 Jie Li
Jie Li Xiaohua Yao
Xiaohua Yao Youhua Yao
Youhua Yao Likun An
Likun An Zongyun Feng
Zongyun Feng Kunlun Wu2,3,4*
Kunlun Wu2,3,4*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 23 June 2022
Sec. Plant Systematics and Evolution
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.924892
This article is part of the Research Topic Evolution of Crop Genomes and Epigenomes View all 13 articles
Drought stress is one of the main factors restricting hulless barley (Hordeum vulgare L. var. nudum Hook. f.) yield. Genome-wide association study was performed using 269 lines of hulless barley to identify single-nucleotide polymorphism (SNP) markers associated with drought-resistance traits. The plants were cultured under either normal or drought conditions, and various quantitative traits including shoot fresh weight, shoot dry weight, root fresh weight, root dry weight, leaf fresh weight, leaf saturated fresh weight, leaf dry weight, ratio of root and shoot fresh weight, ratio of root and shoot dry weight, shoot water loss rate, root water loss rate, leaf water content and leaf relative water content, and field phenotypes including main spike length, grain number per plant, grain weight per plant, thousand grain weight (TGW), main spike number, plant height, and effective spike number of plants were collected. After genotyping the plants, a total of 8,936,130 highly consistent population SNP markers were obtained with integrity > 0.5 and minor allele frequency > 0.05. Eight candidate genes potentially contributed to the hulless barley drought resistance were obtained at loci near significant SNPs. For example, EMB506, DCR, and APD2 genes for effective spike number of plants, ABCG11 gene for main spike number (MEN), CLPR2 gene for main spike length, YIP4B gene for root and shoot dry weight (RSWD), and GLYK and BTS genes for TGW. The SNPs and candidate genes identified in this study will be useful in hulless barley breeding under drought resistance.
Plants live in complex and changeable environmental conditions, often bring huge misfortune on plant growth (Zhu, 2016). As the global climate becomes drier and warmer, more than 15% of the world's population faces severe water shortages (Schewe et al., 2014; Gong et al., 2020). Drylands cover 40% of the global land surface and drought has caused losses in agriculture up to $30 billion over the past decade (Dai, 2013; Gupta et al., 2020). Drought has brought a great strain on the growth of plants, at the meantime, plants also have corresponding effective measures to prevent water loss, maintain cell water content, and help plants to survive the difficult drought period. Understanding drought resistance and water use efficiency of plants will provide guarantee for maintaining normal plant growth and improving agricultural yield under drought (Gupta et al., 2020; Yu et al., 2021).
Hulless barley (Hordeum vulgare L. var. nudum Hook. f.) is an important economic crop (He and Jia, 2008). As the only crop growing at high altitude, the planting area of hulless barley accounts for 43% of the grain crop area on the Qinghai Tibet Plateau (Dai et al., 2012; Zhong et al., 2016). Hulless barley has made great contribution as the main food, fuel, and livestock feed of the Tibetan people, and also is the raw material for beer, medicine, and health care products (Yang et al., 2013; Zhu et al., 2015; Liu et al., 2018). Hulless barley is rich in β-glucan, phenolic acid, and anthocyanins, which has high nutritional and medicinal value and is of great significance to human health (Bonoli et al., 2004; Siebenhandl et al., 2007; Kohyama et al., 2008; Zhao et al., 2015). The climate inside the Qinghai–Tibet Plateau is gradually drying out, and some scientists predict that only plants that can tolerate drought conditions will be able to settle on the plateau's platforms (Meng et al., 2017). Therefore, it is very important to study the drought tolerance of hulless barley.
To predict the important agronomic traits such as drought tolerance, it is necessary to understand the specific loci based on phenotype and the genetic structure of the traits. Genome-wide association study (GWAS) is just such a powerful tool for connecting genotypes–phenotypes (Korte and Farlow, 2013). Genome-wide association study refers to the association analysis of traits through the sequence and the SNP marker information on the whole genome so as to detect the loci significantly associated with the target trait (Li, 2013; Tam et al., 2019). Genome-wide association study provides higher resolution and finer scale association, and has been widely used in the identification of markers associated with desirable traits in crops (Nordborg and Weigel, 2008; Xu et al., 2017).
This study based on the identification results of hulless barley drought tolerance traits in 269 lines, SNP markers were developed by simplified genome sequencing (SLAF) to genotype natural populations. Using linear mixed model (LMM) and EmMax, the association between the quantitative traits of drought tolerance and genotype was analyzed, and the SNP loci and chromosome segments significantly associated with the target traits were screened.
The 269 hulless barley lines with different drought resistance assessment were used as the GWAS panel in this study (Supplementary Table 1). Phenotypic observation was performed on each line, both in the laboratory and in the field. The laboratory experiment was conducted in two growth condition with three biological replicates. The normal culture group was used as control, and the treatment group was applied with PEG-6000 to simulate drought stress. The associated phenotypes including shoot fresh weight SFW (g),shoot dry weight SDW (g), root fresh weight RFW (g), root dry weight RDW (g), leaf fresh weight LFW (g), leaf saturated fresh weight SFW (g), leaf dry weight LDW (g), ratio of root and shoot fresh weight RSFW (%), ratio of root and shoot dry weight RSWD (%), shoot water loss rate SWLR (%), root water loss rate RWLR (%), leaf water content WC (%), and leaf relative water content RWC (%) were measured. Field planting data were collected in 2019–2020 from three different growing environments at two sites, including drought treatment and natural irrigation at two different habitats. The associated phenotypes of different habitats consisted of main spike length MSL (cm), grain number per plant GNPP, grain weight per plant GWPP (g), thousand grain weight TGW(g), main spike number MEN(g), plant height (cm) and effective spike number of plants ESNP.
