 Yueling Li1,2
Yueling Li1,2 Zhongshuai Sun
Zhongshuai Sun Junmin Li
Junmin Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 20 February 2023
Sec. Plant Metabolism and Chemodiversity
Volume 14 - 2023 | https://doi.org/10.3389/fpls.2023.1044581
This article is part of the Research Topic Advances in Metabolism and Chemodiversity – Focus - Anthocyanin and Proanthocyanin: Biosynthesis, Accumulation, Regulation View all 7 articles
Heptacodium miconioides Rehd., commonly known as “seven-son flower,” is an ornamental species with a beautiful flower pattern and persistent sepals. Its sepals are of horticultural value, turning bright red and elongating in the autumn; however, the molecular mechanisms that cause sepal color change remain unclear. We analyzed the dynamic changes in anthocyanin composition in the sepal of H. miconioides at four developmental stages (S1-S4). A total of 41 anthocyanins were detected and classified into 7 major anthocyanin aglycones. High levels of the pigments cyanidin-3,5-O-diglucoside, cyanidin-3-O-galactoside, cyanidin-3-O-glucoside, and pelargonidin-3-O-glucoside were responsible for sepal reddening. Transcriptome analysis revealed 15 differentially expressed genes involved in anthocyanin biosynthesis that were detected between 2 developmental stages. Of these, the high expression of HmANS was considered critical structural gene related to anthocyanin biosynthesis pathway in the sepal through co-expression analysis with anthocyanin content. In addition, a transcription factor (TF)-metabolite correlation analysis revealed that three HmMYB, two HmbHLH, two HmWRKY, and two HmNAC TFs exhibited a strong positive role in the regulation of the anthocyanin structural genes (Pearson’s correlation coefficient > 0.90). Luciferase activity assay showed that HmMYB114, HmbHLH130, HmWRKY6, and HmNAC1 could activate the promoters of HmCHS4 and HmDFR1 genes in vitro. These findings increase our understanding of anthocyanin metabolism in the sepal of H. miconioides and provide a guide for studies involving sepal color conversion and regulation.
Anthocyanin is classified as a flavonoid and is present in land plants. To date, at least 635 anthocyanins have been identified in nature (He and Giusti, 2010). Six categories of anthocyanins were the most common in the blue, red, and purple colors of plants, including pelargonidin, cyanidin, delphinidin, peonidin, petunidin, and malvidin (Tanaka et al., 2008; Yang et al., 2015). In flowering plants, the color changes that occur in floral organs are closely related to anthocyanins (Shen et al., 2021). Thus far, several genes involved in the anthocyanin biosynthesis pathway have been identified in most plants (Boss et al., 1996; Saito et al., 2013; Zheng et al., 2022). These genes mainly include dihydroflavonol 4-reductase (DFR), anthocyanidin synthase (ANS), and UDP-glucose: flavonoid 3-glucosyltransferase (UFGT) (Schaart et al., 2013; Wang et al., 2019). Subsequently, anthocyanins are accumulated and stored through anthocyanin glycosylation, glutathione S- transferase (GST) proteins, multidrug and toxic compound extrusion (MATE) transporters, etc. (Koes et al., 2005). A protein complex composed of MYB TF and bHLH regulator together with a WD40 repeat protein (MYB-bHLH-WD40, MBW) could binds to the promoters of anthocyanin structural genes to activate their expression (Ramsay and Glover, 2005). For example, Carretero-Paulet et al. (2010) showed that MYB123/bHLH42/TTG1 and MYB75/bHLH2/TTG1 are involved in the expression of anthocyanin biosynthesis genes in Arabidopsis thaliana. In addition, other TFs, such as WRKY72, NAC52, HY5 and ERF1B have also been identified to be directly or indirectly involved in the biosynthesis of anthocyanin (An et al., 2017; Zhang et al., 2018; Sun et al., 2019; Hu et al., 2020). The sepals, which play an important role in protecting the other floral organs (stamens, pistils, and petals), generally surround the outside of the flower. In some plants, sepals are uniformly green and provide the energy required for fruit or seed development through photosynthesis (Liu et al., 2020; Li et al., 2020a). In other plants, such as Hydrangea macrophylla, the sepals perform the function of petals. The actual flower petals are replaced by sepals that have the ability to change color by accumulating anthocyanins (Yoshida et al., 2021). Whether the anthocyanins in sepals and their biosynthetic pathways are the same as that in the petals is unclear. To date, little is known regarding the underlying mechanism of color formation in sepals.
The Caprifoliaceae family contains more than 800 species, some containing pink or white flowers with fragrance and persistent sepals, which have a long duration and ornamental period. A representative species of this family, Heptacodium miconioides Rehd., which is commonly known as “seven-son flower” in China and is a deciduous tree from the Heptacodium genus (Coombes, 1990; Bian et al., 2002; Zhang et al., 2022). Although this plant is native to China, it is now widely cultivated in botanical institutions and nurseries throughout the world (Coombes, 1990). The tree is elegant in shape and has beautiful white flowers with a jasmine-like scent. The sepals of its flowers enlarge and change color from green to red, as if undergoing a second blooming. Its beauty has attracted the attention of many horticulturalists (Liu et al., 2006). Therefore, identifying the mechanism of sepal color formation will be valuable for understanding plant evolution and for breeding novel ornamental lines. Currently, there are few systematic studies on the characteristics and coloration of H. miconioides sepals (Liu et al., 2006).
Recently, transcriptomics and metabolomics have been widely used to explore the relationships between genes and metabolites, and to unravel structural genes and TFs that may play a role in secondary metabolic pathways (Dong et al, 2009; Wu et al., 2020). For example, in the flowers of tea (Camellia sinensis L.), the DFR, CHS, F3H, FLS, and LDOX1 genes at five developmental stages were identified by metabolomics and transcriptome sequencing analysis. These genes were found to have a direct relationship with the biosynthesis and accumulation of cyanidin 3-O-glucoside and petunidin 3-O-glucoside (Rothenberg et al., 2019). Chen et al. (2022) performed a metabolome and transcriptome analysis of sweet cherry (Prunus avium L.), which revealed a different temporal expression pattern of anthocyanin accumulation associated with fruit color. They suggested that the MYC2, NAC71, WRKY57, and TCP7 TFs might be participated in the regulation of anthocyanin structural genes. Similarly, Zheng et al. (2022) found that the UFGT gene and two ANS genes were upregulated in the whole developmental stages of Zanthoxylum bungeanum fruit peels, which may represent key functional genes for the accumulation of peonidin 3-O-glucoside and peonidin O-hexoside. Therefore, multiomics analysis is a powerful strategy to explore the regulation of the anthocyanin network at different developmental stages in plants (Liu et al., 2017; Lin et al., 2020).
In this study, we determined the differences in anthocyanin biosynthesis during H. miconioides sepal development. Candidate genes and regulators of anthocyanin biosynthesis were identified using a transcriptomic analysis. The anthocyanin compounds in the different sepal developmental stages were determined using multiple reaction monitoring (MRM). Then, an unbiased network analysis was constructed to detect the relationship between genes and anthocyanin accumulation. Our major objectives were (1) to elucidate the potential key genes of enzyme and TFs involved in the anthocyanin biosynthesis pathway at different sepal developmental stages, and (2) to explore the potential regulatory mechanisms underlying the biosynthesis of anthocyanin in the sepal of H. miconioides. The results provide insight into anthocyanin biosynthesis in the sepal of this ornamental species and increase our understanding of the molecular mechanisms responsible for sepal color development.
The H. miconioides used in this study was cultivated at the Taizhou University botanical garden, Zhejiang Province, China (121°17′E, 28°87′N; 10 m above sea level) under natural conditions. The annual average of temperature and precipitation is approximately 17.10°C and 1,231.40 mm, respectively. The growth soil condition of H. miconioides is mainly yellow loam with a pH of approximately 6.10. Sepal samples from healthy plants grown under natural light were collected from 9:00 to 11:00 in September to October 2018. Sepal samples were collected during four sepal developmental stages, corresponding to lengths of 0.1 cm (S1), 0.3 cm (S2), 0.5 cm (S3), and 0.8 cm (S4), respectively. The samples were frozen in liquid nitrogen immediately after collection and stored at −80°C for further transcriptomic and metabolomic analysis. In addition, to ensure the consistency of the subsequent correlation analysis, samples representing each period were mixed and divided into two parts, which were used for transcriptome sequencing and metabolite analysis. There were three biological replicates for each sepal from each period for a total of 12 samples.
Anthocyanin content was analyzed as previously described, with minor modifications (Lin et al., 2020). Briefly, sepal samples were freeze-dried and powdered in a MM 400 grinder (Retsh Technology, Germany). Then, 50 mg of powdered samples were extracted with 500 μL of 0.1% (v/v) hydrochloric methanol solution for 20 h at 4°C. The pigment extract sample was filtered through an HPLC PTFE syringe filter (0.22 μm). The anthocyanin composition was analyzed using UPLC (ExionLC™ AD) equipped with a reverse Waters ACQUITY BEH C18 column (1.7 µm, 2.1 mm*100 mm) and Tandem mass spectrometry (MS/MS, Applied Biosystems 6500 QTRAP). The UPLC analysis was performed under the following conditions: solvent system, ultrapure water (0.1% formic acid): methanol (0.1% formic acid); gradient program, 95:5 v/v at 0 min, 50:50 v/v at 6.0 min, 5:95 v/v at 2.0 min, and 95:5 v/v at 14.0 min; flow rate, 0.35 mL/min; temperature, 40°C; injection volume, 2 μL. Based on the MetWare database (MetWare Biotechnology Co., Ltd., Wuhan, China), the data detected by mass spectrometry was analyzed qualitatively. The concentration of anthocyanin in the sample was calculated using MRM. The MRM for each sepal sample was measured in triplicate. A metabolite heatmap was generated by complexheatmap R package (version 2.7.1.1009) after unit variance scaling. Differentially accumulated metabolites (DAMs) were identified based on a log2(FoldChange) ≤ 0.5 or ≥2 and a variable importance in the projection (VIP) ≥1.
