e., starch synthesis-related genes). Here, we found Sobic.007G204600, which encodes a DBE (SbISA1), localized within 3.4 Mb on the candidate region (Figure 2A). It should be noted that Sobic.007G204600 is the sorghum homolog of maize Su1 (Figure 2A). Sequencing analysis of the genomic DNA of the Sobic.007G204600 of MS3B and FET revealed that these genes produce proteins with the same amino acid sequences as BTx623, the reference genome (Figure 2B). Conversely, the allele in SUF had five SNPs which resulted in nonsynonymous amino acid substitutions in the protein-coding region (Figure 2B). One of those in SUF (L421) was next to the highly conserved catalytic residue (D420) of ISA1 (Figure 2C) (Sim et al., 2014). A protein structure analysis using Chlamydomonas reinhardtii ISA1 (CrISA1) suggested that L453H (corresponding to L421H in SUF) could sterically hinder the binding of the catalytic residue (Figure 2D). Gene expression analysis by qRT-PCR indicated that Sobic.007G204600 was highly expressed in developing seeds (Figure 2E). From these results, we supposed that this gene might play a role in starch metabolism in endosperm cells and that mutations therein result in their loss of function. Therefore, it is a candidate for the corresponding gene of SUF.
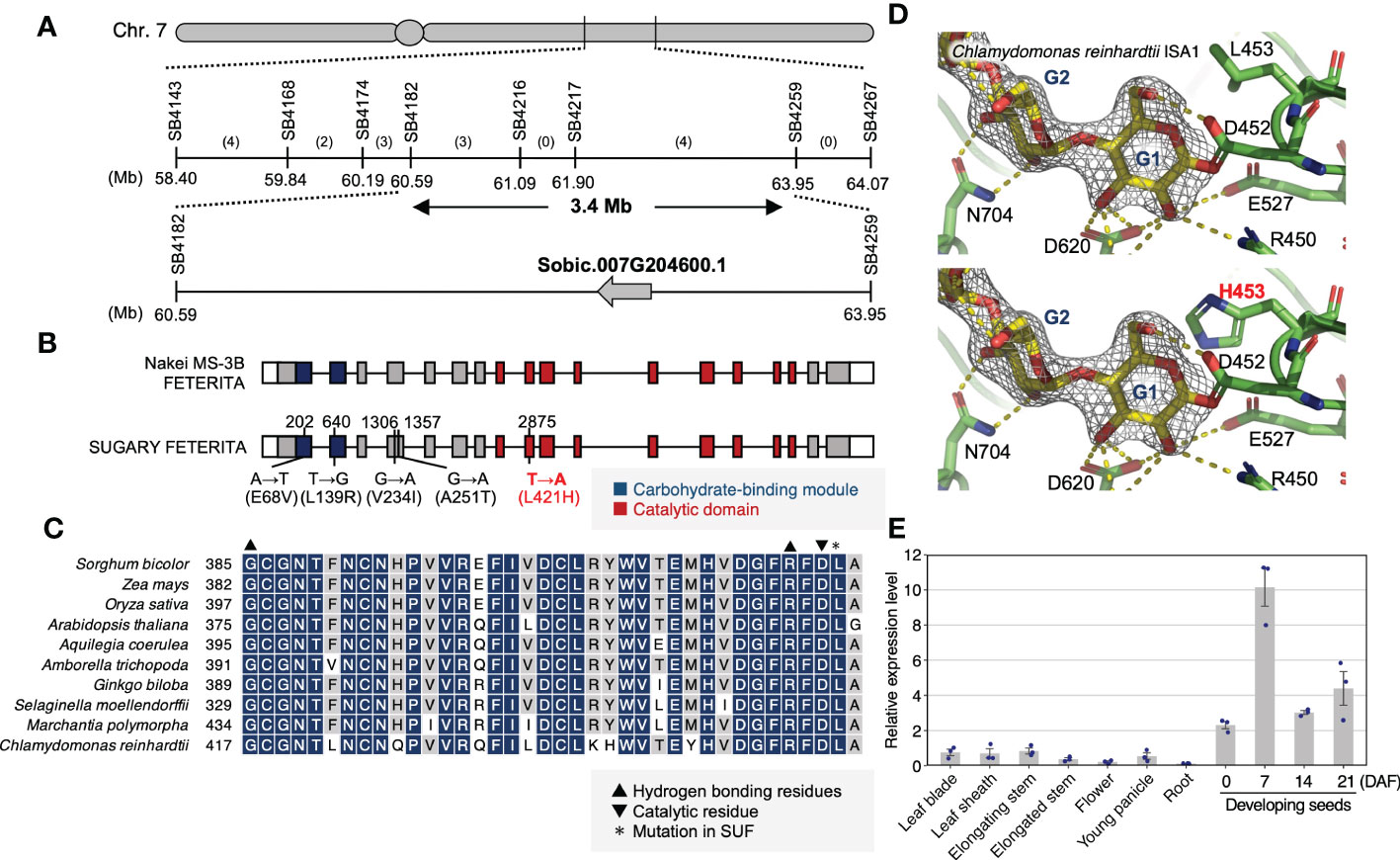
Figure 2 Positional cloning of the corresponding gene for sugary endosperm in ‘SUGARY FETERITA’. (A) A high-resolution map of the candidate locus on chromosome 7. Vertical lines indicate the positions of the SSR markers with their physical positions (Mb) in Phytozome 13 (Sorghum bicolor v3.1.1). The number of recombinants is shown between the markers. The mapping analysis narrowed the candidate region to a 3.4 Mb stretch between SB4182 and SB4259 on the long arm of chromosome 7. Around 400 genes were annotated in this region, including Sobic.007G204600. (B) Gene structures of SbSu according to the three cultivars, ‘Nakei MS-3B’(MS3B), ‘FETERITA’(FET), and ‘SUGARY FETERITA’(SUF). Exons are represented as boxes and introns as the lines between boxes. White, grey, black, and red boxes indicate the 3’- or 5’-untranslated regions (UTR), protein-coding regions, a carbohydrate-binding module, and a catalytic domain, respectively. SUF has five SNPs in its coding region compared to the MS3B and FET alleles. (C) The conservation of L421 among plants and algae is indicated with an asterisk. It is located in the catalytic domain, indicated by an inverted triangle. (D, top) The expanded structure of Chlamydomonas reinhardtii Isoamylase 1 (CrISA1) obtained from PDBj (Protein Data Bank Japan, PDB DOI: 10.2210/pdb4J7R/pdb) and visualized in PyMOL v2.1. The L453 in C reinhardtii, which corresponds to L421 in FET, was substituted to Histidine (bottom, highlighted in red). (E) Expression pattern of SbSu among organs. Error bars indicate the standard error of three biological replicates.
