 Kun-Li Xiang1,2Wei Mao3Huan-Wen Peng2,4
Kun-Li Xiang1,2Wei Mao3Huan-Wen Peng2,4 Andrey S. Erst5,6Ying-Xue Yang1*†
Andrey S. Erst5,6Ying-Xue Yang1*† Wen-Chuang He1*†
Wen-Chuang He1*† Zhi-Qiang Wu1,7*†
Zhi-Qiang Wu1,7*†- 1Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
- 2State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, China
- 3College of Ecology and Environment, Hainan University, Haikou, China
- 4College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 5Central Siberian Botanical Garden, Russian Academy of Sciences, Novosibirsk, Russia
- 6Laboratory Herbarium (TK), Tomsk State University, Tomsk, Russia
- 7Kunpeng Institute of Modern Agriculture at Foshan, Chinese Academy of Agricultural Sciences, Foshan, China
Thalictrum is a phylogenetically and economically important genus in the family Ranunculaceae, but is also regarded as one of the most challengingly difficult in plants for resolving the taxonomical and phylogenetical relationships of constituent taxa within this genus. Here, we sequenced the complete plastid genomes of two Thalictrum species using Illumina sequencing technology via de novo assembly. The two Thalictrum plastomes exhibited circular and typical quadripartite structure that was rather conserved in overall structure and the synteny of gene order. By updating the previously reported plastome annotation of other nine Thalictrum species, we found that the expansion or contraction of the inverted repeat region affect the boundary of the single-copy regions in Thalictrum plastome. We identified eight highly variable noncoding regions—infA-rps8, ccsA-ndhD, trnSUGA-psbZ, trnHGUG-psbA, rpl16-rps3, ndhG-ndhI, ndhD-psaC, and ndhJ-ndhK—that can be further used for molecular identification, phylogenetic, and phylogeographic in different species. Selective pressure and codon usage bias of all the plastid coding genes were also analyzed for the 11 species. Phylogenetic relationships showed Thalictrum is monophyly and divided into two major clades based on 11 Thalictrum plastomes. The availability of these plastomes offers valuable genetic information for accurate identification of species and taxonomy, phylogenetic resolution, and evolutionary studies of Thalictrum, and should assist with exploration and utilization of Thalictrum plants.
Introduction
Thalictrum L., comprising ca. 200 species, is a phylogenetically and economically important genus in the family Ranunculaceae (Tamura, 1995) and is worldwide with main distribution in northern temperate regions. Thalictrum plants are rich in benzylisoquinoline-derived alkaloids; at least 250 such compounds have been isolated from 60 species, and most of them show strong biological activities (Zhu and Xiao, 1991). Thalictrum plants are used in folk medicine for the treatment of many kinds of diseases by various ethnic groups of China, which has a long history (Wang and Xiao, 1979; Zhu and Xiao, 1989; Wu et al., 1998; Wang et al., 2001). In some place, roots of Thalictrum were used as substitutes for Rhizoma coptidis to treat enteritis and dysentery (Wu et al., 1998). Furthermore, bearing luxuriant foliage, extended branches, and attractive flowers, Thalictrum species have previously been mainly applied as perennial garden plants. At present, the horticultural values of Thalictrum plants, such as Thalictrum delavayi, Thalictrum reniforme, and Thalictrum grandiflorum have been widely paid attention with great commercial prospects (Wang and Xiao, 1979).
Thalictrum is taxonomically and phylogenetically regarded as one of the most challengingly difficult taxa in plants. Traditionally, Thalictrum was classified into 14 sections based on morphological traits such as leaf, flower, and fruit characteristics (Tamura, 1995). Molecular phylogenetic analyses have consistently suggested only that Thalictrum is a monophyletic group containing two major clades, based on the nuclear ribosomal internal transcribed spacer (ITS) region (ITS1, ITS2, and 5.8S) and the chloroplast DNA (cpDNA) rpl16 intron (Soza et al., 2012). Then, a revised phylogeny yielded better resolution based on nuclear ribosomal ITS region, external transcribed spacer (ETS) region, and the cpDNA 3’trnV-ndhC (trnV-ndhC) intergenic region (Soza et al., 2013). Nonetheless, none of the sections traditionally assigned to the genus (Tamura, 1995) are monophyletic (Soza et al., 2012, 2013). Moreover, numerous species and varieties in Thalictrum are poorly defined owing to insufficient field studies and lack of consistent characteristics for diagnostic methods in the literature (Wang et al., 2001). Therefore, further exploiting more stable genetic variations and effective molecular markers in Thalictrum species is greatly important for conservation and utilization of the plants from this genus.
The popularity of the ITS region for infrageneric studies within angiosperms is well-known (Baldwin et al., 1995; Hughes et al., 2006; Mort et al., 2007). Levels of ITS sequence divergence within Thalictrum are relatively high (Soza et al., 2012, 2013). However, Thalictrum exhibits an enormous range of ploidy, from 2n = 2x = 14 to 2n = 24x = 168 (Löve, 1982; Tamura, 1995), with very small chromosomes known as the T-type in Ranunculaceae (Langlet, 1927). In Thalictrum, the ITS region is often presented as more than one copy (Soza et al., 2012, 2013). Owing to their haploidy, maternal inheritance, and high conservation in gene content and genome structure, the plastomes have been popular in researches on evolutionary relationships at almost any taxonomic level in plants. Although sequence divergence among the interspecific cpDNAs is generally less than ITS (Hughes et al., 2006; Mort et al., 2007), it is necessary to utilize cpDNA regions that exhibit relatively high rates of substitution in Thalictrum. With the advent of high-throughput sequencing technologies, it is now more practical and inexpensive to obtain plastome sequences and to upgrade cp-based phylogenetics to phylogenomics.
In the present study, we sequenced the complete plastid genomes of two Thalictrum species by using the next-generation sequencing platform and performed the first comprehensive analysis of Thalictrum plastomes by combining these data with previously reported plastomes of other nine species (Park et al., 2015; He et al., 2019, 2021b; Morales-Briones et al., 2019). Our study aims were as follows: (1) to investigate global structural patterns of the 11 Thalictrum plastomes; (2) to identify the most variable regions of these plastomes as prospective DNA barcodes for future species identification; (3) to choose more effective molecular markers via reconstruction of phylogenetic relationships among the 11 Thalictrum species using various makers; and (4) to test for the presence of adaptive evolution in all genes located in the two single-copy regions, and one of the two inverted-repeat (IR) regions by analyses of selective pressure and codon usage bias. The results will provide abundant information for further species identification, phylogenetic, and phylogeographic studies on Thalictrum, and will assist in exploration and utilization of Thalictrum plants.
Materials and Methods
Sample Preparation, Sequencing, Assembly, and Annotation
The sequenced two Thalictrum species (Thalictrum minus var. hypoleucum and Thalictrum simplex) are growing in the Beijing Botanical Garden, Beijing, China. Genomic DNA was extracted from fresh leaves and purified using the Tiangen Isolation/Extraction/Purification Kit [Tiangen Biotech (Beijing) Co., Ltd.]. Short insert of 300–500 bp libraries were prepared for sequencing on the Illumina HiSeq X-Ten platform.
Before assembly of the short reads, plastome original reads were extracted by mapping all short reads to the nine plastomes as reference with BWA (Li and Durbin, 2009) and SAMtools (Danecek et al., 2021). Then the two plastomes were de novo assembled with SPAdes v3.15.2 (Bankevich et al., 2012) as described in He et al. (2021a). Highly accurate annotation of organelle genomes was performed by using the Organellar Genome GeSeq tool (Tillich et al., 2017) with subsequent manual correction. Three chloroplast genomes from Thalictrum coreanum (GenBank accession No. NC_026103), Thalictrum minus (NC_041544), and Thalictrum thalictroides (NC_039433) were used as reference sequences. The circular plastomes were visualized by using OGDRAW v1.3.1 (Greiner et al., 2019), with subsequent manual editing. We also updated the annotation of plastomes for the other 11 species in this study.
