 Ke Chen
Ke Chen Peter Zhurbenko
Peter Zhurbenko Lavrentii Danilov
Lavrentii Danilov Tatiana Matveeva
Tatiana Matveeva Léon Otten
Léon Otten
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 06 December 2022
Sec. Plant Systematics and Evolution
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.997762
This article is part of the Research TopicRhizobiaceae mediated HGT: facts, mechanisms, and evolutionary consequencesView all 6 articles
Introduction: Many higher plants contain cellular T-DNA (cT-DNA) sequences from Agrobacterium and have been called “natural genetically modified organisms” (nGMOs). Among these natural transformants, the tea plant Camellia sinensis var. sinensis cv. Shuchazao contains a single 5.5 kb T-DNA fragment (CaTA) with three inactive T-DNA genes, with a 1 kb inverted repeat at the ends. Camellia plants are allogamous, so that each individual may contain two different CaTA alleles.
Methods: 142 Camellia accessions, belonging to 10 of 11 species of the section Thea, were investigated for the presence of CaTA alleles.
Results discussion: All accessions were found to contain the CaTA insert, showing that section Thea derives from a single transformed ancestor. Allele phasing showed that 82 accessions each contained two different CaTA alleles, 60 others had a unique allele. A phylogenetic tree of these 225 alleles showed two separate groups, A and B, further divided into subgroups. Indel distribution corresponded in most cases with these groups. The alleles of the different Camellia species were distributed over groups A and B, and different species showed very similar CaTA alleles. This indicates that the species boundaries for section Thea may not be precise and require revision. The nucleotide divergence of the indirect CaTA repeats indicates that the cT-DNA insertion took place about 15 Mio years ago, before the emergence of section Thea. The CaTA structure of a C. fangchengensis accession has an exceptional structure. We present a working model for the origin and evolution of nGMO plants derived from allogamous transformants.
The genus Camellia belongs to the Theaceae family and contains several economically and culturally important species. C. sinensis consists of two main varieties: var. sinensis (generally used for green tea) and var. assamica (mainly used for black tea). Camellia systematics is still uncertain (Hoi et al., 2020). Species numbers range from 120 (Min and Bartholomew, 2007, based on Sealy, 1958) to 280 (Vijayan et al., 2009). Min and Bartholomew (2007) propose a Theaceae classification based on morphological criteria. In their system, subfamily Theoideae contains six genera. One of these, Camellia, is divided into subgenera Thea and Camellia. Subgenus Thea is further divided into six sections, among which section Thea. The tea plant, C. sinensis (L.) O. Kuntze, and the wild teas, belong to section Thea, which contains eleven species (according to Min and Bartholomew, 2007). C. sinensis is further divided in varieties sinensis, assamica, pubilimba and dehungensis.
Although the name “Thea” has been used to designate the genus Camellia, it is now only used for the subgenus Thea (which belongs to the genus Camellia) and for the section Thea (which belongs to the subgenus Camellia, itself part of the genus Camellia). Because of the economical and cultural importance of the genus Camellia, many efforts have been made to reconstruct its molecular evolution. The complex genetic relations between cultivated tea plants and their wild ancestors have been studied extensively (Prince and Parks, 2001; Wachira et al., 2001; Chen and Yamaguchi, 2002; Xiao and Parks, 2003; Sharma et al., 2009; Vijayan et al., 2009; Sharma et al., 2010; Liu et al., 2012; Yao et al., 2012; Yang et al., 2013; Huang et al., 2014; Zhang et al., 2014; Xu et al., 2015; Meegahakumbura et al., 2016; Yang et al., 2016; Wang et al., 2020; Xia et al., 2020; Zhang et al., 2020b; Wu et al., 2022); Zhang et al., 2020b.
Nuclear DNA-based Camellia phylogenies have been proposed on the basis of ribosomal DNA (Vijayan et al., 2009; Xu et al., 2015; Zhang M. et al., 2021), single-nucleotide polymorphisms (SNPs) and restriction-site associated (RAD) markers (Yang et al., 2016), transposons (Yao et al., 2017) and transcript sequences (Zhang et al., 2022; Wu et al., 2022). Recently, whole genome sequencing has provided new opportunities for phylogenetic tree construction (Wei et al., 2018; Wang et al., 2020; Xia et al., 2020; Zhang et al., 2020b; Zhang et al., 2020b; Zhang X. et al., 2021). In Next Generation Sequencing (NGS) data, DNA sequences newly acquired as a result of horizontal gene transfer can be found. They can also be used as a tool to study phylogeny (Matveeva et al., 2011). Thus, many dicotyledonous species have been found to harbor one or several Agrobacterium DNA fragments in their genomes. These sequences are called cellular T-DNAs or cT-DNAs, and the resulting plants have been called natural genetically modified organisms (nGMOs) (Matveeva and Otten, 2019; Matveeva, 2021a).
Agrobacterium is a phytopathogenic bacterium with a unique mechanism to transfer well-defined DNA fragments (T-DNAs) into the nuclear genome of a large variety of dicotyledonous plant species (Chilton et al., 1977; Zhu et al., 2000; Nester, 2015; Gelvin, 2017; Lacroix and Citovsky, 2019; Matveeva and Otten, 2019). Normally, the transfers result in Crown gall tumors (in the case of Agrobacterium tumefaciens or A. vitis) or hairy roots (in the case of A. rhizogenes, also called Rhizobium rhizogenes). In both cases, expression of T-DNA genes leads to abnormal growth and to the synthesis of small molecules, called opines. Opines can be degraded by the transforming Agrobacteria; they constitute the “raison d’être” of the transformation process (Petit and Tempé, 1985; Dessaux et al., 1998). It has been experimentally demonstrated that hairy roots can regenerate into fertile plants (Tepfer, 1990; Christey, 2001; Lütken et al., 2012).
After the initial discovery of cT-DNA sequences in N. glauca and other Nicotiana species (White et al., 1983; Furner et al., 1986; Suzuki et al., 2002), further nGMOs were discovered: Linaria (Matveeva et al., 2012), Ipomoea (Kyndt et al., 2015) and Cuscuta (Zhang Y. et al., 2020). Detailed evolutionary studies on the cT-DNAs of Nicotiana (Chen et al., 2014; Chen et al., 2018) revealed 4 different cT-DNA sequences (called TA to TD) in the ancestor of N. tabacum (tobacco), N. tomentosiformis. They occur in different combinations among Nicotiana species of section Tomentosa, and result from independent transformation events during the evolution of section Tomentosa. Additional cT-DNAs were found outside section Tomentosa, showing the widespread occurrence of natural transformation in this genus (Otten, 2020). Interestingly, some cultivars of the nGMO N. tabacum (Chen et al., 2016) and Cuscuta (Zhang Y. et al., 2020) have been shown to produce opines, but the biological significance of this is not yet clear. The frequent presence of inverted repeats in cT-DNAs provides relative dates for their insertion (Chen et al., 2014; Chen et al., 2018; Otten, 2020). Evolutionary data are also available for Linaria (Matveeva, 2018) and Ipomoea (Quispe-Huamanquispe et al., 2019). The number of nGMOs was considerably enlarged when it was discovered that 7-10% of dicotyledonous plants carry cT-DNA sequences. Among these, Camellia sinensis carries a 5.5 kb cT-DNA (Matveeva and Otten, 2019).
In this paper we compare a large number of phased alleles of the cT-DNA sequence CaTA obtained from different Camellia species of section Thea. The results show that CaTA sequences from different Thea section species do not cluster as expected, and therefore require further investigation of the Thea section phylogeny.
Camellia sequences of the SRR/SRX type (as of June 2022) were blasted to the Camellia sinensis var. sinensis G240 cultivar CaTA query sequence, using the discontiguous megablast (Altschul et al., 1990; O'Leary et al., 2016). Recovered reads were assembled and alleles separated using the allele separation function of CodonCode Aligner (version 10.0.2). (CodonCode Corporation). Apart from Default settings, a value of 100% of Minimum Percent Identity was used, in order to separate alleles with sequences as similar as possible (down to about 10 nt differences per 5500 nt). Some descriptions of Camellia sequences investigated here use species names not recognized by Min and Bartholomew (2007), but mentioned by them as synonyms. These are quinquelocularis and tetracocca (synonyms of tachangensis), atrothea and makuanica (crassicolumna), and pubilimba and angustifolia (sinensis).