3.3 Complementation of the rice sugary-1 mutant
To confirm that the SUF allele of Sobic.007G204600 is a loss-of-function allele, we performed a complementation test using the rice sugary-1 (osisa1) mutant line (SG0807), which exhibits thin seeds with distorted starch, resulting in a less stained endosperm following iodine staining (Figure 3). A genomic DNA fragment containing the promoter (2 kb upstream from ATG) and the coding region from MS3B or SUF was introduced into SG0807. The results showed that the endosperm of transgenic lines transformed with coding region of MS3B stained positive with iodine (Figure 3) and almost recovered the seed thickness of SG0807. In contrast, the endosperm of transgenic lines complemented with the genomic fragment derived from SUF did not stain the same level as SG0807 (Figure 3). These results suggested that the corresponding gene is Sobic.007G204600 (SbSu), and that the haplotype of SUF (pronounces E68V, L139R, V234I, A251T and L421H mutations) causes sugary endosperm, and the MS3B and FET alleles of this gene are gain-of-function alleles, whereas SUF allele is a loss-of-function allele. This sugary gene Su/su assigned firstly by Karper and Quinby (1963) was redefined as SbSu in the later sections.
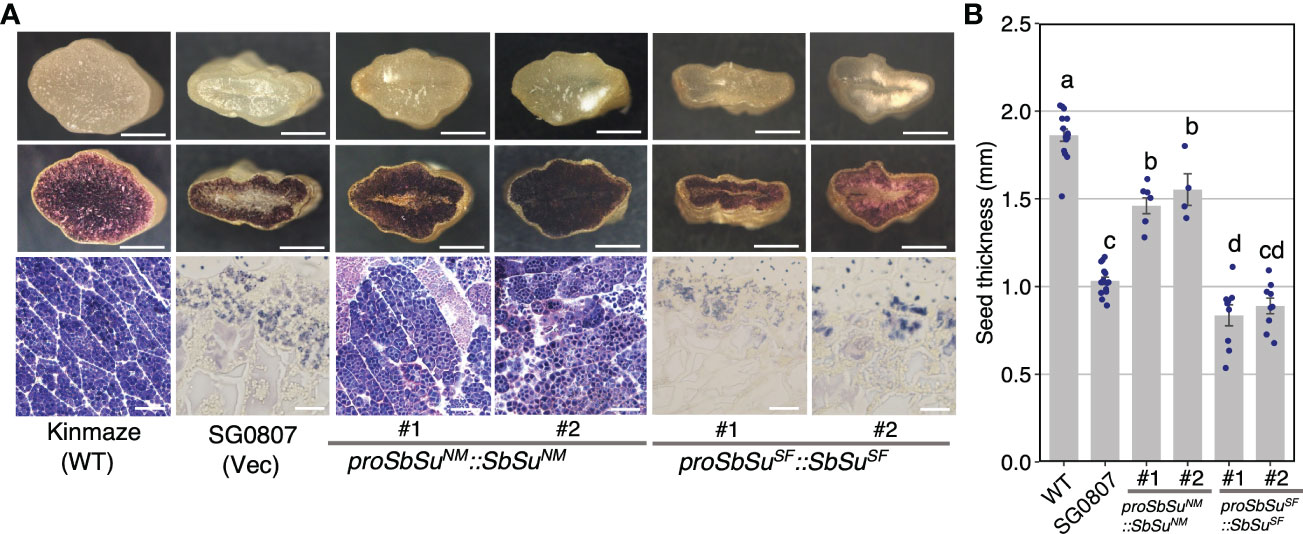
Figure 3 Complementation of the rice su1 mutant with SbSu. A DNA fragment containing a 2 kb promoter fragment followed by the SbSu coding region (NM; Nakei MS-3B, SF; SUGARY FETERITA) was introduced into the rice su1 mutant, SG0807. The SG0807 mutant (vec) and Kinmaze (wild-type) plants transformed with an empty vector were used as controls. (A) Grain morphologies of wild-type rice (Kinmaze), SG0807, and transgenic seeds. Crossed sections shown in the upper pictures were stained with iodine solution (middle) to observe starch structures (bottom). Scale bars: 1 mm (upper and middle) and 20 μm (bottom). (B) Comparison of seed thickness in wild-type, SG0807, and transgenic lines. For statistical analysis, an ANOVA model was followed using Tukey’s HSD test to estimate statistical differences. Different lowercase letters in each plot indicated significant differences in the post-hoc test (P < 0.05).
3.4 Expression profiles of starch synthesis genes
To understand how the mutation (L421H) in SUF affects starch synthesis, we examined the expression of 16 starch synthesis-related genes (Supplementary Figure S4) using qRT-PCR. These genes include four DBEs (SbSu, SbIsa2, SbIsa3, and SbPul1) and four SBEs (SbSbeI, SbSbeIIa, SbSbeIIb, and SbSbeIII). The DBEs catalyze the hydrolysis of α-1,6-glucosidic linkages in α-polyglucans, while SBEs catalyze the formation of α-1,6-glucosidic linkages in amylopectin and glycogen. The other starch synthesis-related genes include an SS (SbSsI), four transporters (SbSweet2-1, SbSweet4-3, SbBt1, SbPho1), an AGPase (SbSh2), and an ortholog of se1 in maize (SbSe1).