Detection and Annotation for Plastid Genomic Variations
Multiple sequence alignments of whole plastome sequences from the 11 Thalictrum species that have the representatives of the two major clades of this genus in previous studies (Soza et al., 2012, 2013), as well as Paraquilegia anemonoides and Leptopyrum fumarioides in Thalictreae as outgroups were implemented using MAFFT v7 (Katoh and Toh, 2010) with standard parameters, and further adjusted manually in Geneious v8.0.4 (Kearse et al., 2012). For comparison, the gene order and structure of the 13 plastomes were compared by using IRscope.1
To identifying hypervariable regions, the sequence alignment of Thalictrum plastomes without outgroups was subjected to a sliding window analysis in DNAsp v6.12.03 (Rozas et al., 2017) to evaluate nucleotide diversity (π) of all genes, genes without introns, and intergenic spacer (IGS) regions. Functional annotations for the nucleotide variations were conducted by using snpEff v5.1 (Cingolani, 2012).
Phylogenetic Analysis
Phylogenetic analyses of Thalictrum were performed with maximum likelihood (ML) method in RAxML v8.2.11 (Stamatakis, 2014) with 1,000 replicates under GTRGAMMA model. The analyses were carried out based on the following nine data sets, including the complete plastid DNA sequences, concatenation of 115 IGS regions, concatenation of 114 gene sequences, and six genes and/or their introns and spacers (rpl16 intron, ndhC-trnVUAC, ndhA intron, trnLUAA-trnFGAA, rpl32-trnLUAG, and rbcL) that have been employed in previous studies on Thalictrum (Soza et al., 2012, 2013; Wang et al., 2019).
Selective Pressure Analysis
Selective pressures were detected throughout the phylogenetic tree of Thalictrum for each plastid gene. Nonsynonymous (dN) and synonymous (dS) substitution rates of each plastid gene were assessed by using the CODEML program in PAML v4.9 (Yang, 2007). We tested different hypotheses via branch models, H0: the one-ratio model (m0), assumes the same dN/dS ratio (ω ratio) for all branches in the phylogeny, HA: the free-ratio model (m1) that assumes an independent ω ratio for each branch. Likelihood ratio tests were used to test each model’s fit. The double log-likelihood difference between the two models (2ΔL) was compared to a chi-square distribution with N–1 degrees of freedom, where N is the number of branches in the phylogeny (Whelan and Goldman, 1999).
Codon Usage Analysis
The program DNAsp v6.12.03 (Rozas et al., 2017) was used to examine the synonymous codon usage of 79 protein-coding genes in the plastome of Thalictrum and to calculate several related parameters such as the effective number codons (ENC), codon bias index (CBI), and relative synonymous codon usage (RSCU). The ENC and CBI are often used to evaluate codon bias at the level of an individual gene (Frank, 1990). RSCU is the observed codon frequency divided by the expected frequency. An RSCU value close to 1.0 indicates that the deviation is not significant (Sharp et al., 1986). Amino acid (AA) frequency was calculated as the percentage of codons encoding the same amino acid divided by the total codons.
Results
Genome Features
The 11 plastomes of the Thalictrum species ranged in size from 154,924 bp (T. thalictroides) to 156,258 bp (T. minus var. hypoleucum). All these plastomes displayed the typical quadripartite structure of nearly all land plants, consisting of a pair of inverted repeats (IRs, 26,273–26,521 bp) separated by a single-copy (LSC) region (84,733–85,700 bp) and a small single-copy (SSC) region (17,479–17,655 bp; Table 1). The average GC content was ~38.39%, which is almost identical with each other among the 11 complete Thalictrum plastomes. In the IR region, the GC content (43.22%) was found to be much higher than that in the LSC (36.62%) and SSC regions (32.45%). Although overall genomic structure including gene number and gene order were well-conserved (Figure 1), the 11 Thalictrum plastomes exhibited obvious differences in the IR-SC boundary regions (Figure 2). The gene ycf1 spanned the SSC-IRB region while a pseudogene fragment ψycf1 was located at the IRA region with a length range of 1,144–1,152 bp. The gene rps19 spanned the LSC-IRA region and a pseudogene fragment ψrps19 (100–122 bp) was located in the IRB region of all Thalictrum species except T. thalictroides. At the junction of IRA and SSC regions in most species, the distance between ψycf1 and ndhF ranged from 0 to 752 bp, except for that of Thalictrum foeniculaceum with an overlap region of 39 bp between ψycf1 and ndhF. At the junction of IRB and LSC regions, the distances between ψrps19 and trnH ranged from 42 to 81 bp.
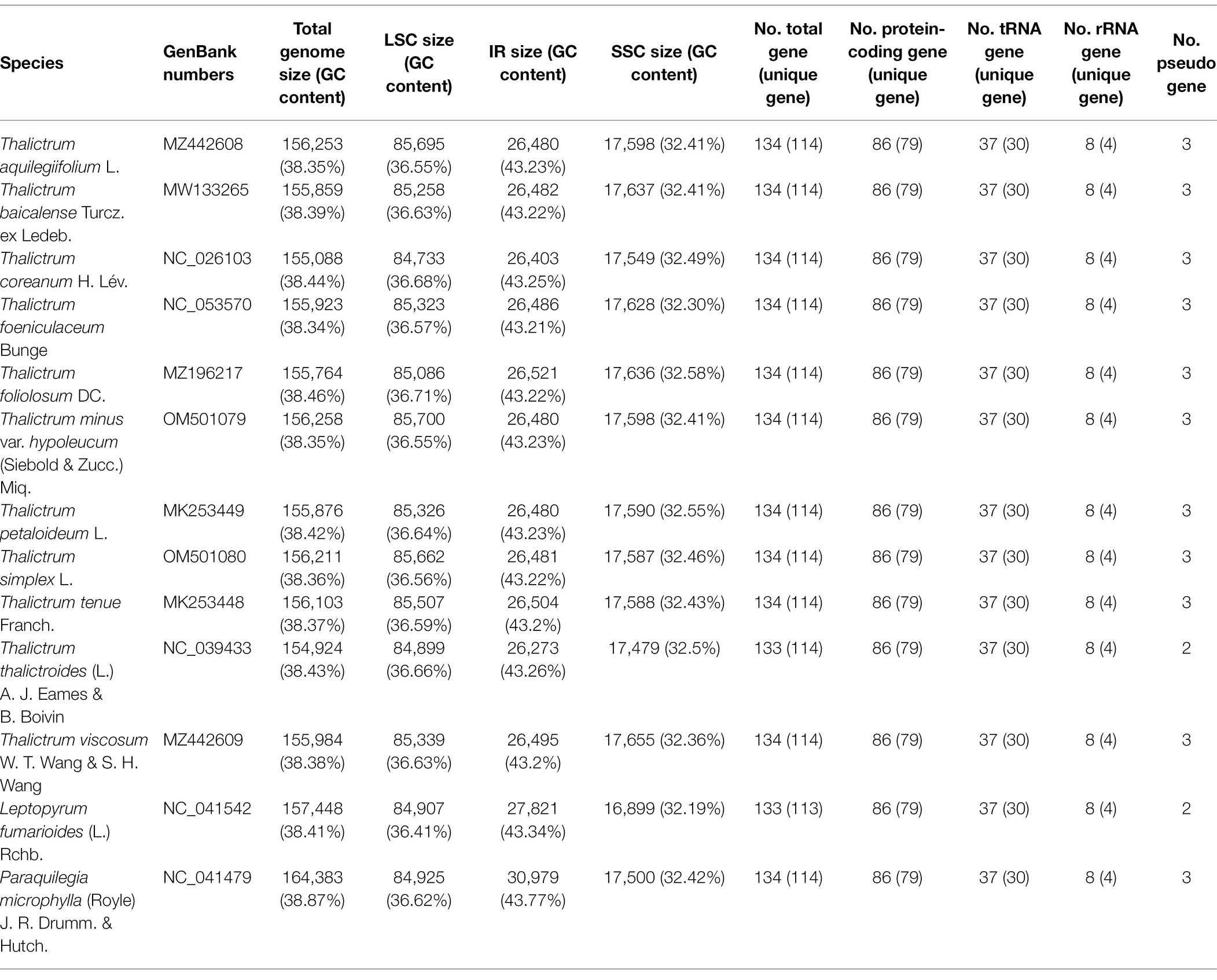
Table 1. Summary of characteristics of plastome sequnences used in the study.
