 Barinder Kaur1†
Barinder Kaur1† Karmvir Singh Garcha1†
Karmvir Singh Garcha1† Dharminder Bhatia2
Dharminder Bhatia2 Jiffinvir Singh Khosa1
Jiffinvir Singh Khosa1 Madhu Sharma1
Madhu Sharma1 Amandeep Mittal3
Amandeep Mittal3 Neha Verma1
Neha Verma1 Ajmer Singh Dhatt4*
Ajmer Singh Dhatt4*- 1Department of Vegetable Science, Punjab Agricultural University, Ludhiana, Punjab, India
- 2Department of Plant Breeding and Genetics, Punjab Agricultural University, Ludhiana, Punjab, India
- 3School of Agricultural Biotechnology, Punjab Agricultural University, Ludhiana, Punjab, India
- 4Directorate of Research, Punjab Agricultural University, Ludhiana, Punjab, India
The hull-less pumpkin (Cucurbita pepo) seed does not require de-hulling before use for human consumption, as a result highly preferred by the oil, nut, and baking industries. In hull-less seeds, a single recessive gene is responsible for the absence of outer thick seed coat layers; however, the genomic region and gene(s) controlling the trait are unclear to date. In this study, four crosses attempted to derive F2 and backcross populations confirmed the single recessive gene inheritance of hull-less seed trait in populations adapted to the sub-tropical climate. The candidate genomic region for hull-less seed trait was identified through the BSA-QTLseq approach using bulks of F2:3 progenies from a cross of HP111 (hulled) and HLP36 (hull-less). A novel genomic region on chromosome 12 ranging from 1.80 to 3.86 Mb was associated with the hull-less seed trait. The re-sequencing data identified a total of 396 SNPs within this region and eight were successfully converted into polymorphic KASP assays. The genotyping of segregating F2 (n = 160) with polymorphic KASP assays resulted in a 40.3 cM partial linkage map and identified Cp_3430407 (10 cM) and Cp_3498687 (16.1 cM) as flanking markers for hull-less locus (Cphl-1). These flanking markers correspond to the 68.28 kb region in the reference genome, and the marker, Cp_3430407 successfully predicted the genotype in 93.33% of the C. pepo hull-less germplasm lines, thus can be used for marker-assisted selection in parents polymorphic for the hull-less seed trait. The Cphl-1-linked genomic region (2.06 Mb) encompasses a total of 182 genes, including secondary cell wall and lignin biosynthesis-related transcriptional factors viz., “NAC” (Cp4.1LG12g04350) and “MYB” (Cp4.1LG12g03120). These genes were differentially expressed in the seeds of hulled and hull-less genotypes, and therefore could be the potential candidate genes governing the hull-less seed trait in pumpkin.
Introduction
Pumpkin (Cucurbita pepo L.) is an important vegetable crop that belongs to the Cucurbitaceae family, which is grown worldwide for its flesh and seeds. Pumpkin seeds are extensively used in the bakery and oil industry due to their high nutritional value (Jafari et al., 2012; De Lamo and Gómez, 2018). Proteins, unsaturated fatty acids (linoleic, oleic, and palmitic acids), minerals (potassium, phosphorous, and zinc), and vitamins (Vitamin E) are the key nutritionally important constituents found in the pumpkin seeds (Dotto and Chacha, 2020). The consumption of pumpkin seeds has positive effects on human health due to their anti-cancer, anti-diabetic, and anti-inflammatory properties (Fruhwirth and Hermetter, 2007; Stevenson et al., 2007; Yoshinari et al., 2009). However, the presence of a thick and leathery seed coat (hull) due to the strong lignification of testa layers requires de-hulling prior to use for human consumption, which proves to be one of the main hindrances in the wide acceptability of the pumpkin seeds (Lelley et al., 2009). A spontaneous mutant called Styrian (hull-less) (C. pepo subsp. pepo var. styriaca) was identified in Austria that lacks the hard seed coat, and as a result, does not require the expensive decortication process (Lelley et al., 2009). Histological characterization of hull-less pumpkin seeds reveals that seed coat development is similar in both hulled and hull-less seeds till 10 days post-anthesis, but after that extent of lignification decreases in hull-less seeds (Stuart and Loy, 1983; Bezold et al., 2005).
Over the years, several studies investigated the genetic inheritance of hull-less seed coat in pumpkins and concluded that a single recessive gene in the homozygous condition (hh) is responsible for the lack of a hard seed coat; however, reports of some modifiers having a minor influence on testa development are also available (Zraidi et al., 2003; Lelley et al., 2009). The availability of hull-less seed trait allowed the breeders to develop pumpkin cultivars for edible seed purposes, especially in Europe and North America (Winkler, 2000). A hull-less seeded variety “Lady Godiva” (EC 664187) has been introduced by Punjab Agricultural University (PAU), Ludhiana, India, and the trait has been transferred into adapted germplasm to release the first Indian hull-less seeded variety “PAU Magaz Kadoo-1” through conventional breeding approaches (Dhatt et al., 2020). However, the conventional breeding methods are time-consuming and labor intensive, which demands dexterous efforts for the identification of hull-less trait from segregating generations and transferring them into different genetic backgrounds. Marker-assisted selection (MAS) can overcome these hurdles but requires the development of tightly linked molecular markers with hull-less seed trait (Gong et al., 2008).
The decrease in the cost of whole-genome sequencing and technological advances in genomics led to the development of a large number of single nucleotide polymorphisms (SNPs) that allow researchers to identify genomic regions governing agronomically important traits more efficiently in comparison to traditional mapping approaches (Varshney et al., 2021). In different crops, it has been demonstrated that QTL-Seq analysis is a fast and efficient approach to identify candidate genomic regions and develop tightly linked SNPs to utilize in marker-assisted selection for traits of economic interest (Takagi et al., 2013; Lu et al., 2014; Illa-Berenguer et al., 2015). QTL-Seq is based on the combination of bulk segregant analysis (BSA) and next-generation sequencing (NGS), in which two extreme phenotypes with respect to the target trait are selected from a segregating population and bulked separately. These bulks are sequenced along with their parents and further subjected to downstream bioinformatic analysis to identify candidate genomic regions (Takagi et al., 2013).
In the present investigation, we studied the genetic inheritance of hull-less seed trait in pumpkin populations adapted to the sub-tropical climate and used QTL-Seq to identify the candidate genomic region responsible for hull-less seed. The genes present in the genomic region were analyzed, and the expression pattern of secondary cell wall/ lignin biosynthesis-related genes was studied in the seeds of hulled and hull-less genotypes at different time intervals (Wyatt et al., 2015). Furthermore, we developed the Kompetitive allele-specific PCR (KASP) markers and demonstrated their potential to follow marker-assisted selection for hull-less seed trait to expedite edible seeded pumpkin breeding programmes.
Materials and methods
Genetic inheritance pattern of hull-less seed trait
Three hull-less genotypes viz., HLP36, HLP53, and HLP8 and two hulled genotypes viz., HP111 and HP112 of C. pepo adapted to the sub-tropical climate of India were used. Crosses were attempted between HP111 × HLP36, HP111 × HLP53, HP112 × HLP8, and HLP8 × HP112 to develop F1, F2, BC1P1, and BC1P2 generations. Seeds of all generations of the above-mentioned cross combinations were sown during subsequent seasons and phenotyped for hulled or hull-less seeds, according to Zraidi et al. (2003). A Chi-square test was used to evaluate the goodness of fit with different testing ratios to determine the genetic control of the hull-less seed trait.
