 Yu Wang
Yu Wang Jia-Xue Ouyang
Jia-Xue Ouyang Dong-Mei Fan
Dong-Mei Fan Shu-Mao Wang
Shu-Mao Wang Yi-Min Xuan
Yi-Min Xuan Xiao-Chang Wang
Xiao-Chang Wang Xin-Qiang Zheng
Xin-Qiang Zheng- 1College of Agriculture and Biotechnology, Tea Research Institute, Zhejiang University, Hangzhou, China
- 2Institute of Dafo Longjing, Xinchang, China
The tea plant is a kind of ammonium-preferring crop, but the mechanism whereby ammonium (NH4+) regulate its growth is not well understood. The current study focused on the effects of NH4+ on tea plants. Transcriptomic analysis was performed to investigate the early- and late-stage NH4+ deprivation and resupply in tea plants shoots. Through short- and long-term NH4+ deficiency, the dynamic response to NH4+ stress was investigated. The most significant effects of NH4+ deficiency were found to be on photosynthesis and gene ontology (GO) enrichment varied with the length of NH4+ deprivation. Enriched KEGG pathways were also different when NH4+ was resupplied at different concentrations which may indicate reasons for tolerance of high NH4+ concentration. Using weighted gene co-expression network analysis (WGCNA), modules related to significant tea components, tea polyphenols and free amino acids, were identified. Hence, NH4+ could be regarded as a signaling molecule with the response of catechins shown to be higher than that of amino acids. The current work represents a comprehensive transcriptomic analysis of plant responses to NH4+ and reveals many potential genes regulated by NH4+ in tea plants. Such findings may lead to improvements in nitrogen efficiency of tea plants.
Introduction
Nitrogen is an indispensable nutrient element for plant growth and represents a major driving force for crop yield improvement (Li et al., 2017). Its absorption and assimilation are similarly important for growth of tea plants. In addition to effects on plant growth and development, nitrogen nutrition also affects their ability to cope with environmental challenges (Vega et al., 2015). Nitrogen affects growth of plant roots and leaves (von Wiren et al., 2000; Menz et al., 2016; Coleto et al., 2017), senescence rate (Vanacker et al., 2006) and flowering time (Castro Marin et al., 2011). The two main sources of inorganic nitrogen for plants are nitrate (NO3–) and ammonium (NH4+) and different plants have different preferences for them. For example, maize and tomato grow better with nitrate, while rice prefers NH4+ (Britto and Kronzucker, 2002). Previous studies have demonstrated the signaling role of NH4+ (Ren et al., 2020), which induces a variety of morphological and physiological responses (Liu and von Wirén, 2017). For example, NH4+ inhibited root growth and affected cell elongation and division in primary roots (Li et al., 2010) but local NH4+ supply increased lateral root initiation and higher-order lateral root branching (Lima et al., 2010). NH4+ also promoted the alleviate of toxicity by hormones, such as auxin (Esteban et al., 2016).
Tea is a favored beverage throughout the world. The harvested leaves contain many primary and secondary metabolites, such as catechins, theanine and caffeine. A great deal of research has investigated regulatory mechanisms behind the biosynthesis of metabolites, including the regulation of nutrient content (Ruan et al., 2010). Tea plants have been reported to be well adapted to high NH4+ concentrations and grow better with a supply of NH4+ (Fan et al., 2015), a characteristic confirmed by the 15N study of hydroponic tea plants (Tang et al., 2020). Previous studies found a better nitrogen performance of NH4+ in tea plants, with increased free amino acids content and reduced secondary metabolites content (Ruan et al., 2007; Yang et al., 2013). NH4+ benefitted tea plant nitrogen metabolism and upregulated expression of ammonium-assimilation genes (Wang et al., 2021). However, knowledge of effects of NH4+ on the biosynthesis of tea components and molecular mechanisms involved has received little attention. To this end, Zhang et al. (2018) have established the limiting and sufficient nitrogen conditions for tea growth in a hydroponic system. This current work explored the effects of NH4+ deficiency and recovery by different ammonium concentrations on physiology and gene regulation in tea plants by the hydroponic method.
Advances in transcriptome analysis technologies have allowed thousands of genes related to nitrogen metabolism in tea plants to be identified. Evaluations of different N sources in tea plants (Liu et al., 2017; Yang et al., 2018; Wang et al., 2021) have found that NH4+ took precedence over NO3– in assimilation. The work of Zhang et al. (2020) investigated the nitrogen uptake in tea roots and identified a group of modules related to low nitrogen treatment using weighted gene co-expression network analysis (WGCNA). Yang et al. (2020) focused on the relationship between nitrogen and amino acid metabolism. The current study explored the effects of NH4+ deficiency time and NH4+ concentration on gene expression in tea plants by hydroponics to reveal using RNA-seq coupled with a time-course experiment. Short- and long-term responses of tea plants to NH4+ deprivation and different concentrations of NH4+ resupply were assessed and a comprehensive and integrated dataset generated.
Materials and methods
Plant materials and experimental treatments
One-year-old “Longjing 43” tea seedlings were hydroponically cultivated in distilled water for 7 days and exposed to 1/4 strength nutrient solution for 1 week. The strength of the nutrient solution was thereafter increased to 1/2 (for around 2 weeks) and full (Zhang et al., 2018). The full nutrient solution contained the following nutrients: 1.5 mM (NH4)2SO4, 0.73 mM KNO3, 0.1 mM KH2PO4, 0.46 mM K2SO4, 0.41 mM MgSO4, 0.5 mM CaCl2, 0.046 mM H3BO3, 0.09 mM MnSO4, 0.0091 mM ZnSO4, 0.002 mM CuSO4, 0.0026 mM Na2MoO4, and 0.032 mM Fe-EDTA (Wang et al., 2022). After preculture for 1 month, tea seedlings with a similar appearance were chosen to be transferred to nutrient solution without N for 6 h (S_UN) or 7 days (L_UN) and controls were incubated at a normal NH4+ level (3 mM NH4+) for 6 h (CK1) or 7 days (CK2). After 1-week nitrogen deficiency, the nutrient solution was resupplied with 3 mM NH4+ (NN) or 10 mM NH4+ (high NH4+ level, HN) for 6 h (Figure 1A). The pH was adjusted to 5.0 ± 0.2 and solutions were refreshed every 4 days. All hydroponic solutions were continuously aerated using air pumps. Shoots with a bud and two leaves (three biological replicates for each treatment) were harvested, immediately snap-frozen in liquid nitrogen and stored at −80°C for analysis.
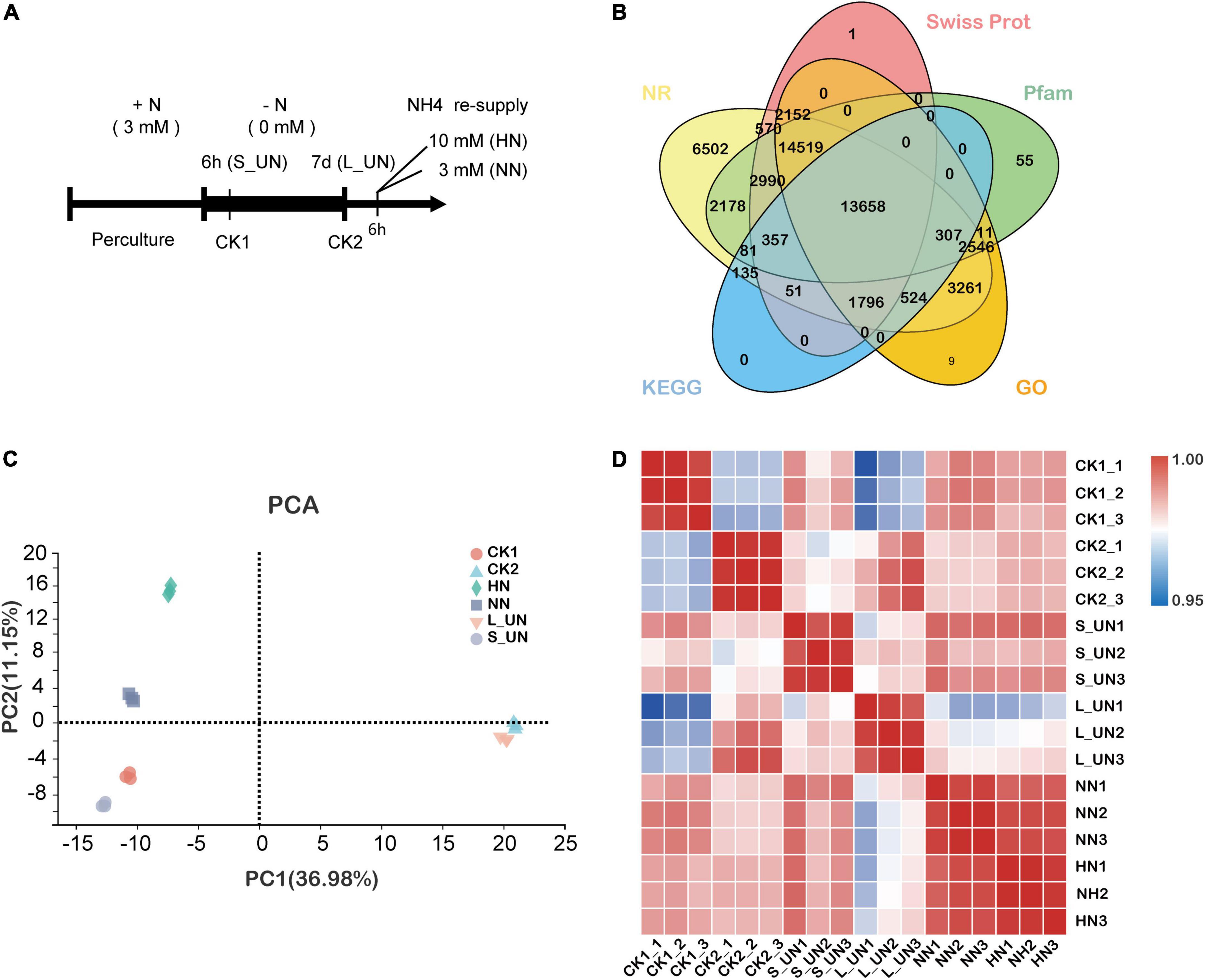
Figure 1. Experimental design and transcriptome relationships. (A) Schematic representation of the experimental design. After preculture, tea plants were transferred to N-deficient media (0 mM) for 6 h (S_UN) or 7 days (L_UN) or maintained in N-sufficient solution for 7 days (CK2). After treatment for 7 days, the N-starved plants were supplied with normal (NN) or high NH4+ (HN) media for up to 6 h. (B) Public gene expression databases. Each database is labeled with a colored oval. (C) Principal component analysis (PCA) of 18 RNA-Seq samples from “Longjing 43.” Each shape represents a different time point. (D) Pearson correlation coefficients of all samples (triplicates).
