 Fatemeh Orooji
Fatemeh Orooji Ghader Mirzaghaderi
Ghader Mirzaghaderi Yi-Tzu Kuo
Yi-Tzu Kuo Jörg Fuchs
Jörg Fuchs
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 09 June 2022
Sec. Plant Systematics and Evolution
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.917310
Nigella is a small genus belonging to the Ranunculaceae family which is presumably originated and distributed in Aegean and the adjacent Western-Irano-Turanian region. Comparative repeat analysis of N. sativa, N. damascena and N. bucharica was performed using low-pass Illumina genomic reads followed by karyotyping and FISH mapping of seven Nigella species using the in silico identified repeats and ribosomal DNA (rDNA) probes. High- and moderate-copy repeat sequences occupy 57.52, 59.01, and 64.73% of N. sativa, N. damascena and N. bucharica genomes, respectively. Roughly, half of the genomes are retrotransposons (class I transposons), while DNA transposons (class II transposons) contributed to only about 2% of the genomes. The analyzed Nigella species possess large genomes of about 7.4 to 12.4 Gbp/1C. Only two satellite repeats in N. sativa, one in N. damascena and four in N. bucharica were identified, which were mostly (peri)centromeric and represented about 1% of each genome. A high variation in number and position of 45S rDNA loci were found among Nigella species. Interestingly, in N. hispanica, each chromosome revealed at least one 45S rDNA site and one of them occurs in hemizygous condition. Based on the chromosome numbers, genome size and (peri)centromeric satellites, three karyotype groups were observed: Two with 2n = 2x = 12 and a karyotype formula of 10m + 2t (including N. sativa, N. arvensis, N. hispanica as the first group and N. damascena and N. orientalis as the second group) and a more distant group with 2n = 2x = 14 and a karyotype formula of 8m + 2st + 4t (including N. integrifolia and N. bucharica). These karyotype groups agreed with the phylogenetic analysis using ITS and rbcL sequences. We conclude that variation in (peri)centromeric sequences, number and localization of rDNA sites as well as chromosome number (dysploidy) are involved in the diversification of the genus Nigella.
Nigella (fennel flower) is a small genus in the tribe Nigelleae (with 18 species) of the Ranunculaceae family (Zohary, 1983; Dönmez et al., 2021) (Supplementary Table 1), native to Southern Europe, North Africa, South Asia, Southwest Asia and Middle East (Tutin, 1964; Zohary, 1983; Raab-Straube et al., 2014) (Supplementary Figure 1). Fourteen species belong to Nigella, among which N. sativa L. (black cumin) is the most popular medical plant and additionally its seeds are used as spices. N. damascena L. and N. arvensis are annual ornamental and medicinal plants (Ghosh and Datta, 2006; Malhotra, 2012; Shaker et al., 2017). Komaroffia bucharica and K. integrifolia belong to the Komaroffia tribe (the sister tribe of Nigelleae), that Zohary accepted as synonyms for Nigella bucharica and N. integrifolia (Zohary, 1983; Heiss et al., 2011; Dönmez et al., 2021). N. bucharica and N. integrifolia are of great importance for beekeeping, as they provide bees with nectar and pollen in south Uzbekistan (Atamuratova et al., 2021). The diploid species, N. sativa, N. damascena, N. arvensis, N. hispanica and N. orientalis (2n = 2x = 12), have five metacentric and one telocentric chromosome pairs, but N. bucharica and N. integrifolia (2n = 2x = 14) have four metacentric, two submetacentric and one subtelocentric chromosome pairs (Gilot-Delhalle et al., 1976). The 1C-values of N. sativa and N. damascena were determined to be 10.39 Gbp (Bennett and Smith, 1976) and 10.29 Gbp (Evans et al., 1972; Kuznetsova et al., 2017; Leitch et al., 2019), respectively. There is little information about the genome composition and cytogenetic characteristics of Nigella species although such information is important to understand the phylogenetic relationship in this genus.
Repetitive DNAs are highly enriched in plant genomes, and repetitive fractions among plant genomes are highly variable, ranging for example from 13–14% in the small genome of Arabidopsis thaliana (157 Mbp/1C) (Bennett et al., 2003), to up to 92% in Allium cepa with a rather large genome (16 Gbp/1C) (Fu et al., 2019). Transposons and tandem repeat DNAs (including satellites and ribosomal DNAs) are major repetitive sequences in eukaryotic genomes (Wicker et al., 2007; Mehrotra and Goyal, 2014; Bao et al., 2015; Piégu et al., 2015; Maumus and Quesneville, 2016). Satellites are commonly used as molecular and cytogenetic markers in studies of the genetic diversity and chromosome evolution due to their species-, or even chromosome-specificity (Elder and Turner, 1995; Ugarkovic and Plohl, 2002; Garrido-Ramos, 2017; Samoluk et al., 2017; Belyayev et al., 2019). Although 45S (18S-5.8S-25S) and 5S rDNA have been widely used as cytological markers for chromosome identification and investigations of chromosomal rearrangements occurring between related species (Mukai et al., 1991; Zoldos et al., 1999; Frello and Heslop-Harrison, 2000; Tagashira and Kondo, 2001; Datson and Murray, 2003), the ITS sequences of 45S rDNA are rather variable between species. In addition to large-scale chromosomal rearrangements, such as inversions and translocations, the high variation in copy number and distribution of tandem repeats can lead to genome divergence and karyotype changes between closely related species (Appels et al., 1980; Mukai et al., 1991; Levin and Donald, 2002).
In this study, we analyzed and compared the repeat composition of N. sativa, N. damascena and N. bucharica using low-coverage genome sequences. Furthermore, we generated karyotypes of seven Nigella species using FISH mapping of major satellite repeats and rDNAs. Types and patterns of satellite repeats and number of chromosomes agreed with the phylogenetic relationships revealed by using ITS and rbcL sequences.
Seeds of seven Nigella species, N. sativa, N. damascena, N. arvensis, N. bucharica, N. hispanica, N. integrifolia and N. orientalis, were provided by the IPK Genebank in Germany (Table 1, Supplementary Table 1, and Supplementary Figure 2). All species were used for phylogenetic analysis and FISH karyotyping. N. sativa, N. damascena and N. bucharica were further used in a comparative analysis of their genome repetitive compositions.
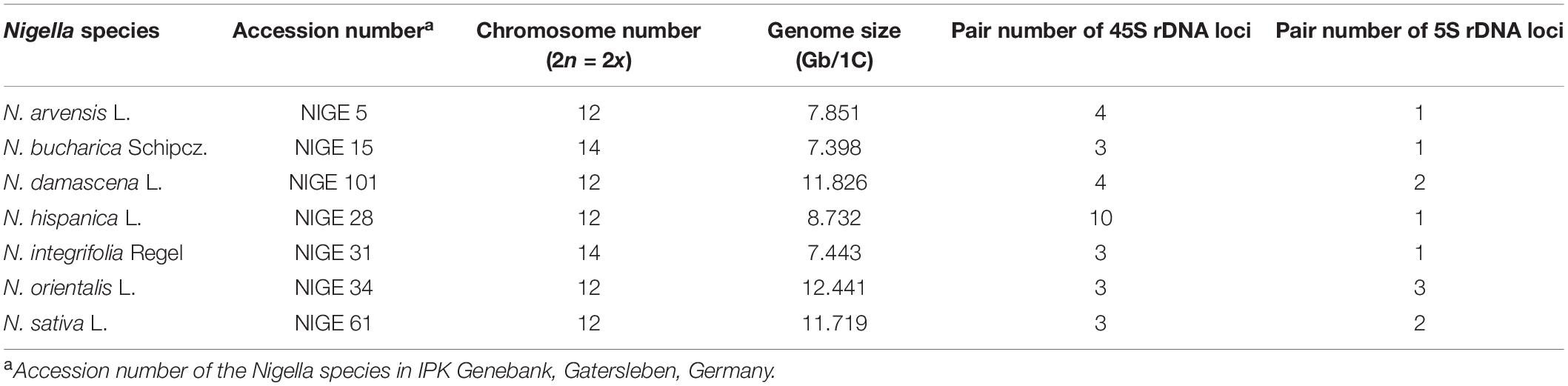
Table 1. Nigella species and accessions used in the present study.
To isolate nuclei, approximately 0.5 cm2 of fresh leaf tissue from a Nigella species and the internal reference standard, Pisum sativum L. subsp. sativum convar. sativum var. ponderosum Alef., Sorte Viktoria, Kifejtö Borsó, Gatersleben Gene Bank accession number: PIS 630, were chopped together in a petri dish using the reagent kit ‘CyStain PI Absolute P’ (Sysmex-Partec) following the manufacturer’s instructions. The nuclei suspension was filtered through a 50-μm CellTrics filter (Sysmex-Partec) and measured on a CyFlow Space flow cytometer (Partec-Sysmex). For each genotype, at least six independent measurements were performed. The absolute DNA content (pg/2C) was calculated based on the values of the G1 peak means and converted to the corresponding genome size (Mbp/1C) according to Dolezel et al. (2003).
Genomic DNAs were extracted from the leaves of N. sativa, N. damascena and N. bucharica using the CTAB method described in Saghai-Maroof et al. (1984), Aboul-Maaty and Oraby (2019). Paired-end (2 × 150 bp) genome sequencing was performed using the Illumina HiSeq 2500 system in a low-coverage scale by Novogene (China). The coverage of sequenced genome was calculated according to the following equation: Coverage = (Number of reads × size of each read)/1C content of the genome.
The quality and GC content of paired-end reads of each species was checked using FastQC (Andrews, 2010) implanted in the RepeatExplorer. The sequence reads were filtered by the quality of 95% of bases equal to or above the quality cut of value of 10. Paired reads were joint using FASTA interlacer tool and pairs with no overlap were selected for the graph-based clustering analysis. The identification and characterization of the repetitive DNA families were then performed using the RepeatExplorer pipeline (Novak et al., 2013; Novák et al., 2017, 2020) with the default setting of 90% similarity over 55% of the read length. Consensus sequences of the identified repeat monomers were reconstructed by TAREAN (TAndem REpeat ANalyzer) (Novák et al., 2017). Comparative RepeatExplorer analysis was performed to identify shared and species-specific repeat clusters. The “Nd,” “Ns,” and “Nb” were used as prefix codes of N. damascena, N. sativa and N. bucharica, respectively. The sequence dataset of each species was then down-sampled to 20% of each genome size (16 million reads for N. sativa and N. damascena and 10 million reads for N. bucharica), followed by a concatenation into a single data file. The settings for comparative clustering analysis were the same as those for individual analysis mentioned above. The sizes of repeat clusters were normalized based on the genome size of analyzed species using optparse package of R version 4.0.2 (The R Project for Statistical Computing, Vienna, Austria).
