
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 25 January 2023
Sec. Plant Abiotic Stress
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1100895
This article is part of the Research TopicSilicon and Heavy Metal Stress in Plants: Current Knowledge and Future ProspectsView all 7 articles
 Ibrahim Khan1†
Ibrahim Khan1† Sajjad Asaf1†
Sajjad Asaf1† Rahmatullah Jan2
Rahmatullah Jan2 Saqib Bilal1
Saqib Bilal1 Lubna1Abdul Latif Khan3Kyung-Min Kim2*Ahmed Al-Harrasi1*
Lubna1Abdul Latif Khan3Kyung-Min Kim2*Ahmed Al-Harrasi1*The WRKY and bHLH transcription factors have been implicated in the regulation of gene expression during various physiological processes in plants, especially in plant stress responses. However, little information about the heavy metal-responsive SlWRKY and SlbHLH in tomato (Solanum lycopersicum) is available. We performed a genome-wide investigation for these two TF families in S. lycopersicum and determined their role in cadmium (Cd) stress tolerance. Furthermore, ortholog analysis with the Arabidopsis genome led to classifying WRKY and bHLH ortholog genes into nine and 11 clusters, respectively. The comparative phylogenetic analysis revealed duplication events and gene loss in Arabidopsis and S. lycopersicum, which occurred during evolution both before and after the last common ancestor of the two species. Orthologous relationships are also supported by additional evidence, such as gene structure, conserved motif compositions, and protein–protein interaction networks for the majority of genes, suggesting their similar functions. A comprehensive transcriptomics analysis revealed that both WRKY and bHLH genes were differentially expressed in response to cadmium stress as compared with control plants. A gene ontology analysis revealed that most WRKYs and bHLHs are DNA-binding essential proteins that regulate gene expression positively and negatively. Analyses of interaction networks revealed that both WRKYs and bHLHs mediate networks implicated in several stress-signaling pathways. The findings of this work may help us to comprehend the intricate transcriptional control of WRKY and bHLH genes and identify potential stress-responsive genes relevant to tomato genetic improvement. Moreover, identifying heavy metal stress-responsive WRKY and bHLH genes in S. lycopersicum will provide fundamental insights for developing new heavy metal stress-tolerant varieties of tomato crops.
Tomato (Solanum lycopersicum) is one of the most economically important cultivated crops worldwide. Its consumption increases annually due to its multiple utilization (Gerszberg et al., 2015). Despite its pivotal importance in breeding and evolutionary studies due to its fruit attractiveness (flavors, colors, shapes, and sizes), genome-wide research studies are limited. The recent release of the complete assembly of the sequenced genome of tomato allowed us to identify and characterize novel gene families genome-wide (Huang et al., 2012). In nature, plants, including tomatoes, constantly face various biotic and abiotic stresses. Heavy metal excess is one of the plants’ most destructive abiotic stresses (Li et al., 2022). Over the last few decades, the rapid increase in human population and anthropogenic activities such as urbanization, industrialization, and modern agricultural practices has resulted in an increase in the quantity of heavy metal toxicity in the surroundings, which causes chronic toxicity to living things (Khalil et al., 2021). Because of pesticides, fertilizers, municipal waste, smelting industries, and metalliferous mining, large areas of land have become contaminated with heavy metals (Singh et al., 2016). Although numerous heavy metals exist naturally in variable amounts in the earth’s crust, the issue arises when they are released into the environment in significant amounts as a result of natural or anthropogenic activities (Duruibe et al., 2007). Based on their density (>5 g/cm3), the 53 d-block elements have been classified as heavy metals (Dutta et al., 2018). Plant cells require only 19 elements, which are further categorized into two groups based on the essentiality for completing a plant’s life cycle, i.e., macro- and micro-elements (Subbarao et al., 2003). Macro-elements include C, H, O, N, Mg, S, P, Cd, and K, while Ni, Zn, Cu, Cl, Fe, Mn, Mo, B, Co, and Br are micro-elements. Different physiological and biochemical processes in plants are significantly influenced by these macro and micro heavy metals, such as the biosynthesis and metabolism of nucleic acids, proteins, and chlorophyll; sugar metabolism; and nitrogen fixation (Rout and Sahoo, 2015). For instance, zinc (Zn) is considered as a multipurpose micro-element due to its capacity to bind to >300 enzymes and 200 transcription factors (TFs) as a co-factor to activate these enzymes and TFs to maintain auxin metabolism, cell membrane integrity, and reproduction (Gondal et al., 2021), and a sufficient supply of nickel (Ni) is essential for several physiological processes including seed germination, optimal vegetative growth and reproductive development, and improved crop yield and quality (Khalil et al., 2021). However, high concentrations and bioavailable forms of heavy metals may adversely affect plant health, eventually resulting in cell death. Some heavy metals such as mercury (Hg), aluminum (Al), cadmium (Cd), lead (Pb), and chromium (Cr) are very toxic at very low concentrations and produce severe toxicity symptoms in plants including biomass accumulation, restriction of growth and photosynthesis, chlorosis, disturbed water balance, and nutrient absorption, all of which eventually lead to plant death (Kalaivanan and Ganeshamurthy, 2016). Heavy metal stress is estimated to be one of the major causes of global crop yield reduction. This situation has deteriorated due to a disruption in the balance between crop productivity and population increase (Shahid et al., 2015). It is therefore crucial to comprehend how plants respond to these stresses to develop novel quantitative and qualitative strategies for enhancing crops. Heavy metal stress signals trigger various physiological and biochemical pathways involving many genes to develop strategies enabling them to cope with the adverse effects of heavy metal toxicity (Singh et al., 2016). In plants, many TFs are involved in regulating the expression of various stress-responsive genes cooperatively or separately, and the genes responsible for TFs can do wonders in modifying the crops such that they can adapt in heavy metal-rich soils (Khan et al., 2021a; Manzoor et al., 2022). Regulatory genes encode biochemical substances such as sugars, alcohols, and amines, which serve an important function in plants against heavy metal stress. Several studies have shown that a single TF can influence the expression of numerous genes by selectively binding to the cis-acting element in the promoter region of its target genes and through a protein–protein or domain–domain interaction that facilitates TF oligomerization with other regulatory proteins (Shiu et al., 2005; Nakashima et al., 2009). From more than 80 different TF families, only a few, such as WRKY, bHLH, GRAS, MYB, AP2/ERF, Dof, bZIP, and DREB, with crucial roles in heavy metal stress responses have been widely studied, and much remains to be discovered about the essential regulatory roles of diverse plant TFs (Khan et al., 2021b). Only a few tomato metal-tolerance proteins (MTPs) have been studied and functionally characterized in tomato (El-Sappah et al., 2021). Therefore, it is necessary to identify genes that are potentially involved in heavy metal stress tolerance. In this regard, the current study has emphasized identifying and characterizing two large and very important families of TFs, i.e., WRKY and bHLH gene families, in tomato plants. The detailed composition and mode of action of both WRKY and bHLH TFs are well-explored (Heim et al., 2003; Chen et al., 2019; Hao et al., 2021; Wani et al., 2021; Guo et al., 2022). Here, we will focus on the comparative genomic studies and functional roles of WRKY and bHLH TFs in tomato, particularly in heavy metal defense.
The WRKY TFs are also called jack-of-all-trades because they regulate many developmental and physiological processes, such as seed dormancy and germination, seed development, root formation, plant growth, senescence, trichome morphogenesis, and response to various biotic and abiotic stress factors (Ülker and Somssich, 2004; Rushton et al., 2010; Mao et al., 2011; Huang et al., 2012; Imran et al., 2019; Guo et al., 2022). To date, 81 WRKY (Zhao et al., 2021) and 161 bHLH (Riaño-Pachón et al., 2007) TFs have been identified from the entire tomato genome through genome-wide analysis. However, very little is known about the specific physiological roles of most WRKY and bHLH genes in response to heavy metal stress in tomato plants. Here, we attempted to predict the role of these uncharacterized TFs in response to heavy metal stress by comparative genomic analysis with the most extensively studied plant species, Arabidopsis thaliana.
The current study will offer evolutionary and functional roles of WRKY and bHLH genes in tomato, which can open new windows for future studies to generate stress-resistant crop cultivars. Several in-silico analyses, such as subcellular locations, phylogenetic relationships, gene structure, conserved domains, gene ontology, protein–protein interactions, co-expression patterns, and expression pattern analysis and functional annotations, were utilized to get insights into the physiological roles of WRKY and bHLH TFs in tomato. To determine the specific regulatory role of WRKY and bHLH genes in various physiological processes, their orthologous and paralogous pairs have also been identified, and the authenticated RNA-seq data with reverse transcription-quantitative PCR (qRT-PCR) were used to evaluate their expression and to elucidate their functional roles.
The nucleotide and amino acid sequences of the AtWRKY and AtbHLH proteins of Arabidopsis were downloaded from The Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org/). Nucleotide sequences of the SlWRKY and SlbHLH proteins of S. lycopersicum were retrieved from Phytozome (https://phytozome-next.jgi.doe.gov/) and Sol Genomics Network (SGN, https://solgenomics.net/), while their corresponding protein sequences were downloaded from the Plant Transcription Factors Database (PlantTFDB, http://planttfdb.gao-lab.org/). The subcellular localizations of SlWRKY and SlbHLH proteins were predicted using CELLO (http://cello.life.nctu.edu.tw/) (Table S1). The protein sequences of putative SlWRKY and SlbHLH members were subjected and analyzed with Expasy ProtParam (https://web.expasy.org/protparam) and SGN (https://solgenomics.net/) to determine their theoretical isoelectric point (pI), molecular weight (Mw), and number of amino acids (AAs).
Neighbor-joining (NJ) phylogenetic trees of the 72 Arabidopsis with 81 S. lycopersicum WRKY and 153 Arabidopsis with 161 S. lycopersicum bHLH proteins were constructed using the software MEGA 11 with 1,000 replicates for bootstrap (BS) analysis for statistical reliability to compare the evolutionary relationships of WRKY and bHLH TFs across the species. We further performed NJ phylogenic tree analysis among WRKY and bHLH protein sequences in S. lycopersicum to test the reliability of the results.
The CDS and genomic sequences of the WRKY and bHLH of Arabidopsis and S. lycopersicum were retrieved from TAIR (https://www.arabidopsis.org) and Phytozome (https://phytozome-next.jgi.doe.gov/), respectively. The Gene Structure Display Server (GSDS, http://gsds.gao-lab.org/) web tool was used to analyze the structure of the members of the WRKY and bHLH gene families and to detect their exon–intron organization, by aligning the CDS sequences with genomic sequences. To identify the conserved motifs in these protein sequences, the Multiple EM for Motif Elicitation (MEME, https://meme-suite.org/meme/tools/meme) online server was used with the following parameters: number of repetitions, any; maximum number of motifs, 15; and optimum motif width set to ≥6 and ≤200 amino acid residues.
