
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 25 April 2022
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.890980
This article is part of the Research TopicApproaches and Applications in Plant Genome Assembly and Sequence AnalysisView all 13 articles
 Bing Zhang1,2Si Chen2Jianxiu Liu3Yong-Bin Yan1Jingbo Chen3Dandan Li3Jin-Yuan Liu1*
Bing Zhang1,2Si Chen2Jianxiu Liu3Yong-Bin Yan1Jingbo Chen3Dandan Li3Jin-Yuan Liu1*Common bermudagrass (Cynodon dactylon L.) is an important perennial warm-season turfgrass species with great economic value. However, the reference genome is still deficient in C. dactylon, which severely impedes basic studies and breeding studies. In this study, a high-quality haplotype-resolved genome of C. dactylon cultivar Yangjiang was successfully assembled using a combination of multiple sequencing strategies. The assembled genome is approximately 1.01 Gb in size and is comprised of 36 pseudo chromosomes belonging to four haplotypes. In total, 76,879 protein-coding genes and 529,092 repeat sequences were annotated in the assembled genome. Evolution analysis indicated that C. dactylon underwent two rounds of whole-genome duplication events, whereas syntenic and transcriptome analysis revealed that global subgenome dominance was absent among the four haplotypes. Genome-wide gene family analyses further indicated that homologous recombination-regulating genes and tiller-angle-regulating genes all showed an adaptive evolution in C. dactylon, providing insights into genome-scale regulation of polyploid genome stability and prostrate growth. These results not only facilitate a better understanding of the complex genome composition and unique plant architectural characteristics of common bermudagrass, but also offer a valuable resource for comparative genome analyses of turfgrasses and other plant species.
Common bermudagrass (Cynodon dactylon L., 2n = 4x = 36) is an important warm-season turfgrass species and is widely used to produce beautiful and uniform turf for public parks, home lawns, golf courses, and sport fields in warm regions around the world (Yang et al., 2018; Zhang et al., 2018a). In some regions, C. dactylon is also used as forage, medicinal, and energy plants (Hill et al., 2001; Nagori and Solanki, 2011; Xu et al., 2011). Since its origination from Africa or Indo-Malaysian, C. dactylon was spread to tropical and subtropical areas worldwidely (Kneebone, 1966; Harlan and de Wet, 1969). As a cross-pollinating plant, wild germplasms of C. dactylon collected at different regions usually show enormous genetical and morphological variations (Wu et al., 2004, 2007; Farsani et al., 2012; Tan et al., 2014; Zheng et al., 2017). Karyotype and molecular marker analyses not only revealed that polyploidy and aneuploidy events exist in C. dactylon but also pointed out the genome of C. dactylon is highly heterozygous (Wu et al., 2006; Chaves et al., 2019; Grossman et al., 2021). These characteristics make C. dactylon an interesting plant material to investigate genome stability, variability, and evolution (Khanal et al., 2017).
Unlike domesticated cereal grasses including rice, wheat, maize, and sorghum, C. dactylon has typical plant architectural characteristics of wild grasses that its stems are differentiated into shoots, stolons, and rhizomes (Dong and de Kroon, 1994; Zhang et al., 2019; Ma et al., 2021). Shoots grow erectly and are widely seen in other plants, whereas stolons and rhizomes are two types of prostrate stems that grow aboveground and underground, respectively (Guo et al., 2021). Through regeneration of new seedlings at stolon nodes, C. dactylon plants are asexually reproduced in a colonial growth mode (Zhang and Liu, 2018). The high efficiency to build turf using commercial C. dactylon cultivars is mainly derived from this virtue. During cold days in winter, the aboveground parts of common bermudagrass plants withered and died, whereas the underground rhizomes remain alive and new plants will regenerate from rhizome nodes at warm days next year (Satorre et al., 1996). By repeating the cycle of growth at aboveground and dormancy at underground, C. dactylon maintains a perennial life style, which also contribute to its usage as an eminent turfgrass. Development of asexual reproductive and perennial versions of important grain crops is an attractive measure to sustainably meet the increasing global food demand (Glover et al., 2010; Ozias-Akins and Conner, 2020). Elucidating the mechanism how C. dactylon possesses its unique plant architectural characteristics could simultaneously provide new insights into turf breeding and crop improvement.