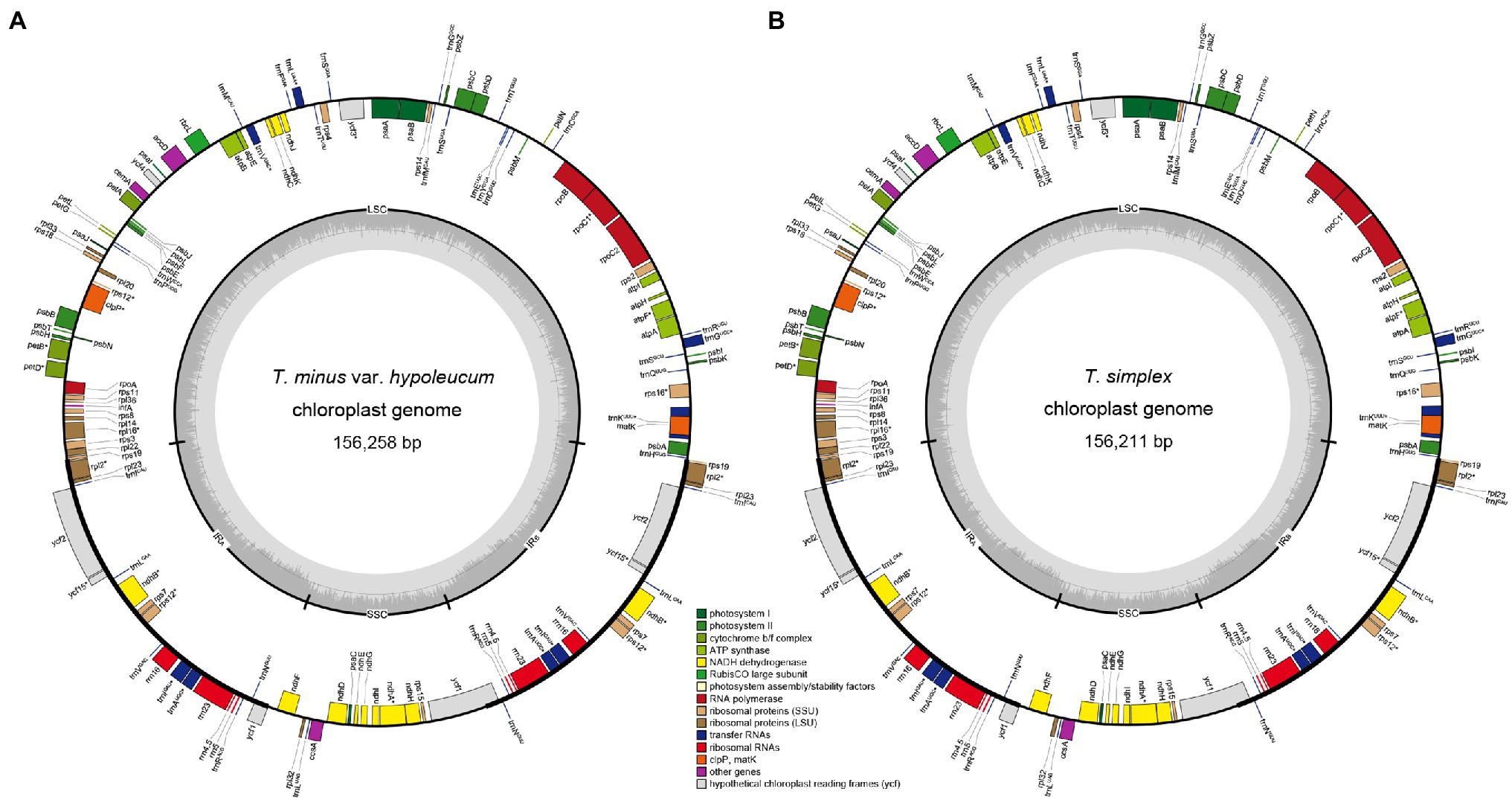
Figure 1. Plastome of Thalictrum minus var. hypoleucum (A) and Thalictrum simplex (B). The genes inside and outside of the circle are transcribed in clockwise and counterclockwise directions, respectively. Genes belonging to different functional groups are shown in different colors. The thick lines indicate the extent of the inverted repeats (IRA and IRB) that separate the genomes into small single-copy (SSC) and large single-copy (LSC) regions.

Figure 2. Comparison of LSC, inverted-repeats (IRs), and SSC junction positions among Thalictrum plastomes.
All the 11 plastomes each identically encoded 131 predicted functional genes and three pseudo genes, of which seven protein-coding genes, seven tRNA genes, four rRNA genes, and two pseudo genes were duplicated in the IR regions (Figure 1). Two introns were detected in each of two protein-coding genes (clpP and ycf3) while a single intron was detected in each of 11 protein-coding genes (atpF, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps12, rps16, and ycf15) and six tRNA genes (trnAUGC, trnGUCC, trnIGAU, trnKUUU, trnLUAA, and trnVUAC; Supplementary Table S1). Among 79 protein-coding genes, 75 contained standard AUG as the initiation codon, while three genes (ndhD, rps19, and ycf15) contained GUG instead, and the rpl2 started with ACG.
Polymorphic Variation and Hypervariable Regions
Nucleotide variations among the complete plastid genomes of the 11 Thalictrum species were identified to elucidate the level of sequence divergence (Figure 3). The aligned matrix of the 11 Thalictrum plastomes (159,334 bp) contained 2,957 single-nucleotide polymorphisms (SNPs) and 1,016 insertion-deletions (indels). The vast majority of SNPs from coding genes were functionally silent (synonymous), while 594 SNPs (43.8%) and six SNPs (0.4%), from altogether 79 coding genes, were missense and nonsense variations (Supplementary Table S2). A total of 549 simple sequence repeats (SSRs) were identified in the 11 Thalictrum plastomes with a range of 39 (Thalictrum petaloideum) to 60 (Thalictrum baicalense) SSRs were detected in each species (Supplementary Table S3), indicating rich polymorphism of the SSRs among plastomes of different species. The SSC regions showed the highest nucleotide diversity (π = 0.01381), followed by the LSC (π = 0.00803) and IR (π = 0.00154) regions. In the 114 unique genes, the nucleotide diversity for each locus ranged from 0 (e.g., rps7, rrn16, and trnCGCA) to 0.02608 (infA) with an average of 0.00438, whereby 10 regions (i.e., infA, rpl32, ycf1, rpl20, ccsA, rpl22, rpl16, rps15, rps16, and accD) had remarkably high values (π > 0.0096; Supplementary Table S1; Figure 3A). For exons in genes, the nucleotide diversity ranged from 0 (e.g., rps7, rrn16, and trnA-UGC) to 0.02608 (infA) with an average of 0.00373, while for the 115 IGS regions it ranged from 0 (e.g., atpE-atpB, rpl23-trnICAU, rrn16-trnIGAU, and trnIGAU-trnAUGC) to 0.03486 (rpoC1-rpoB) with an average of 0.01025, except for the rpoC1-rpoB (π > 0.07171) with a targetable sequence of only 5 bp. Additionally, 10 of those regions showed considerably high values (π > 0.0217; i.e., ndhF-rpl32, infA-rps8, ccsA-ndhD, rpl32-trnLUAG, trnSUGA-psbZ, trnHGUG-psbA, rpl16-rps3, ndhG-ndhI, ndhD-psaC, and ndhJ-ndhK; see Supplementary Table S4; Figure 3B).
