 Feng Liu
Feng Liu Hongshu Wang
Hongshu Wang Wenli Song
Wenli Song
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 05 August 2022
Sec. Aquatic Photosynthetic Organisms
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.937398
Comparative mitogenomics of Ulva species have revealed remarkable variations in genome size due to the integration of exogenous DNA fragments, the proliferation of group I/II introns, and the change of repeat sequences. The genus Ulva is a species-rich taxonomic group, containing a variety of green-tide forming algae. In this study, five complete mitogenomes of the green-tide forming macroalga, Ulva meridionalis R. Horimoto and S. Shimada, were assembled and compared with the available ulvophyceae mtDNAs. The main circular mitogenomes of U. meridionalis ranged from 82.94 to 111.49 kb in size, and its 111.49-kb mitogenome was the largest Ulva mitogenome sequenced so far. The expansion of U. meridionalis mitogenomes is mainly due to the tandem integration of a 5.36-kb mitochondrial circular plasmid (pUme), as well as the proliferation of introns. An intact DNA-directed RNA polymerase gene (rpo) was present in pUme of U. meridionalis and was then detected in two putative plasmids (pUmu1 and pUmu2) found in Ulva mutabilis. The observed integration of the circular plasmid into U. meridionalis mitogenomes seems to occur via homologous recombination, and is a more recent evolutionary event. Many highly homologous sequences of these three putative plasmids can be detected in the other Ulva mtDNAs sequenced thus far, indicating the integration of different mitochondrial plasmid DNA into the mitogenomes is a common phenomenon in the evolution of Ulva mitogenomes. The random incidence of destruction of plasmid-derived rpos and open reading frames (orfs) suggests that their existence is not the original characteristic of Ulva mitogenomes and there is no selective pressure to maintain their integrity. The frequent integration and rapid divergence of plasmid-derived sequences is one of the most important evolutionary forces to shape the diversity of Ulva mitogenomes.
The species of genus Ulva Linnaeus 1753 (Ulvophyceae, Chlorophyta) have attracted much attention not only because of their potential economic value in food and pharmaceutical industry, but also because of their important ecological functions and effects. These green seaweeds widely distributed on the coasts of the world are important indicators to reflect the state of ecological and environmental health. Globally, these opportunistic Ulva species often accumulate large amounts of biomass due to eutrophication, resulting in large-scale green tides (Ye et al., 2011; Liu et al., 2013; Wang et al., 2015, 2019). In the past 5 years from 2017 to 2021, Ulva meridionalis R. Horimoto and S. Shimada has proliferated and grown rapidly every summer in the Sakura Lake, Rongcheng, Shandong Province, China. The Sakura Lake is a semi-enclosed brackish inner lake, located at the tip of Shandong Peninsula, China, and extends to the Sanggou Bay which is connected to the Yellow Sea. This lake receives the freshwater runoffs and effluents from the local largest river, Gu River, and other small rivers, which carry the essential dissolved plant nutrients from agricultural, industrial, and municipal activities and cause a worsening trend of eutrophication. The large U. meridionalis biomass had a strong negative impact on the local landscape and ecosystem, and thousands of tons of biomass could only be salvaged manually. This brackish alga with tubular, winkled or lubricous thalli was taxonomically named in 2011, and mainly inhabits estuaries and marshes by the sea in Japan and China (Horimoto et al., 2011; Xie et al., 2020; Liu J. et al., 2022). This alga has attracted much attention because of its rapid growth ability (e.g., Hiraoka et al., 2020; Tsubaki et al., 2020).
The species-rich macroalgal genus Ulva currently contains at least 99 taxonomically accepted species worldwide (Guiry and Guiry, 2022), as well as some unconfirmed cryptic species (Steinhagen et al., 2019a,b). Ulva species have high morphological diversity and plasticity at the intraspecific level (Gao et al., 2016; Steinhagen et al., 2019c), so accurate and reliable species identification often requires the use of common DNA markers (e.g., ITS, rbcL, tufA, etc.) (Blomster et al., 2002; Liu et al., 2013; Fort et al., 2020). Organelle genome as a molecular marker can make us more accurately understand the concept of Ulva species and more comprehensively understand their genetic diversity and evolutionary relationships, which could not be done by a single or several DNA markers. Recently, organelle genomes of Ulva species showed a variety of obvious dynamic changes involving genome size, integration of exogenous DNA fragments, gene content, acquisition or loss of intron, genome rearrangement, and abundance of repeat sequence, at the interspecific and intraspecific level (Liu and Melton, 2021; Liu F. et al., 2022). Based on the phylogenetic analysis of organelle genome data, Ulva species are obviously divided into two independent genetic lineages (I and II) (Liu and Melton, 2021; Liu F. et al., 2022). In addition, nuclear genomes of two Ulva species, Ulva mutabilis and Ulva prolifera, have been sequenced and deposited in the GenBank database up to now (De Clerck et al., 2018).
In the last 10 years, mitogenomic data on Ulva have accumulated rapidly. Thus far, a total of 32 mitochondrial genomes (mitogenomes or mtDNAs) from 19 Ulva species have been sequenced and deposited in the GenBank database (Liu F. et al., 2022). The complete Ulva mitogenomes are circular molecules with the size ranging from 55.81 to 88.42 kb, and display great changes in genome size (Liu et al., 2017, 2020; Liu F. et al., 2022). The Ulva mitogenomes contain the same set of 62 core genes which are usually coded on one strand, while specific genes and open reading frames (orfs) vary greatly in quantity and sequence. It is worth noting that DNA-directed RNA polymerase genes (rpos) are very commonly present in the Ulva mitogenomes, which are likely to be the origin of mitochondrial plasmid DNA, and many rpos have been split into small segments in varying degrees due to multiple mutations (Liu F. et al., 2022). The introns in Ulva mitogenomes show drastic dynamic changes in the number, distribution and diversity at interspecific and intraspecific level, and none of introns is shared by all sequenced Ulva mitogenomes, indicating that the homing or “jumping” of group I/II introns occurred frequently in Ulva (Liu et al., 2017; Liu F. et al., 2022). The Ulva mitochondrial introns observed are mainly distributed at 29 insertion sites in seven genes (atp1, cox1, cox2, nad3, nad5, rnl, and rns) and usually harbor an intronic orf which encoded an LAGLIDADG homing endonuclease (LHE) or a GIY-YIG homing endonuclease (GHE) or a reverse transcriptase/maturase (RTM). Six types of group I/II introns have been found in Ulva mitogenomes, and in particular, the mitochondrial LHEs in group II introns have close relationships with that in group IB introns (Liu and Melton, 2021).
In addition to the main mitochondrial genomes, mitochondria of some fungi and plants harbor some smaller DNA molecules regarded as plasmid-like elements or true plasmids, which could be autonomously replicated (Handa, 2008; Lang, 2014). The true plasmids can be divided into three different categories: (1) linear plasmids that encode a DNA and/or an RNA polymerase, (2) circular plasmids that encode a DNA polymerase, and (3) linear or circular retroplasmids that encode a reverse transcriptase (Hausner, 2011; Baidyaroy et al., 2012). Mitochondrial plasmids show great diversity in sequence and structure, and many plasmids are species-specific in distribution pattern. Although most plasmids appear to be cryptic in nature, they seem to be involved in the evolution of mitogenomes and are related to mitochondrial instability in fungi and cytoplasmic male sterility (CMS) in plants (Hausner, 2003; Gualberto et al., 2014). More evidences showed that mitochondrial plasmid DNA could be integrated into the mitogenomes in fungi and plants, which caused the increase of mitogenome size (e.g., Allen et al., 2007; Formighieri et al., 2008). However, there is little knowledge on the mitochondrial plasmid in green algae thus far.
To understand the evolution of Ulva mitogenomes and the formation mechanism of mitogenome diversity, in this study, five complete mitochondrial genomes of the green-tide forming macroalga U. meridionalis (Ume) have been sequenced and compared with the available ulvophyceae mtDNAs deposited in the GenBank database.
The drifting algal thalli of Ulva meridionalis R.Horimoto and S.Shimada were collected on 4 August 2021 in the Sakura Lake (37°7′30″–56″’N, 122°27′3″–50″E), Rongcheng, Shandong Province, China, when a green tide occurred in the lake due to the proliferation of U. meridionalis (Ume). These Ulva thalli were transported to laboratory in coolers (5–8°C) after collection. Five algal individuals (LF008, LF010, LF011, LF012, and LF018) were randomly selected from 60 identified samples, and named as Ume1 to Ume5, respectively (Supplementary Figure 1). Algal thallus for each individual was cultured in a 9-cm diameter Petri dish containing 25-mL L1 medium with 0.5‰ GeO2, 50 μg/mL dipterex (Fengcheng Animal Medicine Co., Ltd, China) and a suite of antibiotics (per mL: 50 μg streptomycin, 66.6 μg gentamycin, 20 μg ciprofloxacin, 2.2 μg chloramphenicol, and 100 μg ampicillin) (Shibl et al., 2020). The culture of U. meridionalis was maintained at 18°C, 100–120 μmol photons m–2 s–1 in the photoperiod of 12 h light: 12 h darkness in a GXZ-380C temperature-controlled incubator (Ningbo Jiangnan, China).
Fresh algal tissue from each individual thallus was used for DNA extraction using a Plant Genome DNA Kit (DP305, Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. Species identification was performed based on phylogenetic analyses of two common DNA marker datasets (the nuclear ITS region including the 5.8S rDNA gene, and the chloroplast rbcL gene) (Hayden and Waaland, 2002; Liu et al., 2020). Primers sequences and polymerase chain reaction (PCR) amplification were used according to our previous study (Liu et al., 2013). Sequence datasets of our samples and other data from the GenBank database were aligned using MEGA 7.0 (Kumar et al., 2016). The maximum likelihood (ML) tree was constructed with 1,000 bootstrap replicates based on the Kimura two-parameter model (Tamura and Nei, 1993). The identification results confirmed that these five samples were U. meridionalis (Supplementary Figures 2, 3).