At the early seedling stage (ca. 14 DAF), the expression levels of the two DBEs (SbSu1 and SbPul) were upregulated in SUF, indicating positive feedback due to the loss-of-function of SbSu1 (Figure 4), although th other DBEs (Sbisa2 and Sbisa3) were not upregulated. During the late stage (21 DAF), the expression of SbSbeI, SbSbeIIa, SbSsI, SbSe1, SbBt1, SbSh1, and three DBEs (SbSu1, SbIsa2 and SbPul1) was significantly downregulated in SUF (Figure 4). The expression of SbSweet4-3 was markedly lower in SUF at 7 and 14 DAF. There were no significant changes for SbSbeIIb and SbSh2 (Figure 4). These observations suggest that a fine-tuning activity works for the appropriate expression of DBEs during the early stage, while during the later stage, the activity disappears in the starch synthesis pathway.
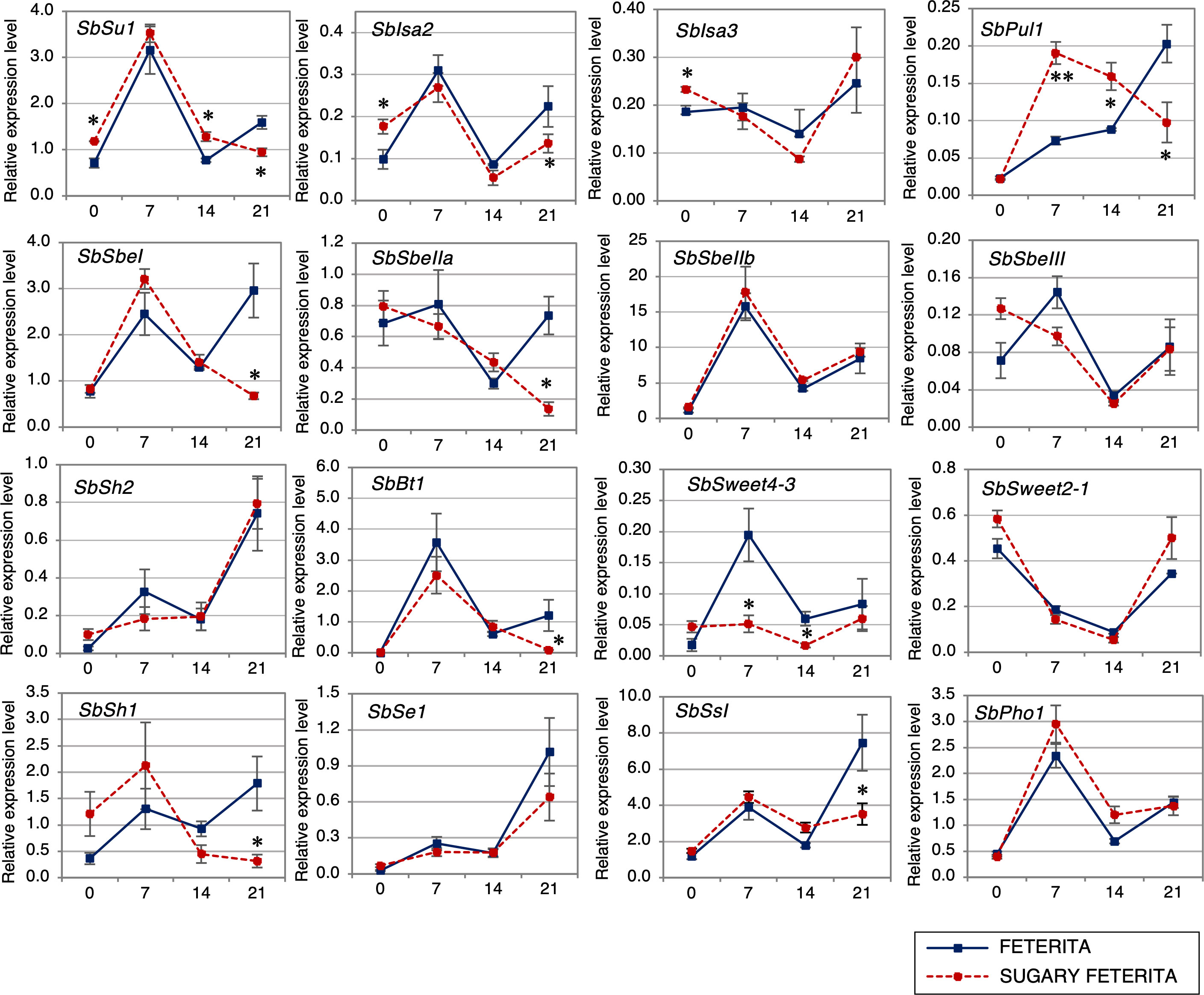
Figure 4 Expression profiles of starch synthesis genes of sorghum during seed development in wild-type ‘FETERITA’ and ‘SUGARY FETERITA’. Total RNA was extracted from seeds at 0, 7, 14, and 21 days after flowering (DAF), and qRT-PCR was conducted for 16 genes; SbSu (Sobic.007G204600), SbIsa2 (Sobic.009G127500), SbIsa3 (Sobic.002G233600), SbPul1 (Sobic.006G015800), SbSbe1 (Sobic.010G273800), SbSbeIIa (Sobic.006G066800), SbSbeIIb (Sobic.004G163700), SbSbeIII (Sobic.003G213800), SbSh2 (Sobic.003G230500), SbBt1 (Sobic.004G085100), SbSweet4-3 (Sobic.004G133600), SbSweet2-1 (Sobic.001G377600), SbSh1 (Sobic.010G072300), SbSe1 (Sobic.005G199300), SbSsI (Sobic.010G047700), SbPhoI (Sobic.001G083900). Expression levels are shown relative to the expression of the sorghum ubiquitin (SbUbi; Sobic.001G311100) gene. All data are shown as means ± SE from three biological replicates. Two-tailed unpaired t-tests were used to determine significant differences. *P < 0.05 and **P < 0.01. More detail about each target gene can be found in Supplementary Figure S4.