Construction of extreme bulks for hull-less seed trait locus identification
Total genomic DNA was extracted from young leaves of two parental lines viz., HP111 (hulled) and HLP36 (hull-less), and their F2:3 population using the CTAB method (Doyle and Doyle, 1987). For Bulked Segregant Analysis, hulled and hull-less seeded F2:3 progenies developed from HP111 × HLP36 along with their parental lines were used (Figure 1). Two DNA bulks were separately constructed by mixing an equal amount of DNA extracted from 14 extremely hulled (B1) and 14 hull-less seeded (B2) F2:3 progenies. Similarly, for parents, DNA from five plants was extracted and bulked. DNA quality was evaluated by running the samples on 0.8% agarose gel and was quantified using NanoDrop 8000 (Thermo Scientific Inc., USA), and the final DNA concentration was adjusted to 100 ng/μl. Subsequently, the genomic DNA of all the four samples (P1, P2, B1, and B2) were outsourced to the NGB Diagnostics Pvt Ltd., Noida, India, for whole genome re-sequencing using the Illumina HiSeq platform.

Figure 1. Seeds of C. pepo parents and bulks used in QTL-seq analysis. (A) Hulled Seeds of HP111; (B) Hull-less seeds of HLP36; (C) F2:3 hulled (1st and 2nd row) and hull-less bulks (3rd and 4th row) of HP111 × HLP36.
QTL-seq to identify the genomic region governing hull-less seed trait
QTL-seq pipeline (QTL-seq_framework1.4.4) was used for the mapping of QTL(s) for hull-less seed trait (Takagi et al., 2013). The raw paired-end sequencing reads were filtered with Phred quality scores (q) < 30 and quality threshold value (p) < 90. Then, the clean reads of both the parents were aligned to the publically available reference genome of C. pepo cv. Zucchini (Montero-Pau et al., 2017) using BWA (Burrows-Wheeler Alignment Tool) version 0.5.9 (Li and Durbin, 2009). Alignment files were converted to SAM/BAM files, and SNPs were identified with Samtools version 0.1.8 (Li et al., 2009) and refined with the Coval software (Kosugi et al., 2013). Finally, the reference-guided parent assemblies were developed for hulled and hull-less parents by substituting the reference bases with alternative bases at the positions of confidence SNPs in the genomic sequences of C. pepo. To identify the genomic regions controlling hull-less seed trait, short reads of both the bulks were first aligned to reference guided parent assemblies, and then SNPs were called. The SNP calling filter, Coval v1.4.1 (cov), was set to a mutation index of 2, 3, and 4 with a reading depth threshold (co) of 5 and 7 to improve the accuracy. SNP-index was computed for both the bulks at each SNP position as a proportion of reads harboring the SNP that are dissimilar from the reference sequence. If all reads matched the reference, the SNP index was 0, and 1 if all reads were different than the reference allele.
Afterwards, the ΔSNP index was calculated for each position as the SNP index of one bulk was subtracted from the SNP index of the other. Sliding window analysis was conducted with 2 and 4 Mb intervals and 50 kb increment, and ΔSNP-index plots were constructed w.r.t confidence intervals (95% and 99%) under the null hypothesis of no QTL. Plots are represented using 2 Mb window size, 50 kb increment with cov 2 and co 7. Only SNPs with ΔSNP index significantly higher than 0.5 or lower than −0.5 at the 95 and/or 99% confidence level were considered as the effective SNPs for hull-less seed trait. Furthermore, the QTLseqr pipeline was also used to calculate p-values at an FDR (q) of 0.001 to identify potential QTLs associated with the hull-less seed trait (Mansfeld and Grumet, 2018). In this approach, variants were called using GATK (Genome Analysis Toolkit) version 4.1.4.1 'HaplotypeCaller' (Van der Auwera et al., 2013) and then filtered according to read depth (>5) and genotype quality (>30) with vcftools version 0.1.13. Further filtering of SNPs was performed using filterSNPs() function based on reference allele frequency = 0.10, minTotalDepth ≥ 10, maxTotalDepth ≤ 400, minSampleDepth ≥14 and genotype quality score (GQ ≥ 30).
Validation of candidate region controlling hull-less seed trait using kompetitive allele-specific PCR assays
Polymorphic SNPs were identified from the candidate genomic region for hull-less seed trait on the basis of high delta SNP index and parental polymorphism. Each SNP site was parsed to retrieve the up- and downstream sequences of 100 bp in the target region using samtools (Semagn et al., 2014) with a criterion of no other SNP and InDels in visualization via IGV software (https://software.broadinstitute.org/software/igv/). For each selected SNP, two allele-specific forward primers and one common reverse primer were designed using the Primer3 software with selection on the basis of relatively unique primer sequences in the C. pepo genome. Target SNPs were used to develop 21 KASP assays (Supplementary Table 1) to validate the candidate genomic region governing the hull-less seed trait.
PCR amplifications were performed using a Veriti 384-Well Thermal Cycler (Applied Biosystems) in a 4 μL reaction volume, comprised of 2 μL of 100 ng/μL genomic DNA, 1.946 μL 2 × low rox KASP™ master mix (LGC Genomics LLC) and 0.056 μL of primer mix. The PCR conditions were set at 95°C for 15 min, followed by touchdown PCR of 10 cycles at 95°C for 20 s, 66°C (reduced by 0.6 °C per cycle) for 25 s, and 72°C for 15 s; subsequently, 35 cycles of 10 s at 95°C, 1 min at 60°C, 15 s at 72°C, and a final extension at 72°C for 5 min. Fluorescent endpoint readings were recorded using an Infinite M200Pro plate reader (Tecan, Group Ltd.), and genotyping calls were made using KlusterCaller™ (LGC Genomics LLC). The fluorescent readings for genotyping a few individuals were ambiguous and considered as missing data that was not included in linkage map construction and marker accuracy prediction.
Linkage analysis, candidate gene identification, and functional annotation
All 21 KASP assays were used to genotype both hulled and hull-less parents along with F1 hybrid plants. The parental polymorphic KASP assays were used to genotype F2:3 progenies of hulled and hull-less seeded used in bulk construction and 160 F2 individuals derived from the HP111 × HLP36 cross. A partial genetic linkage was developed with Join Map 4.1 using the Kosambi mapping function and an LOD score of 5 (Van Ooijen, 2006). The final genetic map was drawn in the MapChart software (Voorrips, 2002).
The linked KASP assays were also tested on the diversity panel using 45 diverse hulled and hull-less genotypes of C. pepo. For the identification of candidate genes, the protein sequences of all the genes present in the QTL-seq identified region were evaluated for their homology with Arabidopsis thaliana (TAIR10) genes involved in the cell wall, cellulose, and lignin biosynthesis.
Expression analysis of genes present in the candidate genomic region and time course differential RNA seq expression between hulled and hull-less seeded genotypes
The expression of genes present in the candidate genomic region was estimated using a publically available RNA-seq dataset [Cucurbit Genomics Database (http://cucurbitgenomics.org/organism/14)] over different developmental stages of hulled and hull-less seeds [at 5, 10, 15, 20, and 40 days after pollination (DAP)] (Wyatt et al., 2015). The time course differential RNA seq expression was used for the differential expression between seeds of hulled (Sweet Reeba) and hull-less (Lady Godiva) genotypes at 15 and 20 DAP, and a total number of differentially expressed genes was calculated using inbuilt edgeR based algorithms with adjusted p-value cutoff at 0.05 and fold change cutoff at 4. Furthermore, gene ontology analysis (Biological processes) with a p-value as good or better than 0.05 was used to calculate gene ontology terms related to cell wall biosynthesis or lignin biosynthesis.
Results
Genetic inheritance pattern of hull-less seed trait in segregating populations
In our breeding programme, hull-less seed trait from the variety Lady Godiva has been transferred into local C. pepo lines adapted to the subtropical climate of North India. To understand the genetic inheritance of hull-less seed traits, two hulled and three hull-less genotypes of C. pepo were used to develop segregating populations. Seeds from all the six generations (P1, P2, F1, F2, BC1P1, and BC1P2) of four crosses were phenotyped at maturity. The F1 plants in all cross combinations developed hulled seeds indicating recessive action of the gene(s) governing hull-less seed trait. Furthermore, chi-square analysis suggested that the hull-less phenotype fit in the Mendelian ratio of 3:1 (hulled: hull-less) in F2 and 1:1 in backcross populations (Table 1). These results conclude that the hull-less seed trait is conferred by a single recessive gene in C. pepo lines adapted to sub-tropical climate.
