
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 02 September 2022
Sec. Plant Systematics and Evolution
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.976669
This article is part of the Research TopicAdaptive Evolution of GrassesView all 8 articles
 Yamei Ma1†
Yamei Ma1† Jian Wang1†
Jian Wang1† Tifeng Yang1Jingfang Dong1Wu Yang1Luo Chen1Lian Zhou1Jiansong Chen1
Tifeng Yang1Jingfang Dong1Wu Yang1Luo Chen1Lian Zhou1Jiansong Chen1 Bin Liu1
Bin Liu1 Shaohong Zhang1*David Edwards2*
Shaohong Zhang1*David Edwards2* Junliang Zhao1*
Junliang Zhao1*High seedling vigor can improve the ability to compete against weeds and flooding at the seedling stage and is essential for the direct seeding of rice. Early shoot length is an important performance index in seedling vigor evaluation. However, information on the identity of rice germplasm with high seedling vigor, and the genetic basis of seedling vigor are not well understood. In this study, we have conducted a genome-wide association study using 302 international diverse rice accessions from the Rice Diversity Panel 2. Six quantitative trait loci (QTLs) were found to associate with shoot length (SL). The locus qSL2 was further analyzed for candidate gene characterization. We identified OsCPS1, which encodes CDP synthase and functions in GA (Gibberellins) biosynthesis in rice, exhibits differential expression between long and short SL accessions. Using the Nipponbare genome sequence as the reference, we identified a 36 bp deletion in the 5’ UTR of OsCPS1 in long SL accessions, which is absent in short SL accessions. GA content analysis showed that the levels of bioactive GA1 and GA4 are considerably higher in long SL accessions than in short SL accessions. Genome-wide gene expression analysis indicated the expression of some photosynthesis genes is higher in long SL accessions than in short SL accessions. In contrast, genes involved in ABA (Abscisic Acid)-activated signal pathway showed lower expression in long SL accessions. Population analysis across wild rice, indica and japonica, suggested that OsCPS1 may be under selection in japonica during domestication. The results suggest that OsCPS1 is a candidate gene underlying qSL2. These data provide a promising source for candidate genetic variation associated with seedling vigor, with practical applications in rice breeding.
Rice is one of the most important crops worldwide. Traditional rice cultivation systems involve the raising of nursery seedlings followed by transplanting the seedlings into paddy fields, requiring significant amounts of water, energy, and labor. Hence, an increasing number of rice farmers are adopting direct-seeding methods that have many advantages over traditional transplanting, including reducing water use, decreased labor, shorter crop duration, reduced methane gas emissions and lower costs (Abe et al., 2012; Mahender et al., 2015; Chakraborty et al., 2017). However, the lack of varieties suitable for direct seeding is a major constraint for large-scale implementation of direct seeding, since most modern cultivars are bred for transplanting and lack some of the traits that are important for direct seeding. Therefore, the breeding of elite varieties suitable for direct seeding is important for the future adoption of direct seeding practices in rice production.
Early seedling vigor (ESV) is one of the most important traits related to direct seeding. ESV reflects the ability of rapid and uniform germination, as well as seedling establishment (Ju et al., 2007). High ESV facilitates nutrient uptake to support shoot growth at the seedling stage, which is required for seedling establishment and weed competition (Rao et al., 2007; Anandan et al., 2016). The mining of important genes regulated ESV, and the breeding of rice varieties with high ESV would support an expansion of rice direct seeding production.
Early seedling vigor is a complex trait, mainly distinguished by the rate of germination and rapid early growth of both the shoot and roots. Several seedling vigor QTLs have previously been identified with biparental populations (Cheng et al., 2013; Zhang et al., 2017; Dimaano et al., 2020), recombinant inbred lines (Huang et al., 2004; Zhou et al., 2007; Xie et al., 2014; Singh et al., 2017) and association mapping studies (Dang et al., 2014; Wang et al., 2018). Seedling vigor QTLs have been detected using several different phenotypes, including germination index and germination rate (Wang et al., 2010), low-temperature vigor of germination (Najeeb et al., 2020), shoot length, root length, root number and weight of germinated seeds (Zhang et al., 2017; Wang et al., 2018; Dimaano et al., 2020). These studies have provided genetic insights into seedling vigor in rice. However, the identification of functional genes is rare, and the mechanism underlying rice early seedling vigor is largely unknown (Zhang et al., 2017).
Among all the traits determining ESV in rice, shoot length (SL) is one of the most important traits related to direct seeding. Rapid, uniform germination and vigorous seedling growth could contribute to early establishment and provide competitive advantage over weeds. Abe et al. (2012) identified QTLs for 14 days old seedling length, and demonstrated the height of seedlings in Dunghan Shali is controlled by a QTL with a major effect, qPHS3-2. Furthermore, they identified the gene OsGA20ox1, which is related to gibberellin (GA) biosynthesis, as a strong candidate gene for qPHS3-2. However, no further detailed results on functional genes or molecular mechanisms for SL have been reported.
In this study, we evaluated the shoot length (SL) phenotype using 302 international diverse rice accessions from the Rice Diversity Panel 2 (McCouch et al., 2016) at the early seedling stage. All accessions were sequenced to ~50X depth, and high-density SNPs were called for genome-wide association (GWAS) analysis. In total, 96 significant SNPs were identified that clustered into 6 regions, which were considered as QTLs, distributed on chromosomes 2, 4, 6, 11 and 12. Among these QTLs, the qSL2 locus on chromosome 2 contributed 3.05% variation across in whole panel and 7.38% variation across the indica subpopulation. Based on gene functional annotation, gene expression and GA content analysis, the OsCPS1 gene, which encodes an enzyme that participates in GA biosynthesis, was identified as the candidate gene for qSL2. Further analysis suggested a 36-bp deletion in the 5' UTR of OsCPS1, which strongly correlated with the expression difference of OsCPS1, may be the functional variation leading to the SL phenotype difference. Analysis of this variation and genomic sequence diversity surrounding OsCPS1 across indica and japonica subpopulations revealed OsCPS1 may be under selection in japonica during domestication. The identification of qSL2 and the candidate gene OsCPS1 provides a promising source of genetic variation for the molecular breeding of rice with high seedling vigor.
A subset of Rice Diversity Panel 2 (RDP2; McCouch et al., 2016) consisting of 302 diverse accessions from 53 countries were used in this study. Seeds were provided by the International Rice Research Institute (IRRI).
Seeds for assessment were harvested at 30–40 days after flowering, air-dried under natural conditions and treated at 49°C for 3 days to eliminate dormancy. After sterilized with 3% sodium hypochlorite solution, the seeds were soaked in distilled water for 24 h, then placed in petri dish with wet filter paper and cultivated at 30°C under artificial illumination. After 5 days, the shoot lengths were measured manually. Each line had three replicate plates, each containing 30 seeds.
DNA was extracted from fresh leaf tissue using the CTAB (hexadecyltrimethylammonium bromide) protocol. DNA sequencing was performed on the Illumina NovaSeq6000 platform (BerryGenomics, China) and data aligned to the Nipponbare reference genome (MSU 7.0;1 Kawahara et al., 2013) using BWA (Li and Durbin, 2009). Nucleotide variants were called using GATK (Van der Auwera and O’Connor, 2020). SNPs were selected for GWAS analysis based on the criteria of missing data <15% and minor allele frequency of >0.05. GWAS was performed using a mixed linear model (MLM) with kinship matrix and principal component analysis in GAPIT 2 (Tang et al., 2016) and a cutoff threshold of –log10(p) = 4. Manhattan plots were produced using the R package ggplot2 (Wickham, 2016). Following GWAS analysis, significant SNPs within a 100 kb interval were classed as a locus, and candidate genes were identified from 100 kb upstream and downstream of the most significant SNP in each QTL.
Three long SL accessions and three short SL accessions were selected for RNA-sequencing based on haplotype analysis. Seeds were germinated in a petri dish in a growth chamber. RNA was extracted from five-day-old seedlings using the HiPure Plant RNA Mini Kit (Magen, Guangzhou, China) with three biological RNA replicates. RNA-Seq was conducted by Annoroad Gene Technology Co., Ltd. (Beijing, China). RNA samples were sequenced using an Illumina HiSeq-2,500, producing 10 Gb of raw sequencing data. Data analysis was conducted using HISAT2-Stringtie-Deseq2 pipeline (Pertea et al., 2016). Raw counts of each sample exported from Stringtie were imported and normalized by DEseq2 (Love et al., 2014). Genes with average read counts less than 10 in all samples were filtered out for further analysis. For each accession, transcript abundance was calculated as the mean normalized counts of three biological replicates. p values between long and short SL accessions were estimated by the Student’s t-test. The genes with corrected p values (false discovery rate or q-value) ≤0.05 were identified the differentially expressed genes. GO enrichment analysis was performed using DAVID (Sherman et al., 2022).