Analysis of tea polyphenols and free amino acids
Each sample (0.15 g) was extracted with 25 mL of 50% methanol at 70°C for 20 min and centrifuged at 5,000 g for 10 min. The supernatants were collected for further analysis. The concentration of total polyphenols was determined using the Folin-Ciocalteu method. The content of total amino acids was determined by ninhydrin colorimetry according to Shao et al. (2008).
RNA isolation, cDNA library construction and Illumina deep sequencing
Total RNA was extracted from the tissue using TRIzol reagent (Plant RNA Purification Reagent for plant tissue, Invitrogen), according to the manufacturer’s instructions and genomic DNA was removed using DNase I (TaKaRa). RNA quality was determined with a the 2100 Bioanalyzer (Agilent) and quantified by NanoDrop 2000. Only high-quality RNA samples (OD260/280 = 1.8∼2.2, OD260/230 ≥ 2.0, RIN ≥ 6.5, 28S:18S ≥ 1.0 > 1 μg) were used to construct the sequencing library.
An RNA-seq transcriptome library was prepared with TruSeq™ RNA sample preparation Kit from Illumina (San Diego, CA) using 1 μg of total RNA. Firstly, messenger RNA was isolated by polyA selection using oligo (dT) beads and then fragmented by fragmentation buffer. Double-stranded cDNA was synthesized using a SuperScript double-stranded cDNA synthesis kit (Invitrogen, CA) with random hexamer primers (Illumina). cDNA was subjected to end-repair, phosphorylation and “A” base addition according to the Illumina library construction protocol. Libraries were size-selected for cDNA target fragments of 300 bp on 2% low range ultra agarose followed by PCR amplification using Phusion DNA polymerase (NEB) for 15 PCR cycles. After quantifying by TBS380, the paired-end RNA-seq sequencing library was sequenced using the Illumina HiSeq XTen/NovaSeq 6000 sequencer (2 × 150 bp read length).
Mapping the RNA-seq reads
The raw paired-end reads were trimmed and quality controlled by using the SeqPrep1 and Sickle software2 with the default parameters. Clean reads were aligned to the tea plant reference genome3 with orientation mode using HISAT2 software4 (Kim et al., 2015). Mapped reads of each sample were assembled using StringTie software5 in a reference-based approach (Pertea et al., 2015).
Differential expression analysis and functional enrichment analysis
In order to identify differentially expressed genes (DEGs) between two different samples, the expression level of each transcript was calculated according to the fragments per kilobase per million reads (FPKM) method. RSEM6 (Li and Dewey, 2011) was used to quantify gene abundance. differential expression analysis was performed using the DESeq2 tool (Love et al., 2014) with FDR<0.05, such that DEGs with |log2FC| ≥ 1 and FDR < 0.05 were considered to have a significantly different expression. In addition, KOBAS7 (Xie et al., 2011) and Goatools8 was used for functional enrichment analyses, including gene ontology (GO) and Kyoto encyclopedia for genes and genomes (KEGG) to identify significantly overrepresented GO terms and metabolic pathways according to the Bonferroni-corrected p-value ≤ 0.05 compared with the whole-transcriptome background. WGCNA was performed as described by Langfelder and Horvath (Langfelder and Horvath, 2008) and data of all DEGs among the different treatments analyzed. The gene co-expression network was visualized using Cytoscape software (version 3.5.1).
RNA-seq data validation using quantitative real-time PCR
Total RNA was used to synthesize the first strand cDNA using PrimeScript™ RT reagent Kit with gDNA Eraser Perfect Real Time (Takara, Japan). RT-qPCR was conducted with SYBR Green reagents (Takara, Japan) using an ABI StepOne Plus real-time PCR machine (Applied Biosystems). The PCR program was as follows: 94°C for 5 min, 40 cycles of 30 s at 94°C, 30 s at 60°C and a final melting curve at 65−95°C. GADPH was used as internal reference gene to quantify cDNA. The threshold cycle (Ct) values of the PCR were averaged and the relative transcript levels were quantified using the 2–△△ Ct method. All primers used for the RT-qPCR are listed in Supplementary Table 1.
Statistical analysis
One-way ANOVA (SPSS 25.0, SPSS Inc., Chicago, IL, United States) and Bonferroni tests (p < 0.05) was used to detect significant differences. All data are presented as means ± SD.
Results and discussion
Experimental design and RNA sequence analysis
Six time points and three biological replicates per condition were used to construct RNA-seq libraries for analysis of the effects of NH4+ status on gene expression in tea shoots. Quality check were performed to remove reads containing adapter, ploy-N and with low quality from the raw data, leaving an average number of clean reads per library above 6.13 Gb and a total of 130.59 Gb of clean reads. Approximately 91.04–92.25% of the clean reads were successfully mapped to the Camellia sinensis genome and 84.50–85.01% of the clean reads matched unique genomic locations. Each of the transcriptomes had 44.40–45.41% GC content. The values of Q30 were between 93.52 and 94.42% (Supplementary Table 2). These results suggested that the RNA sequencing data used in the present study were highly reliable for de novo assembly and expression analysis. The BLAST tool was used to assess sequence similarity against five public databases: GO, KEGG, NR, Swiss Prot, and Pfam to assign potential functions to the assembled unigenes (Figure 1B).
The results of PCA analysis are shown in Figure 1C. PC1 accounted for 36.98% of the total variance, with clearly separated samples according to the duration of nitrogen deficiency. PC2 accounted for 11.15% of the total variance with samples separated according to different NH4+ concentrations, such that the samples with a high concentration showed higher PC2 values (Figure 1C). Three biological replicates of each sample were clustered together, indicating high data reproducibility. Overall, the PCA analysis results showed that both the time and concentration of nitrogen supply could affect the gene expression patterns of tea plants. In addition, development status also affected gene expression. Pearson correlation coefficients of the FPKM distribution among biological replicates of all treatments ranged from 0.958 to 0.997 (Figure 1D), indicating a high reproducibility of the sequencing data.
Analysis of responses to short- and long-term NH4+ starvation
Tea polyphenols and free amino acids are the two most important components of tea plants and are affected by nitrogen supply. The content of tea polyphenols (TP) increased with time, being higher after 7 days. TP content was higher after long-term nitrogen deficiency (L_UN) than after short-term nitrogen deficiency (S_UN) (Figure 2A). Development status had great influence on the content of tea polyphenols and free amino acids. DEGs involved in plant growth were eliminated to enhance the accuracy of experimental results. There were 397 DEGs in short-term nitrogen starvation plants compared with CK1, and most of them (273 genes) were downregulated (Figure 2B). After long-term nitrogen starvation, 172 DEGs were identified, including 92 upregulated and 80 downregulated. Thus, tea plants have a more obvious response to short-term NH4+ deficiency. The number of DEGs after long-term NH4+ deficiency may have been fewer because gene expression tended to stabilize after a long period of NH4+ deprivation.