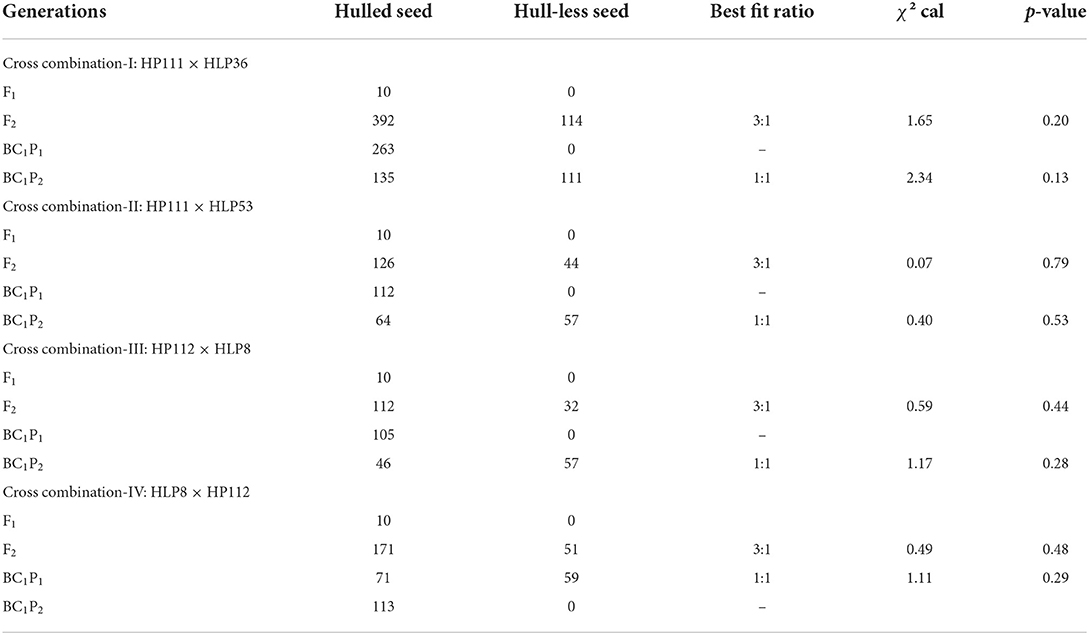
Table 1. Segregation of hulled and hull-less seed trait in different crosses.
Construction and sequencing of bulks
On the basis of the genetic inheritance pattern of the hull-less seed trait, two extreme bulks, which represent hulled seeds (B1) and hull-less seeds (B2), were constituted by pooling the DNA of 14 plants each from the F2:3 population of HP111 (P1, hulled) × HLP36 (P2, hull-less) (Figure 1). These two DNA samples along with their parental lines DNA were used to construct libraries and subjected to whole-genome resequencing. A total of 33.00, 37.84, 33.12, and 39.41 million paired-end clean reads were retained for P1, P2, B1, and B2, respectively. These trimmed reads were then mapped to the C. pepo reference genome. The parents, P1 and P2, aligned 29 and 34 million reads with a reference genome, yielding an overall alignment rate of 89.05 and 91.98%, respectively. Similarly, a total of 29 and 35 million reads of bulk B1 and B2 were aligned against the reference genome, which represents the overall alignment rate as 90.87 and 91.19%, respectively. Variant calling in these mapped reads with reference genome resulted in a total of 2,205,961 SNPs/Indels. The filtering of these variants retained 1,150,511 SNPs, which were used for the analysis. Furthermore, these SNPs were filtered in QTLseqr and resulted in 39,827 SNPs, which were used for the QTL(s) analysis in this pipeline.
QTL-seq predicted candidate genomic region governing hull-less seed trait
QTL-Seq pipeline (Takagi et al., 2013) was used to identify the candidate genomic region governing hull-less seed trait in C. pepo, in which SNP-index was calculated for each identified SNP by comparing to the reference genome assembly in both the bulks. Then, the average SNP-index of each bulk was computed within a sliding window of 2 Mb interval with a 50 kb increment and was plotted against the genomic positions of C. pepo. The highly contrasting patterns of SNP-index graphs for hulled (B1) and hull-less bulk (B2) were observed on chromosome 12 from 1.80 to 2.90 Mb region (Supplementary Figures 1, 2). However, SNP-index graphs on other chromosomes were identical, suggesting these chromosomes might not be relevant to the phenotypic difference between hulled and hull-less bulks. Plotting of SNP-indices between hulled and hull-less bulks using 95 and 99% confidence intervals identified significant genomic positions governing hull-less seed trait. At a 95% statistical level, only one genomic region spanning 1.10 Mb on chromosome 12 from 1.80 to 2.90 Mb had a ΔSNP index value of 0.6 that was significantly different from 0, indicating this region is associated with phenotypic differences between hulled and hull-less bulks (Figure 2A, Supplementary Figure 3).
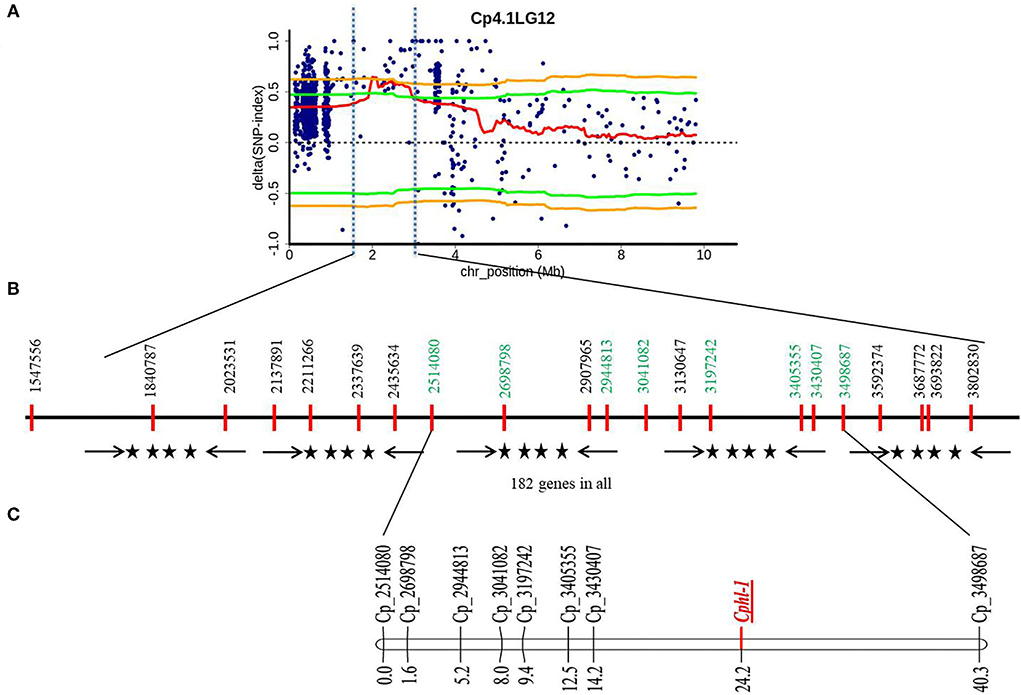
Figure 2. Physical and genetic position of QTL region governing hull-less seed trait in C. pepo on chromosome 12. (A) ΔSNP index graph from hulled and hull-less bulks, Orange line indicates 99% confidence interval and the green line indicates 95% confidence interval upper/lower side; (B) Genomic position of SNPs in QTL region selected for designing KASP assay where SNP shown in green were polymorphic and used for linkage map construction; (C) Partial Genetic linkage map of chromosome 12 from the HP111 × HLP36 F2 population showing the position of the hull-less locus (Cphl-1).