First-strand cDNA was synthesized from 1 μg total RNA using the PrimeScriptTM RT reagent kit (Takara, Dalian, China). The house-keeping ubiquitin gene was used as an internal control. Real-time PCR was performed using the SYBR Premix ExTaqTM kit (Takara, Dalian, China) and the Biorad CFX 96 Real-Time System (BioRad, Hercules, CA, United States). Relative expression levels were calculated with the 2-△△CT method (Livak and Schmittgen, 2001).
The sequences of OsCPS1 of all 302 accessions and rufipogon was used for DNA diversity analysis. Sequences were imported into ClustalW to construct a nucleotide alignment matrix. Nucleotide diversity (π) values were calculated using VCFtools with 500 adjacent nonoverlapping windows and a 100-bp step (Danecek et al., 2011).
The RNAseq datasets are available at NCBI project number: PRJNA839180. The DNA sequencing datasets are available at NCBI project number: PRJNA820969. The wild rice sequencing data was downloaded from https://www.ncbi.nlm.nih.gov/, PRJNA657701.
A phylogenetic tree was constructed by SNPhylo (Lee et al., 2014) using SNP data for all accessions. An LD heatmap was drawn with the R package LD heatmap. Student’s t-test was used to test the significance of difference.
The GAs were extracted from five-day-old rice seedlings and assessed by HPLC-MS/MS. About 1 g of frozen rice sample was ground to a fine powder in liquid nitrogen, and 10 times the volume of acetonitrile solution was added followed by shaking at 4°C for overnight. The supernatant was extracted and the sample treated with another 5 times the volume of acetonitrile solution. The supernatants were combined and subjected to 35 mg solid-phase C18 extraction. Following centrifugation at 10000 rpm for 5 min, the supernatants was collected, dried under nitrogen gas, and redissolved in 400 μl of methanol. HPLC-MS/MS was then used to measure the GA content after passing through a 0.22-μm filter, using a 1,290 HPLC system (Agilent, Santa Clara, CA, United States) with a 6,500 Qtrap MS/MS (AB SCIEX, Framingham, MA, United States).
A panel consisting of 302 diverse rice accessions was selected based on diversity and representativeness from an international rice panel (1,568 accessions; McCouch et al., 2016). Shoot length (SL) was measured and used for GWAS analysis. A wide variations in SL was observed, ranging from 23.3 to 74 mm, with an average of 41 mm (Figure 1A; Supplementary Table 1). The SL distribution was continuous and close to normal distribution. Particularly, two indica accessions, TI KU and MA GU ZI HE had an extreme SL phenotype, longer than 70 mm.
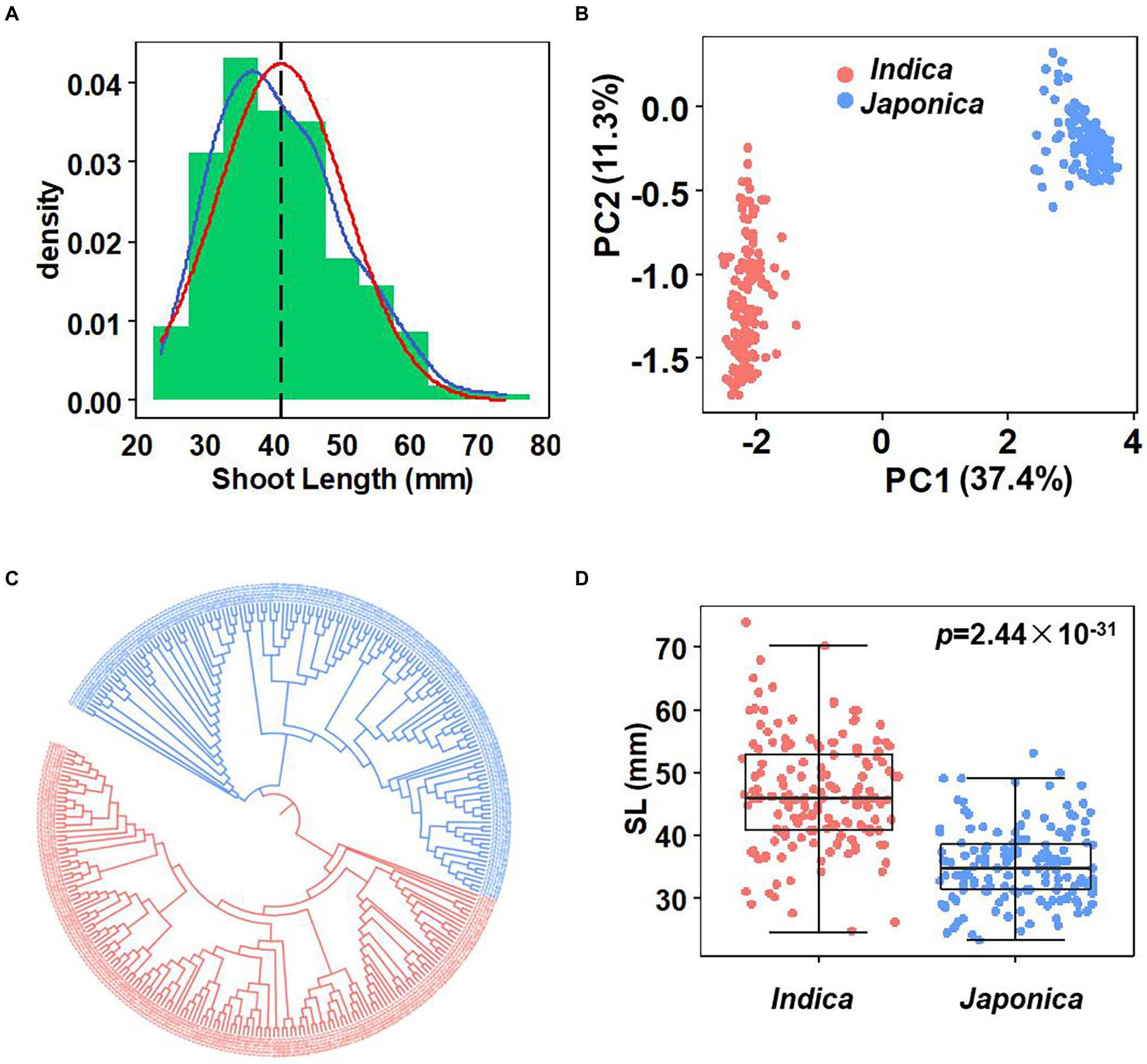
Figure 1. Phenotype variation and population structure. (A) Histogram of the SL. Blue line: Trendline, red line: Normal distribution line, black line: Mean of SL, mean = 41.02 (mm). (B) PCA of the population. (C) Phylogenetic tree of the population. The red and blue shape represent indica and japoncia, respectively. (D) SL variation in two subpopulations. Box edges represent the 0.25 quantile and 0.75 quantile with the median values shown by bold lines. Ymin lower whisker = smallest observation greater than or equal to lower hinge-1.5* IQR (interquartile range). Ymax upper whisker = largest observation less than or equal to upper hinge +1.5* IQR. Student’s t-test was used for statistical analysis.
All 302 accessions were sequenced with the average depth of 50X. Sequencing data was mapped to the Nipponbare reference genome to call the SNPs. After removing loci with more than 15% missing data and minor allele frequency (MAF) less than 5%, we obtained 1,422,101 high-quality SNPs.
The population structure was evaluated by SNP phylogenetic and PCA analysis. The results showed that these accessions clearly clustered into two groups, representing 155 indica accessions and 147 japonica accessions (Figures 1B,C). Phenotype analysis indicated that japonica accessions had shorter shoot length than indica accessions (p < 0.001; Figure 1D). According to the LD decay analysis, the LD distance of the population used in this study is about 150–200 kb on chromosomes level (Supplementary Figure 1).
To determine genetic loci associated with SL, GWAS was performed using a mixed linear model (MLM). According to the LD decay results above, a region was considered as one QTL where it had more than two SNPs with −log10(p) > = 4 within a 200 kb window. The results of GWAS were shown using the Manhattan plot. Six QTLs were identified in the whole population (Figure 2A), distributed across chromosomes 2, 4, 6, 11, 12 and designated as qSL2, qSL4, qSL6, qSL11-1, qSL11-2 and qSL12 (Table 1). Among these QTLs, qSL4 co-localized with a previously identified QTL for seedling vigor (Abe et al., 2012), demonstrating the reliability of the results. The remaining QTLs are newly identified. Due to the observed differences in SL between indica and japonica, GWAS was further conducted on the japonica and indica subpopulations, respectively. Four QTLs were identified in the indica subpopulation, while no QTL were identified in the japonica subpopulation (Figure 2A). Comparisons of the QTLs in different subpopulations demonstrated that qSL2, qSL11-1 and qSL12 can be identified in both the whole population and the indica subpopulation.