In this study, we reported a haplotype-resolved assembly of the highly heterozygous C. dactylon genome through the combined application of Pacific Biosciences (PacBio) single-molecule sequencing, Illumina paired-end sequencing, Bionano optical mapping, and chromosome conformation capture (Hi-C) technologies. With the assembled genome dataset and annotation information, we further analyzed the subgenomic composition and adaptive evolution of C. dactylon. Results of this study not only expand our understanding of genome structure and plant architectural regulation in C. dactylon, but also provide a valuable resource for genetic studies and breeding of turfgrasses.
Cynodon dactylon cultivar Yangjiang was used for genome sequencing and assembly in this study. The bermudagrass turf were grown in turfgrass plots of Yangzhou University (longitude and latitude: 32°35’N, 119°40′E; average annual temperatures: 22.4°C; average annual precipitation: 1, 106 mm; annual average sunshine hours: 1, 960 h; soil type: 80% river sand; and 20% peat soil) under routine management conditions (irrigation: keep the soil moist as required; fertilization: four times/year; and mowing: one times/month) for 3 years. Healthy leaves were randomly collected from the turf plots. Half of the leaf samples were frozen and used for de novo sequencing, whereas another half of fresh leaf samples were used for Bionano and Hi-C sequencing. Oryza sativa subspecies indica cultivar 93–11 was grown in growth chamber at 24°C under 16 h/8 h light/dark conditions.
The genome size of C. dactylon cultivar Yangjiang was estimated using flow cytometry as previously described (Zhang et al., 2020). Specifically, O. sativa cv. 93–11 with a genome size of 430 Mb was used as an internal standard. Young leaves of C. dactylon and O. sativa were homogenized on ice in Galbraith’s buffer (45 mM MgCl2, 30 mM sodium citrate, 20 mM MOPS, and 0.1% (v/v) Triton X-100, pH 7.0) with 50 μg mL−1 propidium iodide. After filtration with 40 μm nylon cell strainer (BD Biosciences, Franklin Lakes, United States), samples were analyzed on a FACSCanto™ II flow cytometer (BD Biosciences). The flow cytometry data were analyzed using BD Spectrum Viewer.
Genomic DNA was isolated from the frozen leave samples using the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). The DNA quality and concentration were tested by 1% agarose gel electrophoresis and Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, United States). Two paired-end libraries with short insert size of 270 bp and 500 bp were constructed using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, United States) and sequenced on the Illumina HiSeq X Ten platform (Illumina, San Diego, United States). The raw Illumina sequencing reads were processed with SOAPnuke v2.1.61 to remove adapters and low-quality reads (Chen et al., 2018). The obtained 161.1 Gb high-quality sequencing reads were used to generate a k-mer depth distribution curve adopting the Jellyfish v2.3.0.2 The obtained peak k-mer number (k = 27) and corresponding peak depth were calculated by GenomeScope v2.03 to estimate the genome size and heterozygosity (Marçais and Kingsford, 2011).
High-molecular weight (HMW) DNA fragments were separated from the extracted genomic DNA samples using BluePippin Size Selection System (Sage Science, Beverly, United States) through pulse-field gel electrophoresis and eight 20-kb sequencing libraries were constructed using SMRTbell Template Prep Kit (Pacific Biosciences, Menlo Park, United States) following the manufacturer’s instructions. The libraries (16 SMRT cells) were sequenced on the PacBio RSII platform (Pacific Biosciences). Contig sequences were assembled from the 151.99 Gb PacBio sequencing reads using Hifiasm v0.124 and polished by Racon v1.4.35 (Cheng et al., 2021). The Illumina sequencing reads were aligned to the assembled contigs using Bwa-mem v2.2.16 and the draft assembly was corrected by the aligned short sequences using Pilon v1.247 (Walker et al., 2014).