The DNA quality and concentration were checked using a NanoPhotometer spectrophotometer (Implen, CA, United States), and a Qubit 2.0 Fluorometer (Life Technologies, CA, United States), respectively. Qualified DNA samples were fragmented into 350 bp by Covaris S220 ultrasonic crater for library construction. The qualified libraries were sequenced on an Illumina NovaSeq platform (Illumina, United States) using paired-end sequencing, yielding about 10 Gb sequencing raw data of paired-end reads with 150 bp in length for each U. meridionalis sample. Clean data were obtained by trimming sequencing adapters and removing short or low-quality reads from the raw data. The complete mitochondrial genomes of U. meridionalis were constructed by the GetOrganelle v1.7.1 (Jin et al., 2020). The mitogenome of Ulva prolifera (KT428794) was used as a reference genome for assembly. The mitogenome assembly was examined by aligning reads using the MEM algorithm of BWA v0.7.17 (Li and Durbin, 2010). The VarScan v2.3.9 (Koboldt et al., 2009) and IGV v2.8.12 (Robinson et al., 2011) were used to examine mutation sites and to verify the assembly results, respectively.
Protein-coding genes (PCGs) were annotated by Open Reading Frame Finder at the National Center for Biotechnology Information (NCBI) website,1 and by aligning the homologous PCGs from the Ulva mtDNAs deposited in the GenBank database with the newly sequenced U. meridionalis mitogenomes. Transfer RNA genes (tRNAs) were searched for by reconstructing their cloverleaf structures using the tRNAscan-SE 2.0 software with default parameters (Chan et al., 2021). Ribosomal RNA genes (rRNAs) were identified by the RNAweasel Tool2 and by aligning the homologous rRNAs. The free-standing and intronic open reading frames (orfs) were found by Open Reading Frame Finder at the NCBI website. Intron insertion-sites were determined manually by aligning the intron-containing homologous genes including atp1, cob, cox1, cox2, nad3, nad5, rnl, and rns. The corresponding genes in the Ulva compressa (KY626327) mitogenome were used as a reference (Liu et al., 2020). Intron name was defined as host gene plus insertion site. The class and core structure of all these introns were determined using the RNAweasel Tool and Mfold (Zuker, 2003). The physical maps of the circular mitogenomes were generated by using Organellar Genome DRAW (OGDRAW) (Greiner et al., 2019).
The DNA-directed RNA polymerase gene (rpo) in mitochondrial circular plasmid of U. meridionalis, which we call pUme, was searched against the database of the 98.5 Mbp haploid genome of Ulva mutabilis Föyn (De Clerck et al., 2018), which has been regarded as a taxonomic synonym of Ulva compressa Linnaeus (Steinhagen et al., 2019a), with tblastn.3 We detected two contigs, WT279 and WT234, which contained only the mitochondrial homologous rpos and orfs with their multiple tandem arrangement. Two putative circular plasmids named pUmu1 and pUmu2 were predicted and annotated using Open Reading Frame Finder at the NCBI website and the tRNAscan-SE 2.0 software. To conduct a thorough search for mitochondrial plasmid-like sequences in Ulva mitogenomes, the putative circular plasmid sequences (pUme, pUmu1, and pUmu2) were searched against the NCBI nucleotide database with blastn.
To avoid phylogenetic artifacts caused by convergent base composition, the phylogenetic tree was constructed based on the amino acid (aa) sequences of full-length rpos found in Ulva mtDNAs and putative plasmids. Multiple sequence alignments of Rpos were conducted using ClustalX 1.83 with the default settings (Thompson et al., 1997). The phylogenetic relationships were inferred by using the Maximum Likelihood (ML) method based on the JTT matrix-based model (Jones et al., 1992) using MEGA 7.0 (Kumar et al., 2016). Initial tree(s) for the heuristic search were obtained by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model. There was a total of 1,022 positions in the final dataset of Rpos.
Base composition of U. meridionalis mitogenomes and plasmid DNA was determined using MEGA 7.0 (Kumar et al., 2016). Differences and identity values of gene sequences were calculated by use of the BioEdit v7.1.9 software (Hall, 1999). Tandem repeats were detected with Tandem Repeats Finder using the default settings (Benson, 1999). Inverted repeats were identified with Inverted Repeats Finder using the default settings.4 The aa sequences of 32 genes including 30 PCGs and two conserved orfs shared by the ulvalean mitogenomes sequenced thus far (Supplementary Table 1) were subjected to concatenated alignments using ClustalX 1.83 with the default settings (Thompson et al., 1997). The evolutionary history was inferred by using the ML method based on the JTT matrix-based model (Jones et al., 1992). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model. There was a total of 10,526 positions in the final aa dataset. Phylogenomic analysis was conducted with 1,000 bootstrap replicates using MEGA 7.0 (Kumar et al., 2016).
Five mitochondrial genomes of U. meridionalis (Ume) had at least 5000 × depth of coverage (Supplementary Table 2 and Supplementary Figures 4–8) and mapped as complete circular molecules with the size ranging from 82.94 kb in Ume4 and Ume5 to 111.49 kb in Ume1 (Table 1), displaying the most obvious genome expansion when compared with other Ulva mitogenomes sequenced (Melton et al., 2015; Suzuki et al., 2018; Liu et al., 2020; Liu F. et al., 2022). Their overall AT content of U. meridionalis mitogenomes is 66.15–66.22%, which is within the range (61.16–67.83%) of the Ulva mitogenomes reported (Liu F. et al., 2022). The 111.49-kb mitogenome of Ume1 is the largest Ulva mitogenome sequenced so far (Figure 1A), and twice the smallest one which is the 55.81 kb mtDNA in Ulva sp. TM708 (Liu F. et al., 2022). The expansion of U. meridionalis mitogenomes is mainly due to the integration of plasmid-derived DNA fragments (Figure 1B), as well as the proliferation of introns (Figure 2).
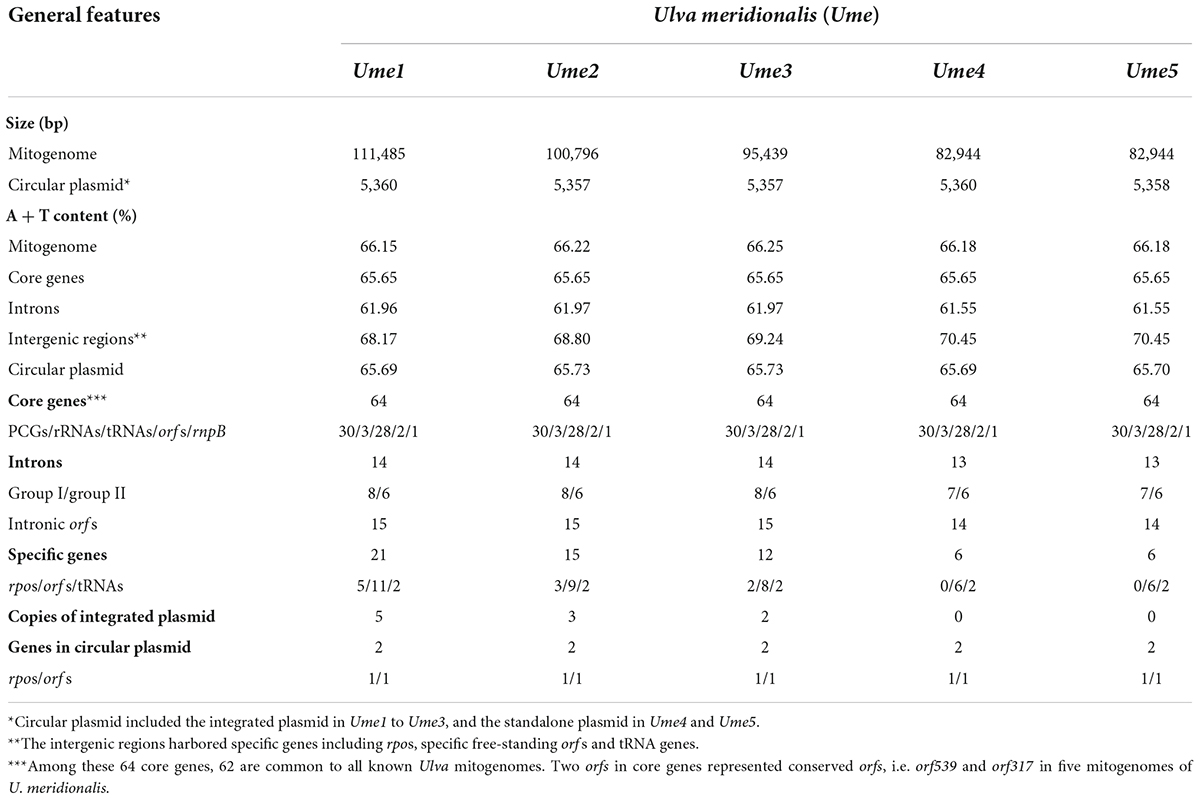
Table 1. General features of mitochondrial genomes and circular plasmids in Ulva meridionalis.