The monomer of (peri)centromeric satellite sequences were aligned using Clustal Omega (Madeira et al., 2019) and viewed in MView (Brown et al., 1998). The 18S, 5.8S, and 26S coding regions of the identified 45S rDNA and the coding region of 5S rDNA were distinguished by referring to the publicly available rDNA coding sequences in NCBI.
To amplify the repeat DNAs, the consensus sequences of satellite repeats and one LTR element identified were used to design primers using Primer3 (Untergasser et al., 2012). The monomer and primer sequences were listed in Supplementary Table 2. The PCR mixture contained 25 ng of genomic DNA as template, 2.5 mM of each dNTP, 2.5 mM MgCl2, 5 pmol of each primer, and 0.5 U Taq DNA polymerase. PCR amplification was performed for 5 min at 94°C, followed by 30 cycles of 30 s at 94°C, 1 min at 52-60°C (depending on primers), 1 min at 72°C and a final extension for 7 min at 72°C. The size of PCR products was checked in 1% agarose gel by electrophoresis.
PCR products were purified by ethanol precipitation. One microgram of each purified PCR products was labeled with Atto488-11-dUTP or Atto550-11-dUTP using a nick translation kit (Jena Bioscience, Germany), recovered by ethanol precipitation and used as FISH probes. For rDNA probes, the 45S rDNA and 5S rDNA containing clones pTa71 (Gerlach and Bedbrook, 1979) and pTa794 (Gerlach and Dyer, 1980), respectively, were labeled with Atto488-11-dUTP and Atto550-11-dUTP by nick translation as mentioned above. To investigate whether Nigella species possess Arabidopsis-like telomeric repeats, FISH was performed using Arabidopsis-type telomere repeats (TTTAGGG)n as a probe, which was generated by non-template PCR according to IJdo et al. (1991) using (TTTAGGG)3 and (CCCTAAA)3 as primers. One microgram of the purified PCR product was labeled with Atto550-11-dUTP as described above, recovered by ethanol precipitation and used as FISH probes.
Nigella seeds were germinated on moist filter paper in petri dishes for 3–6 days at room temperature. Roots were subjected to nitrous oxide (N2O) gas at 10 bar pressure for 2 h to arrest dividing cells at metaphase. Treated roots were fixed in ice-cold 90% acetic acid for 10 min, then transferred to 75% ethanol and stored at −20°C until use. Roots were first washed in ice-cold water, followed by 0.01 M citrate buffer (0.01 M citric acid and 0.01 M sodium citrate, pH 4.8) each for 10 minutes. Root meristems were placed in a microtube containing 30 μl enzyme mixture [0.7% cellulase (CalBiochem 219466), 0.7% cellulase R10 (Duchefa C8001), 1% cytohelicase (Sigma C8274) and 1% pectolyase (Sigma P3026) in 0.01 M citrate buffer] and were digested at 37°C for 60 to 90 minutes. Slides were prepared using the dropping method according to Abdolmalaki et al. (2019). The specimens were fixed in 4% paraformaldehyde in 1 × PBS (3 mM NaH2PO4, 7 mM Na2HPO4, 0.13 M NaCl, pH 7.4) for 10 min at room temperature, followed by washing in 2 × SSC (0.3 M sodium chloride, 0.03 M sodium citrate, pH 7.0) and dehydrating in 96% ethanol.
FISH and reprobing was performed according to Abdolmalaki et al. (2019). Briefly, 20 μl of hybridization mixture, containing 2 × SSC, 50% formamide, 20% dextran sulfate, 1 μg sheared salmon testes DNA and 20–30 ng of each labeled probe, was applied on each slide and covered with a plastic coverslip. Specimens were then denatured at 80°C for 2 min on a hot plate and were incubated in a humidified plastic container at 37°C, overnight. Coverslips were removed and slides were washed in 2 × SSC for 20 minutes in a water bath at 56°C. Slides were dehydrated in 96% ethanol and dried at room temperature. A drop of Vectashield mounting medium (Vector Laboratories) containing 1 μg/ml DAPI (4′, 6-diamidino-2-phenylindole) was added to each slide as counterstain and a glass coverslip was applied. Slides were inspected with a fluorescence Olympus BX51 microscope (Olympus, Japan), and images were captured using a DP72 digital camera (Olympus, Japan).
Chromosomal and karyotypic indices for numerical characterization of mitotic metaphase chromosomes of the Nigella species were measured using IdeoKar software (Mahmoudi and Mirzaghaderi, 2021). The calculated indices include total chromosome length of the haploid complement (HCL); mean chromosome length (CL), and mean centromeric index (CI). Karyotype asymmetry was determined using the A1 (intrachromosomal asymmetry index) and A2 (interchromosomal asymmetry index) indices calculated using Σ(b/B)/n and s/x equations, respectively, where b and B are the mean lengths of the short and long arms of each homologous chromosome pair, respectively; n is the number of homologs, and s and x are standard deviation and mean of the chromosome length, respectively (Romero-Zarco, 1986). Three high-quality FISH-banded metaphase chromosome spreads were traced for each species. The description of chromosome morphology was based on the nomenclature proposed by Levan et al. (1964). Idiograms were generated using the R package “idiogramFISH” (Roa and Telles, 2020).
The ITS1-5.8S-ITS2 region of 45S rDNA in N. sativa, N. damascena and N. bucharica were identified by RepeatExplorer analysis and were extracted using BLASTn at NCBI database. N. damascena complete chloroplast sequence (MN648403.1) was downloaded from NCBI and used as a reference genome to assemble the chloroplast sequence of N. sativa using CLC software. The reference-aided assembled genome was annotated using the GeSeq annotation tool (Tillich et al., 2017). The conserved sequences flanking the ITS1-5.8S-ITS2 and rbcL gene were used to design PCR primers to amplify and sequence the corresponding regions in the other five Nigella genomes (Supplementary Tables 2–4). The ITS1-5.8S-ITS2 and rbcL sequences were used as input for multiple sequence alignment by MUSCLE algorithm using MEGA11 software (Tamura et al., 2021). The concatenated ITS1-5.8S-ITS2 and rbcL sequences were used to build a maximum likelihood tree with 500 bootstrapping replications in MEGA11.
According to flow cytometric estimation of the DNA content, Nigella orientalis has the largest genome with 12.44 Gbp/1C among the seven species, followed by N. damascena and N. sativa with 11.72 Gbp/1C and 11.83 Gbp/1C, respectively (Table 1 and Figure 1). N. hispanica (8732 Mbp/1C), N. arvensis (7851 Mbp/1C), N. integrifolia (7443 Mbp/1C) and N. bucharica (7398 Mbp/1C) have considerably smaller genomes than those of the other three species mentioned above.
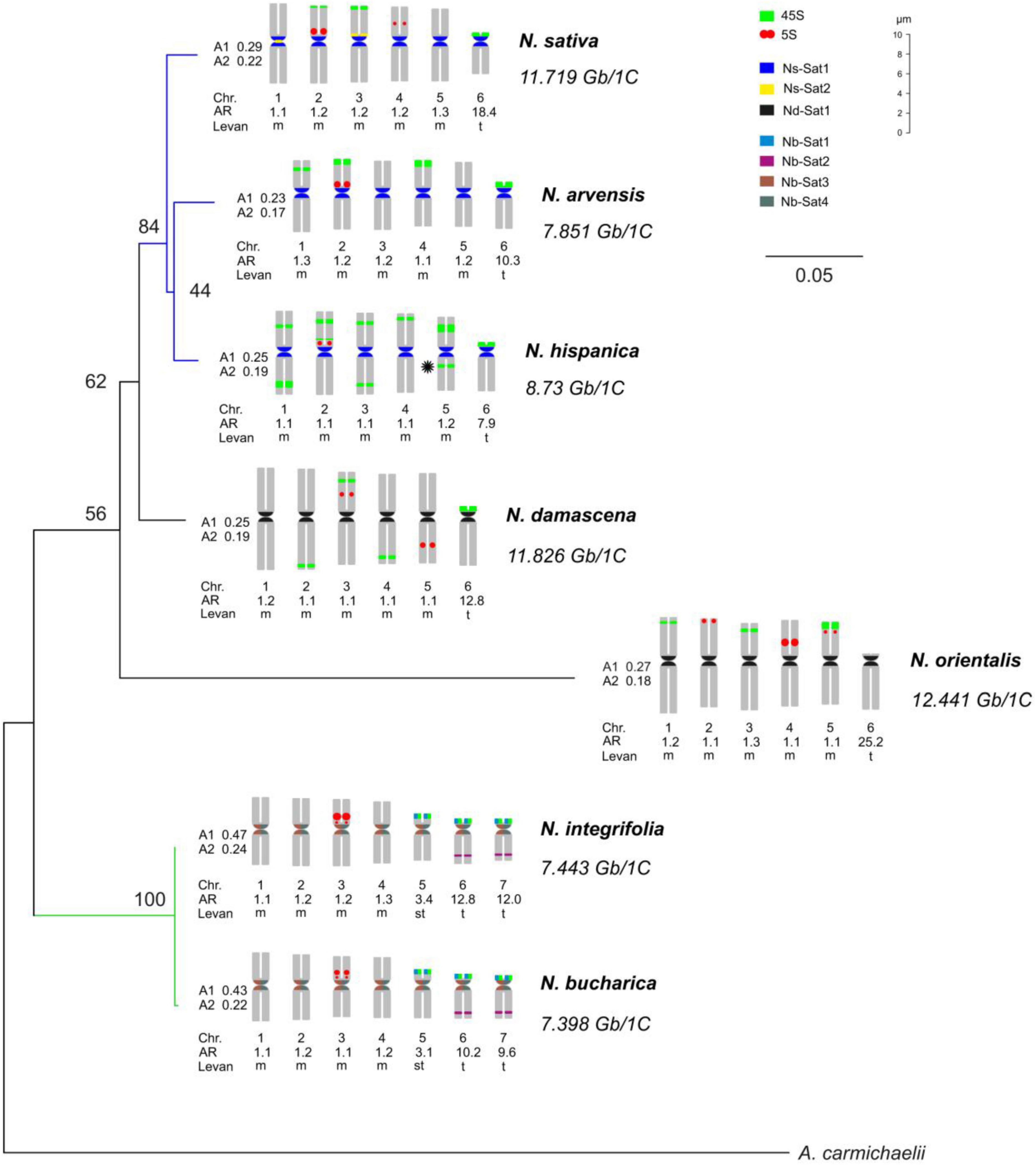
Figure 1. Phylogeny and idiograms of Nigella. A maximum likelihood phylogenetic tree of Nigella species with 500 bootstrap replications inferred from ITS1-5.8S-ITS2 and rbcL sequences. A. carmichaelii has been included as an outgroup. The tree has been annotated with idiograms (showing the locations of the identified satellite repeats and 45S and 5S arrays), karyotypic parameters and the estimated DNA C-values. The asterisk in N. hispanica indicates the hemizygous locus. Chr.: Chromosome; A1: intrachromosomal asymmetry index; and A2: interchromosomal asymmetry index; Levan: the description of chromosome morphology was based on the nomenclature proposed by Levan et al. (1964), AR: arm ratio (long arm/short arm).