QTLseqr pipeline (Mansfeld and Grumet, 2018) also detected a similar genomic region at chromosome 12, spanning 1.89 to 3.86 Mb and covering 1.97 Mb. The peak of ΔSNP index was recorded at 2,615,628 bp at a 99% confidence interval (Supplementary Figure 4), whereas the maximum G' value was predicted at 3,209,540 bp w.r.t FDR (q) of 0.01 (Figure 3A, Supplementary Figure 5). Furthermore, the p-value analysis revealed a significant peak on chromosome 12 with a mean p-value of 4.98e−05 w.r.t FDR (q) of 0.01 (Figure 3B, Supplementary Figure 6). Overall, these results indicated a major QTL related to the hull-less seed trait at chromosome 12 from 1.80 to 3.86 Mb region.
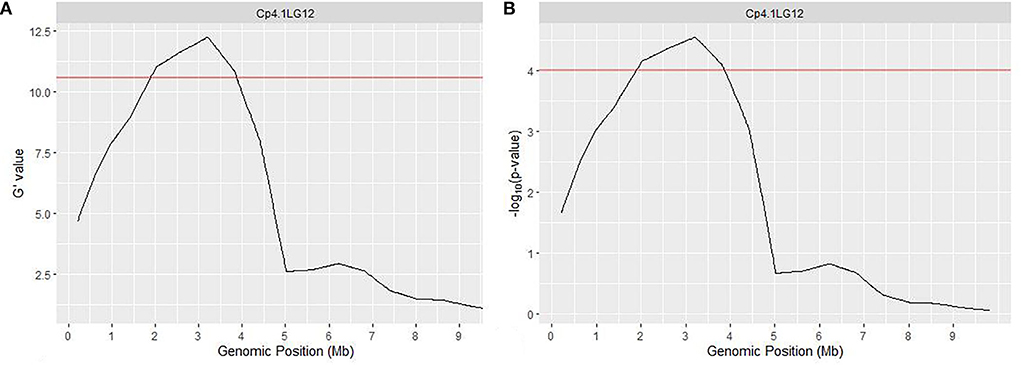
Figure 3. QTL region governing hull-less seed trait in C. pepo on chromosome 12 using G' (A) and p-value (B). Red line indicates a significant threshold for FDR (false discovery rate), q = 0.01.
Molecular mapping of a hull-less locus
To validate QTL-seq results and to further narrow down the candidate region, 21 KASP assays were designed (Figure 2B). However, only eight KASP assays scored reliably on the parents, F1 as well as in the hulled and hull-less bulks (Supplementary Figure 7). These polymorphic markers were used to genotype 160 F2 plants derived from the cross HP111 × HLP36 to study marker trait association for hull-less seed (Figure 4). The KASP assays were able to accurately predict hull-less plants, ranging from 73.47% to 87.23% of the F2 individuals (Table 2). The hull-less locus, Cphl-1 (named as Cucurbita pepo hull-less), was mapped to the partial linkage map of 40.3 cM with closest flanking markers viz., Cp_3430407 and Cp_3498687 placed at 10.0 cM and 16.1 cM genetic distances from Cphl-1 locus, respectively (Figure 4C). The physical locations of these two markers on chromosome 12 of the C. pepo genome were 3430407 bp (Cp_3430407) and 3498687 bp (Cp_3498687), spanning 68.28 kb.
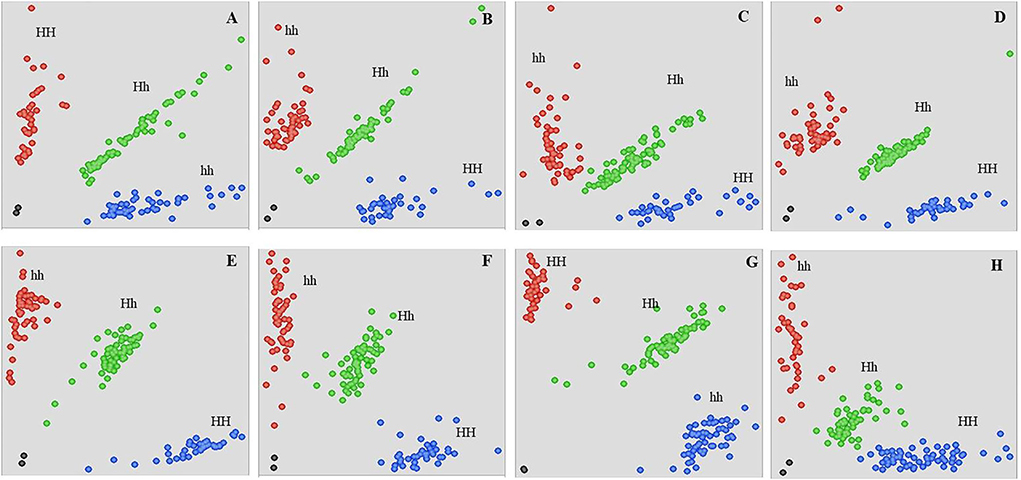
Figure 4. Genotyping of HP111 × HLP36 derived 160 F2 individuals using the KASP assay. Scatter plots for KASP assays show clustering of individual plants on the X- (FAM) and Y- (HEX) axes. Blue and red dots represent the homozygous plants, and green dots represent the heterozygous plants. Black dots represent the NTC (non-template control). HH, Homozygous for hulled parent SNP; hh, Homozygous for hull-less parent SNP; Hh, Heterozygous; (A) Cp_2514080; (B) Cp_2698798, (C) Cp_2944813, (D) Cp_3041082, (E) Cp_3197242, (F) Cp_3405355, (G) Cp_3430407, (H) Cp_3498687.

Table 2. Marker-phenotype association in amplified F2 individuals of HP111 × HLP36 cross.
Furthermore, based on the linkage map, two KASP assays (Cp_3430407 and Cp_3498687) were selected to determine their potential in differentiating hull-less and hulled seeds in genetically diverse populations. Marker Cp_3430407 precisely predicted the phenotype in the evaluated hull-less genotypes except two (28 out of 30), giving an accuracy percentage of 93.33. However, the accuracy was reduced to 60% in hulled genotypes as only nine out of 15 hulled genotypes were accurately predicted (Figure 5). Thus, the results indicate that the marker Cp_3430407 can be used in marker-assisted selection for hull-less seed trait till the gene-based marker can be identified.
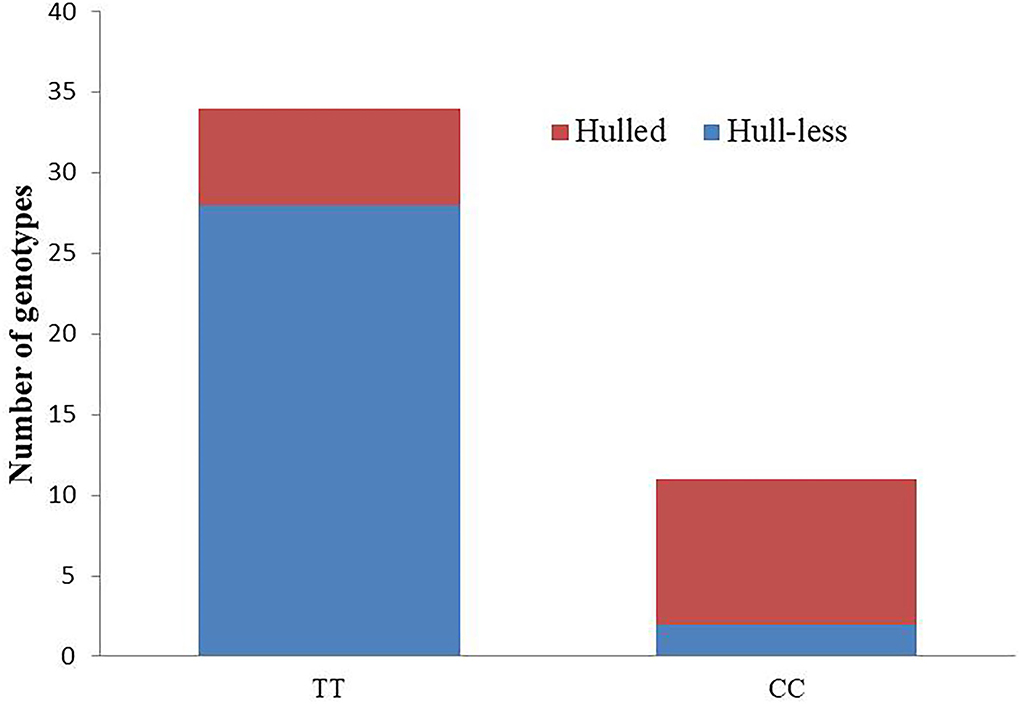
Figure 5. Genotypic and phenotypic data for KASP assay, Cp_3430407 in the diversity panel. (Total number of genotypes = 45; hull-less: 30; hulled: 15); X-axis indicates the genotype of Cp_3430407 KASP assay (TT or CC), and Y-axis indicates a number of genotyped individuals. Red and Blue sections denote the number of individuals with hulled and hull-less seed types, respectively.