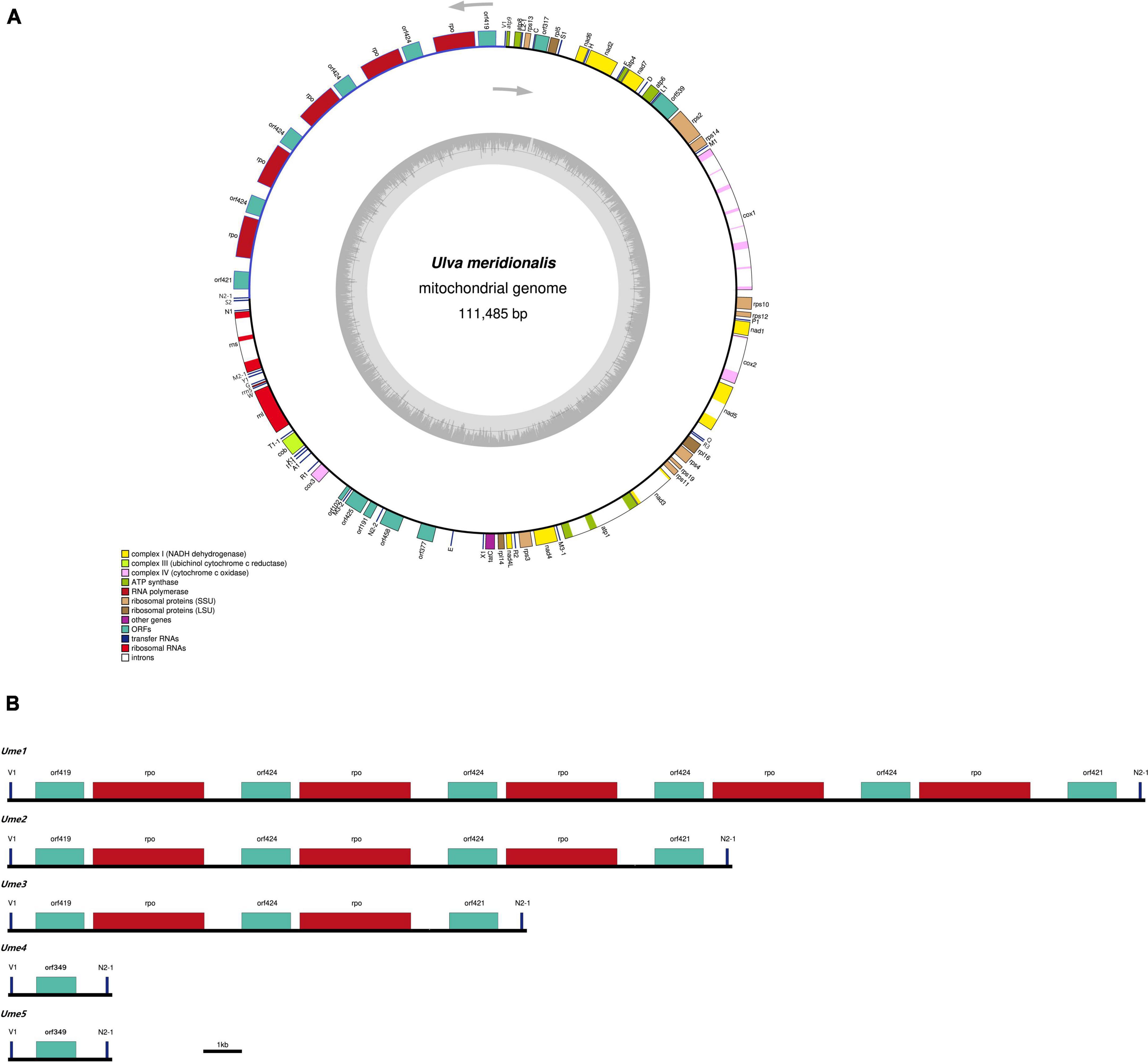
Figure 1. The mitochondrial genomes of Ulva meridionalis. (A) Gene map of the largest U. meridionalis mitochondrial genome (Ume1). Genes (filled boxes) shown on the outside of the map are transcribed counterclockwise. The smaller circle on the inside of the gene mapping shows a graph of the GC content. Genes are color coded for their functional gene group (legend bottom left). (B) Variations in the trnV1-trnN2-1 intergenic region of five U. meridionalis mitogenomes (Ume1–Ume5) due to the tandem integration of circular plasmid (pUme).
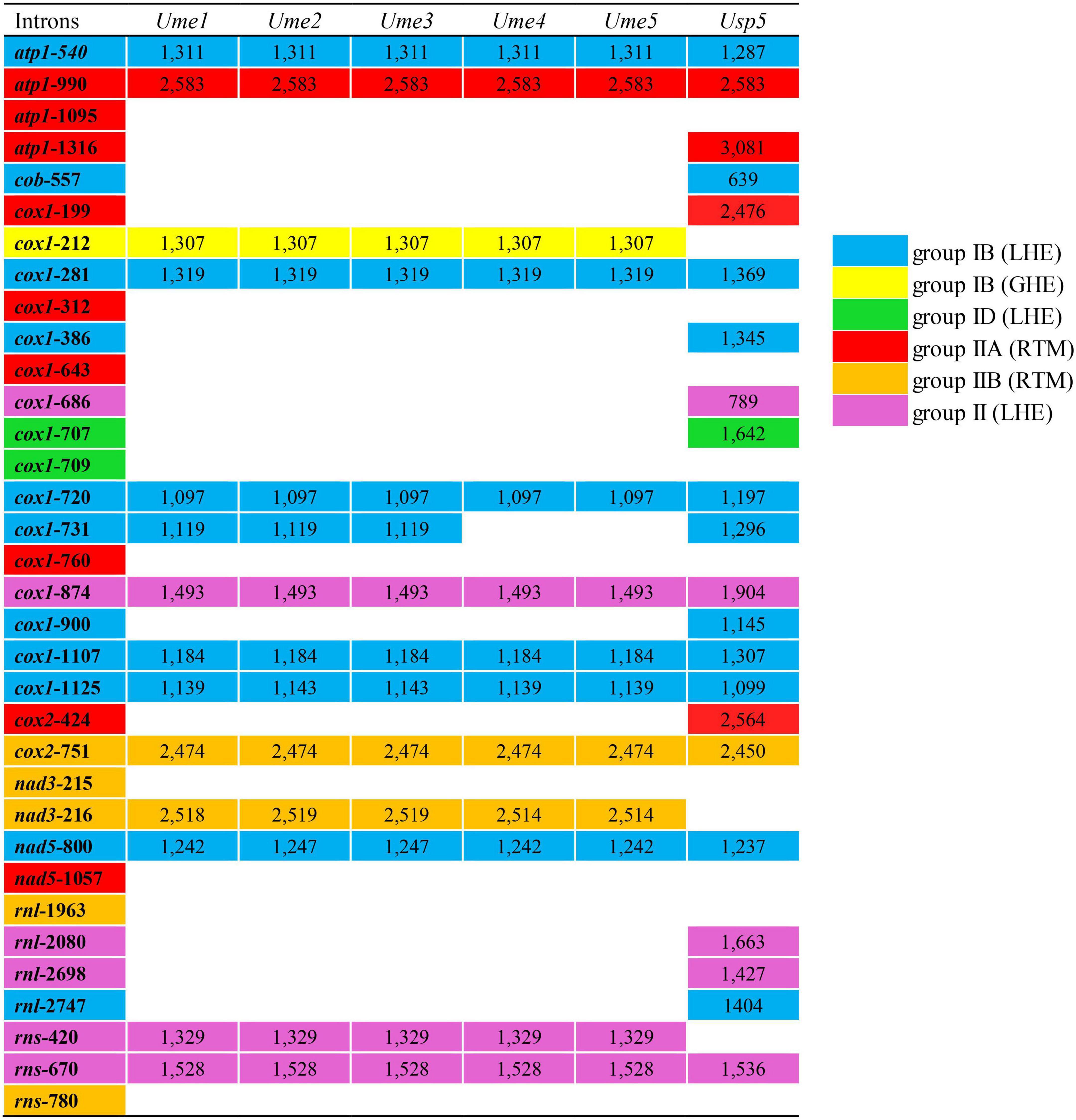
Figure 2. Insertion site, size and group of introns detected in five mitogenomes of Ulva meridionalis (Ume1–Ume5), as well as the mtDNA of Ulva sp. (MN853878) (Usp5) for comparative analysis. Ulva prolifera (MN853878) deposited in the GenBank database was corrected to Ulva sp. (MN853878), due to its wrong species name assignment. Intron name was defined as host gene plus insertion site which was determined by comparing homologous genes relative to the mitogenome of U. compressa (KY626327) (Liu F. et al., 2022). All insertion sites previously detected in the sequenced Ulva mitogenomes were listed in the first column. The intron cox1-214 is corrected to intron cox1-212.
The five U. meridionalis mitogenomes share the same set of 62 core genes as other Ulva mitogenomes (Liu F. et al., 2022), including 30 PCGs, three rRNAs (rnl, rns, and rrn5), 26 tRNAs, two conserved free-standing orfs, and one putative RNA subunit of RNase P (rnpB) (Table 1). One conserved orf (orf219 in U. meridionalis) situated between trnX1 and rpl14 encodes the SecY-independent transporter protein TatC (gene name: tatC or mttB) (Turmel et al., 2016). A conserved EXXDSEL motif at the C-terminal fragment of TatC is shared by Ulvales and Ulotrichales, but not found in Oltmannsiellopsidales and Bryopsidales. The duplication mutation of trnM3(cau) was detected in the U. meridionalis mitogenomes (Table 2), which was not found in other Ulva mtDNAs, and resulted into two perfect copies of trnM3(cau) which located in the nad4-atp1 and cox3-trnE intergenic regions, respectively. The function of trnX1 is unknown, and the structure of trnS3 is seriously degraded to loss of function in U. meridionalis mtDNAs. On top of that, two copies of specific trnN2(guu) found only in the U. meridionalis mitogenomes are a bit different from the core trnN1(guu) in the sequence of loops, especially DHU loop (Table 2), while the core trnN1(guu) is very conserved in all known ulvalean mitogenomes. These facts indicate that the trnN2(guu) is likely to appear in the U. meridionalis mitogenome through horizontal transfer rather than trnN1(guu) duplication.