The sepal samples at four developmental stages used for transcriptomics were the same as the anthocyanin analysis samples. The genome of H. miconioides was used as a reference (Unpublished data). Functional annotation information for all expressed genes was obtained from the H. miconioides genome annotation project using the following public databases: Nr (http://www.ncbi.nlm.nih.gov/protein ), SwissProt (http://www.uniprot.org/ ), Pfam (http://pfam.xfam.org/ ), InterPro (https://www.ebi.ac.uk/interpro/ ) and KEGG (http://www.genome.jp/kegg/ ), with an e-value threshold of 1e-05. We also confirmed the identified candidate genes involved in anthocyanin biosynthesis using knowledge-based pathway enzyme identification (KIPEs, version 3) (Pucker et al., 2020). Total RNA was extracted from the four stages of sepal development with three biological replicates. Libraries were prepared and sequenced using the Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) and paired-end reads were generated. The raw paired-end RNA-Seq reads were filtered into clean data using FASTP (version 0.19.5) (Chen et al., 2018). The RNA clean reads were aligned to the reference genome using HISAT2 (version 2.0.4) (Kim et al., 2015). The FPKM value was used to calculate gene expression levels. DESeq (version 1.10.1) with a model based on the negative binomial distribution was used to identify differentially expressed genes (DEGs) (Anders and Huber, 2010), and the screening threshold was set as padj < 0.05 and the absolute value of log2(FoldChange) ≥1 between different developmental stages (S1 vs. S2, S2 vs. S3, S3 vs. S4, S1 vs. S3, S1 vs. S4, and S2 vs. S4).
The identification of the MYB TF was based on the freely available described in a previous study (https://github.com/bpucker/MYB_annotator , version 0.23) (Pucker, 2022). The bHLHs, WRKYs, NACs, bZIPs, and HSFs TFs families from the transcriptome data were predicted with iTAK software (version 1.2) (Zheng et al., 2016). The parameters of iTAK were set as follows: -s n -m b -a 5, where -s is sequence type and nucleic acid sequence, -m is the type of analysis, and -a is a hmmscan computational resource. The TF homologous protein sequences of A. thaliana that were downloaded from the TAIR database. The phylogenetic tree of the predicted TFs and the known TFs in A. thaliana was generated by the MEGA version 11.0.13 program (Tamura et al., 2021) using the neighbor-joining method with default parameters (1,000 bootstrap replicates). Phylogenetic trees were modified using iTOL (https://itol.embl.de/personal_page.cgi ). H. miconioides TFs homologs were classified on the basis of their relationships with corresponding A. thaliana.
To determine potential key candidate structural genes and TFs involved in anthocyanin biosynthesis pathway in H. miconioides, a joint analysis between transcriptomic and metabolomic datasets was performed. Joint analysis parameters were established by calculating the mean of three biological replicates of the DEGs, differential transcript expression FPKM value of the transcriptomic data, and the mean of the differential anthocyanin components in metabolomic data. The resulting data was log transformed with R (www.r-project.org/ ) and a correlation analysis between differential genes and metabolites was conducted by calculating pearson correlation coefficient (PCC) values in hmisc R package (4.4.0). Significant correlations were considered at |PCC| > 0.80 and p-value < 0.05 (Lin et al., 2020). The network diagrams were visualized by Cytoscape software (version 3.6.1).
The CDS fragments of HmMYB114, HmMYB11, HmbHLH130, HmWRKY6, HmNAC1, and HmNAC2 was cloned into the pCAMBIA1300-35S vector (effector) (pro35S:HmMYB114; pro35S:HmMYB11; pro35S:HmbHLH130; pro35S:HmWRKY6; pro35S:HmNAC1; pro35S:HmNAC2). The promoter sequences of HmCHS4 and HmDFR1 were amplified from the genomic DNA and then transformed into the pLLOOR-Bar luciferase (LUC) vector (reporter) (producing proHmCHS4:LUC). Agrobacterium tumefaciens GV3101 harbouring the effector and reporter vectors was injected into N. benthamiana leaves following the method described in Lou et al., 2018. The LUC activity tests were performed using a Tanon-5500 chemiluminescence imaging system (Tanon, Shanghai, China) according to the manufacturer’s instructions. The LUC signal intensity value was generated by TanonImage (version 201201 1.10) with a quantitation parameter of 1ng standard. The single HmCHS4 and HmDFR1 promoter-LUC recombinant vector was used as the blank control. The experiments were technically repeated three times with similar results. The primer sequences for the LUC assays involved in anthocyanin biosynthesis genes are listed in Supplementary Table 1.
The data was analyzed using SPSS 20.0 statistical software. A one-way analysis of variance was used to test the differences of total anthocyanin contents at different sepal developmental stages and relative LUC signal intensity, followed by LSD or Dunnett’s T3 test with significant differences at 5% level. Origin 8.5 software was used to draw graphs. The data are presented as the mean ± standard deviations (SD).
The color of the sepals of H. miconioides changed from green to dark red at the different developmental stages (Figure 1A). To gain insight into the differences in anthocyanin compounds and biosynthesis, four sepal samples were used for anthocyanin-targeted metabolome analysis. The results indicated that the total anthocyanin content gradually increased during sepal development. The total anthocyanin content of the sepals in S3 and S4 was significantly higher compared with that in S1 and S2, but there was no significant difference between S3 and S4 (Figure 1B).
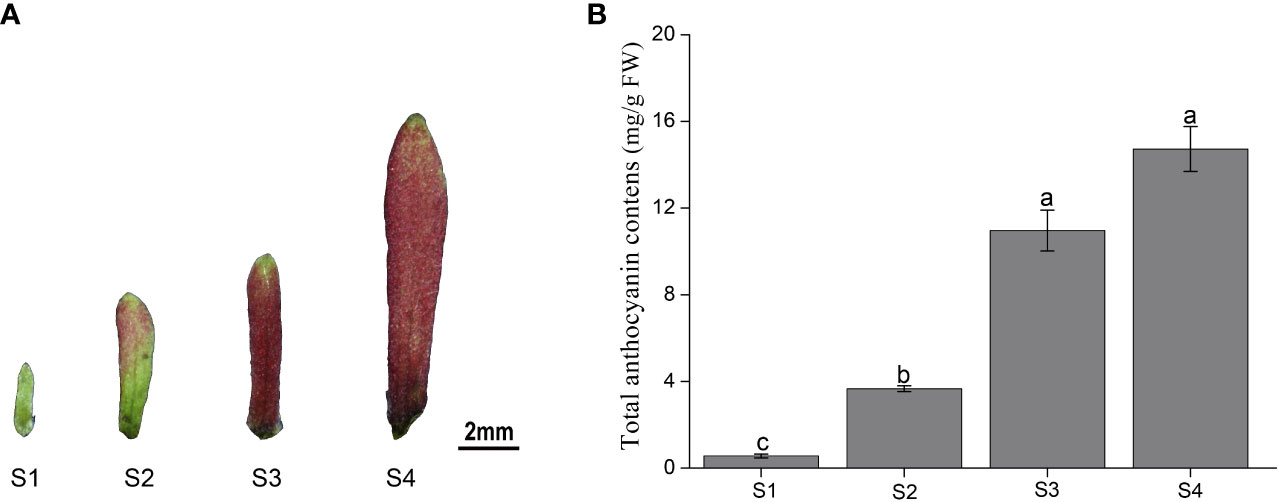
Figure 1 Color and total anthocyanin content of Heptacodium miconioides sepals at four developmental stages (S1-S4). (A) The characteristics of the (H) miconioides sepal at four growth stages. (B) Changes in total anthocyanin content during sepal development. S1, sepal length = 0.1 cm; S2, sepal length = 0.3 cm; S3, sepal length = 0.5 cm; S4, sepal length=0.8 cm. Values are the means ± standard deviation (SD). Data is the mean of three biological replicates. Different lowercase letters indicated significant differences at 0.05 level.
To compare the differences in anthocyanin metabolite content between the S1 to S4 sepals of H. miconioides, we used UPLC/ESI-QTRAP-MS/MS. A total of 41 anthocyanin metabolites were detected in the sepal samples of H. miconioides (Figure 2A and Supplementary Table 2). The 41 anthocyanin metabolites were classified into 7 categories: cyanidin, delphinidin, malvidin, pelargonidin, peonidin, petunidin, and proanthocyanidin. Of these, the main components of anthocyanin content were cyanidin-3,5-O-diglucoside, cyanidin-3-O-galactoside, cyanidin-3-O-glucoside, proanthocyanidin B3, pelargonidin-3-O-glucoside, and peonidin-3-O-galactoside (Figure 2A and Supplementary Table 2). There were 31 DAMs between S1 and S2: 8 cyanidins, 7 delphinidins, 3 malvidins, 4 pelargonidins, 2 peonidins, 3 petunidins, and 4 proanthocyanidins (Figure 2A and Supplementary Table 2). There were 28 DAMs between S2 and S3 including 7 cyanidins, 6 delphinidins, 2 malvidins, 3 pelargonidins, 2 peonidins, 3 petunidins, and 5 proanthocyanidins (Figure 2A and Supplementary Table 2). There were 14 DAMs between S3 and S4. Of these DAMs, most anthocyanins were significantly more abundant at S3 and S4 compared with S1 and S2. A number of DAMs were shared between the two comparisons: three, three, and four DAMs were shared between the S1/S2 and S2/S3, S2/S3 and S3/S4, and S1/S2 and S3/S4 comparisons, respectively (Figure 2B).