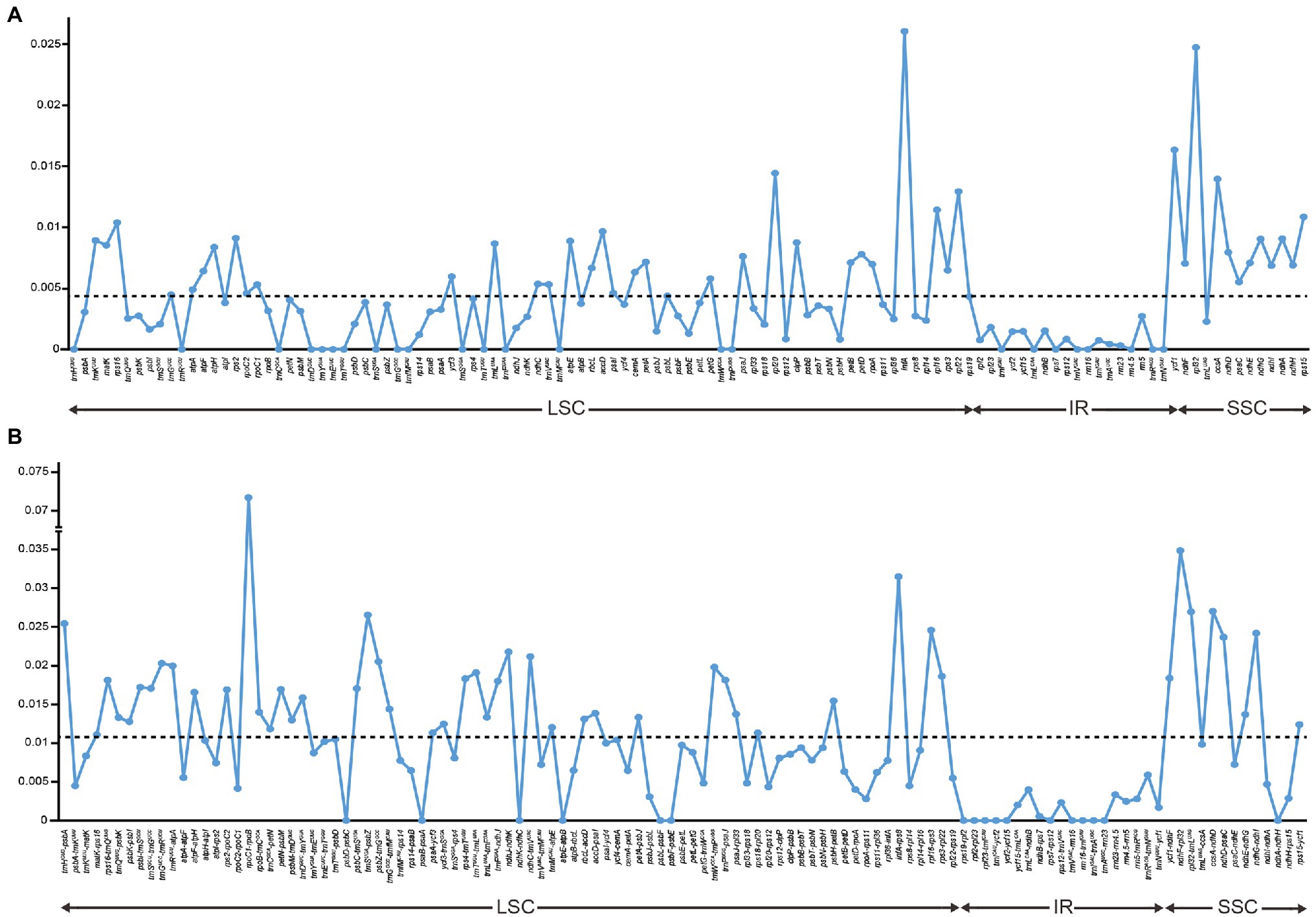
Figure 3. Comparison of nucleotide variability (π) values in Thalictrum plastomes. (A) Pi values among genes, (B) Pi values among intergenic spacer (IGS) regions. The break in the middle of the bars indicated that other regions and genes are omitted here. The dot line denoted the average value.
Phylogenetic Analysis
Three datasets, the whole complete plastid genome sequences, IGS regions, and gene sequences were constructed to investigate the phylogenetic relationships among the 11 Thalictrum species, with P. anemonoides and Leptopyrum fumarioides as two outgroups. By using ML method, three phylogenetic trees were built based on the three respective datasets, whose topologies were found to be highly concordant between one another (Figures 4A–C). The Thalictrum was strongly supported as a monophyletic group [bootstrap support (bs) = 100%], and contained two major clades that are strongly supported as sister groups: clades I (bs = 100%) and II (bs = 100%; Figures 4A–C). The resolution of previously used six molecular fragments was also evaluated for Thalictrum species. Five genes and/or their introns and spacers yielded similar results except for the rpl32-trnLUAG (Figures 4D–I). However, different supporting values were observed from the nodes based on different sequence dataset. For example, two nodes in clades II derived from the dataset of gene sequences both showed weaker supports (bs = 54% and bs = 68%; Figure 4C) than those derived from complete plastid genome sequences (bs = 100% and bs = 95%; Figure 4A) and IGS regions (bs = 100% and bs = 95%; Figure 4B). Additionally, the rpl16 intron had the strongest support within clades II (Figure 4D), while rbcL had the weakest support in them (Figure 4I). These results indicated a much stronger resolving power of complete plastid genome sequences as well as IGS and intron regions as compared to the exon regions, which may serve as a reliable source of phylogenetic information in Thalictrum.
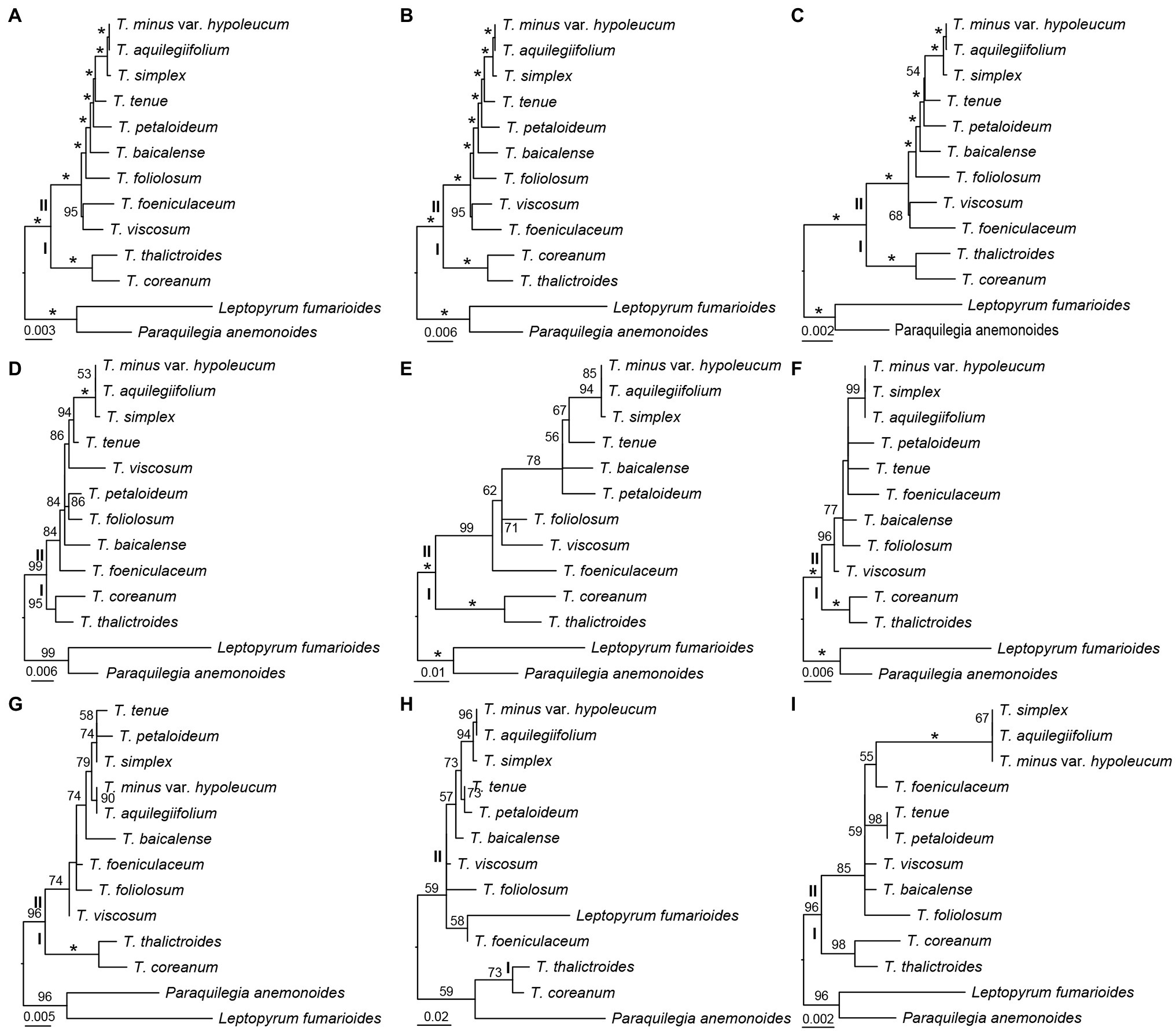
Figure 4. Phylogenetic relationships of Thalictrum inferred from maximum likelihood (ML) analysis. (A) All sequence, (B) concatenation of 115 IGS regions, (C) concatenation of 114 gene sequences, (D) rpl16 (with intron, Soza et al., 2012), (E) ndhC-trnVUAC (Soza et al., 2013), (F) ndhA intron (Wang et al., 2019), (G) trnLUAA-trnFGAA (Wang et al., 2019), (H) rpl32-trnLUAG (Wang et al., 2019), and (I) rbcL (Wang et al., 2019). The numbers above the branches indicate bootstrap support (%), and the asterisk indicates 100% bootstrap support in ML tree.