Chromosomes of Nigella species were mainly metacentric with one or two telocentric chromosome pairs in each species. Based on their basic chromosome number, the seven species can be classified into two groups. The first group, comprising N. arvensis, N. damascena, N. hispanica, N. orientalis and N. sativa, has a basic chromosome number of x = 6 (2n = 2x = 12) with a karyotype formula of 10m + 2t. N. bucharica and N. integrifolia belong to the second group with a basic chromosome number of x = 7 (2n = 2x = 14) and a karyotype formula of 8m + 2st + 4t. All these species fell into the 2A category of Stebbin’s asymmetry indices (Stebbins, 1971; Figure 1). The size of metacentric chromosomes ranged from 5.99 μm (N. bucharica) to 10.14 μm (N. damascena) and the telocentric chromosome size ranged from 4.04 μm (N. bucharica) to 5.51 μm (N. damascena). N. bucharica and N. integrifolia also have a pair of subtelocentric chromosomes with a size range from 4.54 to 4.71 μm. The total metaphase chromosome length was between 38.21 μm in N. bucharica and 53.17 μm in N. damascena (Supplementary Table 5).
To determine the karyotype evolution among the seven Nigella species, FISH mapping of 45S and 5S rDNA loci on mitotic chromosomes was performed (Figure 2). FISH of both ribosomal probes revealed a considerable interspecific variation regarding the number and position of rDNA loci (Figures 1A, 2). While three 45S rDNA-positive chromosome pairs were observed in N. sativa, N. orientalis, N. integrifolia and N. bucharica (Figures 2A–D), four pairs of 45S rDNA loci were present in N. damascena and N. arvensis (Figures 2E,F). N. hispanica revealed ten pairs of 45S rDNA loci, the highest number among the investigated species. Each chromosome of this species harbors at least one 45S rDNA locus. Interestingly, one of the 45S rDNA sites in N. hispanica did not show a signal on its corresponding homologous chromosome representing hemizygosity (Figures 1A, 2G). While in N. sativa, N. arvensis, N. hispanica and N. damascena, 45S rDNA loci were found on metacentric and telocentric chromosomes, 45S rDNA loci were exclusively found on metacentric chromosomes in N. orientalis or on submetacentric and telocentric chromosomes in N. bucharica and N. integrifolia (Figure 2). The 5S rDNA was found on one (N. integrifolia, N. bucharica, N. arvensis and N. hispanica) (Figures 2C,D,F,G), two (N. sativa and N. damascena) (Figures 2A,E) or three (N. orientalis) (Figure 2B) chromosome pairs. 45S rDNA loci are located mainly either in distal or proximal regions of the chromosome arms, while 5S rDNA arrays were also found interstitially. The size of hybridization signals varied between chromosome pairs both within and between species (Figure 2).
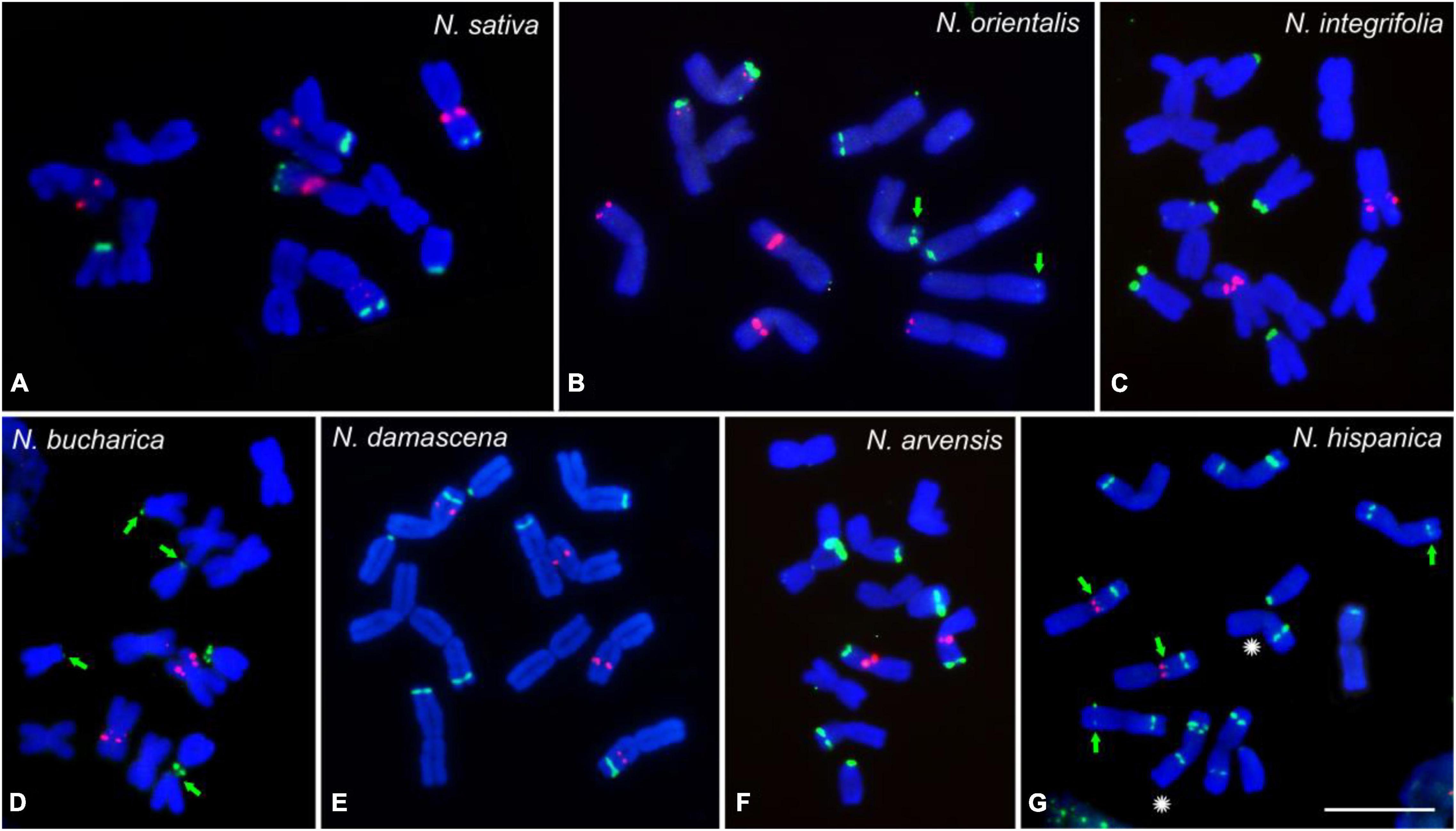
Figure 2. rDNA loci distribution of the studied Nigella species. FISH mapping of 45S rDNA (green) and 5S rDNA (red) on mitotic metaphase chromosomes of N. sativa (A), N. orientalis (B), N. integrifolia (C), N. bucharica (D), N. damascena (E), N. arvensis (F) and N. hispanica (G). Chromosomes were counterstained with DAPI. Arrows indicate weak 45S rDNA signals and the asterisks indicate the homologous chromosomes with hemizygous locus. Scale bar 10 μm.
To determine the phylogenetic relationship among the analyzed Nigella species, the sequences of nuclear ribosomal internal transcribed spacer (ITS) and rbcL gene were used. The sequence length of ITS (ITS1-5.8S-ITS2) varied from 732 to 759 bp (Supplementary Table 3), whereas rbcL sequences ranged from 871 to 1428 bp (Supplementary Table 4) in the seven Nigella species. Aconitum carmichaelii, a distantly related species belonging to the same family, was used as an outgroup, and the resulting consensus had high bootstrap support values (Figure 1). N. bucharica and N. integrifolia formed a robust cluster with 100% bootstrap support, and both of them have a basic chromosome number of x = 7. The other cluster included N. sativa, N. arvensis and N. hispanica (with 84% bootstrap support), to which N. damascena and N. orientalis were jointed with lower support. All members of this cluster possess a chromosome number of 2n = 12.
The clustering of the seven Nigella species based on molecular phylogenetic analysis correlates with their basic chromosome number (x = 6 or 7). The phylogenetically close N. integrifolia and N. bucharica have the same chromosome number, similar genome size and rDNA-based karyotypes. Nevertheless, among the other five species, despite having the same chromosome number, their genome size, the number and chromosomal distribution of rDNA loci are diverse.