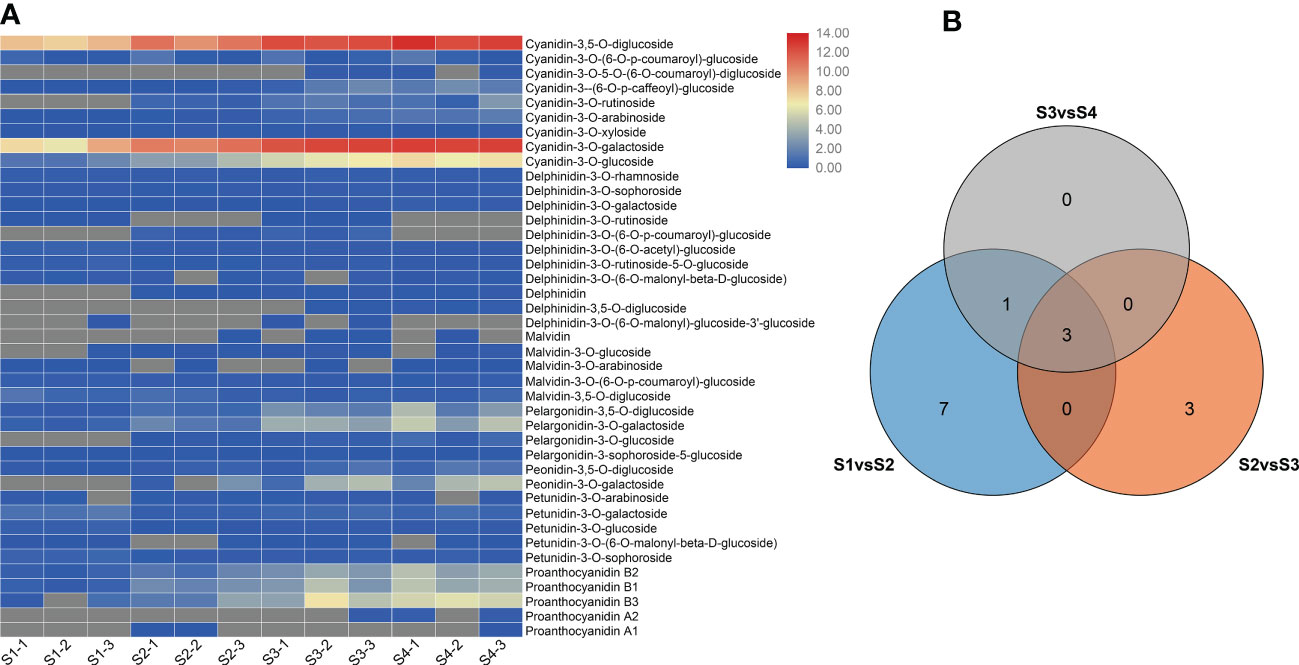
Figure 2 Differential anthocyanin metabolite content in Heptacodium miconioides sepals during development. (A) Heat maps of DAMs in the S1 through S4 developmental stages. (B) Venn diagram of DAMs shared among two or three comparisons.
To elucidate the molecular mechanisms of anthocyanin biosynthesis in the sepal of H. miconioides, a transcriptome analysis was performed at the four developmental stages to identify DEGs. A total of 89.69 GB of clean data were generated and the clean reads ranged from 38471686 to 63799778 for each library, with bases scoring Q30 in an average of 89.63–92.49% (Supplementary Table 3). The clean reads were obtained from RNA-seq raw data by filtering out uncertain reads, adapter related reads, and low-quality reads. These high-quality reads guaranteed the further gene expression analysis. Based on a |log2(FoldChange)| ≥1 and a p-value < 0.05, a total of 185 (27 up- and 158 down-regulated), 51 (40 up- and 11 down-regulated), 920 (490 up- and 430 down-regulated), 230 (39 up- and 191 down-regulated), 1460 (604 up- and 856 down-regulated), and 3106 (1886 up- and 1220 down-regulated) DEGs were identified for the S1 vs. S2, S2 vs. S3, S3 vs. S4, S1 vs. S3, S1 vs. S4, and S2 vs. S4 comparisons, respectively (Figure 3A and Supplementary Figure 1). The results indicated that the gene expression data for S2 and S3 was closer, whereas the expression profiles of S3 vs. S4, S1 vs. S4, and S2 vs. S4 were considerably different. KEGG analysis of the DEGs was conducted for the different stages and showed enrichment in many metabolic pathways including biosynthesis of secondary metabolic pathways, starch and sucrose metabolism pathways, phenylalanine metabolism pathways, flavonoid metabolism pathways, and phenylpropanoid biosynthesis pathways (Figures 3B–D). Of these, the last three metabolic pathways were closely associated with anthocyanin biosynthesis and were significantly enriched in the developmental stages of the sepal.
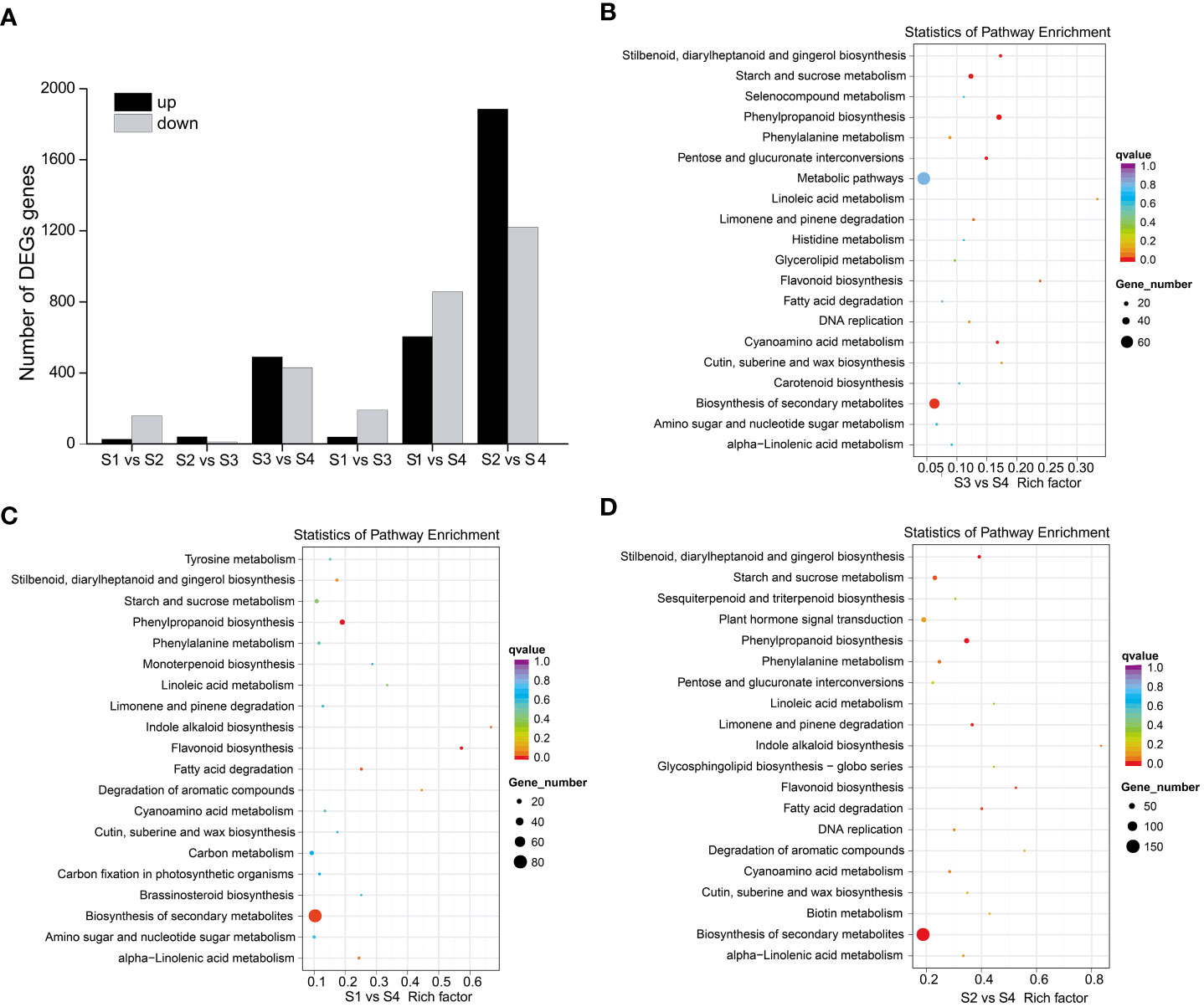
Figure 3 DEGs and KEGG enrichment analyses of sepal color during Heptacodium miconioides sepal development. (A) Number of up- and down-regulated DEGs in six comparisons. (B–D) The top 20 pathways of the DEGs based on a KEGG analysis of S3 vs. S4, S1 vs. S4, and S2 vs. S4.
