 Chainika Gupta
Chainika Gupta Romesh K. Salgotra
Romesh K. Salgotra
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci. , 15 August 2022
Sec. Plant Biotechnology
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.925688
This article is part of the Research Topic Crop Breeding Involving Epigenetic Inheritance View all 7 articles
Climate-resilient crops with improved adaptation to the changing climate are urgently needed to feed the growing population. Hence, developing high-yielding crop varieties with better agronomic traits is one of the most critical issues in agricultural research. These are vital to enhancing yield as well as resistance to harsh conditions, both of which help farmers over time. The majority of agronomic traits are quantitative and are subject to intricate genetic control, thereby obstructing crop improvement. Plant epibreeding is the utilisation of epigenetic variation for crop development, and has a wide range of applications in the field of crop improvement. Epigenetics refers to changes in gene expression that are heritable and induced by methylation of DNA, post-translational modifications of histones or RNA interference rather than an alteration in the underlying sequence of DNA. The epigenetic modifications influence gene expression by changing the state of chromatin, which underpins plant growth and dictates phenotypic responsiveness for extrinsic and intrinsic inputs. Epigenetic modifications, in addition to DNA sequence variation, improve breeding by giving useful markers. Also, it takes epigenome diversity into account to predict plant performance and increase crop production. In this review, emphasis has been given for summarising the role of epigenetic changes in epibreeding for crop improvement.
The increase in the world’s population at an alarming rate as well as the problems posed by global climate change on crop production, urges us to improve agricultural yield and quality in a sustainable manner (Takeda and Matsuoka, 2008; Fedoroff, 2010). This goal can be achieved by genetic crop improvement, which integrates various fields like plant physiology, genetics and biotechnology. The incorporation of genetic material in to a crop of interest is one of its most important prerequisites (Lombardo et al., 2016). Besides genetic variation, a better knowledge of the influence of epigenetic changes on plant phenotype has allowed the crop development process to move even faster (Merce et al., 2020). The word epigenetics was given by Waddington, combining the phrases “epigenesis” and “genetics.” He defined epigenetics as “the study of the interaction between a gene and its product, that produces the phenotype” (Waddington, 1942). Plant developmental processes and phenotypic plasticity, particularly adaptive responses to environmental challenges, are influenced by epigenetic information. This has prompted scientists to reconsider the relationship between genotypes and phenotypes (Pecinka and Mittelsten Scheid, 2012; Springer, 2013; Zhang Y. Y. et al., 2013; Baulcombe and Dean, 2014; Pikaard and Mittelsten Scheid, 2014; Meyer, 2015; Rey et al., 2016). Epigenetic variations have been assessed and quantified with their effect on not only plant developmental processes and plant responses to environmental constraints (Baulcombe and Dean, 2014; Cortijo et al., 2014; Meyer, 2015; Takatsuka and Umeda, 2015) but also important agronomical traits like respiration, energy-use efficiency, yield components and seed quality (Hauben et al., 2009; Shi et al., 2009; Long et al., 2011; Chen and Zhou, 2013). At the molecular level, a gene’s transcription is influenced by its DNA sequence as well as how genes are organised within the chromosome’s intricate architecture. The DNA of eukaryotes is extremely condensed and closely connected with proteins called histones and this combination is referred to as chromatin. To start transcription at a given gene, the chromatin at that location must be open for binding of transcription factors (TF) as well as RNA polymerase. Hence, whether a gene is “on” or “off” is determined by the chromatin state at a specific gene. The accessibility of chromatin to the transcriptional machinery is influenced by a variety of mechanisms like methylation of DNA, posttranslational modifications of histone, chromatin remodelling as well as non-coding RNAs. The chromatin state and expression pattern of genes that have been created can be conserved through numerous generations, thus called epigenetic.
The environmental changes may result in heritable epigenetic changes which are associated with changes in gene expression and variation in phenotype (Kakoulidou et al., 2021). These phenotypic variations are contributing to epibreeding for the improvement of various crop production traits (Figure 1). Although the epigenetic mechanism are well established in model plants, the emphasis has been given to the improvement of phenotypic traits of crops. The epigenetic modifications may be used as markers to allow the use of epigenome diversity in crop improvement programmes. No doubt various difficulties are arising in transferring the epigenetic system from model plants to crops, however numerous high-throughput technologies have been evolved to solve these problems. The epigenetic modifications influence gene expression by changing the state of chromatin, which underpins plant growth and dictates phenotypic responsiveness with respect to extrinsic and intrinsic inputs. Epigenetic modifications, in addition to DNA sequence variation, improve breeding by giving useful markers. Also, it takes epigenome diversity into account to predict plant performance and increase crop production. Plant epibreeding or the utilisation of epigenetic variation for crop development, has a wide range of applications in the field of crop improvement. Epigenetics is the study of changes in gene expression that are heritable and caused by methylation of DNA, post-translational modifications of histones or RNA interference rather than an alteration in the underlying sequence of DNA (Kapazoglou et al., 2018). Epigenetic variants of agronomic importance have been identified in rice (dwarf phenotype), apple (anthocyanin production), oilseed rape (decreased oil content), pigeon pea (high heterosis), pineapple (increased somatic embryogenesis), soybean (enhanced yield and stability), melon (sex determination), and tomato (fruit ripening). The main challenge faced by agriculture in the present century is to boost agricultural productivity. Epigenetics provides essential biological information that could be directly used to enhance crop tolerance and adaptability (Kakoulidou et al., 2021). Keeping in perspective the importance of epigenetic changes in improving the crop species, the present review summarises the cutting-edge knowledge comprehensively on the epigenetic molecular aspects and their utilisation in crop breeding. Moreover, it also presents acumens gleaned on the improvement of agronomic traits of important crops.
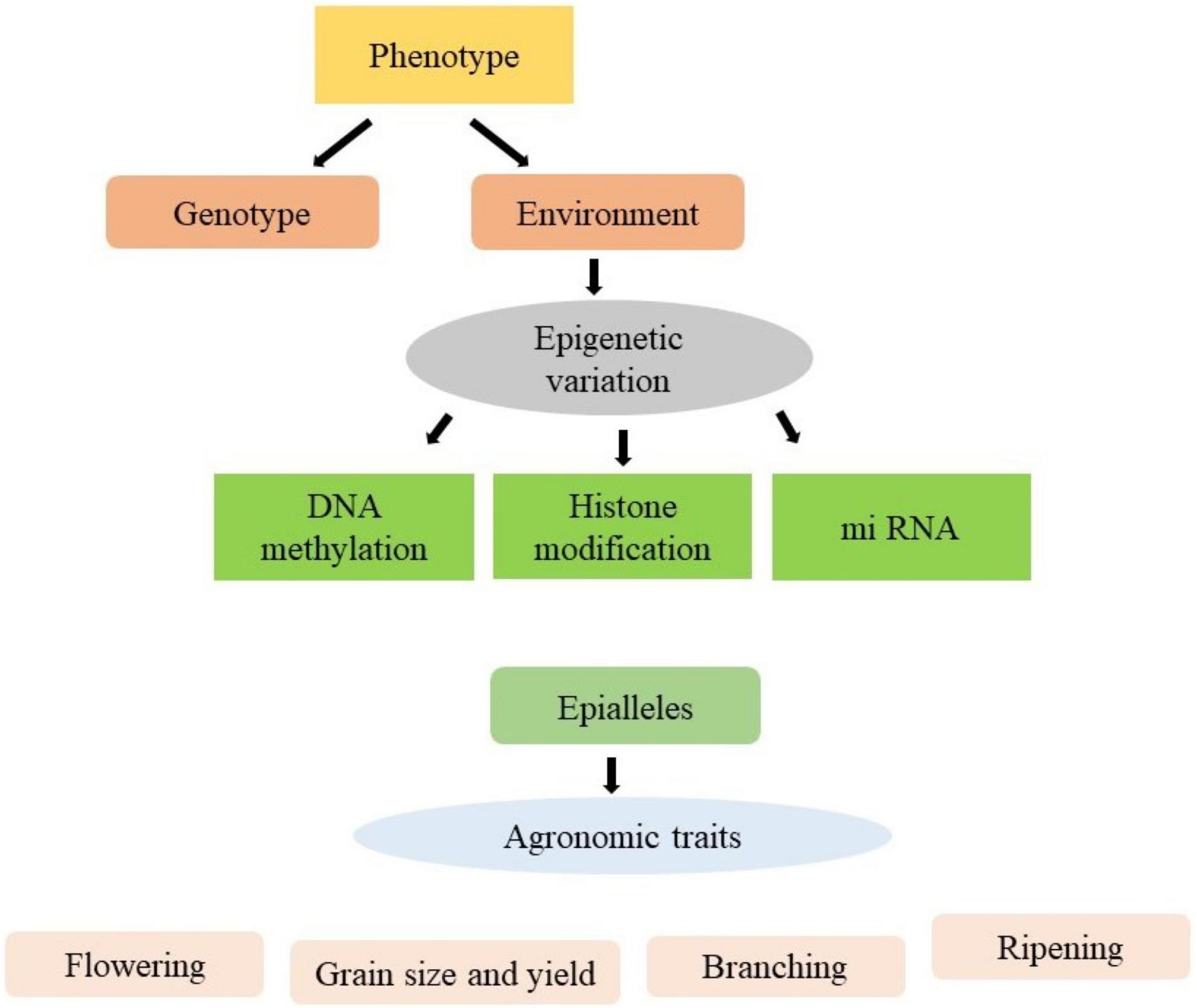
Figure 1. Use of epialleles for improvement of various agronomical traits.
DNA methylation is a heritable process where a methyl group (CH3) is added to 5th carbon of a cytosine base. It governs a variety of functions like an expression of a gene, genomic stability, gene imprinting, inactivation of transposable elements and its disruption can result in developmental abnormalities such as failure in tomato fruit ripening (Baulcombe and Dean, 2014; Iwasaki and Paszkowski, 2014; Lang et al., 2017; Zhang H. et al., 2018; Gallego-Bartolome, 2020). It is passed on in a mendelian fashion (Niederhuth and Schmitz, 2014). However, in paramutation, methylation variations can generate phenotypic diversity because of spontaneous methylation alterations that result in non-mendelian inheritance (Weigel and Colot, 2012). Novel epialleles may result from variations in DNA methylation patterns, which may aid plant improvement and adaptation. Heavy methylation of transposable elements and other repetitive DNA sequences in heterochromatin has been observed in Arabidopsis thaliana during a genome-wide DNA methylation study (Zhang et al., 2006; Henderson and Jacobsen, 2007). Euchromatic chromosomal arms include interspersed transposon-associated DNA methylation (Zhang et al., 2006). Symmetric and asymmetric methylation of cytosine bases also occurs (Henderson and Jacobsen, 2007). The former denotes the presence of cytosine that is methylated in double stranded DNA, whereas the latter denotes the presence of cytosine that is methylated in single-stranded DNA only (Mbichi et al., 2020).
The methylation pattern is usually CG (symmetric), CHG (symmetric) or CHH (H = a nucleotide other than G, asymmetric). It is particularly abundant in heterochromatic transposable elements (TEs) and repetitions. When DNA methylation occurs in gene regulatory areas, it can cause Transcriptional Gene Silencing (TGS). It has been discovered that the DNA methyl-readers SU(VAR)3-9 homologs SUVH1 and SUVH3 have a role in enhancing gene expression in some cases (Harris et al., 2018; Xiao et al., 2019).
DNA methylation is further categorised into
1. De novo methylation
2. maintenance methylation
De novo methylation occurs when previously unmethylated cytosine residues are methylated, resulting in the formation of novel methylation patterns. On the other hand, in maintenance methylation, previous existing methylation patterns are retained after DNA replication (Chen and Li, 2004). The enzymes methyltransferases and demethylases control DNA methylation. The methylation of CG and CHG is carried out by METHYLTRANSFERASE 1 (MET1) and CHROMOMETHYLASE 3 (CMT3), respectively (Zhang H. et al., 2018). Two pathways create De novo CHH methylation. The first one involves RNA-dependent DNA methylation. Here, the small interfering RNAs (siRNAs) are targeted to homologous genomic loci by members of the ARGONAUTE (AGO) family and then methylated by DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2). The second one involves histone H1-rich chromatic areas, where CHROMOMETHYLASE 2 (CMT2) interacts with DECREASE IN DNA METHYLATION1 (DDM1) (Zemach et al., 2013). The methylation in DNA is eliminated by a process involving active or passive demethylation. Active demethylation is mediated by DNA glycosylases like Arabidopsis DEMETER (DME) and REPRESSOR OF SILENCING 1 (ROS1), which play a significant role in the gene expression control. Passive demethylation mainly occurs during DNA replication (Gehring et al., 2009; Zhu, 2009; Tang et al., 2016).
The accessibility of genomic areas to regulatory proteins/protein complexes is influenced by cytosine methylation, which alters chromatin structure and the rate of gene transcription. When found in the promoter and enhancer regions, it has been linked to gene repression (Charlet et al., 2016). However, in the case of gene body methylation (gbM), 5-mC in the transcribed region has the potential to supress or increase transcription (Buck-Koehntop and Defossez, 2013; Spruijt and Vermeulen, 2014). Due to its heredity and potential to influence plant phenotypes, methylation of DNA contributes to crop productivity. The traits of agronomic importance including time of flowering, dormancy in seed, yield, etc., are influenced by the methylation of DNA (Liu and Wendel, 2003; Zhang Y. Y. et al., 2013; Song et al., 2017).
The DNA methylation in plants takes place through the canonical RNA-directed DNA methylation (RdDM) pathway as well as non-canonical RdDM pathways. The RNA polymerase IV (Pol IV) synthesises single-stranded RNAs (ssRNAs), which get transformed into double-stranded RNAs (dsRNAs) with the help of RNA-DEPENDENT RNA POLYMERASE 2 (RDR2), in the canonical RdDM pathway. DICER-LIKE 3 (DCL3) cuts these dsRNAs into small interfering RNAs (siRNAs) of 24-nucleotides in length, which get integrated into ARGONAUTE 4 (AGO4) and ARGONAUTE 6 (AGO6) (Zhang H. et al., 2018). The recruitment of Pol IV to chromatin requires chromatin remodelers like SAWADEE HOMEODOMAIN HOMOLOG 1 (SHH1) and the CLASSY family (Zhang H. et al., 2018; Zhou et al., 2018).
Small RNAs (sRNAs) (from viruses and Pol II transcripts) guide RdDM in non-canonical RdDM pathways (Cuerda-Gil and Slotkin, 2016). The dsRNAs are sliced into 21–24 nucleotide sRNAs by different DCL proteins followed by their incorporation into AGO proteins. The complex binds to complementary RNA and can cleave it or repress translation leading to post-transcriptional gene silencing (PTGS) (Martinez de Alba et al., 2013). When sRNAs are incorporated into AGO4/AGO6, they can cause complementary DNA sequences to be methylated by Pol V and DRM2, potentially leading to the silencing of genes (Cuerda-Gil and Slotkin, 2016).
MicroRNAs (miRNAs) are short non-coding RNA encoded by miRNA genes. They are of 20–24 nucleotides long (Jones-Rhoades et al., 2006). They can influence complex biological processes in plants by regulating gene expression (Chen, 2009; Voinnet, 2009; Pantaleo et al., 2010; Sun, 2012; Djami-Tchatchou and Dubery, 2015). They bind to messenger RNAs with partially complementary sequences (mRNAs). miRNA genes are found in intergenic regions as well as within introns, while some of them are found in clusters throughout the genome and transcribed as long polycistronic RNAs (Djami-Tchatchou et al., 2017).
miRNA biogenesis takes place in the nucleus (Bartel, 2004; Jones-Rhoades et al., 2006). RNA polymerase II is commonly used to transcribe miRNA genes, leading to the formation of primary miRNA (pri-miRNA) (Xie et al., 2005; Pantaleo et al., 2010). It is converted into precursor miRNA (pre-miRNA) with a self-complementary stem-loop on which the Dicer-like 1 (DCL1) enzyme acts. It dices the pre-miRNA in the presence of HYPONASTIC LEAVES 1 (HYL1) and Serrate (SE) and miRNA: miRNA duplex is formed (Tang and Chu, 2017). HUA ENHANCER 1 (HEN1) methylates the duplex which is transported to the cytoplasm by EXPORTIN5, HASTY (Park et al., 2005; Pelaez et al., 2012). The duplexes are then placed in the RNA-induced silencing complex (RISC), which contains the ARGONAUTE (AGO) proteins. After RISC loading, AGO1 unwinds miRNA: miRNA duplexes with one strand sent to the exosome for degradation and the other strand is inserted into the RISC complex (Sun, 2012). The mature miRNA directs RISC to target complementary mRNAs leading to their cleavage or translational repression (Sun, 2012).