Table 2. The aligned sequences of tRNA genes with duplication mutation in U. meridionalis mitogenomes.
The content of mitochondrial introns changed a bit at the intraspecific level in U. meridionalis, i.e., 14 introns in Ume1 to Ume3 and 13 in Ume4 and Ume5. The intron cox1-731 was present in Ume1 to Ume3 but absent in Ume4 and Ume5 (Figure 2). These introns were located in six housekeeping genes, including atp1 (two introns), cox1 (seven or six), cox2 (one), nad3 (one), nad5 (one), and rns (two). Most introns harbored an intact intronic orf encoded an LAGLIDADG homing endonuclease (LHE), or a GIY-YIG homing endonuclease (GHE), or a reverse transcriptase/maturase (RTM) (Dai et al., 2003; Haugen et al., 2005; Lambowitz and Zimmerly, 2011), but in intron cox1-212, the GHE gene was split into two parts, orf152 and orf158. These introns can be divided into five types according to their secondary structure and intronic-encoding proteins, including group IB (LHE), group IB (GHE), group IIA (RTM), group IIB (RTM), and group II (LHE) (Figure 2). Five intron families, including intron atp1-1316, cob-557, cox1-686, cox1-707, and cox1-720, were discovered in Ulva for the first time, and other introns have been reported before (Liu F. et al., 2022).
The total intron length in U. meridionalis mitogenomes reached 20.52–21.65 kb, greatly exceeding the values of all reported Ulva mitogenomes (4.90–19.73 kb). The intronic AT content (61.55–61.97%) was significantly lower than that in mitogenomes (Table 1), indicating that introns as heterogeneous DNA showed distinct characteristics in base composition. Recently, a new record from the GenBank database on the 107.51-kb mitogenome of Ulva sp. (MN853878) showed that it contained a total of 22 introns (Figure 2), with a total intron length of 35.44 kb. The proliferation of introns greatly leads to the expansion of Ulva mitochondrial genomes, as was similar to that observed in mtDNAs of Ulotrichales and Bryopsidales (Pombert et al., 2004; Turmel et al., 2016; Zheng et al., 2018; Repetti et al., 2020).
Based on comparative analysis of these five U. meridionalis mitogenomes, we observed that five copies of a 5,360-bp plasmid-derived sequence were tandemly integrated into the trnV1-trnN2-1 intergenic region in the mitogenome of Ume1 (Figure 1), and three and two copies of its homolog with the size of 5,357 bp were integrated into the same target site in mtDNAs of Ume2 and Ume3, respectively. However, none of such plasmid-derived sequence was present in mtDNAs of Ume4 and Ume5. The copy number of integration plasmid was well supported by the similar depth of coverage between plasmid-derived sequence and the rest of mitochondrial genome (Supplementary Tables 2, 3 and Supplementary Figures 4–6). It is worth noting that this plasmid sequence was not found in mtDNAs of Ume4 and Ume5 (Supplementary Figures 7, 8), but a standalone mitochondrial circular plasmid was discovered in Ume4 and Ume5 (Figure 3A), with the size of 5,360 and 5,358 bp, respectively. This standalone plasmid in Ume4 and Ume5 is highly consistent with the integrated plasmid found in Ume1 to Ume3 in sequence, but slightly different in size mainly due to changes in the two polyC regions. The plasmid contigs have only 350 × and 1,000 × depth of coverage in Ume4 and Ume5, respectively, which are significantly lower than that of mitochondrial contigs (9,100 × in Ume4 and 13,500 × in Ume5) (Supplementary Table 4). These facts indicate that the circular plasmid exists in a free state in Ume4 and Ume5, and the integrated plasmid in Ume1 to Ume3 should be from the free circular plasmid. Undoubtedly, it is reasonable that the circular plasmid exists in mitochondria of Ume 1 to Ume3 in both free and integrated states.
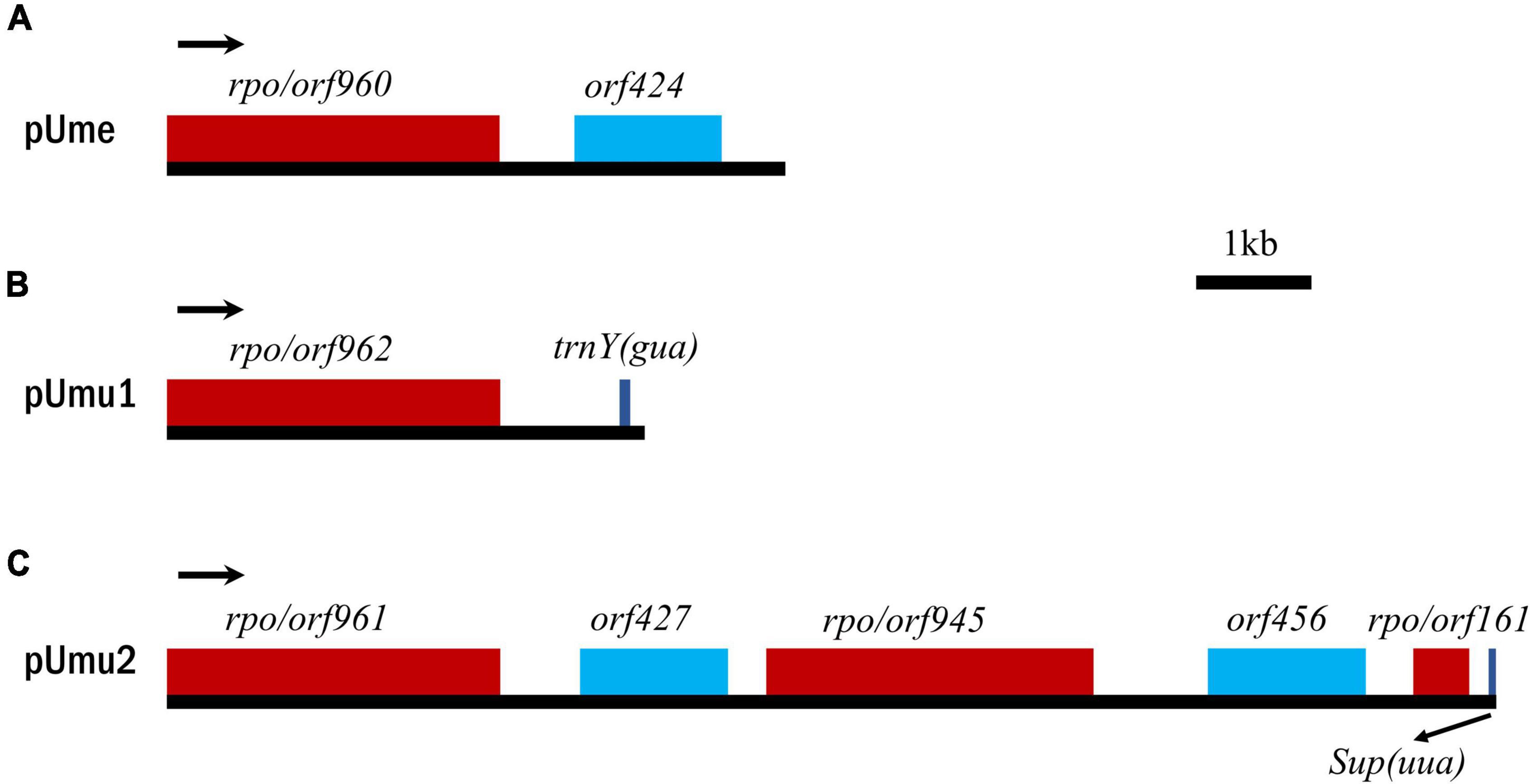
Figure 3. Comparison of gene maps of circular plasmids. (A) Circular plasmid (pUme) in U. meridionalis. (B,C) Two putative plasmids (pUmu1 and pUmu2) in U. mutabilis. Arrows indicate the direction of gene transcription.
The free circular plasmid found in Ume4 and Ume5, as well as the integrated plasmid in Ume1 to Ume3, which we call pUme (Figure 3A), have the AT content of 65.69–65.73%, which is very similar to that of U. meridionalis mitogenomes (66.15–66.22%). The similarity of the AT content between pUme and U. meridionalis mitogenomes might be closely related to their coevolution and plasmid origin, as was markedly different from that observed in plants. Plant mitochondrial plasmids usually exhibit higher AT content than that of mitogenomes, and are suspected to have been acquired from a fungal donor (Handa, 2008; Beaudet et al., 2013; Warren et al., 2016).
The circular plasmids harbored one intact DNA-directed RNA polymerase (rpo) orf960 and a specific orf424 with unknown function based on blastp search. A small tandem repeat with the period size of 35 bp was located between rpo and orf424. The plasmid-encoded Rpo is in the single-chain RNA polymerase family of phage type that executes a specific-promoter transcription process similar to other multichain RNA polymerases (Cermakian et al., 1997; Yin et al., 2010; Warren et al., 2016). Previously, DNA and/or RNA polymerase genes were usually found in linear mitochondrial plasmids in fungi and plants, while some circular mitochondrial plasmids often contained DNA polymerase or reverse transcriptase genes (Hausner, 2003; Gualberto et al., 2014). It is worth noting that our study confirms that the rpo found in U. meridionalis mitogenomes is derived from the mitochondrial circular plasmid, and the rpo-containing circular plasmid represents a new class of mitochondrial plasmids, which expands our understanding of eukaryotic plasmid diversity.