A total of 28 candidate genes (encoding 11 key enzymes) involved in the anthocyanin biosynthetic pathway were identified based on KEGG pathway and Swiss-Prot annotations. Candidate genes were identified as showing high transcriptional activity in at least one sepal developmental stage and their expression levels were high (FPKM>10.0). These putative structural genes included HmPAL (5), HmC4H (2), Hm4CL (2), HmCHS (4), HmCHI (2), HmLAR (1), HmF3H (2), HmANR (2), HmDFR (3), HmANS (1), and HmUFGT (4) (Table 1). The expression patterns of these genes were different in the four developmental stages. A comparison of the four sepal stages revealed 15 DEGs including four HmPAL genes, one HmC4H gene, one Hm4CL gene, four HmCHS genes, one HmLAR gene, one HmF3H gene, one HmANR gene, one HmDFR gene, and one HmANS gene (Supplementary Table 4). The expression levels of three DEGs (HmPAL, HmDFR, and HmLAR) decreased with development and peaked during the S2 stage (Table 1; Figure 4). The other DEGs exhibited varying expression patterns; however, their expression levels were the highest during the S4 stage. In particular, one HmANR gene, one HmDFR gene, three HmCHS genes, and one HmANS gene were upregulated by more than 10-fold in the S1 stage. In addition, we also identified the glycosyltransferases (GTs) genes responsible for further glycosylation modification of anthocyanins. HmMT (3) and Hm3MaT1 (1) showed a higher level of expression increased with development and peaked during the S4 stage. The expression levels of HmRT (3) and HmMATE (1) were the highest during the S2 stage.

Table 1 Candidates in the anthocyanin structural genes of Heptacodium miconioides sepal.
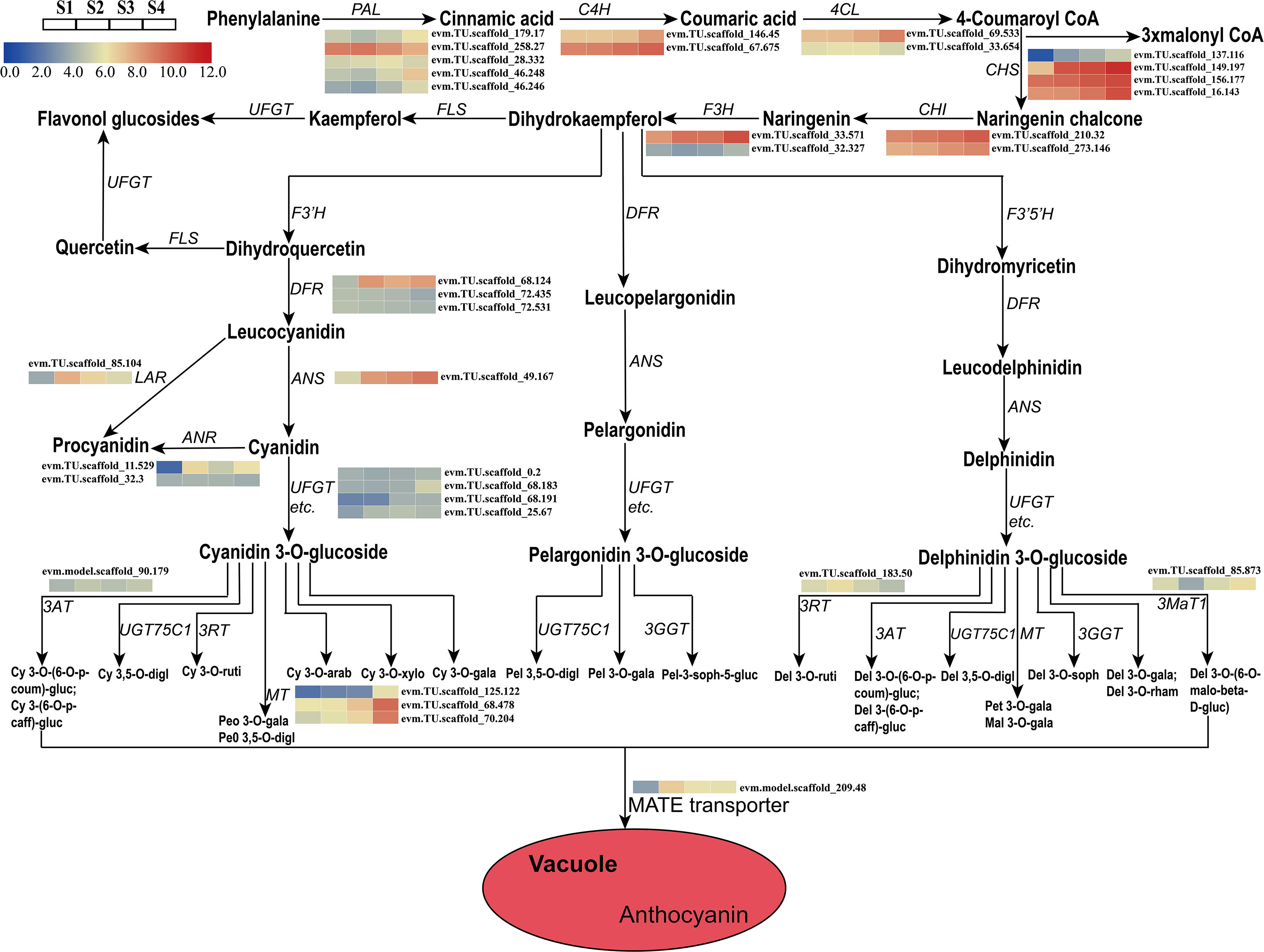
Figure 4 Expression analyses of structural genes involved in the pathway of anthocyanin biosynthesis at four developmental stages in the sepal were evaluated by RNA-Seq. Cy, cyanidin; Peo, Peonidin; digl, diglucoside; gluc, glucoside; coum, coumaroyl; caff, caffeoyl; ruti, rutinoside; gala, galactoside; arab, arabinoside; xylo, xyloside; Pel, Pelargonidin; soph, sophoroside; rham, rhamnoside; malo, malonyl; Pet, Petunidin; Mal, Malvidin; Del, Delphinidin.
To identify regulatory candidate genes involved in anthocyanin biosynthesis in H. miconioides sepals, a network correlation analysis between the expression of 15 DEGs and the amount of 38 DAMs was conducted. All correlations between the DEGs and DAMs are shown in Supplementary Table 5. There were 13 differentially expressed anthocyanin structural genes that exhibited strong correlation coefficients (PCC > 0.9, P < 0.05) with 20 anthocyanins and their interaction networks were organized and presented in Figure 5A. In particular, the relative amount of cyanidin-3,5-O-diglucoside, cyanidin-3-O-galactoside, cyanidin-3-O-glucoside, proanthocyanidin B3, pelargonidin-3-O-glucoside, and peonidin-3-O-galactoside was significantly positively correlated with HmPAL5, Hm4CL1, HmCHS1, HmCHS2, HmCHS3, HmCHS4, and HmANS1 expression (PCC > 0.9, P < 0.05). These anthocyanins were significantly negatively correlated with HmPAL2 (PCC < −0.9, P < 0.05) (Figure 5A). The results indicated that these genes may represent key genes affecting anthocyanin biosynthesis in the sepal.
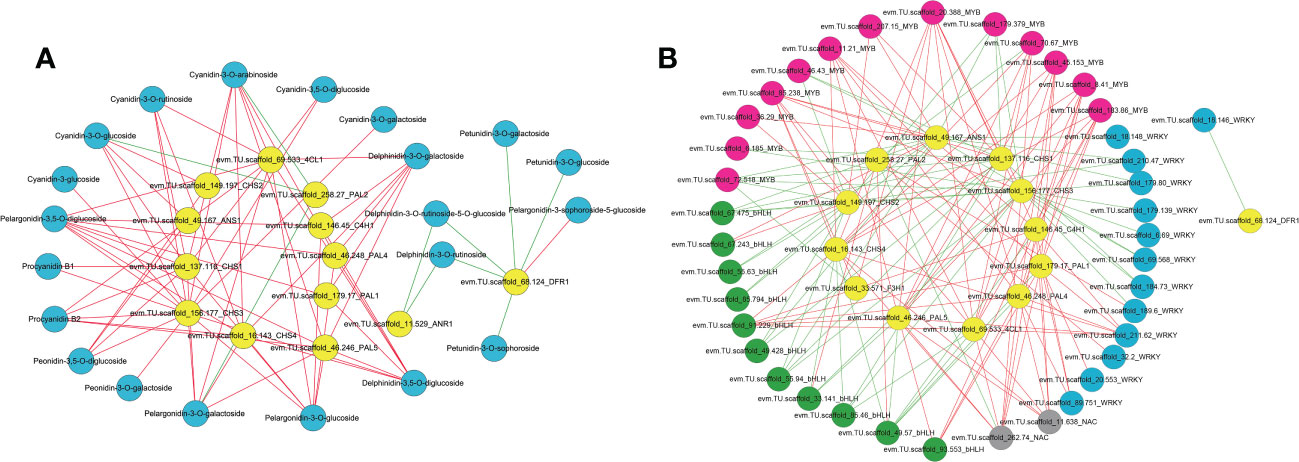
Figure 5 Correlation network analysis of anthocyanin structural genes with anthocyanin components, MYB, bHLH, WRKY and NAC TFs in the sepal of Heptacodium miconioides. (A) Interaction network between selected genes and anthocyanins. (B) Interaction network between selected genes and candidate transcription factors. The red line indicates a positive correlation and the green line indicates a negative correlation.