Most miRNAs work as negative regulators of their target transcripts. They use considerable sequence complementarity to target coding sequences (ORFs). Translational repression occurs in targets with low sequence complementarity. It has been reported that pairing of bases at the initial 2–13 nucleotides of miRNA towards the 5′ end is important because many targets suffer AGO-mediated cleavage at positions 9–11 (Axtell, 2013). Mismatches around the 3′ end of miRNAs are thought to be less “destructive” than mismatches near the 5′ end or in the core regions (Liu N. et al., 2014). Although perfect complementarity between miRNA targets is uncommon, mismatches have been observed frequently at either the extreme 5′ end of miRNAs or toward 3′. In some cases, the interaction of miRNA with a target is complementary at two ends of the region, with a bulge or mismatch in the center area (Pandey et al., 2019). The targets of miRNA are transcription factors (TFs) which are the important regulators of plant development. These include
(i) SQUAMOSA-promoter binding protein-like (SPL) TFs: It plays a role in phase transition or vegetative and reproductive development and is negatively regulated by miR156, miR157, or miR529 (Chuck et al., 2010; Miura et al., 2010; Bhogale et al., 2014; Ferreira e Silva et al., 2014; Chen et al., 2015; Wang et al., 2015).
(ii) APETALA2 (AP2) family of TFs: They are inhibited by miR172 and impact phase transition, organ development and sex determination (Martin et al., 2009a; Houston et al., 2013; Lee et al., 2014; Wang et al., 2015).
(iii) TEOSINTE BRANCHED/CYCLOIDEA/PCF (TCP) TFs: These are regulated by miR319, which impacts tolerance to cold, defence, and morphogenesis in leaves (Ori et al., 2007; Yang et al., 2013; Zhao et al., 2015; Zhang C. et al., 2016).
(iv) MYB TFs: miR159 antagonised these TFs and plays a role in the development of flowers and response to heat or acts along with miR828 in controlling the modulation of fibre growth (Tsuji et al., 2006; Wang et al., 2012; Guan et al., 2014).
(v) NAC TFs: These are expressed by genes targeted by miR164 and are involved in the growth of lateral roots, tolerance to drought and immunological response (Li et al., 2012; Fang et al., 2014; Feng et al., 2014).
(vi) Growth-regulating factors (GRFs): These are controlled by miR396 that regulates the size of grain and yield, development of inflorescence and tolerance to salt–alkali stress (Hewezi et al., 2012; Liu N. et al., 2014; Che et al., 2015; Duan et al., 2015; Gao et al., 2015; Hu et al., 2015).
(vii) Auxin response factors (ARFs): These are responsible for response to auxin, defence responses as well as crucial in developmental stages and are targeted by miR160 or miR167 (Li et al., 2014; Liu N. et al., 2014; Nizampatnam et al., 2015; Wang et al., 2015; Damodharan et al., 2016; Huang et al., 2016).
(viii) DNA-binding with one finger (Dof) TF RDD1, SCARECROW-like (SCL) TF HvSCL, zinc finger domain-containing TF GhCHR, Timing of CAB Expression 1 TF TaTOC1 and the HD-ZIPIII family member ROLLED LEAF1 (RLD1): miR166, miR171, miRNVL5, miR408, and miR166 are all negative regulators of these genes. These are involved in the uptake of ions, determination of floral meristem, salt stress response, and flowering time (Juarez et al., 2004; Curaba et al., 2013; Gao et al., 2016; Iwamoto and Tagiri, 2016; Zhao et al., 2016).
Genomic DNA in eukaryotes is firmly condensed with proteins to form chromatin, which impacts the accessibility of transcription factors and cofactors for modulating gene expression. The most fundamental unit of chromatin is the nucleosome, which consists of DNA wind around a histone octamer (two copies of four histone proteins viz H2A, H2B, H3, and H4) (Luger et al., 1997; Liu et al., 2016). Histone H1 maintains this structure by binding to the nucleosome along with sections including linker DNA. The availability of chromatin to the transcriptional machinery is influenced by DNA methylation, posttranslational histone modifications, histone variant exchange, chromatin remodelling, and the inclusion of non-coding RNAs. Histone proteins undergo post translational modifications (PTMs). Most histone modification sites are found in histone N-terminal tails and include acetylation, methylation, phosphorylation, ubiquitination, and sumoylation (Millar and Grunstein, 2006; Lauria and Rossi, 2011; Yuan et al., 2013; Liu X. et al., 2014). Due to the presence of high lysine and arginine content in histones, their amino (N-terminal) tails are extremely basic. These tails protrude from the nucleosome core, making protein–protein and protein–DNA interactions possible. Reversible covalent changes occur at these histone tails. These changes act as “histone code,” indicating how chromatin functions and transcriptional activities are carried out (Jenuwein and Allis, 2001). These alterations can influence chromatin shape and transcription of genes by changing the interface linking histones and DNA as well as modifying the attachment of regulatory proteins to DNA (Pikaard and Mittelsten Scheid, 2014; Allis and Jenuwein, 2016; Chang et al., 2019). The activation or repression of transcription is linked to histone modifications.
It is required for biological processes like transcriptional control and the formation of heterochromatin. Histone methylation leads to retention of the charge on amino acids and does not affect electrostatic properties of histones (Ueda and Seki, 2020). It occurs mainly at the lysine and arginine residues of H3 and H4 histones resulting in alteration in gene expression that has both activating and repressive effects (Swygert and Peterson, 2014). The lysine residues can be monomethylated, dimethylated or trimethylated (Cloos et al., 2008). Different methylation has effects on gene expression in different ways (Liu et al., 2010; Zhao et al., 2019). In the case of A. thaliana, trimethylation of Lys 27 (H3K27me3) suppresses gene expression while trimethylation of Lys 4 (H3K4me3) increases gene transcription (Berr et al., 2011; Zheng and Chen, 2011). The enzymes that carry out addition, removal or reading out of methylation marks are known as writers, erasers and readers. Writers are the enzymes that add posttranslational modifications to a protein. Erasers are the enzymes that remove a protein’s posttranslational modification. Readers are the proteins that recognise and bind to a post-translationally modified substrate (Xu et al., 2017). The enzymes which carry out the transfer of methyl groups to histones are histone methyltransferases (HMTs). They comprise of histone lysine methyltransferases (HKMTs) and protein arginine methyltransferases (PRMTs). Histone methyltransferases contain the Set (Suppressor of Variegation 3–9, Enhancer of zeste, and Trithorax) Domain, belong to the gene family SDG, and add lysine methylation to histones. In Arabidopsis, 37 SDG genes have been discovered (Thorstensen et al., 2011). The action of many HKMTs bring about lysine methylation, primarily at K4, K9, K27, and K36 of H3 along with K 20 of H4. This changes their interaction with reading proteins, causing structural changes in chromatin that result in transcription activation or repression (Teperino et al., 2010). The methylation in H3K4 and H3K36 can lead to transcriptional activation, whereas H3K9 and H3K27 methylation can lead to transcriptional repression (Ueda and Seki, 2020). Two types of histone demethylases catalyse the removal of histone lysine methylation: Jumonji C (JmjC) domain-containing proteins and Lysine-Specific Demethylase (LSD)-like proteins. Jumonji-C (JmjC) domain–containing proteins remove methyl groups from dimethylated and trimethylated lysines preferentially while Lysine-specific demethylases (LSDs) demethylate monomethylated and dimethylated lysines (Shi et al., 2004; Tsukada et al., 2006). H3 and H4 arginine [R] residues can also be methylated. Arginine methylation can be symmetric or asymmetric and it has been observed in R17 (of H3) and R3 (of H4) (Ueda and Seki, 2020). Methylation of arginine could give an opposing outcome depending on whether it is symmetric or asymmetric. For example, H4R3me2a (asymmetric) is an activation mark while H4R3me2s (symmetric) is a repression mark.
Acetylation on histones is a flexible and dynamic process. It is correlated with transcription activity (Allfrey et al., 1964). The amino terminal tails of histones contain lysine and arginine (Luger and Richmond, 1998). The transfer of an acetyl group to the ε-amine group of the N-terminal lysine residue of histone protein is known as histone acetylation (Pandey et al., 2002; Kouzarides, 2007). It neutralises the positive charge of the histone tails and enhances hydrophobicity (Allis and Jenuwein, 2016; Onufriev and Schiessel, 2019). As a result, the affinity of histone proteins for negatively charged DNA decreases and the chromatin state switches from a closed to an open state (Clayton et al., 2006; Li S. et al., 2019). This leads to the initiation of transcription by attachment of RNA polymerase along with transcription factors to the promoter region of the gene. It has been discovered that an increase in histone acetylation near the transcription start site is positively linked with gene expression (Srivastava et al., 2014; Wang et al., 2017; Kim et al., 2020).
Histone acetylation is mediated by histone acetyltransferases (HATs) and deacetylation by histone deacetylases (HDACs) (Pandey et al., 2002; Kumar et al., 2021). HATs activate genes by adding acetyl groups to lysine residues in histone N-terminal tails as well as globular domains (Bjerling et al., 2002; Pandey et al., 2002; Iwasaki et al., 2011; Pradeepa et al., 2016). HDACs, on the other hand, remove these acetyl groups from histones leading to an increase in interaction between DNA and histones. These counteract the effects of HATs resulting in gene repression (Gallinari et al., 2007; Gu et al., 2017; Yang et al., 2020). HATs and HDACs mainly target lysine residues in histones, such as H3K9, H3K14, H3K36, H4K5, H4K8, H4K12, and H4K16 (Bjerling et al., 2002).
Based on their subcellular distribution, HATs are categorised as: Type A HAT and Type B HAT. The type A HATs are engaged in the acetylation of nuclear histone and consequently regulate chromatin assembly and transcription of genes (Carrozza et al., 2003). It includes GCN5-related acetyltransferases (GNATs), MYST (for MOZ, Ybf2/Sas3, Sas2, and Tip60)-related HATs, p300/CBP HATs and the TFIID subunit TAF250 (Sterner and Berger, 2000; Pandey et al., 2002; Carrozza et al., 2003; Hu et al., 2019). The Arabidopsis genome predicts five p300/CBP-type A HATs, one of which, PCAT2 (p300/CBP acetyltransferase-related protein 2), has HAT activity (Pandey et al., 2002). Specific HATs are responsible for the acetylation of different lysine residues in histones (Earley et al., 2007). H3K14 and H4K12 acetylation are catalysed by two GNAT class HATs, HAG1, and HAG2, respectively. HAM1 and HAM2 are MYST class HATs that redundantly acetylate H4K5. Multiple HATs are involved in the H3K9, H4K8 and H4K16 acetylations (Earley et al., 2007). On the other hand, the Type B HAT proteins are present in the cytoplasm. They catalyse acetylation of histone H4 at lysine 5 and 12 in the cytoplasm, before integration of histone into newly replicated chromatin (Parthun et al., 1996; Verreault et al., 1998). In maize, type B HAT functions as a heterodimeric complex and was detected in the cytoplasm as well as in the nucleus indicating its role in the nucleus also (Eberharter et al., 1996; Lusser et al., 1999). The number of HATs varies in different crops. For example, a tomato has 32 HATs while a litchi has only 6 (Aiese Cigliano et al., 2013; Peng et al., 2017).
Histone deacetylases have been categorised into three families based on sequence similarity and cofactor dependency: Reduced Potassium Dependency 3 (RDP3)/ Histone DeAcetylase 1 (HDA1), Silent Information Regulator 2 (SIR2) and the plant-specific Histone Deacetylase 2 (HD2) (Pandey et al., 2002). The catalytic domain of the SIR2 family (Sirtuins) requires NAD as a cofactor (Haigis and Guarente, 2006), whereas RPD3/HDA1 requires Zn2+ as a cofactor (Yang and Seto, 2007). Furthermore, HD2 isonly found in plants (Pandey et al., 2002). These have a histone deacetylase domain that is conserved and lacks a DNA binding domain (Kumar et al., 2021). Some of the HDACs have zinc finger motifs, which play a role in protein-protein interaction (Aquea et al., 2010; Peng et al., 2017; Kumar et al., 2018). HDACs also vary in different crops. In upland cotton, 30 HDACs were reported while in litchi there were only 11 (Peng et al., 2017; Kumar et al., 2018).
Traditional crop breeding involves crossing followed by the selection of genetic variants containing desirable traits (Palmgren et al., 2015). This resulted in a narrowing of the genetic base as well as a loss of genetic diversity, thereby hampering the crop improvement programme (Esquinas-Alcazar, 2005; Gallusci et al., 2017). Phenotypic traits, however, are influenced by both genetics and epigenetics. The exploitation of epigenetic variants or epigenome alteration could be a feasible breeding method for improving response to changing environmental conditions while ensuring agricultural yield and quality (Pecinka et al., 2010; Tirnaz and Batley, 2019). Many genes or QTLs governing traits of interest have been identified, but missing heritability remains a key barrier that affects phenotype. One of the key causes of missing heritability is epigenetic changes (Slatkin, 2009). The epialleles, also known as epigenetic alleles, are the loci having epigenetic modifications that may arise naturally or be induced artificially and are transferred stably to the next generation (Taudt et al., 2016). In addition to natural genetic variation, which contributes to phenotypic diversity, epialleles impart an additional source of heritable variation (Varotto et al., 2022; Figure 2). There are two ways in which epialleles could be formed: non-genetic (also known as epigenetic) and genetic (Taudt et al., 2016). Nongenetic epialleles originate from developmental factors or environmental signals that result in changes in chromatin state (Zheng et al., 2017). It also arises from spontaneous epimutation due to the inability to maintain an existing methylation state. The rate of spontaneous epimutations at cytosines in Arabidopsis has been observed to be 1,000 times higher as compared to the rate of genetic mutation (Becker et al., 2011). Besides, differentially methylated regions (DMRs), which span many cytosines, are substantially less common. It has been reported in soybean, maize, and tomato (Manning et al., 2006; Shen et al., 2018; Xu et al., 2019). Furthermore, genetic epialleles arise due to the insertion of a transposon in an intergenic region, leading to its inactivation primarily by methylation of DNA (Pecinka et al., 2013). The methylated DNA enhances methylation on all sides of the insertion site, leading to the formation of new epialleles (Varotto et al., 2022). It has been documented in Arabidopsis, rice and maize (Banks et al., 1988; Schmitz et al., 2013; Zhang et al., 2015).
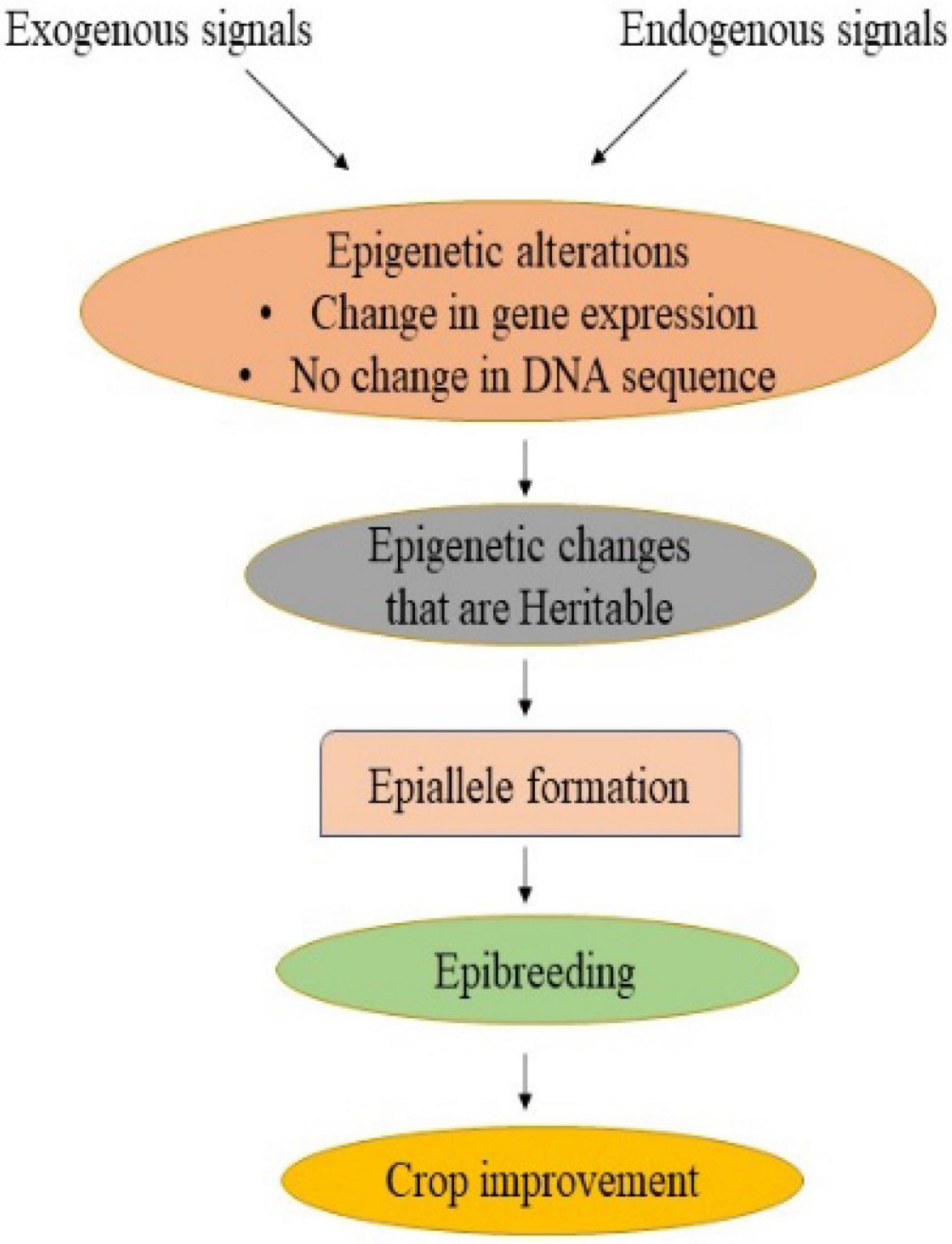
Figure 2. Epigenetic modifications can take place in response to external and internal cues, resulting in a change in gene expression without any change in DNA sequence. Stable and heritable epigenetic changes lead to the formation of epialleles which can be used in epibreeding programmes for crop improvement.