3.5 Haplotype analysis of the SbSu gene
If SbSu is used for modern breeding in grain sorghum, it is estimated that weak alleles were widely used during the establishment of those landraces and varieties. To find weak allels, we did a haplotype analysis using a NUP containing 187 accessions (see Materials Methods). Here, 13 nonsynonymous SNPs were found in the coding region of SbSu, which resulted in 8 haplotypes (Figure 5). Interestingly, the accession containing Hap 8 is the only SUF among the NUP accessions. Subsequently, we measured the Brix values for each haplotype and found that the values were mostly similar except for Hap 8 (SUF), which had a higher score than the rest (Figure 5). These results suggest that although Hap 8 (SUF allele) shows a high Brix value, it is not used or spread in NUP. It is supposed that the sugary endosperm of Hap 8 is too strong of a phenotype, decreasing grain weight and wrinkled seed, thereby decreasing the germination rate. It should be noted that the five amino acid substitutions (E68V, L139R, V234I, A251T and L421H) observed in Hap 8 (Figure 2B) compared with Hap 1; however, four non-synonymous substitutions (E68V, L139R, V234I, and A251T) were the same as in Hap 2. Considerd that the Brix score of Hap 8 was high, although that of Hap 2 was almost as low as that of Hap 1, the L421H substitution (caused by the SNP of T2875A) should be the corresponding mutation for the sugary phenotype of SUF.
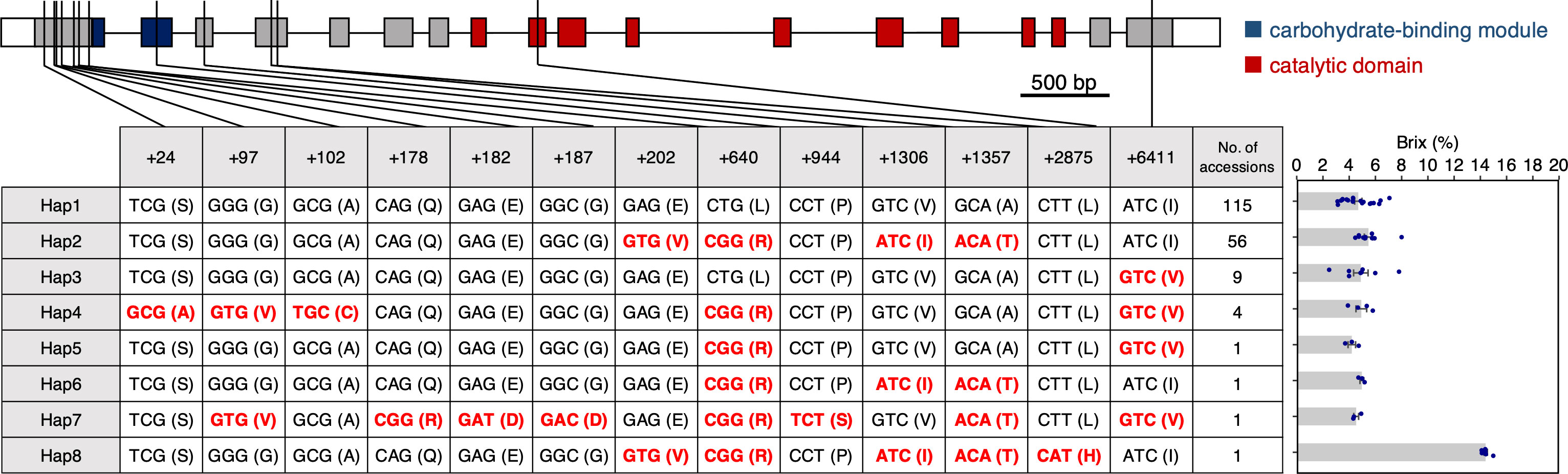
Figure 5 Haplotype analysis of SbSu. Eight haplotypes were observed in 187 accessions based on nonsynonymous SNPs in the coding region. Schematic diagram of SbSu is shown on the top (also, see Figure 2B). The number in the first row of the table indicates the relative physical position from the transcription start site. Nonsynonymous mutations are indicated in red. For Hap5-7 (only one accession), data were obtained from more than three biological replicates.
3.6 Identifying a novel weak allele of SbSu
The previous results reveal the importance of a weak allele for breeding; i.e., showing both a relatively high Brix value and a relatively normal seed shape (Supplementary Figure S5). However, we could not find a weak allele in the NUP accessions. Therefore, we searched the sorghum mutant panel of the Functional Gene Discovery Platform for sorghum (https://www.purdue.edu/sorghumgenomics). The accessions in the platform were mutagenized by EMS, generating 12,000 mutant families in the BTx623. Genome sequences were generated for 586 EMS lines and bioinformatic processing was used to identify all mutations that impact predicted protein sequences. We searched the mutants of Sobic.007G204600 and found three mutants: PI 678144, PI 678119, and PI 678151. The mutations comprised single nonsynonymous amino acid substitutions; D182N (PI678144), P308L (PI678119), and R701Q (PI678151) (Figure 6A). It should be noted that these mutations were located in the highly conserved region (Supplementary Figure S6), and especially, D182 and P308 were located in the catalytic domain and carbohydrate-binding module, respectively. To study how these mutations influence endosperm phenotype, the Brix value and grain weight were analyzed and compared to BTx623 (wild-type). Of the three mutants, PI 678144 is an intermediate phenotype of sugary endosperm, with a Brix value of 9.6 compared with 5.9 in wild-type and 14.4 in SUF (Figure 6B). For grain weight, the relative value of PI 678144 against wild-type was 74.6, which was higher than that of SUF (69.8) (Figure 6C). It was supported by the section stained with an iodine solution of PI 678144, which showed small and distorted starch granules, but seemed a weaker phenotype than that of SUF (Figure 6D). The Brix value of PI678119 was slightly higher than that of the wild-type, and the sections also indicated that it had a weaker phenotype than that of PI 678144. The phenotype of PI678151 was similar to the wild-type (Figure 6B-D). These results suggested that PI678144 (D182N) is an appropriate weak allele, which could make it valuable for the breeding of sugary endosperm.