In order to determine the physical locations of the SlWRKY and SlbHLH genes in the S. lycopersicum genome, the starting and ending positions of all the identified genes on each chromosome were obtained from the Solanaceae family database (Sol Genomics Network). The MapInspect software (http://mapinspect.software.informer.com/) was used to map the physical locations of the SlWRKY and SlbHLH genes on their respective chromosomes. The duplicate chromosomal blocks were retrieved using the Plant Genome Duplication Database (PGDD, accessible at http://chibba.agtec.uga.edu/duplication/), and the WRKY and bHLH genes within the duplication block were identified. This allowed us to find duplicate S. lycopersicum WRKY and bHLH genes (Lee et al., 2012). Genes separated by five or fewer gene loci in a range of 100 kb distance were considered to be tandem duplicates, and those which were co-paralogs and located within duplicated chromosomal blocks in multiple locations as a result of duplication and chromosome rearrangement and shared >90% sequence identity were considered as segmental duplicates (Wei et al., 2007). The PGDD, a public web service database, was used to identify and characterize the genes in terms of intra- or interplant genomic syntenic relationships.
In the current study, tomato seeds (S. lycopersicum cv. Yegwang) were used. First, the seeds were surface-sterilized with 10% hypochlorous acid and 70% ethanol and washed with autoclaved distilled water to remove the impurities. The soaked seeds were germinated on hygiene filter paper in an incubator at 30°C in dark conditions. After successful sprouting, the seeds were planted in plastic pots in a greenhouse. The greenhouse temperature was kept constant at 28°C ± 2°C, 55% ± 5% relative humidity, and 16-h light/8-h dark photoperiod. The experiment was conducted utilizing three groups of plants: a) distilled water-treated plants (control plants), b) 1-mM Cd-treated plants, and c) 2-mM Cd-treated plants. After every 3 days, the plants were treated with their respective treatments under the same growth conditions. Four replicates were prepared per treatment. Leaf samples were collected randomly and immediately placed in liquid nitrogen and stored at –80°C in a fridge until analysis.
RNA was isolated using the RNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). The quantity of total RNA was adjusted to 10 μg, and the NanoDrop ND-1000 (Thermo Scientific, Waltham, MA, USA) was used to evaluate the quality. Sequencing and analysis of libraries from three different biological replicates of each treatment were performed. The Illumina HiSeq 2000 platform was utilized to perform library construction according to the described approach (Liao et al., 2015), resulting in single-end reads with 51 bp. To identify variations in gene expression regulation between the cadmium-treated and non-treated plants, a computational pipeline of optimized tools was employed. For trimming and quality check, Trim Galore (Krueger, 2012) and FastQC (Andrews, 2010) were used, respectively. To align the reads to the reference genome, HISAT2 (Sirén et al., 2014) was used before the read count quantification by using Feature Count (subread_v2.0.2). The R software using the DESeq2 (Love et al., 2014) package was used for differential gene expression analysis.
The STRING protein interaction database version 11.5 (https://string-db.org) was used to identify the interacting protein networks and functional annotations.
The online software agriGO analysis toolbox (http://bioinfo.cau.edu.cn/agriGO/) was used to enrich gene ontology (GO) categories (Du et al., 2010) using the TopGO “elim” algorithm (Alexa et al., 2006) covering the following characteristics: biological processes, molecular processes, and cellular processes.
For qRT-PCR analysis, approximately 10 highly expressed genes (5 WRKY and 5 bHLH) from RNA-seq data were selected to authenticate the RNA-seq results. The Primer3 (https://bioinfo.ut.ee/primer3-0.4.0/) program was used to design primers for each selected gene as listed in Table S3. The standard cDNA was produced using PCR Biosystems’ qPCRBIO cDNA Synthesis Kits after total RNA was diluted to a final concentration of 100 ng/μl, and transcript quantification was done as reported previously (Asaf et al., 2022). Actin (a housekeeping gene) was used as an internal control to normalize gene expression, and the comparative ΔΔCt method of qRT-PCR was utilized to calculate the expression level of the genes in control plants in comparison with Cd-treated ones.
In order to systematically identify and analyze WRKY and bHLH genes in the tomato genome, their sequences were downloaded as described above. Finally, a total of 81 and 161 non-redundant putative WRKY and bHLH genes were confirmed by their specific domain using SMART (http://smart.embl-heidelberg.de) (Table S1). Subsequently, the downloaded gene sequences were compared with their expressed sequence tag (EST) sequences in PlantTFDB (http://planttfdb.gao-lab.org). Arabidopsis WRKY and bHLH genes were obtained from the TAIR (https://www.arabidopsis.org) and PlantTFDB (http://planttfdb.gao-lab.org). A total of 72 WRKY and 161 bHLH annotated TFs were extracted from these sources (Table S1).
In order to determine the evolutionary and phylogenetic relationship among the S. lycopersicum WRKY as well as among bHLH proteins, unrooted neighbor-joining phylogenetic trees were built by a multiple sequence alignment of their amino acid sequences using the ClustalW program. As shown in Figure 1A, WRKY TFs of S. lycopersicum were divided into six main groups (group I to group VI). Notably, the maximum number, i.e., 19 (23.4%) of the WRKY members, was found in group I, followed by groups III and IV with 18 (22.2%) members. The least number, i.e., 6 (7.4%) of the WRKY members, was found in group V. Similarly, bHLH TFs were divided into eight main groups (group I to group VIII) based on the phylogenetic tree. The maximum number, i.e., 32 (19.9%) of the bHLH members, was found in group I, followed by group VIII with 30 (18.6%) and group V with 26 (16%) members. The least number, i.e., 7 (4%) of the bHLH members, was found in group II (Figure 1B). Furthermore, most of the members from the same phylogenetic group have the same number of exons and conserved exon–intron structure (Figures 2, 3). For instance, in groups III and IV, 18 (75%) and 14 (78%) of the SlWRKY members have three exons, respectively. On the other hand, in the phylogenetic tree of SlbHLH TFs, group VIII contained the majority of the genes with 6–13 exons. Our results also showed that most of the evolutionarily related members have similar motif compositions and the same subcellular locations (Figures S1, S2 and Table S1). The similarity in these features may be related to their specific physiological functions in tomato cells. The high bootstrap values (95%–100%) indicate a strong phylogenetic relationship between some pairs of the SlWRKY TFs. For instance, Solyc05g050050–Solyc05g050060, Solyc01g058540–Solyc04g051540, and Solyc05g050330–Solyc05g050340 pairs of SlWRKY genes showed a paralogous relationship, supported by 99% of bootstrap values (Figure S3), while some pairs of SlbHLH TFs, i.e., Solyc01g014910–Solyc09g018130, Solyc01g020170–Solyc01g107970, Solyc03g119390–Solyc12g036470, Solyc09g089870–Solyc10g006510, and Solyc10g006510–Solyc08g075090, had 100% bootstrap values as shown in Figure S4.
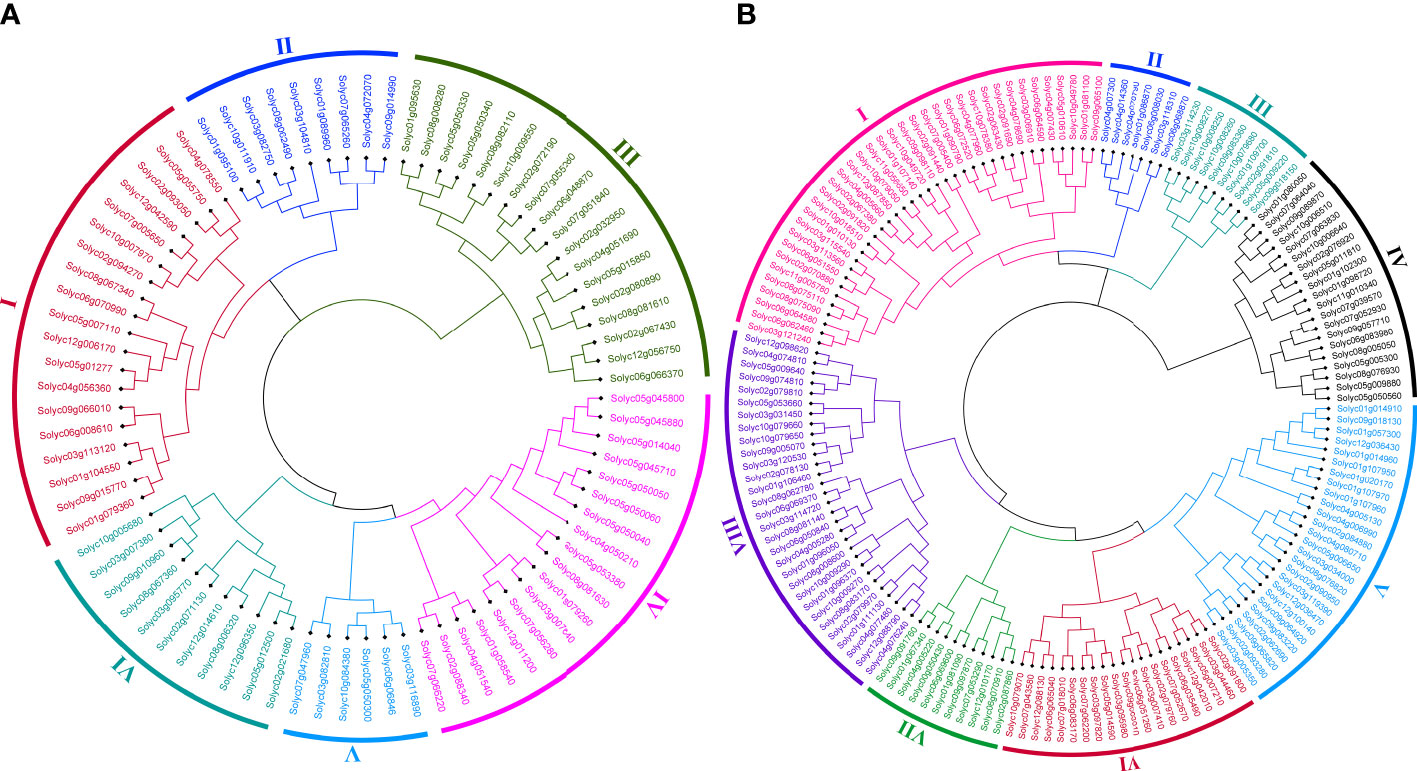
Figure 1 (A) The phylogenetic tree of 81 Solanum lycopersicum WRKY TFs constructed by the neighbor-joining (NJ) method using the MEGA 11 software with 1,000 bootstrap replicates. The six major phylogenetic groups are marked as I to IV, respectively. (B) The phylogenetic tree of 161 S. lycopersicum bHLH TFs constructed by the NJ method using the MEGA 11 software with 1,000 bootstrap replicates. The eight major phylogenetic groups are marked as I to VIII, respectively.