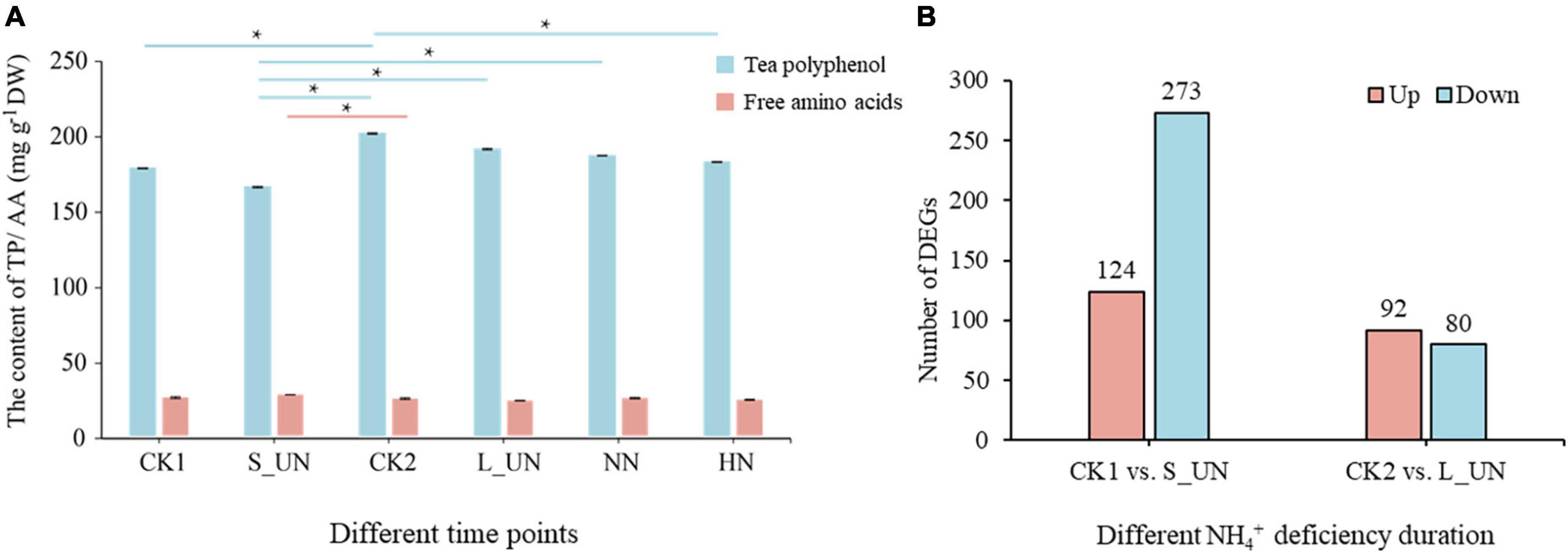
Figure 2. An overview of different periods of NH4+ treatments. (A) The content of tea polyphenols and free amino acids in tea plants at different time points. (B) Differentially expressed genes between different NH4+ deficiency durations (S_UN vs. CK1 and L_UN vs.CK2). * indicates a significant difference (p < 0.05).
A total of 14 upregulated and 36 downregulated genes were common to short- and long-term nitrogen deficiency groups (Figure 3A), indicating that duration of nitrogen deficiency affected the expression of different genes. According to GO enrichment analysis, upregulated genes were involved in photosynthesis and protein-chromophore linkage in both long-term and short-term nitrogen deficiency (Figure 3B), suggesting a link between nitrogen deficiency and photosynthesis. Photosynthesis genes, including those encoding photosystem P700 chlorophyll a apoprotein A2 (psaB) (TEA002548), acetyl-CoA carboxylase transferase (chloroplast) (TEA027086) and ribulose bisphosphate carboxylases (Rubiscos) (TEA020293) were significantly upregulated by N deficiency (Supplementary Table 3). When nitrogen is scarce, photosynthesis becomes limited and the expression of genes related to the phenylpropanoid pathway (PAL, CHS, and CHI) are known to be up-regulated (Dong et al., 2019). Downregulated genes were enriched in the GO term, cell wall macromolecule catabolic (Figure 3B). Cell wall remodeling represents an important characteristic of plant growth and differentiation (Krouk et al., 2010). The results showed that nitrogen deficiency resulted in slow cell wall remodeling. Biological process in GO terms related to photosynthesis, plant hormone and amino acid metabolism was greatly affected during the early stage of the nitrogen deficiency response. The enhancement of these metabolic pathways may result from the lack of nitrogen disrupting the carbon/nitrogen balance. By contrast, regulations of genes involved in ion transport, lipid and terpenoid metabolism were affected by a long period of nitrogen deficiency. Long-term nitrogen deficiency not only affected carbon fixation in photosynthesis (Makino, 2011), but also inhibited the TCA cycle and fatty acid synthesis (Bouché and Fromm, 2004). Previous studies on Arabidopsis thaliana have shown that PII proteins that regulate nitrogen metabolism was also involved in regulating fatty acid synthesis and metabolism through interaction with the biotin carboxyl carrier protein BCCP (Baud et al., 2010). These results indicate the importance of nitrogen in the growth and metabolism of tea plants.
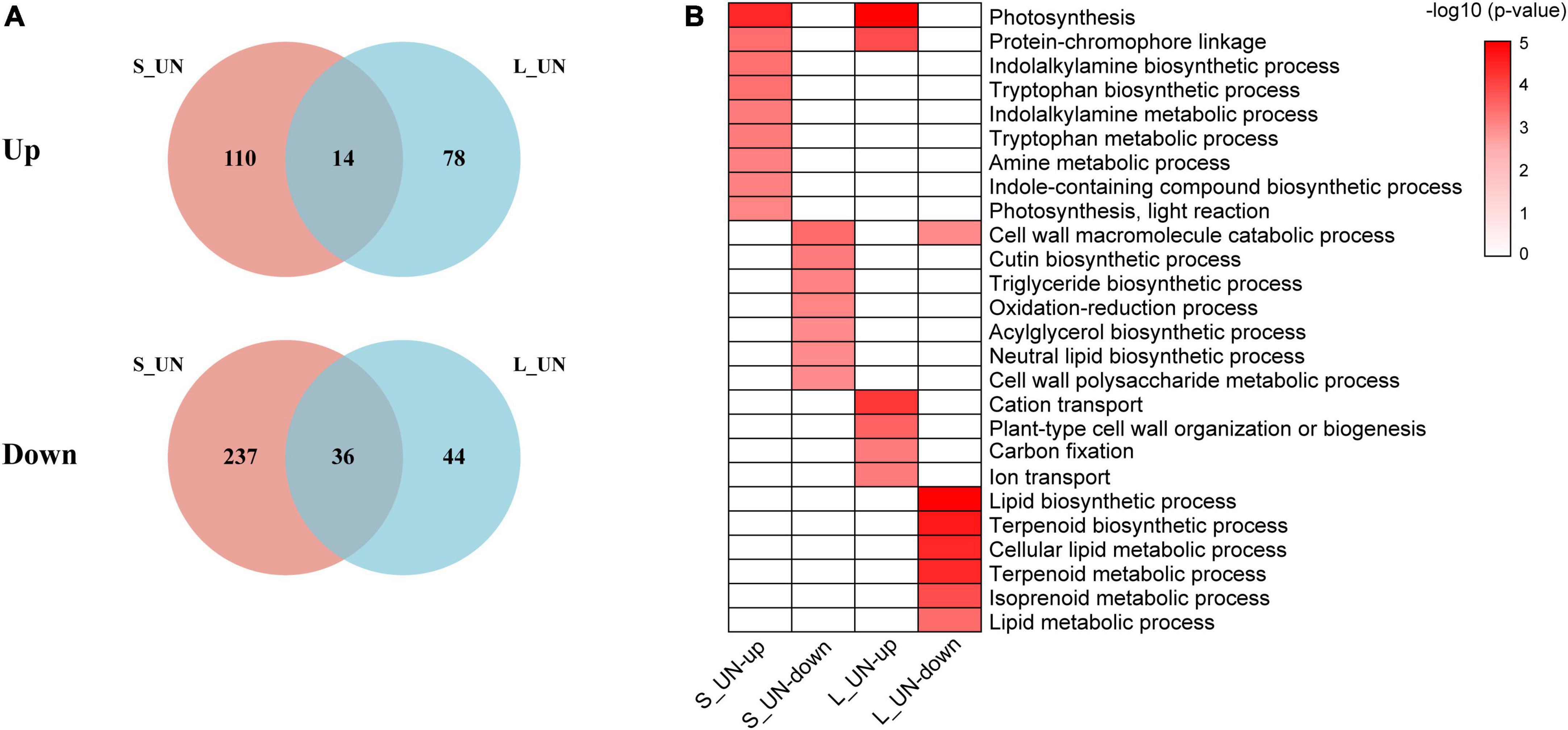
Figure 3. Venn diagram and functional analysis of DEGs of different time periods of NH4+ deficiency. (A) Venn diagram of the number of upregulated and downregulated DEGs between short-term NH4+ deficiency and long-term NH4+ deficiency. (B) Significantly enriched GO terms (biological process) of specific DEGs between different NH4+ deficiency durations (Bonferroni’s test, p < 0.05).
Analysis of normal and high NH4+ concentration recovery
Following nitrogen deficiency, different gene expression patterns with different NH4+ resupply concentrations were found. A total of 3,692 DEGs were found, including 1,268 upregulated and 2,424 downregulated at normal resupply concentration of NH4+ (Figure 4). With high NH4+ resupply concentration, there were 3,063 DEGs, with nearly equal numbers of upregulated and downregulated genes (Figure 4). NH4+ resupply enhanced the changes in gene expression in tea plants, reflecting the rapid and obvious response of tea plants to NH4+.