Based on tblastn search of rpo gene in the 98.5 Mbp haploid genome of U. mutabilis (De Clerck et al., 2018), we detected two contigs, WT279 and WT234, which contained only the mitochondrial homologous rpos and orfs with their multiple tandem arrangement. Their repeat units are rpo/orf962-ψtrnY(gua) and rpo/orf961-orf427-rpo/orf945-orf456-rpo/orf161-ψSupres(uua) in WT279 and WT234, respectively (Figures 3B,C). A total of three full-length rpos, and one incomplete rpo/orf161 which is very similar to the homologous region in rpo/orf945, were detected in U. mutabilis. Considering that the complete sequences of these putative plasmids were not found in the mitogenome (WT177) of U. mutabilis, we infer that the reads assembled into these two contigs should belong to mitochondrial plasmid DNA. Two putative circular plasmids named pUmu1 and pUmu2 were constructed, with the size of 4,145 and 11,556 bp, respectively.
Comparisons of five U. meridionalis mitogenomes allowed us to model the integration process of circular plasmid into mtDNAs. The integration of circular plasmid should be via homologous recombination. A 152-bp homologous sequence is shared by orf416 (or orf349) in U. meridionalis mtDNAs and orf424 in standalone circular plasmid DNA, and these sequences are nearly completely identical, except that there is only one base difference at the integration site (the 43rd base of homologous sequences) (Figure 4A). The gtA(V) in orf416 (or orf349) is instead of gtC(V) in orf424. After the first integration of the circular plasmid, two chimeric orfs (orf419 and orf421) were formed. The subsequent integration of circular plasmid should occur at the same recombination site in orf419 which retained the only differential base (Figure 4B), and generate two chimeric orfs which are identical to orf419 and orf424, respectively. The observed integration of circular plasmid in U. meridionalis mitogenomes is a more recent evolutionary event, considering the sequence consistency and integrity between integrated and standalone plasmids.
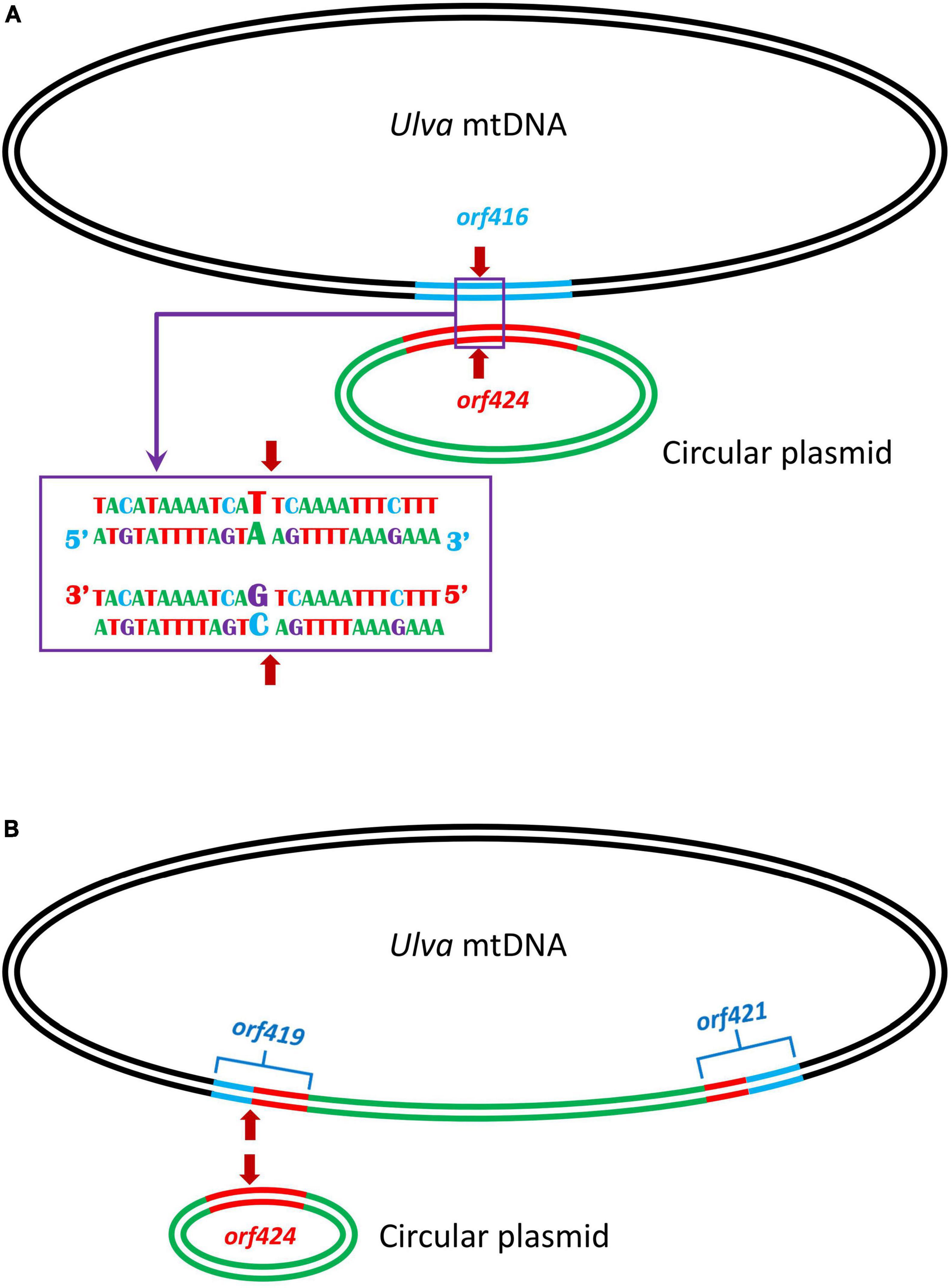
Figure 4. Model of tandem integration of circular plasmid into the Ulva meridionalis mitochondrial genome. (A) Integration of circular plasmid involving homologous recombination. A 152-bp sequence is shared by orf416 in Ulva mtDNA and orf424 in circular plasmid, and the homologous sequences are nearly completely identical, except that there is only one base difference at the integration site (the 43rd base of homologous sequences). The red arrow shows the integration site. (B) After the first integration of the circular plasmid, two chimeric orfs (orf419 and orf421) were formed, and the subsequent integration of circular plasmid is more likely to occur in orf419 which retained the only differential base at the integration site.
Many highly homologous sequences of these plasmids (pUme, pUmu1, and pUmu2) can be detected in mtDNAs of Ulva species but not found in mitogenomes of other ulvophyceae lineages (e.g., Ulotrichales, Bryopsidales, and Oltmannsiellopsidales) based on blastn search, but they showed varying levels of disruption and degradation in DNA sequence and different types of gene arrangement in structure, indicating the high diversity of plasmids and the differentiation of integration sites via homologous recombination. Sequence homology analysis of these rpos and orfs (orf424 in pUme, and orf427 and orf456 in pUmu2) showed that they were highly homologous with plasmid-derived rpos and orfs previously found in Ulva mitogenomes (Figures 5A–C; Liu F. et al., 2022). However, we did not detect the ψtrnY(gua) in all sequenced Ulva mtDNAs, while the homologous suppressor tRNAs of ψSupres(uua) were found in the nad6-trnS1 intergenic region of Ulva rigida mtDNA and in the trnR2-rps3 intergenic region of Ulva flexuosa mtDNAs. These facts showed that the integration of different mitochondrial plasmid DNA into the mitogenome is a common phenomenon in the evolution of Ulva mitogenomes.

Figure 5. Schematic alignments of specific free-standing orfs detected in all sequenced mitogenomes (Supplementary Table 1) and putative circular plasmids of Ulva species. (A) Homologs of plasmid orf424. (B) Homologs of putative plasmid orf427. (C) Homologs of putative plasmid orf456. (D) Homologs of orf102. The solid squares with different colors represent different sources of specific orfs, including different intergenic regions (IRs) and putative plasmid. The orfs in blue font are from Ulva species in lineage I, and the orfs in red font are from Ulva species in lineage II. The orfs transcribed on the same strand as the core genes are marked with a positive sign (+), and the orfs transcribed on the minus strand are marked with a negative sign (–).
Plasmid-derived sequence is one of the most important driving forces for the expansion of Ulva mitogenomes. In U. meridionalis mitogenomes, almost all specific free-standing orfs originate from plasmid. Although there is no evidence that the orf102 which is located at the cox3-trnX1 intergenic region of U. meridionalis mtDNAs is derived from plasmids, its homologs are present in different intergenic regions of other several Ulva mtDNAs (Figure 5D). The plasmid-derived DNA is even up to 33.96 kb in Ume1, accounting for 30% of the mitogenome, and appear at three intergenic regions, including trnV1-trnN2-1, trnM3-2-trnN2-2, and trnN2-2-trnE. Similar phenomena that the integration of circular or linear plasmids has greatly contributed to the increase of mitogenome size have also been reported in plants and fungi (e.g., Robison and Wolyn, 2005; Allen et al., 2007; Formighieri et al., 2008; Himmelstrand et al., 2014).