Candidate gene identification, functional annotation, transcriptional differences, and gene ontology analysis
The candidate genomic region associated with the hull-less seed trait in C. pepo consists of 182 genes (Supplementary Table 2), out of which 19 are directly or indirectly involved in the cell wall, cellulose, and lignin biosynthesis (Table 3). The expression of these 19 genes in seeds of hulled and hull-less C. pepo genotypes assessed from the publically available RNA-seq dataset revealed two genes viz., Cp4.1LG12g04350 (NAC) and Cp4.1LG12g03120 (MYB), which are highly expressed in hull-less genotype compared to the hulled genotype at different time intervals (Figures 6A,B; Supplementary Table 3). These genes share homology with the Arabidopsis secondary cell wall related NAC and MYB transcriptional factors, respectively, which act as master regulators of lignin biosynthesis (Zhong and Ye, 2009). Furthermore, the differential expression analysis at 15 and 20 DAP between seeds of hulled and hull-less genotypes found a total of 3,124 unique differentially expressed genes. The gene ontology analysis (biological process) of these genes linked 482 to the cell wall, cellulose, or lignin biosynthesis that corresponds to 15.42% of the total (Figure 6C). These results imply that the transcriptional profile of genes involved in cell wall development and lignification was altered between hulled and hull-less seeds.
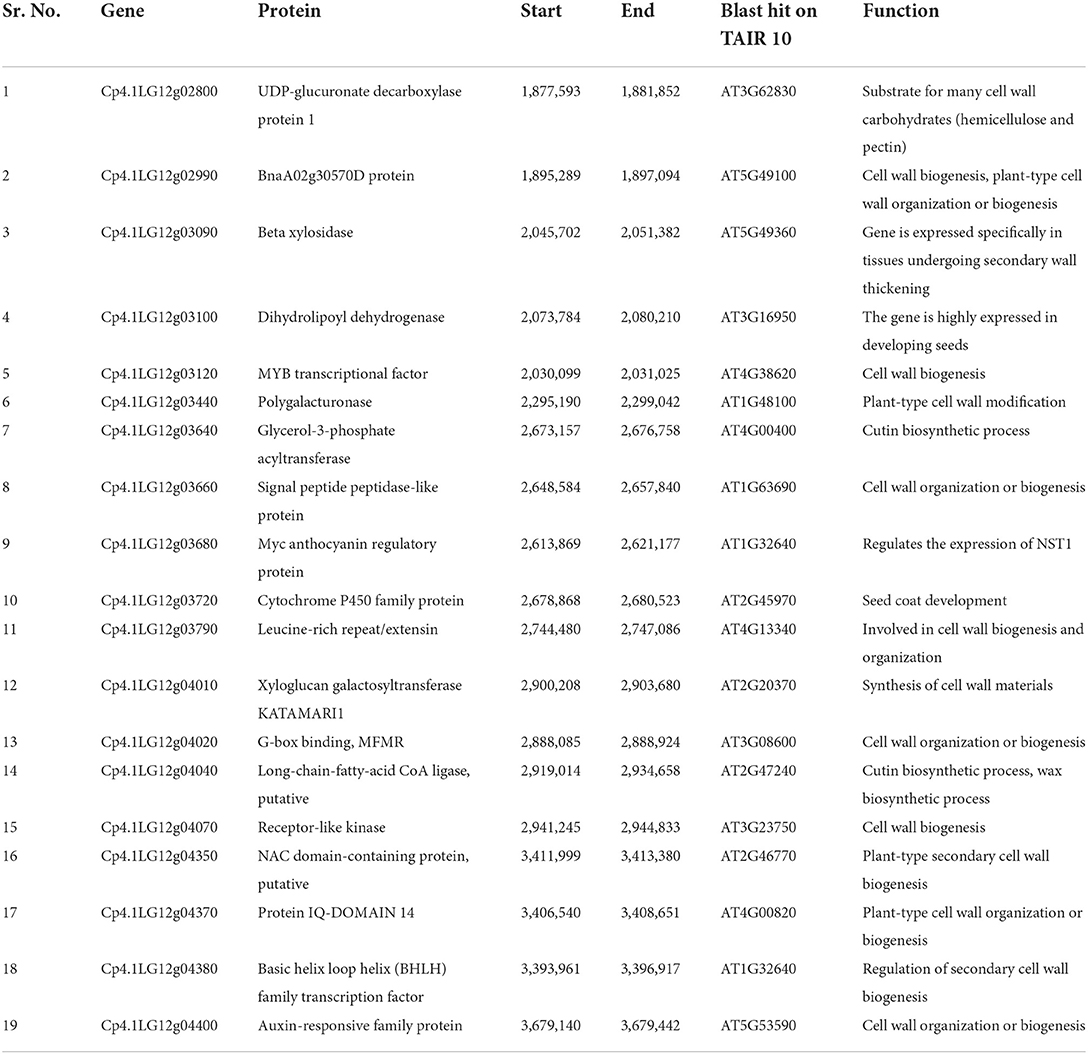
Table 3. List of genes associated with the cell wall, lignin, or cellulose biosynthesis from the candidate region governing hull-less seed trait.