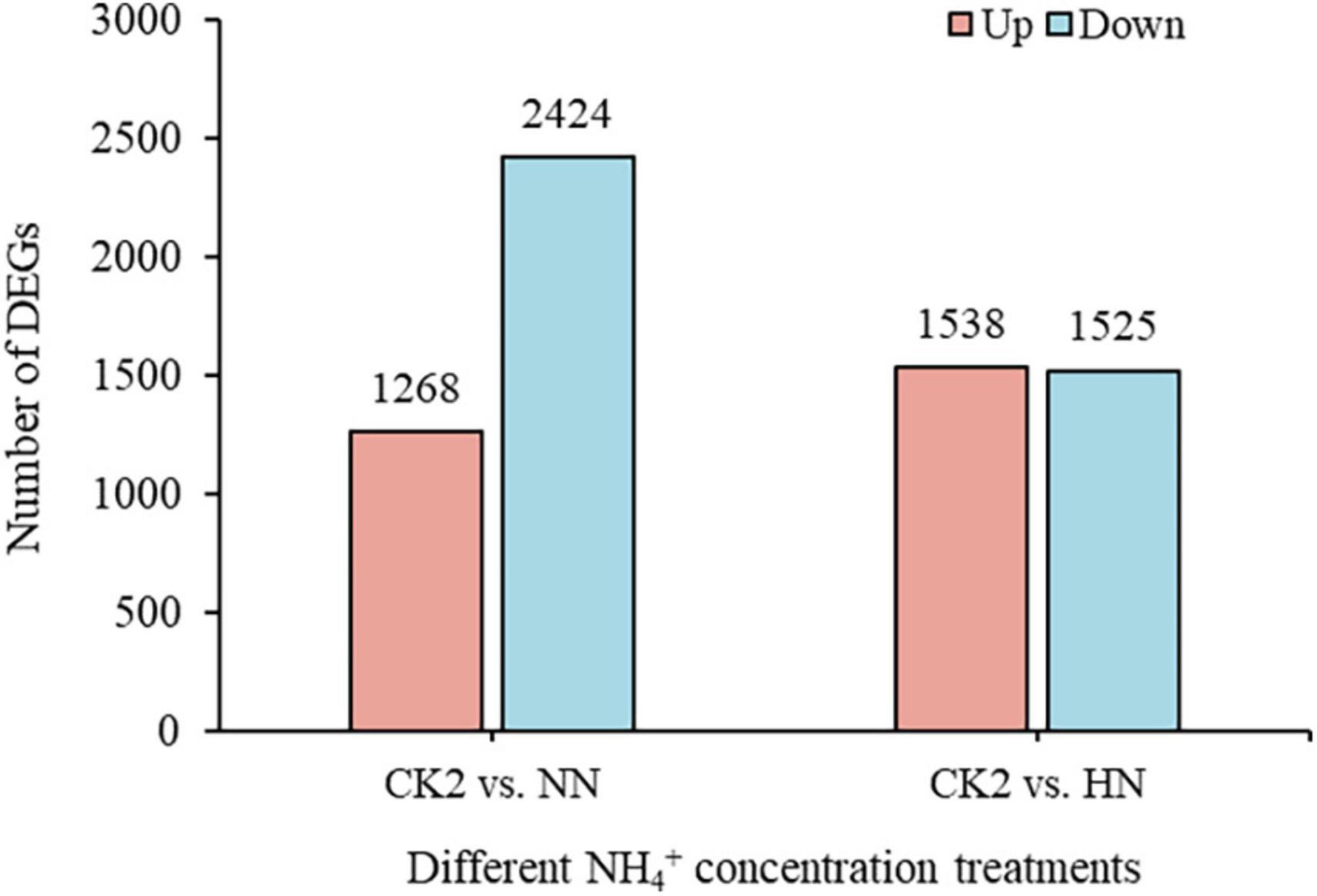
Figure 4. The number of DEGs under different NH4+ resupply concentrations (HH vs. CK2 and NN vs. CK2).
The influence of different NH4+ concentrations on the gene expression patterns of tea plants may be seen in the Venn diagram in Figure 5A. At normal NH4+ concentration, there were 1,428 unique genes (320 upregulated and 1,175 downregulated) while there were 799 unique genes (590 upregulated and 276 downregulated) with high NH4+ concentration resupply. Thus, normal and high NH4+ concentrations may have different gene expression models for tea plants. Regardless of NH4+ concentration, starch and sucrose metabolism, phenylpropanoid biosynthesis and plant-pathogen interaction were the main response pathways after NH4+ resupply (Figure 5B), suggesting an impact on metabolism of nitrogen resupply.
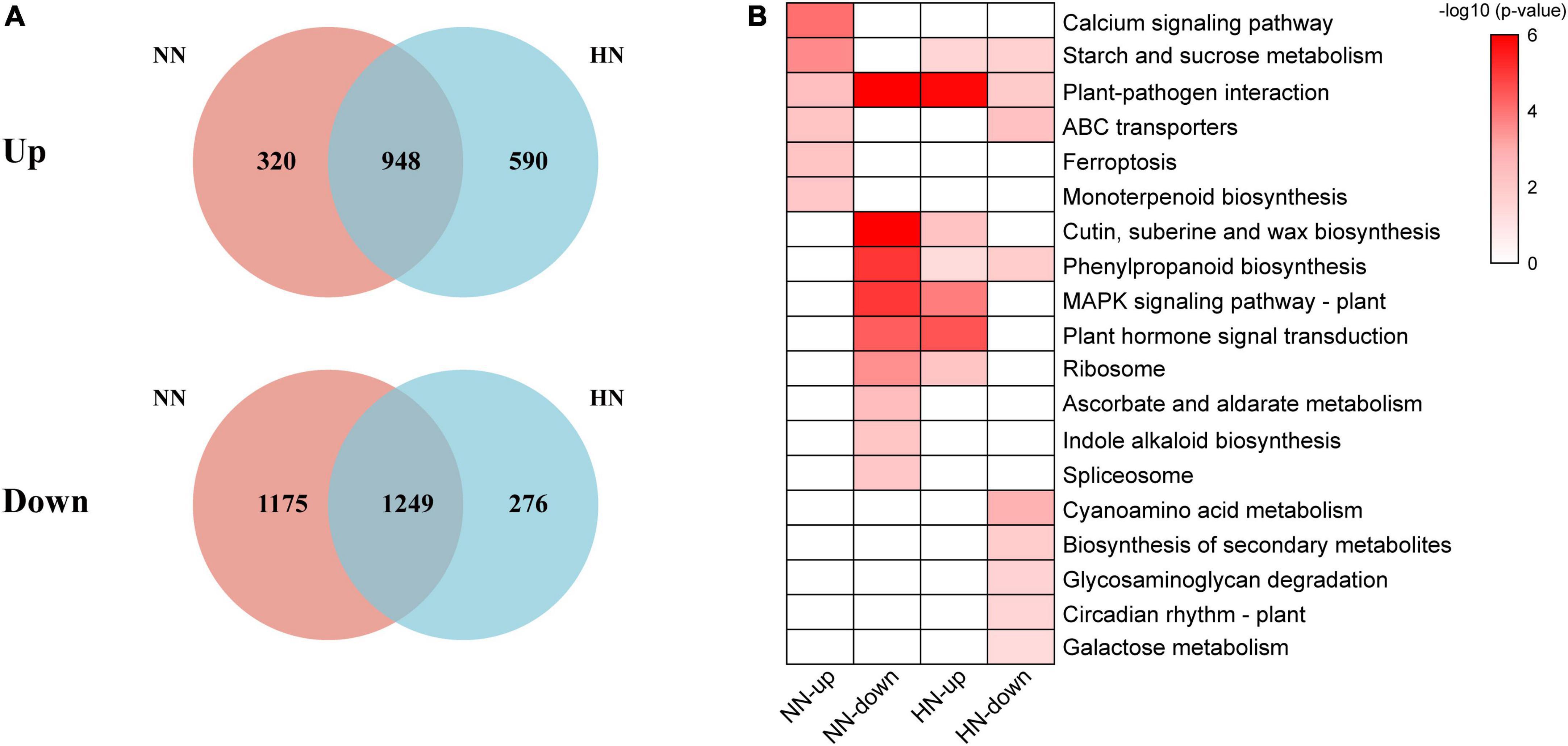
Figure 5. Venn diagram and functional analysis of DEGs under different NH4+ resupply concentrations. (A) Venn diagram of DEGs (upregulated and downregulated genes) under different NH4+ resupply concentrations. (B) Significantly enriched KEGG pathways of specific DEGs under different NH4+ resupply concentrations (Bonferroni’s test, p < 0.05).