Selective Pressure and Codon Usage Analysis
Selective pressure analysis was conducted for CDS of all the 79 plastid protein-coding genes. A total of 66 genes are fit of m1 model in which atpF showed the highest ω ratio (1.13) except for rpl23 (ω = 999), while other 13 genes (psbL, psaC, rps12, rps19, petB, psbN, psbF, psaJ, psbE, rpl36, psbZ, petN, and rps7 are fit of m0 model; Table 2). Among the 66 genes, most (50/66) were located in LSC region following by IR (7/66) and SSC (9/66) regions. The values of ω are significantly different (p < 0.05) between Thalictrum species for ndhG (SSC), petA (LSC), and rpl22 (LSC) gene based on likelihood ratio tests, within some species have positive selection (e.g., ndhG in T. coreanum, T. foeniculaceum, Thalictrum foliolosum, and T. thalictroides; petA in T. minus var. hypoleucum). No genes in IR regions were detected significantly different between different species. However, 12 genes (LSC: atpF, rpl33, rpl20, rps16, rps18, petG, rpl2, petL, psbJ, psbM; IR: rpl23; SSC: rps15) were subject to positive selection in most species (median of ω > 1; see Table 2; Supplementary Tables S6, S7), although their values of ω are not significantly different between different species.
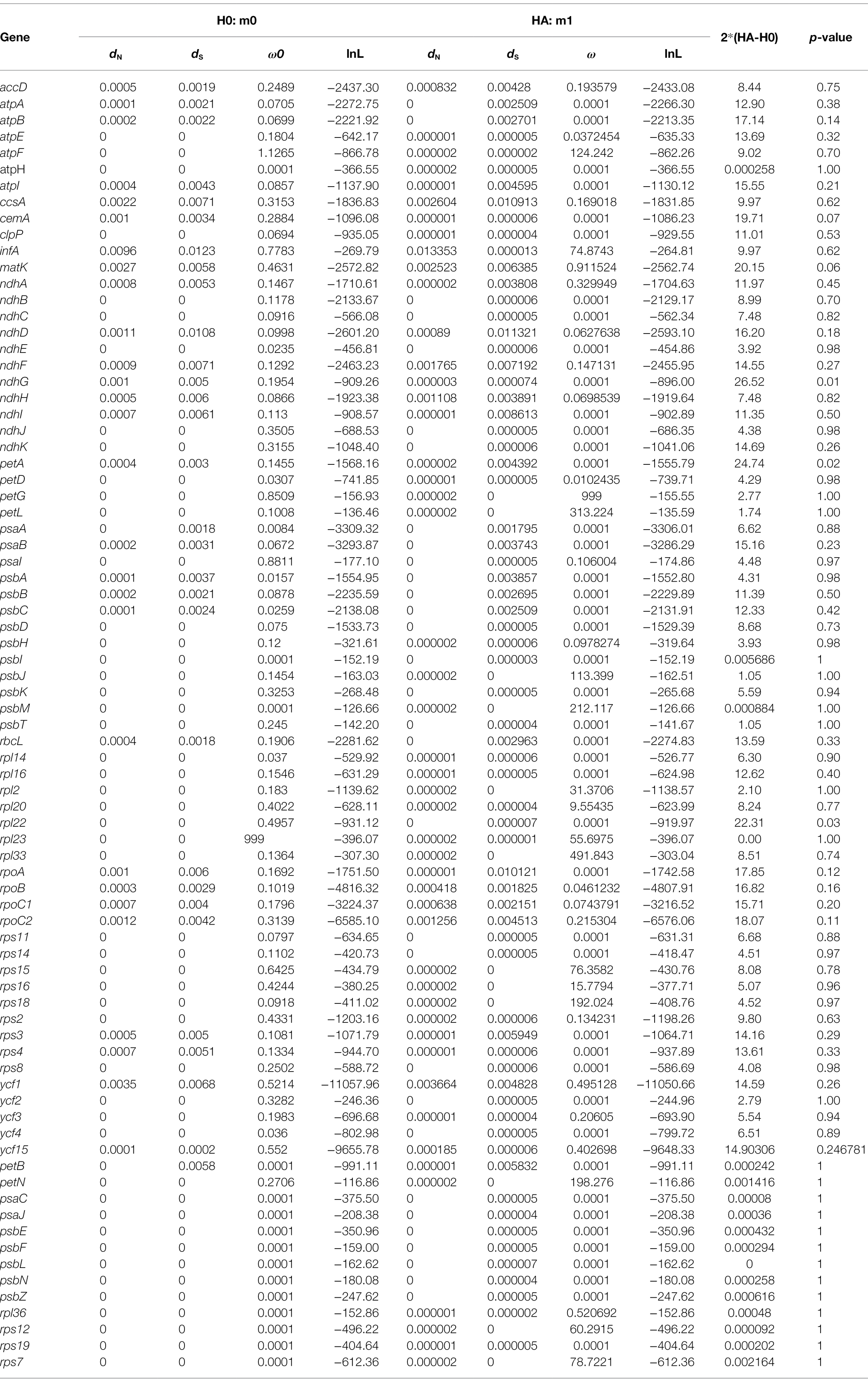
Table 2. Summary of models H0 and HA analyzed in the study. dN, dS, and ω are presented as medians.
We further analyzed the codon usage bias of the 79 protein coding genes in the plastomes of the 11 Thalictrum species. Most codons (55/64) were found to be used without bias or with only a slight bias (0.5 ≤ RSCU ≤ 1.5) in the protein-coding genes (Supplementary Table S8). The effective number of codons (ENC) and codon bias index (CBI) of all the 79 genes varied within a wide range, e.g., from 25.02 to 61.00 and from 0.28 to 0.85, respectively, with a median value of 49.0 and 0.50, respectively (Figure 5; Supplementary Figure S1; Supplementary Table S8). The data indicated that these genes were probably expressed in different levels due to their different usage frequencies of the rare and optimal codons, although they are all highly conserved in the plastomes. Most genes in SSC region (80.0%) showed relatively strong bias in the codon usage (ENC ≤ ENCmedian = 49.0 or CBI ≥ CBImedian = 0.5), while 67.2% of genes in LSC region and 50.0% of genes in IR region performed relatively strong codon usage bias. Notably, almost all genes under positively selective pressures in more than half species performed relatively strong bias in the codon usage (ENC ≤ ENCmedian = 49.0 or CBI ≥ CBImedian = 0.5), e.g., atpF featured a relatively strong codon usage bias with a low ENC of 41.61. This finding suggested that those important genes with higher expression levels may played important roles in the evolution and divergence of Thalictrum plastomes.