Mitogenomes of U. meridionalis are readily distinguished by numerous DNA polymorphisms which have been characterized in this study (Supplementary Table 5), but their intraspecific polymorphisms are extremely low at the core genes. Nearly all core genes are identical in nucleotide sequence, and only two base substitutions were observed in two PCGs, including the 288 ggT → ggG transversion (amino acid: G, same sense mutation) which occurred only in cox3 of Ume5, and the 567 ttA → ttT transversion (amino acid: L → F, missense mutation) which happened in rps10 of Ume1 to Ume3 not in that of Ume4 and Ume5. In addition, a 1-bp (A) deletion mutation occurred in the intronic orf575 (RTM) of intron nad3-216 in Ume1, leading to premature termination of its homolog (orf250). A 2-bp (AT) deletion mutation which happened in the latter part of the free-standing orf416 caused the early termination of its homolog (orf349) in Ume4 and Ume5. Intraspecific polymorphisms of five U. meridionalis mitogenomes are significantly high at the intergenic regions and intronic non-coding regions. The divergence in length and sequence of intergenic regions in U. meridionalis mitogenomes was mainly caused by frequent insertion of foreign DNA fragments (e.g., plasmid DNA), rapid accumulation of multiple mutations, and dynamic fluctuation of repeat sequences (Supplementary Table 5), indicating the intergenic regions showed a rapid rate of DNA sequence evolution. There is a mutation hot spot region in the trnE-trnX1 intergenic region, which can be divided into two DNA polymorphisms, i.e., type I in Ume1 to Ume3 and type II in Ume4 and Ume5 (Supplementary Table 5). The insertion of a specific DNA fragment occurred in the trnA1-trnR1 intergenic region of the mitogenomes of Ume1 to Ume3, but this DNA fragment is not present in mtDNAs of Ume4 and Ume5 as well as all other Ulva mtDNAs. The integrated DNA fragment with the size of 628 bp contains high A + T content (75.64%). A 7-bp sequence (CCTTTGC) is shared by the U. meridionalis mtDNAs and integrated DNA fragment, which should have function in the integration of this fragment.
All genes are coded on one strand in U. meridionalis mitogenomes, and the order of core genes is almost the same as that of other Ulva mitogenomes (Melton et al., 2015; Cai et al., 2018; Liu et al., 2020), except for the transposition of trnE(uuc) from the intergenic region of trnQ-nad5 to that of orf377-trnX1 (Figure 1A). We observed that some plasmid-derived rpos and/or orfs sometimes encoded on another strand (Liu F. et al., 2022), which seems to depend on the integration direction of the plasmid DNA. This rarely occurs in core genes, but there is one exception. We previously found that large differences in the content of small inverted/tandem repeats can be observed in U. compressa mitogenomes at the intraspecific level, and the integration of small mobile inverted repeats has been shown to produce genomic rearrangements, which caused a gene cluster consisting of eight genes coding on another strand in one mitogenome of U. compressa (Liu et al., 2020). In addition to the repeats in the integrated plasmid sequence, five U. meridionalis mitogenomes shared almost the same set of small inverted/tandem repeats, most of which were present in intergenic regions. Only one tandem repeat with the period size of 16 bp appeared only in the orf421-trnN1-2 intergenic region of Ume2 and Ume3, due to a mutation event of this repeat sequence.
The ML phylogenetic analyses were conducted and restricted to full-length plasmid(-derived) Rpos, because many of these split Rpos found in Ulva mtDNAs are too short and divergent to provide comprehensive phylogenetic signals due to their rapid evolution. The Rpo dataset, including the newly discovered Rpos in circular plasmids and mitogenomes of U. meridionalis, and three Rpos found in two putative plasmids of U. mutabilis, as well as three full-length Rpos we previously reported in Ulva mtDNAs, represents a Chlorophyta-specific lineage, which is far away from other eukaryotic lineages (Liu F. et al., 2022). These full-length Rpos were clearly resolved into three different clades (Rpo-I, II and III), and each clade contained one of three copies of Rpo from two putative plasmids in U. mutabilis (Figure 6), indicating that the specific Ulva lineage of Rpo family showed great diversity, even at the intraspecific level. However, our understanding of these rpo genes in green algae is only the tip of the iceberg. Protein homologs of mitochondrial plasmid rpos and orfs are very common in different intergenic regions of Ulva mitogenomes (Figure 5; Liu F. et al., 2022), indicating that the mitochondrial plasmid could be frequently integrated into many different target sites via horizontal transfer in the evolution of Ulva mitogenomes. These plasmid-derived sequences have rapidly diverged in Ulva mtDNAs, leaving numerous plasmid-derived genes or their remnants in the form of full-length or split rpos and orfs, as well as large intergenic regions, due to multiple mutations that introduce internal stop codons, or insertions or deletions that result in frameshifts or loss of conserved domains. The random incidence of destruction of plasmid-derived rpos and orfs suggests that their existence is not the original characteristic of Ulva mitogenomes, and there is no selective pressure to maintain their integrity in mitogenomes (Robison and Wolyn, 2005). These mobile rpos and orfs are significantly different from the core genes in evolution, and could not be used to determine phylogenetic relationships of Ulva species.

Figure 6. Phylogenetic tree based on Maximum Likelihood (ML) analysis of amino acid (aa) sequences of the full length Rpo proteins found in Ulva mitogenomes and putative plasmids. The ML analysis was performed with 1,000 bootstrap replicates using MEGA 7.0. The bootstrap support values greater than 70% were displayed at branches. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees. The tree was rooted with two Rpo proteins from Punica granatum (XP031374084) and Cocos nucifera (YP009315942), respectively.
Based on the ML phylogenomic analysis of the aa sequences of 32 genes including 30 PCGs and two conserved orfs from the 39 ulvalean mtDNAs, Ulva species robustly clustered into two primary branches representing two Ulva genetic lineages, I and II, displaying the evolutionary nature of double crown radiation in the phylogeny and speciation of the Ulva group, as is consistent with our previous findings based on phylogenomics analysis of chloroplast genes (Liu and Melton, 2021; Liu F. et al., 2022). In the Ulva lineage I, U. meridionalis (Ume1–Ume5) first clustered with Ulva sp. (KP720617) and Ulva sp. (MN853878) to form an independent subclade with high support value (100%) (Figure 7). An incomplete mitogenome (MN861072) from one Ulva sample designated as U. meridionalis previously has been sequenced (Kang et al., 2020), which lacks the region from rps10 to cox1. However, our result of phylogenomic analysis showed that this Ulva sample was closely related to U. flexuosa (Figure 7). The mitochondrial nucleotide (nt) sequences of some PCGs (rps14, rpl5, atp9, tatC, and rps11), two conserved orfs (orf505 and orf315) and some intergenic regions in this sample are completely consistent with that in U. flexuosa, while other PCGs appear to be chimeras of U. flexuosa (KX455878) and Ulva sp. (MN853878). These results suggest that the mitogenome of this sample is an abnormal chimera, which may represent a cryptic species which we named Ulva sp. (MN861072) in this study.
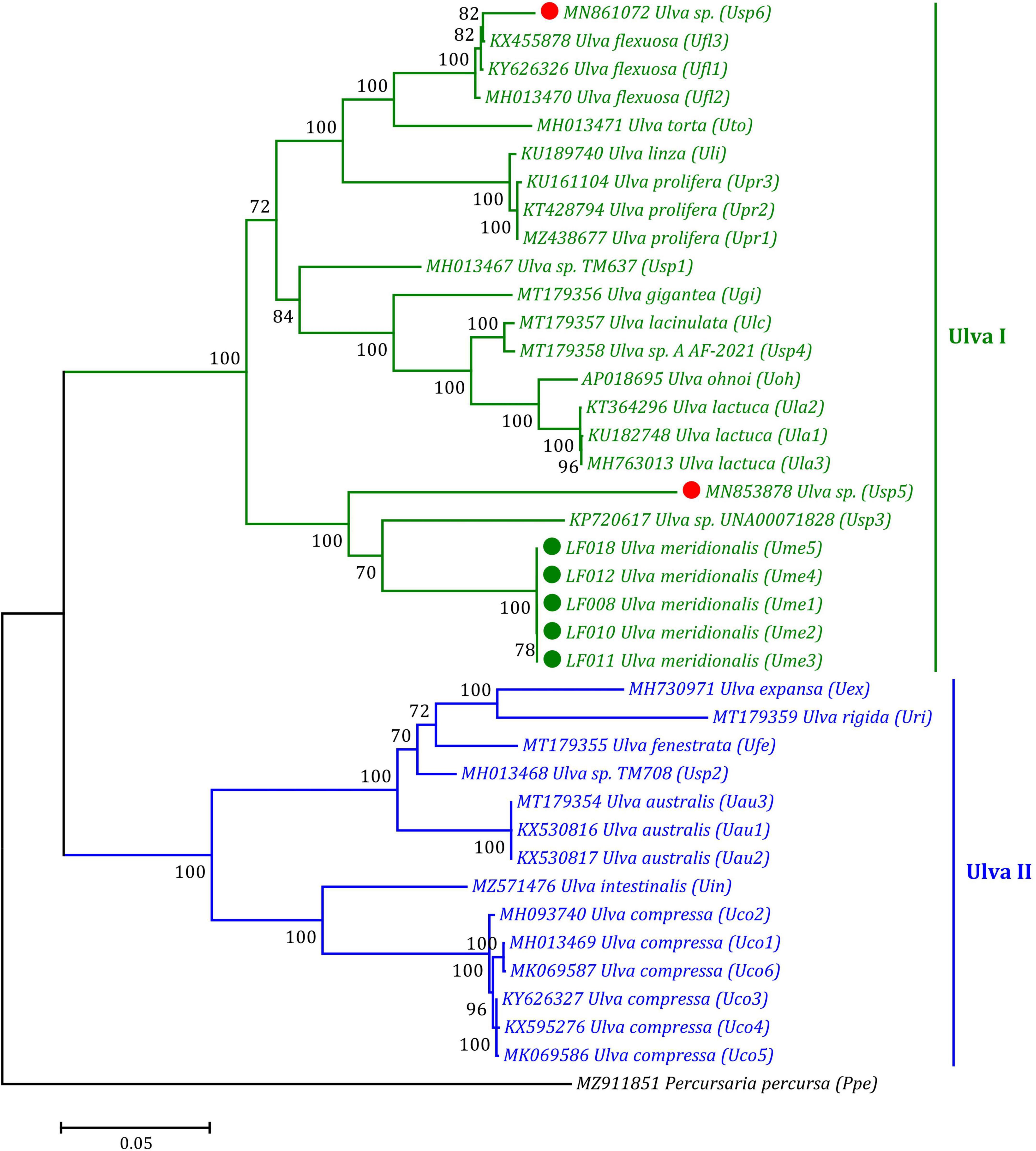
Figure 7. Phylogenomic tree based on Maximum Likelihood (ML) analysis of the amino acid (aa) sequences of 32 genes in 39 ulvalean mitogenomes. The ML analysis was performed with 1,000 bootstrap replicates using MEGA 7.0. The bootstrap support values greater than 70% were displayed at branches. Branch lengths were proportional to the amount of sequence change, which were indicated by the scale bar below the trees. Ulva prolifera (MN853878) and Ulva meridionalis (MN861072) deposited in the GenBank database were corrected to Ulva sp. (MN853878) and Ulva sp. (MN861072), respectively, due to their wrong species name assignment. The tree was rooted with Percursaria percursa (MZ911851) as an outgroup.