In 2021, 269 pieces of hulless barley lines were planted in germinating boxes and cultured in greenhouse to two leaves stage. Whole-genome DNA of each germplasm resource leaves was extracted by CTAB method (Allen et al., 2006). The DNA quality and concentration were detected by 0.1% agarose gel electrophoresis, and whole-genome SNP genotyping was produced by Biomarker technologies company. The SLAF tags were developed by enzyme digestion (RsaI) of the genomic DNA, followed by adaptor ligation, amplification and purification. Then, the SLAF library were sequenced by Illumina Novaseq 6000. The sequencing reads were mapped to the reference genome by BWA software (Li and Durbin, 2009). GATK (McKenna et al., 2010) and samtools (Li et al., 2009) were used to identify SNPs. The intersection of SNP markers obtained by the two methods was used as the final reliable SNP marker dataset, and a total of 5,949,446 SNPs were obtained. The genotypic data obtained were screened as integrity > 0.8 and minor allele frequency (MAF) > 0.05.
Based on the SNPs obtained from the above genotypes, 269 phylogenetic trees of hulless barley was constructed by neighbor-joining (NJ) method (1,000 replicates) with Kimura 2-parameter (K2-P) model using MEGA X software (Kumar et al., 2018). The phylogenetic tree was colored based on the analysis results of STRUCTURE.
Based on the developed high-density SNP molecular markers, GEMMA, FaST-LMM, and EMMAX were used for association analysis. Correlation analysis between phenotypic value of drought-tolerant-resistant traits and genotypes was carried out to obtain the p-value of each SNP. Screened with p < 5 × 10−6, the genetic variation loci most likely to affect the trait was selected. The quantile–quantile (Q–Q) scatter plot and Manhattan plot were made by the qqman package in R software.
To screen the drought-tolerant-resistant genes near the significant associative loci, the genetic information of specific association regions was queried from barley genome in plant whole-genome information database (http://plants.ensembl.org/index.html). All genes with coding regions in the 100–500-kb window were used for subsequent analysis.
The molecular markers of 269 hulless barley lines were developed by Specific-Locus Amplified Fragment Sequencing (LAF-SEQ) to obtain molecular markers in the whole genome. An average of 311,695 SLAF tags were developed per sample for a total of 862,999, including 480,790 polymorphic SLAF tags and 5,532,468 SNP markers. The average sequencing depth of SLAF tags was 10.43 ×, and 1,067.96 Mb reads data were generated. These markers were evenly distributed on the chromosomes of hulless barley (Table 1, Figure 1). The average Q30 of sequences was 94.78%, and the average GC content was 44.23%. A total of 8,936,130 SNP markers with high consistency were obtained from 269 hulless barley lines filtered by integrity > 0.5 and MAF > 0.05. Chromosome 3 had the largest number of SNP markers (1,532,190), with an average label distance of 456 bp. On the contrary, chromosome 1 had the lowest number of SNP markers (914,610), with an average label distance of 610 bp.
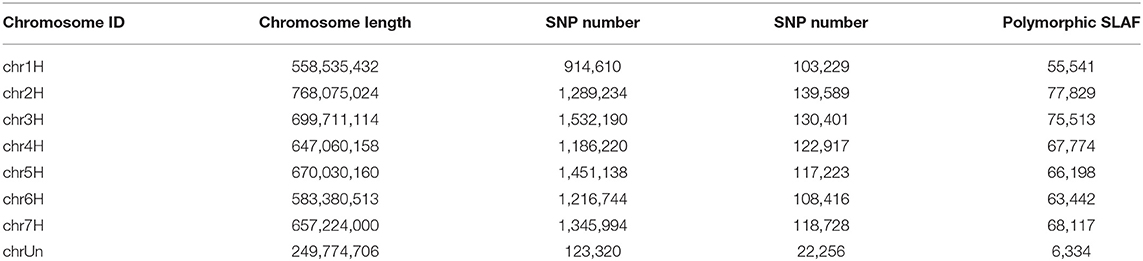
Table 1. Distribution of SLAF markers on chromosomes.
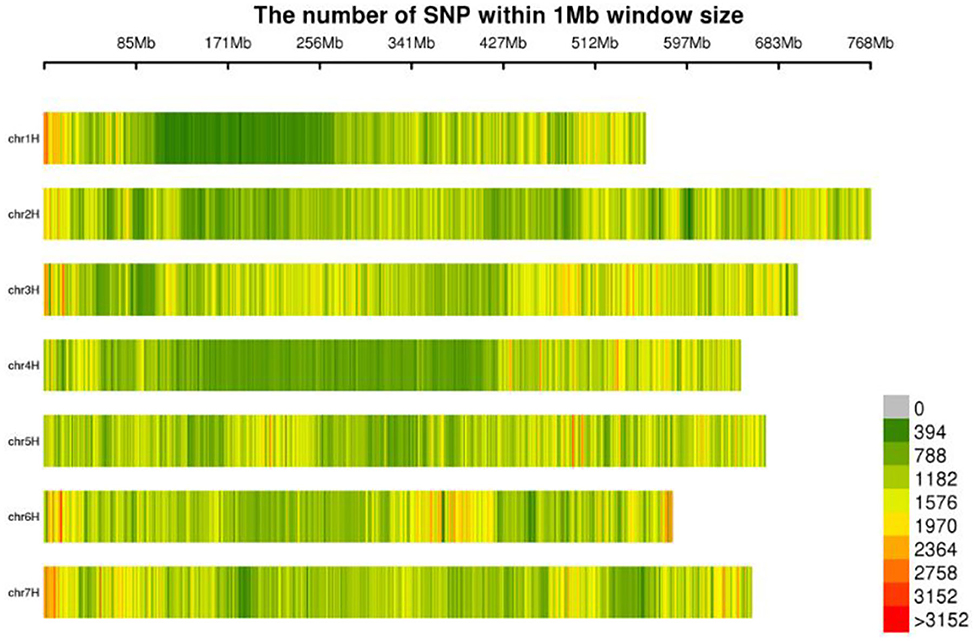
Figure 1. Distribution of SNPs on chromosomes.