Interestingly, the results showed that the downregulated genes with normal NH4+ resupply were similar to those upregulated genes with high NH4+ resupply (Figure 5B). In addition, DEGs were related to plant-pathogen interaction, MAPK signaling, and plant hormone signaling which may account for considerable environmental adaptability. It had also been previously found that the ammonium-specific response is related to biological stress and plant defense (Patterson et al., 2010). High NH4+ content can protect the plants from pathogens, increase the cross-tolerance to other forms of abiotic stresses and improve the quality of crops (Marino and Moran, 2019). The genes involved in the plant-pathogen interaction, including leucine-rich repeat protein, LRR receptor-like serine/threonine-protein kinase, cyclic nucleotide-gated ion channel (CNGC), disease resistance protein, ethylene-responsive transcription factor, and transcription factor WRKY were significantly highly expressed with NH4+ resupply. These results have rarely been reported in tea plants. In summary, our results revealed that the signaling role of the NH4+ molecule.
Identification of gene co-expression modules using weighted gene co-expression network analysis
WGCNA was used to comprehensively understand the gene expression patterns related to NH4+ treatments in tea plants. After removing the genes with low variability, five modules were identified, as shown in Figure 6. After generating a summary profile for each module, the turquoise module, containing 601 genes, was highly correlated with tea polyphenols, while the brown module, containing 232 genes, was correlated with free amino acids (Figure 6). Tea polyphenols and free amino acids are the two most important components of tea plants. Fewer genes were associated with free amino acids than with tea polyphenols. These results suggested that the effect of NH4+ on tea polyphenols was more significant compared with that of free amino acids, consistent with previous research (Liu et al., 2017). The top 30 associated genes according to the weighting values were selected from the turquoise and brown modules to construct a network (Supplementary Table 4). Hub genes are genes with the most connections in the network, as shown by their degree.
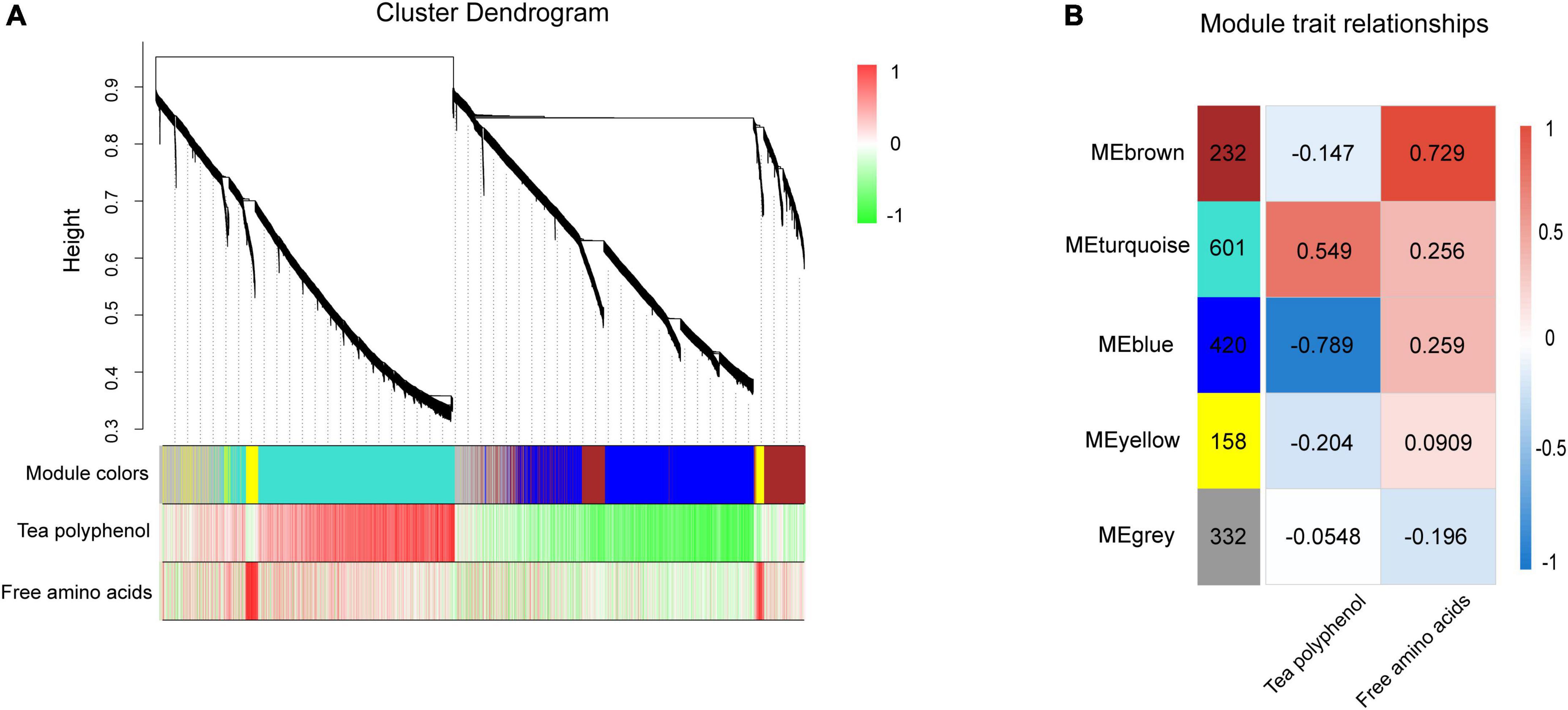
Figure 6. WGCNA of genes in tea leaves under different NH4+ treatments. (A) Gene cluster dendrogram and relationships between the contents of TP, AA and modules. According to the expression trend of transcripts, transcripts were divided into modules, where branches represent a gene, and a color represents a module. (B) Heatmap of correlation between module and trait. The vertical axis represents different modules, the left column of numbers represents the number of genes in the module and each set of data on the right represents the correlation coefficient.
In the turquoise module, 20 genes were enriched in amino acid metabolism. Both carbohydrate metabolism and biosynthesis of other secondary metabolites contained 17 genes according to KEGG analysis, as shown in Figure 7A. Photosynthesis represents the basis of plant metabolism and energy source and influences tea polyphenols formation. Carbonic anhydrase (CA) (TEA013076) is related to carbon utilization and was one of the hub genes in photosynthesis. Phytochrome B (TEA031363) was an important gene. Tea polyphenols assist the resistance of plants to damage by external factors, such as UV-B, low temperature, and drought (Zoratti et al., 2014). Genes encoding for the REF/SRPP-like protein (TEA001749), E3 ubiquitin ligase (TEA022683) and carboxylesterase (CXE) (TEA001625) were involved in plant stress resistance (Tong et al., 2017; Cao et al., 2019; Choi et al., 2021). Glutathione S-transferase (GST) (TEA006546) was also a hub gene and involved in normal metabolism of plant secondary products, such as flavonoids and cinnamic acid (Marrs, 1996). Other hub genes identified included glutathione synthetase (GS) (TEA017688) and altered xyloglucan (AXY) (TEA016707) (Figure 7B). These results suggest that nitrogen deficiency may be an abiotic stress that produces reactive oxygen species (ROS). The accumulation of tea polyphenols with high antioxidant activity may contribute to ROS scavenging (Kovacik et al., 2014). Although these genes are not directly involved in the formation of tea polyphenols, they are related to tea plant growth and metabolism, via processes such as photosynthesis, plant stress and carbon and nitrogen balance (Margaria et al., 2014; Wang et al., 2021).
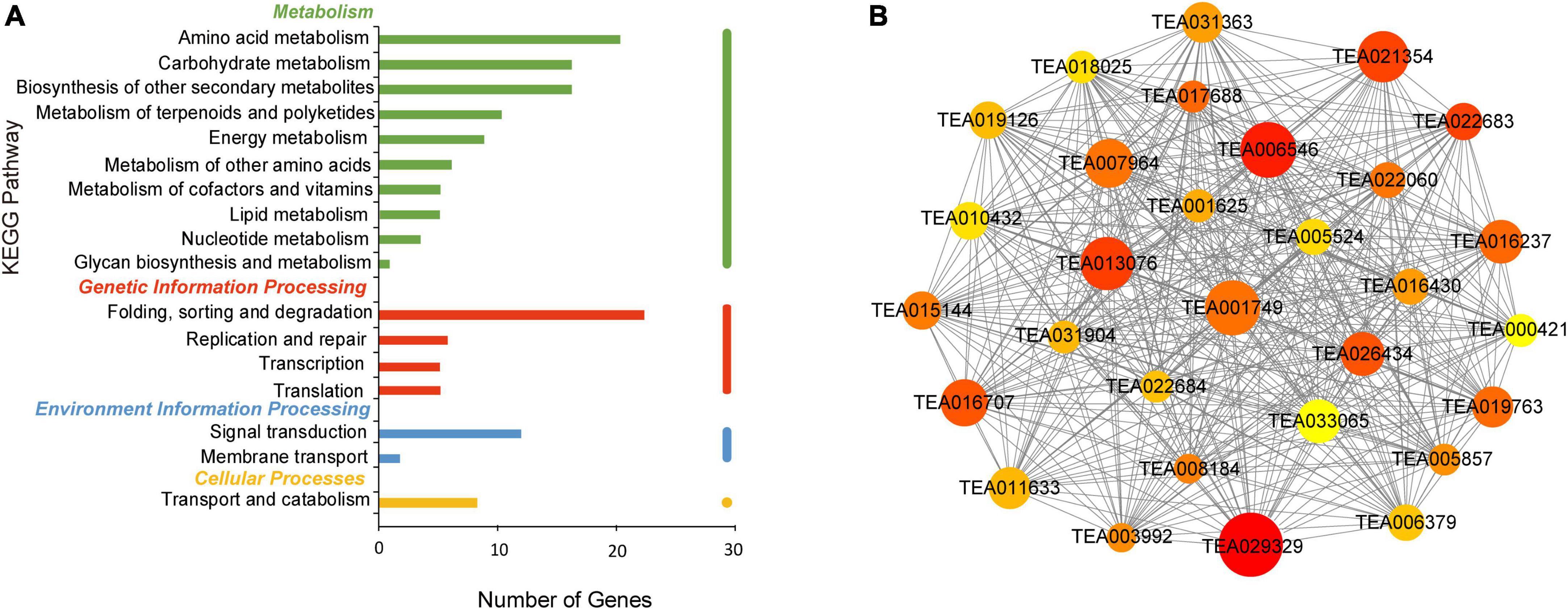
Figure 7. KEGG pathway analysis and network diagram associated with the turquoise module. (A) KEGG pathway annotation of genes in the turquoise module. The genes with higher weight are shown in red, and the size of the node represents the number of genes associated with the node. (B) Gene networks of hub genes of the turquoise module.