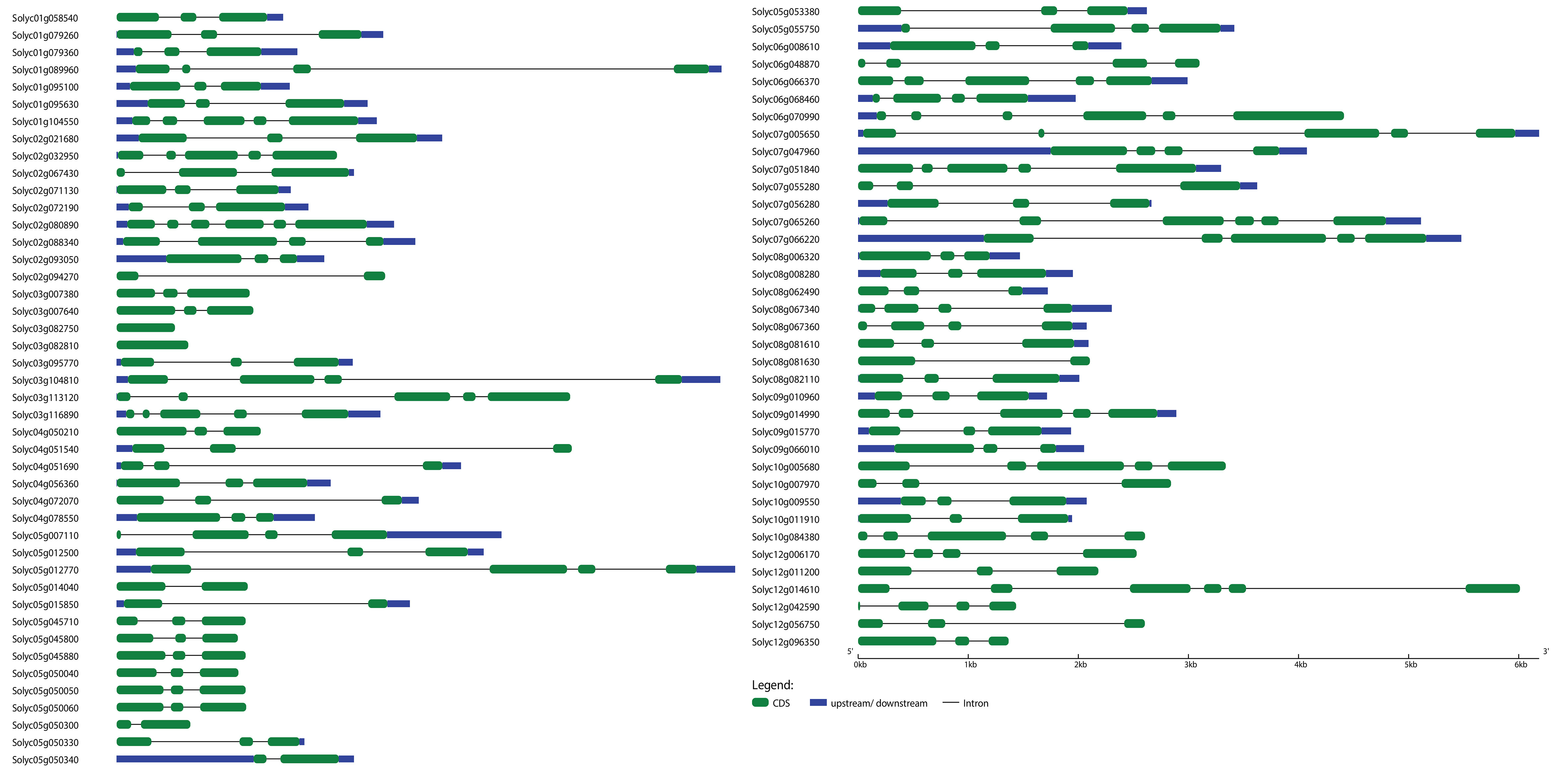
Figure 2 Exon–intron structure analysis of Solanum lycopersicum WRKY genes was performed by the GSDS database. The blue boxes indicate upstream/downstream, the green boxes indicate exons, and the black lines indicate introns.
To identify the evolutionary history and phylogenetic relationship between the SlWRKY (81 members) and AtWRKY (72 members) and between the SlbHLH (161 members) and AtbHLH (153 members), unrooted NJ phylogenetic trees were derived from the alignment of the amino acid sequences of these TFs. The tree inferred from the WRKY TFs of Arabidopsis and S. lycopersicum organized WRKY into nine main groups (group I to group IX) (Figure 3A). Among the nine main groups, group IX is the largest one containing 29 members, followed by group I with 24 members and group IV and group VIII with 20 members, while group VI was the smallest which contained only nine members. For bHLH TFs, 11 groups (group I to group XI) were defined based on their constructed phylogenetic tree. Group V is the largest one with 50 bHLH members, followed by group I with 45 members and group III with 41 members, while group II has the least number (only 10) of members (Figure 3B).
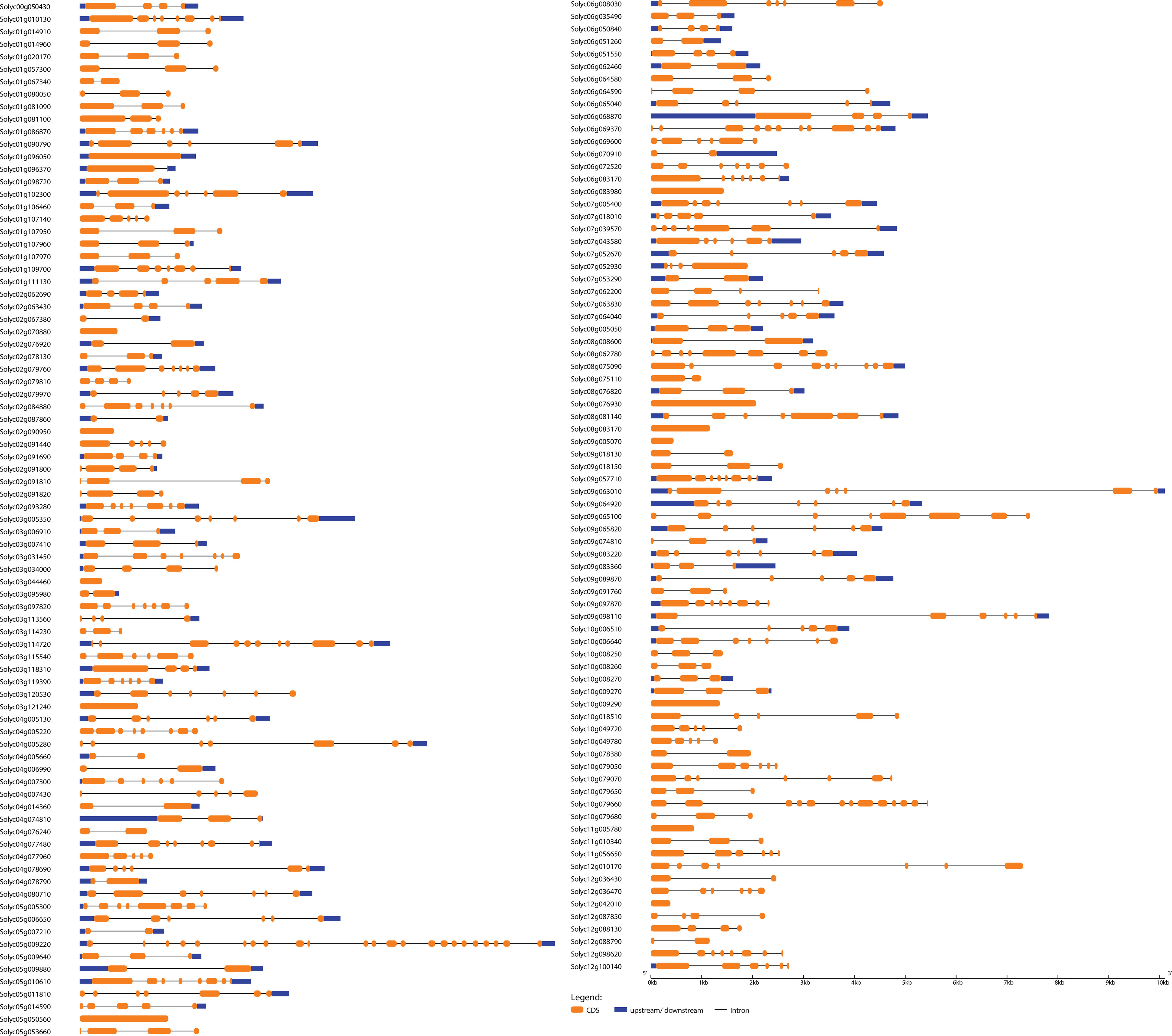
Figure 3 Exon–intron structure analysis of S. lycopersicum bHLH genes was performed by the GSDS database. The yellow boxes indicate upstream/downstream, the blue boxes indicate exons, and the black lines indicate introns.
Genes that descended from a common ancestral gene are likely to have the same functions. Therefore, homological analysis is widely used to predict the function of an uncharacterized gene (Dai et al., 2020). Paralogs are homologous genes evolved by duplication of an ancestral gene which can keep the same or diverge to a different functional role (Gabaldón and Koonin, 2013), and orthologous relationships between genes from different species can reflect the same evolutionary and functional niche in different species (Fang et al., 2010). The tree topology and the organization of most of the groups and subgroups resembled those from the S. lycopersicum and Arabidopsis individual trees. The trees presented in Figures 1, 3 identified putative paralogs and orthologs. A total of 27 and 50 orthologous pairs of WRKY and bHLH genes, respectively, were identified across the species. Most S. lycopersicum and Arabidopsis paralogous genes observed in Figures 3A, B were already displayed as paralogs in the respective trees (Figures 1A, B), for instance, Solyc01g058540–Solyc04g051540, Solyc05g050330–Solyc05g050340, and Solyc05g050050–Solyc05g050060 pairs of the SlWRKY TFs and Solyc09g089870–Solyc10g006510, Solyc08g075090–Solyc08g075110, and Solyc01g020170–Solyc01g107970 pairs of the SlbHLH TFs. The exception of a few TFs for not displaying in the same paralogous pairing in both individual and combined trees may be due to the existence of an apparent ortholog from a different species, providing better support for the new location, e.g., Solyc12g014610–AT1G13960 and Solyc02g021680–AT2G40740 pairs of group VI of the combined phylogenetic tree of SlWRKY and AtWRKY TFs. It is worth noting that most of the described phylogenetic groups and subgroups were supported by additional evidence, such as gene structure, conserved motifs, gene ontology, protein–protein interaction, and similarity in physiological functions of most of the characterized genes.
Analysis of the orthologous and paralogous relationships is widely used to predict the function of an uncharacterized gene because the genes that descended from a common ancestral gene are likely to have the same functions (Dai et al., 2020). When comparing multigene families of different species, it is frequent and interesting to find multiple genes in one species that are orthologous to a single gene in the other, indicating recent duplications specific to the former. The orthologous genes with similar sequences and the same expression patterns in various organisms suggest a possible case of functional redundancy. There are no detailed data on the specific physiological roles of most S. lycopersicum genes. Since both WRKY and bHLH TFs have important roles in several important major developmental and physiological processes and responses to different environmental stimuli, it is critical to conduct extensive research on the WRKY and bHLH gene families in S. lycopersicum. The comparative genome-wide study of S. lycopersicum WRKY and bHLH with WRKY and bHLH TFs of Arabidopsis, which is the most extensively studied plant, will allow us to predict their physiological functions. For instance, AT1G01260 is involved in negatively regulating jasmonic acid during leaf senescence and making an orthologous pair by a strong BS score with the Solyc01g096050 gene of the bHLH family. Similarly, AT1G26945 is involved in ABA and salt stress responses resulting from an orthologous pair by a strong BS score with Solyc06g070910 of the bHLH family (Hao et al., 2021). So, based on these orthologous relationships, we can predict the function of Solyc01g096050 and Solyc06g070910 genes in S. lycopersicum.