The 269 lines were divided into 6 groups according to the geographic location information of the samples, and the group information was used for linkage disequilibrium (LD) and evolutionary tree analysis. The phylogenetic tree was colored by the clustering result of STRUCTURE, basically, each cluster was gathered into one block in the phylogenetic tree, especially Q5 and Q6 (Figure 2).
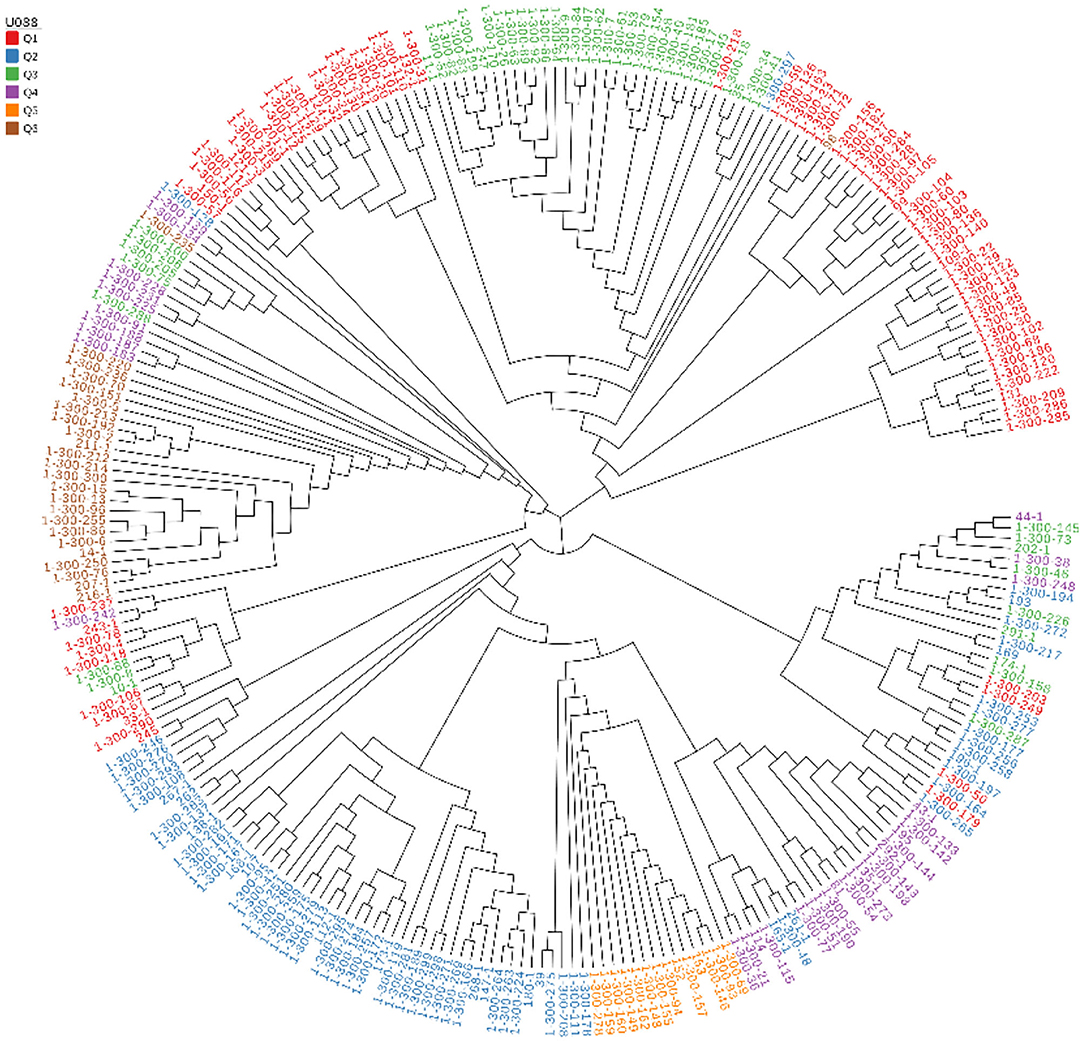
Figure 2. Genetic relatedness among the 269 hulless barley lines estimated by neighbor-joining method and represented as a polar tree diagram. The estimated genetic relatedness is based on 5,949,446 SNPs identified by genotyping-by-sequencing and filtered for MAF of 0.05.
Using the SNP information mentioned above, the principal component analysis (PCA) was conducted. The top three principal components could explain 34.23% of the genomic variations, and principal component 1 could explain 18.3%. Consistent with the phylogenetic tree, Q5 and Q6 were separate from other populations (Figure 3).
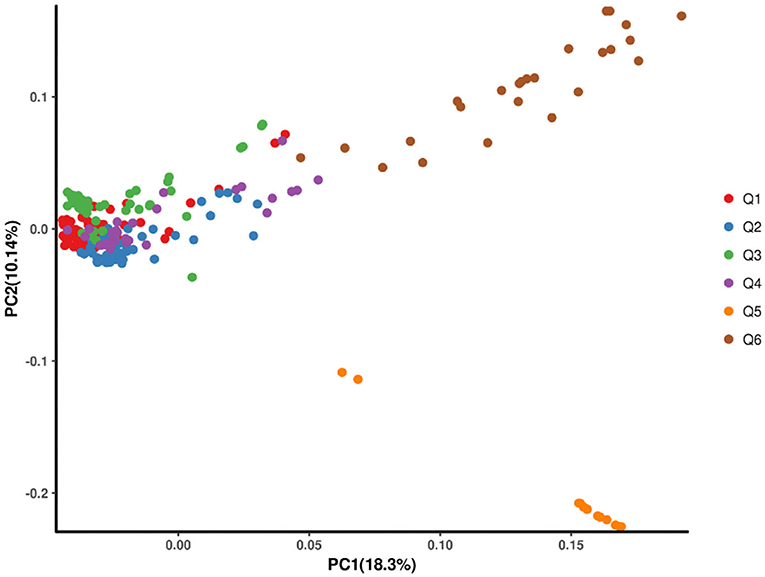
Figure 3. The scatter plots of the first two principal components (PCs) showing the distribution of the 269 hulless barley lines in PC1 vs. PC2.