Peloric mutants in toadflax (Linaria vulgaris) are a classical example of epialleles that are heritable. These have flowers with radial symmetry, whereas wild-type plants have bilateral floral symmetry. The epimutation is due to hypermethylation of the promoter region of the Lcyc gene, which is a CYCLOIDEA homologue of Antirrhinum and controls flower symmetry by silencing the gene (Cubas et al., 1999). An epiallele clark-kent (clk) was reported in Arabidopsis, which led to an increase in the number of stamens and carpels. This was due to hypermethylation of the cytosine base at the SUPERMAN locus, which was associated with flower development (Jacobsen and Meyerowitz, 1997). The switch from male to female flowers in melon was due to hypermethylation of the CmWIP1 TF promoter region (Martin et al., 2009b). The epiallele SP11/SCR locus in Brassica was associated with self-incompatibility (Shiba et al., 2006). The epiallele of the QSS (QuaQuine Starch) gene in Arabidopsis arises due to methylation in the 5’ region and is responsible for starch metabolism (Silveira et al., 2013). It has been reported that 43 percent and 46 percent of epialleles are passed down in successive generations in two different lines of Arabidopsis (Hofmeister et al., 2017). Also, near-isogenic lines that separated from a common ancestor a century ago, displayed persistent methylome inheritance over generations despite variable environments (Hagmann et al., 2015). More than 99% of the methylome in maize and Arabidopsis is preserved between accessions (Li et al., 2015; Hofmeister et al., 2017). Thus, epialleles that are stably heritable across generations are of interest to breeders and can be utilised in crop improvement programmes. These can directly influence a plant’s phenotype. Epigenetic modifications can also be induced by chemical compounds. The methyltransferase inhibiting agents like 5-Azacytidine (5-AzaC), 5-Aza deoxycytidine, and Zebularine reduce methyl group transfer to cytosine and cause hypomethylation. Inhibitors of histone deacetylase include trichostatin-A (TSA), helminthosporium carbonum (HC) toxin, and nicotinamide, which enhance the acetylation of histones, thereby resulting in the activation of genes (Gahlaut et al., 2020). The transmission of induced epialleles has been reported in Arabidopsis in an epigenetic recombinant inbred line (epiRIL) population (Johannes et al., 2009; Reinders et al., 2009). EpiRILs are recombinant inbred lines derived from parents that have identical genetic makeup but they differ in DNA methylation (Johannes et al., 2009). These are produced by crossing mutants having hypomethylated DNA (met1 and ddm1) with wild plants. The generated lines are homozygous for all traits except DNA methylation pattern, which affects traits like plant height, flowering time, biomass, yield and biotic as well as abiotic stress tolerance (Johannes et al., 2009; Reinders et al., 2009; Kooke et al., 2015; Zhang Y. Y. et al., 2018). It has been reported that the hypomethylated state was trans-generationally inherited in A. thaliana, even up to eight generations (Quadrana and Colot, 2016). Until now, two epi-RIL populations have been generated in Arabidopsis (Johannes et al., 2009; Reinders et al., 2009). The first one was produced by a cross of met1 mutant (DNA methyltransferase defective) to its wild type (isogenic) (Kankel et al., 2003; Saze et al., 2003; Reinders et al., 2009). Another one was derived by a cross of a ddm mutant (DDM locus defective, which is responsible for maintaining cytosine methylation) to its wild type (isogenic) (Jeddeloh et al., 1999; Lippman et al., 2004; Johannes et al., 2009). A population with remarkably comparable genomes that differ only in methylation levels is produced by selecting progeny with the wild-type copy of a gene and then allowing them to self-pollinate for several generations. Some of these epiRILs can express information that is generally repressed by DNA methylation in natural variations due to the stripping of methylation at certain chromosomal locations (Kumar, 2019). The genes that are affected by DNA methylation can be mapped using epiRILs to determine their association with phenotypic traits (Cortijo et al., 2014; Kooke et al., 2015).
Rice (Oryza sativa L.) is a key cereal crop that feeds more than half of the world’s population as a staple diet (Priya et al., 2019). The epiallele epid1 codes for a GTP-binding protein, which causes stunted growth. The d1 genes are silenced by DNA methylation together with histone acetylation, resulting in rice plant stature regulation (Miura et al., 2009). OsSPL14 is an epiallele linked to WEALTHY FARMERS PANICLE (WFP). The epigenetic change enhanced OsSPL14 expression, leading to an increase in panicle branching and grain yield (Miura et al., 2010). Flowering time (heading) is the most crucial stage for the production of grain in rice. Early flowering causes decrease in yield while delayed flowering results in a reduction in seed set. It is regulated by epigenetic modifications (He, 2009; Liu et al., 2010). Histone methyltransferase (HMTase) genes such as SET DOMAIN GENE 724 (SDG724) belong to Class II in the SET domain family and promote flowering by methylating histone H3 lysine 36 (H3K36) (Ng et al., 2007; Sun et al., 2012). Under both long day (LD) and short day (SD), the SDG724 loss-of-function mutant lvp1 demonstrated delayed flowering, which was linked to decreased expression of RICE FLOWERING LOCUS T 1 (RFT1) along with Heading date 3a (Hd3a). Even though the two genes are near at a distance of 11.5 kb, only the chromosomal region of RFT1 decreased the transcriptional activation through H3K36me2/3 modification (Sun et al., 2012). In another study, it was reported that the expression of RFT1 can be enhanced in Hd3a-RNAi transgenic plants by acetylation of H3K9 near the start site of transcription (Komiya et al., 2008). SDG725, also belonging to SET domain family class II promotes flowering through H3K36me2/3 (Ng et al., 2007; Berr et al., 2009). OsTrx1, a member of Class III in the SET domain family, activates or conserves the active state of transcribed genes. It also prolongs the time of flowering in LD plants (Ng et al., 2007). WOX11 (Wuschel-related homeobox gene) with the help of ADA2-GCN5 histone acetyltransferase regulates genes associated with crown root development (Zhou et al., 2017). A QTL OsglHAT1 (a new-type GNAT-like protein) was reported to have inbuilt H4 histone acetyltransferase activity and was associated with grain weight. PGL2 (6-PHOSPHOGLUCONOLACTONASE 2), which is critical for grain length, is positively regulated by OsglHAT1. Enhanced expression of OsglHAT1 has a positive effect on agronomic traits like grain length, grain weight, yield, and total biomass (Song et al., 2015). In rice, OsSRT1 is a SIR2-type HDAC that suppresses carbon metabolic flux of the glycolysis pathway while enhancing the accumulation of starch in growing seeds (Huang et al., 2007; Zhang H. et al., 2016). Leaf angle is another agronomic feature that directly impacts the architecture of a plant and grain yield (Zhao et al., 2010; Jang et al., 2017). Plants having upright leaves catch more sunlight to carry out photosynthesis as well as have increased nitrogen storage for grain filling, thereby increasing yield and making them suitable for dense planting (Sakamoto et al., 2006). Leaf angle, grain size and yield potential are regulated by brassinosteroid (BR) phytohormones (Zhang et al., 2014). When the BR biosynthesis genes are overexpressed, the leaves become less upright and have a considerable leaf inclination whereas in BR-deficient or BR-insensitive mutants, the leaves are erect (Yamamuro et al., 2000; Hong et al., 2005; Wu et al., 2008; Li et al., 2009; Tong et al., 2012; Sun et al., 2015). Zhang et al. (2015) identified epiallele Epi-rav6 being associated with a larger lamina inclination and smaller grain size. This is due to hypomethylation in the promoter region of RAV6. The OsPCF7 gene which encodes transcription factors family viz TCP, plays a role in the architecture of the plant. Increased expression of OsPCF7 in transgenic rice seedlings enhances the height of shoot, length of root, number of roots, tillers and heading, resulting in an increase in grain yield per plant (Li et al., 2020). Promoter DNA methylation is predicted to silence OsFIE1 in vegetative tissues. Ectopic OsFIE1 expression, caused by an epimutation with loss of promoter DNA methylation, results in dwarfism, floral abnormalities and changes in H3K27me3 levels in hundreds of genes (Zhang et al., 2012). The hypomethylation of the ESP gene is associated with the regulation of panicle architecture. The plants exhibited short and dense panicles (Luan et al., 2019). The deletion of SE1 or impairment in the function of components of the repressor complex hinders histone deacetylation and H3K27me3 methylation in the Eui1 region, resulting in the switching of chromatin from a closed to an open state, thus increasing Eui1 transcription, reducing gibberellic acid and causes dwarf phenotype (Xie et al., 2018). NGR5 promotes tillering in response to increased nitrogen supply by facilitating the recruitment of PRC2 (Polycomb repressive complex 2) to restrict the expression of shoot branching-inhibitory genes via H3K27me3 (histone H3 lysine 27 trimethylation) (Wu et al., 2020). Hypermethylation of the OsAK1 gene at the promoter region is associated with photosynthetic capacity (Wei et al., 2017). The OsSPL14 gene has been reported to be regulated by microRNA and affects the branching of panicles and increased yield (Miura et al., 2010). miR160 affects auxin signalling by down- regulating OsARF18 expression, thereby affecting growth and development in rice (Huang et al., 2016). miR172 targets AP2-like TFs and affects the development of flowers, flowering time and branching of panicles (Zhu et al., 2009; Lee et al., 2014; Wang et al., 2015). Down-regulation of the OsLAC gene, which produces laccase-like protein by osa-miR397, controls grain size and yield (Zhang Y. C. et al., 2013).
Maize (Zea mays L.) is an important staple crop cultivated worldwide to meet human needs (Xiao et al., 2017). Barbara McClintock discovered transposon silencing in maize (Ravindran, 2012), which is an epigenetic process. It reduces transposition and genomic disruption by transposable elements (TEs). Active TEs are frequently associated with hypomethylation, whereas silenced TEs show significant levels of DNA methylation (Chandler and Walbot, 1986; Okamoto and Hirochika, 2001). Many processes in plants, including development regulation (He et al., 2011; Grossniklaus and Paro, 2014), the response to external stimuli (Eichten and Springer, 2015) and adaptation, is influenced by epigenetic regulation via DNA/RNA methylation, posttranslational histone modifications and ncRNAs (Chinnusamy and Zhu, 2009). The methylation of the ZmMRP4 gene produces a lpa1-241 phenotype with a high amount of inorganic phosphate in the seed (Pilu et al., 2009). In another study, methylation of the p1 gene (which is Myb-homologous and regulates red pigment biosynthesis) is associated with a reduction in pigmentation (Cocciolone et al., 2001). HDA108 is a histone deacetylase, that regulates plant height, leaf development and fertility by reducing H3K9 dimethylation and increasing H3 and H4 histone acetylation in nuclei (Forestan et al., 2018). Downregulation and overexpression of the HDA101 histone deacetylase gene affected the number of acetylated histones, resulting in morphological and developmental defects (Rossi et al., 2007). Interaction of HDA101 with corepressors such as NFC103/MSI1 and SNL1/SIN3-like proteins causes hypoacetylation of lysine 5 in histone H4 (H4K5ac) on targeted genes without affecting their transcript levels (Varotto et al., 2003). It influences the size of the kernel by regulating the transfer of cell-specific gene expression and its excision causes hyperacetylation of histones in the target region without affecting transcript levels. Some inactive genes are also targeted by HDA101 which are associated with hyperacetylation and enhanced expression (Yang et al., 2016). ZmGCN5 advances the development of endosperm during seed maturation by interacting with the adaptor protein ZmADA2 and the bZIP transcriptional factor ZmO2 (Bhat et al., 2004). DNA methyltransferase genes (dmt102 and dmt103) are silenced by DNA demethylation, resulting in apomixis-like phenotypes (Garcia-Aguilar et al., 2010). The Arabidopsis FLOWERING LOCUS T (FT) gene is an ortholog of the ZEA CENTRORADIALIS8 (ZmZCN8) gene in maize, which codes for a phosphatidylethanolamine binding protein of the ZmZCN family (Danilevskaya et al., 2008). In transgenic plants, the expression of ZmZCN8 can lead to early flowering, but suppressing ZmZCN8 using artificial microRNA causes late flowering (Meng et al., 2011). The INDETERMINATE1 (ID1) gene codes for a monocot-specific zinc finger transcriptional regulator that activates ZmZCN8 expression in mature maize leaves (Lazakis et al., 2011). Through epigenetic control in the immature leaf, the ID1 protein controls the transcription of ZmZCN8 and promotes nucleosome remodelling (Mascheretti et al., 2013). ZmZCN7, which is very similar toZmZCN8, is engaged in remodelling regulation of chromatin indicating that it may code for a different maize florigen (Mascheretti et al., 2015). The intake of nutrients from soil like nitrogen (N) and phosphorus (P) is also regulated. Plants growing in less nitrogenous environments activate an adaption process that involves differentially produced miRNAs (Zhao et al., 2013). The miR169, miR399, miR408, and miR528 act in response to severe low nitrogen stress while miRC10 and miRC68 may respond to low soil nitrogen. In the case of a shortage of nitrate, miR528s, miR169s, miR166s and miR408/b cause developmental changes in roots (Xu et al., 2011; Trevisan et al., 2012; Zhao et al., 2012, 2013). miR169s, miR160f-5p, miR156s, and miR171s act by targeting NFY, ARF, SBP, and GRAS transcription factors, respectively, on exposure to cadmium stress (Wang et al., 2019). miR156 negatively regulates SBP-box transcription factor tasselsheath4 (tsh4), allowing the formation of lateral meristems as well as inhibition of initiation of leaves and hence plays a vital role in establishing meristems and the boundary of leaves (Chuck et al., 2010). miR164 negatively regulates NAC1 expression, which influences lateral root growth (Li et al., 2012). The expression of miRNAs can show significant differences during maize seedling development. The majority of miRNA target genes engaged in this mechanism are involved in transcriptional control. miR156, miR396, miR393, miR393 target SPL, GRF, TIR1, LAC, respectively, and may play a crucial role in the regulation of grain filling by governing growth and development (Jin et al., 2015). miR164 plays a regulatory role during seed development (Zheng et al., 2019). The development of the maize kernel is regulated by nine miRNAs belonging to the conserved zma-miR169 family (Xing et al., 2017).