HMW DNA was extracted from the agarose-embedded cell nuclei fractions, which were isolated from fresh leaf samples, using the Bionano Prep™ Plant DNA Isolation Kit (Bionano Genomics, San Diego, United States) following the manufacturer’s instructions. The DNA was digested by the single-stranded nicking endonuclease Nt.BspQI, fluorescently labeled, loaded into a Saphyr Chip®, and imaged on a Saphyr Optical Genome Mapping Instrument (Bionano Genomics). The 395.4 Gb image data were filtered using a molecule length cutoff of 100 kb and a label number cutoff of 6, and assembled to 954 genome maps. To assist genome assembly, contigs obtained from the above-mentioned PacBio sequencing were transformed into in silico BspQI-digested reference genome maps and compared with the optical genome maps. The aligned and merged genome maps were further transformed into scaffold sequences using the Bionano Solve™ v3.6.1.8
Fresh leaf samples were fixed in 1% formaldehyde to maintain the 3-D structure of genome. Genomic DNA was extracted and digested with restriction endonuclease MboI. The digested DNA fragments were biotin-labeled at the ends and ligated to each other randomly. The ligated DNA was sheared into 300–600 bp fragments, blunt-end repaired, and purified using streptavidin pull-down. The purified DNA was also sequenced on the Illumina HiSeq X Ten platform, which yielded 231.38 Gb of data with 771 million paired-end reads. The paired-end reads were mapped to the assembled scaffold sequences using Juicer v1.69 to discriminate valid and invalid interaction pairs (Durand et al., 2016). The obtained 185 million valid interaction pairs (55.5 Gb data) were further used to adjust the relative locations of the scaffolds and cluster the scaffolds into pseudochromosomes using 3D-DNA10 (Kronenberg et al., 2021).
Repetitive sequences were annotated by combining the homology alignment and de novo prediction approaches (Zhang et al., 2021). For the homology alignment approach, the assembled genome sequence was blast searched against the RepBase repeat sequence collection11 using RepeatMasker v4.0.912 (Tempel, 2012). For the de novo prediction approach, five softwares, including RepeatModeler,13 PILER,14 RepeatScout,15 LTR_Finder,16 and Tandem Repeats Finder,17 were used to find the possible repeat sequences (Price et al., 2005; Flynn et al., 2020). The identified repetitive sequences were manually checked and classified according to the nomenclature system of transposons. The insertion time of different families of long-terminal repeat retrotransposons (LTR-RTs) were calculated using the formula T = k/2r, where k is the divergence distance between the 5′ LTR and 3′ LTR of intact LTR-RTs and r is the base substitution rate (1.38 × 10−8 substitutions/site/year for grasses; Ma et al., 2004). The LTR Assembly Index (LAI) scores of assembled pseudo chromosomes and whole genome were calculated using LTR_retriever v2.9.018 with default parameters (Ou et al., 2018). Putative centromeric repeat arrays were specifically identified using Tandem Repeats Finder with searching parameters “1 1 2 80 5 200 2000 -d –h” as previously described (VanBuren et al., 2020). The identified centromeric repeat array sequences were used to construct a maximum likelihood phylogenetic tree using MEGA v10.0.5 with a bootstrap of 1,000.
Protein-coding genes were identified by combining the homology alignment prediction, ab initio prediction, and transcriptome-assisted prediction approaches (Zhang et al., 2021). For the homology alignment approach, protein sequences of Arabidopsis thaliana and five grass species, including O. sativa, Brachypodium distachyon, Zea mays, Sorghum bicolor, and Oropetium thomaeum, were downloaded from the Phytozome database19 and blast searched against the assembled genome sequence to identify the homologous proteins, which were then aligned to the genome by GeneWise20 to annotate gene structures (Birney et al., 2004). Ab initio gene prediction was conducted using five softwares, including Augustus v3.4.0,21 geneid v1.4.4,22 FgeneSH,23 GlimmerHMM v3.0.4,24 and Genscan25 with default parameters (Yao et al., 2005; Nachtweide and Stanke, 2019). For transcriptome-assisted prediction, the PacBio single-molecule transcriptome sequencing data of mixed organ samples (Zhang et al., 2018a) were aligned to the assembled genome using GMAP26 and the gene structures were modeled using PASA,27 whereas Illumina transcriptome sequencing data of six different organs (Chen et al., 2021) were aligned to the genome using TopHat v2.1.128 and the gene structures were modeled using Cufflinks v2.2.129 (Wu and Watanabe, 2005; Ghosh and Chan, 2016). A non-redundant reference gene set was generated by merging the predicted genes using EVidenceModeler v1.1.130 (Haas et al., 2008). Functional annotations of the reference gene set were obtained through orthology assignment of the eggNOG v5.0 database31 using eggNOG-mapper v232 (Cantalapiedra et al., 2021). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation of the reference gene set were obtained through BLAST searching against the GO database33 and the KEGG pathway database,34 respectively. KEGG enrichment analysis were performed using KEGG-Orthology Based Annotation System (KOBAS; Xie et al., 2011).35 Transcription factors (TFs) were annotated using iTAK36 incorporated with PlantTFDB database37 (Zheng et al., 2016).
rRNA and tRNA genes were predicted using the programs Barrnap38 and tRNAscan-SE-2.0,39 respectively (Chan et al., 2021). miRNA, snoRNA, and snRNA genes were all identified by searching against the Rfam database via Infernal v1.1.440 with default parameters (Nawrocki and Eddy, 2013).