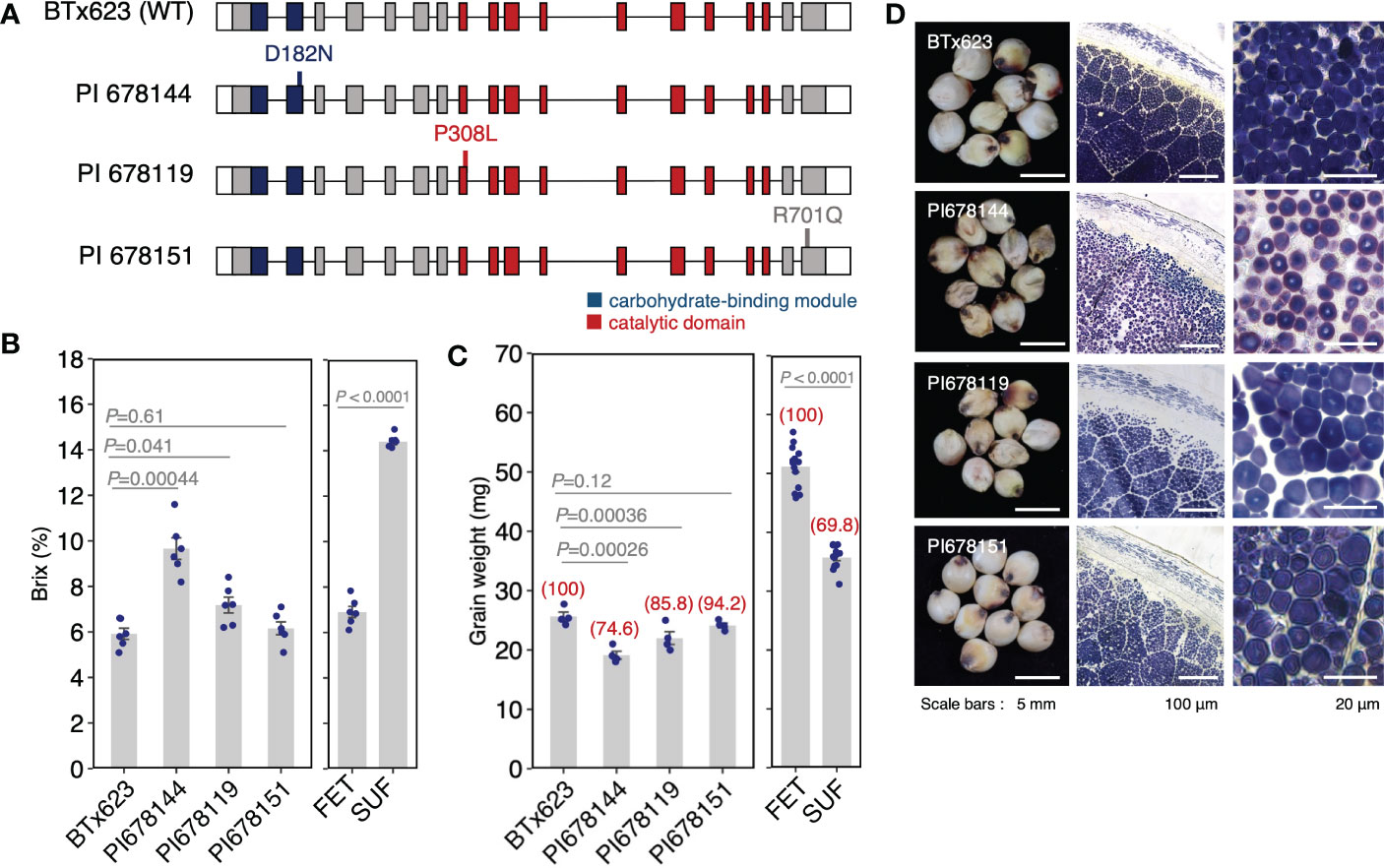
Figure 6 Novel sugary mutants of sorghum. (A) The position of the mutation in SbSu for each mutant. (B, C) Comparison of Brix values (C) and grain weight (D) of the mutants. In panel (C) red numbers on each bar with parentheses indicate the relative ratio of grain weight against wild-type. Data for FET and SUF are the same as in Figure 1 M, N. (D) Cross sections of seeds of SbSu mutants induced by EMS (see Materials and Methods).
4 Discussion
In 1963, Karper and Quinby stated the following: “A sugary endosperm sorghum reached the United States in 1903 when the Office of Grass and Forage Plant Investigations, U. S. Department of Agriculture, received 255 lots of sorghum seed from India.” This might be the oldest recognition of sugary endosperm in the United States. After, two sugary varieties originating from India were described in detail. The first was P.I.14746 (Kachakachi), which was recognized as an abnormal endosperm type in 1933 in the United States. The second was T.S. I7890 (Vani), a variety of the Surat District of India (Patel and Patel, 1928). The seeds were obtained from B. S. Kadam, a Crop Botanist (Government of Bombay, Karjat, District of Kolaba, India) in 1932, and later, was recognized as a sugary endosperm (Karper and Quinby, 1963). In India, the antiquity of the sugary endosperm character is not known, but the people of India have undoubtedly known of its peculiar sweetness for a long time (Karper and Quinby, 1963). During sorghum breeding in the United States, a mutation from normal to sugary endosperm was recognized at the Chillicothe Station in 1934 in a row of a breeding stock (Blackhull Kafir x Sumac; (Quinby and Karper, 1954)). The three sugary sorghums mentioned above are caused by the same gene based on the results of complementarity tests. This gene pair was later designated as Su/su (Karper and Quinby, 1963).
Although a detailed pedigree of SUF was not described, it is possible to estimate that at least one allele (or a maximum of three alleles) existed, based on the literature. Here, however, we found only one allele in NUP in this study. We conducted a molecular genetic analysis of the mutation of SUF, a typical model for sugary sorghum (Figure 1). We found that the segregation ratio of F2 seeds derived from a cross between MS3B (normal endosperm) and SUF showed a 3:1 ratios, indicating the sugary phenotype was caused by a single recessive gene (Supplementary Table 1). These results are consistent with a previous report, that is, the segregation ratio of F2 seeds derived from a cross between Kafir (normal endosperm) and Vani (Karper and Quinby, 1963). A subsequent positional mapping identified Sobic.007G204600 (SbSu), encoding isoamylase 1 (ISA1), as the corresponding gene for sugary endosperm in SUF (Figure 2). Considered the history of sugary sorghum, it is not inconceivable that SbSu is Su (Karper and Quinby, 1963).