The mitochondrial genomes of Ulva species exhibit high diversity at the interspecific and intraspecific level due to their multiple variations which involves the integration of foreign DNA fragments, the acquisition or loss of introns, the dynamic change of repeat sequences, genome rearrangement, and multiple mutations (Liu et al., 2017, 2020; Liu F. et al., 2022). In this study, many new discoveries were unraveled in the evolution of Ulva mitogenomes. First, the expansion of U. meridionalis mitogenomes is mainly due to the tandem integration of mitochondrial circular plasmid as well as the proliferation of introns at the intraspecific level. The 111.49-kb U. meridionalis mitogenome is the largest Ulva mitogenome sequenced so far, even larger than some Ulva chloroplast genomes. Second, a 5.36-kb standalone mitochondrial circular plasmid (pUme) identified in U. meridionalis and two putative circular plasmids (pUmu1 and pUmu2) detected in U. mutabilis all harbor RNA polymerase genes (rpos) which have only been found in mitochondrial linear plasmids in fungi and plants, which expand our understanding of eukaryotic plasmid diversity. Third, many highly homologous sequences of these plasmids (pUme, pUmu1, and pUmu2) can be detected in intergenic regions of Ulva mitogenomes sequenced so far, indicating that the integration of different mitochondrial plasmid DNA into the mitogenome is a common phenomenon in the evolution of Ulva mitogenomes. Fourth, these plasmid-derived rpos and orfs have rapidly diverged and degenerated in Ulva mtDNAs, displaying markedly different evolution patterns from the core genes. The frequent integration and rapid divergence of plasmid-derived sequences greatly shape the diversity of Ulva mitogenomes at the intraspecific level. Finally, the ML phylogenomic analysis clearly depicted the evolutionary nature of double crown radiation in the phylogeny and speciation of the Ulva group.
The data presented in the study are deposited in the GenBank database, accession number ON402236 - ON402240. Data is publicly available by the following links: https://www.ncbi.nlm.nih.gov/nuccore/ON402236, https://www.ncbi.nlm.nih.gov/nuccore/ON402237, https://www.ncbi.nlm.nih.gov/nuccore/ON402238, https://www.ncbi.nlm.nih.gov/nuccore/ON402239, and https://www.ncbi.nlm.nih.gov/nuccore/ON402240.
FL designed the study, performed the analysis, and wrote the manuscript. FL, HW, and WS performed the experiments. All authors have read and approved the final version of the manuscript.
This work was financially supported by the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDA23050302/XDA23050403), the National Natural Science Foundation of China (No. 41876165), the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (No. QYZDB-SSW-DQC023), the Science and Technology Basic Resources Investigation Program of China (No. 2018FY100200), and the Major Scientific and Technological Innovation Project of Shandong Province (No. 2019JZZY020706).
We thank Shanshan Wang and Wei Luan for their assistance in algal collection.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.937398/full#supplementary-material
Allen, J. O., Fauron, C. M., Minx, P., Roark, L., Oddiraju, S., and Lin, G. N. (2007). Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics 177, 1173–1192. doi: 10.1534/genetics.107.073312
Baidyaroy, D., Hausner, G., and Bertrand, H. (2012). In vivo conformation and replication intermediates of circular mitochondrial plasmids in Neurospora and Cryphonectria parasitica. Fungal Biol. 116, 919–931. doi: 10.1016/j.funbio.2012.06.003
Beaudet, D., Terrat, Y., Halary, S., de la Providencia, I. E., and Hijri, M. (2013). Mitochondrial genome rearrangements in Glomus species triggered by homologous recombination between distinct mtDNA haplotypes. Genome Biol. Evol. 5, 1628–1643. doi: 10.1093/gbe/evt120
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Blomster, J., Bäck, S., Fewer, D. P., Kiirikki, M., Lehvo, A., Maggs, C. A., et al. (2002). Novel morphology in Enteromorpha (Ulvophyceae) forming green tides. Am. J. Bot. 89, 1756–1763. doi: 10.3732/ajb.89.11.1756
Cai, C., Liu, F., Jiang, T., Wang, L., Jia, R., and Zhou, L. (2018). Comparative study on mitogenomes of green tide algae. Genetica 146, 529–540. doi: 10.1007/s10709-018-0046-7
Cermakian, N., Ikeda, T. M., Miramontes, P., Lang, B. F., Gray, M. W., and Cedergren, R. (1997). On the evolution of the single-subunit RNA polymerases. J. Mol. Evol. 45, 671–681. doi: 10.1007/PL00006271
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Dai, L., Toor, N., Olson, R., Keeping, A., and Zimmerly, S. (2003). Database for mobile group II introns. Nucleic Acids Res. 31, 424–426. doi: 10.1093/nar/gkg049
De Clerck, O., Kao, S. M., Bogaert, K. A., Blomme, J., Foflonker, F., Kwantes, M., et al. (2018). Insights into the evolution of multicellularity from the sea lettuce genome. Curr. Biol. 28, 2921–2933. doi: 10.1016/j.cub.2018.08.015
Formighieri, E. F., Tiburcio, R. A., Armas, E. D., Medrano, F. J., Shimo, H., Carels, N., et al. (2008). The mitochondrial genome of the phytopathogenic basidiomycete Moniliophthora perniciosa is 109 kb in size and contains a stable integrated plasmid. Mycol. Res. 112, 1136–1152. doi: 10.1016/j.mycres.2008.04.014
Fort, A., McHale, M., Cascella, K., Potin, P., Usadel, B., Guiry, M. D., et al. (2020). Foliose Ulva species show considerable inter-specific genetic diversity, low intra-specific genetic variation, and the rare occurrence of inter-specific hybrids in the wild. J. Phycol. 57, 219–233. doi: 10.1111/jpy.13079
Gao, G., Zhong, Z. H., Zhou, X. H., and Xu, J. T. (2016). Changes in morphological plasticity of Ulva prolifera under different environmental conditions: a laboratory experiment. Harmful Algae 59, 51–58. doi: 10.1016/j.hal.2016.09.004
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res 47, W59–W64. doi: 10.1093/nar/gkz238
Gualberto, J. M., Mileshina, D., Wallet, C., Niazi, A. K., Weber-Lotfi, F., and Dietrich, A. (2014). The plant mitochondrial genome: dynamics and maintenance. Biochimie 100, 107–120. doi: 10.1016/j.biochi.2013.09.016
Guiry, M. D., and Guiry, G. M. (2022). AlgaeBase. World-Wide Electronic Publication. Galway: National University of Ireland.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98.
Handa, H. (2008). Linear plasmids in plant mitochondria: peaceful coexistences or malicious invasions? Mitochondrion 8, 15–25. doi: 10.1016/j.mito.2007.10.002
Haugen, P., Simon, D. M., and Bhattacharya, D. (2005). The natural history of group I introns. Trends Genet. 21, 111–119. doi: 10.1016/j.tig.2004.12.007
Hausner, G. (2003). Fungal mitochondrial genomes, plasmids and introns. Appl. Mycol. Biotechnol. 3, 101–131. doi: 10.1016/S1874-5334(03)80009-6
Hausner, G. (2011). “Introns, mobile elements, and plasmids,” in Organelle Genetics, ed. C. E. Bullerwell (Berlin/Heidelberg: Springer), 329–358. doi: 10.1007/978-3-642-22380-8_13
Hayden, H. S., and Waaland, J. R. (2002). Phylogenetic systematics of the Ulvaceae (Ulvales, Ulvophyceae) using chloroplast and nuclear DNA sequences. J. Phycol. 38, 1200–1212. doi: 10.1046/j.1529-8817.2002.01167.x
Himmelstrand, K., Olson, A., Brandström Durling, M., Karlsson, M., and Stenlid, J. (2014). Intronic and plasmid-derived regions contribute to the large mitochondrial genome sizes of Agaricomycetes. Curr. Genet. 60, 303–313. doi: 10.1007/s00294-014-0436-z
Hiraoka, M., Kinoshita, Y., Higa, M., Tsubaki, S., Monotilla, A. P., Onda, A., et al. (2020). Fourfold daily growth rate in multicellular marine alga Ulva meridionalis. Sci. Rep. 10:12606. doi: 10.1038/s41598-020-69536-4
Horimoto, R., Masakiyo, Y., Ichihara, K., and Shimada, S. (2011). Enteromorpha-like Ulva (Ulvophyeae, Chlorophyta) growing in the Todoroki River, Ishigaki Island, Japan, with species reference to Ulva meridionalis Horimoto et Shimada sp. nov. Bull. Natl. Sci. Mus. Ser. B. Bot. 37, 155–167.