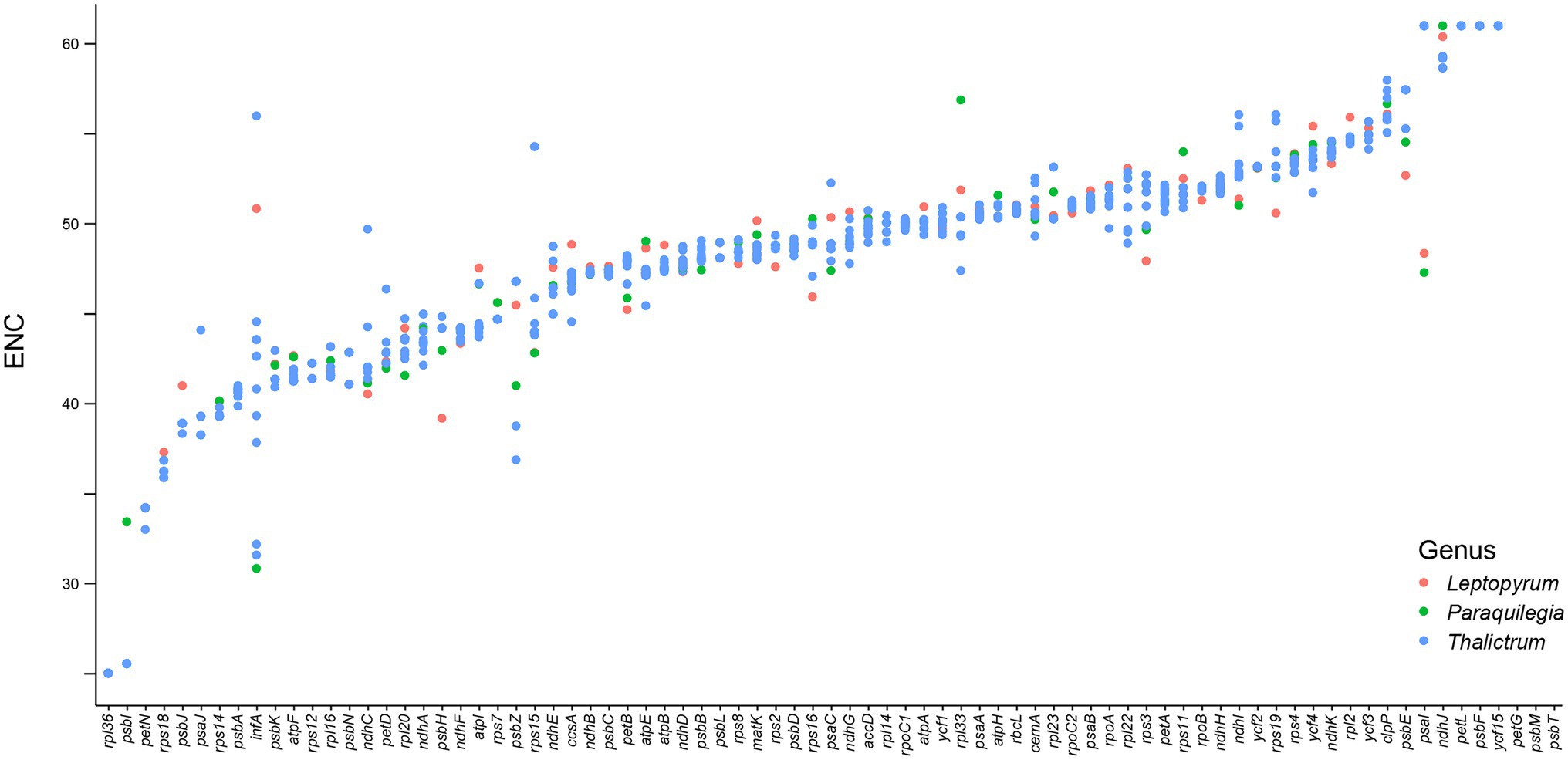
Figure 5. The effective number of codons in the study.
Discussion
Plastome Characteristics of Thalictrum
In the present study, complete plastome sequences were firstly assembled for T. minus var. hypoleucum and T. simplex in the Thalictrum genus, with a total length of 156,211 and 156,258 bp, respectively (Table 1). The two plastomes are also highly similar in overall structure and gene order when compared to the majority of previously published plastomes of other nine species in Thalictrum (Park et al., 2015; He et al., 2019, 2021b; Morales-Briones et al., 2019). However, there was obvious variation in the IR-SC boundary regions among the 11 Thalictrum plastomes (Figure 2). The variations in IR-SC boundary regions in the 11 Thalictrum plastomes led to their length variation of the four regions and whole genome sequences. The expansion and contraction of the IR-SC boundary regions was considered as a primarily mechanism causing the length variation of angiosperm plastomes (Kim and Lee, 2004). In general, such expansions or contractions of the IRs into or out of adjacent single-copy regions are frequently observed in angiosperm plastomes (e.g., Yang et al., 2016; Zhang et al., 2016; Ye et al., 2018).
Nonetheless, there are particular genes, especially ycf1, rps19, ndhF, ycf15, and ψrpl32, which deserve closer scrutiny. For instance, in various members of Thalictrum, ycf1 is duplicated, with a shorter copy (ψycf1, 1,144–1,152 bp) and a larger copy (ycf1, 5,616–5,658 bp) located at the SSC-IRA and SSC-IRB boundaries, respectively (Figure 2). Similarly, the rps19 is present as two copies including ψrps19 (100–122 bp) and rps19 (279 bp) at the SSC-IRB and SSC-IRA boundaries respectively except in T. thalictroides (Figure 2). Both shorter copies apparently resulted from incomplete duplication. Similar pseudogenizations of ycf1 and locations of ψycf1 copies are known from other plants (Yang et al., 2013, Szczecińska and Sawicki, 2015; Ye et al., 2018), and two copies of rps19 have been found in Podophylloideae (Berberidaceae; Ye et al., 2018). As for the ndhF, the coding sequence was unexpectedly terminated by a stop-codon-gained event caused by nucleotide variation of a poly-A region in eight Thalictrum species except for T. coreanum, T. foeniculaceum, and T. thalictroides. For the ycf15, an intact copy and an interrupted gene have been found in other plants, with lengths of c. 150–300 bp (Raubeson et al., 2007, Shi et al., 2013). By contrast, an interrupted ycf15 gene has been annotated in the sequenced chloroplast genomes in Thalictrum species. Additionally, ψrpl32 is incomplete because the rpl32 gene was found to be transferred to the nucleus in the ancestor of the subfamily Thalictroideae (Park et al., 2015).
Regarding the initiation codon, ndhD, rps19, and ycf15 used GUG, while rpl2 used ACG in Thalictrum. The ACG codon may be restored to the canonical start codon (AUG) by RNA editing (Hoch et al., 1991; Takenaka et al., 2013), whereas GUG has been detected in in other plastomes (Kuroda et al., 2007; Gao et al., 2009; Zhang et al., 2016).
Noncoding Regions as a Source of Phylogenetic Information in Thalictrum
Given that the nuclear-genome coded ITS region is often presented as more than one copy in Thalictrum, sequences of cpDNA intergenic spacers have been employed to uncover intraspecific variability in Thalictrum (Soza et al., 2012, 2013). The IRs usually showed lower sequence divergence than the SC regions in most of higher plants and possibly due to copy correction between IR sequences by gene conversion (Khakhlova and Bock, 2006; Zhang et al., 2016). In the present study, the whole genome and IGS regions manifested higher sequence divergence than genes did, and genes with introns showed higher sequence divergence than genes without introns in Thalictrum species (Figure 3). In general, the non-coding regions (introns and spacers) had higher variability proportions than coding regions, which was also true for most higher plants (Shaw et al., 2014; Zhang et al., 2016).
In some studies, eight noncoding regions (ndhF-rpl32, rpl32-trnLUAG, ndhC-trnV-UAC, rps16-trnQUUG, psbE-petL, trnTGGU-psbD, petA-psbJ, and rpl16 intron) have been identified as the best possible choices for low-level phylogenetic studies on angiosperms (Shaw et al., 2014). Among these regions, ndhF-rpl32, rpl32-trnLUAG, and the rpl16 intron were also identified as highly divergent loci among Thalictrum species in the present study. Nonetheless, two IGS regions related to rpl32 are not suitable as molecular markers in Thalictrum because the rpl32 gene is often transferred to the nucleus (Park et al., 2015). Aside from these loci, we also observed high nucleotide diversity in infA-rps8, ccsA-ndhD, trnSUGA-psbZ, trnHGUG-psbA, rpl16-rps3, ndhG-ndhI, and ndhD-psaC regions. Additionally, an intron of rps16 also showed highly variable here, similarly to Podophylloideae (Berberidaceae; Ye et al., 2018). These divergence hotspot regions of the 11 Thalictrum plastid genome sequences provided abundant information for developing effective molecular markers to the phylogenetic analyses and plant identification of Thalictrum species. Besides, the resolution and efficiency of chloroplast markers can be strongly affected by the length of target fragment. The rpoC1-rpoB region has a relatively high nucleotide diversity among different plastomes, but cannot be a good molecular marker as its target length is only 5 bp.