In the starch biosynthesis pathway, ISA1 hydrolyzes α-1,6 glycosidic bonds and removes excess or misplaced branch points in amylopectin introduced by SBEs (Streb et al., 2008). The isa1 (su1) mutants of maize and rice commonly share an endosperm phenotype characterized by wrinkled grains. This phenotype is caused by the accumulation of phytoglycogen, which cannot be stained with iodine solution, and water-soluble sugars in the inner part of the endosperm instead of starch (Pan and Nelson, 1984; James et al., 1995). The previous observations of the isa1 mutation were also observed in the endosperm of SUF (Figure 1), suggesting that a loss of function of SbSu causes a similar defect of the starch biosynthesis pathway, especially, debranching in amylopectin.
Gene expression analysis of SUF showed that a loss-of-function of SbSu downregulated the expression of essential starch biosynthetic genes encoding the SBEs (SbSbe1 and SbSbeIIa) and SSs (SbSsI). Genes encoding DBEs (SbSu, SbIsa2, SbIsa3, and SbPul1) were initially up-regulated during the early stages of seed development (Figure 4). This up-regulation may be due to a mechanism triggered by the decreased ISA1 (SbSU) activity, which may partake in a fine-tuning system for balancing the expression levels of DBEs; this has not been reported before. In the rice isa1 mutant, the lower expression of most starch synthesis-related genes was observed (Shufen et al., 2019). Similarly, Zhang et al. (2019) reported that the se1 mutant, a recessive modifier of su1 in maize, exhibits decreased expression of many known starch biosynthetic genes (Figure 4). Thus, our results indicated that the mutation of SbSu induces negative feedback to other DBEs including itself, and subsequently the activity is disappeared in starch synthesis. This may lead to the reduced activity of the pathway, resulting in the accumulation of soluble sugars and wrinkled grains as an end phenotype in SUF (Figure 1).
In India, sugary sorghum landraces have been cultivated, however, sugary sorghum varieties from those have not been cultivated outside of India. Two reasons are considered. Firstly, the wrinkled phenotype causes a significant reduction in grain weight (Figure 1). Secondly, sugary grains easily become moldy because of the high sugar content and the wrinkled seeds, when it is grown in humid regions. This will lead to decreased grain quality and germination rate. A major reason for these troubles is that only one allele (or some alleles) has been used in the breeding history of sugary sorghum. This could be solved by obtaining moderate to weak alleles for the sugary trait (Supplementary Figure S5). In this study, although no haplotype was found showing a sugary trait except for the SUF haplotype in the NUP (Figure 5), we found a moderate allele (PI678144) in the EMS mutant panel. This mutation (D182N) is located in a highly conserved region, highlighting the importance of the region for the activity of this DBE (Supplementary Figure S6). Moreover, this allele may have potential value in future breeding. In this context, allele mining for other genes related to the sugary trait should also be important. For example, sorghum orthologs of Sh2 (Sobic.003G230500) and Se1 (Sobic.005G199300) are reasonable targets, which encode the large subunit of AGPase (Hannah and Nelson, 1976; Bhave et al., 1990) and a monocot-specific protein containing a FANTASTIC FOUR domain with unknown function (Zhang et al., 2019), respectively. Both genes have been incorporated into many sweet corn varieties, including super-sweet corn with improved fresh market quality (Tracy et al., 2019).
Recently, a methodology to create beneficial alleles selected from a continuum of variations in the expression level by genome editing of promoter regions using CRISPR/Cas9 was proposed (Rodriguez-Leal et al., 2017; Cui et al., 2020; Kawai et al., 2020). It is valuable for sorghum breeding to select optimal sugary alleles from a continuum of variations. The findings of this study have aided our understanding of the mechanisms of starch biosynthesis in sorghum and will accelerate breeding using sugary alleles in grain sorghum.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material.
Author contributions
SH, SA-N, and KO-S performed genotyping and plasmid constructions; SH performed rice transformation, histological analysis, and haplotype analysis; SH, HK, and CO performed gene expression analyses; SH, SO, KM, SK, and TS performed field experiments; SH and TS wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by the JST-Mirai Program (Grant No. JPMJMI17EG), the Moonshot Research and Development Program (Grant No. JPNP18016), commissioned by the New Energy and Industrial Technology Development Organization (NEDO), and JSPS KAKENHI (Grant Nos. JP21J13351 and JP22K14868).
Acknowledgments
We thank Drs. A. Yoshimura, T. Kumamaru, and H. Yasui (Kyushu Univ.), and the Functional Gene Discovery Platform for Sorghum (https://www.purdue.edu/sorghumgenomics/) for the courtesy of providing the rice sugary-1 mutant and the sorghum EMS mutants, respectively. We also thank Drs. M. Matsuoka and H. Kitano for their in-depth discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1114935/full#supplementary-material
References
Awika, J. M., Rooney, L. W. (2004). Sorghum phytochemicals and their potential impact on human health. Phytochemistry 65, 1199–1221. doi: 10.1016/j.phytochem.2004.04.001
Ball, S., Guan, H. P., James, M., Myers, A., Keeling, P., Mouille, G., et al. (1996). From glycogen to amylopectin: A model for the biogenesis of the plant starch granule. Cell 86, 349–352. doi: 10.1016/S0092-8674(00)80107-5
Ball, S. G., Morell, M. K. (2003). From bacterial glycogen to starch: understanding the biogenesis of the plant starch granule. Annu. Rev. Plant Biol. 54, 207–233. doi: 10.1146/annurev.arplant.54.031902.134927
Bhave, M. R., Lawrence, S., Barton, C., Hannah, L. C. (1990). Identification and molecular characterization of shrunken-2 cDNA clones of maize. Plant Cell 2, 581–588. doi: 10.1105/tpc.2.6.581
Burton, R. A., Jenner, H., Carrangis, L., Fahy, B., Fincher, G. B., Hylton, C., et al. (2002). Starch granule initiation and growth are altered in barley mutants that lack isoamylase activity. Plant J. 31, 97–112. doi: 10.1046/j.1365-313X.2002.01339.x
Ciacci, C., Maiuri, L., Caporaso, N., Bucci, C., Del Giudice, L., Rita Massardo, D., et al. (2007). Celiac disease: in vitro and in vivo safety and palatability of wheat-free sorghum food products. Clin. Nutr. 26, 799–805. doi: 10.1016/j.clnu.2007.05.006
Creech, R. G. (1965). Genetic control of carbohydrate synthesis in maize endosperm. Genetics 52, 1175–1186. doi: 10.1093/genetics/52.6.1175
Cui, Y., Hu, X., Liang, G., Feng, A., Wang, F., Ruan, S., et al. (2020). Production of novel beneficial alleles of a rice yield-related QTL by CRISPR/Cas9. Plant Biotechnol. J. 18, 1987–1989. doi: 10.1111/pbi.13370
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
de Mesa-Stonestreet, N. J., Alavi, S., Bean, S. R. (2010). Sorghum proteins: the concentration, isolation, modification, and food applications of kafirins. J. Food Sci. 75, R90–R104. doi: 10.1111/j.1750-3841.2010.01623.x
FAOSTAT (2019). Available at: https://www.fao.org/faostat/en/#data.