To further understand the evolutionary patterns and gene duplication events, structural and compositional analyses of genes can be used as supporting evidence (Khan et al., 2021a). The WRKY and bHLH genes’ exon–intron patterns and conserved motifs were studied to provide insight into the evolution of these two gene families in the S. lycopersicum genome. The results showed that the number of exons and introns in WRKY genes ranged from 6 to 1 and 0 to 5, respectively, whereas the number of exons and introns in bHLH genes ranged from 23 to 1 and 0 to 22, respectively (Figures 2, 4). The majority of the genes (45 WRKY and 35 bHLH) from both families have three exons and two introns. Genes belonging to the same group of phylogenetic trees almost had parallel structures apart from a few genes. Among the SlWRKYs and SlbHLHs, Solyc07g005650 and Solyc09g063010 possess the longest structures, respectively. Among all SlbHLHs, a few genes have a complex structure, such as Solyc05g009220, Solyc10g079660, and Solyc03g114720. The exon–intron arrangement of the SlbHLH genes showed that there are exons lost or gained during the evolution process of the S. lycopersicum genome.
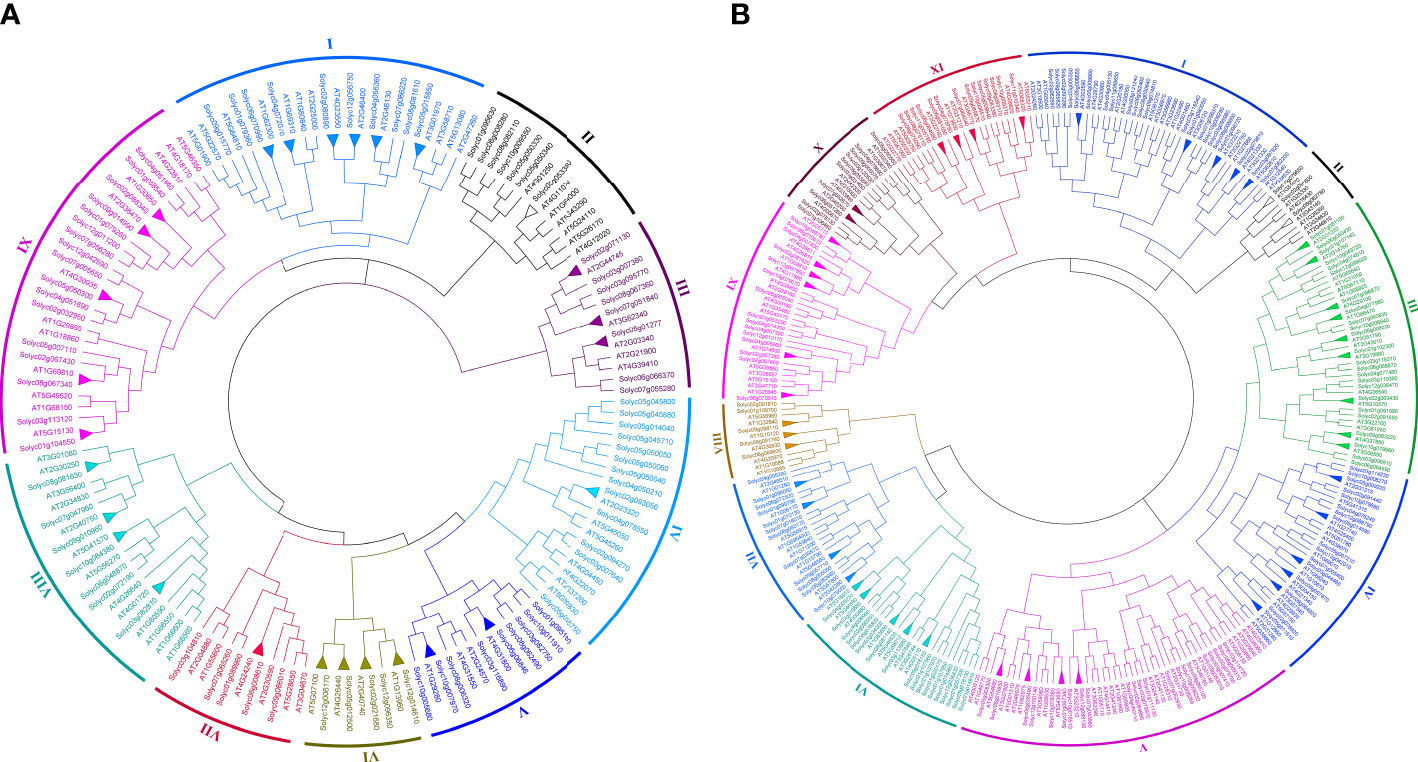
Figure 4 (A) Joined phylogenetic tree constructed from an alignment of 81 Solanum lycopersicum (SlWRKYs) and 72 Arabidopsis thaliana (AtWRKYs) protein sequences by the NJ method with bootstrapping (1,000 replicates) using the MEGA 11 software. The resulting nine groups are shown in different colors. (B) Joined phylogenetic tree constructed from an alignment of 161 S. lycopersicum (SlbHLHs) and 153 A. thaliana (AtbHLHs) protein sequences by the NJ method with bootstrapping (1,000 replicates) using the MEGA 11 software. The resulting 11 groups are shown in different colors.
The MEME software was used to recognize conserved motifs to better understand the divergence and possible function of the WRKY and bHLH gene families (Figures S1, S2). Like gene structure, the motif distributions were similar within the same phylogenetic group. For instance, most of the members of group IV of the individual phylogenetic tree of SlWRKY have eight conserved motifs, and most of the members of group V of the SlbHLH individual tree have seven conserved motifs. The aqua blue-colored motifs were uniformly found in almost all of the WRKY and bHLH TFs, so these motifs, i.e., DPSIVITTYEGEHNH and KTDKASMLDEAINYIKELQKQ, significantly represented the conserved WRKY and bHLH domains, respectively. We did not identify a potentially conserved motif (aqua blue) in only two members (Solyc10g008250 and Solyc10g008270) of the bHLH family possibly due to lack of homology, sequence repetition, rearrangements, or disruption of alignment (Gribskov, 2019). These results of the gene structures and conserved motifs suggested that TFs clustered in the same group might be meaningful for gene evolution and their physiological roles.
We map the chromosomal locations of the SlWRKY and SlbHLH genes and illustrate that majority of the SlWRKYs and SlbHLHs are clustered at the chromosomal ends. The 81 SlWRKY genes are distributed in all S. lycopersicum chromosomes except chromosome 11. As represented in Figure 5, most of the WRKY genes (19.7%) were located on chromosome 5, followed by chromosome 2 with 11.1% and chromosomes 3 and 8 with 9.8% of WRKY genes. In S. lycopersicum, out of 161, 160 bHLH genes were mapped from chromosomes 1 to 12, while the precise location of only one SlbHLH gene could not be determined. Chromosome 1 contains the highest number (13.7%) of bHLH genes, followed by chromosome 2 with 11.2% and chromosomes 3, 6, and 10 with 10% of bHLH genes, while chromosome 11 contains only 1.8% of the genes.

Figure 5 Chromosomal localizations of SlWRKY and SlbHLH genes on the 12 chromosomes of Solanum lycopersicum.
We further determined the tandem duplications of both WRKY and bHLH genes along the 12 S. lycopersicum chromosomes. Tandemly duplicated genes were characterized as an array of two or more homologous genes separated by 100 kb (Khan et al., 2021a). The gene duplication study showed that more than 50% of SlWRKY and SlbHLH genes were tandemly duplicated. As shown in Figure 6, approximately 25 SlWRKY genes were tandemly duplicated unevenly on distinct chromosomes. Chromosome 2 had the most with eight SlWRKY gene pairs, followed by chromosome 5 with six SlWRKY gene pairs. Only one SlWRKY gene pair was discovered on chromosome 9, and chromosomes 8 and 11 had no WRKY gene pairs, while the Solyc05g045800 (SlWRKY67) and Solyc05g045710 (SlWRKY65) genes mapped on chromosome 5 had two pairs. The findings showed that segmental duplications assisted in the expression of SlWRKY genes. Similarly, approximately 28 SlbHLH gene pairs were discovered on distinct chromosomes in the S. lycopersicum genome. The maximum number of tandemly duplicated SlbHLH genes on chromosome 1 was 13, showing that a large number of SlWRKY genes on chromosome 1 were partly attributable to tandem gene duplication events.
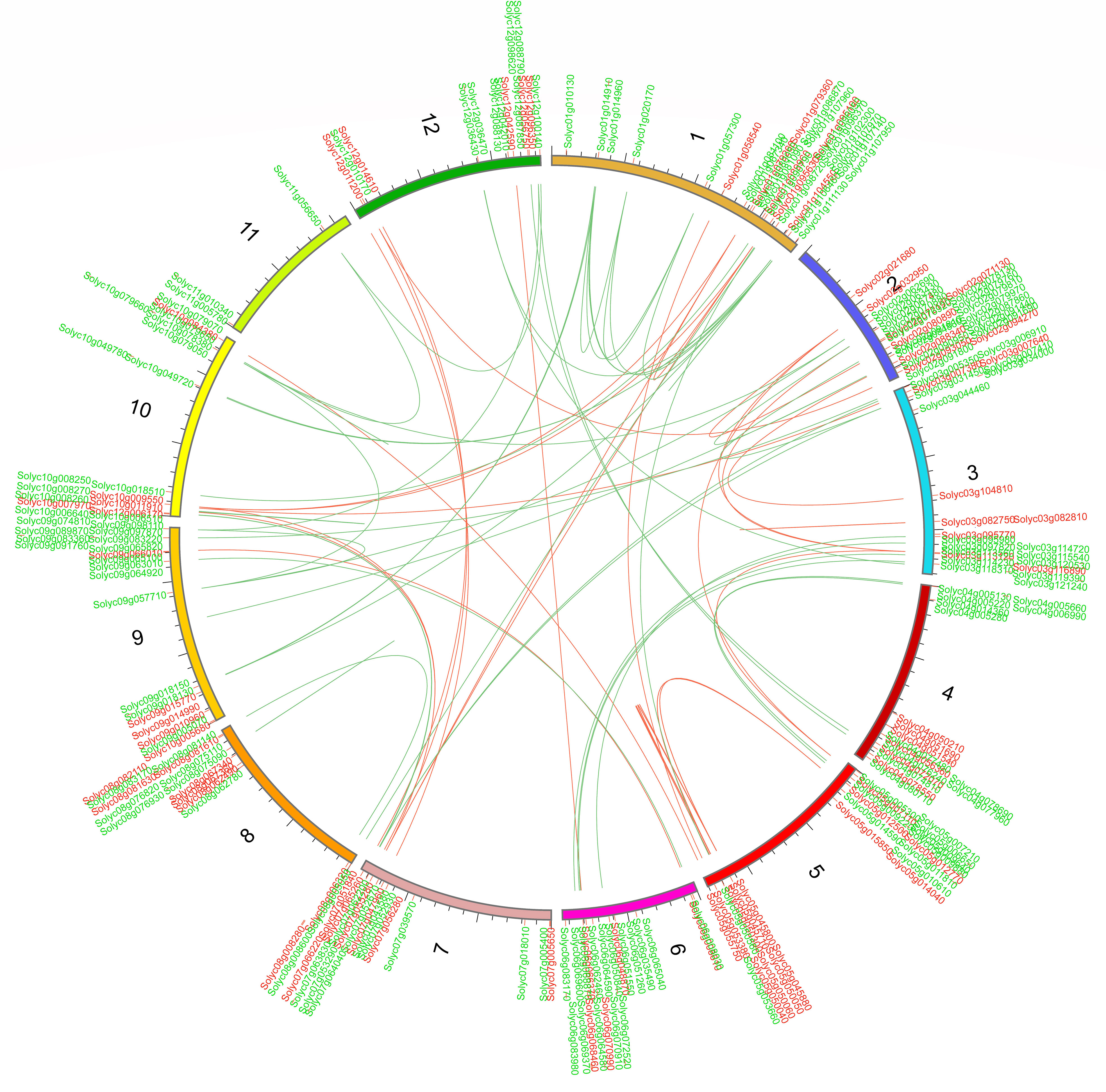
Figure 6 Chromosomal positions and interchromosomal groups of duplicated SlWRKY and SlbHLH gene pairs were mapped on the 12 Solanum lycopersicum chromosomes (Chr1–Chr12). The red and green lines represent the segmental or tandem duplication network zones among WRKY and bHLH genes, respectively.