Phylogenetic Relationships
The plastid genome sequences have been utilized successfully for the phylogenetic studies on angiosperms (Jansen et al., 2007; Huang et al., 2014; Kim et al., 2015; Li et al., 2019). Our phylogenetic trees based on whole complete plastid genome sequences, 116 IGS regions, and 114 gene sequences revealed that Thalictrum contains two major clades that is consistent with previous studies (Figures 4A–C; Soza et al., 2012, 2013; Morales-Briones et al., 2019; Wang et al., 2019). However, the relationships along the backbone of the clades are not well-supported in their studies. None of the sections traditionally circumscribed for this genus (Tamura, 1995) is monophyletic. It is necessary to apply more samplings and find more efficient molecular markers for Thalictrum.
Our phylogenetic trees indicated that 116 IGS regions had stronger support than 114 gene sequences (Figures 4B,C). Additionally, the rpl16 intron—that was used by Soza et al. (2012) with high sequence divergence in the studies—showed also strong support in clades II here (Figure 4D). While the coding regions of rbcL employed by Wang et al. (2019) showed lower supports within clades II in our analysis (Figure 4I). The non-coding regions (introns and spacers) are more variable molecular markers. For the ML tree of rpl32-trnLUAG used by Wang et al. (2019), the outgroups are embedded in Thalictrum probably because the matrix of rpl32-trnLUAG contained lots of indels (Figure 4H). The rpl32 gene is often transfers to the nucleus (Park et al., 2015) that make the ndhF-rpl32, rpl32-trnLUAG, and rpl32 regions not reliable to be markers for phylogeny in Thalictrum.
Positive Selection in Different Genes
It is believed that selection is the most probable components of the evolutionary forces acting on most highly expressed genes, although all genes are basically subjected to a certain degree of natural selection (Gouy and Gautier, 1982; Sueoka, 1999; Sharp et al., 2010). And the degeneracy of genetic code leads to the expression of variation contained in a gene through its manifestation in protein, which varied among different species (Edelman and Gally, 2001; Wan et al., 2004; Chakraborty et al., 2020). In the present study, we observed different codon usage frequency on different genes under positive pressure. For example, 12 plastid genes (atpF, rpl33, rps15, rpl20, rps16, rps18, petG, rpl2, petL, psbJ, psbM, and rpl23) were observed under positive selective pressure in most of the 11 Thalictrum species among which 11 showed relatively higher CBI values (>0.5) suggesting high expression level in vivo; while three plastid genes that are relative with NADH oxidoreductase (ndhG), cytochrome b6/f complex (petA), and ribosomal proteins (rpl22) were observed under significantly strong positive selective pressure (p < 0.05 based on likelihood ratio tests) in only 1–4 Thalictrum species, showing relatively lower CBI (<0.5). The former and latter genes performed different codon usage bias suggesting different expression levels due to different usage frequency of the rare and optimal codons, which could further affected the functional patterns of those genes during their evolution process. Additionally, it also indicated potential functional divergence among plastid genomes of different Thalictrum species, according to abundant differences observed between selective pressures and usage codon frequencies for different plastid genes in these species.
Conclusion
This is the first report to describe a comprehensive landscape of plastomic variations among Thalictrum species on the basis of 11 complete plastomes. Comparison between these plastomes uncovered not only high similarities in overall structure, gene order, and content but also some structural variations caused by the expansion or contraction of the IR regions into or out of adjacent single-copy regions. DNA sequence divergence across 11 Thalictrum plastomes revealed that infA-rps8, ccsA-ndhD, trnSUGA-psbZ, trnHGUG-psbA, rpl16-rps3, ndhG-ndhI, ndhD-psaC, and ndhJ-ndhK are among the fastest-evolving loci and are promising molecular markers. Therefore, these highly variable loci should be valuable for future phylogenetic and phylogeographic studies on Thalictrum. Our phylogenomic analyses based on whole complete plastid genome sequences, 116 IGS regions and 114 gene sequences were all supported the monophyly of Thalictrum and two major clades within this genus. Furthermore, among 79 plastome-derived protein-coding genes (CDSs), 15 genes were identified as fast evolving genes, which were all proved to be under positive selection but showed different bias in their codon usage frequencies. Overall, our results demonstrate the ability of plastid phylogenomics to improve phylogenetic resolution, and will expand the understanding of plastid gene evolution in Thalictrum.
Data Availability Statement
All raw sequencing reads generated in the study have been deposited in NCBI under the BioProject accession PRJNA817687. The complete sequences and annotations of plastomes have also been deposited at GenBank under the accessions OM501079 and OM501080. The updated annotations of plastomes for the other 11 species in this study have been deposited to the Figshare online database (https://doi.org/10.6084/m9.figshare.19108097.v1).
Author Contributions
W-CH and Z-QW conceived the research. H-WP carried out taxon sampling and generated all the data. K-LX and W-CH performed the data analyses. K-LX, W-CH, and WM wrote the manuscript with help from Z-QW. Y-XY revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the China Postdoctoral Science Foundation (grant number 2021M703540), the Training of Excellent Science and Technology Innovation talents in Shenzhen-Basic Research on Outstanding Youth (grant number RCYX20200714114538196), the National Natural Science Foundation of China (grant number 32011530072), the Initial fund of Shenzhen Agricultural Genome Research Institute, Chinese Academy of Agricultural Sciences (grant number SJXW19073), and the Russian Science Foundation [grant number 19-74-10082 (preparation of material)], within state assignments for CSBG SB RAS [grant number АААА-А21-121011290024-5 (study of herbarium collections)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful for Jian-Fei Ye for kind advices on identification of the species used in the present study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.897843/full#supplementary-material
Footnotes
References
Baldwin, B. G., Sanderson, M. J., Porter, J. M., Wojciechowski, M. F., Campbell, C. S., and Donoghue, M. J. (1995). The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann. Missouri Bot. Gard. 82, 247–277. doi: 10.2307/2399880
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Chakraborty, S., Yengkhom, S., and Uddin, A. (2020). Analysis of codon usage bias of chloroplast genes in Oryza species. Planta 252:67. doi: 10.1007/s00425-020-03470-7
Cingolani, P. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Flying 6, 80–92. doi: 10.4161/fly.19695
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, 1–4. doi: 10.1093/gigascience/giab008
Edelman, G. M., and Gally, J. A. (2001). Degeneracy and complexity in biological systems. Proc. Natl. Acad. Sci. U. S. A. 98, 13763–13768. doi: 10.1073/pnas.231499798
Frank, W. (1990). The ‘effective number of codons’ used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Gao, L., Yi, X., Yang, Y. X., Su, Y. J., and Wang, T. (2009). Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 9:130. doi: 10.1186/1471-2148-9-130
Gouy, M., and Gautier, C. (1982). Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 10, 7055–7074. doi: 10.1093/nar/10.22.7055
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
He, W., Chen, C., Xiang, K., Wang, J., Zheng, P., Tembrock, L. R., et al. (2021a). The history and diversity of rice domestication as resolved from 1464 complete plastid genomes. Front. Plant Sci. 12:781793. doi: 10.3389/fpls.2021.781793