Low-pass sequencing of N. sativa, N. damascena and N. bucharica genomes resulted in 4,232,251, 7,553,644, and 15,352,348 Illumina 150 bp paired-end reads corresponding to 0.24×, 0.43×, and 1.50× genome coverage, respectively. The GC content for N. sativa and N. damascena genomes showed a value of 38%, while this value was 42% for N. bucharica. The repeat compositions were inferred from the paired-end reads corresponding to approximately ∼0.2× of the genome for each analyzed species. The proportions of individual repeat types are presented in Table 2. About 57.52, 59.01, and 64.73% of N. sativa, N. damascena and N. bucharica genomes are composed of high- or moderate-copy repeats, respectively. The majority of the repeats (47.91% in N. sativa, 39.47% in N. damascena, and 51.25% in N. bucharica) are retroelements, followed by unclassified repeats (6.95, 17.30, and 10.10%) and tandem repeats (0.75, 0.39, and 0.74% of rDNAs and 0.75, 1.21, and 1.45% of satellites). The proportions of 45S rDNA repeats in N. sativa, N. damascena and N. bucharica genomes were 0.72, 0.38, and 0.65%, respectively, while the 5S rDNA proportions were 0.03%, 0.01% and 0.09% as determined by RepeatExplorer analysis (Table 2). The consensus monomers of the rDNA sequences in N. sativa, N. damascena and N. bucharica identified by TAREAN are listed in Supplementary Table 6. Among the retroelements, LTR retroelements are the most abundant in the N. sativa (47.91%), N. damascena (39.47%) and N. bucharica (51.25%) genomes. LTRs in N. sativa include Ty3–gypsy and Ty1–copia super families with a proportion of 44.01 and 3.76% in the genome, respectively, while they compose 37.77 and 1.64% in N. damascena and 48.55 and 2.58% in N. bucharica. A major part (34.89% in N. sativa, 26.19% in N. damascena and 30.99% in N. bucharica) of Ty3–gypsy belongs to the retrotransposon chromoviral Tekay clade (Table 2). In contrast, DNA transposons contribute to only 1.13, 0.62, and 1.19% of the N. sativa, N. damascena and N. bucharica genomes, respectively, and only three common DNA transposons, EnSpm_CACTA, MuDR_Mutator and PIF_Harbinger, were identified. EnSpm_CACTA composes 0.72% of the N. sativa genome, but its proportion was much lower in N. damascena (0.24%) and N. bucharica (0.16%). MuDR_Mutator comprises about 0.32% of N. sativa, 0.34% of N. damascena and 0.71% of N. bucharica genome. Also, PIF_Harbinger composes 0.32% of the N. bucharica genome, but its proportion was lower than 0.1% in N. sativa (0.06%) and N. damascena (0.04%). The DNA transposon hAT was only detected in N. sativa (Table 2).
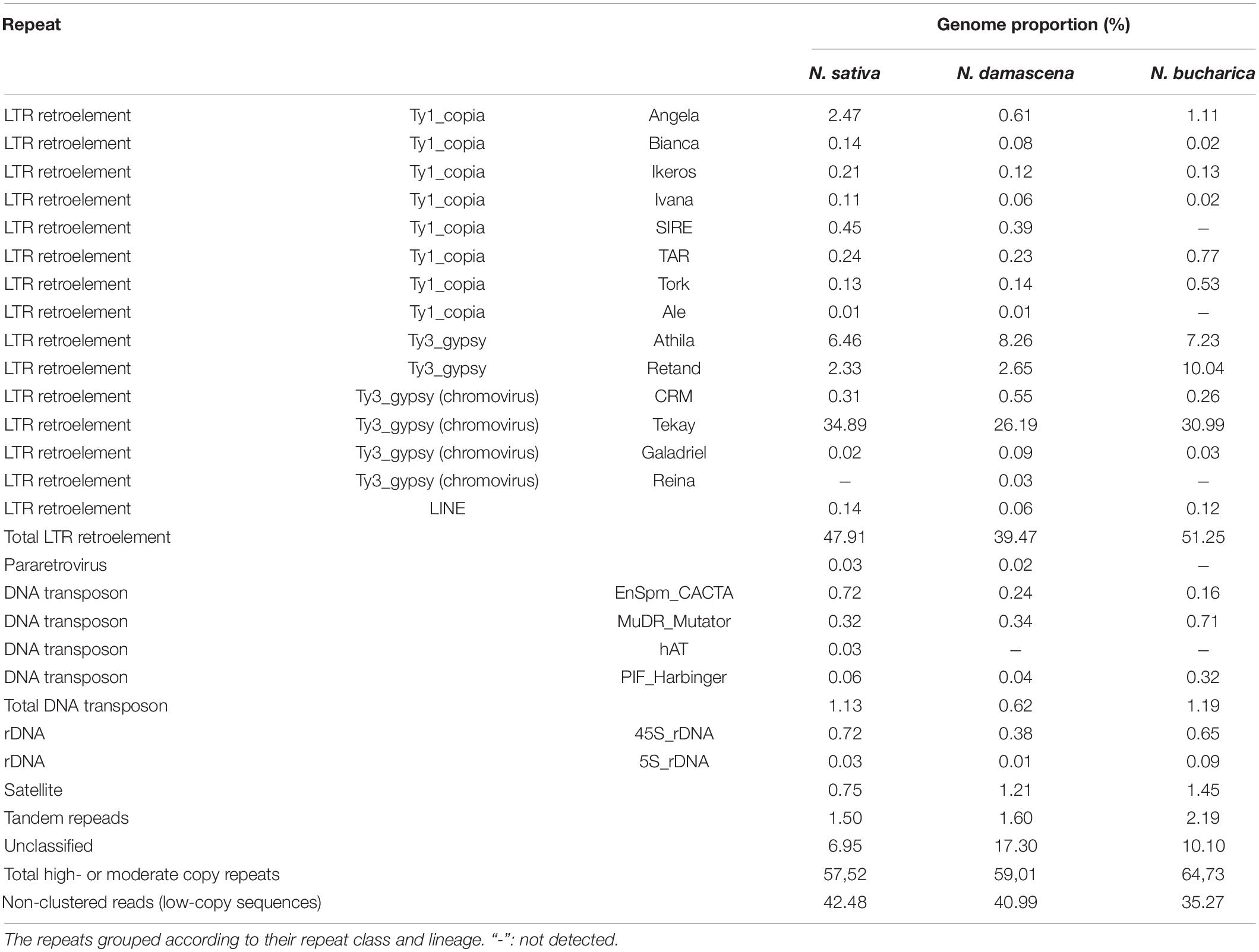
Table 2. Types and proportions of highly-repetitive sequences in N. sativa, N. damascena and N. bucharica characterized by RepeatExplorer2.
To compare the repeat compositions between the genomes of N. sativa, N. damascena and N. bucharica, a comparative clustering analysis was performed. About a quarter of the top clusters (Figure 3) are shared between the species. Not all of these clusters had similar abundance in the genomes. Out of the in total 272 major repeat clusters, only 16 clusters (5.88%) were relatively evenly shared between the three genomes, and they were annotated as Ty1_copia-TAR and Tork, Ty3_gypsy-Athila, DNA transposon-EnSpm CACTA and rDNAs (Figure 3). Up to 97 clusters (35.66%) were almost N. bucharica specific, and shared clusters between N. bucharica and either N. damascena or N. sativa were barely detectable. N. damascena and N. sativa contributed to 123 and 120 clusters, respectively, of which 77 clusters were shared between the two genomes, whereas 61 and 37 of them were highly enriched or specific to N. damascena and N. sativa, respectively. The comparative analysis demonstrated that N. bucharica is relatively more distinct from N. damascena and N. sativa. This result is in line with their phylogenetic relationships inferred based on ITS and rbcL sequences. The monomer length and cluster proportion of satellites and high copy retrotransposons identified by TAREAN is listed in Supplementary Table 7. Most of retrotransposons were common between N. sativa and N. damascena, (Supplementary Table 7).
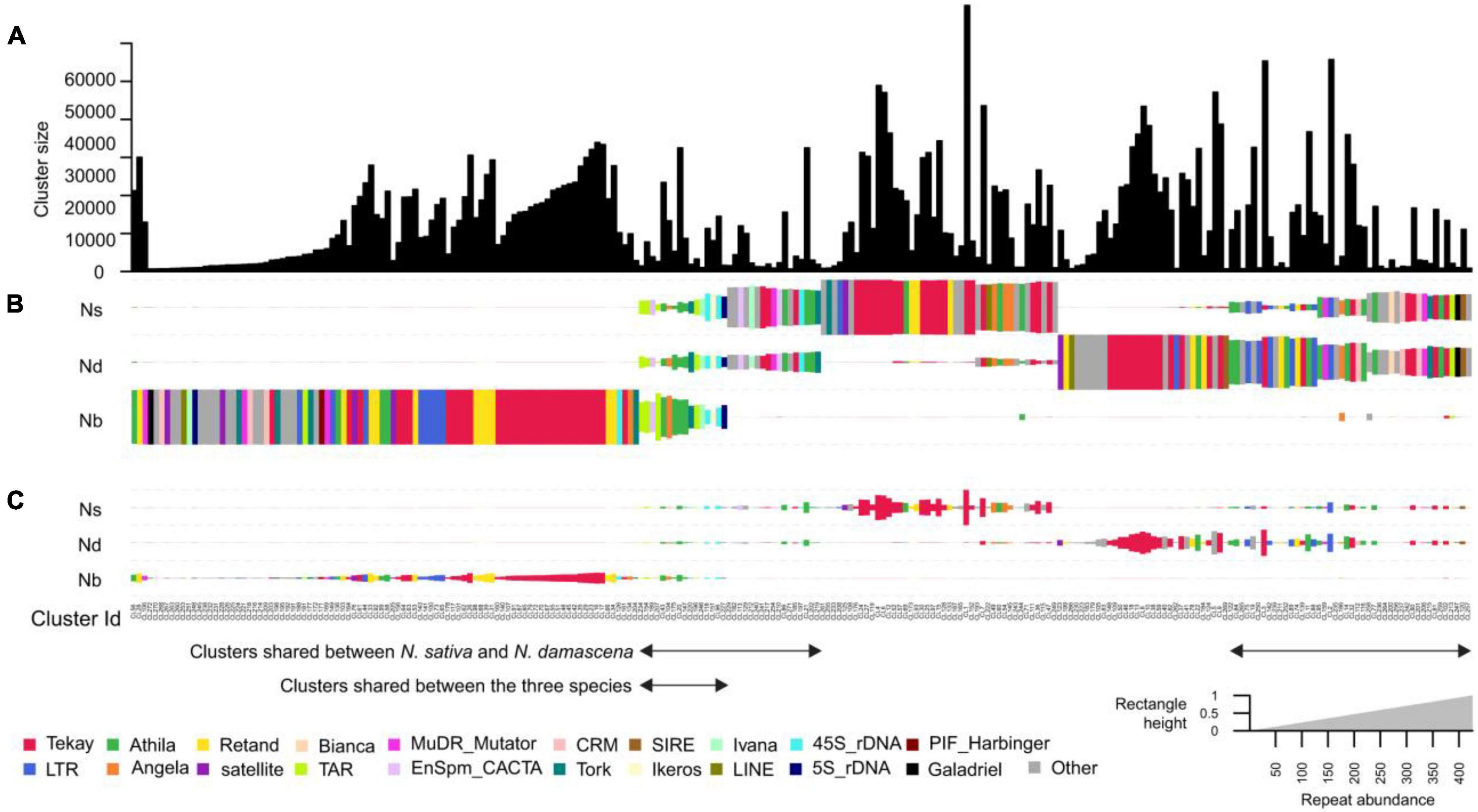
Figure 3. Comparative analysis of genome repetitive composition among three Nigella species. (A) Bar plot of N. damascena, N. sativa and N. bucharica showing the sizes (numbers of reads) of individual clusters. (B) Rectangle size is proportional to the number of reads in a cluster for each species. Clusters were sorted by using hierarchical clustering and the rectangles are colored based on their cluster annotation. (C) The size of the rectangles in (B) was normalized based on the genome size of analyzed species. Species codes: ND, N. damascena; NS, N. sativa; Nb, N. bucharica.