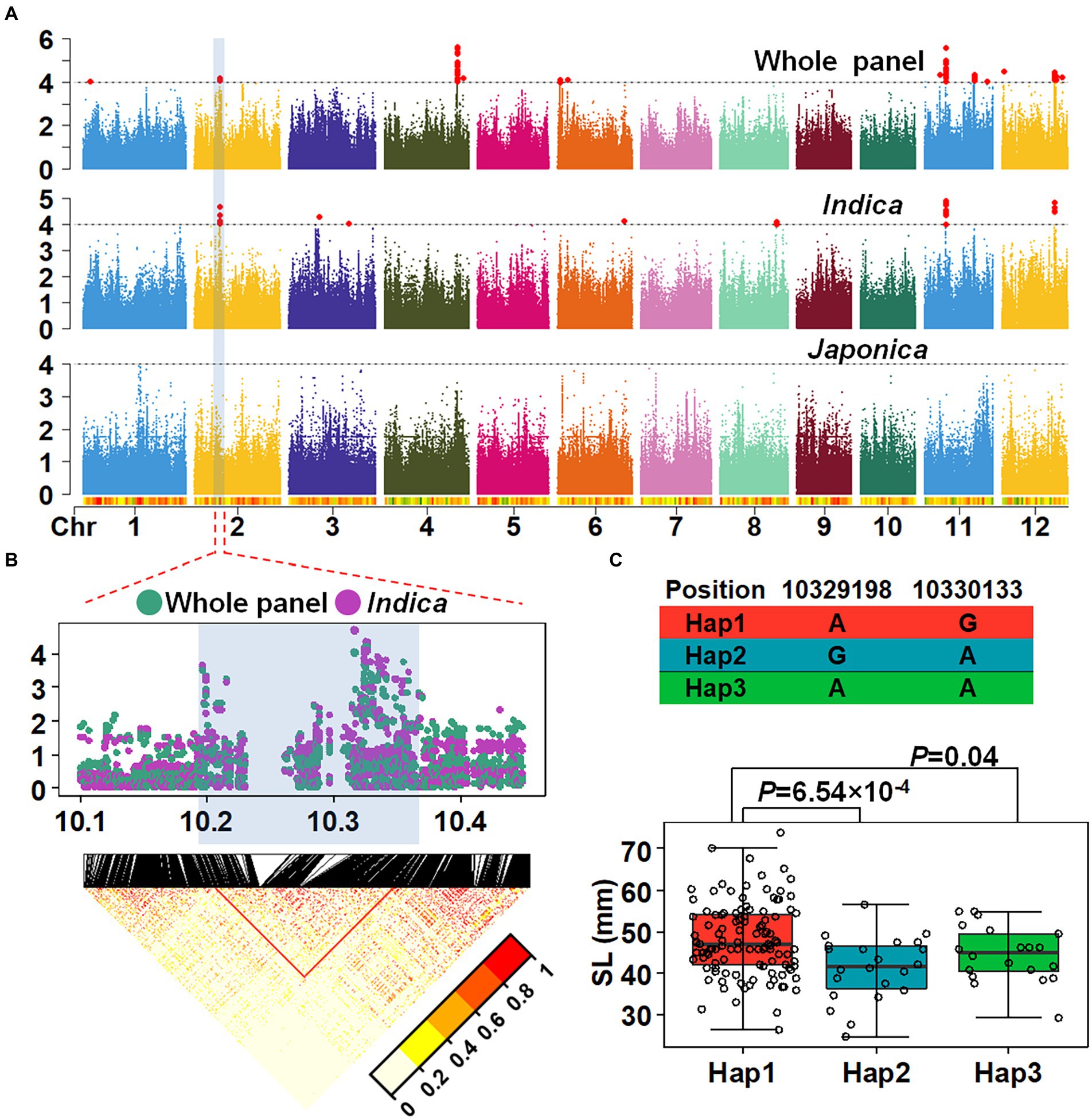
Figure 2. Identification and Mapping of QTL for SL through GWAS. (A) GWAS with shoot length with different population. (B) Local manhattan plots merged by the whole panel and Indica population. (C) Haplotype analysis with SL in Indica subpopulation. Student’s t-test was used for statistical analysis.
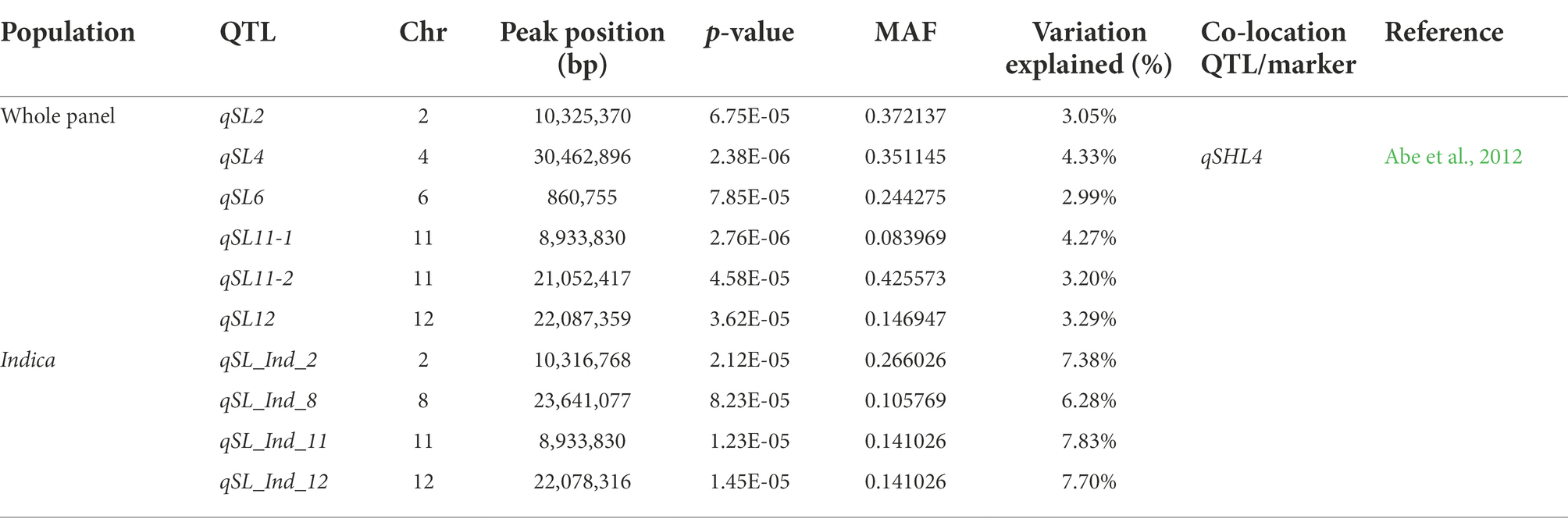
Table 1. QTLs for shoot length identified in the present study and their co-location QTLs identified in the previous studies.
A QTL on chromosome 2 (qSL2) was further analyzed. The most significant SNP within qSL2 interval was located at 10,325,370 bp on chromosome 2, explaining 3.05 and 7.38% of the phenotypic variation in the whole panel and indica subpopulation, respectively. According to the LD decay in the region, we delimited qSL2 into a ~ 200 kb region (10.2–10.4 Mb on chromosome 2; Figure 2B; Supplementary Figure 1). Haplotype analysis identified three haplotypes based on the two most significant SNPs in the interval. Further analysis showed that 72.73% of the accessions harbor Hap1, 14.29% for Hap2 and 12.99% for Hap3 in the indica subpopulation (Supplementary Figure 2A). However, in the japonica subpopulation, 99.07% of the accessions harbored Hap1, 0% for Hap2 and 0.93% Hap3 (Supplementary Figure 2B). A similar result was obtained using SNP data from 3,000 rice accessions (Supplementary Figures 2C,D; Mansueto et al., 2017). Phenotypic variation related to haplotype was assessed and the results showed that in indica accessions with Hap1 showed longer SL than accessions with Hap2 (p value: 6.54 × 10−4) or Hap3 (p value: 0.04; Figure 2C).