The completeness and accuracy of the assembled genome and predicted reference gene set were both assessed using the embryophyta_odb10 core gene collect (1,375 genes) of the Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.2.2 database41 (Simão et al., 2015). The number of single-copy and duplicated genes with complete coverage, genes with fragment coverage, and missing genes were all counted.
The protein sequences of A. thaliana, O. sativa, B. distachyon, Z. mays, S. bicolor, O. thomaeum, Panicum hallii, Setaria viridis, Hordeum vulgare, and Tritcum urartu were downloaded from the Phytozome database. Orthologous gene families were clustered using OrthoFinder v2.5.442 through all-against-all blast alignment of these protein sequences and predicted protein sequences of C. dactylon (Emms and Kelly, 2015). The identified 112 single-copy orthologous gene families were aligned using MUSCLE43 and the alignments of each gene family were concatenated to a super-alignment matrix. A phylogenetic tree was then constructed using OrthoFinder with A. thaliana as the outgroup. PAML v4.944 was used to estimate the divergence time of C. dactylon using recorded divergence times of other 10 species in the TimeTree database45 as calibrations (Yang, 2007).
Homologous pairs of C. dactylon proteins were identified using the all-to-all search in BLASTP v2.12.046 with an E-value cutoff of 10−5. Syntenic blocks with at least 50 collinear gene pairs were then identified using MCScanX47 with default parameters (Wang et al., 2012). The same method was used to identify the collinear blocks between C. dactylon and O. thomaeum/B. distachyon. Synonymous substitutions per site values (Ks) of syntenic gene pairs were calculated using PAML v4.9 and the distribution of Ks values was plotted to infer the time for speciation or whole-genome duplication (WGD) events using the formula T = Ks/2λ, where Ks is peak Ks value and λ is the average substitution rate (6.5 × 10−9 substitutions/site/year for grasses; Gaut et al., 1996).
The Illumina transcriptome sequencing data of six different organs of bermudagrass cultivar Yangjiang were aligned to the assembled genome using HISAT v2.1.148 with default parameters (Kim et al., 2015). The numbers of mapped reads for each genes were converted to RPKM (reads per kilobase of transcript per million mapped fragments) values. The log2 transformed RPKM values were applied to perform Hierarchical clustering using Pearson’s correlation distance (Chen et al., 2021). The significantly expressed genes were defined as RPKM value > 1, the organ-enhanced genes were defined as RPKM value is 5-fold above the average RPKM values of other organs, whereas organ-enriched genes were defined as RPKM value is 5-fold above the RPKM values of any other organs (Uhlén et al., 2016; Nautiyal et al., 2020).
To obtain ZMM (acronym for Zip1-4, Msh4-5, and Mer3) protein-coding genes in C. dactylon and other 10 plant species, ZMM genes from A. thaliana were used as baits to search against the assembled genome of C. dactylon and other plant species recorded in Phytozome database or Ensembl Plants database49 using BLASTP v2.12.0 with an E-value cutoff of 10−5. The gene copy numbers and chromosome locations of different genes were manually summarized based on their identities. For PROG1, LA1, and TAC1 genes, PROG1, LA1, and TAC1 proteins from O. sativa were used as baits to search against the assembled genome of C. dactylon and other seven species of Oryza genus recorded in Ensembl Plants database as described above. The amino acid sequences of proteins encoded by each gene families were searched against the Pfam database50 for domain comparisons (Mistry et al., 2021).
The C. dactylon cultivar Yangjiang was used for genome sequencing. As a national authorized C. dactylon cultivar., cultivar Yangjiang is a typical turf-type common bermudagrass and is widely used for turf planting in China (Zhang et al., 2018a; Supplementary Figure S1). Based on the K-mer genome survey result, the estimated genome size of C. dactylon cultivar Yangjiang is approximately 984 Mb, which is in line with the flow cytometry genome size estimation result of 1.02 Gb (Supplementary Figure S2). K-mer analysis also revealed that the genome of C. dactylon cultivar Yangjiang has a high heterozygosity (1.92%) with a repeat frequency of 56.91%.