Fujita, N., Hanashiro, I., Suzuki, S., Higuchi, T., Toyosawa, Y., Utsumi, Y., et al. (2012). Elongated phytoglycogen chain length in transgenic rice endosperm expressing active starch synthase IIa affects the altered solubility and crystallinity of the storage alpha-glucan. J. Exp. Bot. 63, 5859–5872. doi: 10.1093/jxb/ers235
Girard, A. L., Awika, J. M. (2018). Sorghum polyphenols and other bioactive components as functional and health promoting food ingredients. J. Cereal Sci. 84, 112–124. doi: 10.1016/j.jcs.2018.10.009
Hannah, L. C., Nelson, O. E. J. (1976). Characterization of ADP-glucose pyrophosphorylase from shrunken-2 and brittle-2 mutants of maize. Biochem. Genet. 14, 547–560. doi: 10.1007/BF00485834
Hashimoto, S., Wake, T., Nakamura, H., Minamiyama, M., Araki-Nakamura, S., Ohmae-Shinohara, K., et al. (2021). The dominance model for heterosis explains culm length genetics in a hybrid sorghum variety. Sci. Rep. 11, 4532. doi: 10.1038/s41598-021-84020-3
James, M. G., Robertson, D. S., Myers, A. M. (1995). Characterization of the maize gene sugary1, a determinant of starch composition in kernels. Plant Cell. 7, 417–429. doi: 10.1105/tpc.7.4.417
Karper, R. E., Quinby, J. R. (1963). Sugary endosperm in sorghum. J. Hered. 54, 121–126. doi: 10.1093/oxfordjournals.jhered.a107240
Kawagoe, Y., Kubo, A., Satoh, H., Takaiwa, F., Nakamura, Y. (2005). Roles of isoamylase and ADP-glucose pyrophosphorylase in starch granule synthesis in rice endosperm. Plant J. 42, 164–174. doi: 10.1111/j.1365-313X.2005.02367.x
Kawai, K., Ueguchi-Tanaka, M., Matsuoka, M. (2020). Future strategy of breeding: learn by two important genes of miracle rice. Mol. Plant 13, 823–824. doi: 10.1016/j.molp.2020.05.001
Kubo, A., Fujita, N., Harada, K., Matsuda, T., Satoh, H., Nakamura, Y. (1999). The starch-debranching enzymes isoamylase and pullulanase are both involved in amylopectin biosynthesis in rice endosperm. Plant Physiol. 121, 399–410. doi: 10.1104/pp.121.2.399
Kubo, A., Rahman, S., Utsumi, Y., Li, Z., Mukai, Y., Yamamoto, M., et al. (2005). Complementation of sugary-1 phenotype in rice endosperm with the wheat isoamylase1 gene supports a direct role for isoamylase1 in amylopectin biosynthesis. Plant Physiol. 137, 43–56. doi: 10.1104/pp.104.051359
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Moraes, É.A., Marineli, R. D. S., Lenquiste, S. A., Steel, C. J., Menezes, C. B., Queiroz, V. A. V., et al. (2015). Sorghum flour fractions: Correlations among polysaccharides, phenolic compounds, antioxidant activity and glycemic index. Food Chem. 180, 116–123. doi: 10.1016/j.foodchem.2015.02.023
Mouille, G., Maddelein, M. L., Libessart, N., Talaga, P., Decq, A., Delrue, B., et al. (1996). Preamylopectin processing: A mandatory step for starch biosynthesis in plants. Plant Cell. 8, 1353–1366. doi: 10.1105/tpc.8.8.1353
Murray, M. G., Thompson, W. F. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325. doi: 10.1093/nar/8.19.4321
Myers, A. M., Morell, M. K., James, M. G., Ball, S. G. (2000). Recent progress toward understanding biosynthesis of the amylopectin crystal. Plant Physiol. 122, 989–997. doi: 10.1104/pp.122.4.989
Nakamura, Y. (2002). Towards a better understanding of the metabolic system for amylopectin biosynthesis in plants: rice endosperm as a model tissue. Plant Cell Physiol. 43, 718–725. doi: 10.1093/pcp/pcf091
Nakamura, Y. (2015). Biosynthesis of reserve starch, in starch: metabolism and structure. Ed. Nakamura, Y. (Tokyo: Springer Japan), 161–209.