The qSL2 genomic interval contains 24 annotated genes based on release 7 of the MSU Rice Annotation Project (Kawahara et al., 2013). To further assess these candidate genes, three long SL accessions (accession No. 690, 620 and 684) and three short SL accessions (accession No. 632, 56 and 463) were selected for whole genome expression analysis by RNA-seq (Figure 3A). 11 of 24 genes were expressed in shoot tissue, with LOC_Os02g17780 (OsCPS1) showing a significant difference (p < 0.01) in expression between long and short SL accessions (Figure 3B; Supplementary Figure 3). To further verify this result, we further performed qRT-PCR to determine the expression level of OsCPS1 using six long SL accessions (accession NO. 690, 620, 684, 692, 565 and 643) and six short SL accessions (accession NO. 632, 56, 463, 967, 562 and 1,400). OsCPS1 levels were significantly higher in long SL accessions than that in short SL accessions (Figure 3C). Previous studies have shown that OsCPS1 encodes an ent-CDP synthase and functions in gibberellin biosynthesis (Otomo et al., 2004; Prisic et al., 2004; Sakamoto et al., 2004). Since GA is an important plant hormone that plays a central role in regulating growth and development, we hypothesized OsCPS1 may be the functional gene of qSL2.
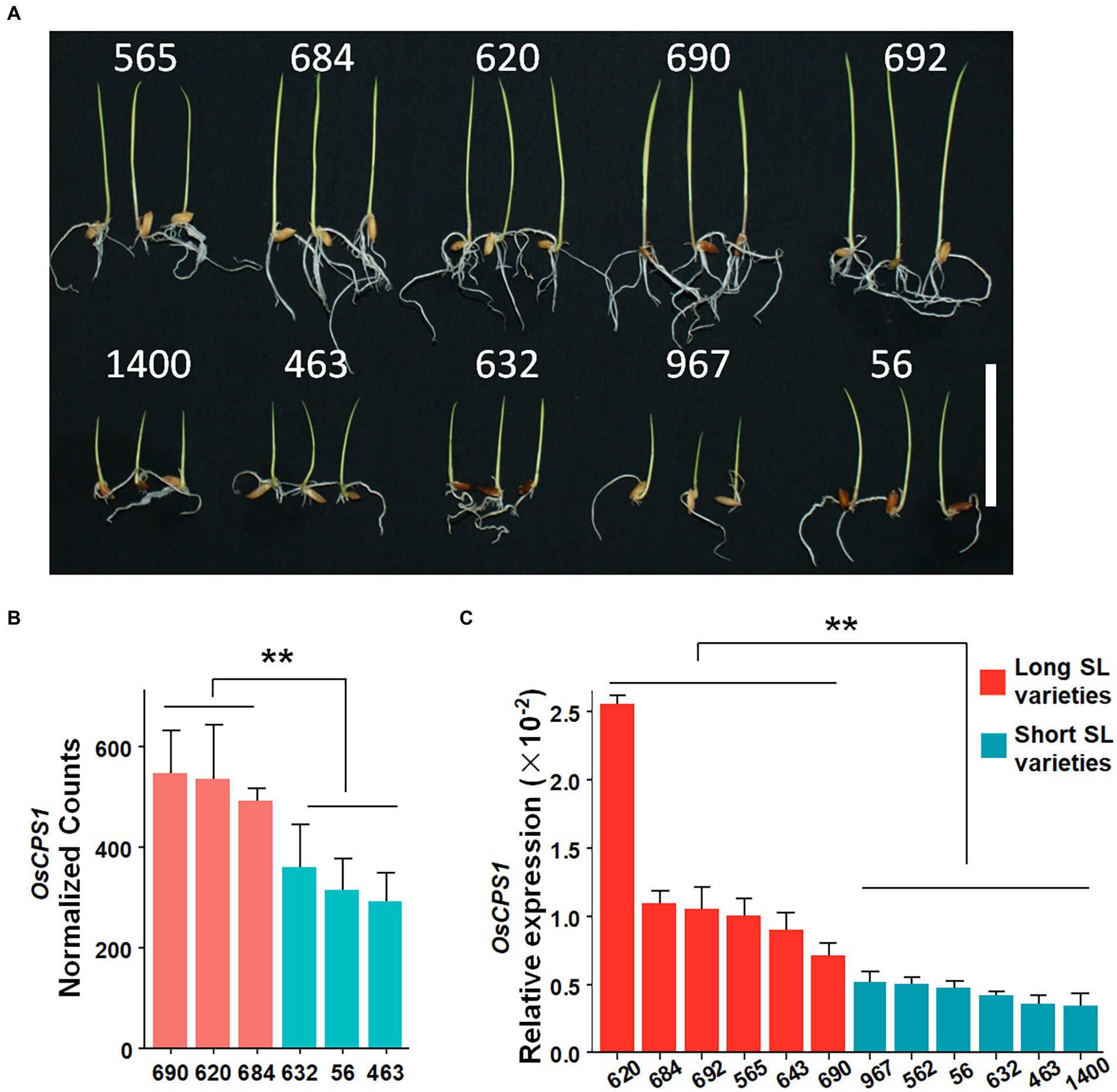
Figure 3. The SL phenotype and the candidate gene OsCPS1 expression in different varieties. (A) The phenotype of representative varieties after 5 days germination. Scale bar 50 mm. (B) OsCPS1 expression analysis by RNA-seq. Red bars represent long SL varieties, blue bars represent short SL varieties. Data are shown as means ± SD (n = 3). **p < 0.01. t-test. (C) OsCPS1 expression analysis by qRT-PCR. Red bars represent long SL varieties, blue bars represent short SL varieties. Data are shown as means ± SD. **p < 0.01. t-test.
To find the cause of differential expression of OsCPS1 between long SL accessions and short SL accessions, we compared the sequence differences of OsCPS1 between 16 long SL accessions and 16 short SL accessions, which were selected based on phenotype and haplotype analysis. As shown in Figures 4B,C, a 36 bp Indel in the 5' UTR that was located 9 bp upstream of the start codon of OsCPS1 was found in long SL accessions (designed as OsCPS1-L), but not in short SL accessions (designed as OsCPS1-S). This sequence variation could be the cause for their differential expression in long and short SL accessions.
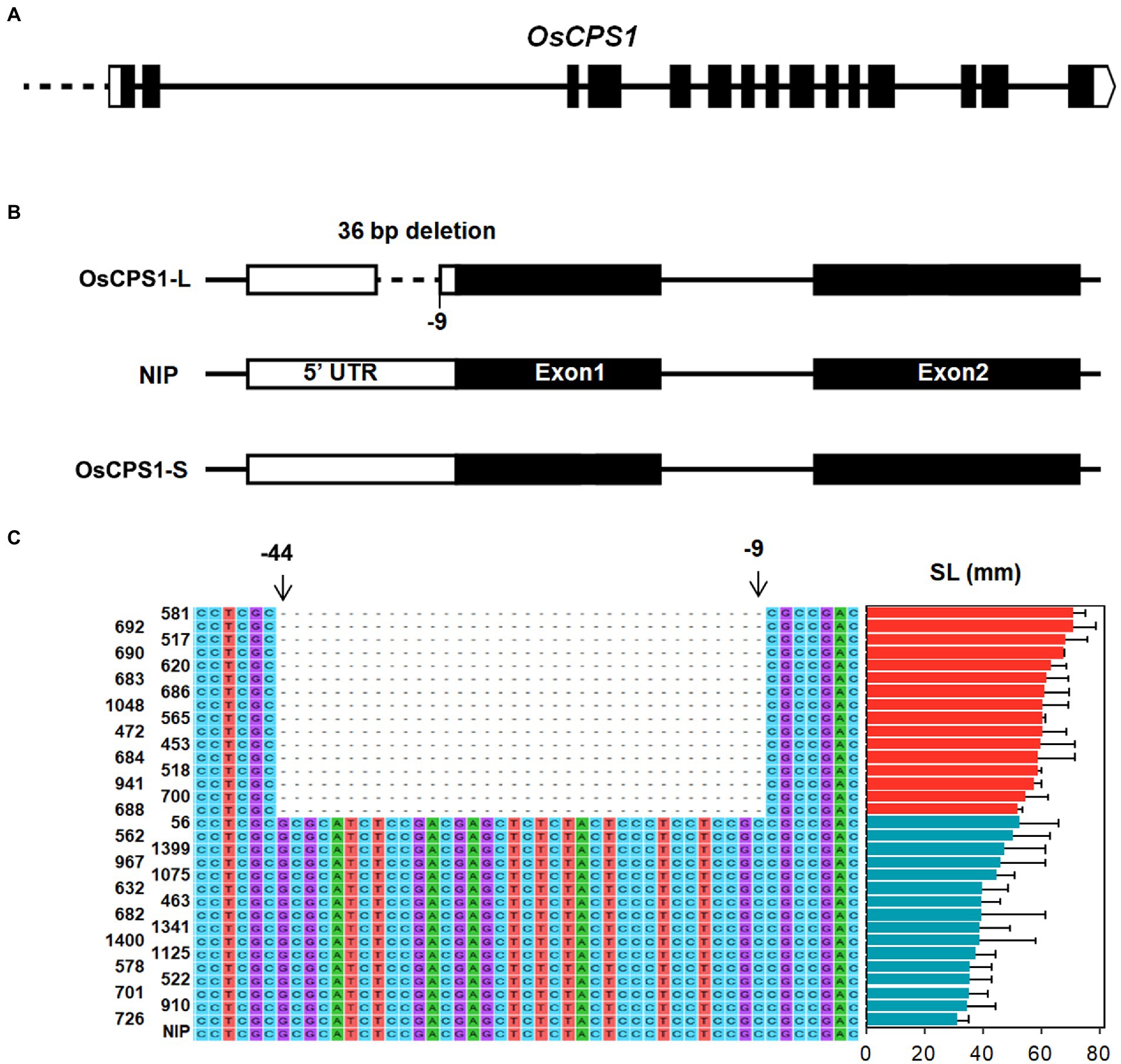
Figure 4. Sequence variations of OsCPS1 in long and short SL varieties. (A) Gene structure of OsCPS1. Black rectangles and white rectangles indicate the exons, 3′ and 5′ UTR, respectively. (B) Indels in OsCPS1-L and OsCPS1-S comparing with Nipponbare (NIP). –9 represents the 9 bp upstream of start codon. (C) Sequence alignment with 36-bp Indel between long and short SL varieties. The sequence of OsCPS1 in Nipponbare (NIP) was taken as reference. The sequences from −9 to −44 were deleted in long SL varieties.