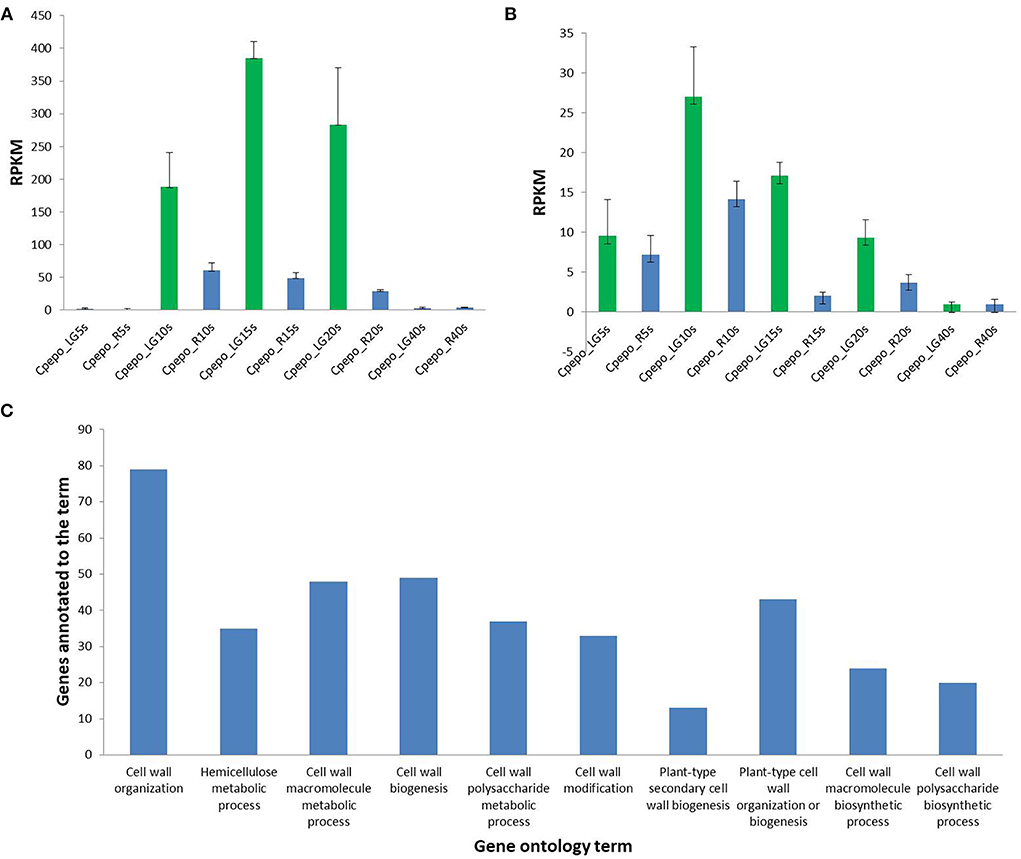
Figure 6. Differential expression of genes, NAC (Cp4.1 LG12g04350) (A) and MYB, (Cp4.1LG12g03120) (B) in seeds of hull-less and hulled C. pepo; where, RPKM, Reads per Kilobase of transcript per million mapped read; LG, hull-less seeded variety “Lady Godiva”; R, Hulled variety “Sweet Reeba”; 5, 10, 15, 20, and 40 s corresponds to 5, 10, 15, 20, and 40 DAP (C): Gene ontology analysis (Biological processes) from transcriptional differences between hull-less and hulled seeds at 15 and 20 DAP.
Discussion
Pumpkin (C. pepo) seeds have a high nutritive value that imparts numerous health benefits (Dotto and Chacha, 2020). However, these are covered by a thick and leathery seed coat due to the lignification of different cell wall layers, and therefore, require its removal prior to use for human consumption. On the contrary, hull-less pumpkin mutant, such as Styrian, lacks the hard seed coat and, as a result, it does not require de-hulling. Hull-less seed trait was transferred from a USA cultivar, “Lady Godiva” into local C. pepo lines adapted to the subtropical climate of India. Genetic inheritance in different hulled and hull-less populations suggested that lack of seed coat lignification is governed by a single recessive gene in C. pepo lines. In the past, different studies in European germplasm demonstrated that a single recessive gene (hh) is responsible for the hull-less seed trait in pumpkin. Although hull-less seeds exhibit variation for a degree of lignification, which might be governed by unspecified minor genes (Lelley et al., 2009).
The selection for hulled and hull-less seeds at the phenotypic level is performed after the maturation of fruits on a visible, sensory, and microscopic basis, but these methods are very time consuming and labor intensive. These limitations can be overcome by developing molecular markers that breeders can use in marker-assisted breeding programmes in a cost and time-effective manner (Collard and Mackill, 2008). Bulk segregant analysis (BSA) is a rapid method to identify markers linked to the trait of interest (Michelmore et al., 1991) and when combined with NGS technologies can be used as a fast-track approach to locate candidate genomic regions more rapidly. This approach is known as QTL-seq and involves the selection of extreme phenotypic values, pooling in equivalent concentration, followed by sequencing of pools for downstream sequence analysis in different crops (Takagi et al., 2013; Bhat et al., 2016; Li and Xu, 2022). The utilization of this approach accelerated the identification of genomic regions and candidate genes governing traits of economic importance and the development of molecular markers that could be used in MAS (Grover and Sharma, 2016; Tan et al., 2018). The availability of the C. pepo genome opens up new avenues to discover the genomic region controlling hull-less seed trait.
In this study, we utilized NGS-based BSA of hulled and hull-less genotypes followed by analysis with QTL-seq and QTLseqr bioinformatics pipelines to determine the genomic region responsible for hull-less seed in C. pepo. The power of the QTL-seq method typically depends upon population size and proportion used in the bulk generation. In general, bulk sizes of 15–20% from a F2 population of 200–300 individuals are considered sufficient to detect both major and minor QTLs (Magwene et al., 2011; Takagi et al., 2013; Tiwari et al., 2016). However, a single major QTL can be detected using an F2 population of 100 individuals (Takagi et al., 2013; Illa-Berenguer et al., 2015). BSA coupled with the NGS approach has been extensively used in the mapping of major effect traits controlled by one or two loci. Thus, this is the effective approach for mapping hull-less loci, as it is governed by a single recessive gene. Moreover, the various mapping studies reported the bulk sizes in the range of 3–11.9% (Takagi et al., 2013; Singh et al., 2016; Wei et al., 2016; Ramos et al., 2020). The filial populations produced through selfing can significantly increase the power and precision, especially from F2 to F3, and have been used in the present study (Huang et al., 2022).
The hull-less locus was mapped on chromosome 12 with a genomic region extending from 1.80 to 3.86. Earlier, the hull-less seed locus was mapped to the linkage group LG09 (Gong et al., 2008); however, its physical position was not known. The BLAST results of the linked SSR markers (CMTm239, CMTp182) from this study revealed significant hits on chromosome 12 of the C. pepo genome, as of the identified region in our study. While we were preparing this manuscript, Meru et al. (2022) identified a large genomic region of 4.43 Mb (1.25–5.68 Mb) on chromosome 12 for the hull-less seed trait; however, in our study, it was narrowed down to ~2 Mb (1.80–3.86 Mb), which might be due to higher number of individuals used for bulk construction.
Over the years, SNPs-based markers have become the first choice for researchers due to their high genomic abundance, co-dominant inheritance, high-throughput analysis, and relatively low genotyping error rates (Mammadov et al., 2012). Among the different platforms, the KASP assay is a promising technology for high throughput SNP genotyping (Ayalew et al., 2019). The SNP-based KASP assays for MAS have been developed for many traits in cucurbits (Paudel et al., 2019; Cao et al., 2021; Kahveci et al., 2021). The polymorphic KASP assays have been used in the present investigation to develop a partial genetic linkage map of 40.3 cM and the Cphl-1 locus was delimited between two markers Cp_3430407 and Cp_3498687. A KASP assay viz., Cp_3430407 can be utilized for MAS with 93.33% accuracy in hull-less germplasm of C. pepo. The physical positions of this KASP assay are also close to recently identified SNP markers (Ch12_3412046 and Ch12_3417142) reported by Meru et al. (2022).
The molecular mechanism of cell wall lignification has been well-studied in Arabidopsis, and over the years, this knowledge has been translated to understand the lignification mechanism in crops (Wang and Dixon, 2012; Barros et al., 2015). In this study, we identified a candidate genomic region associated with hull-less seed trait in C. pepo that tends to have limited lignification on seed coat in comparison to hulled seeds. Furthermore, two C. pepo genes from this region share homology with NAC and MYB transcription factors which act as master regulators of lignification (Wang et al., 2011; Yoon et al., 2015). These genes were highly expressed in hull-less seeds between 10 and 20 days after anthesis in comparison to hulled genotypes (Wyatt et al., 2015). Similarly, a high proportion of cell wall related genes (15.42%) was differentially expressed between hulled and hull-less seeds at 15 and 20 DAP. Based on earlier studies, it has been demonstrated that the extent of lignification starts increasing in hulled seeds 10 days after pollination in comparison to hull-less seeds (Bezold et al., 2005; Xue et al., 2022). In the future, it will be fascinating to study the functional role of candidate genes differentially expressed between hulled and hull-less seeds of pumpkin to understand the molecular mechanism of seed coat lignification.