The application of the TAREAN pipeline (Novák et al., 2017) allowed the identification of tandem repeat clusters in Nigella species. Two satellite repeats in N. sativa, i.e., Ns-Sat1 (CL21) and Ns-Sat2 (CL144) were detected, representing 0.52 and 0.01% of the genome, respectively (Table 3). Only one satellite repeat, named Nd-Sat1 (CL23), was identified in N. damascena which corresponds to 0.52% of the genome. On the other hand, four satellite repeats, Nb-Sat1 (CL21), Nb-Sat2 (CL129), Nb-Sat3 (CL64) and Nb-Sat4 (CL144), were identified in N. bucharica, representing 0.86, 0.08, 0.42, and 0.03% of the genome, respectively. All these repeats represented satellite-typical globular graph layouts, and their consensus monomer sequences are available in Supplementary Table 2. The monomers of Ns-Sat-1, Ns-Sat-2 and Nd-Sat-1 are all 178 bp in length and AT-rich (e.g., 68% AT for Ns-Sat1) (Table 3 and Supplementary Figure 3). Their sequence similarity ranged from 78.8% (between Ns-Sat1 and Nd-Sat1) to 71.8% (between Ns-Sat1 and Ns-Sat2). In addition, the monomer sequence of the Ty3_gypsy LTR-annotated retrotransposon, Ns-CL6, was reconstructed. To determine the chromosomal distribution of the identified repeats, the corresponding DNA fragments were PCR amplified using the respective primers and labeled as FISH probes (Table 3).
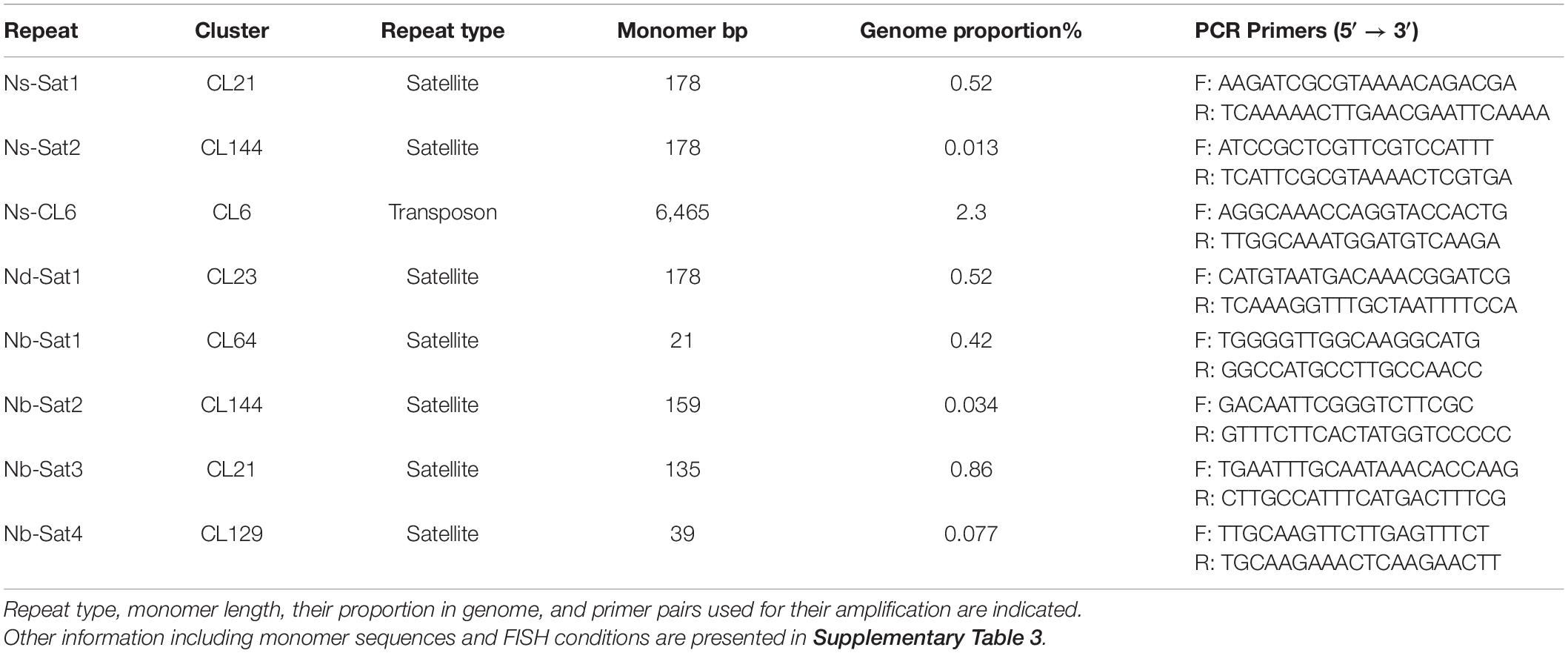
Table 3. Repeats used as probes in FISH experiments on N. sativa, N. damascena and N. bucharica chromosomes.
After FISH, all metaphase chromosomes of N. sativa revealed (peri)centromeric Ns-Sat1 signals while Ns-Sat2 localized in the (peri)centromeric regions of only chromosomes 1 and 4 (Figures 4A,B). The Ns-CL6 probe which is a Ty3_gypsy LTR retrotransposon, resulted in evenly distributed signals, although with a lower density toward the distal chromosome regions (Figure 4A). Nd-Sat1-specific signals were found in the (peri)centromeric regions of all N. damascena chromosomes (Figure 4C). Ns-Sat1 also cross-hybridized to the (peri)centromeric regions of N. arvensis and N. hispanica (Figures 4D,E). The Nd-Sat1 of N. damascena also cross-hybridized to the (peri)centromeric regions of N. orientalis (Figure 4F). None of the Nd-Sat1, Ns-Sat1 and Ns-Sat2 probes cross-hybridized with N. integrifolia or N. bucharica. The observed clustering of (peri)centromeric repeats at one pole of the nuclei indicates a Rabl-like chromosome configuration in interphase nuclei of Nigella (Figures 4A,E).
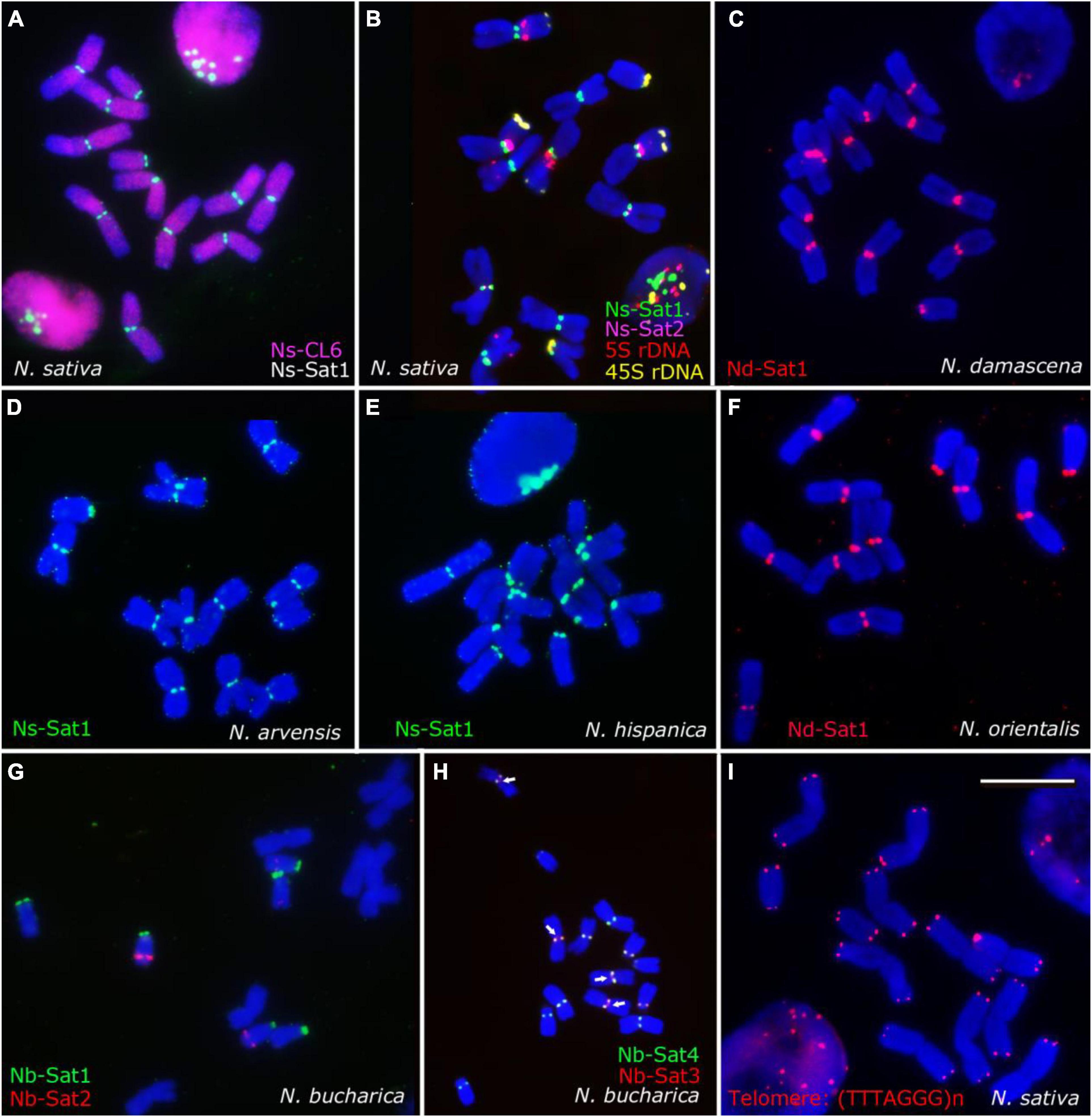
Figure 4. Chromosomal distribution of the identified high-copy repeats of Nigella. FISH results using the identified satellite repeats and Arabidopsis-type telomeric repeat probe (TTTAGGG)n probes on mitotic metaphase chromosomes of N. sativa (A,B,I), N. damascena (C), N. arvensis (D), N. hispanica (E), N. orientalis (F) and N. bucharica (G,H). The Ns-CL6 (red) repetitive sequence is dispersed throughout the genome (A), while the satellite sequences are mostly (peri)centromeric. Scale bar 10 μm.