As OsCPS1 is predicted to participate in GA synthesis, the GA content in shoot was determined using six rice accessions that were used for RNA-seq. The long SL accessions showed increased GA1 and GA4 level compared to the short SL accessions (Figure 5A). Since GA1 and GA4 are two major active GAs involved in regulating vegetative growth (Magome et al., 2013), these results further suggested that GA content may be affected by OsCPS1 and responsible for the SL phenotype.
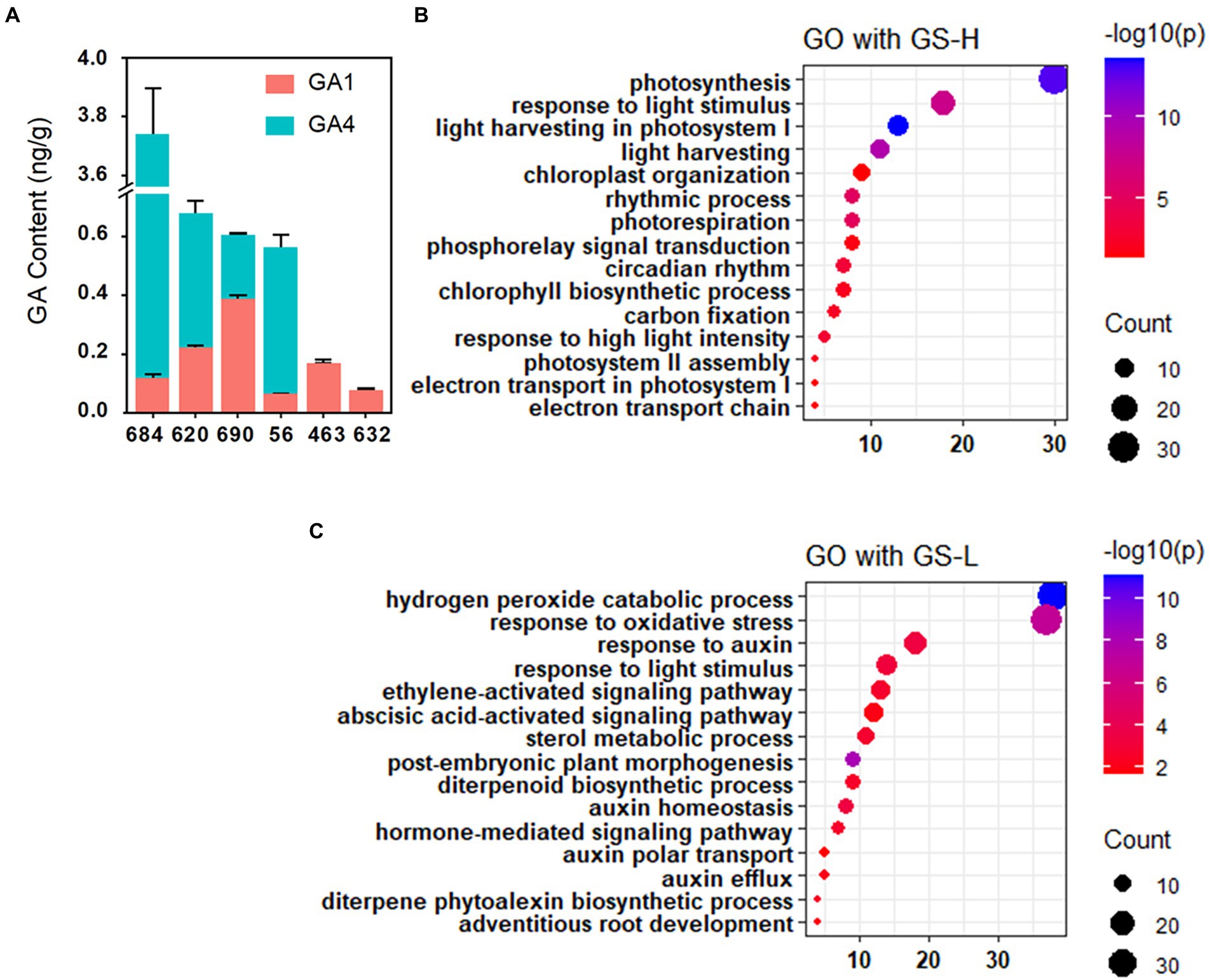
Figure 5. GA content analysis between long and short SL varieties and GO enrichment analysis. (A) GA1 and GA4 content analysis between long and short SL varieties. The results showing higher GA1 and GA4 levels in long SL varieties than short SL varieties. Data are shown as means ± SD (n = 3). (B) GO enrichment analysis with GS-H. GS-H: gene set with higher expression in long SL varieties than in short SL varieties. (C) GO enrichment analysis with GS-L. GS-L: gene set with lower expression in long SL varieties than in short SL varieties.
To further confirm that OsCPS1 is the candidate gene, we analyzed differentially expressed genes between long and short SL accessions by RNA-seq data. As shown in Figure 5B, genes with higher expression in long SL accessions than those in short SL accessions were significantly enriched for GO terms related to photosynthesis (GO:0015979), light harvesting (GO:0009765), chlorophyll biosynthetic process (GO:0015995) and carbon fixation (GO:0015977). In contrast, gene involved in abscisic acid-activated signaling pathway (GO:009738) showed lower expression in long SL accessions than in short SL accessions (Figure 5C). It has been reported photosynthesis-related genes can be induced by GA treatment, while ABA functions as an antagonist of GA (Gómez-Cadenas et al., 2001; Biemelt et al., 2004; Xie et al., 2016). The above results together suggested OsCPS1-mediated GA signaling may participate in regulating the SL phenotypes conferred by qSL2.
To further evaluate the possible function of the 36-bp deletion in the 5' UTR of OsCPS1, the rufipogon sequencing data was downloaded from NCBI.2 All sequencing data of the 302 accessions and rufipogon was analyzed. The results identified 2 haplotypes (OsCPS1-L and OsCPS1-S) of OsCPS1 based on the deletions in the 5′ UTR. In total, 17.18% of indica accessions, 0.88% of japonica accessions and 7.98% of rufipogon accessions harbored the deletion (Figure 6A). We further analyzed the effect of the deletions on shoot length. As shown in Figure 6B, the shoot length differed significantly among the 302 accessions, with the OsCPS1-L type accessions showing longer shoot length (average 53.4 mm) than that OsCPS1-S type accessions (average 45.4 mm; p = 1.15 × 10−9). Interestingly, the height of mature plants demonstrated no difference between OsCPS1-L and OsCPS1-S accessions (Figure 6C), suggesting that this indel correlates with early seedling vigor, but not with adult plant height. Genealogical networks and geographical distribution of haplotypes were analyzed in a larger international panel contained 509 accessions from 51 countries worldwide. The result showed OsCPS1-S only exists in accessions from a few countries in East or Southeast Asia, including China, Indonesia, Malaysia, Nepal, Philippines and Sri Lanka (Figure 6D).