To further determine the candidate TFs involved in the regulation of anthocyanin biosynthesis in sepals, we analyzed MYB, bHLH, WRKY, NAC, bZIP and HSF TFs with significant differences in expression in the sepal at each developmental stage (Supplementary Figure 2 and Supplementary Table 6). After screening DEGs with FPKM <1.0 at the four sepal developmental stages, 69 TFs including 30 MYB, 14 bHLH, 15 WRKY, 5 NAC, 3 bZIP, and 2 HSF TFs remained (Supplementary Figure 2). A correlation network was established between the anthocyanin biosynthetic pathway genes and the 69 differential TFs. Seven previously screened anthocyanin biosynthesis genes (PAL, 4CL, C4H, CHS, ANR, DFR, and ANS) were selected as “guide genes” to analyze co-expression correlations. Of the differentially expressed TFs, 9 MYB, 3 bHLH, 4 WRKY, and 2 NAC TFs were significantly positively correlated with anthocyanin biosynthesis genes (Supplementary Table 7). Subsequently, the MYB, bHLH, and WRKY TFs of H. miconioides were used to construct a phylogenetic tree with A. thaliana (Supplementary Figures 3–5). Several MYB, bHLH, and WRKY TFs were clustered into the subgroup of the confirmed anthocyanin biosynthesis-regulating A. thaliana MYB, bHLH, and WRKY families, which were also significantly correlated with anthocyanin. HmMYB114 (evm.TU.scaffold 70.67, AtMYB114) and HmMYB11 (evm.TU.scaffold 85.238, AtMYB11) were positively correlated with five (HmPAL5, Hm4CL1, HmCHS1, HmCHS3, and HmCHS4) and seven (Hm4CL1, HmCHS1, HmCHS2, HmCHS3, HmCHS4, HmF3H1, and HmANS1) anthocyanin structural genes, respectively (Figure 5B and Supplementary Table 6). HmMYB12 (evm.TU.scaffold 36.29, AtMYB12) was positively correlated with two (HmCHS2, HmANS1) structural genes. HmbHLH42 (evm.TU.scaffold 67.243, AtbHLH42/AtTT8) was positively correlated with two (HmCHS2, HmANS1) anthocyanin structural genes (Figure 5B and Supplementary Table 6). HmbHLH130 (evm.TU.scaffold_91.229, AtbHLH130) was positively correlated with five (HmPAL1, HmPAL4, HmPAL5, HmC4H1, and HmCHS4) anthocyanin structural genes. In contrast, HmbHLH1 (evm.TU.scaffold 49.57, AtHLH1/AtHLH2) was negatively correlated with these structural genes. HmWRKY6-1 (evm.TU.scaffold_211.62, AtWRKY6) was positively correlated with six (HmPAL1, HmPAL4, HmPAL5, HmC4H1, Hm4CL1, and HmCHS4) structural genes. HmWRKY6-2 (evm.TU.scaffold_89.751, AtWRKY6) was positively correlated with three (HmPAL1, HmPAL4, and HmC4H1) structural genes. In addition, there were also two NAC TFs (evm.TU.scaffold 262.74, evm.TU.scaffold 11.638) that were significantly positively correlated with several anthocyanin structural genes.
To validate the correlation between TFs and structural genes, HmMYB114, HmMYB11, HmbHLH130, HmWRKY6, HmNAC1, HmNAC2, HmCHS4, and HmDFR1 were selected for subsequent functional analysis. We sequenced the upstream promoter region of HmCHS4 (1820bp), and analyzed its cis-acting elements. Many cis-acting elements related to MYB, bHLH, WRKY, and NAC TFs were identified, including the four core MYB binding sites (5′-AACC-3′), one bHLH-binding site (5′-CATGTG-3′), one G-box (5′-CACGTG-3′), two WRKY binding sites [5′-(C/T)TGAC(T/C)-3′], and three NAC binding sites [5′-CGT(A/G)-3′] (Figure 6A). Analysis of the upstream promoter region of HmDFR1 (1960bp) also found several cis-acting elements related to MYB, bHLH, WRKY, and NAC TFs, including six core MYB binding sites, two G-box binding sites, one WRKY binding site, and three NAC binding sites (Figure 6B). The results suggested that these TFs may have potential interaction with HmCHS4 and HmDFR1 promoters.

Figure 6 Transactivation assays of HmCHS4 and HmDFR1 promoters by HmMYB114, HmMYB11, HmbHLH130, HmWRKY6, HmNAC1, and HmNAC2 transcription factors. (A, B) Schematic overview of HmCHS4 and HmDFR1 promoters. (C, D) Imaging of luciferase enzyme activity of HmCHS4 and HmDFR1 promoters. Luciferase activity was detected in the tobacco leaves. The 35S-nos construct is used as a negative control. (E, F) The relative luciferase signal intensity of HmCHS4 and HmDFR1 promoters were measured in the effector and reporter vector. Values are the means ± standard deviation (SD). Data is the mean of three biological replicates. Different lowercase letters indicated significant differences at 0.05 level.
To further confirm this hypothesis, HmCHS4 and HmDFR1 were selected for subsequent transient expression assay in N. benthamiana leaves. We generated constructs including the LUC reporter gene individually driven by HmCHS4 promoter (proHmCHS4:LUC) as reporter, constructs including HmMYB114, HmMYB11, HmbHLH130, HmWRKY6, HmNAC1, and HmNAC2 CDS driven by the cauliflower mosaic virus (CaMV35S) as effectors (pro35S:HmMYB114, pro35S:HmMYB11, pro35S:HmbHLH130, pro35S:HmWRKY6-1, pro35S:HmNAC1, and pro35S:HmNAC2, respectively), and a negative control containing only the CaMV35S promoter and the terminator (pro35S:nos). As shown in Figure 6C, the amount of LUC luminescence was significantly higher when each effector (pro35S:HmMYB114, pro35S:HmMYB11, pro35S:HmbHLH130, pro35S:HmWRKY6, pro35S:HmNAC1, and pro35S:HmNAC2) was co-transformed with the proHmCHS4:LUC reporter compared with the corresponding controls. The LUC luminescence was significantly higher when each effector (pro35S:HmMYB114, pro35S:HmbHLH130, pro35S:HmMYB114+HmbHLH130, pro35S:HmWRKY6, and pro35S:HmNAC1) was co-transformed with the proHmDFR1:LUC reporter (Figures 6D, F). These results indicated that the HmMYB114, HmbHLH130, HmWRKY6, and HmNAC1 could activate the promoters of HmCHS4 and HmDFR1 in vitro.
Studies on color changes and anthocyanins in flowering plants have been primarily focused on petal organs and less attention has been given to sepal color changes. The molecular mechanisms of color formation in this species remain unclear. Anthocyanins are a class of water-soluble pigments present in the vacuoles of plant epidermal cells, which impart various colors to plant organs and tissues. Anthocyanin accumulation in floral organs is a key trait that affects the quality and ornamental value of plants (Zhao et al., 2012; Luo et al., 2021). In the present study, an integrated metabolome and transcriptome analysis was performed to examine the anthocyanin components and identify the genes involved in anthocyanin biosynthesis during sepal development. In recent years, the components and anthocyanin content of different plant sepals have been identified. For example, the major anthocyanin enriched in Rhyncholaeliocattleya Beauty Girl “KOVA” (KOVA) sepals is cyanidin O-acetylhexoside (Li et al., 2020a). Delphinidin-3-sambubioside and cyanidin-3-sambubioside are the two major anthocyanins in the calyx of Hibiscus sabdariffa (Hinojosa-Gómez et al., 2020). Hydrangea is a unique flower composed of sepals rather than true petals, and the main anthocyanin in the sepals is 3-O-glucosyldelphinidin (Yoshida et al., 2021). Similarly, we further identified 41 anthocyanins based on an anthocyanin-targeted metabolome assay and found that cyanidin-3,5-O-diglucoside, cyanidin-3-O-galactoside, cyanidin-3-O-glucoside, proanthocyanidin, pelargonidin-3-O-glucoside, and peonidin-3-O-galactoside are the major anthocyanins in the H. miconioides sepal. By analyzing the anthocyanin components in the developing sepal, we found that the content of most delphinidin, malvidin, and petunidin derivatives decreased with the development of the sepal, indicating that the delphinidin, malvidin, and petunidin components were not the main factors in the color change of the sepals. In addition, the cyanidin-3,5-O-diglucoside, cyanidin-3-O-galactoside, and cyanidin-3-O-glucoside content exhibited an increasing trend with the development of the sepal, suggesting that the high content of the cyanidin derivatives may be essential for the bright red color of the H. miconioides sepal. This is consistent with previous studies on ornamental plants, such as that by Shen et al. (2021), in which the red-colored flower in wintersweet was associated with cyanidin-3-O-glucoside, cyanidin-3-O-rutinoside, and cyanidin-3-O-galactoside content.