To overcome the impact of high heterozygosity on the genome assembly, we adopted an integrated assembly strategy incorporating PacBio sequencing, Illumina sequencing, and Bionano and Hi-C techniques with the haplotype-resolving Hifiasm algorithm (Cheng et al., 2021; Supplementary Figure S3). Firstly, 151.99 Gb PacBio long reads (about 150× coverage of the genome) were de novo assembled into contigs, which were polished by 161.1 Gb Illumina paired-end reads (about 160× coverage of the genome; Supplementary Tables S1 and S2). Totally, 3,703 contigs with a N50 contig length of 2.65 Mb and a sum contig length of 1.295 Gb were obtained (Table 1). Secondly, 395.4 Gb Bionano optical maps (about 390× coverage of the genome) were used to integrate the contigs into scaffolds (Supplementary Figure S4; Supplementary Table S3). This procedure generated 241 scaffolds with a N50 scaffold length of 9.38 Mb and a sum scaffold length of 1.26 Gb (Table 1). Lastly, 231.3 Gb Hi-C data (24% useful information, about 55× coverage of the genome) were used to further cluster the scaffolds into pseudo chromosomes (Supplementary Figure S5; Supplementary Table S4). The finally obtained genome assembly (1.01 Gb) contained 36 chromosome-level superscaffolds, among which the longest and the shortest are 52.77 Mb and 14.32 Mb, respectively (Figure 1; Table 1). The assembly size was consistent with the estimated genome size. Furthermore, BUSCO analysis against the 1,375 Embryophyta gene sets indicated that 96.2% complete genes were successfully identified in the genome assembly, among which 88.1% were duplicated genes (Supplementary Table S5). These results collectively suggested that the assembled C. dactylon genome is high quality and complete.
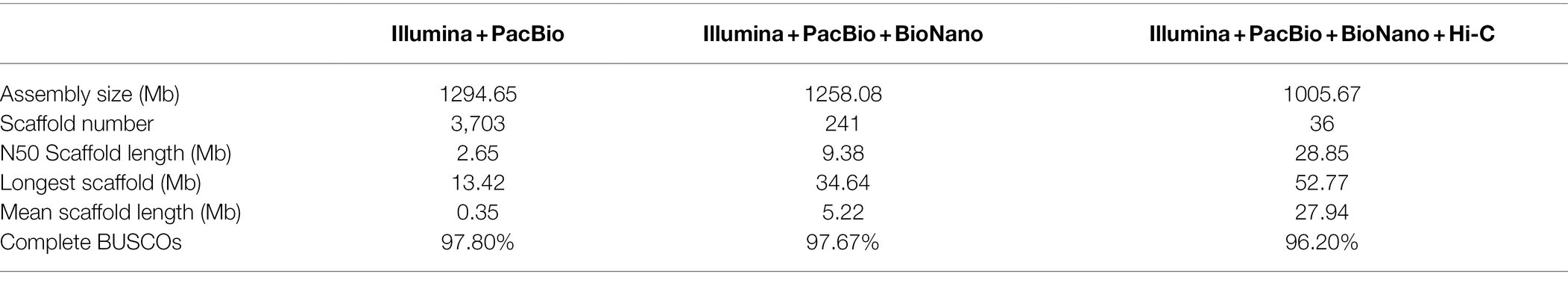
Table 1. Statistics of Cynodon dactylon genome assembly.