Conclusion
Hull-less seed trait in pumpkin has shown monogenic recessive inheritance in C. pepo. QTL-seq analysis has also shown a single major QTL on chromosome 12 spanning 2.06 Mb associated with hull-less seed, which was further delimited to ~68 kb using KASP assays. Further analysis of candidate genes spanning the QTL region has identified lignin-related genes, NAC, and MYB as potential candidate genes governing hull-less seed trait in C. pepo. Finally, the linked KASP assay generated in the current investigation can further be used in breeding programmes for marker-assisted selection of hull-less trait. The study has also laid the foundation for research on molecular mechanisms related to hull-less seed trait in pumpkin.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/PRJNA849145.
Author contributions
AD conceived the research. AD, DB, JK, AM, and MS conceptualized the experiments. BK and KG conducted experiments and analyzed the data. All authors have prepared and approved the final manuscript.
Acknowledgments
We gratefully acknowledge the Department of Science and Technology, Government of India, for providing lab facilities under the PURSE project to Punjab Agricultural University, Ludhiana. The authors also acknowledge DST-SERB, CII, and Mr. Avtar Singh Dhindsa, Proprietor of Beauscape Farms for supporting doctoral research through Prime Minister Fellowship to BK and KG.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.948106/full#supplementary-material
Supplementary Figure 1. SNP-index distribution for hulled bulk sample (B1). Green dots: SNP-index of hulled bulk B1's; Red line: Sliding window average of SNP-index.
Supplementary Figure 2. SNP-index distribution for hull-less bulk sample (B2). Orange dot: SNP-index of hull-less bulk B2's; Red line: Sliding window average of SNP- index.
Supplementary Figure 3. ΔSNP-index distribution in hulled and hull-less bulks. Blue dots: ΔSNP-index; Red line: Sliding window average of ΔSNP-index; Green line: Sliding window average of 95%-confidence interval upper/lower side; Orange line: Sliding window average of 99%-confidence interval upper/lower side.
Supplementary Figure 4. Tricube smoothed ΔSNP-index distribution across 20 chromosome of C. pepo. Red line: 95% confidence interval; Blue line: 95% confidence interval; X-axis: Physical position (Mb) of C. pepo genome cv. Zucchini across the respective chromosome number; Y-axis: Designates the ΔSNP-index.
Supplementary Figure 5. G' distribution across 20 chromosomes of C. pepo. Red line: Significant threshold for FDR (false discovery rate), q=0.01; X-axis: Physical position (Mb) of C. pepo genome cv. Zucchini across the respective chromosome number; Y-axis: Designates the G' value.
Supplementary Figure 6. p-value distribution across 20 chromosomes of C. pepo. Red line: Significant threshold for FDR (false discovery rate), q = 0.01; X-axis: Physical position (Mb) of C. pepo genome cv. Zucchini across the respective chromosome number; Y-axis: Designates the -log10 (p-value).
Supplementary Figure 7. Genotyping of parents, F1, and bulk individual F2:3 lines using the Kompetitive Allele Specific PCR (KASP) assay. Scatter plots for selected KASP assays show clustering on the X- (FAM) and Y-axes (HEX). The blue and red dots represent the homozygous bulk individuals, and the green dots represent heterozygous plants. The black dots represent the NTC (non-template control). The pink dots represent the non-amplified plants. HH, Homozygous for hulled parent SNP; hh, Homozygous for hull-less parent SNP; Hh, Heterozygous; (A) Cp_2514080, (B) Cp_2698798, (C) Cp_2944813, (D) Cp_3041082, (E) Cp_3197242, (F) Cp_3405355, (G) Cp_3430407, (H) Cp_3498687.
Supplementary Table 1. List of KASP markers used in the study.
Supplementary Table 2. List of genes present in the candidate region.
Supplementary Table 3. Variance for RNA-seq expression between seeds of hull-less and hulled genotypes at different time intervals.
References
Ayalew, H., Tsang, P. W., Chu, C., Wang, J., Liu, S., Chen, C., et al. (2019). Comparison of TaqMan, KASP and rhAmp SNP genotyping platforms in hexaploid wheat. PLoS ONE 14:e0217222. doi: 10.1371/journal.pone.0217222
Barros, J., Serk, H., Granlund, I., and Pesquet, E. (2015). The cell biology of lignification in higher plants. Ann. Bot. 115, 1053–1074. doi: 10.1093/aob/mcv046
Bezold, T. N., Mathews, D., Loy, J. B., and Minocha, S. C. (2005). Molecular analysis of the hull-less seed trait in pumpkin: expression profiles of genes related to seed coat development. Seed Sci. Res. 15, 205–217. doi: 10.1079/SSR2005211
Bhat, J. A., Ali, S., Salgotra, R. K., Mir, Z. A., Dutta, S., and Jadon, V. (2016). Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 7, 221. doi: 10.3389/fgene.2016.00221
Cao, M., Li, S., Deng, Q., Wang, H., and Yang, R. (2021). Identification of a major-effect QTL associated with pre-harvest sprouting in cucumber (Cucumis sativus L.) using the QTL-seq method. BMC Genom. 22, 249. doi: 10.1186/s12864-021-07548-8
Collard, B. C., and Mackill, D. J. (2008). Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 557–572. doi: 10.1098/rstb.2007.2170
De Lamo, B., and Gómez, M. (2018). Bread enrichment with oilseeds. A review. Foods 7, 191. doi: 10.3390/foods7110191
Dhatt, A. S., Sharma, M., and Kaur, B. (2020). “Advances in improvement of pumpkin and squashes,” in Accelerated Plant Breeding, Volume 2, eds S. Gosal, and S. Wani (New York, NY: Springer), 301–335. doi: 10.1007/978-3-030-47298-6_11
Dotto, J. M., and Chacha, J. S. (2020). The potential of pumpkin seeds as a functional food ingredient: a review. Sci. Afr. 10, e00575. doi: 10.1016/j.sciaf.2020.e00575
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Fruhwirth, G. O., and Hermetter, A. (2007). Seeds and oil of the Styrian oil pumpkin: components and biological activities. Eur. J. Lipid Sci. Technol. 109, 1128–1140. doi: 10.1002/ejlt.200700105
Gong, L., Stift, G., Kofler, R., Pachner, M., and Lelley, T. (2008). Microsatellites for the genus Cucurbita and an SSR-based genetic linkage map of Cucurbita pepo L. Theor. Appl. Genet. 117, 37–48. doi: 10.1007/s00122-008-0750-2
Grover, A., and Sharma, P. C. (2016). Development and use of molecular markers: past and present. Crit. Rev. Biotechnol. 36, 290–302. doi: 10.3109/07388551.2014.959891
Huang, L., Tang, W., and Wu, W. (2022). Optimization of BSA-seq experiment for QTL mapping. G3 12, jkab370. doi: 10.1093/g3journal/jkab370
Illa-Berenguer, E., Van Houten, J., Huang, Z., and Van der Knaap, E. (2015). Rapid and reliable identification of tomato fruit weight and locule number loci by QTL-seq. Theor. Appl. Genet. 128, 1329–1342. doi: 10.1007/s00122-015-2509-x
Jafari, M., Goli, S. A. H., and Rahimmalek, M. (2012). The chemical composition of the seeds of Iranian pumpkin cultivars and physicochemical characteristics of the oil extract. Eur. J. Lipid Sci. Technol. 114, 161–167. doi: 10.1002/ejlt.201100102
Kahveci, E., Devran, Z., Özkaynak, E., Hong, Y., Studholme, D. J., and Tör, M. (2021). Genomic-assisted marker development suitable for CsCvy-1 selection in cucumber breeding. Front. Plant Sci. 12, 691576. doi: 10.3389/fpls.2021.691576
Kosugi, S., Natsume, S., Yoshida, K., MacLean, D., Cano, L., Kamoun, S., et al. (2013). Coval: improving alignment quality and variant calling accuracy for next-generation sequencing data. PLoS ONE 8, e75402. doi: 10.1371/journal.pone.0075402
Lelley, T., Loy, B., and Murkovic, M. (2009). “Hull-less oil seed pumpkin,” in Oil Crops, Handbook of Plant Breeding 4, eds J. Vollmann, and I. Rajcan (Berlin: Springer Science + Business Media, LLC), 469–492. doi: 10.1007/978-0-387-77594-4_16
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). 1000 genome project data processing subgroup the sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Z., and Xu, Y. (2022). Bulk segregation analysis in the NGS era: a review of its teenage years. Plant J. 109, 1355–1374. doi: 10.1111/tpj.15646
Lu, H., Lin, T., Klein, J., Wang, S., Qi, J., Zhou, Q., et al. (2014). QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor. Appl. Genet. 127, 1491–1499. doi: 10.1007/s00122-014-2313-z
Magwene, P. M., Willis, J. H., and Kelly, J. K. (2011). The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput. Biol. 7, e1002255. doi: 10.1371/journal.pcbi.1002255
Mammadov, J., Aggarwal, R., Buyyarapu, R., and Kumpatla, S. (2012). SNP markers and their impact on plant breeding. Int. J. Plant Genom. 728398, 1–11. doi: 10.1155/2012/728398
Mansfeld, B. N., and Grumet, R. (2018). QTLseqr: an R package for bulk segregant analysis with next-generation sequencing. Plant Genome 11, 180006. doi: 10.3835/plantgenome2018.01.0006
Meru, G., Fu, Y., Shrestha, S., Michael, V. N., Dorval, M., and Mainviel, R. (2022). Genomic position and markers associated with the hull-less seed trait in pumpkin. Plants 11, 1238. doi: 10.3390/plants11091238
Michelmore, R. W., Paran, I., and Kesseli, R. (1991). Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. U. S. A. 88, 9828–9832.