The observed hybridization signals of all four Nb satellite probes showed similar intensities, locations and numbers in N. bucharica and N. integrifolia (Figure 1). Nb-Sat1 seems to co-localize with the 45S-rDNA loci, since it is found in terminal positions on the short arms of the two telocentric and the submetacentric chromosomes. Nb-Sat2 revealed signals on the distal ends of the long arm of the telocentric chromosomes (Figure 4G), while Nb-Sat3 and Nb-Sat4 showed signals in (peri) centromeric positions of all chromosomes (Figures 1A, 4H). It seems that at least in some of the chromosomes Nb-Sat3 is extended toward the inner part of the centomeres compared with Nb-Sat4 (arrows in Figure 4H). FISH with the Arabidopsis-type telomere repeat (TTTAGGG)n detected corresponding signals exclusively at both ends of all Nigella chromosomes (Figure 4I).
The genome-wide repetitive analysis in the three Nigella species indicated that retroelements, especially Ty_gypsy LTRs, are the main contributors to the relatively large genomes of Nigella. On the contrary, the abundance and diversity of satellite DNAs are relatively low. Most of these satellites locate at (peri)centromeric regions. The (peri)centromeric satellite repeats of N. sativa (Ns-Sat1), N. damascena (Nd-Sat1) and N. bucharica (Nb-Sat3 and Nb-Sat4) are highly distinct and cross-hybridized only to the closely related genomes as indicated in Figure 1.
We studied the phylogenetic relationship and karyotype structure of seven Nigella species by using sequences of ITS and rbcL gene, analyzing the repeatome and FISH mapping. Except for N. sativa and N. damascena, whose genome sizes were previously reported, the genome size of the other species was estimated for the first time. N. orientalis, N. damascena, and N. sativa have roughly 1.5 times larger genomes than N. arvensis, N. hispanica, N. bucharica and N. integrifolia. The DNA C-values estimated for N. sativa and N. damascena were quite similar to the previous estimations based on Feulgen densitometry [10.30 and 10.58 Gbp/1C for N. damascena (Evans et al., 1972; Olszewska and Osiecka, 1983) and 10.39 Gbp/1C for N. sativa (Bennett and Smith, 1976). The slight differences might be explained by the different methods used (Feulgen densitometry versus flow cytometry) and/or the different reference standards used (P. sativum versus Allium cepa).
In spite of the smaller genome sizes, N. bucharica and N. integrifolia have with a genome formula of 2n = 2x = 14 one additional chromosome pair more than the other species (basic chromosome number x = 6). A previous study using Giemsa C-banding on Nigella chromosomes (Gilot-Delhalle et al., 1976) suggested that the telocentric chromosomes originated from a centromeric fission event of a metacentric chromosome. Subsequent structural rearrangements might have formed the submetacentric chromosome in N. bucharica and N. integrifolia (Gilot-Delhalle et al., 1976).
The morphology of the studied species is rather similar except for N. bucharica and N. integrifolia, which have distinct flower and leaf morphology (Supplementary Figure 3). Both species, in turn, show substantial morphological, karyological and sequence similarities, which raises the question if they should be classified as varieties of a single species instead of two independent species.
N. sativa, N. damascena, N. arvensis, N. hispanica and N. orientalis showed similar karyotypes in terms of chromosome numbers and morphology, but the patterns of 5S and 45S rDNA loci differ between species, suggesting that the evolution of these species was accompanied by chromosomal segment rearrangements such as inversions, translocations and Robertsonian fission or mobility of rDNA loci without noticeably affecting the arm ratios. Similarly, in a cytogenetic survey of the Ranunculaceae, the number, location and intensity of rDNA signals varied between various species of Pulsatilla and Anemone genera. Most of the 45S rDNA loci in these genus are located at distal regions of the short arms of acrocentric chromosomes, while 5S rDNA loci don’t show preferential chromosomal positions. Such a rDNA mobility might be the result of homologous and non-homologous recombination mechanisms and retroelement-mediated rDNA transpositions (Mlinarec et al., 2006, 2012; Sramkó et al., 2019). Variation in the number and position of rDNA loci has also been reported among other species such as e.g., legumes (Abirached-Darmency et al., 2005). rDNA might be moved by transposition, as previously shown in Allium species and their hybrids (Schubert, 1984). In fact, rDNA sequences are conserved, but their chromosomal distribution is a source of species differentiation and evolution (Raskina et al., 2008). In Nigella, the number of 45 rDNA loci varied from three to ten. However, no positive correlation between the loci number and genome size was observed. While the three larger genomes (N. sativa, N. damascena and N. orientalis) all showed only three loci, the highest number was found in N. hispanica which has a rather small genome. Also in diploid lineages of Brassicaceae (Hasterok et al., 2006), Cyperaceae (Da Silva et al., 2010), Iris (Martinez et al., 2010), and Rosaceae (Mishima et al., 2002) no positive correlation exists between the number of rDNA arrays and the number of chromosomes or genome size.
The presence of additional 45S rDNA loci in N. damascena (4 loci), N. arvensis (4 loci) and N. hispanica (10 loci) compared with N. sativa, N. orientalis, N. integrifolia and N. bucharica (all 3 loci) may be due to independent formation of a separate 45S rDNA array after the divergence of these species. N. hispanica also showed hemizygosity for one 45S rDNA site. rDNA site hemizygosity has been reported in other genera such as Anacyclus (Rosato et al., 2017), Vicia (Li et al., 2001), Chrysanthemum (He et al., 2021) and Lilium (Wang et al., 2012). In most cases such a heterozygosity is related to hybridization events. The reason for the detected hemizygous locus in Nigella remains to be elucidated. Similarly, it is not clear if this heterozygosity is only occurring in the investigated genotype or if it is a general feature of N. hispanica.
Overall, 45S rDNA composed about 0.38, 0.72, and 0.65% of the N. damascena, N. sativa and N. bucharica genomes, respectively (Table 2). The relative amount and size of rDNA units in the nuclear genome can be highly variable, and the rDNA copy number can vary between 150 to 26,048 copies in plants (Prokopowich et al., 2003; Wicke et al., 2011).
In Nigella, the majority (47.91% in N. sativa, 39.47% in N. damascena and 51.25% in N. bucharica) of the repeats are retrotransposons (class I transposons), while DNA transposons (class II transposons) contributed to only 1.13, 0.62, and 1.19% of the genomes. Although the three Nigella species possess rather large genomes of about 7.4 to 12.4 Gb/C, only one to four satellite repeats were found in these species, and no correlation was found between the number of satellites and the size of the genome in the studied Nigella species. Interestingly, N. bucharica and N. integrifolia with smaller genome sizes contained a higher number of satellite sequences than the larger genome of N. damascena. The most abundant satellites in the three species were found in (peri)centromeric position on all chromosomes.
Ns-CL6 is a retrotransposon distributed over all chromosomes of N. sativa although with a reduced density at distal regions. The reduced frequency of Ns-CL6 at chromosome ends could be explained by the potential enrichment of coding sequences in this region. In many plant species, especially well investigated in cereals, the terminal and subterminal chromosomal regions are often enriched in coding sequences1.
The rDNA probes alone or in combination with chromosome-specific satellite sequences are useful markers to identify individual chromosomes. While in N. damascena, N. hispanica and N. orientalis the 5S rDNA and 45S rDNA were sufficient to characterize the complete chromosome set, in N. sativa additionally NS-Sat2 was required. In N. arvensis 4 out of 6 and in N. integrifolia and N. bucharica 2 out of 7 chromosome pairs could be unequivocally identified by using the rDNA probes.
Sequence alignment indicated that the Ns-Sat1, Ns-Sat2 and Nd-Sat1 (peri)centromeric repeats are similar, suggesting they might share a common origin. However, the retained identity between Ns-Sat1 and Nd-Sat1 (78.8%, Supplementary Figure 2) was not enough for each of them to cross-hybridize on the other species, indicating their sequence divergence after specification. Ns-Sat1, Ns-Sat2 and Nd-Sat1 are all 178 bp long AT-rich satellite repeats. Due to their localization patterns and their length similarity with described centromeric satellites in other species such as Arabidopsis (Copenhaver et al., 1999), human (Choo et al., 1991) and the fish Pungitius pungitius (Varadharajan et al., 2019) it is tempting to speculate that these sequences indeed represent centromeric repeats of Nigella, although a functional proof is still missing. The 178 bp satellite unit is consistent with the 150–180 bp length DNA required to wrap around a single nucleosome (Henikoff et al., 2001). However, the (peri)centromeric satellites in N. bucharica and N. integrifolia (Nb-Sat3 and Nb-Sat4) have a deviating monomer length of only 135 and 39 bp.