In the brown module, enriched genes were involved in carbohydrate and lipid metabolism and biosynthesis of other secondary metabolites (Figure 8A). The co-expression network is shown in Figure 8B. Cytochrome P450 86A22 (TEA020004) was found to be a hub gene and is a key fatty acid acyl coenzyme essential for the synthesis of esters in petunia (Han et al., 2010). Hub genes included 3-ketoacyl-CoA synthase (KCS) (TEA005835), a rate-limiting enzyme involved in the elongation of very long fatty acid chains (Yang et al., 2021). In rice, the germin-like protein regulates plant height and disease resistance (Banerjee and Maiti, 2010). GDSL esterases/lipases (TEA032710) have also been identified as hub genes and are mainly involved in the regulation of plant development, synthesis of secondary metabolites and the defense response (Chepyshko et al., 2012). Moreover, pathogenesis related protein and germin-like protein are thought to be involved in the response to abiotic stress in plants (Kapoor et al., 2019). In addition, laccase (LAC) (TEA004738) is a hub gene related to lignin synthesis, disease resistance and pigment synthesis (Li and Steffens, 2002; Ranocha et al., 2002; Pourcel et al., 2005). These results demonstrated the complex network system connecting synthesis of free amino acids with many pathways in tea plants.
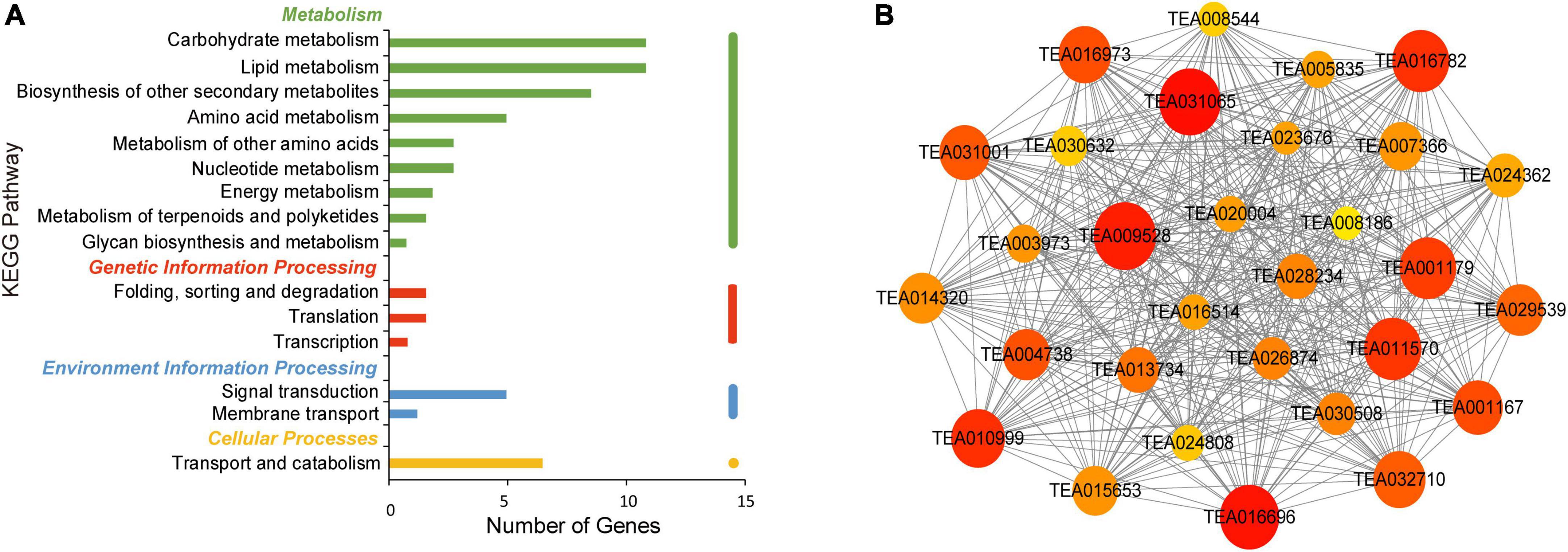
Figure 8. KEGG pathway analysis and network diagram associated with the brown module. (A) KEGG pathway annotation of genes in the brown module. The genes with higher weight are shown in red, and the size of the node represents the number of genes associated with the node. (B) Gene networks of hub genes of the brown module.
Validation of the differentially expressed genes by RT-qPCR
Twenty genes from different expression profiles were chosen for RT-qPCR analysis to validate the quality of the RNA-Seq data. Genes belonged to different metabolic pathways, including photosynthesis (photosystem II, chlorophyllase 1), polyphenol synthesis [leucoanthocyanidin reductase (LAR), phalcone synthase (CHS), flavone synthase II], amino acid synthesis (terpene synthase), anti-resistant genes (pathogenesis-related protein, disease resistance protein) and some transporters and transcription factors. The expression value of RT-qPCR was positively correlated with log2 fold change of RNA sequencing results (R2 = 0.8338), indicating the reliability of our transcriptional data (Figure 9). The expression levels of sugar transporter (TEA027183), disease resistance protein (TEA023317) and flavone synthase (TEA012257) genes were increased under high concentration of NH4+. High NH4+ concentrations, therefore, had a stress effect on tea plants. In summary, tea plants prefer NH4+ within a defined concentration range (Ruan et al., 2007).
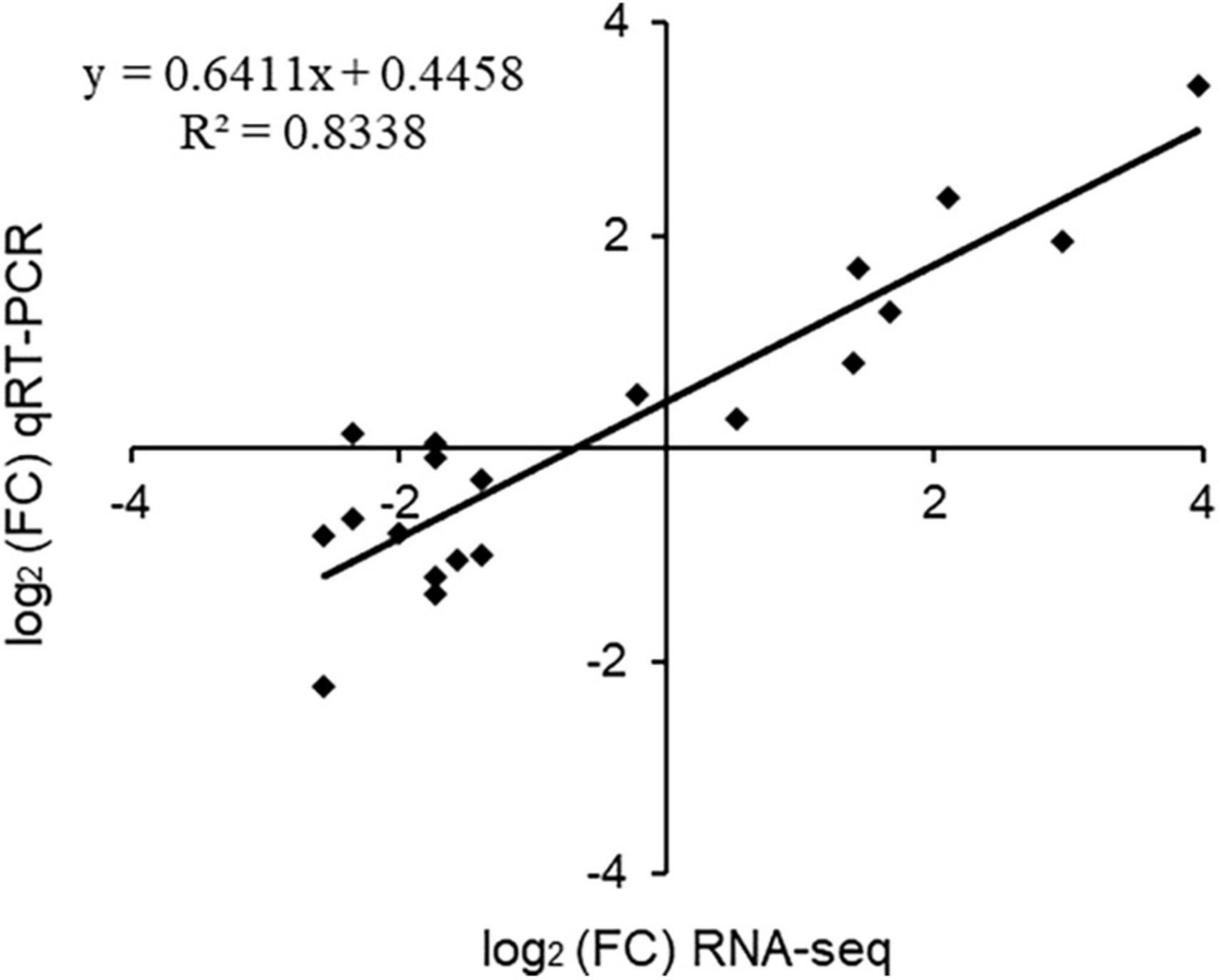
Figure 9. Scatterplot of RNA-seq transcript abundance fold change values in RNA-Seq (on the x axis) against RT-PCR expression values (on y axis) for the 20 selected genes by RT-qPCR. The GADPH gene (XM_002263109) was used as the reference gene.