Montero-Pau, J., Blanca, J., Esteras, C., Martínez-Pérez, E. M., Gómez, P., Monforte, A. J., et al. (2017). An SNP-based saturated genetic map and QTL analysis of fruit-related traits in Zucchini using genotyping-by-sequencing. BMC Genom. 18, 94. doi: 10.1186/s12864-016-3439-y
Paudel, L., Clevenger, J., and McGregor, C. (2019). Refining of the egusi locus in watermelon using KASP assays. Sci. Hortic. 257, 108665. doi: 10.1016/j.scienta.2019.108665
Ramos, A., Fu, Y., Michael, V., and Meru, G. (2020). QTL-seq for identification of loci associated with resistance to Phytophthora crown rot in squash. Sci. Rep. 10, 1–8. doi: 10.1038/s41598-020-62228-z
Semagn, K., Babu, R., Hearne, S., and Olsen, M. (2014). Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): overview of the technology and its application in crop improvement. Mol. Breed. 33, 1–14. doi: 10.1007/s11032-013-9917-x
Singh, V. K., Khan, A. W., Jaganathan, D., Thudi, M., Roorkiwal, M., Takagi, H., et al. (2016). QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotechnol. J. 14, 2110–2119. doi: 10.1111/pbi.12567
Stevenson, D. G., Eller, F. J., Wang, L., Jane, J. L., Wang, T., and Inglett, G. E. (2007). Oil and tocopherol content and composition of pumpkin seed oil in 12 cultivars. J. Agril. Food Chem. 55, 4005–4013. doi: 10.1021/jf0706979
Stuart, S. G., and Loy, J. B. (1983). Comparison of testa development in normal and hull-less seeded strains of Cucurbita pepo L. Bot. Gazette 144, 491–500.
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013). QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Tan, J., Guo, J. J., Yin, M. Y., Wang, H., Dong, W. P., Zeng, J., et al. (2018). Next generation sequencing-based molecular marker development: a case study in Betulaalnoides. Molecules 23, 2963. doi: 10.3390/molecules23112963
Tiwari, S., Sl, K., Kumar, V., Singh, B., Rao, A. R., Mithra, S. V., et al. (2016). Mapping QTLs for salt tolerance in rice (Oryza sativa L.) by bulked segregant analysis of recombinant inbred lines using 50K SNP chip. PLoS ONE 11, e0153610. doi: 10.1371/journal.pone.0153610
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 11–10. doi: 10.1002/0471250953.bi1110s43
Van Ooijen, J (2006). JoinMap 4: Software for the Calculation of Genetic Linkage MapsinExperimental Populations. Wageningen: Kyazma B.V.
Varshney, R. K., Bohra, A., Yu, J., Graner, A., Zhang, Q., and Sorrells, M. E. (2021). Designing future crops: genomics-assisted breeding comes of age. Trends Plant Sci. 26, 631–649. doi: 10.1016/j.tplants.2021.03.010
Voorrips, R (2002). MapChart: software for the graphical presentation of linkage mapsand QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Wang, H., Zhao, Q., Chen, F., Wang, M., and Dixon, R. A. (2011). NAC domain function and transcriptional control of a secondary cell wall master switch. Plant J. 68, 1104–1114. doi: 10.1111/j.1365-313X.2011.04764.x
Wang, H. Z., and Dixon, R. A. (2012). On–off switches for secondary cell wall biosynthesis. Mol. Plant 5, 297–303. doi: 10.1093/mp/ssr098
Wei, Q. Z., Fu, W. Y., Wang, Y. Z., Qin, X. D., Wang, J., Li, J., et al. (2016). Rapid identification of fruit length loci in cucumber (Cucumis sativus L.) using next-generation sequencing (NGS)-based QTL analysis. Sci. Rep. 6, 1–11. doi: 10.1038/srep27496
Winkler, J (2000). Breeding of hull-less seeded pumpkins (Cucurbita pepo) for the use of the oil. Acta Hortic. 510, 123–128. doi: 10.17660/ActaHortic.2000.510.20
Wyatt, L. E., Strickler, S. R., Mueller, L. A., and Mazourek, M. (2015). An acorn squash (Cucurbita pepo ssp. ovifera) fruit and seed transcriptome as a resource for the study of fruit traits in Cucurbita. Hortic. Res. 2, 14070. doi: 10.1038/hortres.2014.70
Xue, Y., Shen, Z., Tao, F., Zhou, J., and Xu, B. (2022). Transcriptomic analysis reveal the molecular mechanisms of seed coat development in Cucurbita pepo L. Front. Plant Sci. 13, 772685. doi: 10.3389/fpls.2022.772685
Yoon, J., Choi, H., and An, G. (2015). Roles of lignin biosynthesis and regulatory genes in plant development. J. Integr. Plant Biol. 57, 902–912. doi: 10.1111/jipb.12422
Yoshinari, O., Sato, H., and Igarashi, K. (2009). Anti-diabetic effects of pumpkin and its components, trigonelline and nicotinic acid on Goto-Kakizaki rats. Biosci. Biotechnol. Biochem. 73, 1033–1041. doi: 10.1271/bbb.80805
Zhong, R., and Ye, Z. H. (2009). Transcriptional regulation of lignin biosynthesis. Plant Signal. Behav. 4, 1028–1034. doi: 10.4161/psb.4.11.9875
Keywords: hull-less seed, Cucurbita pepo, BSA, QTL-seq, MAS
Citation: Kaur B, Garcha KS, Bhatia D, Khosa JS, Sharma M, Mittal A, Verma N and Dhatt AS (2022) Identification of single major QTL and candidate gene(s) governing hull-less seed trait in pumpkin. Front. Plant Sci. 13:948106. doi: 10.3389/fpls.2022.948106
Received: 19 May 2022; Accepted: 12 July 2022;
Published: 11 August 2022.
Edited by:
Baohua Wang, Nantong University, ChinaReviewed by:
Atefeh Sabouri, University of Guilan, IranWei-Li Guo, Henan Institute of Science and Technology, China
Copyright © 2022 Kaur, Garcha, Bhatia, Khosa, Sharma, Mittal, Verma and Dhatt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ajmer Singh Dhatt, YWptZXJkaGF0dEBwYXUuZWR1
†These authors have contributed equally to this work and share first authorship