Some WRKY, as well as bHLH proteins of S. lycopersicum, regulate the transcription process of the target genes when they form homodimer or heterodimer complexes to determine the DNA binding sites, and as a result, protein–protein interactions are critical in gene expression. The protein sequences in FASTA format were submitted to the STRING server. The protein–protein interaction network of differentially expressed proteins was constructed with the default setting. The retrieve included networks of the TFs (WRKY and bHLH), which highlight several hub proteins (Table S2). WRKY proteins that have co-expression supported by a high confidence score (0.700) include WRKY70 (Solyc03g095770), which form a network with SlWRKY33A (Solyc06g066370), Solyc09g014990, Solyc03g116890, Solyc05g050300, and SlWRKY40 (Solyc06g068460), while Solyc05g014040 has co-expression with Solyc07g005650 and Solyc09g010960 (Figure 7A). In the bHLH family, the TFs with a higher co-expression score include Solyc07g043580–Solyc01g102300, Solyc08g075090–Solyc05g009220, and ICE1a (Solyc06g068870)–Solyc08g005050, while Solyc08g076820 has co-expression with Solyc05g053660 and Solyc09g091760 (Figure 7B).
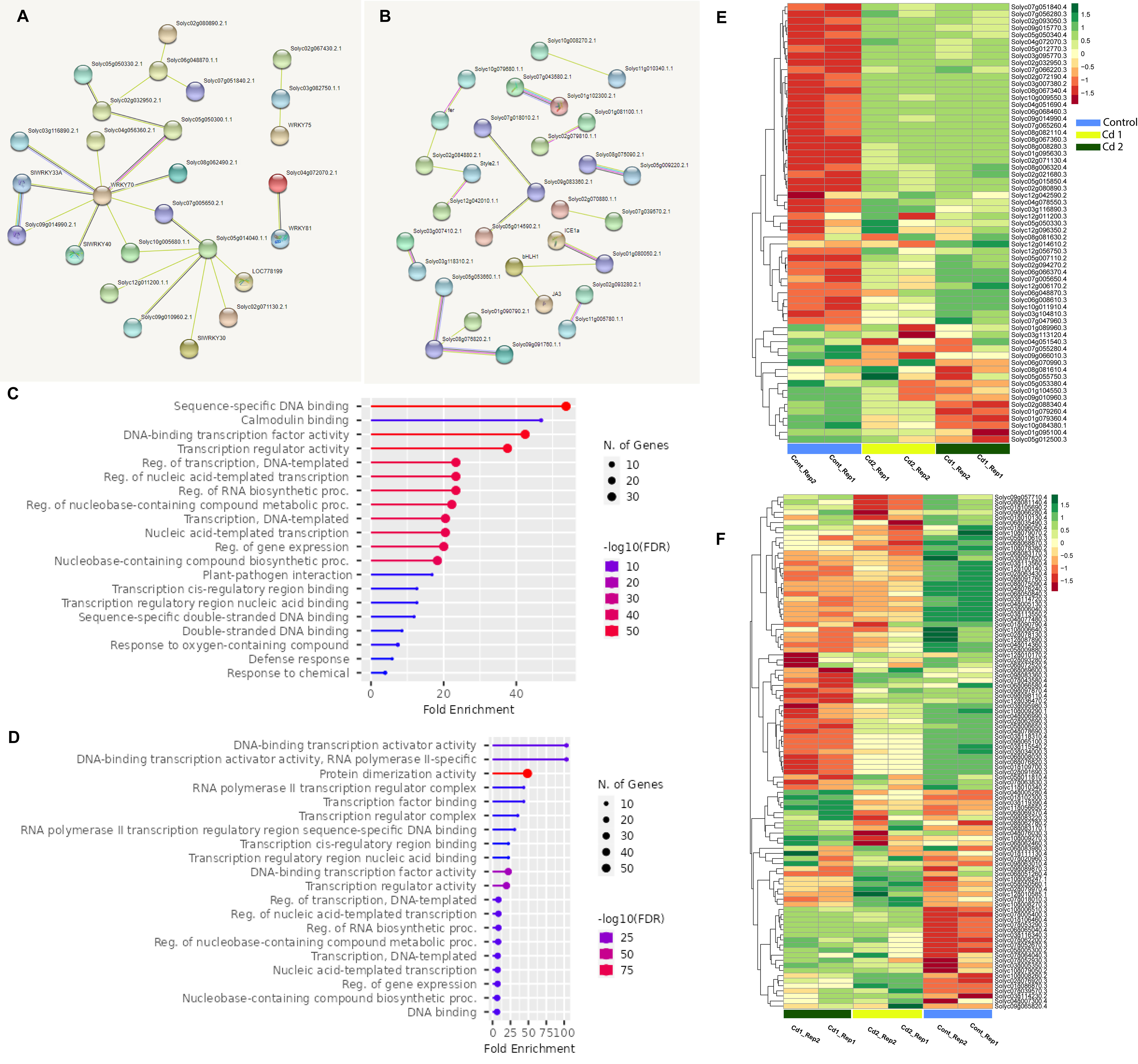
Figure 7 (A) Protein–protein association network of the SlWRKY genes based on their available information. The online tool STRING was used to predict the entire network. Different line colors represent the type of evidence for the associations, which are shown in the legend. (B) Protein–protein association network of the SlbHLH genes based on their available information. The online tool STRING was used to predict the entire network. Different line colors represent the type of evidence for the associations, which are shown in the legend. (C) Gene ontology (GO) enrichment of the SlWRKY genes. The size of the circles represents the number of genes in each category. (D) GO enrichment of the SbHLH genes. The size of the circles represents the number of genes in each category. Heatmap showing the expression patterns of transcriptome-wide differentially expressed (E) SlWRKY and (F) SlbHLH genes. The expression levels are quantified as log2 FPKM. Red and green colors indicate up- and downregulated, respectively. Two biological replicates were used for the control, 1 mM of Cd stress, and 2 mM of Cd stress.
To characterize the functions of SlWRKY and SlbHLH genes, GO enrichment analysis was conducted using the GO online tool. The results showed that the majority of the genes of both families were primarily involved in molecular functions and biological processes such as DNA binding (GO:0003677), regulation of DNA-templated transcription (GO:0006355), regulation of gene expression (GO:0010468), protein dimerization process (GO:0046983), regulation of RNA biosynthesis (GO:2001141), and nucleobase-containing compound biosynthetic process (GO:0034654) (Figures 7C, D).
WRKY and bHLH transcription factors play an important role in plants’ heavy metal stress defense (Guo et al., 2022). However, little information is available about the Cd-responsive WRKYs and bHLHs in tomato (S. lycopersicum). To determine the expression levels of the target genes in tomato plants under heavy metal stress, RNA-seq analysis was taken into consideration. The overall analysis of downregulated and upregulated genes was based on the differentially expressed genes in both Cd concentrations (1 and 2 mM of Cd) (Figures 7E, F). Genes were downregulated and upregulated based on log2 fold-change >2 and log2 fold-change <−2, respectively, with an adjusted p-value <0.05. Overall, 54 and 47 SlWRKY genes were differentially expressed in 1 and 2 mM of Cd stress, respectively, in which 43 and 40 genes were upregulated while 11 and 7 SlWRKY genes were found to be downregulated (Figure 8A). Similarly, approximately 46 common SlWRKYs were differentially expressed in both 1 and 2 mM of Cd stress (Figure 8B). On the other hand, a total of 70 and 69 SlbHLH genes were differentially expressed in 1 and 2 mM of Cd stress, respectively. However, most of the SlbHLH genes were to be downregulated in both 1 and 2 mM of Cd stress (Figures 8A–C). These findings suggest significant variations in both WRKY and bHLH genes between the control and Cd stress plants (Figures 8D, E). As shown in Figure 8D, our results revealed that SlWRKYs have almost the same expression levels under 1 and 2 mM of cadmium stress. Cadmium stress highly enhanced the expression of the following WRKY genes: Solyc05g007110 (SlWRKY76), Solyc02g094270 (SlWRKY38), Solyc08g067340 (SlWRKY46), Solyc06g048870 (SlWRKY19), Solyc09g014990 (SlWRKY33), Solyc02g021680 (SlWRKY35), Solyc08g067360 (SlWRKY45), Solyc04g051690 (SlWRKY51), and Solyc04g072070 (SlWRKY55). Contrastingly, cadmium stress downregulated the expression of Solyc01g104550 (SlWRKY9), Solyc10g084380 (SlWRKY44), Solyc09g010960 (SlWRKY49), Solyc08g081610 (SlWRKY29), Solyc04g051540 (SlWRKY13), and Solyc06g070990 (SlWRKY74). The results demonstrated that most of the SlbHLH genes’ overall expression level was downregulated during 1 and 2 mM of Cd stress, respectively (Figure 8E). The expression pattern of a few genes, including Solyc05g005300 (SlbHLH084), Solyc07g062200 (SlbHLH088), Solyc08g083170 (SlbHLH056), Solyc07g052930 (SlbHLH141), Solyc07g039570 (SlbHLH051), Solyc10g008260 (SlbHLH093), Solyc03g114230 (SlbHLH082), Solyc02g076920 (SlbHLH013), and Solyc01g106460 (SlbHLH007), was found to be upregulated. On the other hand, the expression of Solyc04g078690 (SlbHLH035), Solyc04g076240 (SlbHLH033), Solyc03g118310 (SlbHLH083), and Solyc08g076820 (SlbHLH146) was found to be significantly downregulated when S. lycopersicum plants were treated with cadmium which shows the importance of bHLH transcriptional factors in Cd stress tolerance.