Tomatoes (Solanum lycopersicum L.) are a globally important crop cultivated worldwide and are one of the most consumed vegetables (Kim et al., 2021). The silencing of the MutS HOMOLOG1 (MSH1) gene by RNAi affects growth and development. These are due to DNA methylation as the observed phenotypes get reversed on treatment with 5-AzaC, which is a methylation inhibitor (Yang et al., 2015). The graft-mediated epigenetic changes are determined using the MSH1 system in the rootstock. The msh1 rootstock mutants have hampered siRNA formation. The progenies have enhanced growth vigor that is heritable over five generations and is RdDM-dependent (Kundariya et al., 2020). Hypermethylation of the promoter region of the colourless non-ripening (cnr) gene at CG and CHG regions silences the cnr, resulting in ripening defects (Manning et al., 2006). In wild fruits during ripening, a similar region is demethylated. Zhong et al. (2013) observed a loss of 5mC in the promoters of more than 200 ripening-related genes in a genome-wide investigation of DNA methylation in tomatoes. A decrease in DNA methylation is associated with the suppression of genes responsible for ripening, which also includes genes that participate in photosynthesis as well as the organisation of the cell wall (Lang et al., 2017). The DEMETER-like DNA demethylase (DML) SlDML2 plays a critical role in controlling DNA methylation in ripening tomatoes. SlDML2 inhibition by RNAi caused ripening defects, which were linked to increased methylation in the promoter region that supresses genes responsible for ripening and softening of fruit (Liu et al., 2015). Numerous differentially methylated genes in ethylene or carotenoid pathways encode transcription factors and important enzymes that may be targeted by differently produced non-coding RNAs. ACO2 was targeted by MSTRG.59396.1 and miR396b, CTR1 by MSTRG.43594.1 and miR171b, ERF2 by MSTRG.183681.1, ERF5 by miR9470-3p, PSY1 by MSTRG.95226.7, ZISO by 12:66127788| 66128276, and NCED by MSTRG.181568.2 (Zuo et al., 2020). Fruit softening is influenced by histone post-translational changes and chromatin structural remodelling. To regulate tomato ripening, several genes producing histone deacetylases have been found, albeit genes from different subfamilies may play diverse roles. SlHDA3 and SlHDA1 from the RPD3/HDA1 subfamily, suppress genes involved in cell wall metabolism thereby acting as negative regulators of fruit softening. SlHDT3 of the HD2 subfamily, on the other hand, performed a beneficial function in fruit softening regulation by activating a similar set of genes that are regulated by SlHDA1/3 (Guo et al., 2017a,b, 2018). SlERF.F12 [belongs to the ERF.F subfamily consisting of Ethylene-responsive element-binding factor-associated Amphiphilic Repression (EAR) motifs] along with TOPLESS 2 (TPL2) and histone deacetylases HDA1/HDA3, reduces histone acetylation marks H3K9Ac and H3K27Ac at the promoter region of ripening related genes, resulting in transcription repression (Deng et al., 2022). The integration of a SINE retrotransposon in the promoter and hypermethylation of the inserted SINE has been linked to decreased Vitamin E (VTE3) gene expression (Quadrana et al., 2014). The upregulation of VTE3 expression and an increase in fruit VTE content is caused by spontaneous demethylation of the VTE3 promoter (Quadrana et al., 2014). miRNA156 regulates plant height, leaf size and number as well as fruit size by targeting six SQUAMOSA PROMOTER BINDING PROTEIN (SBP)-box transcription factor genes (Zhang et al., 2011). miRNA156 also controls ovary and fruit development in tomatoes by targeting SPL transcription factors (Ferreira e Silva et al., 2014). miR157 modulates ripening in tomatoes by targeting the LeSPL-CNR gene, leading to mRNA degradation and translation repression. miR159 targets the SGN-U567133 gene and plays a role in leaf and flower development. miR167 causes shorter petals, stamens and styles, as well as reduced leaf size and internode length by targeting ARF6 and ARF8 (Liu N. et al., 2014). miR4376 plays a role in the reproductive growth of tomatoes by targeting Ca2+-ATPase (Wang et al., 2011). miR168 causes phase transition, leaf epinasty and fruit development by targeting AGO1 (Xian et al., 2014).
Soybean (Glycine max (L.) Merr.) is an important seed crop. It supplies both protein and oil, mostly for feed and human consumption. The effect of epigenetics in domestication was investigated in 45 diverse accessions of soybean. Methylome analysis indicated that about 75% of DMRs were due to factors other than genetic variations (Shen et al., 2018). A breeding system for enhancing yield and stability in soybean was reported by Raju et al. (2018). By crossing wild-type and MSH1-acquired soybean memory lines, the MSH1 system was exploited to create epi-lines with a wide variety for numerous yield-related parameters in both greenhouse and field trials. Additionally, obtained epitypes had little epitype–environment interaction, which indicated improved yield stability and less of an impact from the environment. The MSH1 suppression-induced epigenetic variation can be passed down for at least three generations, and it can be bred for crop improvement through a few rounds of selection to improve and stabilise crop yield. DNA methylation is an evolutionarily conserved modification particularly in the symmetric CG context and causes the silencing of genes affecting leaf morphology, flowering time, floral organ identity, fertility and embryogenesis (Jacobsen et al., 2000; Miura et al., 2001; Choi et al., 2002; Xiao et al., 2006; Butenko and Ohad, 2011; Rea et al., 2012). Seed development is also affected by mutation in met1 and ddm1, an ATP-dependent SWI2/SNF2 chromatin-remodeling factor (Lister et al., 2008; Lafos et al., 2011; Baubec et al., 2014; Hossain et al., 2017). Significant CG methylation in soybeans and DMRs occur between the methylomes of cotyledons, leaves, stems and roots (Song et al., 2013). An et al. (2017) reported role of DNA methylation during seed development. They observed methylation in CG (66%), CHG (45%), and CHH (9%) regions of cotyledons. CHH methylation levels increased dramatically from 6% in the initial stages to 11% in late stages. Also, transcribed genes were two times more differentially methylated than non-transcribed genes. In the CG, CHG and CHH contexts, 40, 66, and 2,136 genes were found with DMRs and a negative correlation between their expression as well as methylation was observed. Most of the DMR genes during seed maturation in the CHH context were transcriptionally down-regulated and were associated with DNA replication and cell division. In soybean seed coat, methylation in DNA was found to alter the transposition and splicing of a TE element from a MYB transcription factor that regulates anthocyanin synthase genes (Zabala and Vodkin, 2014).
Rapeseed (Brassica napus L.) is another principal oilseed crop and a major source of protein-rich livestock feed (Wang et al., 2020). Energy consumption efficiency is a distinctive characteristic of plant yield and vigour, and it has an epigenetic component that can be influenced by artificial selection. Populations with distinct physiological and agronomical traits were produced from an isogenic rapeseed population in which the individual plants and their self-fertilised progeny were repeatedly chosen for respiration intensity. Recurrent selection of isogenic lines of B. napus produces stable epigenotypes with better energy usage efficiency. Crossing these epigenetically different but genetically identical lines resulted in hybrids with a 5% yield enhancement (Hauben et al., 2009). Mutation in BrSDG8 (which encodes a histone methyltransferase that affects H3K4 trimethylation in FLOWERING LOCUS C chromatin) is associated with early bolting that governs traits like leafy head formation and seed yield (Fu et al., 2020). In another study, insertion mutations in the Bra032169 gene (encoding histone methyltransferase) are linked to early bolting (Huang et al., 2020). Recently, the role of Jumonji H3K27me3 demethylases (BraA.REF6 and BraA.ELF6) in controlling flower transition has been reported. BraA.REF6 is more important than BraA.ELF6 in regulating H3K27me3 levels. In braA.ref6 mutants, the expression of FLC genes remains unchanged and there is a delay in flowering, while in braA.elf6 mutants, H3K27me3 levels are high at FLC, leading to early flowering (Poza-Viejo et al., 2022). Through an epigenetic change of upstream FLC components, BrcuHAC1 may influence the bolting and flowering time (Si et al., 2021). The demethylating drug 5-AzaC causes DNA hypomethylation in targeted regions, resulting in enhanced amount of seed protein and linoleic acid (Amoah et al., 2012). Solis et al. (2012) observed that decrease in methylation of DNA can initiate microspore embryogenesis. On the other hand, an increase in DNA methylation in a cell-type-specific manner is associated with pollen and embryo differentiation (Solis et al., 2012). 5-AzaC enhances microspore reprogramming, totipotency acquisition, and embryogenesis initiation and prevents embryo differentiation indicating role of methylation in the repression of microspore reprogramming and totipotency acquisition (Solis et al., 2015). A novel small molecule, BIX-01294, reduces histone H3K9 methylation thereby promoting microspore reprogramming and initiation of embryogenesis (Berenguer et al., 2017). During cell reprogramming and embryo development, H3K9me2 and HKMT are engaged in embryo cell differentiation and heterochromatinisation events, whereas H3Ac, H4Ac and HAT are involved in transcriptional activation, totipotency and proliferation events (Rodriguez-Sanz et al., 2014).
The improvement in agronomic traits through epigenetic modification in some other crops is given in Table 1.
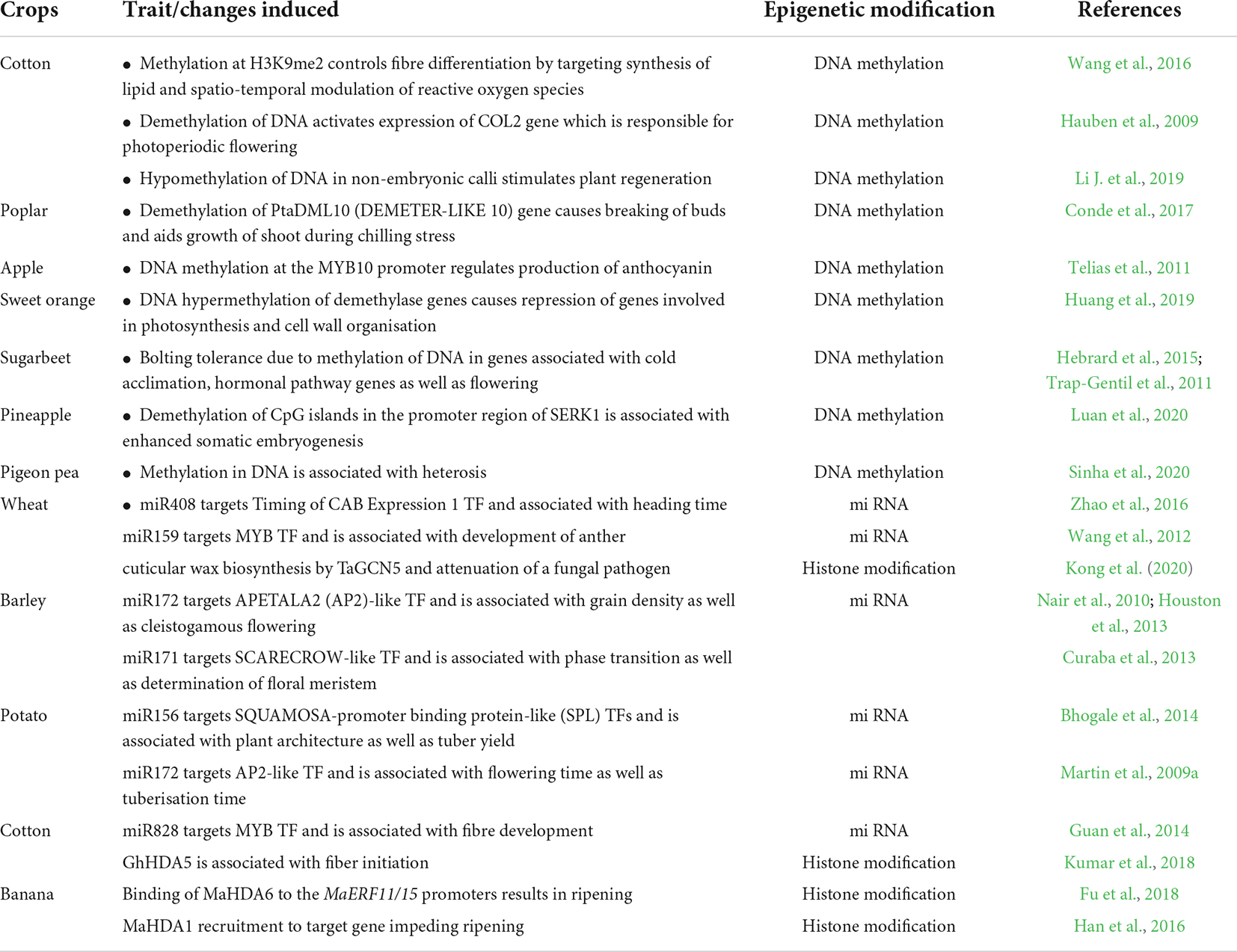
Table 1. Epigenetic changes in crops associated with agronomic traits.
Epigenetics contributes to phenotypic variation. Thus, understanding epigenetics and epigenomics can aid in elucidating the mechanisms through which environmental factors influence plant phenotypes (Agarwal et al., 2020). Breeders may be able to combat the ongoing issue of genetic erosion and uncover cryptic variation through the use of the existing epigenetic variability or epigenome modification. Epibreeding programmes could produce a wide range of phenotypic variability in just one generation and some of these alterations may be passed down from one generation to next (Dalakouras and Vlachostergios, 2021). It should eventually overcome all limitations and constraints that reduce the effectiveness of any crop breeding programme. Novel epialleles would be especially important in breeding populations with low genetic diversity (House and Lukens, 2019). As current breeding techniques mostly concentrate on genetics and disregard epigenetic factors, using epigenetic information at the epialleles level may provide new opportunities for crop development. However, further research on a wider variety of plant species is required to develop a more thorough knowledge of the processes that trigger and maintain epigenetic diversity in crops (Varotto et al., 2020).
The development of high yielding crop varieties is the need of the hour so as to fulfil the demands of an ever-increasing population. Yield is a quantitative trait that is regulated by a number of loci as well as environment. Epigenetic mechanisms like DNA methylation, histone modification and chromatin remodelling have played a major role in controlling agronomic traits. These can increase the phenotypic variation that breeders can use to generate climate-resistant crops. There is also a need to capture and identify the epiallelic variations across the genome sequences of different crop species and even across species. The improvement in agronomic traits through epigenetics is found in many crops including rice, Arabidopsis, maize, soybean, rapeseed, etc. Existing breeding approaches generally focus on genetics and disregard epigenetic components, therefore using epigenetic information could bring new opportunities for crop development.