After allele phasing, full-length CaTA sequences were aligned using MUSCLE (Edgar, 2004) with subsequent manual indel deletion in MEGA v.11 (Tamura et al., 2021). The phylogenetic analysis was carried out by IQ-TREE v.1.6.1 (Nguyen et al., 2015) using the maximum likelihood method with 1000 bootstrap replicates and C. fangchengensis CaTA as outgroup. The tree was visualized using iTOL (Letunic and Bork, 2021).
Matveeva and Otten (2019) reported a cT-DNA in C. sinensis var. sinensis commercial variety Shuchazao (accession SDRB01002054.1, gene coordinates 1834755-1838742). In the present paper, we used the 98% identical cT-DNA sequence from C. sinensis var. sinensis G240 (Zhang et al., 2020a) as the model sequence. We will call this cT-DNA “CaTA” (for Camellia TA sequence). CaTA is located on contig JACBKZ010000009.1 (chromosome 9), coordinates 78660407-78665693. Here we investigate this cT-DNA sequence and its surrounding 200 nt flanking plant sequences (together 5687 nt) in various Camellia accessions, using G240-CaTA coordinates as a reference.
G240-CaTA carries an inverted repeat on the left (TGCTCGCGTG….GTCGCTTGGC, 201-1148) and right (GCCAAGCGAC….CGCTCGAGCA, 4528-5487). The 948 nt and 960 nt repeats are 90% identical. The 5287 nt G240-CaTA sequence is flanked by a moderately repeated plant sequence on the left, and a highly repeated plant sequence on the right. G240-CaTA (Figure 1A) shows regions with homology to agrocinopine synthase (acs) genes (CaTA-acs, 642-1840), starting in the left repeat, an L,L-succinamopine synthase (susL) gene (CaTA-susL, 2677-3657) in the central unique part, and a truncated root locus B (rolB) sequence (CaTA-rolB, 4218-4532) on the right, interrupted by the right repeat. Thus, CaTA is derived from a 4327 nt partial T-DNA fragment, followed by a second fragment similar to its left end, but linked to it in an inverted orientation (right arm). We assume the repeats were originally identical.
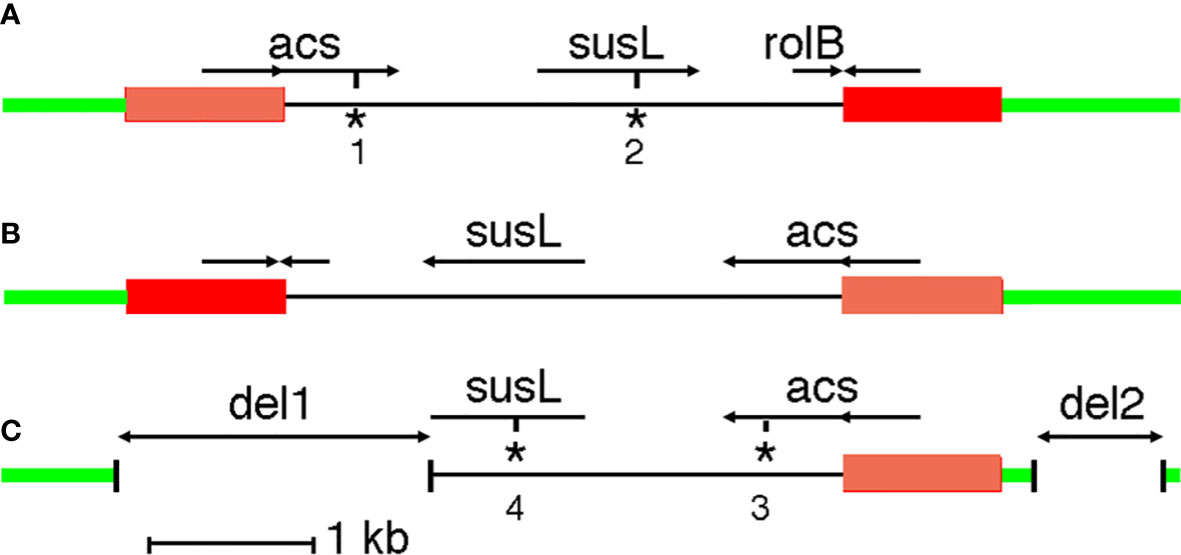
Figure 1 CaTA maps. (A) CaTA map common to all accessions, except C. fangchengensis. (B) First putative step in origin of C. fangchengensis CaTA. Inversion of the central part. (C) Second putative step in origin of C. fangchengensis CaTA: two deletions (del1 and del2). The plant DNA surrounding the CaTA insert is marked in green, the CaTA inverted repeats are marked in orange (left repeat) and red (right repeat). acs: agrocinopine synthase gene, susL: L,L-succinamopine synthase gene, rolB: root locus B gene. *1: stop codon at 1556, *2: 32 nt deletion at 3182 and frameshift, *3: stop codon at 1538, *4: 1 nt deletion at 3147 and frameshift.
The closest Agrobacterium relative of CaTA-acs is found in A. tumefaciens strain Kerr14, but the corresponding proteins are only 31% identical. R. rhizogenes strains also carry acs genes (Otten, 2021), these are even less similar. However, the nGMO Parasponia andersonii (Matveeva and Otten, 2019) potentially encodes much more similar Acs sequences (PON34801, PON34802, PON34803, about 75% identity). The same difference is found for CaTA-SusL: 37% identity for SusL from R. rhizogenes strain A4, but 52% for P. andersonii PON40029.1 SusL. The protein fragment deduced from the partial rolB gene has 46% identity to R. rhizogenes K599 RolB. CaTA-acs and CaTA-susL are interrupted by premature stop codons. The gene order acs-susL-rolB has not been found earlier in Agrobacterium and represents a new type of T-DNA.
The original, unmodified CaTA insertion site might be preserved in Camellia species without CaTA. As the CaTA insert is surrounded by repeated sequences, no short SRR/SRX sequences from other Camellia species could be used to reconstruct this site. We therefore used the assembled genome from C. oleifera isolate CON1 (JAKJMZ), which does not carry CaTA, to search for the insertion site. A 45 kb fragment of G240 containing G240-CaTA and surrounding sequences (JACKBKZ010000009.1:78640000-78685000) was used as a query sequence. This detected a colinear region on C. oleifera chromosome 5 (JAKJMZ010000005.1:112492914-112526157), with an expected gap at the position of the CaTA DNA. The alignment of a 3.7 kb C. oleifera CON1 region with the corresponding G240 region is shown in Figure 2. From left to right the aligment shows a 1219 nt common region, followed by a 1042 nt insert in CON1 (insert A). Insert A has an 10 nt inverted repeat TGCAACTAAG at its ends, and a direct repeat of the original target sequence GTCCAGCT. Insert A is absent in two other CaTA-less accessions, C. oleifera SRX10143503, and C. sasanqua SRX8770629. It is followed by a small 88 nt region, also found in G240. After this, G240 shows the 5286 nt CaTA region, and to the right of it, a 872 nt plant sequence (insert B). A sequence similar to insert B (94% identity) is found in chromosome 3 of G240 (located at JACBKZ010000003.1:79203545-79204449) and could have been ligated to the CaTA fragment before insertion. A 73 nt CON1 fragment (112515620-112515692) seems to have been lost in G240. Insert B is followed by a 250 nt sequence common to G240 and CON1, a 162 nt CON1-specific sequence (marked in blue), and a 930 nt common sequence.
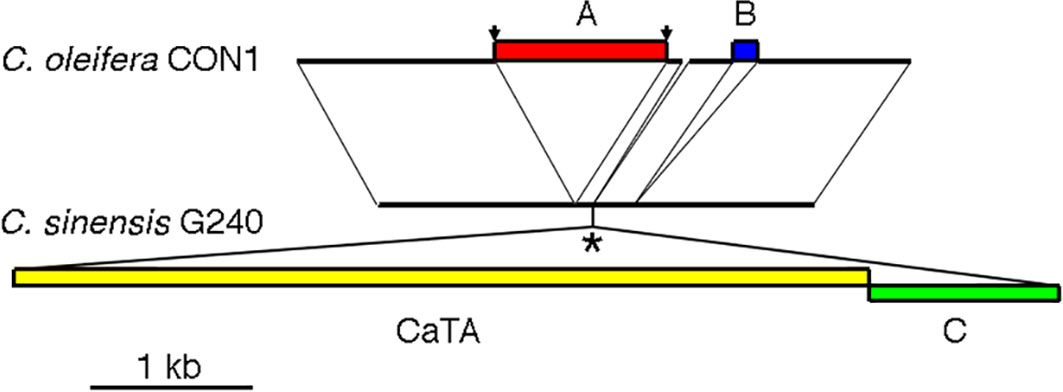
Figure 2 Sequence of the CaTA-less species C. oleifera isolate CON1 (top) at the CaTA insertion site of C. sinensis G240 (bottom). (A) insert in C. oleifera CON1, flanked by inverted repeats TGCAACTAAG at its ends, and a direct repeat of the original target sequence GTCCAGCT (arrows). (B) insert in C. oleifera. *: putative site of insertion of the C. sinensis G240 CaTA insert ligated to an adjacent plant sequence (indicated by (C).