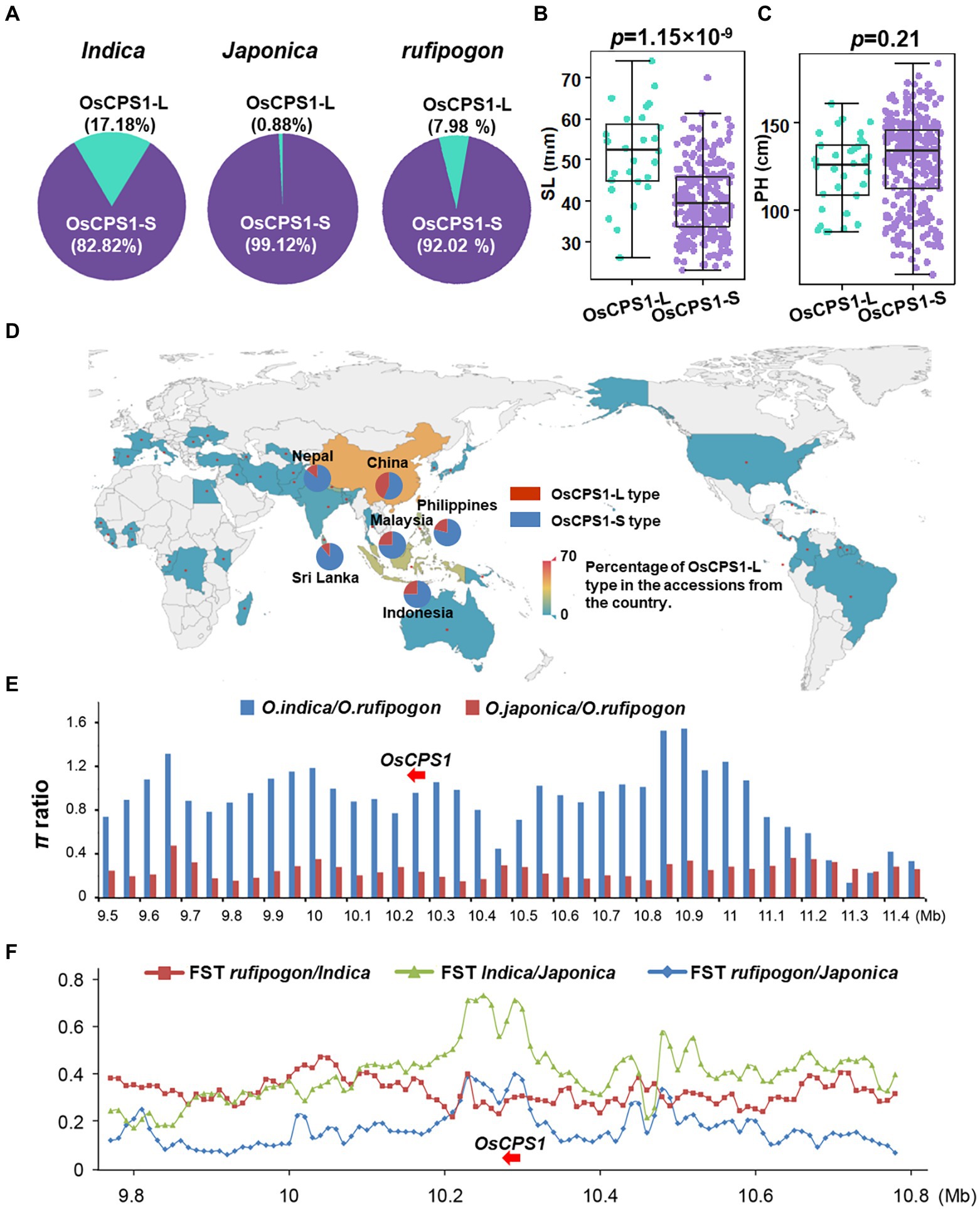
Figure 6. Haplotype analysis and domestication analysis of OsCPS1. (A) The percentage of 36-bp deletion in indica, japonica and rufipogon. (B) Shoot length of OsCPS1-L type accessions and OsCPS1-S type accessions. (C) Plant height of OsCPS1-L type accessions and OsCPS1-S type accessions. (D) Geographical distribution analysis of OsCPS1-L type accessions and OsCPS1-S type accessions in the world. (E) π (nucleotide diversity) ratio in genome region surrounding OsCPS1. Blue bar means π ratio between indica and rufipogon, red bar means π ratio between japonica and rufipogon. (F) FST (fixation index) in the region surrounding OsCPS1 among rufipogen, indica and japonica.
To identify potential selective signals during domestication or breeding selection of the genome interval surrounding OsCPS1, the nucleotide diversity (π) and fixation index (Fst) were employed as indicators. A significant decrease in nucleotide diversity was observed in the region surrounding OsCPS1 between japonica and rufipogon. In contrast, similar diversity was observed between indica and rufipogon (Figure 6E). In accordance with these results, a significant Fst peak was identified in the same region between indica and japonica (Figure 6F). These findings revealed significant genetic divergence in the OsCPS1 region between indica and japonica.
Seedling shoot length is an important performance indicator for seedling vigor evaluation, and it is an important agronomic trait in direct seeding of rice. Understanding genetic variations controlling shoot length is valuable for breeding varieties suitable for direct seeding. Here, we used a diverse panel consisting of 302 accessions from 53 countries, presenting phenotypic variance in shoot length (SL) to identify novel QTLs controlling SL.
Six QTLs were found to associate with shoot length and all QTLs were under further investigation. In this study, a QTL located in chromosome 2 (qSL2) was studied in detail to find possible causative variations related to shoot length. OsCPS1(LOC_Os02g17780), which encodes an enzyme for gibberellin (GA) synthesis (Toyomasu et al., 2015), was identified as candidate gene by sequence comparisons and gene expression analysis. OsCPS1 showed significant expression differences between long and short SL accessions (Figures 3B,C). Furthermore, these expression level differences are strongly correlated with a 36-bp Indel located in the 5' UTR of OsCPS1. Assay of the GA content between long and short SL accessions revealed higher expression of OsCPS1 is associated with increased GA content (Figure 5A). From these results, we hypothesized that OsCPS1 may be the candidate functional gene underlying qSL2, and the 36-bp may be the functional variation leading to the SL phenotype, by modulating the expression of OsCPS1 and resulting in the differences in GA content.
GA plays an important role in regulating plant growth and development, including seed germination and stem elongation. Bioactive GAs are synthesized from trans-geranylgeranyl diphosphate (GGDP), and GGDP is converted to ent-kaurene by CDP synthase (CPS) and ent-kaurene synthase (KS; Yamaguchi, 2008). Four CPS genes (OsCPS1-4) have been identified in the rice genome, including a pseudogene (OsCPS3; Sakamoto et al., 2004). OsCPS1 is involved in growth related GA biosynthesis, while OsCPS2 and OsCPS4 are involved in the biosynthesis of phytoalexins for defense (Otomo et al., 2004; Prisic et al., 2004; Sakamoto et al., 2004). The oscps1 mutant exhibited a severely dwarfed phenotype with a decreased level of ent- kaurene compared to wild type plants (Sakamoto et al., 2004). Moreover, no bioactive GA1 was detected in the oscps1 mutant (Sakamoto et al., 2004). These data suggested that OsCPS1 acts as a regulator of GA synthesis. We found that the expression of OsCPS1 is significant higher in long SL accessions (Figures 3B,C). In addition, the content of GA1 and GA4 in long SL accessions were higher than that in short SL accessions (Figure 5A). These results suggested that the differences of SL between long and short SL accessions may be due to the difference in OsCPS1-mediated GA content.
Recently, Toyomasu et al. (2015) reported that OsCPS1 and OsCPS2 exhibited different tissue-specific expression patterns. The OsCPS2 transcript level was much lower than that of OsCPS1 in the basal part of second-leaf sheaths in third-leaf stage rice seedlings. Furthermore, qRT-PCR suggested that OsCPS1 transcripts mainly localized in vascular bundle tissues, whereas OsCPS2 transcripts mainly localized in epidermal cells that address environmental stressors. More importantly, OsCPS2 expression under the OsCPS1 promoter, but not its native promoter, rescued the oscps1 mutant dwarfed phenotype, suggesting that tissue-specific expression of OsCPS genes is important for regulating growth. Our results showed that, compared with OsCPS1-S type accessions, OsCPS1-L type accessions that harbored a 36-bp deletion in the 5' UTR, showed longer shoot length (Figures 4, 6B), suggesting that the 36-bp deletion may cause differential expression of OsCPS1 between OsCPS1-S and OsCPS1-L type accessions, leading to the difference in shoot length. Interestingly, the height of mature plants was not different between OsCPS1-L and OsCPS1-S type accessions (Figure 6C), suggesting that this locus may be involved in regulating early seedling vigor, but not adult plant height, which is one of the main factors affecting lodging resistance. Collectively, these data indicated this locus has significant potential in breeding rice varieties suitable for direct seeding.