Both authors contributed equally in writing and editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agarwal, G., Kudapa, H., Ramalingam, A., Choudhary, D., Sinha, P., Garg, V., et al. (2020). Epigenetics and epigenomics: Underlying mechanisms, relevance, and implications in crop improvement. Funct. Integr. Genom. 20, 739–761. doi: 10.1007/s10142-020-00756-7
Aiese Cigliano, R., Sanseverino, W., Cremona, G., Ercolano, M. R., Conicella, C., and Consiglio, F. M. (2013). Genome-wide analysis of histone modifiers in tomato: Gaining an insight into their developmental roles. BMC Genom. 14:57. doi: 10.1186/1471-2164-14-57
Allfrey, V. G., Faulkner, R., and Mirsky, A. E. (1964). Acetylation and methylation of histones and their possible role in regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794. doi: 10.1073/pnas.51.5.786
Allis, C., and Jenuwein, T. (2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500. doi: 10.1038/nrg.2016.59
Amoah, S., Kurup, S., Rodriguez Lopez, C. M., Welham, S. J., Powers, S. J., Hopkins, C. J., et al. (2012). A hypomethylated population of Brassica rapa for forward and reverse epi-genetics. BMC Plant. Biol. 12:193. 193 doi: 10.1186/1471-2229-12-
An, Y. Q. C., Goettel, W., Han, Q., Bartels, A., Liu, Z., and Xiao, W. (2017). Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci. Rep. 7:12263. doi: 10.1038/s41598-017-12510-4
Aquea, F., Timmermann, T., and Arce-Johnson, P. (2010). Analysis of histone acetyltransferase and deacetylase families of Vitis vinifera. Plant Physiol. Biochem. 48, 194–199. doi: 10.1016/j.plaphy.2009.12.009
Axtell, M. J. (2013). Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 64, 137–159. doi: 10.1146/annurev-arplant-050312-120043
Banks, J. A., Masson, P., and Fedoroff, N. (1988). Molecular mechanisms in the developmental regulation of the maize Suppressor-mutator transposable element. Genes Dev. 2:1364–1380. doi: 10.1101/gad.2.11.1364
Bartel, D. P. (2004). MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Baubec, T., Finke, A., Scheid, O. M., and Pecinka, A. (2014). Meristem-specific expression of epigenetic regulators safeguards transposon silencing in Arabidopsis. EMBO Rep. 15, 446–452. doi: 10.1002/embr.201337915
Baulcombe, D. C., and Dean, C. (2014). Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 6:a019471. doi: 10.1101/cshperspect.a019471
Becker, C., Hagmann, J., Muller, J., Koenig, D., Stegle, O., Borgwardt, K., et al. (2011). Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480, 245–249. doi: 10.1038/nature10555
Berenguer, E., Barany, I., Solis, M. T., Perez-Perez, Y., Risueno, M. C., and Testillano, P. S. (2017). Inhibition of histone H3K9 methylation by BIX-01294 promotes stress-induced microspore totipotency and enhances embryogenesis initiation. Front. Plant Sci. 8:1161. doi: 10.3389/fpls.2017.01161
Berr, A., Shafiq, S., and Shen, W. H. (2011). Histone modifications in transcriptional activation during plant development. BBA Gene Regul. Mech. 1809, 567–576. doi: 10.1016/j.bbagrm.2011.07.001
Berr, A., Xu, L., Gao, J., Cognat, V., Steinmetz, A., Dong, A., et al. (2009). SET DOMAIN GROUP25 encodes a histone methyltransferase and is involved in FLOWERING LOCUS C activation and repression of flowering. Plant Physiol. 151, 1476–1485. doi: 10.1104/pp.109.143941
Bhat, R., Borst, J., Riehl, M., and Thompson, R. (2004). Interaction of maize Opaque-2 and the transcriptional co-activators GCN5 and ADA2, in the modulation of transcriptional activity. Plant Mol. Biol. 55, 239–252. doi: 10.1007/s11103-004-0553-z
Bhogale, S., Mahajan, A. S., Natarajan, B., Rajabhoj, M., Thulasiram, H. V., and Banerjee, A. K. (2014). MicroRNA156: A potential graft-transmissible microRNA that modulates plant architecture and tuberization in Solanum tuberosum ssp. andigena. Plant Physiol. 164, 1011–1027. doi: 10.1104/pp.113.230714
Bjerling, P., Silverstein, R. A., Thon, G., Caudy, A., Grewal, S., and Ekwall, K. (2002). Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol. Cell Biol. 22, 2170–2181. doi: 10.1128/mcb.22.7.2170-2181.2002
Buck-Koehntop, B. A., and Defossez, P. A. (2013). On how mammalian transcription factors recognize methylated DNA. Epigenetics 8, 131–137. doi: 10.4161/epi.23632
Butenko, Y., and Ohad, N. (2011). Polycomb-group mediated epigenetic mechanisms through plant evolution. Biochim. Biophys. Acta. 1809, 395–406. doi: 10.1016/j.bbagrm.2011.05.013
Carrozza, M. J., Utley, R. T., Workman, J. L., and Cote, J. (2003). The diverse functions of histone acetyltransferase complexes. Trends Genet. 19, 321–329. doi: 10.1016/S0168-9525(03)00115-X
Chandler, V., and Walbot, V. (1986). DNA modification of a maize transposable element correlates with loss of activity. Proc. Natl. Acad. Sci. U.S.A. 83, 1767–1771. doi: 10.1073/pnas.83.6.1767
Chang, Y. N., Zhu, C., Jiang, J., Zhang, H., Zhu, J. K., and Duan, C. G. (2019). Epigenetic regulation in plant abiotic stress responses. J. Integr. Plant Biol. 62, 563–580. doi: 10.1111/jipb.12901
Charlet, J., Duymich, C. E., Lay, F. D., Mundbjerg, K., Dalsgaard Sorensen, K., Liang, G., et al. (2016). Bivalent regions of cytosine methylation and H3K27 acetylation suggest an active role for DNA methylation at enhancers. Mol. Cell 62, 422–431. doi: 10.1016/j.molcel.2016.03.033
Che, R., Tong, H., Shi, B., Liu, Y., Fang, S., Liu, D., et al. (2015). Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat. Plants 2:15195. doi: 10.1038/nplants.2015.195
Chen, T., and Li, E. (2004). Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol. 60, 55–89. doi: 10.1016/S0070-2153(04)60003-2
Chen, W., Kong, J., Lai, T., Manning, K., Wu, C., Wang, Y., et al. (2015). Tuning LeSPL–CNR expression by SlymiR157 affects tomato fruit ripening. Sci. Rep. 5:7852. doi: 10.1038/srep07852
Chen, X. (2009). Small RNAs and their roles in plant development. Annu. Rev. Cell Dev. Biol. 25, 21–44. doi: 10.1146/annurev.cellbio.042308.113417
Chen, X., and Zhou, D. X. (2013). Rice epigenomics and epigenetics: Challenges and opportunities. Curr. Opin. Plant Biol. 16, 164–169. doi: 10.1016/j.pbi.2013.03.004
Chinnusamy, V., and Zhu, J. K. (2009). Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 12, 133–139. doi: 10.1016/j.pbi.2008.12.006
Choi, Y., Gehring, M., Johnson, L., Hannon, M., Harada, J. J., Goldberg, R. B., et al. (2002). DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 110, 33–42. doi: 10.1016/s0092-8674(02)00807-3
Chuck, G., Whipple, C., Jackson, D., and Hake, S. (2010). The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development 137, 1243–1250. doi: 10.1242/dev.048348
Clayton, A. L., Hazzalin, C. A., and Mahadevan, L. C. (2006). Enhanced histone acetylation and transcription: A dynamic perspective. Mol. Cell 23, 289–296. doi: 10.1093/embo-reports/kvf053
Cloos, P. A., Christensen, J., Agger, K., and Helin, K. (2008). Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 22, 1115–1140. doi: 10.1101/gad.1652908
Cocciolone, S. M., Chopra, S., Flint-Garcia, S. A., McMullen, M. D., and Peterson, T. (2001). Tissue-specific patterns of a maize Myb transcription factor are epigenetically regulated. Plant J. 27, 467–478. doi: 10.1046/j.1365-313x.2001.01124.x
Conde, D., Le Gac, A. L., Perales, M., Dervinis, C., Kirst, M., Maury, S., et al. (2017). Chilling-responsive DEMETER-LIKE DNA demethylase mediates in poplar bud break. Plant Cell Environ. 40, 2236–2249. doi: 10.1111/pce.13019
Cortijo, S., Wardenaar, R., Colome-Tatche, M., Gilly, A., Etcheverry, M., Labadie, K., et al. (2014). Mapping the epigenetic basis of complex traits. Science 343, 1145–1148. doi: 10.1126/science.1248127
Cubas, P., Vincent, C., and Coen, E. (1999). An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161. doi: 10.1038/43657
Cuerda-Gil, D., and Slotkin, R. K. (2016). Non-canonical RNA-directed DNA methylation. Nat. Plants 2:16163. doi: 10.1038/nplants.2016.163
Curaba, J., Talbot, M., Li, Z. Y., and Helliwell, C. (2013). Over-expression of microRNA171 affects phase transitions and floral meristem determinancy in barley. BMC Plant Biol. 13:6. doi: 10.1186/1471-2229-13-6
Dalakouras, A., and Vlachostergios, D. (2021). Epigenetic approaches to crop breeding: Current status and perspectives. J. Exp. Bot. 72, 5356–5371. doi: 10.1093/jxb/erab227
Damodharan, S., Zhao, D., and Arazi, T. (2016). A common miRNA160-based mechanism regulates ovary patterning, floral organ abscission and lamina outgrowth in tomato. Plant J. 86, 458–471. doi: 10.1111/tpj.13127
Danilevskaya, O. N., Meng, X., Hou, Z., Ananiev, E. V., and Simmons, C. R. (2008). A genomic and expression compendium of the expanded PEBP gene family from maize. Plant Physiol. 146, 250–264. doi: 10.1104/pp.107.109538
Deng, H., Chen, Y., Liu, Z., Liu, Z., Shu, P., Wang, R., et al. (2022). SlERF.F12 modulates the transition to ripening in tomato fruit by recruiting the co-repressor TOPLESS and histone deacetylases to repress key ripening genes. Plant Cell 34, 1250–1272. doi: 10.1093/plcell/koac025
Djami-Tchatchou, A. T., and Dubery, I. A. (2015). Lipopolysaccharide perception leads to dynamic alterations in the microtranscriptomes of Arabidopsis thaliana cells and leaf tissues. BMC Plant Biol. 15:79. doi: 10.1186/s12870-015-0465-x
Djami-Tchatchou, A. T., Sanan-Mishra, N., Ntushelo, K., and Dubery, I. A. (2017). Functional roles of microRNAs in agronomically important plants—potential as targets for crop improvement and protection. Front. Plant Sci. 8:378. doi: 10.3389/fpls.2017.00378
Duan, P., Ni, S., Wang, J., Zhang, B., Xu, R., Wang, Y., et al. (2015). Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat. Plants 2:15203. doi: 10.1038/nplants.2015.203
Earley, K. W., Shook, M. S., Brower-Toland, B., Hicks, L., and Pikaard, C. S. (2007). In vitro specificities of Arabidopsis co-activator histone acetyltransferases: Implications for histone hyperacetylation in gene activation. Plant J. 52, 615–626. doi: 10.1111/j.1365-313X.2007.03264.x
Eberharter, A., Lechner, T., Goralik-Schramel, M., and Loidl, P. (1996). Purification and characterization of the cytoplasmic histone acetyltransferase B of maize embryos. FEBS Lett. 386, 75–81. doi: 10.1016/0014-5793(96)00401-2
Eichten, S. R., and Springer, N. M. (2015). Minimal evidence for consistent changes in maize DNA methylation patterns following environmental stress. Front. Plant Sci. 6:308. doi: 10.3389/fpls.2015.00308
Esquinas-Alcazar, J. (2005). Science and society: Protecting crop genetic diversity for food security: Political, ethical and technical challenges. Nat. Rev. Genet. 6, 946–953. doi: 10.1038/nrg1729
Fang, Y., Xie, K., and Xiong, L. (2014). Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 65, 2119–2135. doi: 10.1093/jxb/eru072
Fedoroff, N. V. (2010). The past, present and future of crop genetic modification. N. Biotechnol. 27, 461–465. doi: 10.1016/j.nbt.2009.12.004
Feng, H., Duan, X., Zhang, Q., Li, X., Wang, B., Huang, L., et al. (2014). The target gene of tae-miR164, a novel NAC transcription factor from the NAM subfamily, negatively regulates resistance of wheat to stripe rust. Mol. Plant Pathol. 15, 284–296. doi: 10.1111/mpp.12089
Ferreira e Silva, G. F., Silva, E. M., Azevedo Mda, S., Guivin, M. A., Ramiro, D. A., Figueiredo, C. R., et al. (2014). microRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 78, 604–618. doi: 10.1111/tpj.12493
Forestan, C., Farinati, S., Rouster, J., Lassagne, H., Lauria, M., Dal Ferro, N., et al. (2018). Control of maize vegetative and reproductive development, fertility, and rRNAs silencing by histone deacetylase 108. Genetics 208, 1443–1466. doi: 10.1534/genetics.117.300625/-/DC1.1
Fu, C. C., Han, Y. C., Guo, Y. F., Kuang, J. F., Chen, J. Y., Shan, W., et al. (2018). Differential expression of histone deacetylases during banana ripening and identification of MaHDA6 in regulating ripening-associated genes. Postharvest Biol. Technol. 141, 24–32. doi: 10.1016/j.postharvbio.2018.03.010
Fu, W., Huang, S., Gao, Y., Zhang, M., Qu, G., Wang, N., et al. (2020). Role of BrSDG8 on bolting in Chinese cabbage (Brassica rapa). Theor. Appl. Genet. 133, 2937–2948. doi: 10.1007/s00122-020-03647-4
Gahlaut, V., Zinta, G., Jaiswal, V., and Kumar, S. (2020). Quantitative epigenetics: A new avenue for crop improvement. Epigenomes. 4:25. doi: 10.3390/epigenomes4040025
Gallego-Bartolome, J. (2020). DNA methylation in plants: Mechanisms and tools for targeted manipulation. N. Phytol. 227, 38–44. doi: 10.1111/nph.16529
Gallinari, P., Di Marco, S., Jones, P., Pallaoro, M., and Steinkuhler, C. (2007). HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 17, 195–211. doi: 10.1038/sj.cr.7310149
Gallusci, P., Dai, Z., Genard, M., Gauffretau, A., Leblanc-Fournier, N., Richard-Molard, C., et al. (2017). Epigenetics for plant improvement: Current knowledge and modeling avenues. Trends Plant Sci. 22, 610–623. doi: 10.1016/j.tplants.2017.04.009
Gao, F., Wang, K., Liu, Y., Chen, Y., Chen, P., Shi, Z., et al. (2015). Blocking miR396 increases rice yield by shaping inflorescence architecture. Nat. Plants 2:15196. doi: 10.1038/nplants.2015.196
Gao, S., Yang, L., Zeng, H. Q., Zhou, Z. S., Yang, Z. M., Li, H., et al. (2016). A cotton miRNA is involved in regulation of plant response to salt stress. Sci. Rep. 6:19736. doi: 10.1038/srep19736
Garcia-Aguilar, M., Michaud, C., Leblanc, O., and Grimanelli, D. (2010). Inactivation of a DNA methylation pathway in maize reproductive organs results in apomixis-like phenotypes. Plant Cell 22, 3249–3267. doi: 10.1105/tpc.109.072181
Gehring, M., Reik, W., and Henikoff, S. (2009). DNA demethylation by DNA repair. Trends Genet. 25, 82–90. doi: 10.1016/j.tig.2008.12.001
Grossniklaus, U., and Paro, R. (2014). Transcriptional silencing by polycomb group proteins. Cold Spring Harb. Perspect. Biol. 6:a019331. doi: 10.1101/cshperspect.a019331
Gu, D., Chen, C. Y., Zhao, M., Zhao, L., Duan, X., Duan, J., et al. (2017). Identification of HDA15-PIF1 as a key repression module directing the transcriptional network of seed germination in the dark. Nucleic Acids Res. 45, 7137–7150. doi: 10.1093/nar/gkx283
Guan, X., Pang, M., Nah, G., Shi, X., Ye, W., Stelly, D. M., et al. (2014). miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat. Commun. 5:3050. doi: 10.1038/ncomms4050
Guo, J. E., Hu, Z., Li, F., Zhang, L., Yu, X., Tang, B., et al. (2017b). Silencing of histone deacetylase SlHDT3 delays fruit ripening and suppresses carotenoid accumulation in tomato. Plant Sci. 265:2938. doi: 10.1016/j.plantsci.2017.09.013
Guo, J. E., Hu, Z., Yu, X., Li, A., Li, F., Wang, Y., et al. (2018). A histone deacetylase gene, SlHDA3, acts as a negative regulator of fruit ripening and carotenoid accumulation. Plant Cell Rep. 37, 125–135. doi: 10.1007/s00299-017-2211-3
Guo, J. E., Hu, Z., Zhu, M., Li, F., Zhu, Z., Lu, Y., et al. (2017a). The tomato histone deacetylase SlHDA1 contributes to the repression of fruit ripening and carotenoid accumulation. Sci. Rep. 7:7930. doi: 10.1038/s41598-017-08512-x
Hagmann, J., Becker, C., Muller, J., Stegle, O., Meyer, R. C., Wang, G., et al. (2015). Century-scale methylome stability in a recently diverged Arabidopsis thaliana lineage. PLoS Genet 11:e1004920. doi: 10.1371/journal.pgen.1004920
Haigis, M. C., and Guarente, L. P. (2006). Mammalian sirtuins - emerging roles in physiology, aging, and calories restriction. Genes Dev. 20, 2913–2921. doi: 10.1101/gad.1467506
Han, Y. C., Kuang, J. F., Chen, J. Y., Liu, X. C., Xiao, Y. Y., Fu, C. C., et al. (2016). Banana transcription factor MaERF11 recruits histone deacetylase MaHDA1 and represses the expression of MaACO1 and expansions during fruit ripening. Plant Physiol. 171, 1070–1084. doi: 10.1104/pp.16.00301
Harris, C. J., Scheibe, M., Wongpalee, S. P., Liu, W., Cornett, E. M., Vaughan, R. M., et al. (2018). A DNA methylation reader complex that enhances gene transcription. Science 362, 1182–1186. doi: 10.1126/science.aar7854
Hauben, M., Haesendonckx, B., Standaert, E., Kelen, K. V. D., Azmi, A., Akpo, H., et al. (2009). Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. U.S.A. 106, 20109–20114. doi: 10.1073/pnas.0908755106
He, G., Elling, A. A., and Deng, X. W. (2011). The epigenome and plant development. Annu. Rev. Plant Biol. 62, 411–435. doi: 10.1146/annurev-arplant-042110-103806
He, Y. (2009). Control of the transition to flowering by chromatin modifications. Mol. Plant 2, 554–564. doi: 10.1093/mp/ssp005
Hebrard, C., Peterson, D., Willems, G., Delaunay, A., Jesson, B., Lefebvre, M., et al. (2015). Epigenomics and bolting tolerance in sugar beet genotypes. J. Exp. Bot. 67, 207–225. doi: 10.1093/jxb/erv449
Henderson, I. R., and Jacobsen, S. E. (2007). Epigenetic inheritance in plants. Nature 447, 418–424. doi: 10.1038/nature05917
Hewezi, T., Maier, T. R., Nettleton, D., and Baum, T. J. (2012). The Arabidopsis microRNA396-GRF1/GRF3 regulatory module acts as a developmental regulator in the reprogramming of root cells during cyst nematode infection. Plant Physiol. 159, 321–335. doi: 10.1104/pp.112.193649
Hofmeister, B. T., Lee, K., Rohr, N. A., Hall, D. W., and Schmitz, R. J. (2017). Stable inheritance of DNA methylation allows creation of epigenotype maps and the study of epiallele inheritance patterns in the absence of genetic variation. Genome Biol. 18:155. doi: 10.1186/s13059-017-1288-x
Hong, Z., Ueguchi-Tanaka, M., Fujioka, S., Takatsuto, S., Yoshida, S., Hasegawa, Y., et al. (2005). The rice brassinosteroid-deficient dwarf2 mutant, defective in the rice homolog of Arabidopsis DIMINUTO/DWARF1, is rescued by the endogenously accumulated alternative bioactive brassinosteroid, dolichosterone. Plant Cell 17, 2243–2254. doi: 10.1105/tpc.105.030973
Hossain, S., Kawakatsu, T., Kim, K. D., Zhang, N., Nguyen, C. T., Khan, S. M., et al. (2017). Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. N. Phytol. 214, 808–819. doi: 10.1111/nph.14421
House, M., and Lukens, L. (2019). “The role of germinally inherited epialleles in plant breeding: an update,” in Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications, eds R. Alvarez-Venegas, C. Dela Pena, and J. Casas-Mollano (Cham: Springer), 115–128.