Since the Agrobacterium-mediated insertion of T-DNA sequences in plant DNA is random, every insert is unique and marks the origin of a new clade (Matveeva et al., 2011; Chen and Otten, 2017). In other words, the presence and distribution of a particular cT-DNA insert among related nGMO species reflects their origin from a single transformant, and can be used to reconstruct the evolution of such a group. We therefore investigated the occurrence of CaTA sequences in C. sinensis and other species of section Thea. For this, we not only used whole genome sequences (as in Matveeva and Otten, 2019), but also investigated short (150 nt) sequence read runs (SRR) from hundreds of Camellia accessions. This considerably enlarged the search for cT-DNA sequences, but required assembly of individual reads into full CaTA sequences. SRR reads homologous to G240-CaTA were recovered from accessions belonging to 9 Thea section species. CaTA alleles of each accession were phased, with manual correction when necessary (Materials and Methods). After selection of 142 accessions with sufficient coverage for phasing (Supplementary Table 1), 60 showed unique alleles, 82 had two different alleles. In all, 225 alleles were identified (Supplementary Table 2, Materials and Methods, see below). The most diverse sequences showed 7.3% divergence. The identity values for the left and right repeat of each CaTA sequence were remarkably similar: 90.3 (SD: +/-0.6%). This suggests the absence of gene conversion between the repeats, and provided a good starting point to calculate the age of the CaTA insertion (see below).
The CaTA sequence of C. fangchengensis (SRX10143457, yielding two alleles with 98% identity) is inserted in the same site as the other CaTAs, but its structure is unusual. It is best explained as a derivative of the more common CaTA form, generated by an inversion of the unique region between the two repeats (Figure 1B), followed by two deletions (Figure 1C). The inversion of the central region could result from recombination between the flanking inverted repeats. One of the alleles carries a plant insert (ins2824, Table 1). Overall identity values of the two C. fangchengensis CaTA alleles with other CaTAs are relatively low, the best matches, sin8233452_2, pubi8770663_1 and assam8233492_2 show only 92.7% identity. Thus, C. fangchengensis CaTA separated at an early stage from the other CaTAs. Both fangchengensis alleles carry an additional 32 nt sequence after CaTA3182 (within susL). This sequence is found in all Agrobacterium susL sequences (Otten, 2021), suggesting that the 32 nt fragment was lost in the ancestor of section Thea, but retained in C. fangchengensis CaTA.

Table 1 Summary of indels in different CaTA alleles.
For C. costata, only one accession was available (SRR17596358) with 19 CaTA reads. C. danzaiensis (SRR19266665, 16 reads) and C. kwangtungensis (SRR19266666, 10 reads) are both considered to be synonyms for C. costata (Min and Bartholomew, 2007). The altogether 45 reads are insufficient for allele phasing, but show the presence of a CaTA in C. costata. No sequences were available for C. grandibracteata. Thus, our data show that at least 10 of the 11 Camellia species of section Thea contain the CaTA insert.
None of the 225 CaTA alleles has an open reading frame. All acs sequences have a TGA at CaTA position 1556 (ATCGCGAAATGA), except C. fangchengensis CaTA. However, the latter has a TAA at 1538 (GATTCAACTTAA), where all others have TCA. All susL sequences, except the one from C. fangchengensis, have a 32 nt deletion at 3182 (see above), leading to a frame shift. C. fangchengensis susL has a deletion at the 3’ end, due to CaTA rearrangements (Figure 1C), and an insertion of a G (TCATGGTGCG) at 3147, leading to a frame shift.
Indels provide interesting markers for evolution. We investigated the occurrence and distribution of indels longer than 2 nucleotides in the 225 CaTA alleles (overview in Table 1, details in Supplementary Table 3). The location of each modification is indicated by the coordinate of the G240-CaTA sequence after which the sequence starts to be different. Six different plant inserts were found at positions 255, 626, 1946, 2748, 2824, and 5347. Those at 1946 and 2748 were found in 15 and 8 accessions respectively, the others are unique. All are flanked by short direct repeats at the insertion site and could be related to transposons. These inserts are highly repeated in the plant genome, and therefore could only partially be reconstructed in the CaTA assembly process. Further reconstruction will require longer reads. Two locations (438-442d and 2767-2781b) are highly polymorphic (Figure 3).

Figure 3 Structures of two variable CaTA regions. (A) Region around G240-CaTA coordinates 438-449, showing 6 different types of deletion, the region starting from 449 shows three types of insertion. (B) Region around 2767-2781. 10 different deletions and one insertion were found in this region. Coordinates on the left indicate the positions after which the sequence diverges from the standard sequence, the latter indicated by 000. Insertions are shown below the normal sequence and start behind the underlined nucleotide. See also Table 1 and Supplementary Table 1.
The phased CaTA sequences (Supplementary Table 2) were used to construct a phylogenetic tree (see Materials and Methods). Camellia species (according to Min and Bartholomew, 2007) were color-coded in order to visualize their distribution. Information on various indels was added to the tree (colored dots) in order to investigate whether indel distribution corresponded to tree topology. Two main groups can be distinguished (Figure 4), called A (mainly with del441) and B (mainly without del441). Group A is subdivided in A1 (mainly with C. sinensis var. assamica, sa), A2 with del441 and del3962a (mainly with C. sinensis var. sinensis, ss), A2a also has del4440, A2b does not. A3 is a mixed group. Group B can be divided in B1, with del2152 (mainly C. quinquelocularis, qu), B2: with del4183 (all sa), B3: ins2748 (mixed group), B4: mixed group, B5: del2779, with B5a (mainly C. taliensis, tl), B5b (qu), and B5c (C. tachangensis var. tachangensis (ta), C. tachangensis var. remotiserrata (tr), and ss). A few indels occur in unexpected branches: del441 in B1, B3, B4, and B5a, and del2779 in A1, A2b, and A3.