Nakamura, Y., Kubo, A., Shimamune, T., Matsuda, T., Harada, K., Satoh, H. (1997). Correlation between activities of starch debranching enzyme and α-polyglucan structure in endosperms of sugary-1 mutants of rice. Plant J. 12, 143–153. doi: 10.1046/j.1365-313X.1997.12010143.x
Ozawa, K. (2012). A high-efficiency Agrobacterium-mediated transformation system of rice (Oryza sativa l.). Methods Mol. Biol. 847, 51–57. doi: 10.1007/978-1-61779-558-9_5
Pan, D., Nelson, O. E. (1984). A debranching enzyme deficiency in endosperms of the sugary-1 mutants of maize. Plant Physiol. 74, 324–328. doi: 10.1104/pp.74.2.324
Patel, M. L., Patel, G. B. (1928). “Studies of the jowars of Gujarat,” in Memoirs of the department of agriculture in India: Botanical series. Nabu Press: Charleston SC, USA
Pfister, B., Zeeman, S. C. (2016). Formation of starch in plant cells. Cell. Mol. Life Sci. 73, 2781–2807. doi: 10.1007/s00018-016-2250-x
Quinby, J. R., Karper, R. E. (1954). Sorghum improvement. Adv. Agron. 6, 305–359. doi: 10.1016/S0065-2113(08)60388-0
Rahman, A., Wong, K., Jane, J., Myers, A. M., James, M. G. (1998). Characterization of SU1 isoamylase, a determinant of storage starch structure in maize. Plant Physiol. 117, 425–435. doi: 10.1104/pp.117.2.425
Rodriguez-Leal, D., Lemmon, Z. H., Man, J., Bartlett, M. E., Lippman, Z. B. (2017). Engineering quantitative trait variation for crop improvement by genome editing. Cell 171, 470–480.e478. doi: 10.1016/j.cell.2017.08.030
Salunkhe, D. K., Kadam, S. S., Chavan, J. K. (1977). Nutritional quality of proteins in grain sorghum. Plant Food Hum. Nutr. 27, 187–205. doi: 10.1007/BF01092359
Shehzad, T., Okuizumi, H., Kawae, M., Okuno, K. (2009). Development of SSR-based sorghum (Sorghum bicolor (L.) moench) diversity research set of germplasm and its evaluation by morphological traits. Genet. Resour. Crop Evol. 56, 809–827. doi: 10.1007/s10722-008-9403-1
Shen, S., Huang, R., Li, C., Wu, W., Chen, H., Shi, J., et al. (2018). Phenolic compositions and antioxidant activities differ significantly among sorghum grains with different applications. Molecules 23, 1203. doi: 10.3390/molecules23051203
Shufen, C., Yicong, C., Baobing, F., Guiai, J., Zhonghua, S., Ju, L., et al. (2019). Editing of rice isoamylase gene ISA1 provides insights into its function in starch formation. Rice Sci. 26, 77–87. doi: 10.1016/j.rsci.2018.07.001
Sim, L., Beeren, S. R., Findinier, J., Dauvillee, D., Ball, S. G., Henriksen, A., et al. (2014). Crystal structure of the chlamydomonas starch debranching enzyme isoamylase ISA1 reveals insights into the mechanism of branch trimming and complex assembly. J. Biol. Chem. 289, 22991–23003. doi: 10.1074/jbc.M114.565044
Smith, A. M., Denyer, K., Martin, C. (1997). The synthesis of the starch granule. Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 67–87. doi: 10.1146/annurev.arplant.48.1.67
Streb, S., Delatte, T., Umhang, M., Eicke, S., Schorderet, M., Reinhardt, D., et al. (2008). Starch granule biosynthesis in arabidopsis is abolished by removal of all debranching enzymes but restored by the subsequent removal of an endoamylase. Plant Cell. 20, 3448–3466. doi: 10.1105/tpc.108.063487
Tracy, W. F. (1997). History genetics and bredding of supersweet (shrunken2) sweet corn. Plant Breed. Rev. 14, 189–236. doi: 10.1002/9780470650073.ch7
Tracy, W. F., Shuler, S. L., Dodson-Swenson, H. (2019). “The use of endosperm genes for sweet corn improvement,” in Plant breed. rev, 215–241. John Wiley & Sons: New York, USA
Tsai, C. Y., Nelson, O. E. (1966). Starch-deficient maize mutant lacking adenosine dephosphate glucose pyrophosphorylase activity. Science 151, 341–343. doi: 10.1126/science.151.3708.341
USDA-NASS (2019). United states department of agriculture–national agricultural statistics service QuickStats. Available at: https://www.nass.usda.gov/
Utsumi, Y., Utsumi, C., Sawada, T., Fujita, N., Nakamura, Y. (2011). Functional diversity of isoamylase oligomers: The ISA1 homo-oligomer is essential for amylopectin biosynthesis in rice endosperm. Plant Physiol. 156, 61–77. doi: 10.1104/pp.111.173435
Wang, M. L., Zhu, C., Barkley, N. A., Chen, Z., Erpelding, J. E., Murray, S. C., et al. (2009). Genetic diversity and population structure analysis of accessions in the US historic sweet sorghum collection. Theor. Appl. Genet. 120, 13–23. doi: 10.1007/s00122-009-1155-6
Yonemaru, J., Ando, T., Mizubayashi, T., Kasuga, S., Matsumoto, T., Yano, M. (2009). Development of genome-wide simple sequence repeat markers using whole-genome shotgun sequences of sorghum (Sorghum bicolor (L.) moench). DNA Res. 16, 187–193. doi: 10.1093/dnares/dsp005
Zeeman, S. C., Umemoto, T., Lue, W.-L., Au-Yeung, P., Martin, C., Smith, A. M., et al. (1998). A mutant of arabidopsis lacking a chloroplastic isoamylase accumulates both starch and phytoglycogen. Plant Cell. 10, 1699–1711. doi: 10.1105/tpc.10.10.1699
Keywords: sorghum, sugary endosperm, debranching enzyme, starch synthesis, novel alleles
Citation: Hashimoto S, Okada S, Araki-Nakamura S, Ohmae-Shinohara K, Miura K, Kawaguchi H, Ogino C, Kasuga S and Sazuka T (2023) An analysis of sugary endosperm in sorghum: Characterization of mutant phenotypes depending on alleles of the corresponding starch debranching enzyme. Front. Plant Sci. 14:1114935. doi: 10.3389/fpls.2023.1114935
Received: 03 December 2022; Accepted: 23 January 2023;
Published: 13 February 2023.
Edited by:
Dongcheng Liu, Hebei Agricultural University, ChinaReviewed by:
Changquan Zhang, Yangzhou University, ChinaZhanguo Xin, Agricultural Research Service, United States Department of Agriculture (USDA), United States
Kurniawan Rudi Trijatmiko, International Rice Research Institute (IRRI), Philippines
Copyright © 2023 Hashimoto, Okada, Araki-Nakamura, Ohmae-Shinohara, Miura, Kawaguchi, Ogino, Kasuga and Sazuka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Sazuka, c2F6dWthQGFnci5uYWdveWEtdS5hYy5qcA==