Our molecular phylogeny using rbcL and ITS1-5.8S-ITS2 sequences grouped the seven Nigella species into three different clades, two groups with x = 6 and the third one with x = 7. These results are in agreement with the morphological classifications reported earlier (Zohary, 1983: Yao et al., 2019). Significant variation observed in the sites and numbers of 45S rDNA loci might be involved in shaping Nigella karyotypes. The more asymmetric karyotype of the third group with additional telo- or subtelocentric chromosomes and the presence of terminal 45S rDNA sites in almost all telo- and subtelocentrics suggest that chromosomal rearrangements might play a role in changing the basic chromosome number (dysploidy) in the genus Nigella. DNA breakage and repair, rDNA mobility and Robertsonian fusions/fissions are suggested as the possible mechanisms during this process (Sramkó et al., 2019).
Overall, our analyses based on the molecular phylogeny, DNA C-value analysis, genomic repeat composition and FISH-karyotyping shed light on the genome organization and evolution of seven Nigella species and supports a classification into three different groups of which two are closer to each other than the third one. The two phylogenetically closer groups (N. sativa, N. arvensis and N. hispanica and accordingly N. damascena and N. orientalis) share the same basic chromosome number (x = 6), and a similar karyotype formula. N. integrifolia and N. bucharica, in contrast, differ with x = 7 from the other five species. The repeatome analysis demonstrated that the genomes of Nigella species increased in size due to the preferential accumulation of Ty3_gypsy retroelements, especially of the Ty3_gypsy-Tekay lineage. In contrast, satellite repeats comprise only a small proportion of the Nigella genomes and are predominantly located at the (peri)centromeric regions. These sequences are only cross-hybridizing within the closely related species and support the proposed grouping. Surprisingly, despite the low total genome proportion of 5S and 45S rDNA, their diverse loci number and patterns on chromosomes of the analyzed species indicated the potential importance of rDNAs in driving the Nigella genome divergence and specification. Additionally, 5S and 45S rDNAs can be further applied as cytogenetic markers for chromosome discrimination and karyotype analysis in the genus Nigella.
The datasets presented in this study can be found in online repositories. The low coverage genomic DNA sequencing data of N. sativa, N. damascena and N. bucharica have been submitted to the Sequence Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra/) under accession number: PRJNA686272.
FO conducted the experiments and data analysis and assisted in the manuscript writing. GM conceived and designed the research and wrote the manuscript. Y-TK contributed to the data analysis, critical discussions, and manuscript revisions. JF contributed to the flow cytometry analysis and critical discussions. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Andreas Houben (IPK, Germany) for the assistance in genomic DNA sequencing, contribution to critical discussions and manuscript revisions, and Kerstin Wolf for technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.917310/full#supplementary-material
Abdolmalaki, Z., Mirzaghaderi, G., Mason, A. S., and Badaeva, E. D. (2019). Molecular cytogenetic analysis reveals evolutionary relationships between polyploid Aegilops species. Plant. Syst. Evol. 305, 459–475. doi: 10.1007/s00606-019-01585-3
Abirached-Darmency, M., Prado-Vivant, E., Chelysheva, L., and Pouthier, T. (2005). Variation in rDNA locus number and position among legume species and detection of 2 linked rDNA loci in the model Medicago truncatula by FISH. Genome 48, 556–561. doi: 10.1139/g05-015
Aboul-Maaty, N. A.-F., and Oraby, H. A.-S. (2019). Extraction of high-quality genomic DNA from different plant orders applying a modified CTAB-based method. Bull. Natl. Res. Cent. 43:25. doi: 10.1186/s42269-019-0066-1
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Cambridge: The Babraham Institute.
Appels, R., Gerlach, W., Dennis, E., Swift, H., and Peacock, W. (1980). Molecular and chromosomal organization of DNA sequences coding for the ribosomal RNAs in cereals. Chromosoma 78, 293–311. doi: 10.1007/s00239-004-0244-z
Atamuratova, N., Mukhamatzanova, R., and Ch, B. K. (2021). Honey significance of forest lands in south Uzbekistan. IOP Conf. Ser. Earth Environ. Sci. 775: 012013. doi: 10.1088/1755-1315/775/1/012013
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6:11. doi: 10.1186/s13100-015-0041-9
Belyayev, A., Josefiová, J., Jandová, M., Kalendar, R., Krak, K., and Mandák, B. (2019). Natural history of a satellite DNA family: from the ancestral genome component to species-specific sequences, concerted and non-concerted evolution. Int. J. Mol. Sci. 20:1201. doi: 10.3390/ijms20051201
Bennett, M. D., and Smith, J. (1976). Nuclear DNA amounts in angiosperms. Philos. Transac. R. Soc. Lond. B Biol. Sci. 274, 227–274. doi: 10.1098/rstb.1976.0044
Bennett, M. D., Leitch, I. J., Price, H. J., and Johnston, J. S. (2003) Comparisons with Caenorhabditis (∼100 Mb) and Drosophila (∼175 Mb) using flow cytometry show genome size in Arabidopsis to be ∼157 Mb and thus ∼25% Larger than the Arabidopsis genome initiative estimate of ∼125 Mb. Ann. Bot. 91, 547–557. doi: 10.1093/aob/mcg057
Brown, N. P., Leroy, C., and Sander, C. (1998). MView: a web-compatible database search or multiple alignment viewer. Bioinformatics 14, 380–381. doi: 10.1093/bioinformatics/14.4.380
Choo, K., Vissel, B., Nagy, A., Earle, E., and Kalitsis, P. (1991). A survey of the genomic distribution of alpha satellite DNA on all the human chromosomes, and derivation of a new consensus sequence. Nucleic Acids Res. 19:1179. doi: 10.1093/nar/19.6.1179
Copenhaver, G. P., Nickel, K., Kuromori, T., Benito, M.-I., Kaul, S., Lin, X., et al. (1999). Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286, 2468–2474. doi: 10.1126/science.286.5449.2468
Da Silva, C., Quintas, C. C., and Vanzela, A. L. (2010). Distribution of 45S and 5S rDNA sites in 23 species of Eleocharis (Cyperaceae). Genetica 138, 951–957. doi: 10.1007/s10709-010-9477-5
Datson, P., and Murray, B. (2003). The use of in situ hybridisation to investigate plant chromosome diversity. Plant Genome 1:298Y318.
Dolezel, J., Bartos, J., Voglmayr, H., and Greilhuber, J. (2003). Nuclear DNA content and genome size of trout and human. Cytometry A 51, 127–8; author reply 129. doi: 10.1002/cyto.a.10013
Dönmez, A. S., Aydin, Z., and Uand Dönmez, E. O. (2021). Taxonomic monograph of the tribe Nigelleae (Ranunculaceae): a group including ancient medicinal plants. Turkish J. Bot. 45, 468–502. doi: 10.3906/bot-2105-39
Elder, J. F., and Turner, B. J. (1995). Concerted evolution of repetitive DNA sequences in eukaryotes. Q. Rev. Biol. 70, 297–320. doi: 10.1086/419073
Evans, G., Rees, H., Snell, C., and Sun, S. (1972). The relationship between nuclear DNA amount and the duration of the mitotic cycle. Chromosomes Today 3, 24–31.
Frello, S., and Heslop-Harrison, J. (2000). Chromosomal variation in Crocus vernus Hill (Iridaceae) investigated by in situ hybridization of rDNA and a tandemly repeated sequence. Ann. Bot. 86, 317–322.
Fu, J., Zhang, H., Guo, F., Ma, L., Wu, J., Yue, M., et al. (2019). Identification and characterization of abundant repetitive sequences in Allium cepa. Sci. Rep. 9:16756. doi: 10.1038/s41598-019-52995-9
Gerlach, W., and Bedbrook, J. (1979). Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucleic Acids Res. 7, 1869–1885. doi: 10.1093/nar/7.7.1869
Gerlach, W., and Dyer, T. (1980). Sequence organization of the repeating units in the nucleus of wheat which contain 5S rRNA genes. Nucleic Acids Res. 8, 4851–4865. doi: 10.1093/nar/8.21.4851
Ghosh, A., and Datta, A. K. (2006). Karyotyping of Nigella sativa L.(black cumin) and Nigella damascena L.(love-in-a-mist) by image analyzing system. Cytologia 71, 1–4. doi: 10.1508/cytologia.71.1
Gilot-Delhalle, J., Degraeve, N., and Moutschen, J. (1976). Cytotaxonomic investigation of the genus Nigella (Helleboreae) with C-banding techniques. Caryologia 29, 139–154. doi: 10.1080/00087114.1976.10796656
Hasterok, R., Wolny, E., Hosiawa, M., Kowalczyk, M., Kulak-Ksiazczyk, S., Ksiazczyk, T., et al. (2006). Comparative analysis of rDNA distribution in chromosomes of various species of Brassicaceae. Ann. Bot. 97, 205–216. doi: 10.1093/aob/mcj031
He, J., Lin, S., Yu, Z., Song, A., Guan, Z., Fang, W., et al. (2021). Identification of 5S and 45S rDNA sites in Chrysanthemum species by using oligonucleotide fluorescence in situ hybridization (Oligo-FISH). Mol. Biol. Rep. 48, 21–31. doi: 10.1007/s11033-020-06102-1
Heiss, A. G., Kropf, M., Sontag, S., and Weber, A. (2011). Seed morphology of Nigella sl (Ranunculaceae): identification, diagnostic traits, and their potential phylogenetic relevance. Int. J. Plant Sci. 172, 267–284. doi: 10.1086/657676
Henikoff, S., Ahmad, K., and Malik, H. S. (2001). The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293, 1098–1102. doi: 10.1126/science.1062939
IJdo, J. W., Wells, R. A., Baldini, A., and Reeders, S. T. (1991). Improved telomere detection using a telomere repeat probe (TTAGGG)n generated by PCR. Nucleic Acids Res. 19:4780. doi: 10.1093/nar/19.17.4780
Kuznetsova, M. A., Chaban, I. A., and Sheval, E. V. (2017). Visualization of chromosome condensation in plants with large chromosomes. BMC Plant Biol. 17:153. doi: 10.1186/s12870-017-1102-7
Leitch, I., Johnston, E., Pellicer, J., Hidalgo, O., and Bennett, M. (2019). Plant DNA C-Values Database (Release 7.1, April 2019). Available online at: https://cvalues.science.kew.org/
Levan, A., Fredga, K., and Sandberg, A. A. (1964). Nomenclature for centromeric position on chromosomes. Hereditas 52, 201–220.