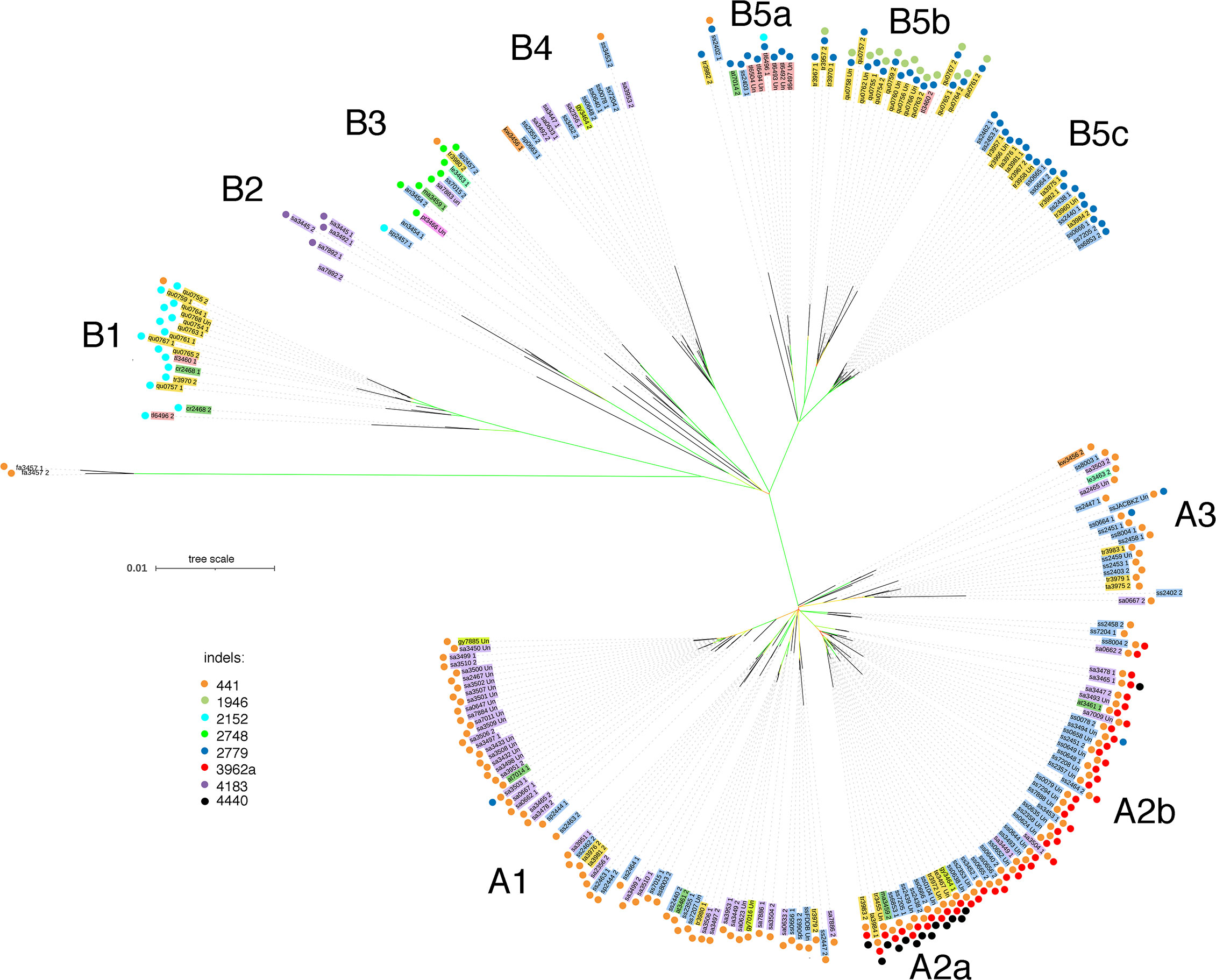
Figure 4 Phylogenetic tree of 225 CaTA alleles of 9 Camellia species of section Thea. CaTA from C. fangchengensis was used as outgroup. Accessions are marked by abbreviations corresponding to their full names given in Supplementary Table 1 and the last 4 digits of the SRX number. Color codes indicate the different species (according to Min and Bartholomew, 2007). C. sinensis var. assamica belongs to C. sinensis, but was given a special color code (purple). C. sinensis: blue (these comprise C. sinensis var. sinensis, C. pubilimba, C. angustifolia). C. tachangensis (C. tachangensis var. tachangensis, C. tachangensis var. remotiserrata, C. quinquelocularis, C. tetracocca): dark yellow, C. crassicolumna (C. crassicolumna, C. atrothea, C. makuanica): green, C. gymnogyna: yellow, C. taliensis: pink, C. leptophylla: dark green, C. kwangsiensis: orange, C. ptilophylla: red, C. fangchengensis: no color. On the outside, the indels are marked with colored dots. Orange: deletion at G240-CaTA coordinate 441. Dark green: insertion at 1946. Light blue: insertion at 2152. Light green: insertion at 2748. Dark blue: deletion at 2779. Red: deletion at 3962a. Purple: deletion at 4183. Black: deletion at 4440. The various groups cluster into 2 main groups, A and B. A can be further subdivided into A1-A3, A2 into A2a and 2b. Group B is subdivided into B1-B5, B5 into B5a-c. In general, indel distribution corresponds to overall tree topology, but exceptions occur. Group B3 contains CaTAs of 5 different species, all have the 2748 plant sequence insert. Bootstrap values are color-coded: green 85-100%, yellow around 50-85%, red: less than 20%.
CaTA sequences of different Thea section species are interspersed. A striking example is B3 which contains sequences from sinensis (var. pubilimba, angustifolia, assamica), makuanica, tachangensis var. remotiserrata, leptophylla, and ptilophylla. These sequences also share the same plant insert (ins2748, Table 1), confirming their close relationship.
We also investigated the distribution of allele pairs on the tree. Many pairs have one member on branch A, the other on branch B. An overview for the cultivated tea varieties C. sinensis var. sinensis and assamica is shown in Figure 5A, for the other Thea section species in Figure 5B. Interestingly, the nine quinquelocularis accessions with two dissimilar alleles (plus one C. taliensis accession) each have one allele on branch B1, the other on B5b. The distribution of these alleles suggests that the quinquelocularis group constitutes a bona fide subgroup derived from a founder plant with two different CaTA alleles, each of which diverged separately.
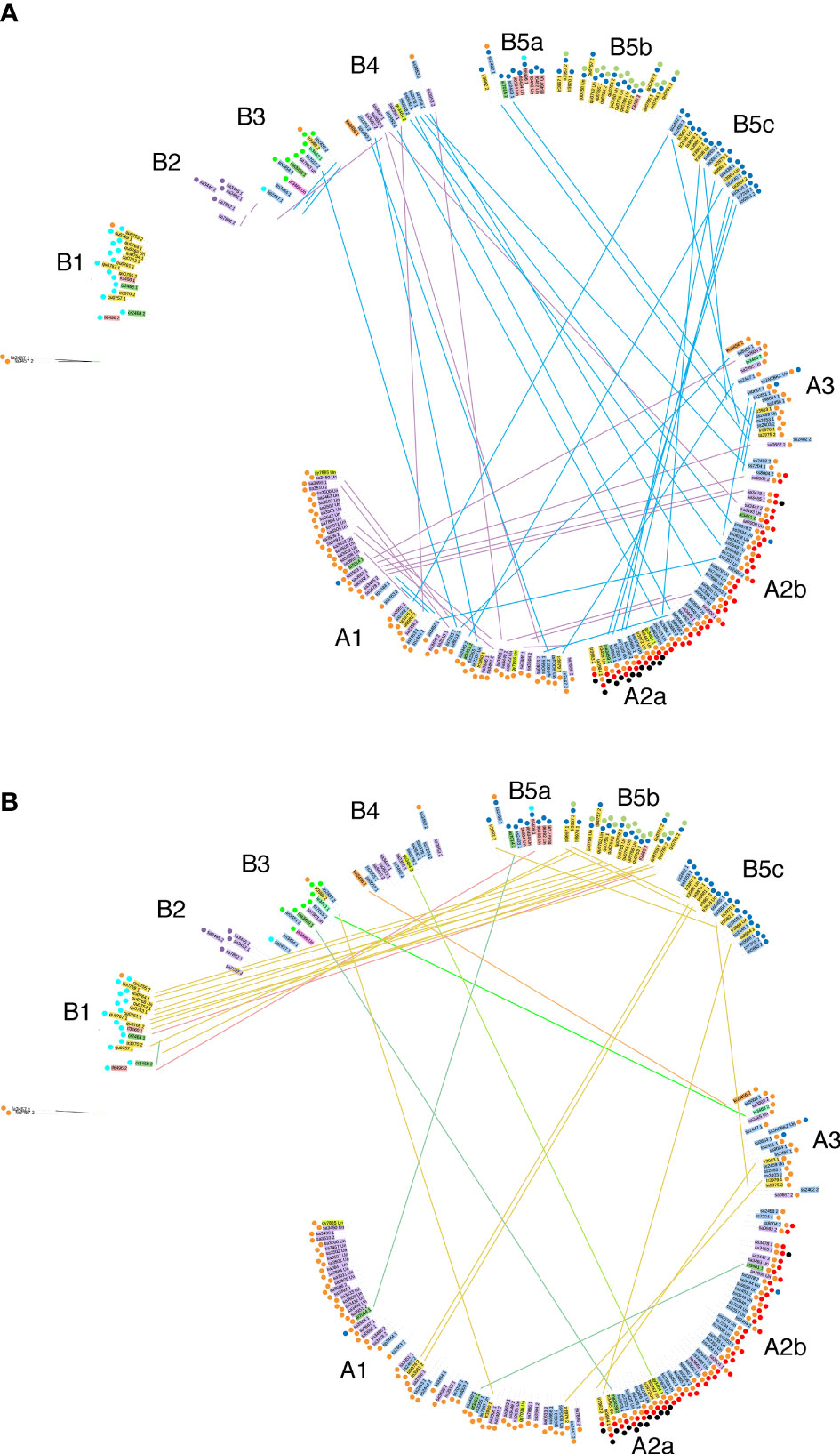
Figure 5 Localisation of paired alleles on the phylogenetic tree (same as in Figure 4) Paired alleles are connected by lines, with the same species color code as in Figure 4. Several pairs have one member in group (A), the other one in group (B).