The regulatory mechanism for color formation is closely related to the expression of anthocyanin structural genes and regulatory genes (Forkmann and Martens, 2001; Nakatsuka et al., 2019). Previous studies have shown that several key genes and enzymes are required for anthocyanin accumulation and sepal color formation in many ornamental plants including H. macrophylla (Yoshida et al., 2021) and Rhyncholaeliocattleya Beauty Girl “KOVA” (Li et al., 2020a). However, the mechanism involved in anthocyanin biosynthesis in H. miconioides sepals is unclear. Anthocyanin biosynthesis is a branch of flavonoid synthesis pathway starting from phenylalanine, which involves a variety of enzymes encoded by early biosynthesis genes (PAL, 4CL, C4H, CHS, CHI, F3H, F3’H, and F3’5’H) and anthocyanin biosynthesis genes (DFR, ANS, and UFGT) (Winkel-Shirley, 2001). In the present study, we identified 15 DEGs associated with the anthocyanin biosynthetic pathway of H. miconioides sepals including 11 early biosynthesis genes, 2 proanthocyanidins biosynthesis gene, and 2 anthocyanin biosynthesis genes, with almost all of them expressed at high levels during S3 and S4 compared with extremely low levels at S1–S2. These results indicate that these DEGs may play a role in the accumulation of anthocyanins at S3 to S4. Intriguingly, the expression profiles changed significantly, whereas there was no significant difference in anthocyanin product levels between S3 and S4. It is probable that the biosynthesis and accumulation of anthocyanins were closely related to environmental factors, structural genes, and TFs. The environmental factors such as temperature and light might affect the gene expression which leads to the change in anthocyanins biosynthesis and accumulation (Xue et al., 2021). Previous studies have shown that there was no significant difference in the expression of early biosynthesis genes in purple pepper and green pepper, whereas the expression of anthocyanin biosynthesis genes was significantly different (Borovsky et al., 2004; Jung et al., 2019). In addition, it was reported that the yellow color of Paeonia lactiflora cultivar “Huangjinlun” results from the accumulation of chalcone induced by low expression of PlCHI (Zhao et al., 2012). In our study, a transcriptome and metabolome association analysis results revealed that the accumulation of anthocyanins was significantly positively correlated with the expression levels of several candidate genes. HmPAL, Hm4CL, HmC4H, HmCHS, HmF3H, and HmANS expression exhibited a strong positive correlation with anthocyanin content during the four sepal stages and these expression patterns were consistent with the results of previous studies. For example, in Cymbidium hybrid flowers, the expression of ANS and DFR was closely associated with the patterns of anthocyanin accumulation (Nakatsuka et al., 2019). The expression of CHS and other anthocyanin genes (UFGT2 and DFR) was also implicated in the accumulation of anthocyanin in mature fruits from different strawberry cultivars (Shen et al., 2021). Of these, the expression of the HmCHS HmF3H1, and HmANS genes was upregulated and highly expressed in the whole developmental stages, which contributed to the production of anthocyanin derivatives. Of note, we identified more than one anthocyanin biosynthesis-related HmCHS gene (four), suggesting that these genes have multiple transcripts or different gene family members. These results suggest that the high copy numbers and expression levels of key genes involved in the anthocyanidin biosynthesis pathway drive the production of these major anthocyanidin compounds in the sepal.
Colourful pigments were closely related to the biosynthesis of anthocyanins and proanthocyanidin. Proanthocyanidin are biopolymers composed primarily of epicatechin and catechin units (Dixon et al., 2005). LAR and ANR are considered two key enzymes in proanthocyanidin biosynthesis, which is in competition with the anthocyanidin formation catalysed by ANS and 3-glucosyltransferase (3GT), respectively (Pucker et al., 2020). It has been reported that the low expression levels of LAR and ANR in Trifolium repens leaves eventually results in a lack of proanthocyanidin biosynthesis (Ma et al., 2022). The suppression of ANR function caused a reduction in proanthocyanidin biosynthesis in the in Glycine max seed coat, with increased anthocyanin accumulation (Kovinich et al., 2012). In our study, the HmANR1 gene was highly expressed at the S2 and S4 stages and contributed to the increase of proanthocyanidin products. This spatiotemporal pattern of proanthocyanidin might play a vital role in the growth and development of sepals and be related to potential antioxidants (Bagchi et al., 2000; Dixon et al., 2005). So far, the anthocyanidin biosynthesis pathway of mechanism in the sepal of H. miconioides is not clear. In this study, we discovered that cyanidin’s derivative is the main factor affecting H. miconioides sepal color. By analyzing the transcriptome data of four developmental sepal stages with different sepal colors, we hypothesized the putative pathway of anthocyanin biosynthesis in the sepal of H. miconioides. The putative pathway contains 15 protein families, including PAL, 4CL, C4H, CHS, CHI, F3H, ANS, LAR, ANR, UFGT, 3MaT, 3RT, 3AT, MT, and MATE. These results will further improve understanding of this anthocyanin biosynthesis and accumulation in H. miconioides sepals.
In addition to structural genes, TFs also play an important role in the regulation of anthocyanin biosynthesis. In the present study, the MYB, bHLH, and other TFs, such as WRKYs, bZIPs NACs, HY5s, and ERFs were classified and identified. Previous studies have shown that the MYB-bHLH-WD40 complex represents a major class of TFs regulating the structural genes of the anthocyanin pathway including those in A. thaliana (Nesi et al., 2001), grapes (Deluc et al., 2006), persimmons (Akagi et al., 2009), and strawberries (Schaart et al., 2013). Thus far, several R2R3-MYB genes have been identified that control the expression of the key CHS, CHI, and F3H genes in early anthocyanin biosynthesis (Mehrtens et al., 2005; Stracke et al., 2007). In the present study, four candidate MYB TFs were identified from the DEG data, which exhibited a strong positive correlation with the expression of structural genes. One of these, HmMYB114 (evm.TU.scaffold_70.67), was in the same clade as MYB75, MYB90, MYB113, and MYB114 in A. thaliana. Previous studies demonstrated that AtMYB75, AtMYB90, AtMYB113, and AtMYB114 are involved in the MYB-bHLH-WD40 complex, which regulates the expression of anthocyanidin biosynthesis-related genes and enhances anthocyanin content (Mondal and Roy, 2018). The other two HmMYBs (evm.TU.scaffold_85.238; evm.TU.scaffold_36.29) were assigned to a large clade with AtMYB11, AtMYB12, and AtMYB111, which are associated with flavonol biosynthesis genes (CHS, CHI, F3H, and FLS) as transcriptional activators (Stracke et al., 2007; Naik et al., 2021). Both of HmMYBs are strongly expressed in H. miconioides sepals and are positively correlated with structural gene expression. In addition, bHLH family members frequently interact with MYB TFs to regulate anthocyanin biosynthesis through downstream gene expression (Spelt et al., 2002). The bHLH TFs can also directly bind to the promoter regions of structural genes to regulate the expression of the CHS, DFR, and UFGT anthocyanin genes in Capsicum annuum (Stommel et al., 2009). Most bHLHs involved in regulating anthocyanin biosynthesis are relatively conserved. In the present study, as shown in the phylogenetic tree, two HmbHLHs (evm.TU.scaffold_67.243; evm.TU.scaffold_49.57) were in the same clade as AtTT8, AtbHLH1, and AtbHLH2, which are involved in anthocyanin biosynthesis (Wang et al., 2019). However, TTG1 (a WD40 TF, evm.TU.scaffold_93.62) showed no significant differences at the transcriptional level at the four sepal developmental stages of H. miconioides in our dataset.
Aside from the MYB-bHLH-WD40 complex, other TFs have also been shown to play a role in controlling anthocyanin accumulation, such as the AP3- and AGL6-like genes (an MADS domain TF) in Cattleya hybrid “KOVA” (Li et al., 2020a), NAC56a/b (an NAC TF) in sweet potato (Wei et al., 2020), MdERF1B (an AP2/ERF TF) in apple (Zhang et al., 2018), PyWRKY26 (a WRKY TF) in pear (Li et al., 2020b), and SlHY5 (a bZIP TF) in tomato (Liu et al., 2018). Li et al. (2020a) found that the AP3- and AGL6-like MADS-box genes have multiple functions and are involved in the differential shape and color of sepals. In the present study, eight differentially expressed MADS-box TFs showed no correlation with the expression of structural genes, indicating that the color changes in the sepals of H. miconioides are different from that of orchids (data not show). Previous studies have shown that PyWRKY26 and PyWRKY31 regulate color formation in red-skinned pears and occurs through the interaction of PyWRKY26 and PybHLH3 co-targeting the PyMYB114 promoter (Li et al., 2020b). Of these, PyWRKY31 exhibits high homology with AtWRKY6. In the present study, two HmWRKY (evm.TU.scaffold_211.62; evm.TU.scaffold_89.751) TFs were identified that are highly homologous to AtWRKY6. Here, two HmWRKYs and two HmNACs exhibited a strong positive correlation with the expression of structural genes. Previous studies have shown that the recognition sequences of MYB, bHLH, WRKY and NAC proteins binding motif are mainly AACC, CANNTG, (C/T)TGAC(T/C) and CGT[A/G], respectively (Xiong et al., 2016; Wang et al., 2017; Luo et al., 2021). In this study, we analyzed the HmCHS4 and HmDFR1 promoter regions for binding sites of these TFs, indicating potential transcriptional regulation. Luciferase activity assays further confirmed that HmMYB114, HmbHLH130, HmWRKY6, and HmNAC1 significantly increased the transcriptional activity of the promoters of the structural genes HmCHS4 and HmDFR1 in N. benthamiana leaves. In addition, co-transformation with HmMYB114 and HmbHLH130 had a stronger activation effect on the HmDFR1 promoter sequence than other TFs. The results indicated that these TFs can be potentially regarded as candidate genes for the regulation of anthocyanin biosynthesis. However, most of the differentially expressed TFs in the sepal might be connected to the development of floral organs. Future work is required to further isolate anthocyanin-related TFs from floral organs development. In this study, the development of sepal may be accompanied by changes in environmental factors such as temperature. Previous studies have shown that low temperature will increase anthocyanin accumulation during grape fruit ripening, while high temperature will lead to anthocyanin degradation (Yamane and Shibayama, 2006). The anthocyanin accumulation in sepal was separately affected by environment factors, which needed to be distinguished by further experiments.
Taken together, our study elucidated anthocyanin metabolites that contribute to the color conversion in H. miconioides sepals. A potential detailed pathway of anthocyanin biosynthesis in sepals of H. miconioides was proposed. We also showed that the altered expression of the anthocyanin pathway genes and regulatory TFs account for the difference in anthocyanin accumulation in the sepal at different developmental stages. Furthermore, the precise functional mechanism of stable transgenic requires further investigation to verify their role in regulation of anthocyanin biosynthesis.
The H. miconioides sepal RNA sequencing data have been submitted to NCBI under the accession no. PRJNA723161 (accession: SRX10718801; SRX10718800; SRX10718799; SRX10718798; SRX10718797; SRX10718796; SRX10718795; SRX10718794; SRX10718793; SRX10718792; SRX10718791; SRX10718790). The genome FASTA assembly and annotations files are available on FigShare at the link: https://doi.org/10.6084/m9.figshare.16803631.v1. The raw data of the metabolome analyses have been identified in MetaboLights as MTBLS6277.