GA regulates stem elongation by promoting both cell elongation and cell division (Rademacher, 2000). In this study, we identified OsCPS1, a gene involved in GA synthesis, as the candidate gene of qSL2 (Figures 3, 4). Long SL accessions showed an increase in GA1 and GA4 content (Figure 5A). To answer whether OsCPS1-mediated GA signaling contributes to SL phenotype, RNA-seq was conducted to evaluate gene expression differences between long and short SL accessions.
Chen et al. (2020) reported that photosynthesis, metabolic pathways and biosynthesis of secondary metabolites, and cell wall components were differentially expressed in the sugarcane internodes of the GA-treated plants, indicating their involvement in GA-mediated internode elongation. Consistent with Chen’s report, our results demonstrated genes with higher expression in long SL accessions than those in short SL accessions were significantly enriched for GO terms related photosynthesis (Figure 5B). Several studies have found that GA could increase expression of genes involved in photosynthesis, as well as the photosynthesis rate (Yuan and Xu, 2001; Tian et al., 2016; Chen et al., 2020). We speculated the higher expression of genes associated with photosynthesis may result from higher GA content in long shoot length accessions.
In contrast, genes involved ABA activated signaling pathways showed lower expression in long SL accession than in short SL accessions (Figure 5C). ABA and GA have central and antagonistic roles in regulating rice growth. The antagonistic action between GA and ABA was an important factor regulating the developmental transition from embryogenesis to seed germination (Gómez-Cadenas et al., 2001). However, the crosstalk between GA and ABA, as well as how this crosstalk regulates shoot length needs further investigation. Collectively, the gene expression profile results suggested that qSL2 may promote shoot growth by OsCPS1-mediated GA signaling at an early stage.
Since the 36-bp deletion may be the functional variation underlying qSL2 conferring SL phenotypes, the origin and evolution of the 36-bp deletion was characterized from wild, indica and japonica rice. Our results indicated that the 36-bp deletion originated from wild rice and was retained in indica accessions (Figure 6A). Surprisingly, the deletion was almost absent from japonica accessions, suggesting a strong selective sweep during domestication or/and breeding of japonica rice (Figure 6A). Further analysis revealed that the 36-bp deletion only exists in accessions from six Asian countries (Figure 6D), suggesting that this deletion was intentionally selected and retained in these countries. Since this deletion is associated with early seedling growth, we hypothesized that this trait may be an important requirement for rice planting in this region, or for rice environment adaption in this region. In contrast, the lack of this deletion in japonica suggested that this phenotype is not suitable for japonica or results from a domestication bottleneck.
We determined the nucleotide diversity and fixation index in the genome region surrounding OsCPS1 among wild rice, indica and japonica accessions. Nucleotide diversity analysis results revealed a significant decrease in diversity in japonica compared to indica or wild rice in the region (Figure 6E). However, no similar phenomenon was found in indica and wild rice (Figure 6E). Fst analysis also demonstrated a peak between indica and japonica in this region (Figure 6F). These results suggested that the region surrounding OsCPS1 is under strong selection in japonica, which is in consistent with the haplotype analysis results. While we provided evidence that OsCPS1, and the 5' UTR deletion may be responsible for the SL trait, more detailed investigation on the molecular function of the 36-bp deletion and OsCPS1 itself is required to fully dissect the process. In future, transgenic methods, including overexpression and CRISPR knockout mutations, could be used to verify OsCPS1 function.
In summary, by using an international diverse rice panel, we successfully identified germplasm with long shoot length in the early seedling stage of rice growth. This germplasm may be useful as parents for breeding of high seedling vigor varieties. GWAS analysis with deep sequencing data identified novel QTLs controlling SL, and whole genome gene expression profiling by RNA-seq further helped to identify OsCPS1 as the candidate gene for one of the major QTLs (qSL2). These multi-omic data and methods helped to elucidate the genomic variation and molecular basis of modulating shoot length and seedling vigor. A 36-bp deletion in the 5' UTR of OsCPS1 was characterized as the possible functional variation underlying qSL2. The identification of qSL2 and the candidate gene OsCPS1 has a potential to accelerate the breeding of superior varieties suitable for direct seeding.
The datasets presented in this study can be found in online repositories. The RNAseq data presented in the study are deposited in the NCBI repository (Bioproject: PRJNA839180, Accession number: SRR19259017, SRR19259020, SRR19259023, etc). The DNA sequencing datasets are deposited in the NCBI repository (Bioproject: PRJNA820969, Accession number: SRR19146044, SRR19146039, SRR19146035, etc.) The wild rice sequencing data were downloaded from NCBI (Bioproject: PRJNA657701). The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
YM, JW, DE, SZ, and JZ designed the experiments and wrote and edited the manuscript. YM and JW performed most of experiments and analyzed the data. Other authors assisted in experiments and discussed the results. All authors contributed to the article and approved the submitted version.
This work was funded by Guangdong Provincial International Cooperation Project of Science & Technology (2021A0505030048), the Key Areas Research Projects of Guangdong Province (2020B0202090003), Science and Technology Program of Guangzhou (202002030375 and 201804020078), National Natural Science Foundation of China (31901441), Special Fund for Scientific Innovation Strategy-Construction of High Level Academy of Agriculture Science (R2021PY-QF001), the evaluation and operation funds of Guangdong key laboratories (2020B1212060047), the “YouGu” Plan of Rice Research Institute of Guangdong Academy of Agricultural Sciences (2021YG001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.976669/full#supplementary-material
ESV, Early seedling vigor; GWAS, Genome-wide association study; LD, Linkage disequilibrium; MAF, Minor allele frequency; QTLs, Quantitative trait loci; RDP2, Rice Diversity Panel 2; SL, Shoot length; SNP, Single-nucleotide polymorphism.
Abe, A., Takagi, H., Fujibe, T., Aya, K., Kojima, M., Sakakibara, H., et al. (2012). OsGA20ox1, a candidate gene for a major QTL controlling seedling vigor in rice. Theor. Appl. Genet. 125, 647–657. doi: 10.1007/s00122-012-1857-z
Anandan, A., Anumalla, M., Pradhan, S. K., and Ali, J. (2016). Population structure, diversity and trait association analysis in rice (Oryza sativa L.) germplasm for early seedling vigor (ESV) using trait linked SSR markers. PLoS One 11:e152406. doi: 10.1371/journal.pone.0152406
Biemelt, S., Tschiersch, H., and Sonnewald, U. (2004). Impact of altered gibberellin metabolism on biomass accumulation, lignin biosynthesis, and photosynthesis in transgenic tobacco plants. Plant Physiol. 135, 254–265. doi: 10.1104/pp.103.036988
Chakraborty, D., Ladha, J. K., Rana, D. S., Jat, M. L., Gathala, M. K., Yadav, S., et al. (2017). A global analysis of alternative tillage and crop establishment practices for economically and environmentally efficient rice production. Sci. Rep. 7, 1–11. doi: 10.1038/s41598-017-09742-9
Chen, R. F., Fan, Y. G., Yan, H. F., Zhou, H. W., Zhou, Z. F., Weng, M. L., et al. (2020). Enhanced activity of genes associated with photosynthesis, phytohormone metabolism and cell wall synthesis is involved in gibberellin-mediated sugarcane internode growth. Front. Genet. 11:570094. doi: 10.3389/fgene.2020.570094
Cheng, X., Cheng, J., Huang, X., Lai, Y., Wang, L., Du, W., et al. (2013). Dynamic quantitative trait loci analysis of seed reserve utilization during three germination stages in rice. PLoS One 8:e80002. doi: 10.1371/journal.pone.0080002
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and vcftools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Dang, X., Thi, T. G. T., Dong, G., Wang, H., Edzesi, W. M., and Hong, D. (2014). Genetic diversity and association mapping of seed vigor in rice (Oryza sativa L.). Planta 239, 1309–1319. doi: 10.1007/s00425-014-2060-z
Dimaano, N. G. B., Ali, J., Mahender, A., Baltazar, A. M., Diaz, M. G. Q., Pang, Y. L., et al. (2020). Identification of quantitative trait loci governing early germination and seedling vigor traits related to weed competitive ability in rice. Euphytica 216, 1–20. doi: 10.1007/s10681-020-02694-8
Gómez-Cadenas, A., Zentella, R., Walker-Simmons, M. K., and Ho, T. D. (2001). Gibberellin/abscisic acid antagonism in barley aleurone cells: site of action of the protein kinase PKABA1 in relation to gibberellin signaling molecules. Plant Cell 13, 667–679. doi: 10.1105/tpc.13.3.667
Huang, Z., Yu, T., Su, L., Yu, S. B., Zhang, Z. H., and Zhu, Y. G. (2004). Identification of chromosome regions associated with seedling vigor in rice. Yi Chuan xue bao=. Acta Genet. Sin. 31, 596–603.