Camellia sinensis var. sinensis cultivar Shuchazao was earlier found to contain a 5.5 kb cT-DNA insert (Matveeva and Otten, 2019). At the time it was unknown whether this represented an exceptional case or not. The results reported here show that at least 10 out of 11 Camellia species of section Thea (according to Min and Bartholomew, 2007) contain this insert, CaTA. The exception is C. grandibracteata, for which no sequences are available, this species needs further study. The original CaTA insertion site could be reconstructed using an assembled C. oleifera sequence, which lacks CaTA. The CaTA acs-susL-rolB structure is unusually small for a T-DNA, and has only low similarity to T-DNAs from Agrobacterium (Otten, 2021). The closest relatives are two cT-DNAs from P. andersonii, Pa3 and Pa6 (Matveeva and Otten, 2019). Interestingly, the C. fangchengensis CaTA structure is different from the other CaTAs, and its overall sequence is more diverged. This seems to set C. fangchengensis apart from other Thea section species. A recent study also found C. fangchengensis at the base of section Thea (Zhang X. et al., 2021). (Note that in this study C. fangchengensis, named after the city of Fangcheng in the province of Guangxi,was indicated as C. fengchengensis). The phylogenetic position of C. fangchengensis merits further analysis, using additional fangchengensis accessions and further nuclear sequences.
About 70% of Angiosperms species are significantly heterozygous (Wright et al., 2013). The use of unphased mosaic sequences in molecular phylogeny leads to imprecise trees. So far, no attempts have been made to separate cT-DNA alleles of heterozygous nGMO species. In the case of Nicotiana, this was not necessary because Nicotiana is largely autogamous and homozygous. However, published cT-DNAs sequences from heterozygous nGMO species like Linaria (Matveeva et al., 2012) and Ipomoea (Kyndt et al., 2015; Quispe-Huamanquispe et al., 2019) are unphased. Allele separation algorithms are now commercially available, but still require manual adjustments, and a sufficiently high coverage. In the present study, we were able to isolate and phase 225 CaTA alleles from SRR/SRX data. Other Camellia sequences are available, but coverage values were insufficient for phasing. The 225 CaTA alleles provide a detailed view of CaTA variability and evolution. They can therefore be used as preliminary indicators for the taxonomic position of Camellia species of section Thea.
The phylogeny of Camellia might be difficult to unravel, as different Camellia species have been found to hybridize (Wight, 1962; Takeda, 1990; Sharma et al., 2010; Wang et al., 2020; Zhang et al., 2020a). C. taliensis wild type plants have been cultivated in tea gardens or were recently domesticated, as they are considered to be interesting sources for tea plant improvement. Having a 5-locule ovary, they are easily differentiated from the 3-locule ovary sinensis/assamica group (Zhao et al., 2014). Possibly, some 5-locule species considered as wild tea species, might in fact carry sequences from cultivated species, due to man-made crosses. This has been shown for C. taliensis var. bangwei (Yang et al., 2016), but other cases may go unnoticed if the genetic contribution from the cultivated species is small. It has also been suggested that some wild tea trees could in fact be of feral origin (Zhang et al., 2020a).
A study based on restriction-site-associated DNA (RAD) sequencing (Yang et al., 2016) found a cluster for the cultivated species C. sinensis var. sinensis (6 accessions), and one for C. sinensis var. assamica (3). Non-cultivated species C. tachangensis (1), C. taliensis (3) and C. crassicolumna (4) clustered together, separately from the cultivated species. Another study (Vijayan et al., 2009) showed clear separation between taliensis and kwangsiensis on the one hand, and assamica, leptophylla, angustifolia, atrothea, ptilophylla and sinensis on the other hand. These two studies did not use allele phasing. Our study yielded a tree composed of two different parts, A and B, with subgroups A1-A3 and B1-B5. Although the tree shows a concentration of C. sinensis var. sinensis accessions in A2 and A3, of C. sinensis var. assamica in A1, and of wild species in B1 and B5, there are several exceptions. Most importantly, when the distribution of paired alleles is taken into account, many accessions can be seen to have one allele in group A and one in group B. Thus, there is no strict separation of CaTA sequences between the different accessions and species. The A and B group may result from expansion of two dissimilar alleles. The origin of this combination may be due to a unique hybridization event between members of two already diverged populations. The distribution of the C. quinquelocularis allele pairs over group B1 and B5b indicates the presence of another founding ancestor plant with a specific combination of alleles. A recent study based on transcript analysis (Wu et al., 2022) obtained a subtree for 9 species of the Thea section (according to Min and Bartholomew, 2007). Different species were interspersed with C. sinensis, and C. taliensis (a single accession) was found at the base of section Thea. This study did not attempt to separate the alleles. The impact of this is difficult to estimate, also because the authors used other accessions as we did.
Recent papers report assembled Camellia sequences and their use in phylogenetic tree construction. Comparison between an unphased CSA and CSS genome yielded a sequence identity of 92,4% and a 0.38-1.54 Mya divergence time (Wei et al., 2018). However, our comparison of several CaTA alleles from CSA and CSS shows no clear separation, as CSA alleles occur in A1, A2a, A2b, A3, B2, B3, B4, and CSS alleles in A1, A2a, A2b, A3, B3, B4, B5a, B5c (Figure 4).
Another study, from BioProject PRJNA597714 (Xia et al., 2020) reported the unphased sequence of the heterozygous CSS Shuchazao genome. SNP’s were used to construct a phylogenetic tree showing clustering of CSA, CSS and wild tea plants, the latter including ancient Laos tea plants. CaTA sequences from CSA and CSS sequences recovered from PRJNA597714 do not cluster separately, and the CaTA sequences from Laos tea plants sa3445-1, sa3445-2, sa3447-1, sa3447-2, sa3492-1, sa3492-2, and sa3493-un are found in B2, B2, B4, A2b, B2, B4, and A2b respectively.
A further study reported the unphased sequence of an ancient wild tea tree, called DASZ (Zhang et al., 2020a). The CaTA alleles from DASZ1 are found in B5a (ss2402-1 and ss2403-1) and A3 (ss2402-2 and ss2403-2). The same authors also obtained unphased RNA sequences from 217 accessions (BioProject PRJNA595795) and used them for tree construction. They found no distinct separation between ancient trees and cultivars, nor between plants with sinensis and assamica characteristics. This fits well with our findings. Interestingly, they labeled all their accessions as “C. sinensis”. The same group (Zhang et al., 2020b) also obtained a phased genome for CSS cultivar Fudingdabai, using sperm cells. Using this sequence, they established a kinship analysis with a subset of the 217 accessions. Unfortunately, the PRJNA595795 RNA sequences contain very few CaTA reads, preventing assembly and phasing, and further comparison with our CaTA data.
A recent paper (Zhang X. et al., 2021) reported the haplotype-resolved genome of the highly heterozygous CSS cultivar Tieguanyin (TGY). SNP-based trees of 190 accessions were constructed, in which 8 C. sinensis accessions clustered in one group, whereas other Thea section species clustered in a separate group. When comparing their results with ours (obtained with the same sequence data), we noted important differences. The authors found that C. tetracocca and C. gymnogyna were very closely related, whereas C. leptophylla, C. angustifolia and C. kwangsiensis formed another highly related group. Based on CaTA comparisons from the same sequences, we found that CaTA from te3467-un, gy3464-1, and gy3464-2 were in groups A2a, A2a and B4 respectively, whereas le3463-1, le3463-2, an3454-1, an3454-2, kw3456-1, kw3456-2 were in B3, A3, B3, B3, B4 and A3. The C. taliensis CaTA sequences tl3460-1 and tl-3460-2 were in two different groups, B1 and B5b. Finally, the unique C. ptilophylla CaTA allele pt3466-un was found in B3, which also contains sequences from CSS and CSA. These differences in clustering may be due to the use of unphased versus phased sequences and requires further analysis.