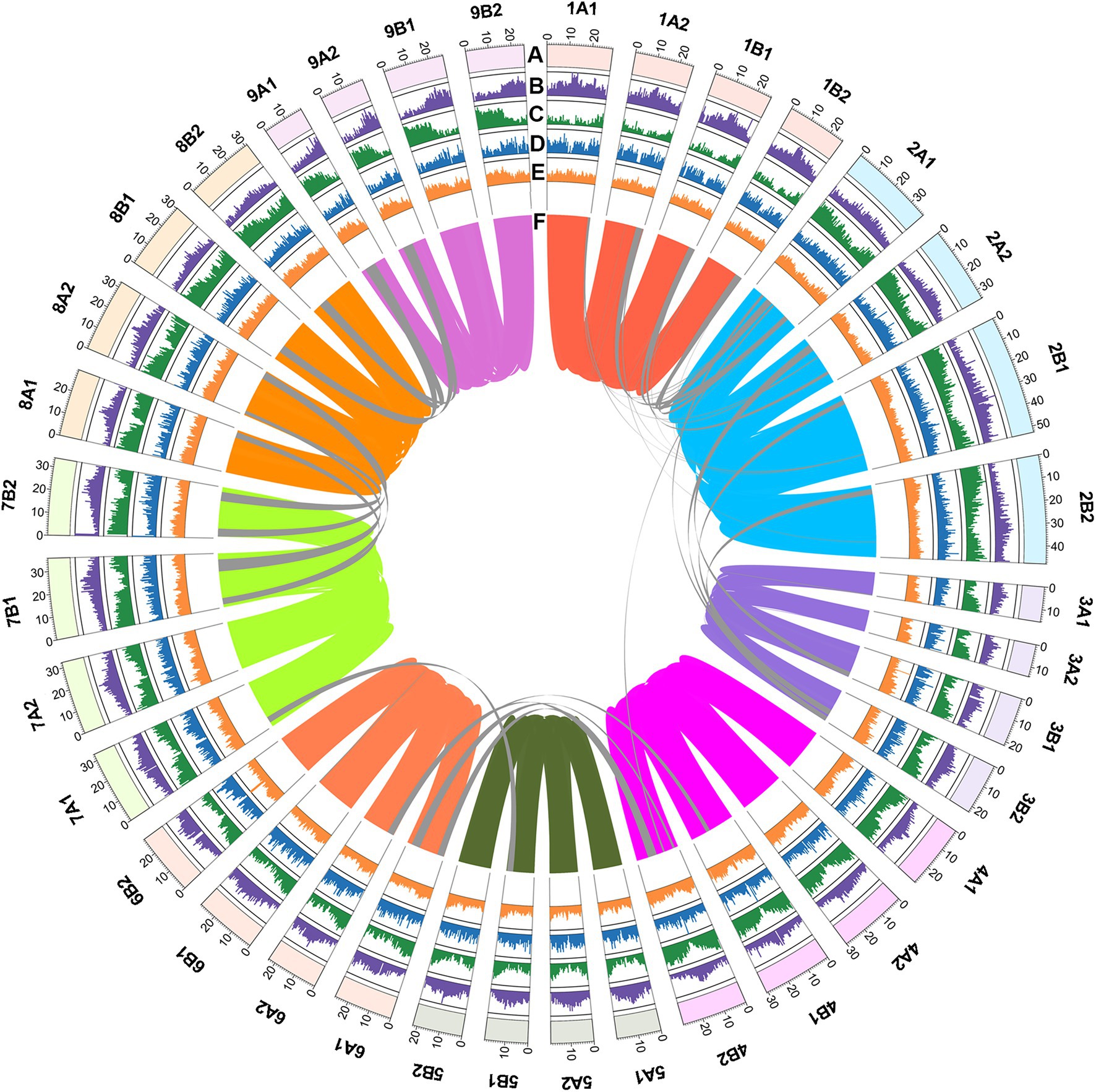
Figure 1. Genome features of C. dactylon cultivar Yangjiang. (A) Circular representation of the 36 pseudo chromosomes with scale mark labeling each 10 Mb. The density of (B) long-terminal repeat retrotransposons (LTR-RT), (C) protein-coding genes, (D) tandem repeat sequences, and (E) GC contents were calculated using 500 kb non-overlap window. (F) Inter-chromosomal synteny was illustrated with color lines.