Data availability statement
The data presented in this study are deposited in the NCBI repository, accession number PRJNA844353 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA844353).
Author contributions
YW and J-XO: data curation. YW, Y-MX, and D-MF: formal analysis. X-CW and X-QZ: funding acquisition, writing—review, and editing. X-CW: project administration. YW: resources and writing—original draft. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Science and Technology Project of Zhejiang Province (2022C02075) and the China Agriculture Research System of MOF and MARA (CARS-19).
Acknowledgments
We appreciate the support of the Science and Technology Project of Zhejiang Province, China Agriculture Research System (Tea).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.963269/full#supplementary-material
Supplementary Table 4 | Top 30 genes in WGCNA modules.
Footnotes
- ^ https://github.com/jstjohn/SeqPrep
- ^ https://github.com/najoshi/sickle
- ^ http://tpia.teaplant.org/index.html
- ^ http://ccb.jhu.edu/software/hisat2/index.shtml
- ^ https://ccb.jhu.edu/software/stringtie/
- ^ http://deweylab.biostat.wisc.edu/rsem/
- ^ http://kobas.cbi.pku.edu.cn/home.do
- ^ https://github.com/tanghaibao/Goatools
References
Banerjee, J., and Maiti, M. K. (2010). Functional role of rice germin-like protein1 in regulation of plant height and disease resistance. Biochem. Biophys. Res. Commun. 394, 178–183. doi: 10.1016/j.bbrc.2010.02.142
Baud, S., Feria Bourrellier, A. B., Azzopardi, M., Berger, A., Dechorgnat, J., Daniel-Vedele, F., et al. (2010). PII is induced by WRINKLED1 and fine-tunes fatty acid composition in seeds of Arabidopsis thaliana. Plant J. 64, 291–303. doi: 10.1111/j.1365-313X.2010.04332.x
Bouché, N., and Fromm, H. (2004). GABA in plants: Just a metabolite? Trends Plant Sci. 9, 110–115. doi: 10.1016/j.tplants.2004.01.006
Britto, D. T., and Kronzucker, H. J. (2002). NH4+ toxicity in higher plants: A critical review. J. Plant Physiol. 159, 567–584. doi: 10.1078/0176-1617-0774
Cao, X., Duan, W., Wei, C., Chen, K., Grierson, D., and Zhang, B. (2019). Genome-wide identification and functional analysis of carboxylesterase and methylesterase gene families in peach (Prunus persica L. Batsch). Front. Plant Sci. 10:1511. doi: 10.3389/fpls.2019.01511
Castro Marin, I., Loef, I., Bartetzko, L., Searle, I., Coupland, G., Stitt, M., et al. (2011). Nitrate regulates floral induction in Arabidopsis, acting independently of light, gibberellin and autonomous pathways. Planta 233, 539–552. doi: 10.1007/s00425-010-1316-5
Chepyshko, H., Lai, C. P., Huang, L. M., Liu, J. H., and Shaw, J. F. (2012). Multifunctionality and diversity of GDSL esterase/lipase gene family in rice (Oryza sativa L. japonica) genome: New insights from bioinformatics analysis. BMC Genomics 13:309. doi: 10.1186/1471-2164-13-309
Choi, J., Lee, W., An, G., and Kim, S. R. (2021). OsCBE1, a substrate receptor of cullin4-based E3 ubiquitin ligase, functions as a regulator of abiotic stress response and productivity in rice. Int. J. Mol. Sci. 22:2487. doi: 10.3390/ijms22052487
Coleto, I., de la Pena, M., Rodriguez-Escalante, J., Bejarano, I., Glauser, G., Aparicio-Tejo, P. M., et al. (2017). Leaves play a central role in the adaptation of nitrogen and sulfur metabolism to ammonium nutrition in oilseed rape (Brassica napus). BMC Plant Biol. 17:157. doi: 10.1186/s12870-017-1100-9
Dong, F., Hu, J. H., Shi, Y. Z., Liu, M. Y., Zhang, Q. F., and Ruan, J. Y. (2019). Effects of nitrogen supply on flavonol glycoside biosynthesis and accumulation in tea leaves (Camellia sinensis). Plant Physiol. Biochem. 138, 48–57. doi: 10.1016/j.plaphy.2019.02.017
Esteban, R., Ariz, I., Cruz, C., and Moran, J. F. (2016). Review: Mechanisms of ammonium toxicity and the quest for tolerance. Plant Sci. 248, 92–101. doi: 10.1016/j.plantsci.2016.04.008
Fan, K., Fan, D. M., Ding, Z. T., Su, Y. H., and Wang, X. C. (2015). Cs-miR156 is involved in the nitrogen form regulation of catechins accumulation in tea plant (Camellia sinensis L.). Plant Physiol. Biochem. 97, 350–360. doi: 10.1016/j.plaphy.2015.10.026
Han, J., Clement, J. M., Li, J., King, A., Ng, S., and Jaworski, J. G. (2010). The cytochrome P450 CYP86A22 is a fatty acyl-CoA omega-hydroxylase essential for Estolide synthesis in the stigma of Petunia hybrida. J. Biol. Chem. 285, 3986–3996. doi: 10.1074/jbc.M109.050765
Kapoor, R., Kumar, G., Arya, P., Jaswal, R., Jain, P., Singh, K., et al. (2019). Genome-wide analysis and expression profiling of rice hybrid proline-rich proteins in response to biotic and abiotic stresses, and hormone treatment. Plants 8:343. doi: 10.3390/plants8090343
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kovacik, J., Klejdus, B., Babula, P., and Jarosova, M. (2014). Variation of antioxidants and secondary metabolites in nitrogen-deficient barley plants. J. Plant Physiol. 171, 260–268. doi: 10.1016/j.jplph.2013.08.004
Krouk, G., Mirowski, P., LeCun, Y., Shasha, D. E., and Coruzzi, G. M. (2010). Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 11:R123. doi: 10.1186/gb-2010-11-12-r123
Langfelder, P., and Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Li, B., and Dewey, C. N. (2011). RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, H., Hu, B., and Chu, C. (2017). Nitrogen use efficiency in crops: Lessons from Arabidopsis and rice. J. Exp. Bot. 68, 2477–2488. doi: 10.1093/jxb/erx101
Li, L., and Steffens, J. C. (2002). Overexpression of polyphenol oxidase in transgenic tomato plants results in enhanced bacterial disease resistance. Planta 215, 239–247. doi: 10.1007/s00425-002-0750-4
Li, Q., Li, B. H., Kronzucker, H. J., and Shi, W. M. (2010). Root growth inhibition by NH4+ in Arabidopsis is mediated by the root tip and is linked to NH4+ efflux and GMPase activity. Plant Cell Environ. 33, 1529–1542. doi: 10.1111/j.1365-3040.2010.02162.x
Lima, J. E., Kojima, S., Takahashi, H., and von Wirén, N. (2010). Ammonium triggers lateral root branching in Arabidopsis in an AMMONIUM TRANSPORTER1;3-dependent manner. Plant Cell 22, 3621–3633. doi: 10.1105/tpc.110.076216
Liu, M. Y., Burgos, A., Zhang, Q. F., Tang, D. D., Shi, Y. Z., Ma, L. F., et al. (2017). Analyses of transcriptome profiles and selected metabolites unravel the metabolic response to NH4+ and NO3– as signaling molecules in tea plant (Camellia sinensis L.). Sci. Hortic. 218, 293–303. doi: 10.1016/j.scienta.2017.02.036
Liu, Y., and von Wirén, N. (2017). Ammonium as a signal for physiological and morphological responses in plants. J. Exp. Bot. 68, 2581–2592. doi: 10.1093/jxb/erx086
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Makino, A. (2011). Photosynthesis, grain yield, and nitrogen utilization in rice and wheat. Plant Physiol. 155, 125–129. doi: 10.1104/pp.110.165076
Margaria, P., Ferrandino, A., Caciagli, P., Kedrina, O., Schubert, A., and Palmano, S. (2014). Metabolic and transcript analysis of the flavonoid pathway in diseased and recovered Nebbiolo and Barbera grapevines (Vitis vinifera L.) following infection by Flavescence doree phytoplasma. Plant Cell Environ. 37, 2183–2200. doi: 10.1111/pce.12332