Our results show that the Thea section species are derived from a single Agrobacterium-transformed ancestor. The CaTA transformation event seems to predate the origin of section Thea because the divergence values for the inverted CaTA repeat (9.7% divergence since insertion in the ancestor) and the maximal divergence values for the two CaTA alleles (7.3%) differ. Using the 9.7% divergence of the CaTA repeat and an estimated universal substitution rate of 6.5x10-9 mutations per site per year (Gaut et al., 1996), the transformation event would have happened 15 Mio years ago. This estimate places the ancestor of the CaTA-carrying plants well before the origin of section Thea, 6.7 Mio years ago, and close to the origin of the Camellia group, 14.3 Mio years ago (Wu et al., 2022). Interestingly, in the CaTA-less oleifera and sasanqua species, the sequences around the CaTA insertion site seem to be intact, suggesting that these species split off at an early phase, before the transformation event.
The precise sequence of events leading from the initial transformant to the CaTA-carrying plants is still unknown. A likely scenario is the following. An R. rhizogenes bacterium transformed a single cell of a Camellia-like ancestor plant, introducing the CaTA insert into chromosome 9. This cell spontaneously regenerated into a fertile R0 plant with one CaTA copy. In order to explain how a single transformed individual gave rise to a large group of descendants, we (Chen et al., 2018; Matveeva, 2021b) and others (Martin-Tanguy et al., 1996) have proposed that strong phenotypic effects of the cT-DNA genes may lead to reproductive isolation, thus generating a new species. Based on experimental evidence, Martin-Tanguy et al. suggested (Martin-Tanguy et al., 1996) that cT-DNA expression could have deleterious effects in the 1-copy state, but lose these in the 2-copy state by silencing. This could lead to rapid selection for phenotypically normal 2-copy plants, and would prevent back-crosses with untransformed plants, as this would restore the one-copy situation, thus favoring speciation. We have proposed an alternative speciation model in which growth effects of the cT-DNA genes would be sufficient to prevent back-crosses to the ancestor plants, but would still allow survival of the transformants. This model was based on the study of the TE-6b genes from a N. otophora cT-DNA, which cause considerable growth changes in Arabidopsis and N. tabacum with a delay in flowering time, but do not prevent growth and reproduction of the transformants (Chen et al., 2018; Potuschak et al., 2020).
The CaTA cT-DNA does not carry growth modification genes which could interfere with reproduction. We therefore prefer an third model for this cT-DNA, in which the acs and susL genes provided initially a selective advantage, which led to fixation of the CaTA sequence. Later the selective advantage was lost (possibly by external changes, like changes in climate or environment), and both genes acquired premature stop codons. Interestingly, the premature stop codon and frameshift mutations in the CaTA acs and susL genes are common to all Thea section sequences, except those from C. fangchengensis, which show other types of mutations. We conclude that the CaTA genes were still active at the time of emergence of section Thea, but lost their function after the separation of C. fangchengensis from the other species, in both groups by independent processes.
The mechanisms by which groups of nGMOs, like Nicotiana plants from section Tomentosa or Camellia plants of section Thea, developed from a single transformed cell, will to a large extent determine their overall genetic make-up. On the one extreme, selfing of R0 plants in the case of autogamous species, and rapid selection of 2-copy plants could lead to a strong reduction in genetic diversity. On the other extreme, continued hybridization of transformed plants with untransformed plants, as in the case of allogamous species, would maintain a much larger diversity. Analysis of the variability of other, carefully phased nuclear alleles of the Camellia accessions investigated here, might shed more light on these processes.
Our results may be of interest for genetic engineering of tea plants as they demonstrate the natural capacity of R. rhizogenes for Camellia transformation. Transformation of tea plants with disarmed A. tumefaciens vectors has been reported (Mondal et al., 2001; Singh et al., 2015; Lü et al., 2018). R. rhizogenes has been used to induce Hairy Roots, but no plants were obtained (Zhang et al., 2006; Rana et al., 2016; Alagarsamy et al., 2018), contrary to what has been reported for other plant species (Tepfer, 1990; Christey, 2001; Lütken et al., 2012). Thus, results are still quite limited, in spite of the potential of genetic engineering for the genetic improvement of tea plants. It would be worthwhile to further optimize HR induction, for example by using special gene constructs allowing inactivation of the HR-inducing genes after induction of transformed hairy roots.
Based on the present study, we believe that cT-DNAs have several advantages to explore the origin and evolution of nGMO species. First, cT-DNAs are well-defined, highly specific and recognizable DNA fragments, unrelated to plant sequences. They do not occur in non-transformed ancestors, and their integration at a particular chromosomal site constitutes a founder event which creates a clean start for a new clade. Second, unlike transposable elements, cT-DNA inserts do not amplify and disseminate in the genome after insertion. This avoids the generation of multiple paralogs, which is a significant advantage over classical nuclear markers. Third, cT-DNAs can be sufficiently large and old to generate a range of variants, thereby allowing the construction of detailed trees. Finally, repeat sequences in cT-DNAs can be used to estimate time since transformation, or as relative time markers. Whatever the precise course of events that led to the present-day section Thea, it is interesting to realize that the tea plant Camellia sinensis and its close relatives are all descendants of a single cell, transformed by the natural engineer Agrobacterium.
The various species of the Thea section of Camellia are derived from a single plant, transformed by Agrobacterium, about 15 Mio years ago, well before the origin of section Thea. The descendants of the transformant show various modifications of the introduced cT-DNA, among which insertions of plant sequences and structural rearrangements. The cT-DNA genes are inactive. The distribution of the CaTA sequences among 143 different accessions belonging to 10 species does not correlate with the proposed separation of section Thea into different species, but shows that different species share very similar CaTA alleles.
The original contributions presented in this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
LO and TM designed the study. KC provided sequence data. KC, PZ, and LD contributed to data analysis. LO wrote the first draft of the manuscript. LO, TM, KC, PZ, and LD contributed to the interpretation of the results and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Russian Science Foundation, with grant number 21-14-00050 to TM. It was also supported with a special fund for scientific research of Shanghai landscaping and city appearance administrative bureau (G222410), and a 中科院青年英才培育计划 (Zhongkeyuan qingnian yingcai peiyu jihua) grant from the Chinese Academy of Sciences to KC.
We would like to acknowledge the help of Peter Richterich from CodonCode Corporation, for developing software for allele phasing in collaboration with LO.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.997762/full#supplementary-material
Supplementary Table 1 | List of accessions and their indels, indicated by positions of the G240-CaTA. First column: composite numbers referring to species (as provided in the sequence description) and SRX accession number. Other columns: positions of indels according to G240-CaTA sequence, the position is the last nucleotide before the modification. For type of modifications, see Table 1.
Supplementary Table 2 | Phased alleles of Camellia accessions from section Thea.
Supplementary Table 3 | Details for indels.