JML and ZXJ conceived and designed the experiments. YLL and ZSS collected the samples and performed the experiments. JYL provided technical assistance to YLL YLL wrote the draft manuscript and then ZXJ and JML revised the manuscript. All authors contributed to the article and approved the submitted version.
The work was supported by the Zhejiang Provincial Natural Science Foundation of China (LQ19C030001), the National Natural Science Foundation of China (32001113), and the Zhejiang Provincial Natural Science Foundation of China (LTY22C030002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1044581/full#supplementary-material
Akagi, T., Ikegami, A., Tsujimoto, T., Sato, A., Kono, A., Yonemori, K. (2009). DkMyb4 is a myb transcription factor involved in proanthocyanidin biosynthesis in persimmon fruit. Plant Physiol. 151, 2028–2045. doi: 10.1104/pp.109.146985
Anders, S., Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi: 10.1186/gb-2010-11-10-r106
An, J. P., Qu, F. J., Yao, J. F., Wang, X. N., You, C. X., Wang, X. F., et al. (2017). The bZIP transcription factor MdHY5 regulates anthocyanin accumulation and nitrate assimilation in apple. Hortic. Res. 4 (1), 17023. doi: 10.1038/hortres.2017.23
Bagchi, D., Bagchi, M., Stohs, S. J., Das, D. K., Ray, S. D., Kuszynski, C. A., et al. (2000). Free radicals and grape seed proanthocyanidin extract: importance in human health and disease prevention. Toxicology 148, 187–197. doi: 10.1016/s0300-483x(00)00210-9
Bian, C. M., Jin, Z. X., Li, J. M. (2002). A study on the reproductive biology of Heptacodium miconioides. Acta Bot. Yunanica 24, 613–618. doi: cnki:sun:yoke.0.2002-05-011
Borovsky, M., Oren-Shamir, R., Ovadia, W. D., De Jong, W., Paran, I. (2004). The a locus thatcontrols anthocyanin accumulation in pepper encodes a MYB transcription factor homologous to Anthocyanin2 of petunia. Theor. Appl. Genet. 109 (1), 23–29. doi: 10.1007/s00122-004-1625-9
Boss, P. K., Davies, C., Robinson, S. P. (1996). Analysis of the expression of anthocyanin pathway genes in developing Vitis vinifera l. cv Shiraz grape berries and the implications for pathway regulation. Plant Physiol. 111 (4), 1059–1066. doi: 10.1104/pp.111.4.1059
Carretero-Paulet, L., Galstyan, A., Roig-Villanova, I., Martínez-García, J. F., Bilbao-Castro, J. R., Robertson, D. L. (2010). Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 153, 1398–1412. doi: 10.1104/pp.110.153593
Chen, C. Q., Chen, H. X., Yang, W. L., Li, J., Tang, W. J., Gong, R. G. (2022). Transcriptomic and metabolomic analysis of quality changes during sweet cherry fruit development and nining of related genes. Int. J. Mol. Sci. 23, 7402. doi: 10.3390/ijms23137402
Chen, S. F., Zhou, Y. Q., Chen, Y. R., Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890. doi: 10.1093/bioinformatics/bty560
Coombes, A. J. (1990). Heptacodium jasminoides the Chinese seven-son flower in Britain. Curtis’s Bot. Mag. 7, 133–138. doi: 10.1111/j.1467-8748.1990.tb00159.x
Deluc, L., Barrieu, F., Marchive, C., Lauvergeat, V., Decendit, A., Richard, T., et al. (2006). Characterization of a grapevine R2R3-MYB transcription factor that regulates the phenylpropanoid pathway. Plant Physiol. 140, 499–511. doi: 10.1104/pp.105.067231
Dixon, R. A., Xie, D. Y., Sharma, S. B. (2005). Proanthocyanidins–a final frontier in flavonoid research? New Phytol. 165, 9–28. doi: 10.1111/j.1469-8137.2004.01217.x
Dong, T. T., Han, R. P., Yu, J. W., Zhu, M. K., Zhang, Y., Gong, Y., et al. (2009). Anthocyanin accumulation and molecular analysis of correlated genes by metabolome and transcriptome in green and purple asparaguses (Asparagus officinalis, l). Food Chem. 271, 18–28. doi: 10.1016/j.foodchem.2018.07.120
Forkmann, G., Martens, S. (2001). Metabolic engineering and applications of flavonoids. Curr. Opin. Biotech. 12, 155–160. doi: 10.1016/S0958-1669(00)00192-0
He, J., Giusti, M. M. (2010). Anthocyanins: natural colorants with health-promoting properties. Annu. Rev. Food Sci. Technol. 1, 163–187. doi: 10.1146/annurev.food.080708.100754
Hinojosa-Gómez, J., Martín-Hernández, C. S., Heredia, J. B., León-Félix, J., Osuna-Enciso, T., Muy-Rangel, M. D. (2020). Anthocyanin induction by drought stress in the calyx of roselle cultivars. Molecules 25, 1555. doi: 10.3390/molecules25071555
Hu, J. F., Fang, H. C., Wang, J., Yue, X. X., Su, M. Y., Mao, Z. L., et al. (2020). Ultraviolet b-induced MdWRKY72 expression promotes anthocyanin synthesis in apple. Plant Sci. 292, 110377. doi: 10.1016/j.plantsci.2019.110377
Jung, S., Venkatesh, J., Kang, M. Y., Kwon, J. K., Kang, B. C. (2019). A non-LTR retrotransposon activates anthocyanin biosynthesis by regulating a MYB transcription factor in capsicum annuum. Plant Sci. 287, 110181. doi: 10.1016/j.plantsci.2019.110181
Kim, D., Langmead., B., Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357. doi: 10.1038/NMETH.3317
Koes, R., Verweij, W., Quattrocchio, F. (2005). Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 10, 236–242. doi: 10.1016/j.tplants.2005.03.002
Kovinich, N., Saleem, A., Rintoul, T. L., Brown, D. C. W., Arnason, J. T., Miki, B. (2012). Coloring genetically modified soybean grains with anthocyanins by suppression of the proanthocyanidin genes ANR1 and ANR2. Transgenic Res. 21, 757–771. doi: 10.1007/s11248-011-9566-y
Lin, W., Li, Y., Lu, Q., Lu, H., Li, J. (2020). Combined analysis of the metabolome and transcriptome identified candidate genes involved in phenolic acid biosynthesis in the leaves of Cyclocarya paliurus. Int. J. Mol. Sci. 21 (4), 1337. doi: 10.3390/ijms21041337
Liu, C. C., Chi, C., Jin, L. J., Zhu, J., Yu, J. Q., Zhou, Y. H. (2018). The bZip transcription factor HY5 mediates CRY1a-induced anthocyanin biosynthesis in tomato. Plant Cell Environ. 41, 1762–1775. doi: 10.1111/pce.13171
Liu, J., Shi, M., Wang, J., Zhang, B., Liang, Y. (2020). Comparative transcriptomic analysis of the development of sepal morphology in tomato (Solanum lycopersicum l.). Int. J. Mol. Sci. 21 (16), 5914. doi: 10.3390/ijms21165914
Liu, F., Wang, Y., Ding, Z. T., Zhao, L., Xiao, J., Wang, L. J., et al. (2017). Transcriptomic analysis of flower development in tea (Camellia sinensis (L.)). Gene (Basel) 631, 39–51. doi: 10.1016/j.gene.2017.08.013
Liu, P., Yang, Y. S., Xu, G. D., Hao, C. Y. (2006). Physiological response of rare and endangered seven-son-flower (Heptacodium miconioides) to light stress under habitat fragmentation. Environ. Exp. Bot. 57, 32–40. doi: 10.1016/j.envexpbot.2005.04.003
Li, C., Wu, J., Hu, K. D., Wei, S. W., Sun, H. Y., Hu, L. Y., et al. (2020b). PyWRKY26 and PybHLH3 cotargeted the PyMYB114 promoter to regulate anthocyanin biosynthesis and transport in red-skinned pears. Hortic. Res. 7, 37. doi: 10.1038/s41438-020-0254-z
Li, B. J., Zheng, B. Q., Wang, J. Y., Tsai, W. C., Lu, H. C., Zoi, L. H., et al. (2020a). New insight into the molecular mechanism of colour differentiation among floral segments in orchids. Commun. Biol. 3, 89. doi: 10.1038/s42003-020-0821-8
Lou, Y., Zhou, H. S., Han, Y., Zeng, Q. Y., Zhu, J., Yang, Z. N. (2018). Positive regulation of AMS by TDF1 and the formation of a TDF1–AMS complex are required for anther development in Arabidopsis thaliana. New Phytol. 217, 378–391. doi: 10.1111/nph.14790
Luo, X. N., Sun, D. Y., Wang, S., Luo, S., Fu, Y. Q., Niu, L. X., et al. (2021). Integrating full-length transcriptomics and metabolomics reveals the regulatory mechanisms underlying yellow pigmentation in tree peony (Paeonia suffruticosa andr.) flowers. Hortic. Res. 8, 235. doi: 10.1038/s41438-021-00666-0
Ma, S. N., Hu, R. H., Ma, J. W., Fan, J. W., Wu, F. F., Wang, Y., et al. (2022). Integrative analysis of the metabolome and transcriptome provides insights into the mechanisms of anthocyanins and proanthocyanidins biosynthesis in trifolium repens. Ind. Crops Prod. 187, 115529. doi: 10.1016/j.indcrop.2022.115529
Mehrtens, F., Kranz, H., Bednarek, P., Weisshaar, B. (2005). The arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 138, 1083–1096. doi: 10.1104/pp.104.058032