He, Y., Wang, R., Gai, X., Lin, P., and Wang, J. (2021b). The complete chloroplast genome of Thalictrum baicalense turcz. ex ledeb. Mitochondrial DNA B Resour. 6, 437–438. doi: 10.1080/23802359.2020.1870896
He, J., Yao, M., Lyu, R. D., Lin, L. L., Liu, H. J., Pei, L. Y., et al. (2019). Structural variation of the complete chloroplast genome and plastid phylogenomics of the genus Asteropyrum (Ranunculaceae). Sci. Rep. 9:15285. doi: 10.1038/s41598-019-51601-2
Hoch, B., Maier, R. M., Appel, K., Igloi, G. L., and Kössel, H. (1991). Editing of a chloroplast mRNA by creation of an initiation codon. Nature 353, 178–180. doi: 10.1038/353178a0
Huang, H, Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Hughes, C. E., Eastwood, R. J., and Bailey, C. D. (2006). From famine to feast? Selecting nuclear DNA sequence loci for plant species-level phylogeny reconstruction. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 361, 211–225. doi: 10.1098/rstb.2005.1735
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Leebens-Mack, J., Müller, K. F., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Katoh, K., and Toh, H. (2010). Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900. doi: 10.1093/bioinformatics/btq224
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Khakhlova, O., and Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, K., Lee, S.-C., Lee, J., Yu, Y., Yang, K., Choi, B.-S., et al. (2015). Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 5:15655. doi: 10.1038/srep15655
Kuroda, H., Suzuki, H., Kusumegi, T., Hirose, T., Yukawa, Y., and Sugiura, M. (2007). Translation of psbC mRNAs starts from the downstream GUG, not the upstream AUG, and requires the extended Shine–Dalgarno sequence in tobacco chloroplasts. Plant Cell Physiol. 48, 1374–1378. doi: 10.1093/pcp/pcm097
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H. T., Yi, T. S., Gao, M. L., Ma, P. F., Zhang, T., Yang, J. B., et al. (2019). Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants. 5, 461–470. doi: 10.1038/s41477-019-0421-0
Löve, Á. (1982). IOPB chromosome number reports LXXIV. Taxon 31, 119–128. doi: 10.1002/j.1996-8175.1982.tb02346.x
Morales-Briones, D. F., Arias, T., Di Stilio, V. S., and Tank, D. C. (2019). Chloroplast primers for clade-wide phylogenetic studies of Thalictrum. Appl. Plant Sci. 7:e11294. doi: 10.1002/aps3.11294
Mort, M. E., Archibald, J. K., Randle, C. P., Levsen, N. D., O’Leary, T. R., Topalov, K., et al. (2007). Inferring phylogeny at low taxonomic levels: utility of rapidly evolving cpDNA and nuclear ITS loci. Am. J. Bot. 94, 173–183. doi: 10.3732/ajb.94.2.173
Park, S., Jansen, R. K., and Park, S. (2015). Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol. 15:40. doi: 10.1186/s12870-015-0432-6
Raubeson, L. A., Peery, R., Chumley, T., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8, 174–200. doi: 10.1186/1471-2164-8-174
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Librado, P. G., Ramos-Onsins, S. E., and Sánchez-Gracia, A. (2017). DnaSP v6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sharp, P. M., Emery, L. R., and Zeng, K. (2010). Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 365, 1203–1212. doi: 10.1098/rstb.2009.0305
Sharp, P. M., Tuohy, T. M., and Mosurski, K. R. (1986). Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14, 5125–5143. doi: 10.1093/nar/14.13.5125
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M., and Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Shi, C., Liu, Y., Huang, H., Xia, E. H., Zhang, H. B., and Gao, L. Z. (2013). Contradiction between plastid gene transcription and function due to complex posttranscriptional splicing: an exemplary study of ycf15 function and evolution in angiosperms. PLoS One 8:e59620. doi: 10.1371/journal.pone.0059620
Soza, V. L., Brunet, J., Liston, A., Smith, P. S., and Di Stilio, V. S. (2012). Phylogenetic insights into the correlates of dioecy in meadow-rues (Thalictrum, Ranunculaceae). Mol. Phylogenet. Evol. 63, 180–192. doi: 10.1016/j.ympev.2012.01.009
Soza, V. L., Haworth, K. L., and Di Stilio, V. S. (2013). Timing and consequences of recurrent polyploidy in meadow-rues (Thalictrum, Ranunculaceae). Mol. Biol. Evol. 30, 1940–1954. doi: 10.1093/molbev/mst101
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sueoka, N. (1999). Two aspects of DNA base composition: G + C content and translation-coupled deviation from intra-strand rule of A = T and G = C. J. Mol. Evol. 49, 49–62. doi: 10.1007/PL00006534
Szczecińska, M., and Sawicki, J. (2015). Genomic resources of three Pulsatilla species reveal evolutionary hotspots, species-specific sites and variable plastid structure in the family Ranunculaceae. Int. J. Mol. Sci. 16, 22258–22279. doi: 10.3390/ijms160922258
Takenaka, M., Zehrmann, A., Verbitskiy, D., Härtel, B., and Brennicke, A. (2013). RNA editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352. doi: 10.1146/annurev-genet-111212-133519
Tamura, M. (1995). “Ranunculaceae,” in Die Naturlichen Pflanzenfamilien. Vol. 17a. ed. P. Hiepko (Germany: Duncker & Humblot, Berlin), 223–497.
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Wan, X. F., Xu, D., Kleinhofs, A., and Zhou, J. (2004). Quantitative relationship between synonymous codon usage bias and GC composition across unicellular genomes. BMC Evol. Biol. 4:19. doi: 10.1186/1471-2148-4-19
Wang, T. N., Clifford, M. R., Martínez-Gómez, J., Johnson, J. C., Riffell, J. A., and Di Stilio, V. S. (2019). Scent matters: differential contribution of scent to insect response in flowers with insect vs. wind pollination traits. Ann. Bot. 123, 289–301. doi: 10.1093/aob/mcy131
Wang, W. C., Fu, D. Z., Li, L. Q., Bartholomew, B., Brach, A. R., Dutton, B. E., et al. (2001). “Ranunculaceae,” in Flora of China. Vol. 6. eds. Z. Y. Wu, P. H. Raven, and D. Y. Hong (Beijing: Science Press), 133–438.
Wang, W. C., and Xiao, P. G. (1979). “Ranunculaceae,” in Flora Reipublicae Popularis Sinicae. Vol. 27. (Beijing: Science Press), 502–592.
Whelan, S., and Goldman, N. (1999). Distributions of statistics used for the comparison of models of sequence evolution in phylogenetics. Mol. Biol. Evol. 16, 1292–1299. doi: 10.1093/oxfordjournals.molbev.a026219
Wu, Z. Y., Zhou, T. Y., and Xiao, P. G. (1998). Xin Hua Compendium of Materia Medica. Shanghai: Science and Technology Press. 1, 133–139.
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, J. B., Yang, S. X., Li, H. T., Yang, J., and Li, D. Z. (2013). Comparative chloroplast genomes of Camellia species. PLoS One 8:e73053. doi: 10.1371/journal.pone.0073053
Yang, Y., Zhou, T., Duan, D., Yang, J., Feng, L., and Zhao, G. (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 7:959. doi: 10.3389/fpls.2016.00959
Ye, W., Yap, Z., Li, P., Comes, H. P., and Qiu, Y. X. (2018). Plastome organization, genome-based phylogeny and evolution of plastid genes in Podophylloideae (Berberidaceae). Mol. Phylogenet. Evol. 127, 978–987. doi: 10.1016/j.ympev.2018.07.001
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zhu, M., and Xiao, P. G. (1989). Study on resource utilization of germander (Thalictrum). Chin. Trad. Herb Drugs 20, 29–31.
Keywords: Thalictrum, plastid genome, genome structure, molecular markers, phylogeny
Citation: Xiang K-L, Mao W, Peng H-W, Erst AS, Yang Y-X, He W-C and Wu Z-Q (2022) Organization, Phylogenetic Marker Exploitation, and Gene Evolution in the Plastome of Thalictrum (Ranunculaceae). Front. Plant Sci. 13:897843. doi: 10.3389/fpls.2022.897843
Edited by:
Jianyu Zhou, Nankai University, ChinaReviewed by:
Xiaoguo Xiang, Nanchang University, ChinaWeishu Fan, Kunming Institute of Botany (CAS), China
Copyright © 2022 Xiang, Mao, Peng, Erst, Yang, He and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Qiang Wu, d3V6aGlxaWFuZ0BjYWFzLmNu; Wen-Chuang He, aGV3ZW5jaHVhbmdAY2Fhcy5jbg==; Ying-Xue Yang, eXl4dWUzMkAxNjMuY29t
†These authors have contributed equally to this work and share last authorship