Plink2 software (Chen et al., 2019) was used to calculate the linkage disequilibrium (LD) between two SNP pairs within a certain distance (1,000 kb) on the same chromosome, and the linkage disequilibrium intensity was represented by r2. The closer r2 is to 1, the stronger the linkage disequilibrium intensity. The distance between SNPs and r2 was fitted, and the curve of r2 changes with distance was presented. Generally speaking, the closer the distance between SNPs is, the larger r2 is and vice versa. The LD decay (LDD) distance was used as the distance traveled when the maximum r2 value dropped to half. The longer LDD, the lower the probability of recombination within the same physical distance. It should be noted that some regions of Q5 and Q6 had very strong linkage, but the length of the strong linkage was short, indicating that these two groups were subjected to some artificial selection pressure and some loci were selected, leading to linkage in some regions (Figure 4).
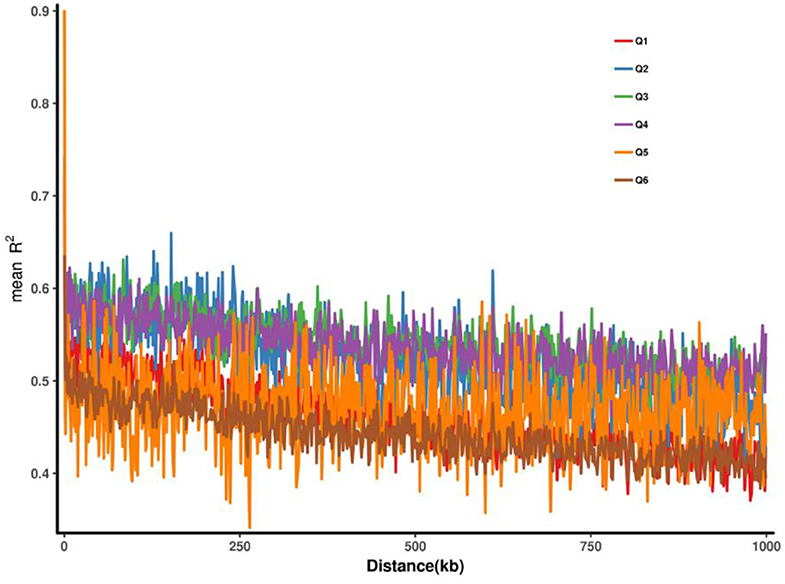
Figure 4. Linkage disequilibrium decay based on six groups.
This analysis was based on SNP data from mutation detection, filtered by secondary allele frequency (MAF > 0.05) and locus integrity (integrity > 0.8) to obtain highly consistent SNP loci for GWAS analysis. Genome-wide association analysis was performed using LMM, EMMAX, and FaST-LMM models, respectively. The following table showed the number of significant SNP markers obtained for each phenotype corresponding to each mode and the number of common SNP markers in each model (Table 2).
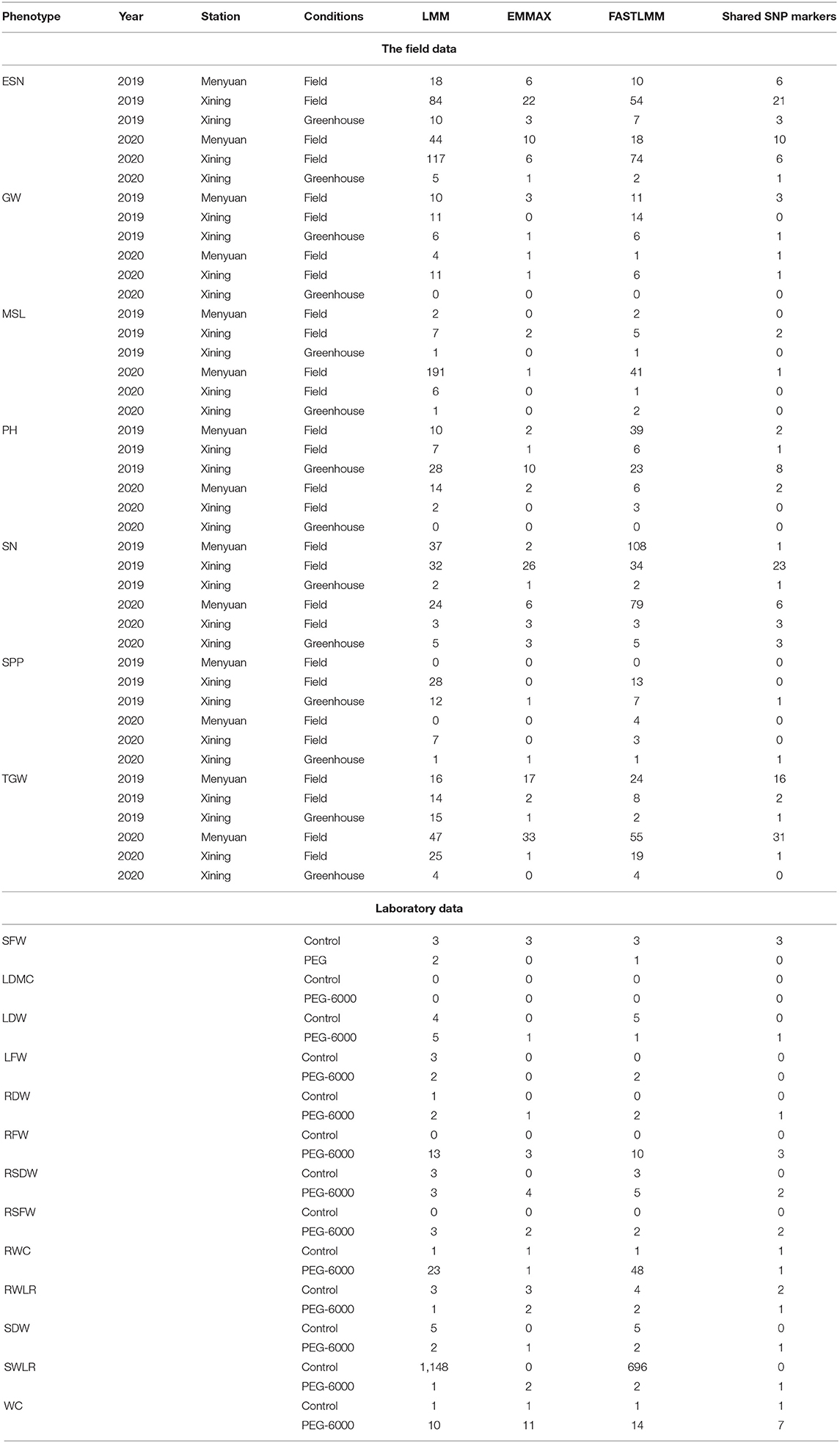
Table 2. SNP markers for each phenotype based on different GWAS analysis models.