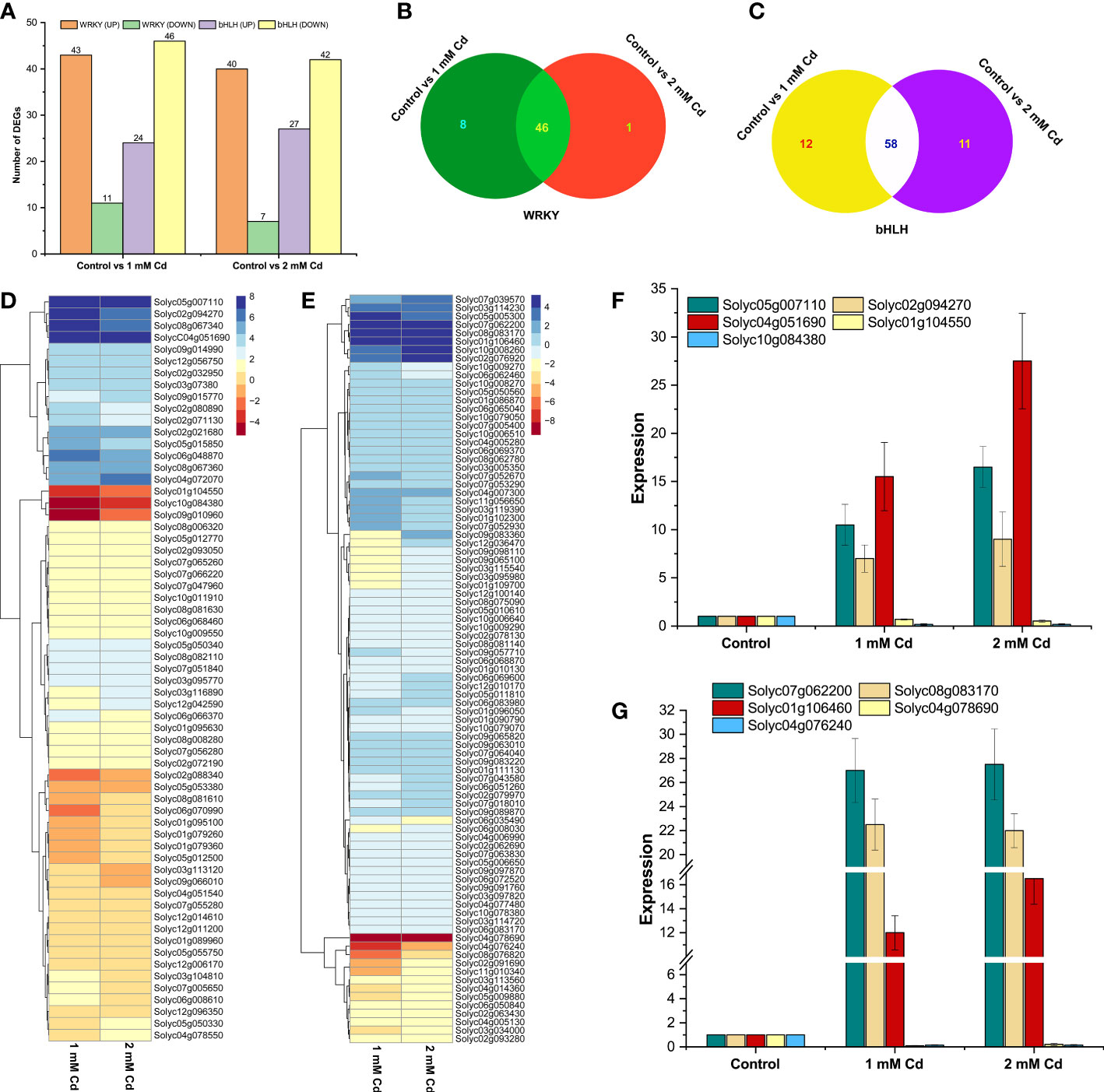
Figure 8 (A) Statistics of DEG (upregulated and downregulated) gene count comparisons of 1 and 2 mM of Cd stress with control. (B) Venn diagrams of the changes in differentially expressed SlWRKY genes in 1 and 2 mM of Cd stress vs. control. (C) Venn diagrams of the changes in differentially expressed SlbHLH genes in 1 and 2 mM of Cd stress vs. control. (D) Expression profiling of SlWRKY genes under Cd stress and (E) expression profiling of SlbHLH genes under Cd stress after DESeq2 analysis. Quantitative real-time PCR (qRT-PCR) verification of the expression level of (F) five SlWRKY genes and (G) five SlbHLH genes obtained by RNA-seq under 1 and 2 mM of Cd stress conditions. The X-axis represents the treatments, and the Y-axis shows the relative expression levels of the genes validated by qRT-PCR.
To check the RNA-seq data accuracy, the relative expression levels of five SlWRKY (Solyc05g007110, Solyc02g094270, Solyc04g051690, Solyc01g104550, and Solyc10g084380) and five SlbHLH (Solyc07g062200, Solyc08g083170, Solyc01g106460, Solyc04g078690, and Solyc04g076240) genes were determined using qRT-PCR with specific primers (Table S3). The expression patterns followed the same pattern as the RNA-seq data, and a substantial relationship was established between the RNA-seq and RT-qPCR results, demonstrating the reliability of RNA-seq data (Figures 8F, G).
The threat of increased heavy metal toxicity is expected to escalate as anthropogenic activities expand over the world (Maanan et al., 2015). Heavy metal stress can induce serious issues that affect the plant’s physiology and causes harmful effects leading to large yield loss and, through the food chain, affects consumers (Khan et al., 2015). Plants acquired different molecular and cellular adaptations to protect themselves from heavy metal toxicity (Ghori et al., 2019). However, most plants, including tomatoes, still need to be elucidated regarding their response to heavy metal stress. Various transcription factors play a vital role in response to heavy metal stress (Manara, 2012; Manzoor et al., 2022). Characterizing the core underlying regulatory network may advance our understanding of the TFs’ functions in heavy metal stress tolerance. Relatively little information is available regarding tomato WRKY and bHLH genes. The information generated from the comparative genome-wide analysis will provide insight into the identification and comprehensive functional characterization of the WRKY and bHLH gene families in S. lycopersicum. We provided the first genome-wide study of tomatoes’ WRKY and bHLH gene families and their possible role in heavy metal stress tolerance. In the current study, we identified 81 and 161 genes encoding WRKY and bHLH transcription factors in the S. lycopersicum genome, respectively. The phylogenetic relationship, subcellular locations, conserved motifs, gene structure, and exon–intron organization were executed to increase our understanding of WRKY and bHLH genes in S. lycopersicum, especially in heavy metal stress tolerance. The results suggested that the same phylogenetic group members were closely related during evolution. Ortholog genes are related genes with the same gene function that may have evolved through speciation processes. The occurrence of a greater number of orthologous gene pairs in the Arabidopsis–S. lycopersicum WRKY and bHLH TF families may indicate ancestral interactions between Arabidopsis and S. lycopersicum prior to separation during evolution. In Arabidopsis, AT4G01250 (AtWRKY22) has a close orthologous relationship with Solyc05g050340 (SlWRKY58) and Solyc05g050330 (SlWRKY59), and AT2G30250 (AtWRKY25) and AT4G23550 (AtWRKY29) have orthologous pairs with Solyc08g081630 (SlWRKY56) and Solyc02g080890 (SlWRKY6), respectively, and are expressed in the roots and leaves where they induced tolerance to Cu2+ stress (Li et al., 2020). The AT1G62300 (AtWRKY6) has an orthologous pair with Solyc06g070990 (SlWRKY74), which is involved to modulate heavy metal uptake (Castrillo et al., 2013). Similarly, AT2G30250 (AtWRKY25) and AT4G23550 (AtWRKY29) are orthologous partners of Solyc08g081630 (SlWRKY56) and Solyc02g080890 (SlWRKY6) and are involved in tolerance against Cu and Cd (Opdenakker et al., 2012). In the bHLH family, AT5G04150 (AtbHLH101) is an ortholog of Solyc09g097870 (SlbHLH062) and plays a crucial role in Fe deficiency responses (Sivitz et al., 2012). AT3G23210 (AtbHLH34) is an ortholog of Solyc12g088130 and positively regulates Fe homeostasis in Arabidopsis (Li et al., 2016). Gene structure analysis explains evolutionary processes such as duplication events (Abdullah-Zawawi et al., 2021). In the current study, two different TF orthologous gene families from Arabidopsis and S. Lycopersicum displayed various exon and intron numbers and organization of conserved motifs, implying possible roles in the diversification events of the two angiosperms. For instance, the Arabidopsis AT1G29280 (AtWRKY65) gene consists of two exons, while its counterpart ortholog, the S. lycopersicum Solyc10g007970 (SlWRKY77), contains three exons. These findings show that some TF family genes may have lost introns during evolutionary processes, resulting in functional changes in Arabidopsis and S. lycopersicum. The majority of Arabidopsis–S. lycopersicum orthologous gene pairs in the WRKY and bHLH TF families contain the same number of exons and conserved motifs, implying identical gene function acquisition during stable evolution (Dai et al., 2007). Functional similarities between WRKY and bHLH genes within the S. lycopersicum genome can be predicted by analyzing their co-expression networks. Important clues and deep insights regarding the physiological functions of the unexplored WRKY and bHLH TFs of S. lycopersicum were obtained from their co-expression patterns and interaction networks. This profiling can aid in predicting the functions of the uncharacterized partner. For instance, Solyc06g066370 is involved in inducing an immune response against Botrytis cinerea fungi in tomato plants (Liu et al., 2014) and has a strong co-expression link with uncharacterized Solyc03g095770, Solyc09g014990, Solyc03g116890, Solyc05g050300, and Solyc06g068460. The bHLH Solyc06g068870 gene is expressed highly in the aerial organs of tomato plants and is probably involved in stomata development (Ortega et al., 2019), while its partner Solyc08g005050 is still uncharacterized for its physiological function. Furthermore, the GO analysis revealed that the majority of these transcriptional factor genes were involved in transcriptional regulating activities such as DNA binding (GO:0003677), regulation of DNA-templated transcription (GO:0006355), and regulation of gene expression (GO:0010468). Transcriptomic studies of genome-wide RNA expression have paved the way for a comprehensive understanding of how genes are expressed in different physiological conditions. Thus, we investigated the expression profile of the WRKY and bHLH gene families in S. lycopersicum under 1 and 2 mM of cadmium stress conditions. Upon exposure to harmful cadmium stress, both up- and downregulation were found in the expression of most of the WRKY and some bHLH genes, suggesting that heavy metal stress can alter gene expression, which further leads to an increase in stress-protecting metabolites for the plant to cope with stress, for instance, Solyc05g007110 (SlWRKY76), Solyc02g094270 (SlWRKY38), Solyc08g067340 (SlWRKY46), Solyc04g051690 (SlWRKY51), Solyc04g072070 (SlWRKY55), Solyc12g056750 (SlWRKY61), Solyc02g071130 (SlWRKY71), Solyc05g015850 (SlWRKY75), Solyc05g005300 (SlbHLH084), Solyc07g062200 (SlbHLH088), Solyc08g083170 (SlbHLH056), Solyc01g106460 (SlbHLH007), Solyc10g008260 (SlbHLH093),Solyc02g076920 (SlbHLH013), and Solyc08g062780 (SlbHLH089). Our findings are consistent with previous studies that SlWRKY76 is a putative regulator in response to various biotic and abiotic stresses (Huang et al., 2012). SlWRKY46 suppressed the salicylic acid (SA) and jasmonic acid (JA) marker genes (Shu et al., 2021). In our investigation, we determined that the tomato SlWRKY3 (Solyc02g088340) gene, which encodes a regulator of tolerance to osmotic stimuli (Hichri et al., 2017), was downregulated. This gene is activated by a variety of osmotic stressors, including SA. These findings are consistent with the SA analysis, which revealed a considerable reduction in Cd-stressed plants (Asaf et al., 2022). SlWRKY51 forms a complex with the SlJAV1 gene and suppresses JA biosynthesis (Tang et al., 2022), while the other highly expressed genes during the Cd stress have been reported for the first time in this study. In addition, previous studies have reported that the gene Solyc01g104550 (SlWRKY9), which is highly downregulated in Cd stress in the current study, is involved in increasing plant biomass and improved salt tolerance (Kissoudis et al., 2016) and that the Solyc09g010960 (SlWRKY49) responds to heat stress (Wang et al., 2020). AT1G69310 (AtWRKY57), the ortholog of Solyc04g072070 (SlWRKY55), plays a key role in the convergence of auxin signaling and jasmonic acid-mediated signaling during jasmonic acid-induced foliar senescence (Caicedo et al., 2021); AT2G46400 (ATWRKY46), the ortholog of Solyc12g056750 (SlWRKY61), mediates cold tolerance (Zhu et al., 2004); AT2G44745 (AtWRKY13), the ortholog of Solyc02g071130 (SlWRKY71), is negatively regulated during Pectobacterium carotovorum infiltration (Tang and Liu, 2021); and AT3G01970 (AtWRKY45), the ortholog of Solyc05g015850 (SlWRKY75), enhances tolerance to salt stress and phosphate starvation (Li et al., 2019). Similarly, in the bHLH family, Solyc01g086870 (SlbHLH076) is highly expressed during Cd stress and its ortholog in Arabidopsis (AT4G29100) is a defense-related gene (Vargas-Salinas et al., 2021). AT2G43140, the ortholog of Solyc08g062780 (SlbHLH089), is highly expressed during Cd stress, which is in agreement with previous investigations that AT2G43140 (AtbHLH129) is most likely to be involved in stress response (Meng et al., 2020) and that AT3G19860 (AtbHLH121), the ortholog of the highly expressed Solyc01g102300 (SlbHLH006) gene, is found to act as a key regulator of the iron-deficient signaling pathway (Ying, 2021). Based on the above analysis, it can be speculated that these upregulated WRKY and bHLH genes during Cd treatment might play a crucial role in S. lycopersicum in response to Cd stress. However, functional analyses still need to verify further the possible positive or negative roles of these WKRY and bHLH genes in plant responses to abiotic stresses.