Alagarsamy, A. K., Shamala, L. F., Wei, S. (2018). Protocol: High-efficiency in-planta agrobacterium-mediated transgenic hairy root induction of camellia sinensis var. sinensis. Plant Methods 4, 17–22. doi: 10.1186/s13007-018-0285-8
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Chen, K., Dorlhac de Borne, F., Julio, E., Obszynski, J., Pale, P., Otten, L. (2016). Root-specific expression of opine genes and opine accumulation in some cultivars of the naturally occurring GMO nicotiana tabacum. Plant J. 87, 258–269. doi: 10.1111/tpj.13196
Chen, K., Dorlhac de Borne, F., Sierro, N., Ivanov, N. V., Alouia, M., Koechler, S., et al. (2018). Organization of the TC and TE cellular T-DNA regions in nicotiana otophora and functional analysis of three diverged TE-6b genes. Plant J. 94, 274–287. doi: 10.1111/tpj.13853
Chen, K., Dorlhac de Borne, F., Szegedi, E., Otten, L. (2014). Deep sequencing of the ancestral tobacco species nicotiana tomentosiformis reveals multiple T-DNA inserts and a complex evolutionary history of natural transformation in the genus nicotiana. Plant J. 80, 669–682. doi: 10.1111/tpj.12661
Chen, K., Otten, L. (2017). Natural agrobacterium transformants: Recent results and some theoretical considerations. Front. Plant Sci. 8, e1600. doi: 10.3389/fpls.2017.01600
Chen, L., Yamaguchi, S. (2002). Genetic diversity and phylogeny of tea plant (Camellia sinensis) and its related species and varieties in the section thea genus camellia determined by randomly amplified polymorphic DNA analysis. J. Hortic. Sci. Biotech. 77, 729–732. doi: 10.1080/14620316.2002.11511564
Chilton, M. D., Drummond, M., Merlo, D., Sciaky, D. (1977). Stable incorporation of plasmid DNA into higher plant cells: The molecular basis of crown gall tumorigenesis. Cell 11, 263–272. doi: 10.1016/0092-8674(77)90043-5
Christey, M. C. (2001). Use of ri-mediated transformation for production of transgenic plants. Vitro Cell. Dev. Biol. Plant 37, 687–700. doi: 10.1007/s11627-001-0120-0
Dessaux, Y., Petit, A., Farrand, S., Murphy, P. (1998). “Opines and opine-like molecules involved in plant-rhizobiaceae interactions,” in The rhizobiaceae. Eds. Spaink, H. P., Kondorosi, A., Hooykaas, P. J. J. (Dordrecht, The Netherlands: Kluwer Academic Publishers), 173–197. doi: 10.1007/978-94-011-5060-6_9
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Furner, I. J., Huffman, G. A., Amasino, R. M., Garfinkel, D. J., Gordon, M. P., Nester, E. W. (1986). An agrobacterium transformation in the evolution of the genus nicotiana. Nature 319, 422–427. doi: 10.1038/319422a0
Gaut, B., Morton, B., McCaig, B., Clegg, M. (1996). Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene adh parallel rate differences at the plastid gene rbcL. Proc. Nat. Acad. Sci. U. S. A. 93, 10274–10279. doi: 10.1073/pnas.93.19.10274
Gelvin, S. B. (2017). Integration of agrobacterium T-DNA into the plant genome. Annu. Rev. Genet. 51, 195–217. doi: 10.1146/annurev-genet-120215-035320
Hoi, Q. V., Thin, D. B., Thinh, B. B. (2020). Some research on the genus camellia l. (Theaceae) with representatives in Vietnam Plant Ecol. Vestnik N.V.G.U. 2, 5–11. doi: 10.36906/2311-4444/21-2/01
Huang, H., Shi, C., Liu, Y., Mao, S. Y., Gao, L. Z. (2014). Thirteen camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14, 151. doi: 10.1186/1471-2148-14-151
Kyndt, T., Quispe, D., Zhai, H., Jarret, R., Ghislain, M., Liu, Q., et al. (2015). The genome of cultivated sweet potato contains agrobacterium T-DNAs with expressed genes: an example of a naturally transgenic food crop. Proc. Natl. Acad. Sci. U.S.A. 112, 5844–5849. doi: 10.1073/pnas1419685112
Lacroix, B., Citovsky, V. (2019). Pathways of DNA transfer to plants from agrobacterium tumefaciens and related bacterial species. Ann. Rev. Phytopath. 57, 231–251. doi: 10.1146/annurev-phyto-082718-100101
Letunic, I., Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucl. Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Liu, Y., Yang, S. X., Ji, P. Z., Gao, L. Z. (2012). Phylogeography of camellia taliensis (Theaceae) inferred from chloroplast and nuclear DNA: Insights into evolutionary history and conservation. BMC Evol. Biol. 12, 92. doi: 10.1186/1471-2148-12-92
Lü, Q., Chen, C., Xu, Y., Hu, S., Wang, L., Sun, K., et al. (2018). Optimization of agrobacterium tumefaciens-mediated transformation systems in tea plant (Camellia sinensis). Horticult. Plant J. 3, 105–109. doi: 10.1016/j.hpj.2017.03.001
Lütken, H., Clarke, J. L., Müller, R. (2012). Genetic engineering and sustainable production of ornamentals: current status and future directions. Plant Cell Rep. 31, 1141–1157. doi: 10.1007/s00299-012-1265-5
Martin-Tanguy, J., Sun, L.-Y., Burtin, D., Vernoy, R., Rossin, N., Tepfer, D. (1996). Attenuation of the phenotype caused by the root-inducing, left-hand, transferred DNA and its rolA gene. Plant Physiol. 111, 259–267. doi: 10.1104/pp.111.1.259
Matveeva, T. V. (2018). Agrobacterium-mediated transformation in the evolution of plants. Curr. Top. Microbiol. Immun. 418, 421–441. doi: 10.1007/82_2018_80
Matveeva, T. V. (2021a). New naturally transgenic plants: 2020 up-date. Biol. Commun. 66, 36–46. doi: 10.21638/spbu03.2021.105
Matveeva, T. V. (2021b). Why do plants need agrobacterial genes? Ecol. Genet. 19, 365–375. doi: 10.17816/ecogen89905
Matveeva, T. V., Bogomaz, D. I., Pavlova, O. A., Nester, E. W., Lutova, L. A. (2012). Horizontal gene transfer from genus agrobacterium to the plant linaria in nature. Mol. Plant Microbe Interact. 25, 1542–1551. doi: 10.1094/MPMI-07-12-0169-R
Matveeva, T. V., Otten, L. (2019). Widespread occurrence of natural transformation of plants by agrobacterium. Plant Mol. Biol. 101, 415–437. doi: 10.1007/s11103-019-00913-y
Matveeva, T. V., Pavlova, O. A., Bogomaz, D. I., Demkovich, A. E., Lutova, L. A. (2011). Molecular markers for plant species identification and phylogenetics. Ecol. Genet. 9, 32–43. doi: 10.17816/ecogen9132-43
Meegahakumbura, M. K., Wambulwa, M. C., Thapa, K. K., Möller, M., Xu, J. C., Yang, J. B., et al. (2016). Indications for three independent domestication events for the tea plant (Camellia sinensis (L.) o. kuntze) and new insights into the origin of tea germplasm in China and India revealed by nuclear microsatellites. PloS One 11, e0155369. doi: 10.1371/journal.pone.0155369
Min, T. L., Bartholomew, B. (2007). “Theaceae,” in Flora of China, vol. 2007 . Eds. Wu, Z. Y., Raven, P. H., Hong, D. Y. (Beijing/St Louis, MO: Science Press & Missouri Botanical Garden Press), 367.
Mondal, T. K., Bhattacharya, A., Ahuja, P. S., Chand, P. K. (2001). Transgenic tea [Camellia sinensis (L.) o. kuntze cv. kangra jat] plants obtained by agrobacterium-mediated transformation of somatic embryos. Plant Cell Rep. 20, 712–716. doi: 10.1007/s002990100382
Nester, E. W. (2015). Agrobacterium: nature's genetic engineer. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00730
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
O'Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Otten, L. (2020). “Natural agrobacterium-mediated transformation in the genus nicotiana,” in The tobacco plant genome. Eds. Ivanov, N., Sierro, N., Peitsch, M. C. (Berlin: Springer). doi: 10.1007/978-3-030-29493-9_12
Otten, L. (2021). T-DNA Regions from 350 agrobacterium genomes: maps and phylogeny. Plant Mol. Biol. 106 (6495), 239–258. doi: 10.1007/s11103-021-01140-0
Petit, A., Tempé, J. (1985). “The function of T-DNA in nature,” in Molecular form and function of the plant genome. Eds. van Vloten-Doting, L., Groot, G., Hall, T. (New York: Plenum Press), 625.
Potuschak, T., Palatnik, J., Schommer, C., Sierro, N., Ivanov, N. V., Kwon, Y., et al. (2020). Inhibition of arabidopsis thaliana CIN-like TCP transcription factors by agrobacterium T-DNA-encoded 6B proteins. Plant J. 101, 1303–1317. doi: 10.1111/tpj.14591
Prince, L. M., Parks, C. R. (2001). Phylogenetic relationships of theaceae inferred from chloroplast DNA sequence data. Am. J. Bot. 88, 2309–2320. doi: 10.2307/3558391
Quispe-Huamanquispe, D. G., Gheysen, G., Yang, J., Jarret, R., Rossel, G., Kreuze, J. F. (2019). The horizontal gene transfer of agrobacterium T-DNAs into the series batatas (Genus ipomoea) genome is not confined to hexaploid sweetpotato. Sc. Rep. 9 (1), 13p. doi: 10.1038/s41598-019-48691-3
Rana, M. M., Han, Z., Song, D., Liu, G., Li, D., Wan, X., et al. (2016). Effect of medium supplements on agrobacterium rhizogenes mediated hairy root induction from the callus tissues of camellia sinensis var. sinensis. Int. J. Mol. Sc. 10, 213–218. doi: 10.3390/ijms17071132
Sealy, J. R. (1958). A revision of the genus camellia (London: The Royal Horticultural Society Press).