In the following part, we made a detailed explanation of some phenotypes which have shared SNP markers in the three relational models. The Manhattan plots showed significant correlation between SNP markers on multiple chromosomes and traits, while the Q–Q plots showed the relationship between observed p-values and expected p-values for each SNP marker (Figure 5, Supplementary Table 2).
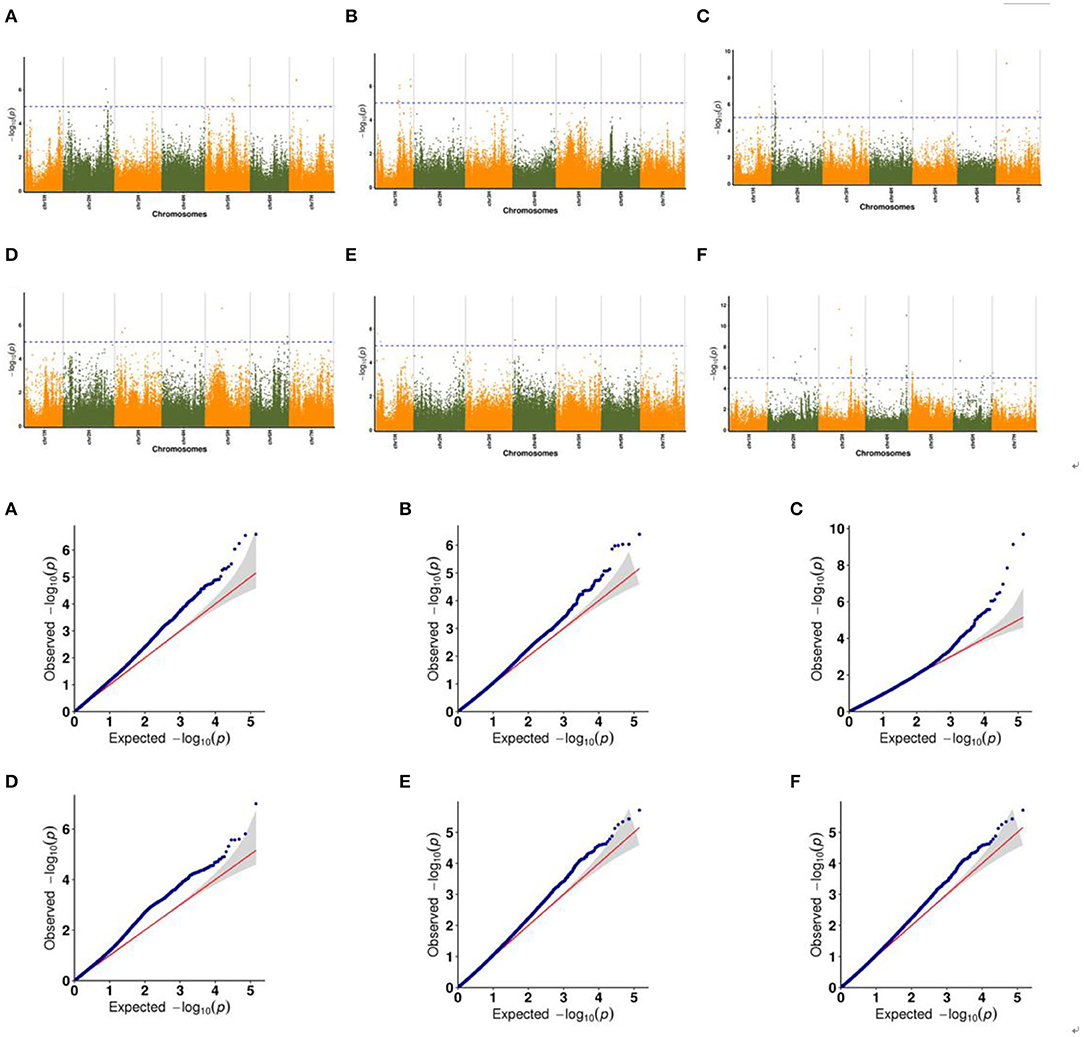
Figure 5. Manhattan maps and Q–Q plots representing the SNP markers associated with drought resistant in multiple trials of GWAS associated with spike development. (A,B) ESNP, (C) MEN, (D) MSL, (E) RSWD, and (F) TGW.