Houston, K., McKim, S. M., Comadran, J., Bonar, N., Druka, I., Uzrek, N., et al. (2013). Variation in the interaction between alleles of HvAPETALA2 and microRNA172 determines the density of grains on the barley inflorescence. Proc. Natl. Acad. Sci. U.S.A. 110, 16675–16680. doi: 10.1073/pnas.1311681110
Hu, J., Wang, Y., Fang, Y., Zeng, L., Xu, J., Yu, H., et al. (2015). A rare allele of GS2 enhances grain size and grain yield in rice. Mol. Plant 8, 1455–1465. doi: 10.1016/j.molp.2015.07.002
Hu, Y., Lu, Y., Zhao, Y., and Zhou, D. X. (2019). Histone acetylation dynamics integrates metabolic activity to regulate plant response to stress. Front. Plant Sci. 10:1236. doi: 10.3389/fpls.2019.01236
Huang, H., Liu, R., Niu, Q., Tang, K., Zhang, B., Zhang, H., et al. (2019). Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. U.S.A. 116, 1430–1436. doi: 10.1073/pnas.1815441116
Huang, J., Li, Z., and Zhao, D. (2016). Deregulation of the osmiR160 target gene OsARF18 causes growth and developmental defects with an alteration of auxin signaling in rice. Sci. Rep. 6:29938. doi: 10.1038/srep29938
Huang, L., Sun, Q., Qin, F., Li, C., Zhao, Y., and Zhou, D. X. (2007). Down-regulation of a silent information regulator2-related histone deacetylase gene, OsSRT1, induces DNA fragmentation and cell death in rice. Plant Physiol. 144, 1508–1519. doi: 10.1104/pp.107.099473
Huang, S., Hou, L., Fu, W., Liu, Z., Li, C., Li, X., et al. (2020). An insertion mutation in Bra032169 encoding a histone methyltransferase is responsible for early bolting in chinese cabbage (Brassica rapa L. ssp. pekinensis). Front. Plant Sci. 11:547. doi: 10.3389/fpls.2020.00547
Iwamoto, M., and Tagiri, A. (2016). MicroRNA-targeted transcription factor gene RDD1 promotes nutrient ion uptake and accumulation in rice. Plant J. 85, 466–477. doi: 10.1111/tpj.13117
Iwasaki, M., and Paszkowski, J. (2014). Epigenetic memory in plants. EMBO J. 33, 1987–1998. doi: 10.15252/embj.201488883
Iwasaki, W., Tachiwana, H., Kawaguchi, K., Shibata, T., Kagawa, W., and Kurumizaka, H. (2011). Comprehensive structural analysis of mutant nucleosomes containing lysine to glutamine (KQ) substitutions in the H3 and H4 histone-fold domains. Biochemistry 50, 7822–7832. doi: 10.1021/bi201021h
Jacobsen, S. E., and Meyerowitz, E. M. (1997). Hypermethylated superman epigenetic alleles in Arabidopsis. Science 277, 1100–1103. doi: 10.1126/science.277.5329.1100
Jacobsen, S. E., Sakai, H., Finnegan, E. J., Cao, X., and Meyerowitz, E. M. (2000). Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol. 10, 179–186. doi: 10.1016/s0960-9822(00)00324-9
Jang, S., An, G., and Li, H. Y. (2017). Rice leaf angle and grain size are affected by the OsBUL1 transcriptional activator complex. Plant Physiol. 173, 688–702. doi: 10.1104/pp.16.01653
Jeddeloh, J. A., Stokes, T. L., and Richards, E. J. (1999). Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet. 22, 94–97. doi: 10.1038/8803
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jin, X., Fu, Z., Lv, P., Peng, Q., Ding, D., Li, W., et al. (2015). Identification and characterization of microRNAs during maize grain filling. PLoS One 10:e0125800. doi: 10.1371/journal.pone.0125800
Johannes, F., Porcher, E., Teixeira, F. K., Saliba-Colombani, V., Simon, M., Agier, N., et al. (2009). Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 5:e1000530. doi: 10.1371/journal.pgen.1000530
Jones-Rhoades, M. W., Bartel, D. P., and Bartel, B. (2006). MicroRNAs and their regulatory roles in plants. Ann. Rev. Plant Biol. 57, 19–53. doi: 10.1146/annurev.arplant.57.032905.105218
Juarez, M. T., Kui, J. S., Thomas, J., Heller, B. A., and Timmermans, M. C. P. (2004). microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 428, 84–88. doi: 10.1038/nature02363
Kakoulidou, I., Avramidou, E. V., Baranek, M., Brunel-Muguet, S., Farrona, S., Johannes, F., et al. (2021). Epigenetics for crop improvement in times of global change. Biology 10:766. doi: 10.3390/biology10080766
Kankel, M. W., Ramsey, D. E., Stokes, T. L., Flowers, S. K., Haag, J. R., Jeddeloh, J. A., et al. (2003). Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163, 1109–1122. doi: 10.1093/genetics/163.3.1109
Kapazoglou, A., Ganopoulos, I., Tani, E., and Tsaftaris, A. (2018). “Epigenetics epigenomics and crop improvement,” in Advances in Botanical Research, ed. M. Kuntz (San Diego, CA: Academic Press), 287–324.
Kim, M., Nguyen, T. T. P., Ahn, J. H., Kim, G. J., and Sim, S. C. (2021). Genome-wide association study identifies QTL for eight fruit traits in cultivated tomato (Solanum lycopersicum L.). Hortic. Res. 8:203. doi: 10.1038/s41438-021-00638-4
Kim, S., Piquerez, S. J. M., Ramirez-Prado, J. S., Mastorakis, E., Veluchamy, A., Latrasse, D., et al. (2020). GCN5 modulates salicylic acid homeostasis by regulating H3K14ac levels at the 5’ and 3’ ends of its target genes. Nucleic Acids Res. 48, 5953–5966. doi: 10.1093/nar/gkaa369
Komiya, R., Ikegami, A., Tamaki, S., Yokoi, S., and Shimamoto, K. (2008). Hd3a and RFT1 are essential for flowering in rice. Development 135, 767–774. doi: 10.1242/dev.008631
Kong, L., Zhi, P., Liu, J., Li, H., Zhang, X., Xu, J., et al. (2020). Epigenetic activation of Enoyl-CoA reductase by an acetyltransferase complex triggers wheat wax biosynthesis. Plant Physiol. 183, 1250–1267. doi: 10.1104/pp.20.00603
Kooke, R., Johannes, F., Wardenaar, R., Becker, F., Etcheverry, M., Colot, V., et al. (2015). Epigenetic basis of morphological variation and phenotypic plasticity in Arabidopsis thaliana. Plant Cell 27, 337–348. doi: 10.1105/tpc.114.133025
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128:693705. 02.005 doi: 10.1016/j.cell.2007
Kumar, S. (2019). Epigenetics and epigenomics for crop improvement. Curr. Opin. Adv. Biotech Micro. 14:555879. doi: 10.19080/AIBM.2019.14.555879
Kumar, V., Singh, B., Singh, S. K., Rai, K. M., Singh, S. P., Sable, A., et al. (2018). Role of GhHDA5 in H3K9 deacetylation and fiber initiation in Gossypium hirsutum. Plant J. 95, 1069–1083. doi: 10.1111/tpj.14011
Kumar, V., Thakur, J. K., and Prasad, M. (2021). Histone acetylation dynamics regulating plant development and stress responses. Cell. Mol. Life Sci. 78, 4467–4486. doi: 10.1007/s00018-021-03794-x
Kundariya, H., Yang, X., Morton, K., Sanchez, R., Axtell, M. J., Hutton, S. F., et al. (2020). MSH1-induced heritable enhanced growth vigor through grafting is associated with the RdDM pathway in plants. Nat. Commun. 11:5343. doi: 10.1038/s41467-020-19140-x
Lafos, M., Kroll, P., Hohenstatt, M. L., Thorpe, F. L., Clarenz, O., and Schubert, D. (2011). Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 7:e1002040. doi: 10.1371/journal.pgen.1002040
Lang, Z., Wang, Y., Tang, K., Tang, D., Datsenka, T., Cheng, J., et al. (2017). Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. U.S.A. 114, E4511–E4519. doi: 10.1073/pnas.1705233114
Lauria, M., and Rossi, V. (2011). Epigenetic control of gene regulation in plants. Biochim. Biophys. Acta 1809, 369–378. doi: 10.1016/j.bbagrm.2011.03.002
Lazakis, C. M., Coneva, V., and Colasanti, J. (2011). ZCN8 encodes a potential orthologue of Arabidopsis FT florigen that integrates both endogenous and photoperiod flowering signals in maize. J. Exp. Bot. 62, 4833–4842. doi: 10.1093/jxb/err129
Lee, Y. S., Lee, D. Y., Cho, L. H., and An, G. (2014). Rice miR172 induces flowering by suppressing OsIDS1 and SNB, two AP2 genes that negatively regulate expression of Ehd1 and florigens. Rice 7:31. doi: 10.1186/s12284-014-0031-4
Li, D., Wang, L., Wang, M., Xu, Y. Y., Luo, W., Liu, Y. J., et al. (2009). Engineering OsBAK1 gene as a molecular tool to improve rice architecture for high yield. Plant Biotechnol. J. 7, 791–806. doi: 10.1111/j.1467-7652.2009.00444.x
Li, J., Guo, G., Guo, W., Guo, G., Tong, D., Ni, Z., et al. (2012). miRNA164-directed cleavage of ZmNAC1 confers lateral root development in maize (Zea mays L.). BMC Plant Biol. 12:220. doi: 10.1186/1471-2229-12-220
Li, J., Wang, M., Li, Y., Zhang, Q., Lindsey, K., Daniell, H., et al. (2019). Multi-omics analyses reveal epigenomics basis for cotton somatic embryogenesis through successive regeneration acclimation process. Plant Biotechnol. J. 17, 435–450. doi: 10.1111/pbi.12988
Li, Q., Song, J., West, P. T., Zynda, G., Eichten, S. R., Vaughn, M. W., et al. (2015). Examining the causes and consequences of context-specific differential DNA methylation in maize. Plant Physiol. 168, 1262–1274. doi: 10.1104/pp.15.00052
Li, S., Lin, Y. C. J., Wang, P., Zhang, B., Li, M., Chen, S., et al. (2019). The AREB1 transcription factor influences histone acetylation to regulate drought responses and tolerance in populus trichocarpa. Plant Cell 31, 663–686. doi: 10.1105/tpc.18.00437
Li, W., Chen, G., Zhu, S., Zhu, P., Zhang, Q., and Hu, T. (2020). Overexpression of TCP transcription factor OsPCF7 improves agronomic trait in rice. Mol. Breed. 40:48. doi: 10.1007/s11032-020-01129-5
Li, Y., Lu, Y., Shi, Y., Wu, L., Xu, Y. J., Huang, F., et al. (2014). Multiple rice microRNAs are involved in immunity against the blast fungus Magnaporthe oryzae. Plant Physiol. 164, 1077–1092. doi: 10.1104/pp.113.230052
Lippman, Z., Gendrel, A. V., Black, M., Vaughn, M. W., Dedhia, N., McCombie, W. R., et al. (2004). Role of transposable elements in heterochromatin and epigenetic control. Nature 430, 471–476. doi: 10.1038/nature02651
Lister, R., Omalley, R., Tonti-Filippini, J., Gregory, B. D., Berry, C. C., Millar, A. H., et al. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536. doi: 10.1016/j.cell.2008.03.029
Liu, B., and Wendel, J. F. (2003). Epigenetic phenomena and the evolution of plant allopolyploids. Mol. Phylogenet. Evol. 29, 365–379. doi: 10.1016/s1055-7903(03)00213-6
Liu, C. Y., Lu, F. L., Cui, X., and Cao, X. F. (2010). Histone methylation in higher plants. Annu. Rev. Plant Biol. 61, 395–420. doi: 10.1146/annurev.arplant.043008.091939
Liu, N., Wu, S., Van Houten, J., Wang, Y., Ding, B., Fei, Z., et al. (2014). Down-regulation of AUXIN RESPONSE FACTORS 6 and 8 by microRNA 167 leads to floral development defects and female sterility in tomato. J. Exp. Bot. 65, 2507–2520. doi: 10.1093/jxb/eru141
Liu, R., How-Kit, A., Stammitti, L., Teyssier, E., Rolin, D., Mortain-Bertrand, A., et al. (2015). A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc. Natl. Acad. Sci. U.S.A. 112, 10804–10809. doi: 10.1073/pnas.1503362112
Liu, X., Yang, S., Yu, C. W., Chen, C. Y., and Wu, K. (2016). Chapter six – histone acetylation and plant development. Enzymes 40, 173–199. doi: 10.1016/bs.enz.2016.08.001
Liu, X., Yang, S., Zhao, M., Luo, M., Yu, C. W., Chen, C. Y., et al. (2014). Transcriptional repression by histone deacetylases in plants. Mol. Plant 7, 764–772. doi: 10.1093/mp/ssu033
Lombardo, L., Coppola, G., and Zelasco, S. (2016). New technologies for insect-resistant and herbicide-tolerant plants. Trends Biotechnol. 34, 49–57. doi: 10.1016/j.tibtech.2015.10.006
Long, Y., Xia, W., Li, R., Wang, J., Shao, M., Feng, J., et al. (2011). Epigenetic QTL mapping in Brassica napus. Genetics 189, 1093–1102. doi: 10.1534/genetics.111.131615
Luan, A., Chen, C., Xie, T., He, J., and He, Y. (2020). Methylation analysis of CpG islands in pineapple SERK1 promoter. Genes 11:425. doi: 10.3390/genes11040425
Luan, X., Liu, S., Ke, S., Dai, H., Xie, X. M., Hsieh, T. F., et al. (2019). Epigenetic modification of ESP, encoding a putative long noncoding RNA, affects panicle architecture in rice. Rice 12:20. doi: 10.1186/s12284-019-0282-1
Luger, K., and Richmond, T. J. (1998). The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 8, 140–146. doi: 10.1016/s0959-437x(98)80134-2
Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., and Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. doi: 10.1038/38444
Lusser, A., Eberharter, A., Loidl, A., Goralik-Schramel, M., Horngacher, M., Haas, H., et al. (1999). Analysis of the histone acetyltransferase B complex of maize embryos. Nucleic Acids Res. 27, 4427–4435. doi: 10.1093/nar/27.22.4427
Manning, K., Tor, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Martin, A., Adam, H., Díaz-Mendoza, M., Zurczak, M., Gonzalez-Schain, N. D., and Suarez-Lopez, P. (2009a). Graft-transmissible induction of potato tuberization by the microRNA miR172. Development 136, 2873–2881. doi: 10.1242/dev.031658
Martin, A., Troadec, C., Boualem, A., Rajab, M., Fernandez, R., Morin, H., et al. (2009b). A transposon-induced epigenetic change leads to sex determination in melon. Nature 461, 1135–1138. doi: 10.1038/nature08498
Martinez de Alba, A. E., Elvira-Matelot, E., and Vaucheret, H. (2013). Gene silencing in plants: A diversity of pathways. Biochim. Biophys. Acta 1829, 1300–1308. doi: 10.1016/j.bbagrm.2013.10.005