Ju, L., Tang, S., Hu, P., Louis, A., Jiao, G., and Jian, T. (2007). Analysis on factors affecting seedling establishment in rice. Rice Sci. 14, 27–32. doi: 10.1016/S1672-6308(07)60005-5
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., McCombie, W. R., Ouyang, S., et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 1–10. doi: 10.1186/1939-8433-6-4
Lee, T., Guo, H., Wang, X., Kim, C., and Paterson, A. H. (2014). SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15, 1–6. doi: 10.1186/1471-2164-15-162
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-△△CT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. doi: 10.1186/s13059-014-0550-8
Magome, H., Nomura, T., Hanada, A., Takeda-Kamiya, N., Ohnishi, T., Shinma, Y., et al. (2013). CYP714B1 and CYP714B2 encode gibberellin 13-oxidases that reduce gibberellin activity in rice. PNAS 110, 1947–1952. doi: 10.1073/pnas.1215788110
Mahender, A., Anandan, A., and Pradhan, S. K. (2015). Early seedling vigour, an imperative trait for direct-seeded rice: an overview on physio-morphological parameters and molecular markers. Planta 241, 1027–1050. doi: 10.1007/s00425-015-2273-9
Mansueto, L., Fuentes, R. R., Borja, F. N., Detras, J., Abriol-Santos, J. M., Chebotarov, D., et al. (2017). Rice SNP-seek database update: new SNPs, indels, and queries. Nucleic Acids Res. 45, D1075–D1081. doi: 10.1093/nar/gkw1135
McCouch, S. R., Wright, M. H., Tung, C., Maron, L. G., McNally, K. L., Fitzgerald, M., et al. (2016). Open access resources for genome-wide association mapping in rice. Nat. Commun. 7, 1–14. doi: 10.1038/ncomms10532
Najeeb, S., Ali, J., Mahender, A., Pang, Y. L., Zilhas, J., Murugaiyan, V., et al. (2020). Identification of main-effect quantitative trait loci (QTLs) for low-temperature stress tolerance germination-and early seedling vigor-related traits in rice (Oryza sativa L.). Mol. Breed. 40, 1–25. doi: 10.1007/s11032-019-1090-4
Otomo, K., Kenmoku, H., Oikawa, H., Konig, W. A., Toshima, H., Mitsuhashi, W., et al. (2004). Biological functions of ent- and syn-copalyl diphosphate synthases in rice: key enzymes for the branch point of gibberellin and phytoalexin biosynthesis. Plant J. 39, 886–893. doi: 10.1111/j.1365-313X.2004.02175.x
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Prisic, S., Xu, M., Wilderman, P. R., and Peters, R. J. (2004). Rice contains two disparate ent-copalyl diphosphate synthases with distinct metabolic functions. Plant Physiol. 136, 4228–4236. doi: 10.1104/pp.104.050567
Rademacher, W. (2000). Growth retardants: effects on gibberellin biosynthesis and other metabolic pathways. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 501–531. doi: 10.1146/annurev.arplant.51.1.501
Rao, A. N., Johnson, D. E., Sivaprasad, B., Ladha, J. K., and Mortimer, A. M. (2007). Weed management in direct-seeded rice. Adv. Agron. 93, 153–255. doi: 10.1016/S0065-2113(06)93004-1
Sakamoto, T., Miura, K., Itoh, H., Tatsumi, T., Ueguchi-Tanaka, M., Ishiyama, K., et al. (2004). An overview of gibberellin metabolism enzyme genes and their related mutants in rice. Plant Physiol. 134, 1642–1653. doi: 10.1104/pp.103.033696
Sherman, B. T., Hao, M., Qiu, J., Jiao, X., Baseler, M. W., Lane, H. C., et al. (2022). DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. doi: 10.1093/nar/gkac194
Singh, U. M., Yadav, S., Dixit, S., Ramayya, P. J., Devi, M. N., Raman, K. A., et al. (2017). QTL hotspots for early vigor and related traits under dry direct-seeded system in rice (Oryza Sativa L.). Front. Plant Sci. 8:286. doi: 10.3389/fpls.2017.00286
Tang, Y., Liu, X., Wang, J., Li, M., Wang, Q., Tian, F., et al. (2016). Gapit version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome 9, 1–9. doi: 10.3835/plantgenome2015.11.0120
Tian, J. X., Song, Y. P., Du, Q. Z., Yang, X. H., Ci, D., Chen, J. H., et al. (2016). Population genomic analysis of gibberellin-responsive long non-coding RNAs in populus. J. Exp. Bot. 67, 2467–2482. doi: 10.1093/jxb/erw057
Toyomasu, T., Usui, M., Sugawara, C., Kanno, Y., Sakai, A., Takahashi, H., et al. (2015). Transcripts of two ent-copalyl diphosphate synthase genes differentially localize in rice plants according to their distinct biological roles. J. Exp. Bot. 66, 369–376. doi: 10.1093/jxb/eru424
Van der Auwera, G. A., and O’Connor, B. D. (2020). Genomics in the cloud: using Docker, GATK, and WDL in terra. United States: O’Reilly Media.
Wang, F., Longkumer, T., Catausan, S. C., Calumpang, C. L. F., Tarun, J. A., Cattin-Ortola, J., et al. (2018). Genome-wide association and gene validation studies for early root vigour to improve direct seeding of rice. Plant Cell Environ. 41, 2731–2743. doi: 10.1111/pce.13400
Wang, Z., Wang, J., Bao, Y., Wang, F., and Zhang, H. (2010). Quantitative trait loci analysis for rice seed vigor during the germination stage. J Zhejiang Univ-Sc B 11, 958–964. doi: 10.1631/jzus.B1000238
Xie, L., Tan, Z., Zhou, Y., Xu, R., Feng, L., and Xing, Y. (2014). Identification and fine mapping of quantitative trait loci for seed vigor in germination and seedling establishment in rice. J. Integr. Plant Biol. 56, 749–759. doi: 10.1111/jipb.12190
Xie, J., Tian, J., Du, Q., Chen, J., Li, Y., Yang, X. H., et al. (2016). Association genetics and transcriptome analysis reveal a gibberellin-responsive pathway involved in regulating photosynthesis. J. Integr. Plant Biol. 67, 3325–3338. doi: 10.1093/jxb/erw151
Yamaguchi, S. (2008). Gibberellin metabolism and its regulation. Annu. Rev. Plant Biol. 59, 225–251. doi: 10.1146/annurev.arplant.59.032607.092804
Yuan, L., and Xu, D. Q. (2001). Stimulation effect of gibberellic acid short-term treatment on leaf photosynthesis related to the increase in Rubisco content in broad bean and soybean. Photosynth. Res. 68, 39–47. doi: 10.1023/A:1011894912421
Zhang, A., Liu, C., Chen, G., Hong, K., Gao, Y., Tian, P., et al. (2017). Genetic analysis for rice seedling vigor and fine mapping of a major QTL qSSL1b for seedling shoot length. Breed. Sci. 67, 307–315. doi: 10.1270/jsbbs.16195
Keywords: rice, shoot length, genome-wide association study, OsCPS1, selection
Citation: Ma Y, Wang J, Yang T, Dong J, Yang W, Chen L, Zhou L, Chen J, Liu B, Zhang S, Edwards D and Zhao J (2022) Genome-wide association mapping and gene expression analysis identify OsCPS1 as a new candidate gene controlling early seedling length in rice. Front. Plant Sci. 13:976669. doi: 10.3389/fpls.2022.976669
Edited by:
Chao Zhang, Northwest A&F University, ChinaReviewed by:
Wei Zong, Indiana University, United StatesCopyright © 2022 Ma, Wang, Yang, Dong, Yang, Chen, Zhou, Chen, Liu, Zhang, Edwards and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaohong Zhang, c3poYW5nZ3pAdG9tLmNvbQ==; David Edwards, ZGF2ZS5lZHdhcmRzQHV3YS5lZHUuYXU=; Junliang Zhao, emhhb19qdW5saWFuZ0BnZGFhcy5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.