Figure 5 showed the association between SNP markers and effective spike number of plants (ESNP) phenotype with Manhattan map and corresponding Q–Q plot. For plants cultured in greenhouse in Xining city during 2019, two SNP markers chr7H_102929775 and chr7H_102929728 with known functions were obtained. Both of them locate on the gene Ankyrin repeat domain-containing protein (EMB506). Gene EMB506 is expressed at flowering and heading stage and closely associated with the character (spike number) (Despres et al., 2001), so it is likely to be the effector gene. There were another two SNP markers detected in plants cultured in filed in Menyuan city during 2019. One was chr1H_12690373, located on the Protein Dicer (DCR) gene, which is required for cutin polyester formation (Panikashvili et al., 2009). The other was chr1H_512690373, located on APD2 gene involved in male gametophyte development (Luo et al., 2012). For main spike number (MEN) phenotype, 23 SNP markers were detected on chromosomes 1H, 2H, and 7H in plants which were cultured in filed in Xining city during 2019. On chromosome 2H, 18 SNP markers were found, including one aldehyde reductase gene Neuroplastin (SDR1), one gene interrelated with chloroplast development and plant growth named Probable GTP-binding protein (OBGC1), and three bacterial infection related genes Peroxidase 2 (PRX112), Probable acyl-CoA dehydrogenase (IBR3) and Ethylene-responsive transcription factor (RAP2-3). The ABC transporter G family member 11 (ABCG11) gene located on chromosome 2H was highly expressed in flowers and young seeds and was closely related to spike number (Panikashvili et al., 2010) so that ABCG11 was likely to be the effector gene of the trait. Our results showed that two SNP markers (chr3H_152206655 and chr5H_250095923) connected with main spike length (MSL) phenotype. Thereinto, ATP-dependent Clp protease proteolytic subunit-related protein 1 (CLPR) is considered to regulate chloroplast and plant development. Deletion of CLPR alleles resulted in embryonic development delay and leaf albinism (Kim et al., 2009). Root and shoot dry weight (RSWD) phenotype was represented by chr1H_64014764 on Ypt Interacting Protein 4b (YIP4B) gene. The YIP4B regulates cell wall composition and participate in root and hypocotyl elongation (Gendre et al., 2013). As for thousand grain weight (TGW) phenotype, the first SNP peak was found at chr3H_482958549, and mapped to photosynthesis related gene D-glycerate 3-kinase, chloroplastic (GLYK). The second SNP peak was found at chr3H_489630701, and mapped to iron accumulation associated gene Geranylgeranyl pyrophosphate synthase (BTS).
Hulless barley is rich in nutrients and is the main food source for Tibetan people (Bonoli et al., 2004; Siebenhandl et al., 2007; Kohyama et al., 2008; Zhao et al., 2015). It grows on the Qinghai–Tibet Plateau and is the only crop that can grow at high altitude of 4,200–4,500 m (Dai et al., 2012; Zhong et al., 2016). However, at present, with the aggravation of drought on the Qinghai–Tibet Plateau (Meng et al., 2017), the selection of drought-tolerant hulless barley strains has become an urgent affair. However, the genetic resource that could be used to assist hulless barley molecular breeding was scarce. In this study, GWAS was used to map SNP markers related to drought tolerance in hulless barley. The SNP markers identified in this study will be used to analyze drought tolerance of hulless barley and facilitate the selection of drought-tolerant strains.
In this study, 269 lines of hulless barley were selected for drought treatment under laboratory and field conditions. Significant phenotypic variation in effective spike number of plants (ESNP), main spike number (MEN), main spike length (MSL), root and shoot dry weight (RSWD), and thousand grain weight (TGW) have been identified under drought conditions. These results indicated that the selected lines could play an important role in exploring the drought-tolerance genes of hulless barley. Hulless barley has great plasticity in adapting to drought stress, which will provide reference for the breeding process of superior hulless barley strains and improve the drought tolerance of hulless barley.
Using the hulless barley GWAS panel, 29 SNPs loci and five candidate genes connected with all spike traits (including ESNP MEN and MSL) were identified. As for ENSP, markers distributed on chromosomes 7H and chromosomes 1H were correlated, a total of 4 SNPs loci on three genes (EMB506, DCR, and APD2) were identified. ABCG11 and CLPR2 are two effector genes for MEN and MSL traits, respectively. Among the genes related to spike traits, DCR and ABCG11 plays a key role in cuticle formation (Panikashvili et al., 2010; Rani et al., 2010). As the contact zone between the plant and the environment, cuticle has been well-characterized for its multiple roles in the regulation of gas exchange, epidermal permeability, and non-stomatal water loss (Sieber et al., 2000). So, it is not surprising that dcr mutants show increased water loss and increased sensitivity to drought conditions (Panikashvili et al., 2009). Besides, EMB506 and CLPR2 genes are associated with chloroplast and plant growth (Despres et al., 2001; Rudella et al., 2006). The lack of CLPR2 gene causes leaf albinism and undoubtedly affects photosynthetic efficiency and crop yield (Kim et al., 2009). For RSWD at the dehydrated growth condition, YIP4B gene represented by chr1H_64014764 SNP was identified. In Arabidopsis thaliana, YIP4B affects root and hypocotyl growth through elongation rather than cell division (Gendre et al., 2013). Two genes located on Chr3H were identified for TGW trait. Among them, GLYK catalyzes the termination of the C2 cycle in photosynthesis, which is an indispensable auxiliary metabolic pathway for the C3 cycle of photosynthesis. The presence of this gene ensures the normal growth of terrestrial plants in an oxygen-containing atmosphere and avoids photoinhibition (Boldt et al., 2005). As iron sensors, BTS gene plays a vital role in modulating iron homeostasis (Zhang et al., 2015).
The mutation of these loci under drought conditions and the resulting phenotypic changes undisputedly gives us huge inspiration. Further development of these SNPs and genes will provide new insights into improving crop phenotypic traits and make plants develop in an environment-adapted direction. For instance, drought-tolerant, higher-yielding plants could be created by genetically modifying these loci. These findings will simplify the tedious process of hybridization and culture, and turn to use molecular methods for seedling breeding, which will reduce our experimental time greatly. What is more exciting is that it also provides direction for drought-tolerance selection of other economic crops besides hulless barley.