Mascheretti, I., Battaglia, R., Mainieri, D., Altana, A., Lauria, M., and Rossi, V. (2013). The WD40-repeat proteins NFC101 and NFC102 regulate different aspects of maize development through chromatin modification. Plant Cell 25, 404–420. doi: 10.1105/tpc.112.107219
Mascheretti, I., Turner, K., Brivio, R. S., Hand, A., Colasanti, J., and Rossi, V. (2015). Florigen-encoding genes of day-neutral and photoperiod sensitive maize are regulated by different chromatin modifications at the floral transition. Plant Physiol. 168, 1351–1363. doi: 10.1104/pp.15.00535
Mbichi, R. W., Wang, Q. F., and Wan, T. (2020). RNA directed DNA methylation and seed plant genome evolution. Plant Cell Rep. 39, 983–996. 020-02558-4 doi: 10.1007/s00299-
Meng, X., Muszynski, M. G., and Danilevskaya, O. N. (2011). The FT-like ZCN8 gene functions as a floral activator and is involved in photoperiod sensitivity in maize. Plant Cell 23, 942–960. doi: 10.1105/tpc.110.081406
Merce, C., Bayer, P. E., Fernandez, C. T., Batley, J., and Edwards, D. (2020). Induced methylation in plants as a crop improvement tool: Progress and perspectives. Agronomy 10:1484. doi: 10.3390/agronomy10101484
Meyer, P. (2015). Epigenetic variation and environmental change. J. Exp. Bot. 66, 3541–3548. doi: 10.1093/jxb/eru502
Millar, C. B., and Grunstein, M. (2006). Genome-wide patterns of histone modifications in yeast. Nat. Rev. Mol. Cell Biol. 7, 657–666. doi: 10.1038/nrm1986
Miura, A., Yonebayashi, S., Watanabe, K., Toyama, T., Shimada, H., and Kakutani, T. (2001). Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411, 212–214. doi: 10.1038/35075612
Miura, K., Agetsuma, M., Kitano, H., Yoshimura, A., Matsuoka, M., Jacobsen, S. E., et al. (2009). A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc. Natl. Acad. Sci. U.S.A. 106, 11218–11223. doi: 10.1073/pnas.0901942106
Miura, K., Ikeda, M., Matsubara, A., Song, X. J., Ito, M., Asano, K., et al. (2010). OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 42, 545–549. doi: 10.1038/ng.592
Nair, S. K., Wang, N., Turuspekov, Y., Pourkheirandish, M., Sinsuwongwat, S., Chen, G., et al. (2010). Cleistogamous flowering in barley arises from the suppression of microRNA-guided HvAP2 mRNA cleavage. Proc. Natl. Acad. Sci. U.S.A. 107, 490–495. doi: 10.1073/pnas.0909097107
Ng, D. W., Wang, T., Chandrasekharan, M. B., Aramayo, R., Kertbundit, S., and Hall, T. C. (2007). Plant SET domain-containing proteins: Structure, function and regulation. Biochim. Biophys. Acta 1769, 316–329. doi: 10.1016/j.bbaexp.2007.04.003
Niederhuth, C. E., and Schmitz, R. J. (2014). Covering your bases: Inheritance of DNA methylation in plant genomes. Mol. Plant 7, 472–480. doi: 10.1093/mp/sst165
Nizampatnam, N. R., Schreier, S. J., Damodaran, S., Adhikari, S., and Subramanian, S. (2015). microRNA160 dictates stage-specific auxin and cytokinin sensitivities and directs soybean nodule development. Plant J. 84, 140–153. doi: 10.1111/tpj.12965
Okamoto, H., and Hirochika, H. (2001). Silencing of transposable elements in plants. Trends Plant Sci. 6, 527–534. doi: 10.1016/s1360-1385(01)02105-7
Onufriev, A. V., and Schiessel, H. (2019). The nucleosome: From structure to function through physics. Curr. Opin. Struct. Biol. 56, 119–130. doi: 10.1016/j.sbi.2018.11.003
Ori, N., Cohen, A., and Etzioni, A. (2007). Regulation of LANCEOLATE by miR319 is required for compound-leaf development in tomato. Nat. Genet. 39, 787–791. doi: 10.1038/ng2036
Palmgren, M. G., Edenbrandt, A. K., Vedel, S. E., Andersen, M. M., Landes, X., Osterberg, J. T., et al. (2015). Are we ready for back-to-nature crop breeding? Trends Plant Sci. 20, 155–164. doi: 10.1016/j.tplants.2014.11.003
Pandey, P., Srivastava, P. K., and Pandey, S. P. (2019). Prediction of plant miRNA targets. Methods Mol. Biol. 1932, 99–107. doi: 10.1007/978-1-4939-9042-9_7
Pandey, R., Muller, A., Napoli, C. A., Selinger, D. A., Pikaard, C. S., Richards, E. J., et al. (2002). Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 30, 5036–5055. doi: 10.1093/nar/gkf660
Pantaleo, V., Szittya, G., Moxon, S., Miozzi, L., Moulton, V., Dalmay, T., et al. (2010). Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 62, 960–976. doi: 10.1111/j.0960-7412.2010.04208.x
Park, M. Y., Wu, G., Gonzalez-Sulser, A., Vaucheret, H., and Poethig, R. S. (2005). Nuclear processing and export of microRNAs in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 102, 3691–3696. doi: 10.1073/pnas.0405570102
Parthun, M. R., Widom, J., and Gottschling, D. E. (1996). The major cytoplasmic histone acetyltransferase in yeast: Links to chromatin replication and histone metabolism. Cell 87, 85–94. doi: 10.1016/s0092-8674(00)81325-2
Pecinka, A., Abdelsamad, A., and Vu, G. T. (2013). Hidden genetic nature of epigenetic natural variation in plants. Trends Plant Sci. 18, 625–632. doi: 10.1016/j.tplants.2013.07.005
Pecinka, A., and Mittelsten Scheid, O. (2012). Stress-induced chromatin changes: A critical view on their heritability. Plant Cell Physiol. 53, 801–808. doi: 10.1093/pcp/pcs044
Pecinka, A., Dinh, H. Q., Baubec, T., Rosa, M., Lettner, N., and Mittelsten Scheid, O. (2010). Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 22, 3118–3129. doi: 10.1105/tpc.110.078493
Pelaez, P., Trejo, M. S., Iniguez, L. P., Estrada-Navarrete, G., Covarrubias, A. A., Reyes, J. L., et al. (2012). Identification and characterization of microRNAs in Phaseolus vulgaris by high-throughput sequencing. BMC Genom. 13:83. doi: 10.1186/1471-2164-13-83
Peng, M., Ying, P., Liu, X., Li, C., Xia, R., Li, J., et al. (2017). Genome-wide identification of histone modifiers and their expression patterns during fruit abscission in litchi. Front. Plant Sci. 8:639. doi: 10.3389/fpls.2017.00639
Pikaard, C. S., and Mittelsten Scheid, O. (2014). Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 6:a019315. doi: 10.1101/cshperspect.a019315
Pilu, R., Panzeri, D., Cassani, E., Cerino Badone, F., Landoni, M., and Nielsen, E. A. (2009). Paramutation phenomenon is involved in the genetics of maize low phytic acid1–241 (lpa1–241) trait. Heredity 102, 236–245. doi: 10.1038/hdy.2008.96
Poza-Viejo, L., Paya-Milans, M., San Martin-Uriz, P., Castro-Labrador, L., Lara-Astiaso, D., Wilkinson, M. D., et al. (2022). Conserved and distinct roles of H3K27me3 demethylases regulating flowering time in Brassica rapa. Plant Cell Environ. 45, 1428–1441. doi: 10.1111/pce.14258
Pradeepa, M. M., Grimes, G. R., Kumar, Y., Olley, G., Taylor, G. C., Schneider, R., et al. (2016). Histone H3 globular domain acetylation identifies a new class of enhancers. Nat. Genet. 48, 681–686. doi: 10.1038/ng.3550
Priya, T. S. R., Nelson, A. R. L. E., Ravichandran, K., and Antony, U. (2019). Nutritional and functional properties of coloured rice varieties of South India: A review. J. Ethn. Foods 6:11. doi: 10.1186/s42779-019-0017-3
Quadrana, L., Almeida, J., Asis, R., Duffy, T., Dominguez, P. G., Bermudez, L., et al. (2014). Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 5:4027. doi: 10.1038/ncomms5027
Quadrana, L., and Colot, V. (2016). Plant transgenerational epigenetics. Annu. Rev. Genet. 50, 467–491. doi: 10.1146/annurev-genet-120215-035254
Raju, S. K. K., Shao, M. R., Sanchez, R., Xu, Y. Z., Sandhu, A., Graef, G., et al. (2018). An epigenetic breeding system in soybean for increased yield and stability. Plant Biotechnol. J. 16, 1836–1847. doi: 10.1111/pbi.12919
Ravindran, S. (2012). Barbara McClintock and the discovery of jumping genes. Proc. Natl. Acad. Sci. U.S.A. 109, 20198–20199. doi: 10.1073/pnas.1219372109
Rea, M., Zheng, W., Chen, M., Braud, C., Bhangu, D., Rognan, T. N., et al. (2012). Histone H1 affects gene imprinting and DNA methylation in Arabidopsis. Plant J. 71, 776–786. doi: 10.1111/j.1365-313X.2012.05028.x
Reinders, J., Wulff, B. B. H., Mirouze, M., Mari-Ordonez, A., Dapp, M., Rozhon, W., et al. (2009). Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 23, 939–950. doi: 10.1101/gad.524609
Rey, O., Danchin, E., Mirouze, M., Loot, C., and Blanchet, S. (2016). Adaptation to global change: A transposable element-epigenetics perspective. Trends Ecol. Evol. 31, 514–526. doi: 10.1016/j.tree.2016.03.013
Rodriguez-Sanz, H., Moreno-Romero, J., Solis, M. T., Kohler, C., Risueno, M. C., and Testillano, P. S. (2014). Changes in histone methylation and acetylation during microspore reprogramming to embryogenesis occur concomitantly with Bn HKMT and Bn HAT expression and are associated with cell totipotency, proliferation, and differentiation in Brassica napus. Cytogenet. Genome Res. 143, 209–218. doi: 10.1159/000365261
Rossi, V., Locatelli, S., Varotto, S., Donn, G., Pirona, R., Henderson, D. A., et al. (2007). Maize histone deacetylase hda101 is involved in plant development, gene transcription, and sequence specific modulation of histone modification of genes and repeats. Plant Cell 19, 1145–1162. doi: 10.1105/tpc.106.042549
Sakamoto, T., Morinaka, Y., Ohnishi, T., Sunohara, H., Fujioka, S., Ueguchi-Tanaka, M., et al. (2006). Erect leaves caused by brassinosteroid deficiency increase biomass production and grain yield in rice. Nat. Biotechnol. 24, 105–109. doi: 10.1038/nbt1173
Saze, H., Mittelsten Scheid, O., and Paszkowski, J. (2003). Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat. Genet. 34, 65–69. doi: 10.1038/ng1138
Schmitz, R. J., He, Y., Valdes-Lopez, O., Khan, S. M., Joshi, T., Urich, M. A., et al. (2013). Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 23, 1663–1674. doi: 10.1101/gr.152538.112
Shen, Y., Zhang, J., Liu, Y., Liu, L., Liu, Z., Duan, Z., et al. (2018). DNA methylation footprints during soybean domestication and improvement. Genome Biol. 19:128. doi: 10.1186/s13059-018-1516-z
Shi, J., Li, R., Qiu, D., Jiang, C., Long, Y., Morgan, C., et al. (2009). Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182, 851–861. doi: 10.1534/genetics.109.101642
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. doi: 10.1016/j.cell.2004.12.012
Shiba, H., Kakizaki, T., Iwano, M., Tarutani, Y., Watanabe, M., Isogai, A., et al. (2006). Dominance relationships between self-incompatibility alleles controlled by DNA methylation. Nat. Genet. 38, 297–299. doi: 10.1038/ng1734
Si, S., Zhang, M., Hu, Y., Wu, C., Yang, Y., Luo, S., et al. (2021). BrcuHAC1 is a histone acetyltransferase that affects bolting development in Chinese flowering cabbage. J. Genet. 100:56. doi: 10.1007/s12041-021-01303-4
Silveira, A. B., Trontin, C., Cortijo, S., Barau, J., Vieira Del Bem, L. E., Loudet, O., et al. (2013). Extensive natural epigenetic variation at a de novo originated gene. PLoS Genet. 9:e1003437. doi: 10.1371/journal.pgen.1003437
Sinha, P., Singh, V. K., Saxena, R. K., Kale, S. M., Li, Y., Garg, V., et al. (2020). Genome-wide analysis of epigenetic and transcriptional changes associated with heterosis in pigeonpea. Plant Biotechnol. J. 18, 1697–1710. doi: 10.1111/pbi.13333
Slatkin, M. (2009). Epigenetic inheritance and the missing heritability problem. Genetics 182, 845–850. doi: 10.1534/genetics.109.102798
Solis, M. T., El-Tantawy, A. A., Cano, V., Risueno, M. C., and Testillano, P. S. (2015). 5-azacytidine promotes microspore embryogenesis initiation by decreasing global DNA methylation, but prevents subsequent embryo development in rapeseed and barley. Front. Plant. Sci. 6:472. doi: 10.3389/fpls.2015.00472
Solis, M. T., Rodriguez-Serrano, M., Meijon, M., Canal, M. J., Cifuentes, A., Risueno, M. C., et al. (2012). DNA methylation dynamics and MET1a-like gene expression changes during stress-induced pollen reprogramming to embryogenesis. J. Exp. Bot. 63, 6431–6444. doi: 10.1093/jxb/ers298
Song, Q. X., Lu, X., Li, Q. T., Chen, H., Hu, X. Y., Ma, B., et al. (2013). Genome-wide analysis of DNA methylation in soybean. Mol. Plant 6, 1961–1974. doi: 10.1093/mp/sst123
Song, Q., Zhang, T., Stelly, D. M., and Chen, Z. J. (2017). Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 18:99. doi: 10.1186/s13059-017-1229-8
Song, X. J., Kuroha, T., Ayano, M., Furuta, T., Nagai, K., Komeda, N., et al. (2015). Rare allele of a previously unidentifed histone H4 acetyltransferase enhances grain weight, yield, and plant biomass in rice. Proc. Natl. Acad. Sci. U.S.A. 112, 76–81. doi: 10.1073/pnas.1421127112
Spruijt, C. G., and Vermeulen, M. (2014). DNA methylation: Old dog, new tricks? Nat. Struct. Mol. Biol. 21, 949–954. doi: 10.1038/nsmb.2910
Srivastava, R., Rai, K. M., Srivastava, M., Kumar, V., Pandey, B., Singh, S. P., et al. (2014). Distinct role of core promoter architecture in regulation of light-mediated responses in plant genes. Mol. Plant 7, 626–641. doi: 10.1093/mp/sst146
Sterner, D. E., and Berger, S. L. (2000). Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459.