Levin, D. A., and Donald, A. (2002). The Role of Chromosomal Change in Plant Evolution. Oxford: Oxford University Press.
Li, R., Taylor, S., and Jenkins, G. (2001). Unravelling the phytogeny of tetraploid vicia amoena (Fabaceae) and its diploid relatives using chromosomal landmarks. Hereditas 134, 219–224. doi: 10.1111/j.1601-5223.2001.00219.x
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Malhotra, S. (2012). “Nigella,” in Handbook of Herbs and Spices, Ed K. V. Peter (Amsterdam: Elsevier), 391–416. doi: 10.1533/9780857095688.391
Martinez, J., Vargas, P., Luceno, M., and Cuadrado, A. (2010). Evolution of Iris subgenus Xiphium based on chromosome numbers, FISH of nrDNA (5S, 45S) and trnL-trnF sequence analysis. Plant Syst. Evol. 289, 223–235. doi: 10.1007/s00606-010-0345-7
Maumus, F., and Quesneville, H. (2016). Impact and insights from ancient repetitive elements in plant genomes. Curr. Opin. Plant Biol. 30, 41–46. doi: 10.1016/j.pbi.2016.01.003
Mehrotra, S., and Goyal, V. (2014). Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function. Genomics Proteomics Bioinformatics 12, 164–171. doi: 10.1016/j.gpb.2014.07.003
Mahmoudi, S., and Mirzaghaderi, G. (2021). Tools for drawing informative idiograms. BioRxiv. doi: 10.1101/2021.09.29.459870
Mishima, M., Ohmido, N., Fukui, K., and Yahara, T. (2002). Trends in site-number change of rDNA loci during polyploid evolution in Sanguisorba (Rosaceae). Chromosoma 110, 550–558. doi: 10.1007/s00412-001-0175-z
Mlinarec, J., Papeš, D. A., and Besendorfer, V. (2006). Ribosomal, telomeric and heterochromatin sequences localization in the karyotype of Anemone hortensis. Bot. J. Linnean Soc. 150, 177–186. doi: 10.1111/j.1095-8339.2006.00467.x
Mlinarec, J., Šatović, Z., Mihelj, D., Malenica, N., and Besendorfer, V. (2012). Cytogenetic and phylogenetic studies of diploid and polyploid members of tribe Anemoninae (Ranunculaceae). Plant Biol. 14, 525–536. doi: 10.1111/j.1438-8677.2011.00519.x
Mukai, Y., Endo, T., and Gill, B. (1991). Physical mapping of the 18S. 26S rRNA multigene family in common wheat: identification of a new locus. Chromosoma 100, 71–78. doi: 10.1007/BF00418239
Novák, P., Ávila Robledillo, L., Koblížková, A., Vrbová, I., Neumann, P., and Macas, J. (2017). TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 45:e111. doi: 10.1093/nar/gkx257
Novák, P., Neumann, P., and Macas, J. (2020). Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 15, 3745–3776. doi: 10.1038/s41596-020-0400-y
Novak, P., Neumann, P., Pech, J., Steinhaisl, J., and Macas, J. (2013). RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–793. doi: 10.1093/bioinformatics/btt054
Olszewska, M. J., and Osiecka, R. (1983). The relationship between 2 C DNA content, life cycle type, systematic position and the dynamics of DNA endoreplication in parenchyma nuclei during growth and differentiation of roots in some dicotyledonous herbaceous species. Biochem. Physiol. Pflanzen 178, 581–599. doi: 10.1016/S0015-3796(83)80073-0
Piégu, B., Bire, S., Arensburger, P., and Bigot, Y. (2015). A survey of transposable element classification systems–a call for a fundamental update to meet the challenge of their diversity and complexity. Mol. Phylogenet. 86, 90–109. doi: 10.1016/j.ympev.2015.03.009
Prokopowich, C. D., Gregory, T. R., and Crease, T. J. (2003). The correlation between rDNA copy number and genome size in eukaryotes. Genome 46, 48–50. doi: 10.1139/g02-103
Raab-Straube, E., von Hand, R., Hörandl, E., and Nardi, E. (2014). Ranunculaceae. Euro+Med Plantbase. Available at: http://ww2. bgbm. org/EuroPlusMed
Raskina, O., Barber, J., Nevo, E., and Belyayev, A. (2008). Repetitive DNA and chromosomal rearrangements: speciation-related events in plant genomes. Cytogenet. Genome Res. 120, 351–357. doi: 10.1159/000121084
Roa, F., and Telles, M. P. C. (2020). idiogramFISH: Idiograms with Marks and Karyotype Indices. R-Package. Version 1.15.3. Goiânia: Universidade Federal de Goiás.
Romero-Zarco, C. (1986). A new method for estimating karyotype asymmetry. Taxon 35, 526–530. doi: 10.2307/1221906
Rosato, M., Álvarez, I., Nieto Feliner, G., and Rosselló, J. A. (2017). High and uneven levels of 45S rDNA site-number variation across wild populations of a diploid plant genus (Anacyclus, Asteraceae). PLoS One 12:e0187131. doi: 10.1371/journal.pone.0187131
Saghai-Maroof, M. A., Soliman, K. M., Jorgensen, R. A., and Allard, R. (1984). Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc. Natl. Acad. Sci. U.S.A. 81, 8014–8018. doi: 10.1073/pnas.81.24.8014
Samoluk, S. S., Robledo, G., Bertioli, D., and Seijo, J. G. (2017). Evolutionary dynamics of an at-rich satellite DNA and its contribution to karyotype differentiation in wild diploid Arachis species. Mol. Genet. Genomics 292, 283–296. doi: 10.1007/s00438-016-1271-3
Schubert, I. (1984). Mobile nucleolus organizing regions (NORs) in Allium (Liliaceae s. lat.)?—Inferences from the specifity of silver staining. Plant Syst. Evol. 144, 291–305. doi: 10.1007/BF00984139
Shaker, S. S., Mohammadi, A., and Shahli, M. K. (2017). Cytological studies on some ecotypes of Nigella sativa L. in Iran. Cytologia 82, 123–126. doi: 10.1508/cytologia.82.123
Sramkó, G., Laczkó, L., Volkova, P. A., Bateman, R. M., and Mlinarec, J. (2019). Evolutionary history of the Pasque-flowers (Pulsatilla, Ranunculaceae): molecular phylogenetics, systematics and rDNA evolution. Mol. Phylogenet. Evol. 135, 45–61. doi: 10.1016/j.ympev.2019.02.015
Stebbins, G. L. (1971). Chromosomal Evolution in Higher Plants. London: Edward Arnold (Publishers) Ltd.
Tagashira, N., and Kondo, K. (2001). Chromosome phylogeny of Zamia and Ceratozamia by means of Robertsonian changes detected by fluorescence in situ hybridization (FISH) technique of rDNA. Plant Syst. Evol. 227, 145–155. doi: 10.1007/s006060170045
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tutin, T. G. (1964). “Nigella,” in Flora Europaea1, ed. T. G. Tutin, V. H. Heywood, N. A. Burges, D. H. Valentine, S. M. Walters, and D. A. Webb (Cambridge: University Press), 209–210.
Ugarkovic, D., and Plohl, M. (2002). Variation in satellite DNA profiles-causes and effects. EMBO 2, 5955–5959. doi: 10.1093/emboj/cdf612
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Varadharajan, S., Rastas, P., Löytynoja, A., Matschiner, M., Calboli, F. C. F., Guo, B., et al. (2019). Genome sequencing of the nine-spined stickleback (Pungitius pungitius) provides insights into chromosome evolution. Genome Biol. Evol. 11, 3291–3308. doi: 10.1101/741751
Wang, X., Xie, S., Zhang, Y., and Niu, L. (2012). Chromosome analysis and mapping of ribosomal genes by fluorescence in situ hybridization (FISH) in four endemic lily species (Lilium) in Qinling Mountians, China. Pak. J. Bot 44, 1319–1323.
Wicke, S., Costa, A., Muñoz, J., and Quandt, D. (2011). Restless 5S: the re-arrangement(s) and evolution of the nuclear ribosomal DNA in land plants. Mol. Phylogenet. Evol. 61, 321–332. doi: 10.1016/j.ympev.2011.06.023
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8:973. doi: 10.1038/nrg2165
Yao, X., Zhang, W., Duan, X., Yuan, Y., Zhang, R., Shan, H., et al. (2019). The making of elaborate petals in Nigella through developmental repatterning. N. Phytol. 223, 385–396. doi: 10.1111/nph.15799
Zohary, M. (1983). The genus Nigella (Ranunculaceae) — a taxonomic revision. Plant Syst. Evol. 142, 71–105. doi: 10.1007/BF00989605
Keywords: repetitive sequences, satellites, karyotype evolution, Nigella genus, repeatome analysis
Citation: Orooji F, Mirzaghaderi G, Kuo Y-T and Fuchs J (2022) Variation in the Number and Position of rDNA Loci Contributes to the Diversification and Speciation in Nigella (Ranunculaceae). Front. Plant Sci. 13:917310. doi: 10.3389/fpls.2022.917310
Received: 11 April 2022; Accepted: 16 May 2022;
Published: 09 June 2022.
Edited by:
Francesco Sunseri, Mediterranea University of Reggio Calabria, ItalyReviewed by:
Chuanliang Deng, Henan Normal University, ChinaCopyright © 2022 Orooji, Mirzaghaderi, Kuo and Fuchs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ghader Mirzaghaderi, Z2gubWlyemFnaGFkZXJpQHVvay5hYy5pcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.