In summary, we used 5,532,468 SNP markers from 269 hulless barley lines to analyze the association between phenotypic values and genotypes of drought-tolerance traits in this study. The SNP markers association with spike traits (chr7H_102929775, chr7H_102929728, chr1H_512690373, and chr1H_512690373, chr1H_349621827, chr1H_349622062, chr2H_39562071, chr2H_47246481, chr2H_47623192, chr2H_47623303, chr2H_48148956, chr2H_48534579, chr2H_48534763, chr2H_48796138, chr2H_496935399, chr2H_496935424, chr2H_54436239, chr2H_54436346, chr2H_55411310, chr2H_55768675, chr2H_57397060, chr2H_57539586, chr2H_62893696, chr2H_64170225, chr7H_149587366, chr7H_158720411, chr7H_621337028, chr3H_152206655, and chr5H_250095923), RSWD trait (chr1H_64014764), and TGW trait (chr3H_482958549, chr3H_489630701) were identified. Under drought conditions, the mutation of these SNPs loci possibly lead to phenotypic changes and improve the adaptation of hulless barley to drought environment. In conclusion, the SNPs identified in this study can be used in drought-tolerance gene analysis, and can provide valuable information for further improvement of crop yield, quality, and adaptability.
All the raw genome data can be found in the National Genomics Data Center (NGDC) BioProject database with the accession number PRJCA009869.
ZF and KW contributed to the conceptualization and methodology. JL contributed to the experimentation, data analysis, and first draft preparation. XY, YY, JL, and LA contributed to the experimentation and data analysis. JL, ZF, and KW contributed to the supervision and manuscript editing. All authors have read and agreed to the published version of the manuscript.
This work was supported by funds from the National Key R & D Program of China (2018YFD1000705 and 2018YFD1000700), National Natural science Foundation of China (32060480), the China Agriculture Research System (CARS-05), the International Science and Technology Cooperation in Sichuan Province of China (Grant No. 2021YFH0113), Qinghai Provincial Academy of Agriculture and Forestry Innovation Fund (2018-NKY-12), and the Double Support Program for Discipline Construction of Sichuan Agricultural University in China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.924892/full#supplementary-material
Allen, G. C., Flores-Vergara, M., Krasynanski, S., Kumar, S., and Thompson, W. (2006). A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325. doi: 10.1038/nprot.2006.384
Boldt, R., Edner, C., Kolukisaoglu, U., Hagemann, M., Weckwerth, W., Wienkoop, S., et al. (2005). D-GLYCERATE 3-KINASE, the last unknown enzyme in the photorespiratory cycle in Arabidopsis, belongs to a novel kinase family. Plant Cell. 17, 2413–2420. doi: 10.1105/tpc.105.033993
Bonoli, M., Verardo, V., Marconi, E., and Caboni, M. F. (2004). Antioxidant phenols in barley (Hordeum vulgare L.) flour: comparative spectrophotometric study among extraction methods of free and bound phenolic compounds. J. Agric. Food Chem. 52, 5195–5200. doi: 10.1021/jf040075c
Chen, Z. L., Meng, J. M., Cao, Y., Yin, J. L., Fang, R. Q., Fan, S. B., et al. (2019). A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 10:3404. doi: 10.1038/s41467-019-11337-z
Dai, A. (2013). Increasing drought under global warming in observations and models. Nat. Clim. Chang. 3, 52–58. doi: 10.1038/nclimate1633
Dai, F., Nevo, E., Wu, D., Comadran, J., Zhou, M., Qiu, L., et al. (2012). Tibet is one of the centers of domestication of cultivated barley. Proc. Natl. Acad. Sci. U.S.A. 109, 16969–16973. doi: 10.1073/pnas.1215265109
Despres, B., Delseny, M., and Devic, M. (2001). Partial complementation of embryo defective mutations: a general strategy to elucidate gene function. Plant J. 27, 149–159. doi: 10.1046/j.1365-313x.2001.01078.x
Gendre, D., McFarlane, H. E., Johnson, E., Mouille, G., Sjödin, A., Oh, J., et al. (2013). Trans-Golgi network localized ECHIDNA/Ypt interacting protein complex is required for the secretion of cell wall polysaccharides in Arabidopsis. Plant Cell. 25, 2633–2646. doi: 10.1105/tpc.113.112482
Gong, Z., Xiong, L., Shi, H., Yang, S., Herrera-Estrella, L., Xu, G., et al. (2020). Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 63, 635–674. doi: 10.1007/s11427-020-1683-x
Gupta, A., Rico-Medina, A., and Caño-Delgado, A. I. (2020). The physiology of plant responses to drought. Science 368, 266–269. doi: 10.1126/science.aaz7614
He, T., and Jia, J. F. (2008). High frequency plant regeneration from mature embryo explants of highland barley (Hordeum vulgare L. var. nudum Hk. f.) under endosperm-supported culture. Plant Cell Tissue Organ Cult. 95, 251–254. doi: 10.1007/s11240-008-9437-2
Kim, J., Rudella, A., Ramirez Rodriguez, V., Zybailov, B., Olinares, P. D., and van Wijk, K. J. (2009). Subunits of the plastid ClpPR protease complex have differential contributions to embryogenesis, plastid biogenesis, and plant development in Arabidopsis. Plant Cell 21, 1669–1692. doi: 10.1105/tpc.108.063784
Kohyama, N., Ono, H., and Yanagisawa, T. (2008). Changes in anthocyanins in the grains of purple waxy hull-less barley during seed maturation and after harvest. J. Agric. Food Chem. 56, 5770–5774. doi: 10.1021/jf800626b
Korte, A., and Farlow, A. (2013). The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9:29. doi: 10.1186/1746-4811-9-29
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, W. (2013). “Genome-wide association study,” in Encyclopedia of Systems Biology, eds W. Dubitzky, O. Wolkenhauer, K.-H. Cho, and H. Yokota (New York, NY: Springer New York), 834.