Several transcription factors significantly enhance resistance to heavy metal tolerance and homeostasis. However, most of the TFs are not explored for their specific physiological roles in S. lycopersicum. Hence, we conducted a genome-wide study of 81 SlWRKY and 161 SlbHLH genes. Phylogenetic analysis identified nine and 11 major clusters of WRKY and bHLH genes, respectively, and nine and 11 major clusters of WRKY and bHLH genes across both species (Arabidopsis and S. lycopersicum). The higher number of ortholog gene pairs in the Arabidopsis–S. lycopersicum WRKY and bHLH TFs represents their common ancestor before the taxonomic splitting of the angiosperms. Furthermore, similarity in gene structure, subcellular locations, conserved motifs, and co-expression interaction among WRKY and bHLH TFs of the S. lycopersicum predict their functional similarity. Overall, our findings presented a standpoint on the evolution of WRKY and bHLH TFs in S. lycopersicum and paved the way for additional functional characterization under heavy metal toxicity.
The data presented in the study are deposited in the National Center for Biotechnology Information (NCBI) repository, accession number PRJNA913645.
IK, RJ, SA, and AL performed the analysis. SB performed the simple sequence repeats and phylogenetic analysis. K-MK and AA-H edited and drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Research Foundation funded by the Korean Government (NRF-2021M3E5E6022715).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1100895/full#supplementary-material
Supplementary Figure 1 | The most conserved common motifs of SlWRKY TFs family were identified by MEME database with the complete amino acids sequences. The aqua-blue colored motif signifies the WRKY motif.
Supplementary Figure 2 | The most conserved common motifs of SlbHLH TFs family were identified by MEME database with the complete amino acids sequences. The aqua-blue colored motif signifies the bHLH motif.
Supplementary Figure 3 | Phylogenetic tree of the SlWRKY genes with bootstrap values of the paralogous gene pairs.
Supplementary Figure 4 | Phylogenetic tree of the SlbHLH genes with bootstrap values of the paralogous gene pairs.
Supplementary Table 1 | The data of 81 WRKY and 161 bHLH genes identified in S. lycopersicum genome
Supplementary Table 2 | The Accession IDs and STRING Identifier of the WRKY and bHLH TFs in S. lycopersicum
Supplementary Table 3 | Genes and primers selected for RT-qPCR.
Abdullah-Zawawi, M.-R., Ahmad-Nizammuddin, N.-F., Govender, N., Harun, S., Mohd-Assaad, N., Mohamed-Hussein, Z.-A. (2021). Comparative genome-wide analysis of WRKY, MADS-box and MYB transcription factor families in arabidopsis and rice. Sci. Rep. 11 (1), 1–18. doi: 10.1038/s41598-021-99206-y
Alexa, A., Rahnenführer, J., Lengauer, T. (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22 (13), 1600–1607. doi: 10.1093/bioinformatics/btl140
Andrews, S. (2010). “FastQC: A quality control tool for high throughput sequence data,” in Babraham bioinformatics (Cambridge, United Kingdom: Babraham Institute).
Asaf, S., Jan, R., Khan, M. A., Khan, A. L., Asif, S., Bilal, S., et al. (2022). Unraveling the mutualistic interaction between endophytic curvularia lunata CSL1 and tomato to mitigate cadmium (Cd) toxicity via transcriptomic insights. Sci. Total Environ. 861, 160542. doi: 10.1016/j.scitotenv.2022.160542
Caicedo, M., Munaiz, E. D., Malvar, R. A., Jiménez, J. C., Ordas, B. (2021). Precision mapping of a maize MAGIC population identified a candidate gene for the senescence-associated physiological traits. Front. Genet. 12. doi: 10.3389/fgene.2021.716821
Castrillo, G., Sánchez-Bermejo, E., de Lorenzo, L., Crevillén, P., Fraile-Escanciano, A., Tc, M., et al. (2013). WRKY6 transcription factor restricts arsenate uptake and transposon activation in arabidopsis. Plant Cell 25 (8), 2944–2957. doi: 10.1105/tpc.113.114009
Chen, X., Li, C., Wang, H., Guo, Z. (2019). WRKY transcription factors: Evolution, binding, and action. Phytopathol. Res. 1 (1), 1–15. doi: 10.1186/s42483-019-0022-x
Dai, X., Xu, Z., Liang, Z., Tu, X., Zhong, S., Schnable, J. C., et al. (2020). Non-homology-based prediction of gene functions in maize (Zea mays ssp. mays). Plant Genome 13 (2), e20015. doi: 10.1002/tpg2.20015
Dai, X., Xu, Y., Ma, Q., Xu, W., Wang, T., Xue, Y., et al. (2007). Overexpression of an R1R2R3 MYB gene, OsMYB3R-2, increases tolerance to freezing, drought, and salt stress in transgenic arabidopsis. Plant Physiol. 143 (4), 1739–1751. doi: 10.1104/pp.106.094532
Duruibe, J. O., Ogwuegbu, M., Egwurugwu, J. (2007). Heavy metal pollution and human biotoxic effects. Int. J. Phys. Sci. 2 (5), 112–118.
Dutta, S., Mitra, M., Agarwal, P., Mahapatra, K., De, S., Sett, U., et al. (2018). Oxidative and genotoxic damages in plants in response to heavy metal stress and maintenance of genome stability. Plant Signaling Behav. 13 (8), e1460048. doi: 10.1080/15592324.2018.1460048
Du, Z., Zhou, X., Ling, Y., Zhang, Z., Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
El-Sappah, A. H., Elrys, A. S., Desoky, E.-S. M., Zhao, X., Bingwen, W., El-Sappah, H. H., et al. (2021). Comprehensive genome wide identification and expression analysis of MTP gene family in tomato (Solanum lycopersicum) under multiple heavy metal stress. Saudi J. Biol. Sci. 28 (12), 6946–6956. doi: 10.1016/j.sjbs.2021.07.073
Fang, G., Bhardwaj, N., Robilotto, R., Gerstein, M. B. (2010). Getting started in gene orthology and functional analysis. PloS Comput. Biol. 6 (3), e1000703. doi: 10.1371/journal.pcbi.1000703
Gabaldón, T., Koonin, E. V. (2013). Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 14 (5), 360–366. doi: 10.1038/nrg3456
Gerszberg, A., Hnatuszko-Konka, K., Kowalczyk, T., Kononowicz, A. K. (2015). Tomato (Solanum lycopersicum l.) in the service of biotechnology. Plant Cell Tissue Organ Culture (PCTOC) 120 (3), 881–902. doi: 10.1007/s11240-014-0664-4
Ghori, N.-H., Ghori, T., Hayat, M., Imadi, S., Gul, A., Altay, V., et al. (2019). Heavy metal stress and responses in plants. Int. J. Environ. Sci. Technol. 16 (3), 1807–1828. doi: 10.1007/s13762-019-02215-8
Gondal, A. H., Zafar, A., Zainab, D., Toor, M., Sohail, S., Ameen, S., et al. (2021). A detailed review study of zinc involvement in animal, plant and human nutrition. Indian J. Pure Appl. Biosci. 9 (2), 262–271. doi: 10.18782/2582-2845.8652
Gribskov, M. (2019). Identification of sequence patterns, motifs and domains. Bioinform. Comput. Biol. 1, 332.–340. doi: 10.1016/B978-0-12-809633-8.20498-6
Guo, X., Ullah, A., Siuta, D., Kukfisz, B., Iqbal, S. (2022). Role of WRKY transcription factors in regulation of abiotic stress responses in cotton. Life 12 (9), 1410. doi: 10.3390/life12091410
Hao, Y., Zong, X., Ren, P., Qian, Y., Fu, A. (2021). Basic helix-Loop-Helix (bHLH) transcription factors regulate a wide range of functions in arabidopsis. Int. J. Mol. Sci. 22 (13), 7152. doi: 10.3390/ijms22137152
Heim, M. A., Jakoby, M., Werber, M., Martin, C., Weisshaar, B., Bailey, P. C. (2003). The basic helix–loop–helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 20 (5), 735–747. doi: 10.1093/molbev/msg088
Hichri, I., Muhovski, Y., Žižková, E., Dobrev, P. I., Gharbi, E., Franco-Zorrilla, J. M., et al. (2017). The solanum lycopersicum WRKY3 transcription factor SlWRKY3 is involved in salt stress tolerance in tomato. Front. Plant Sci. 8 1343. doi: 10.3389/fpls.2017.01343
Huang, S., Gao, Y., Liu, J., Peng, X., Niu, X., Fei, Z., et al. (2012). Genome-wide analysis of WRKY transcription factors in solanum lycopersicum. Mol. Genet. Genomics 287 (6), 495–513. doi: 10.1007/s00438-012-0696-6
Imran, Q. M., Lee, S.-U., Mun, B.-G., Hussain, A., Asaf, S., Lee, I.-J., et al. (2019). WRKYs, the jack-of-various-Trades, modulate dehydration stress in populus davidiana–a transcriptomic approach. Int. J. Mol. Sci. 20 (2), 414. doi: 10.3390/ijms20020414
Kalaivanan, D., Ganeshamurthy, A. N. (2016). “Mechanisms of heavy metal toxicity in plants,” in Abiotic stress physiology of horticultural crops (Springer), 85–102.