Sharma, R. K., Bhardwaj, R., Negi, R., Mohapatra, T., Ahuja, P. S. (2009). Identification, characterization and utilization of unigene derived microsatellite markers in tea (Camellia sinensis l.). BMC Plant Biol. 9, 53. doi: 10.1186/1471-2229-9-53
Sharma, R. K., Negi, M. S., Sharma, S., Bhardwaj, P., Kumar, R., Bhattachrya, E., et al. (2010). AFLP-based genetic diversity assessment of commercially important tea germplasm in India. Biochem. Genet. 48, 549–564. doi: 10.1007/s10528-010-9338-z
Singh, H. R., Deka, M., Das, S. (2015). Enhanced resistance to blister blight in transgenic tea (Camellia sinensis [L.] o. kuntze) by overexpression of class I chitinase gene from potato (Solanum tuberosum). Funct. Integr. Gen. 13, 144–147. doi: 10.1007/s10142-015-0436-1
Suzuki, K., Yamashita, I., Tanaka, N. (2002). Tobacco plants were transformed by agrobacterium rhizogenes infection during their evolution. Plant J. 32, 775–787. doi: 10.1046/j.1365-313x.2002.01468.x
Takeda, Y. (1990). Cross compatibility of tea (Camellia sinensis) and its allied species in the genus camellia. Jap. Agric. Res. Q. 24, 111–116.
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tepfer, D. (1990). Genetic transformation using agrobacterium rhizogenes. Physiol. Plant 79, 140–146. doi: 10.1111/j.1399-3054.1990.tb05876.x
Vijayan, K., Zhang, W., Tsou, C. (2009). Molecular taxonomy of camellia (Theaceae) inferred from nrITS sequences. Am. J. Bot. 96, 1348–1360. doi: 10.3732/ajb.0800205
Wachira, F. N., Tanaka, J., Takeda, Y. (2001). Genetic variation and differentiation in tea (Camellia sinensis) germplasm revealed by RAPD and AFLP variation. J. Hortic. Sci. Biotech. 76, 557–563. doi: 10.1080/14620316.2001.11511410
Wang, X., Feng, H., Chang, Y., Ma, C., Wang, L., Hao, X., et al. (2020). Population sequencing enhances understanding of tea plant evolution. Nat. Commun. 11, 4447. doi: 10.1038/s41467-020-18228-8
Wei, C., Yang, H., Wang, S., Zhao, J., Liu, C., Gao, L., et al. (2018). Draft genome sequence of camellia sinensis var. sinensis provides insight into the evolution of the tea genome and tea quality. Proc. Nat. Acad. Sc. U. S. A. 115, E4151–E4158. doi: 10.1073/pnas.1719622115
White, F. F., Garfinkel, D. J., Huffman, G. A., Gordon, M. P., Nester, E. W. (1983). Sequence homologous to agrobacterium rhizogenes T-DNA in the genomes of uninfected plants. Nature 301, 348–350. doi: 10.1038/301348a0
Wright, S. I., Kalisz, S., Slotte, T. (2013). Evolutionary consequences of self-fertilization in plants. Proc. Biol. Sci. 280 (1760), 20130133. doi: 10.1098/rspb.2013.0133
Wu, Q., Tong, W., Zhao, H., Ge, R., Li, R., Huang, J., et al. (2022). Comparative transcriptomic analysis unveils the deep phylogeny and secondary metabolite evolution of 116 camellia plants. Plant J 111, 406–421. doi: 10.1011/tpj.15799
Xiao, T. J., Parks, C. R. (2003). Molecular analysis of the genus camellia. Int. Camellia J. 35, 57–65.
Xia, E., Tong, W., Hou, Y., An, Y., Chen, L., Wu, Q., et al. (2020). The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant 13, 1013–1026. doi: 10.1016/j.molp.2020.04.010
Xu, J., Xu, Y., Yonezawa, T., Li, L., Hasegawa, M., Lu, F., et al. (2015). Polymorphism and evolution of ribosomal DNA in tea (Camellia sinensis, theaceae). Mol. Phylogen. Evol. 89, 63–72. doi: 10.1016/j.ympev.2015.03.020
Yang, H., Wei, C. L., Liu, H. W., Wu, J. L., Li, Z. G., Zhang, L., et al. (2016). Genetic divergence between camellia sinensis and its wild relatives revealed via genome-wide SNPs from RAD sequencing. PloS One 11 (3), e0151424. doi: 10.1371/journal.pone.0151424
Yang, J. B., Yang, S. X., Li, H. T., Yang, J., Li, D. Z. (2013). Comparative chloroplast genomes of camellia species. PloS One 8 (8), e73053. doi: 10.1371/journal.pone.0073053
Yao, M. Z., Ma, C. L., Qiao, T. T., Jin, J. Q., Chen, L. (2012). Diversity distribution and population structure of tea germplasms in China revealed by EST-SSR markers. Tree Genet. Genomes 8, 205–220. doi: 10.1007/s11295-011-0433-z
Yao, J., Xiaoyu, L., Cheng, P., Yeyun, L., Jiayue, J., Changjun, J. (2017). Cloning and analysis of reverse transcriptases from Ty1-copia retrotransposons in camellia sinensis. Biotech. Biotechn. Equip. 31; 663–669. doi: 10.1080/13102818.2017.1332492
Zhang, X., Chen, S., Shi, L., Gong, D., Zhang, S., Zhao, Q., et al. (2021). Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant camellia sinensis. Nat. Genet. 53, 1250–1259. doi: 10.1038/s41588-021-00895-y
Zhang, G., Liang, Y., Lu, J. (2006). Agrobacterium rhizogenes mediated high frequency induction and genetic transformation of tea hairy roots. J. Tea Sc. 26, 1–10. doi: 10.13305/j.cnki.jts.2009.01.001
Zhang, W., Luo, C., Scossa, F., Zhang, Q., Usadel, B., Fernie, A., et al. (2020b). A phased genome based on single sperm sequencing reveals crossover pattern and complex relatedness in tea plants. Plant J. 105, 197–208. doi: 10.1111/tpj.15051
Zhang, M., Tang, Y.-W., Xu, Y., Yonezawa, T., Shao, Y., Wang, Y.-G., et al. (2021). Concerted and birth-and-death evolution of 26S ribosomal DNA in camellia l. Ann. Bot. London 127, 63–73. doi: 10.1093/aob/mcaa169
Zhang, Y., Wang, D., Wang, Y., Dong, H., Yuan, Y., Yang, W., et al. (2020). Parasitic plant dodder (Cuscuta spp.): A new natural agrobacterium-to-plant horizontal gene transfer species. Sci. China Life Sc. 63, 312–316. doi: 10.1007/s11427-019-1588-x
Zhang, C. C., Wang, L. Y., Wei, K., Cheng, H. (2014). Development and characterization of of single nucleotide polymorphism markers in camellia sinensis (Theaceae). Gen. Mol. Res. 13, 5822–5831. doi: 10.4238/2014.April.14.10
Zhang, W., Zhang, Y., Qiu, H., Guo, Y., Wan, H., Zhang, X., et al. (2020a). Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat. Commun. 11, 3719. doi: 10.1038/s41467-020-17498-6
Zhang, Q., Zhao, L., Folk, R. A., Zhao, J. L., Zamora, N. A., Yang, S. X., et al. (2022). Phytotranscriptomics of theaceae: Generic-level relationships, reticulation and whole-genome duplication. Ann. Bot. London 129, 457–471. doi: 10.1093/aob/mcac007
Zhao, D. W., Yang, J. B., Yang, S. X., Kato, K., Luo, J. P. (2014). Genetic diversity and domestication origin of tea plant camellia taliensis (Theaceae) as revealed by microsatellite markers. BMC Plant Biol. 14, 14. doi: 10.1186/1471-2229-14-14
Keywords: natural genetically transformed organisms, cT-DNA, allele phasing, Camellia sinensis, Thea section, tea plant evolution
Citation: Chen K, Zhurbenko P, Danilov L, Matveeva T and Otten L (2022) Conservation of an Agrobacterium cT-DNA insert in Camellia section Thea reveals the ancient origin of tea plants from a genetically modified ancestor. Front. Plant Sci. 13:997762. doi: 10.3389/fpls.2022.997762
Received: 20 July 2022; Accepted: 16 November 2022;
Published: 06 December 2022.
Edited by:
Shu-Miaw Chaw, Academia Sinica, TaiwanReviewed by:
Gagandeep Singh, The University of Queensland, AustraliaCopyright © 2022 Chen, Zhurbenko, Danilov, Matveeva and Otten. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Léon Otten, bGVvbi5vdHRlbkBpYm1wLWNucnMudW5pc3RyYS5mcg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.