Liu, H., Chen, X., Zhang, D., Wang, J., Wang, S., and Sun, B. (2018). Effects of highland barley bran extract rich in phenolic acids on the formation of N(ε)-carboxymethyllysine in a biscuit model. J. Agric. Food Chem. 66, 1916–1922. doi: 10.1021/acs.jafc.7b04957
Luo, G., Gu, H., Liu, J., and Qu, L. J. (2012). Four closely-related RING-type E3 ligases, APD1-4, are involved in pollen mitosis II regulation in Arabidopsis. J. Integr. Plant Biol. 54, 814–827. doi: 10.1111/j.1744-7909.2012.01152.x
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meng, L. H., Yang, J., Guo, W., Tian, B., Chen, G. J., Yang, Y. P., et al. (2017). Differentiation in drought tolerance mirrors the geographic distributions of alpine plants on the Qinghai-Tibet Plateau and adjacent highlands. Sci. Rep. 7:42466. doi: 10.1038/srep42466
Nordborg, M., and Weigel, D. (2008). Next-generation genetics in plants. Nature 456, 720–723. doi: 10.1038/nature07629
Panikashvili, D., Shi, J. X., Bocobza, S., Franke, R. B., Schreiber, L., and Aharoni, A. (2010). The Arabidopsis DSO/ABCG11 transporter affects cutin metabolism in reproductive organs and suberin in roots. Mol. Plant. 3, 563–575. doi: 10.1093/mp/ssp103
Panikashvili, D., Shi, J. X., Schreiber, L., and Aharoni, A. (2009). The Arabidopsis DCR encoding a soluble BAHD acyltransferase is required for cutin polyester formation and seed hydration properties. Plant Physiol. 151, 1773–1789. doi: 10.1104/pp.109.143388
Rani, S. H., Krishna, T. H., Saha, S., Negi, A. S., and Rajasekharan, R. (2010). Defective in cuticular ridges (DCR) of Arabidopsis thaliana, a gene associated with surface cutin formation, encodes a soluble diacylglycerol acyltransferase. J. Biol. Chem. 285, 38337–38347. doi: 10.1074/jbc.M110.133116
Rudella, A., Friso, G., Alonso, J. M., Ecker, J. R., and van Wijk, K. J. (2006). Downregulation of ClpR2 leads to reduced accumulation of the ClpPRS protease complex and defects in chloroplast biogenesis in Arabidopsis. Plant Cell. 18, 1704–1721. doi: 10.1105/tpc.106.042861
Schewe, J., Heinke, J., Gerten, D., Haddeland, I., Arnell, N. W., Clark, D. B., et al. (2014). Multimodel assessment of water scarcity under climate change. Proc. Natl. Acad. Sci. U.S.A. 111, 3245–3250. doi: 10.1073/pnas.1222460110
Siebenhandl, S., Grausgruber, H., Pellegrini, N., Del Rio, D., Fogliano, V., Pernice, R., et al. (2007). Phytochemical profile of main antioxidants in different fractions of purple and blue wheat, black barley. J. Agric. Food Chem. 55, 8541–8547. doi: 10.1021/jf072021j
Sieber, P., Schorderet, M., Ryser, U., Buchala, A., Kolattukudy, P., Métraux, J. P., et al. (2000). Transgenic Arabidopsis plants expressing a fungal cutinase show alterations in the structure and properties of the cuticle and postgenital organ fusions. Plant Cell. 12, 721–738. doi: 10.1105/tpc.12.5.721
Tam, V., Patel, N., Turcotte, M., Bossé, Y., Paré, G., and Meyre, D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484. doi: 10.1038/s41576-019-0127-1
Xu, Y., Li, P., Yang, Z., and Xu, C. (2017). Genetic mapping of quantitative trait loci in crops. Crop J. 5, 175–184. doi: 10.1016/j.cj.2016.06.003
Yang, L., Christensen, D., McKinnon, J., Beattie, A., and Yu, P. (2013). Effect of altered carbohydrate traits in hulless barley (Hordeum vulgare L.) on nutrient profiles and availability and nitrogen to energy synchronization. J. Cereal Sci. 58, 182–190. doi: 10.1016/j.jcs.2013.05.005
Yu, L., Zhao, X., Gao, X., Jia, R., and Siddique, K. (2021). Effect of natural factors and management practices on agricultural water use efficiency under drought: a meta-analysis of global drylands. J. Hydrol. 594:125977. doi: 10.1016/j.jhydrol.2021.125977
Zhang, J., Liu, B., Li, M., Feng, D., Jin, H., Wang, P., et al. (2015). The bHLH transcription factor bHLH104 interacts with IAA-LEUCINE RESISTANT3 and modulates iron homeostasis in Arabidopsis. Plant Cell 27, 787–805. doi: 10.1105/tpc.114.132704
Zhao, C., Wang, X., Wang, X., Wu, K., Li, P., Chang, N., et al. (2015). Glucose-6-phosphate dehydrogenase and alternative oxidase are involved in the cross tolerance of highland barley to salt stress and UV-B radiation. J. Plant Physiol. 181, 83–95. doi: 10.1016/j.jplph.2015.03.016
Zhong, Z.-M., Shen, Z.-X., and Fu, G. (2016). Response of soil respiration to experimental warming in a highland barley of the Tibet. Springerplus 5:137. doi: 10.1186/s40064-016-1761-0
Zhu, F., Du, B., and Xu, B. (2015). Superfine grinding improves functional properties and antioxidant capacities of bran dietary fibre from Qingke (hull-less barley) grown in Qinghai-Tibet Plateau, China. J. Cereal Sci. 65, 43–47. doi: 10.1016/j.jcs.2015.06.006
Keywords: hulless barley, GWAS, drought resistance, high throughput sequencing, quantitative traits, SNP
Citation: Li J, Yao X, Yao Y, An L, Feng Z and Wu K (2022) Genome-Wide Association Mapping of Hulless Barely Phenotypes in Drought Environment. Front. Plant Sci. 13:924892. doi: 10.3389/fpls.2022.924892
Received: 20 April 2022; Accepted: 18 May 2022;
Published: 23 June 2022.
Edited by:
Zhe Liang, Biotechnology Research Institute (CAAS), ChinaCopyright © 2022 Li, Yao, Yao, An, Feng and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zongyun Feng, enlmZW5nNDlAMTI2LmNvbQ==; Kunlun Wu, d2tscWFhZkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.