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Kang, X., Liu, J., Yang, X., Cui, J., Zhao, L., Wen, Q., et al. (2020). The complete mitochondrial genome of a green macroalgae species: Ulva meridionalis (Ulvales: Ulvaceae). Mitochondrial DNA B Resour. 5, 760–761. doi: 10.1080/23802359.2020.1715856
Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., McLellan, M. D., Mardis, E. R., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285. doi: 10.1093/bioinformatics/btp373
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lambowitz, A. M., and Zimmerly, S. (2011). Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb. Perspect. Biol. 3:a003616. doi: 10.1101/cshperspect.a003616
Lang, B. F. (2014). “Mitochondrial genomes in fungi,” in Molecular Life Sciences, ed. E. Bell (New York, NY: Springer). doi: 10.1007/978-1-4614-6436-5_113-2
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Liu, F., and Melton, J. T. (2021). Chloroplast genomes of the green-tide forming alga Ulva compressa: comparative chloroplast genomics in the genus Ulva (Ulvophyceae, Chlorophyta). Front. Mar. Sci. 8:668542. doi: 10.3389/fmars.2021.66854
Liu, F., Melton, J. T., and Bi, Y. (2017). Mitochondrial genomes of the green macroalga Ulva pertusa (Ulvophyceae, Chlorophyta): novel insights into the evolution of mitogenomes in the Ulvophyceae. J. Phycol. 53, 1010–1019. doi: 10.1111/jpy.12561
Liu, F., Melton, J. T., Lopez-Bautista, J. M., and Chen, N. (2020). Multiple intraspecific variations of mitochondrial genomes in the green-tide forming alga, Ulva compressa Linnaeus (Ulvophyceae, Chlorophyta). Front. Mar. Sci. 7:714. doi: 10.3389/fmars.2020.00714
Liu, F., Melton, J. T., Wang, H., Wang, J., and Lopez-Bautista, J. M. (2022). Understanding the evolution of mitochondrial genomes in the green macroalgal genus Ulva (Ulvophyceae, Chlorophyta). Front. Mar. Sci. 9:850710. doi: 10.3389/fmars.2022.850710
Liu, F., Pang, S., Chopin, T., Gao, S., Shan, T., Zhao, X., et al. (2013). Understanding the recurrent large-scale green tide in the Yellow Sea: temporal and spatial correlations between multiple geographical, aquacultural and biological factors. Mar. Environ. Res. 83, 38–47. doi: 10.1016/j.marenvres.2012.10.007
Liu, J., Tong, Y., Xia, J., Sun, Y., Zhao, X., Sun, J., et al. (2022). Ulva macroalgae within local aquaculture ponds along the estuary of Dagu River, Jiaozhou Bay, Qingdao. Mar. Pollut. Bull. 174:113243. doi: 10.1016/j.marpolbul.2021.113243
Melton, J. T., Leliaert, F., Tronholm, A., and Lopez-Bautista, J. M. (2015). The complete chloroplast and mitochondrial genomes of the green macroalga Ulva sp. UNA00071828 (Ulvophyceae, Chlorophyta). PLoS One 10:e0121020. doi: 10.1371/journal.pone.0121020
Pombert, J.-F., Otis, C., Lemieux, C., and Turmel, M. (2004). The complete mitochondrial DNA sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) highlights distinctive evolutionary trends in the Chlorophyta and suggests a sister-group relationship between the Ulvophyceae and Chlorophyceae. Mol. Biol. Evol. 21, 922–935. doi: 10.1093/molbev/msh099
Repetti, S. I., Jackson, C. J., Judd, L. M., Wick, R. R., Holt, K. E., and Verbruggen, H. (2020). The inflated mitochondrial genomes of siphonous green algae reflect processes driving expansion of noncoding DNA and proliferation of introns. Peer J. 8:e8273. doi: 10.7717/peerj.8273
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Robison, M. M., and Wolyn, D. J. (2005). A mitochondrial plasmid and plasmid-like RNA and DNA polymerases encoded within the mitochondrial genome of carrot (Daucus carota L.). Curr. Genet. 47, 57–66. doi: 10.1007/s00294-004-0549-x
Shibl, A. A., Isaac, A., Ochsenkühn, M. A., Cárdenas, A., Fei, C., Behringer, G., et al. (2020). Diatom modulation of select bacteria through use of two unique secondary metabolites. Proc. Natl. Acad. Sci. U.S.A. 117, 27445–27455. doi: 10.1073/pnas.2012088117
Steinhagen, S., Barco, A., Wichard, T., and Weinberger, F. (2019a). Conspecificity of the model organism Ulva mutabilis and Ulva compressa (Ulvophyceae, Chlorophyta). J. Phycol. 55, 25–36. doi: 10.1111/jpy.12804
Steinhagen, S., Karez, R., and Weinberger, F. (2019b). Cryptic, alien and lost species: molecular diversity of Ulva sensu lato along the German coasts of the North and Baltic Seas. Eur. J. Phycol. 54, 466–483. doi: 10.1080/09670262.2019.1597925
Steinhagen, S., Weinberger, F., and Karez, R. (2019c). Molecular analysis of Ulva compressa (Chlorophyta, Ulvales) reveals its morphological plasticity, distribution and potential invasiveness on German North Sea and Baltic Sea coasts. Eur. J. Phycol. 54, 102–114. doi: 10.1080/09670262.2018.1513167
Suzuki, S., Yamaguchi, H., Hiraoka, M., and Kawachi, M. (2018). Mitochondrial and chloroplast genome sequences of Ulva ohnoi, a green-tide forming macroalga in the Southern coastal regions of Japan. Mitochondrial DNA B Resour. 3, 765–767. doi: 10.1080/23802359.2018.1483778
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The ClustalX windows interface flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Tsubaki, S., Nishimura, H., Imai, T., Onda, A., and Hiraoka, M. (2020). Probing rapid carbon fixation in fast-growing seaweed Ulva meridionalis using stable isotope 13C-labelling. Sci. Rep. 10:20399. doi: 10.1038/s41598-020-77237-1
Turmel, M., Otis, C., and Lemieux, C. (2016). Mitochondrion-to-chloroplast DNA transfers and intragenomic proliferation of chloroplast group II introns in Gloeotilopsis green algae (Ulotrichales, Ulvophyceae). Genome Biol. Evol. 8, 2789–2805. doi: 10.1093/gbe/evw190
Wang, Y., Liu, F., Liu, X., Shi, S., Bi, Y., and Moejes, F. W. (2019). Comparative transcriptome analysis of four co-occurring Ulva species for understanding the dominance of Ulva prolifera in the Yellow Sea green tides. J. Appl. Phycol. 31, 3303–3316. doi: 10.1007/s10811-019-01810-z
Wang, Z., Xiao, J., Fan, S., Li, Y., Liu, X., and Liu, D. (2015). Who made the world’s largest green tide in China?—an integrated study on the initiation and early development of the green tide in Yellow Sea. Limnol. Oceanogr. 60, 1105–1117. doi: 10.1002/lno.10083
Warren, J. M., Simmons, M. P., Wu, Z., and Sloan, D. B. (2016). Linear plasmids and the rate of sequence evolution in plant mitochondrial genomes. Genome Biol. Evol. 8, 364–374. doi: 10.1093/gbe/evw003
Xie, W., Wu, C., Zhao, J., Lin, X., and Jiang, P. (2020). New records of Ulva spp. (Ulvophyceae, Chlorophyta) in China, with special reference to an unusual morphology of U. meridionalis forming green tides. Eur. J. Phycol. 55, 412–425. doi: 10.1080/09670262.2020.1740946
Ye, N., Zhang, X., Mao, Y., Liang, C., Xu, D., Zou, J., et al. (2011). ‘Green tides’ are overwhelming the coastline of our blue planet: taking the world’s largest example. Ecol. Res. 26, 477–485. doi: 10.1007/s11284-011-0821-8
Yin, C., Richter, U., Börner, T., and Weihe, A. (2010). Evolution of plant phage-type RNA polymerases: the genome of the basal angiosperm Nuphar advena encodes two mitochondrial and one plastid phage-type RNA polymerases. BMC Evol. Biol. 10:379. doi: 10.1186/1471-2148-10-379
Zheng, F., Liu, H., Jiang, M., Xu, Z., Wang, Z., Wang, C., et al. (2018). The complete mitochondrial genome of the Caulerpa lentillifera (Ulvophyceae, Chlorophyta): Sequence, genome content, organization structure and phylogenetic consideration. Gene 673, 225–238. doi: 10.1016/j.gene.2018.06.050
Keywords: mitochondrial genome, circular plasmid, DNA-directed RNA polymerase, Ulva meridionalis, Ulvophyceae, green tide
Citation: Liu F, Wang H and Song W (2022) Tandem integration of circular plasmid contributes significantly to the expanded mitochondrial genomes of the green-tide forming alga Ulva meridionalis (Ulvophyceae, Chlorophyta). Front. Plant Sci. 13:937398. doi: 10.3389/fpls.2022.937398
Received: 06 May 2022; Accepted: 05 July 2022;
Published: 05 August 2022.
Edited by:
Peter J. Lammers, Arizona State University, United StatesReviewed by:
Yoshihisa Hirakawa, University of Tsukuba, JapanCopyright © 2022 Liu, Wang and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Liu, bGl1ZmVuZ0BxZGlvLmFjLmNu, cHJjbGl1ZmVuZ0BzaW5hLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.