Sun, C., Fang, J., Zhao, T., Xu, B., Zhang, F., Liu, L., et al. (2012). The histone methyltransferase SDG724 mediates H3K36me2/3 deposition at MADS50 and RFT1 and promotes flowering in rice. Plant Cell 24, 3235–3247. doi: 10.1105/tpc.112.101436
Sun, G. (2012). MicroRNAs and their diverse functions in plants. Plant Mol. Biol. 80, 17–36. doi: 10.1007/s11103-011-9817-6
Sun, S., Chen, D., Li, X., Qiao, S., Shi, C., Li, C., et al. (2015). Brassinosteroid signaling regulates leaf erectness in Oryza sativa via the control of a specific U-type cyclin and cell proliferation. Dev. Cell 34, 220–228. doi: 10.1016/j.devcel.2015.05.019
Swygert, S. G., and Peterson, C. L. (2014). Chromatin dynamics: Interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta 1839, 728–736. doi: 10.1016/j.bbagrm.2014.02.013
Takatsuka, H., and Umeda, M. (2015). Epigenetic control of cell division and cell differentiation in the root apex. Front. Plant Sci. 6:1178. doi: 10.3389/fpls.2015.01178
Takeda, S., and Matsuoka, M. (2008). Genetic approaches to crop improvement: Responding to environmental and population changes. Nat. Rev. Genet. 9, 444–457. doi: 10.1038/nrg2342
Tang, J., and Chu, C. (2017). MicroRNAs in crop improvement: Fine-tuners for complex traits. Nat. Plants 3:17077. doi: 10.1038/nplants.2017.77
Tang, K., Lang, Z., Zhang, H., and Zhu, J. K. (2016). The DNA demethylase ROS1 targets genomic regions with distinct chromatin modifications. Nat. Plants 2:16169. doi: 10.1038/nplants.2016.169
Taudt, A., Tatche, M. C., and Johannes, F. (2016). Genetic sources of population epigenomic variation. Nat. Rev. Genet. 17, 319–332. doi: 10.1038/nrg.2016.45
Telias, A., Lin-Wang, K., Stevenson, D. E., Cooney, J. M., Hellens, R. P., Allan, A. C., et al. (2011). Apple skin patterning is associated with differential expression of MYb10. BMC Plant Biol. 11:93. doi: 10.1186/1471-2229-11-93
Teperino, R., Schoonjans, K., and Auwerx, J. (2010). Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab. 12, 321–327. doi: 10.1016/j.cmet.2010.09.004
Thorstensen, T., Grini, P. E., and Aalen, R. B. (2011). SET domain proteins in plant development. BBA Gene Regul. Mech. 1809, 407–420. doi: 10.1016/j.bbagrm.2011.05.008
Tirnaz, S., and Batley, J. (2019). Epigenetics: Potentials and challenges in crop breeding. Mol. Plant 12, 1309–1311. doi: 10.1016/j.molp.2019.09.006
Tong, H., Liu, L., Jin, Y., Du, L., Yin, Y., Qian, Q., et al. (2012). DWARF AND LOW-TILLERING acts as a direct downstream target of a GSK3/SHAGGY-like kinase to mediate brassinosteroid responses in rice. Plant Cell 24, 2562–2577. doi: 10.1105/tpc.112.097394
Trap-Gentil, M. V., Hebrard, C., Lafon-Placette, C., Delaunay, A., Hagege, D., Joseph, C., et al. (2011). Time course and amplitude of DNA methylation in the shoot apical meristem are critical points for bolting induction in sugar beet and bolting tolerance between genotypes. J. Exp. Bot. 62, 2585–2597. doi: 10.1093/jxb/erq433
Trevisan, S., Nonis, A., Begheldo, M., Manoli, A., Palme, K., Caporale, G., et al. (2012). Expression and tissue-specific localization of nitrate-responsive miRNAs in roots of maize seedlings. Plant Cell Environ. 35, 1137–1155. doi: 10.1111/j.1365-3040.2011.02478.x
Tsuji, H., Aya, K., Ueguchi-Tanaka, M., Shimada, Y., Nakazono, M., Watanabe, R., et al. (2006). GAMYB controls different sets of genes and is differentially regulated by microRNA in aleurone cells and anthers. Plant J. 47, 427–444. doi: 10.1111/j.1365-313X.2006.02795.x
Tsukada, Y. I., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. doi: 10.1038/nature04433
Ueda, M., and Seki, M. (2020). Histone modifications form epigenetic regulatory networks to regulate abiotic stress response. Plant Physiol. 182, 15–26. doi: 10.1104/pp.19.00988
Varotto, S., Krugman, T., Cigliano, R. A., Kashkush, K., Kondic-Spika, A., Aravanopoulos, F. A., et al. (2022). Exploitation of epigenetic variation of crop wild relatives for crop improvement and agrobiodiversity preservation. Theor. Appl. Genet. 1–17. doi: 10.1007/s00122-022-04122-y
Varotto, S., Locatelli, S., Canova, S., Pipal, A., Motto, M., and Rossi, V. (2003). Expression profile and cellular localization of maize Rpd3-type histone deacetylases during plant development. Plant Physiol. 133, 606–617. doi: 10.1104/pp.103.025403
Varotto, S., Tani, E., Abraham, E., Krugman, T., Kapazoglou, A., Melzer, R., et al. (2020). Epigenetics: Possible applications in climate-smart crop breeding. J. Exp. Bot. 71, 5223–5236. doi: 10.1093/jxb/eraa188
Verreault, A., Kaufman, P. D., Kobayashi, R., and Stillman, B. (1998). Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr. Biol. 8, 96–108. doi: 10.1016/s0960-9822(98)70040-5
Voinnet, O. (2009). Origin, biogenesis, and activity of plant microRNAs. Cell 136, 669–687. doi: 10.1016/j.cell.2009.01.046
Wang, B., Cheng, D., Chen, Z., Zhang, M., Zhang, G., Jiang, M., et al. (2019). Bioinformatic exploration of the targets of Xylem Sap miRNAs in Maize under cadmium stress. Int. J. Mol. Sci. 20:1474. doi: 10.3390/ijms20061474
Wang, L., Sun, S., Jin, J., Fu, D., Yang, X., Weng, X., et al. (2015). Coordinated regulation of vegetative and reproductive branching in rice. Proc. Natl. Acad. Sci. U.S.A. 112, 15504–15509. doi: 10.1073/pnas.1521949112
Wang, L., Zhang, F., Rode, S., Chin, K. K., Ko, E. E., Kim, J., et al. (2017). Ethylene induces combinatorial effects of histone H3 acetylation in gene expression in Arabidopsis. BMC Genom. 18:538. doi: 10.1186/s12864-017-3929-6
Wang, M., Wang, P., Tu, L., Zhu, S., Zhang, L., Li, Z., et al. (2016). Multi-omics maps of cotton fibre reveal epigenetic basis for staged single-cell differentiation. Nucleic Acids Res. 44, 4067–4079. doi: 10.1093/nar/gkw238
Wang, T., Wei, L., Wang, J., Xie, L., Li, Y. Y., Ran, S., et al. (2020). Integrating GWAS, linkage mapping and gene expression analyses reveals the genetic control of growth period traits in rapeseed (Brassica napus L.). Biotechnol. Biofuels 13:134. doi: 10.1186/s13068-020-01774-0
Wang, Y., Itaya, A., Zhong, X., Wu, Y., Zhang, J., van der Knaap, E., et al. (2011). Function and evolution of a microRNA that regulates a Ca2+-ATPase and triggers the formation of phased small interfering RNAs in tomato reproductive growth. Plant Cell 23, 3185–3203. doi: 10.1105/tpc.111.088013
Wang, Y., Sun, F., Cao, H., Peng, H., Ni, Z., Sun, Q., et al. (2012). TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS One 7:e48445. doi: 10.1371/journal.pone.0048445
Wei, X., Song, X., Wei, L., Tang, S., Sun, J., Hu, P., et al. (2017). An epiallele of rice AK1 affects photosynthetic capacity. J. Integr. Plant Biol. 59, 158–163. doi: 10.1111/jipb.12518
Weigel, D., and Colot, V. (2012). Epialleles in plant evolution. Genome Biol. 13:249. doi: 10.1186/gb-2012-13-10-249
Wu, C. Y., Trieu, A., Radhakrishnan, P., Kwok, S. F., Harris, S., Zhang, K., et al. (2008). Brassinosteroids regulate grain filling in rice. Plant Cell 20, 2130–2145. doi: 10.1105/tpc.107.055087
Wu, K., Wang, S., Song, W., Zhang, J., Wang, Y., Liu, Q., et al. (2020). Enhanced sustainable green revolution yield via nitrogen-responsive chromatin modulation in rice. Science 367:eaaz2046. doi: 10.1126/science.aaz2046
Xian, Z., Huang, W., Yang, Y., Tang, N., Zhang, C., Re, M., et al. (2014). miR168 influences phase transition, leaf epinasty, and fruit development via SlAGO1s in tomato. J. Exp. Bot. 65, 6655–6666. doi: 10.1093/jxb/eru387
Xiao, W., Custard, K. D., Brown, R. C., Lemmon, B. E., Harada, J. J., Goldberg, R. B., et al. (2006). DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 18, 805–814. doi: 10.1105/tpc.105.038836
Xiao, X., Zhang, J., Li, T., Fu, X., Satheesh, V., Niu, Q., et al. (2019). A group of SUVH methyl-DNA binding proteins regulate expression of the DNA demethylase ROS1 in Arabidopsis. J. Integr. Plant Biol. 61, 110–119. doi: 10.1111/jipb.12768
Xiao, Y. J., Liu, H. J., Wu, L. J., Warburton, M., and Yan, J. B. (2017). Genome-wide association studies in maize: Praise and stargaze. Mol. Plant 10, 359–374. doi: 10.1016/j.molp.2016.12.008
Xie, Y., Zhang, Y., Han, J., Luo, J., Li, G., Huang, J., et al. (2018). The Intronic cis Element SE1 Recruits trans-Acting Repressor Complexes to Repress the Expression of ELONGATED UPPERMOST INTERNODE1 in Rice. Mol Plant. 11, 720–735. doi: 10.1016/j.molp.2018.03.001
Xie, Z., Allen, E., Fahlgren, N., Calamar, A., Givan, S. A., and Carrington, J. C. (2005). Expression of Arabidopsis MIRNA genes. Plant Physiol. 138, 2145–2154. doi: 10.1104/pp.105.062943
Xing, L., Zhu, M., Zhang, M., Li, W., Jiang, H., Zou, J., et al. (2017). High-throughput sequencing of small RNA transcriptomes in maize kernel identifies miRNAs involved in embryo and endosperm development. Genes 8:385. doi: 10.3390/genes8120385
Xu, J., Chen, G., Hermanson, P. J., Xu, Q., Sun, C., Chen, W., et al. (2019). Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 20:243. doi: 10.1186/s13059-019-1859-0
Xu, Y., Zhang, S., Lin, S., Guo, Y., Deng, W., Zhang, Y., et al. (2017). WERAM: A database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 45, D264–D270. doi: 10.1093/nar/gkw1011
Xu, Z., Zhong, S., Li, X., Li, W., Rothstein, S. J., Zhang, S., et al. (2011). Genome-wide identification of microRNAs in response to low nitrate availability in maize leaves and roots. PLoS One 6:e28009. doi: 10.1371/journal.pone.0028009
Yamamuro, C., Ihara, Y., Wu, X., Noguchi, T., Fujioka, S., Takatsuto, S., et al. (2000). Loss of function of a rice brassinosteroid insensitive1 homolog prevents internode elongation and bending of the lamina joint. Plant Cell 12, 1591–1606. doi: 10.1105/tpc.12.9.1591
Yang, C., Li, D., Mao, D., Liu, X., Ji, C., Li, X., et al. (2013). Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.). Plant Cell Environ. 36, 2207–2218. doi: 10.1111/pce.12130
Yang, C., Shen, W., Yang, L., Sun, Y., Li, X., Lai, M., et al. (2020). HY5-HDA9 module transcriptionally regulates plant autophagy in response to light-to-dark conversion and nitrogen starvation. Mol. Plant 13, 515–531. doi: 10.1016/j.molp.2020.02.011
Yang, H., Liu, X., Xin, M., Du, J., Hu, Z., Peng, H., et al. (2016). Genome-wide mapping of targets of Maize histone deacetylase HDA101 reveals its function and regulatory mechanism during seed development. Plant Cell. 28, 629–645. doi: 10.1105/tpc.15.00691
Yang, X. J., and Seto, E. (2007). HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318. doi: 10.1038/sj.onc.1210599
Yang, X., Kundariya, H., Xu, Y. Z., Sandhu, A., Yu, J., Hutton, S. F., et al. (2015). MutS HOMOLOG1-derived epigenetic breeding potential in tomato. Plant Physiol. 168, 222–232. doi: 10.1104/pp.15.00075
Yuan, L., Liu, X., Luo, M., Yang, S., and Wu, K. (2013). Involvement of histone modifications in plant abiotic stress responses. J. Integr. Plant Biol. 55, 892–901. doi: 10.1111/jipb.12060
Zabala, G., and Vodkin, L. O. (2014). Methylation affects transposition and splicing of a large CACTA transposon from a MYB transcription factor regulating anthocyanin synthase genes in soybean seed coats. PLoS One 9:e111959. doi: 10.1371/journalpone.0111959
Zemach, A., Kim, M. Y., Hsieh, P. H., Coleman-Derr, D., Eshed-Williams, L., Thao, K., et al. (2013). The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153, 193–205. doi: 10.1016/j.cell.2013.02.033
Zhang, C., Bai, M. Y., and Chong, K. (2014). Brassinosteroid-mediated regulation of agronomic traits in rice. Plant Cell Rep. 33, 683–696. doi: 10.1007/s00299-014-1578-7
Zhang, C., Ding, Z., Wu, K., Yang, L., Li, Y., Yang, Z., et al. (2016). Suppression of jasmonic acid-mediated defense by viral-inducible microRNA319 facilitates virus infection in rice. Mol. Plant 9, 1302–1314. doi: 10.1016/j.molp.2016.06.014
Zhang, H., Lang, Z., and Zhu, J. K. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506. doi: 10.1038/s41580-018-0016-z
Zhang, H., Lu, Y., Zhao, Y., and Zhou, D. X. (2016). OsSRT1 is involved in rice seed development through regulation of starch metabolism gene expression. Plant Sci. 248, 28–36. doi: 10.1016/j.plantsci.2016.04.004
Zhang, L., Cheng, Z., Qin, R., Qiu, Y., Wang, J., Cui, X., et al. (2012). Identification and characterization of an epi-allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 24, 4407–4421. doi: 10.1105/tpc.112.102269
Zhang, X., Sun, J., Cao, X., and Song, X. (2015). Epigenetic mutation of RAV6 affects leaf angle and seed size in rice. Plant Physiol. 169, 2118–2128. doi: 10.1104/pp.15.00836
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W., Chen, H., et al. (2006). Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 126, 1189–1201. doi: 10.1016/j.cell.2006.08.003
Zhang, X., Zou, Z., Zhang, J., Zhang, Y., Han, Q., Hu, T., et al. (2011). Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant. FEBS Lett. 585, 435–439. doi: 10.1016/j.febslet.2010.12.036
Zhang, Y. C., Yu, Y., Wang, C. Y., Li, Z. Y., Liu, Q., Xu, J., et al. (2013). Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat. Biotechnol. 31, 848–852. doi: 10.1038/nbt.2646
Zhang, Y. Y., Fischer, M., Colot, V., and Bossdorf, O. (2013). Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol. 197, 314–322. doi: 10.1111/nph.12010
Zhang, Y. Y., Latzel, V., Fischer, M., and Bossdorf, O. (2018). Understanding the evolutionary potential of epigenetic variation: A comparison of heritable phenotypic variation in epiRILs, RILs, and natural ecotypes of Arabidopsis thaliana. Heredity 121, 257–265. doi: 10.1038/s41437-018-0095-9
Zhao, M., Tai, H., Sun, S., Zhang, F., Xu, Y., and Li, W. X. (2012). Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS One 7:e29669. doi: 10.1371/journal.pone.0029669
Zhao, S. Q., Hu, J., Guo, L. B., Qian, Q., and Xue, H. W. (2010). Rice leaf inclination2, a VIN3-like protein, regulates leaf angle through modulating cell division of the collar. Cell Res. 20, 935–947. doi: 10.1038/cr.2010.109
Zhao, T., Zhan, Z., and Jiang, D. (2019). Histone modifications and their regulatory roles in plant development and environmental memory. J. Genet. Genom. 46, 467–476. doi: 10.1016/j.jgg.2019.09.005
Zhao, W., Li, Z., Fan, J., Hu, C., Yang, R., Qi, X., et al. (2015). Identification of jasmonic acid-associated microRNAs and characterization of the regulatory roles of the miR319/TCP4 module under root-knot nematode stress in tomato. J. Exp. Bot. 66, 4653–4667. doi: 10.1093/jxb/erv238
Zhao, X. Y., Hong, P., Wu, J. Y., Chen, X. B., Ye, X. G., Pan, Y. Y., et al. (2016). The tae-miR408-mediated control of TaTOC1 genes transcription is required for the regulation of heading time in wheat. Plant Physiol. 170, 1578–1594. doi: 10.1104/pp.15.01216
Zhao, Y., Xu, Z., Mo, Q., Zou, C., Li, W., Xu, Y., et al. (2013). Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann. Bot. 112, 633–642. doi: 10.1093/aob/mct133
Zheng, B., and Chen, X. (2011). Dynamics of histone H3 lysine 27 trimethylation in plant development. Curr. Opin. Plant Biol. 14, 123–129. doi: 10.1016/j.pbi.2011.01.001
Zheng, L., Zhang, X., Zhang, H., Gu, Y., Huang, X., Huang, H., et al. (2019). The miR164-dependent regulatory pathway in developing maize seed. Mol. Genet. Genom. 294, 501–517. doi: 10.1007/s00438-018-1524-4
Zheng, X., Chen, L., Xia, H., Wei, H., Lou, Q., Li, M., et al. (2017). Transgenerational epimutations induced by multigeneration drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 7:39843. doi: 10.1038/srep39843
Zhong, S., Fei, Z., Chen, Y. R., Zheng, Y., Huang, M., Vrebalov, J., et al. (2013). Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 31, 154–159. doi: 10.1038/nbt.2462
Zhou, M., Palanca, A. M. S., and Law, J. A. (2018). Locus-specific control of the de novo DNA methylation pathway in Arabidopsis by the CLASSY family. Nat. Genet. 50, 865–873. doi: 10.1038/s41588-018-0115-y
Zhou, S., Jiang, W., Long, F., Cheng, S., Yang, W., Zhao, Y., et al. (2017). Rice homeodomain protein WOX11 recruits a histone acetyltransferase complex to establish programs of cell proliferation of crown root meristem. Plant Cell 29, 1088–1104. doi: 10.1105/tpc.16.00908
Zhu, J. K. (2009). Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 43, 143–166. doi: 10.1146/annurev-genet-102108-134205
Zhu, Q. H., Upadhyaya, N. M., Gubler, F., and Helliwell, C. A. (2009). Over-expression of miR172 causes loss of spikelet determinacy and floral organ abnormalities in rice (Oryza sativa). BMC Plant Biol. 9:149. doi: 10.1186/1471-2229-9-149
Keywords: epigenetic, epialleles, agronomic traits, plant species, improvement
Citation: Gupta C and Salgotra RK (2022) Epigenetics and its role in effecting agronomical traits. Front. Plant Sci. 13:925688. doi: 10.3389/fpls.2022.925688
Received: 21 April 2022; Accepted: 11 July 2022;
Published: 15 August 2022.
Edited by:
Hong Luo, Clemson University, United StatesCopyright © 2022 Gupta and Salgotra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Romesh K. Salgotra, cmtzXzI5NTlAcmVkaWZmbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.