Khalil, R., Haroun, S., Bassyoini, F., Nagah, A., Yusuf, M. (2021). Salicylic acid in combination with kinetin or calcium ameliorates heavy metal stress in phaseolus vulgaris plant. J. Agric. Food Res. 5, 100182. doi: 10.1016/j.jafr.2021.100182
Khan, A., Khan, S., Khan, M. A., Qamar, Z., Waqas, M. (2015). The uptake and bioaccumulation of heavy metals by food plants, their effects on plants nutrients, and associated health risk: A review. Environ. Sci. pollut. Res. 22 (18), 13772–13799. doi: 10.1007/s11356-015-4881-0
Khan, I., Khan, S., Zhang, Y., Zhou, J. (2021a). Genome-wide analysis and functional characterization of the dof transcription factor family in rice (Oryza sativa l.). Planta 253 (5), 1–14. doi: 10.1007/s00425-021-03627-y
Khan, I., Khan, S., Zhang, Y., Zhou, J., Akhoundian, M., Jan, S. A. (2021b). CRISPR-cas technology based genome editing for modification of salinity stress tolerance responses in rice (Oryza sativa l.). Mol. Biol. Rep. 48 (4), 3605–3615. doi: 10.1007/s11033-021-06375-0
Kissoudis, C., Gao, D., Pramanik, D., Birhanu, M., Visser, R., Bai, Y. (2016). Supplementary data: Roles and contribution of tomato WRKY genes to salt stress and powdery mildew resistance. doi: 10.18174/369640
Krueger, F. (2012). Trim galore: A wrapper tool around cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files, with some extra functionality for MspI-digested RRBS-type (Reduced representation bisufite-seq) libraries.
Lee, T.-H., Tang, H., Wang, X., Paterson, A. H. (2012). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res. 41 (D1), D1152–D1158. doi: 10.1093/nar/gks1104
Liao, J.-L., Zhou, H.-W., Peng, Q., Zhong, P.-A., Zhang, H.-Y., He, C., et al. (2015). Transcriptome changes in rice (Oryza sativa l.) in response to high night temperature stress at the early milky stage. BMC Genomics 16 (1), 1–14. doi: 10.1186/s12864-015-1222-0
Li, S., Han, X., Lu, Z., Qiu, W., Yu, M., Li, H., et al. (2022). MAPK cascades and transcriptional factors: Regulation of heavy metal tolerance in plants. Int. J. Mol. Sci. 23 (8), 4463. doi: 10.3390/ijms23084463
Li, C., Liu, X., Ruan, H., Zhang, J., Xie, F., Gai, J., et al. (2019). GmWRKY45 enhances tolerance to phosphate starvation and salt stress, and changes fertility in transgenic arabidopsis. Front. Plant Sci. 10 1714. doi: 10.3389/fpls.2019.01714
Liu, B., Hong, Y.-B., Zhang, Y.-F., Li, X.-H., Huang, L., Zhang, H.-J., et al. (2014). Tomato WRKY transcriptional factor SlDRW1 is required for disease resistance against botrytis cinerea and tolerance to oxidative stress. Plant Sci. 227, 145–156. doi: 10.1016/j.plantsci.2014.08.001
Li, X., Zhang, H., Ai, Q., Liang, G., Yu, D. (2016). Two bHLH transcription factors, bHLH34 and bHLH104, regulate iron homeostasis in arabidopsis thaliana. Plant Physiol. 170 (4), 2478–2493. doi: 10.1104/pp.15.01827
Li, J., Zhang, M., Sun, J., Mao, X., Wang, J., Liu, H., et al. (2020). Heavy metal stress-associated proteins in rice and arabidopsis: Genome-wide identification, phylogenetics, duplication, and expression profiles analysis. Front. Genet. 11, 477. doi: 10.3389/fgene.2020.00477
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi: 10.1186/s13059-014-0550-8
Maanan, M., Saddik, M., Maanan, M., Chaibi, M., Assobhei, O., Zourarah, B. (2015). Environmental and ecological risk assessment of heavy metals in sediments of nador lagoon, Morocco. Ecol. Indic. 48, 616–626. doi: 10.1016/j.ecolind.2014.09.034
Manara, A. (2012). “Plant responses to heavy metal toxicity,” in Plants and heavy metals (Netherlands: Springer), 27–53.
Manzoor, Z., Hassan, Z., Ul-Allah, S., Khan, A. A., Sattar, A., Shahzad, U., et al. (2022). “Transcription factors involved in plant responses to heavy metal stress adaptation,” in Plant perspectives to global climate changes (Pakistan: Elsevier), 221–231.
Mao, G., Meng, X., Liu, Y., Zheng, Z., Chen, Z., Zhang, S. (2011). Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in arabidopsis. Plant Cell 23 (4), 1639–1653. doi: 10.1105/tpc.111.084996
Meng, X., Liu, S., Dong, T., Xu, T., Ma, D., Pan, S., et al. (2020). Comparative transcriptome and proteome analysis of salt-tolerant and salt-sensitive sweet potato and overexpression of IbNAC7 confers salt tolerance in arabidopsis. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.572540
Nakashima, K., Ito, Y., Yamaguchi-Shinozaki, K. (2009). Transcriptional regulatory networks in response to abiotic stresses in arabidopsis and grasses. Plant Physiol. 149 (1), 88–95. doi: 10.1104/pp.108.129791
Opdenakker, K., Remans, T., Keunen, E., Vangronsveld, J., Cuypers, A. (2012). Exposure of arabidopsis thaliana to cd or Cu excess leads to oxidative stress mediated alterations in MAPKinase transcript levels. Environ. Exp. Bot. 83, 53–61. doi: 10.1016/j.envexpbot.2012.04.003
Ortega, A., De Marcos, A., Illescas-Miranda, J., Mena, M., Fenoll, C. (2019). The tomato genome encodes SPCH, MUTE, and FAMA candidates that can replace the endogenous functions of their arabidopsis orthologs. Front. Plant Sci. 10, 1300. doi: 10.3389/fpls.2019.01300
Riaño-Pachón, D. M., Ruzicic, S., Dreyer, I., Mueller-Roeber, B. (2007). PlnTFDB: An integrative plant transcription factor database. BMC Bioinf. 8 (1), 1–10.
Rout, G. R., Sahoo, S. (2015). Role of iron in plant growth and metabolism. Rev. Agric. Sci. 3, 1–24. doi: 10.7831/ras.3.1
Rushton, P. J., Somssich, I. E., Ringler, P., Shen, Q. J. (2010). WRKY transcription factors. Trends Plant Sci. 15 (5), 247–258. doi: 10.1016/j.tplants.2010.02.006
Shahid, M., Khalid, S., Abbas, G., Shahid, N., Nadeem, M., Sabir, M., et al. (2015). “Heavy metal stress and crop productivity,” in Crop production and global environmental issues (Pakistan: Springer), 1–25.
Shiu, S.-H., Shih, M.-C., Li, W.-H. (2005). Transcription factor families have much higher expansion rates in plants than in animals. Plant Physiol. 139 (1), 18–26. doi: 10.1104/pp.105.065110
Shu, P., Zhang, S., Li, Y., Wang, X., Yao, L., Sheng, J., et al. (2021). Over-expression of SlWRKY46 in tomato plants increases susceptibility to botrytis cinerea by modulating ROS homeostasis and SA and JA signaling pathways. Plant Physiol. Biochem. 166, 1–9. doi: 10.1016/j.plaphy.2021.05.021
Singh, S., Parihar, P., Singh, R., Singh, V. P., Prasad, S. M. (2016). Heavy metal tolerance in plants: role of transcriptomics, proteomics, metabolomics, and ionomics. Front. Plant Sci. 6, 1143. doi: 10.3389/fpls.2015.01143
Sirén, J., Välimäki, N., Mäkinen, V. (2014). HISAT2-fast and sensitive alignment against general human population. IEEE/ACM Trans. Comput. Biol. Bioinforma 11 (2), 375–388. doi: 10.1109/tcbb.2013.2297101
Sivitz, A. B., Hermand, V., Curie, C., Vert, G. (2012). Arabidopsis bHLH100 and bHLH101 control iron homeostasis via a FIT-independent pathway. PloS ONE 7. doi: 10.1371/journal.pone.0044843
Subbarao, G., Ito, O., Berry, W., Wheeler, R. (2003). Sodium–a functional plant nutrient. Crit. Rev. Plant Sci. 22 (5), 391–416. doi: 10.1080/07352680390243495
Tang, H., Liu, H. (2021). Roles of single gene in plant hypoxia and pathogen responses. Plant Signal Behav. 16 (10), 1934295. doi: 10.1080/15592324.2021.1934295
Tang, B., Tan, T., Chen, Y., Hu, Z., Xie, Q., Yu, X., et al. (2022). SlJAZ10 and SlJAZ11 mediate dark-induced leaf senescence and regeneration. PloS Genet. 18 (7), e1010285. doi: 10.1371/journal.pgen.1010285
Ülker, B., Somssich, I. E. (2004). WRKY transcription factors: From DNA binding towards biological function. Curr. Opin. Plant Biol. 7 (5), 491–498. doi: 10.1016/j.pbi.2004.07.012
Vargas-Salinas, M., Medina-Hernández, D., Arcos-Ortega, G. F., Luis-Villaseñor, I. E., Holguín-Peña, R. J. (2021). RNAi activation with homologous and heterologous sequences that induce resistance against the begomovirus pepper golden mosaic virus (PepGMV). 3 Biotech. 11 (3), 114. doi: 10.1007/s13205-021-02653-7
Wang, Y., Zhuang, K., Meng, Q., Meng, C. (2020). Characterization and analysis of some chilling-response WRKY transcription factors in tomato. Plant Physiol. J. 57 (6), 1349–1362. doi: 10.21203/rs.3.rs-75411/v1
Wani, S. H., Anand, S., Singh, B., Bohra, A., Joshi, R. (2021). WRKY transcription factors and plant defense responses: latest discoveries and future prospects. Plant Cell Rep. 40 (7), 1071–1085. doi: 10.1007/s00299-021-02691-8
Wei, F., Coe, E., Nelson, W., Bharti, A. K., Engler, F., Butler, E., et al. (2007). Physical and genetic structure of the maize genome reflects its complex evolutionary history. PloS Genet. 3 (7), e123. doi: 10.1371/journal.pgen.0030123
Ying, S. (2021). Genome-wide identification and transcriptional analysis of arabidopsis DUF506 gene family. Int. J. Mol. Sci. 22 (21), 11442. doi: 10.3390/ijms222111442
Zhao, W., Li, Y., Fan, S., Wen, T., Wang, M., Zhang, L., et al. (2021). The tomato WRKY32 transcription factor affects ripe fruit color by regulating YFT1, a core component of ethylene signal transduction. J. Exp. Botany. 72 (12), 4269–4282. doi: 10.1093/jxb/erab113
Keywords: Solanum lycopersicum, phylogenetic analysis, heavy metal stress, expression pattern, WRKY and bHLH
Citation: Khan I, Asaf S, Jan R, Bilal S, Lubna, Khan AL, Kim K-M and Al-Harrasi A (2023) Genome-wide annotation and expression analysis of WRKY and bHLH transcriptional factor families reveal their involvement under cadmium stress in tomato (Solanum lycopersicum L.). Front. Plant Sci. 14:1100895. doi: 10.3389/fpls.2023.1100895
Received: 17 November 2022; Accepted: 04 January 2023;
Published: 25 January 2023.
Edited by:
Muhammad Ansar Farooq, National University of Sciences and Technology (NUST), PakistanReviewed by:
Jianwei Gao, Shandong Academy of Agricultural Sciences, ChinaCopyright © 2023 Khan, Asaf, Jan, Bilal, Lubna, Khan, Kim and Al-Harrasi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Al-Harrasi, YWhhcnJhc2lAdW5pendhLmVkdS5vbQ==; Kyung-Min Kim, a2ttQGtudS5hYy5rcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.