Mondal, S. K., Roy, S. (2018). Genome-wide sequential, evolutionary, organizational and expression analyses of phenylpropanoid biosynthesis associated MYB domain transcription factors in arabidopsis. J. Biomol. Struct. Dyn. 36, 1577–1601. doi: 10.1080/07391102.2017.1329099
Naik, J., Rajput, R., Pucker, B., Stracke, R., Pandey, A. (2021). The R2R3-MYB transcription factor MtMYB134 orchestrates flavonol biosynthesis in Medicago truncatula. Plant Mol. Biol. 106, 157–172. doi: 10.1007/s11103-021-01135-x
Nakatsuka, T., Suzuki, T., Harada, K., Kobayashi, Y., Ohno, H. (2019). Floral organ- and temperature-dependent regulation of anthocyanin biosynthesis in Cymbidium hybrid flowers. Plant Sci. 287, 110173. doi: 10.1016/j.plantsci.2019.110173
Nesi, N., Jond, C., Debeaujon, I., Caboche, M., Lepiniec, L. (2001). The arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell 13, 2099–2114. doi: 10.1105/TPC.010098
Pucker, B. (2022). Automatic identification and annotation of MYB gene family members in plants. BMC Genomics 23, 220. doi: 10.1186/s12864-022-08452-5
Pucker, B., Reiher, F., Schilbert, H. M. (2020). Automatic identification of players in the flavonoid biosynthesis with application on the biomedicinal plant croton tiglium. Plants (Basel) 9, 1103. doi: 10.3390/plants9091103
Ramsay, N. A., Glover, B. J. (2005). MYB–bHLH–WD40 protein complex and the evolution of cellular diversity. Trends Plant Sci. 10 (2), 63–70. doi: 10.1016/j.tplants.2004.12.011
Rothenberg, D. O., Yang, H. J., Chen, M. B., Zhang, W. T., Zhang, L. Y. (2019). Metabolome and transcriptome sequencing analysis reveals anthocyanin metabolism in pink flowers of anthocyanin-rich tea (Camellia sinensis). Molecules 24, 1064. doi: 10.3390/molecules24061064
Saito, K., Yonekura-Sakakibara, K., Nakabayashi, R., Higashi, Y., Yamazaki, M., Tohge, T., et al. (2013). The flavonoid biosynthetic pathway in arabidopsis: structural and genetic diversity. Plant Physiol. Bioch. 72, 21–34. doi: 10.1016/j.plaphy.2013.02.001
Schaart, J. G., Dubos, C., de la Fuente, I. R., van Houwelingen, A. M. M. L., de Vos, R. C. H., Jonker, H. H., et al. (2013). Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria x ananassa) fruits. New Phytol. 197, 454–467. doi: 10.1111/nph.12017
Shen, Z., Li, W., Li, Y., Liu, M., Cao, H., Provart, N., et al. (2021). The red flower wintersweet genome provides insights into the evolution of magnoliids and the molecular mechanism for tepal color development. Plant J. 108, 1662–1678. doi: 10.1111/tpj.15533
Spelt, C., Quattrocchio, F., Mol, J., Koes, R. (2002). ANTHOCYANIN1 of petunia controls pigment synthesis, vacuolar pH, and seed coat development by genetically distinct mechanisms. Plant Cell 14, 2121–2135. doi: 10.1105/tpc.003772
Stommel, J. R., Lightbourn, G. J., Winkel, B. S., Griesbach, R. J. (2009). Transcription factor families regulate the anthocyanin biosynthetic pathway in capsicum annuum. J. Hortic. Sci. Biotech. 134, 244–251. doi: 10.1051/fruits/2009006
Stracke, R., Ishihara, H., Huep, G., Barsch, A., Mehrtens, F., Niehaus, K., et al. (2007). Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 50, 660–677. doi: 10.1111/j.1365-313X.2007.03078.x
Sun, Q. G., Jiang, S. H., Zhang, T. L., Xu, H. F., Fang, H., Zhang, J., et al. (2019). Apple NAC transcription factor MdNAC52 regulates biosynthesis of anthocyanin and proanthocyanidin through MdMYB9 and MdMYB11. Plant Sci. 289, 110286. doi: 10.1016/j.plantsci.2019.110286
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi: 10.1093/molbev/msab120
Tanaka, Y., Sasaki, N., Ohmiya, A. (2008). Biosynthesis of plant pigments: anthocyanins, betalains and caroten. Plant J. 54, 733–749. doi: 10.1111/j.1365-313X.2008.03447.x
Wang, L., Li, Z., Lu, M., Wang, Y. (2017). ThNAC13, a NAC transcription factor from Tamarix hispida, confers salt and osmotic stress tolerance to transgenic Tamarix and Arabidopsis. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00635
Wang, L. H., Tang, W., Hu, Y. W., Zhang, Y. B., Sun, J. Q., Guo, X. H., et al. (2019). A MYB/bHLH complex regulates tissue-specific anthocyanin biosynthesis in the inner pericarp of red-centered kiwifruit actinidia chinensis cv. hongyang. Plant J. 99, 359–378. doi: 10.1111/tpj.14330
Wei, Z. Z., Hu, K. D., Zhao, D. L., Tang, J., Huang, Z. (2020). MYB44 competitively inhibits the formation of the MYB340-bHLH2-NAC56 complex to regulate anthocyanin biosynthesis in purple-fleshed sweet potato. BMC Plant Biol. 20, 258. doi: 10.1186/s12870-020-02451-y
Winkel-Shirley, B. (2001). Flavonoid biosynthesis. a colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 126, 485–493. doi: 10.1104/pp.126.2.485
Wu, Y. Q., Guo, J., Wang, T. L., Cao, F. L., Wang, G. B. (2020). Metabolomic and transcriptomic analyses of mutant yellow leaves provide insights into pigment synthesis and metabolism in ginkgo biloba. BMC Genomics 21, 858. doi: 10.1186/s12864-020-07259-6
Xiong, S. X., Lu, J. Y., Lou, Y., Teng, X. D., Gu, J. N., Zhang, C., et al. (2016). The transcription factors MS188 and AMS form a complex to activate the expression of CYP703A2 for sporopollenin biosynthesis in Arabidopsis thaliana. Plant J. 88, 936–946. doi: 10.1111/tpj.13284
Xue, X., Duan, Y., Wang, J., Ma, F., Li, P. (2021). Nighttime temperatures and sunlight intensities interact to influence anthocyanin biosynthesis and photooxidative sunburn in “Fuji” apple. Front. Plant Sci. 12, 6944954. doi: 10.3389/fpls.2021.694954
Yamane, T., Shibayama, K. (2006). Effects of changes in the sensitivity to temperature on skin coloration in ‘Aki queen’ grape berries. J. Jpn. Soc Hortic. Sci. 75, 458–462. doi: 10.2503/jjshs.75.458
Yang, Y. X., Chen, X. X., Xu, B., Li, Y. X., Ma, Y. H., Wang, G. D. (2015). Phenotype and transcriptome analysis reveals chloroplast development and pigment biosynthesis together influenced the leaf color formation in mutants of Anthurium andraeanum 'Sonate'. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00139
Yoshida, K., Ito, D., Miki, N., Kondo, T. (2021). Single-cell analysis clarifies mosaic color development in purple hydrangea sepal. New Phytol. 229, 3549–3557. doi: 10.1111/nph.17099
Zhang, Y. F., Chen, C., Jin, Z. X., Yang, Z. N., Li, Y. L. (2022). Leaf anatomy, photosynthesis, and chloroplast ultrastructure of Heptacodium miconioides seedlings reveal adaptation to light environment. Environ. Exp. Bot. 195, 104780. doi: 10.1016/j.envexpbot.2022.104780
Zhang, J., Xu, H. F., Wang, N., Jiang, S. H., Fang, H. C., Zhang, Z. Y., et al. (2018). The ethylene response factor MdERF1B regulates anthocyanin and proanthocyanidin biosynthesis in apple. Plant Mol. Biol. 98, 205–218. doi: 10.1007/s11103-018-0770-5
Zhao, D. Q., Tao, J., Han, C. X., Ge, J. T. (2012). Flower color diversity revealed by differential expression of flavonoid biosynthetic genes and flavonoid accumulation in herbaceous peony (Paeonia lactiflora pall.). Mol. Biol. Rep. 39, 11263–11275. doi: 10.1007/s11033-012-2036-7
Zheng, T., Han, J., Su, K. X., Sun, B. Y., Liu, S. M. (2022). Regulation mechanisms of flavonoids biosynthesis of hancheng dahongpao peels (Zanthoxylum bungeanum maxim) at different development stages by integrated metabolomics and transcriptomics analysis. BMC Plant Biol. 22, 251. doi: 10.1186/s12870-022-03642-5
Keywords: Heptacodium miconioides, sepal development, transcriptomic, metabolomic, anthocyanin
Citation: Li Y, Sun Z, Lu J, Jin Z and Li J (2023) Integrated transcriptomics and metabolomics analysis provide insight into anthocyanin biosynthesis for sepal color formation in Heptacodium miconioides. Front. Plant Sci. 14:1044581. doi: 10.3389/fpls.2023.1044581
Received: 14 September 2022; Accepted: 09 February 2023;
Published: 20 February 2023.
Edited by:
Prashant Misra, Indian Institute of Integrative Medicine (CSIR), IndiaReviewed by:
Ashish Sharma, University of Copenhagen, DenmarkCopyright © 2023 Li, Sun, Lu, Jin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zexin Jin, anp4QHR6Yy5lZHUuY24=; Junmin Li, bGlqbXR6Y0AxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.