Marino, D., and Moran, J. F. (2019). Can ammonium stress be positive for plant performance? Front. Plant Sci. 10:1103. doi: 10.3389/fpls.2019.01103
Marrs, K. A. (1996). The functions and regulation of glutathione S-transferases in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47, 127–158. doi: 10.1146/annurev.arplant.47.1.127
Menz, J., Li, Z., Schulze, W. X., and Ludewig, U. (2016). Early nitrogen-deprivation responses in Arabidopsis roots reveal distinct differences on transcriptome and (phospho-) proteome levels between nitrate and ammonium nutrition. Plant J. 88, 717–734. doi: 10.1111/tpj.13272
Patterson, K., Cakmak, T., Cooper, A., Lager, I., Rasmusson, A. G., and Escobar, M. A. (2010). Distinct signalling pathways and transcriptome response signatures differentiate ammonium- and nitrate-supplied plants. Plant Cell Environ. 33, 1486–1501. doi: 10.1111/j.1365-3040.2010.02158.x
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). Stringtie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Pourcel, L., Routaboul, J. M., Kerhoas, L., Caboche, M., Lepiniec, L., and Debeaujon, I. (2005). TRANSPARENT TESAT10 encodes a laccase-like enzyme involved in oxidative polymerization of flavonoids in Arabidopsis seed coat. Plant Cell 17, 2966–2980. doi: 10.1105/tpc.105.035154
Ranocha, P., Chabannes, M., Chamayou, S., Danoun, S., Jauneau, A., Boudet, A. M., et al. (2002). Laccase down-regulation causes alterations in phenolic metabolism and cell wall structure in poplar. Plant Physiol. 129, 145–155. doi: 10.1104/pp.010988
Ren, Q., Zhou, Y., and Zhou, X. (2020). Combined transcriptome and proteome analysis of masson pine (Pinus massoniana Lamb.) seedling root in response to nitrate and ammonium supplementations. Int. J. Mol. Sci. 21:7548. doi: 10.3390/ijms21207548
Ruan, J., Haerdter, R., and Gerendas, J. (2010). Impact of nitrogen supply on carbon/nitrogen allocation: A case study on amino acids and catechins in green tea [Camellia sinensis (L.) O. Kuntze] plants. Plant Biol. 12, 724–734. doi: 10.1111/j.1438-8677.2009.00288.x
Ruan, J. Y., Gerendas, J., Hardter, R., and Sattelmacher, B. (2007). Effect of root zone pH and form and concentration of nitrogen on accumulation of quality-related components in green tea. J. Sci. Food Agric. 87, 1505–1516. doi: 10.1002/jsfa.2875
Shao, J. L., Dong, B. S., Liu, H. C., and Shu, J. H. (2008). Determination of total free-amino acid in tea by Nihydrin colorimetry. China Food Addit. 2, 162–165. doi: 10.3969/j.issn.1006-2513.2008.02.039
Tang, D., Liu, M. Y., Zhang, Q., Ma, L., Shi, Y., and Ruan, J. (2020). Preferential assimilation of NH4+ over NO3– in tea plant associated with genes involved in nitrogen transportation, utilization and catechins biosynthesis. Plant Sci. 291:110369. doi: 10.1016/j.plantsci.2019.110369
Tong, Z., Wang, D., Sun, Y., Yang, Q., Meng, X., Wang, L., et al. (2017). Comparative proteomics of Rubber Latex revealed multiple protein species of REF/SRPP family respond diversely to ethylene stimulation among different rubber tree clones. Int. J. Mol. Sci. 18:958. doi: 10.3390/ijms18050958
Vanacker, H., Sandalio, L., Jimenez, A., Palma, J. M., Corpas, F. J., Meseguer, V., et al. (2006). Roles for redox regulation in leaf senescence of pea plants grown on different sources of nitrogen nutrition. J. Exp. Bot. 57, 1735–1745. doi: 10.1093/jxb/erl012
Vega, A., Canessa, P., Hoppe, G., Retamal, I., Moyano, T. C., Canales, J., et al. (2015). Transcriptome analysis reveals regulatory networks underlying differential susceptibility to Botrytis cinerea in response to nitrogen availability in Solanum lycopersicum. Front. Plant Sci. 6:911. doi: 10.3389/fpls.2015.00911
von Wiren, N., Lauter, F. R., Ninnemann, O., Gillissen, B., Walch-Liu, P., Engels, C., et al. (2000). Differential regulation of three functional ammonium transporter genes by nitrogen in root hairs and by light in leaves of tomato. Plant J. 21, 167–175. doi: 10.1046/j.1365-313x.2000.00665.x
Wang, Y., Wang, Y. M., Lu, Y. T., Qiu, Q. L., Fan, D. M., Wang, X. C., et al. (2021). Influence of different nitrogen sources on carbon and nitrogen metabolism and gene expression in tea plants (Camellia sinensis L.). Plant Physiol. Biochem. 167, 561–566. doi: 10.1016/j.plaphy.2021.08.034
Wang, Y., Xuan, Y. M., Wang, S. M., Fan, D. M., Wang, X. C., and Zheng, X. Q. (2022). Genome-wide identification, characterization, and expression analysis of the ammonium transporter gene family in tea plants (Camellia sinensis L.). Physiol. Plant. 174:e13646. doi: 10.1111/ppl.13646
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Yang, T., Li, H., Tai, Y., Dong, C., Cheng, X., Xia, E., et al. (2020). Transcriptional regulation of amino acid metabolism in response to nitrogen deficiency and nitrogen forms in tea plant root (Camellia sinensis L.). Sci. Rep. 10:6868. doi: 10.1038/s41598-020-63835-6
Yang, T., Li, Y., Liu, Y., He, L., Liu, A., Wen, J., et al. (2021). The 3-ketoacyl-CoA synthase WFL is involved in lateral organ development and cuticular wax synthesis in Medicago truncatula. Plant Mol. Biol. 105, 193–204. doi: 10.1007/s11103-020-01080-1
Yang, Y. Y., Li, X. H., Ratcliffe, R. G., and Ruan, J. Y. (2013). Characterization of ammonium and nitrate uptake and assimilation in roots of tea plants. Russ. J. Plant Physiol. 60, 91–99. doi: 10.1134/S1021443712060180
Yang, Y. Y., Wang, F., Wan, Q., and Ruan, J. Y. (2018). Transcriptome analysis using RNA-Seq revealed the effects of nitrogen form on major secondary metabolite biosynthesis in tea (Camellia sinensis) plants. Acta Physiol. Plant. 40:127. doi: 10.1007/s11738-018-2701-0
Zhang, F., Liu, Y., Wang, L., Bai, P. X., Ruan, L., Zhang, C. C., et al. (2018). Molecular cloning and expression analysis of ammonium transporters in tea plants (Camellia sinensis (L.) O. Kuntze) under different nitrogen treatments. Gene 658, 136–145. doi: 10.1016/j.gene.2018.03.024
Zhang, F., Wang, L., Bai, P., Wei, K., Zhang, Y., Ruan, L., et al. (2020). Identification of regulatory networks and hub genes controlling nitrogen uptake in tea plants [Camellia sinensis (L.) O. Kuntze]. J. Agric. Food Chem. 68, 2445–2456. doi: 10.1021/acs.jafc.9b06427
Keywords: transcriptome, ammonium, tea plants, RNA-sequencing, nitrogen, WGCNA
Citation: Wang Y, Ouyang J-X, Fan D-M, Wang S-M, Xuan Y-M, Wang X-C and Zheng X-Q (2022) Transcriptome analysis of tea (Camellia sinensis) leaves in response to ammonium starvation and recovery. Front. Plant Sci. 13:963269. doi: 10.3389/fpls.2022.963269
Received: 07 June 2022; Accepted: 04 August 2022;
Published: 30 August 2022.
Edited by:
Zhaoliang Zhang, Anhui Agriculture University, ChinaReviewed by:
Mei-Ya Liu, Tea Research Institute (CAAS), ChinaLu Wang, Tea Research Institute (CAAS), China
Copyright © 2022 Wang, Ouyang, Fan, Wang, Xuan, Wang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Chang Wang, eGN3YW5nQHpqdS5lZHUuY24=; Xin-Qiang Zheng, eHF6aGVuZ0B6anUuZWR1LmNu