A total of 76,879 protein-coding genes with an average gene length of 3,535 bp and an average transcript number per gene of 1.9 were successfully predicted from the assembled genome (Table 2). The predicted gene model was also evaluated by BUSCO analysis. The result indicated that 1,324 (96.3%) complete core Embryophyta genes were identified and the majority (1,272, 96.07%) was duplicated genes (Supplementary Table S5). Among the predicted 146,743 transcripts, 87.89% (128, 966) were annotated by various functional database (Supplementary Figure S6; Supplementary Table S6). Functional classification further indicated that signal transduction mechanism, post-translation modification/protein turnover/chaperones, and transcription are the top three categories containing the largest number of transcripts (Supplementary Figure S7). Specifically, 4,888 transcription factors (TFs) belonging to 65 classes were successfully identified. Compared with other grass species, gene numbers of HSF, WRKY, NF-X1, NF-YA, NF-YC, CPP, GARP-G2-like, and DDT TF families were greatly increased in common bermudagrass (Supplementary Table S7). In addition, 6,265 non-protein-coding genes were also identified, including 1349 rRNAs, 2047 tRNAs, 1025 miRNAs, 1441 snoRNAs, and 403 snRNAs (Table 2).
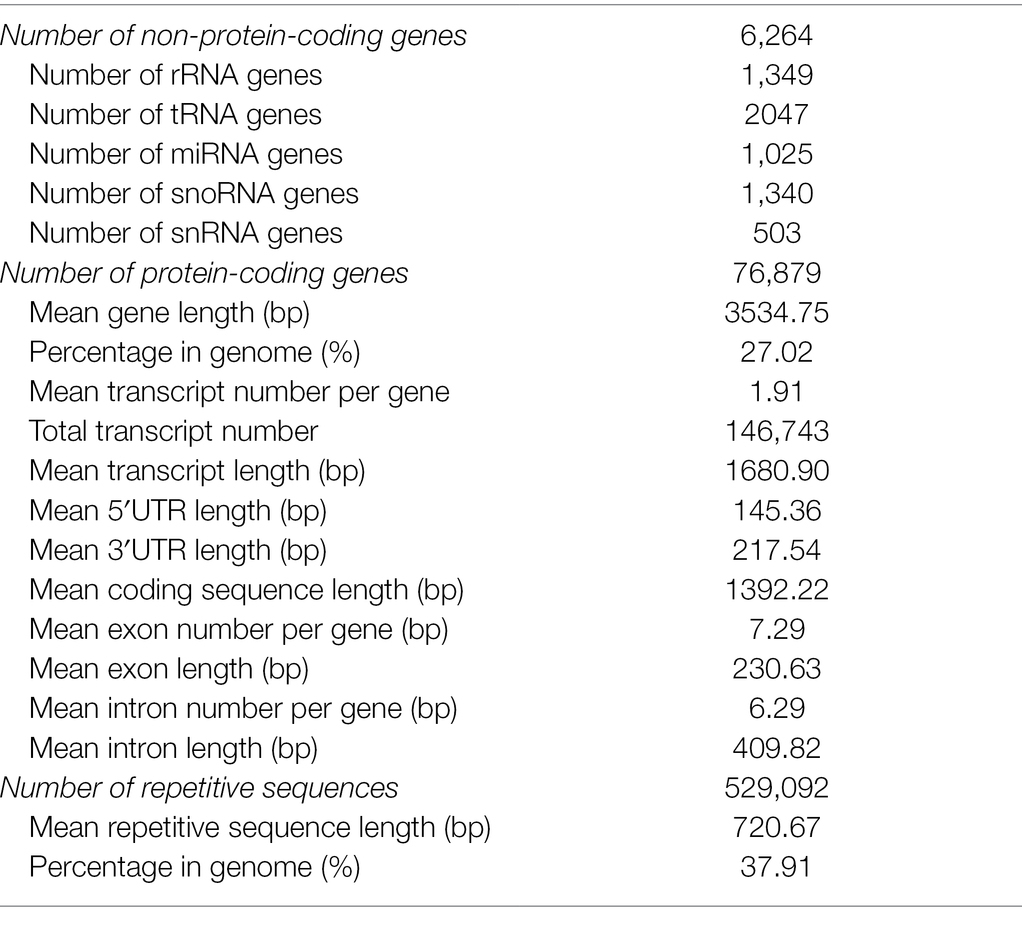
Table 2. Statistics of C. dactylon genome annotation.
Orthologous clustering of protein-coding genes of C. dactylon with other ten plant species totally identified 32,695 orthologous gene families, including 7,792 commonly shared gene families and 3,173 bermudagrass-specific gene families consisting of 9,152 genes (Supplementary Tables S8 and S9). KOBAS enrichment analysis indicated that these bermudagrass-specific genes were enriched in glutathione metabolism, zeatin biosynthesis, ubiquitin mediated proteolysis, and other eight pathways (q value <0.05; Supplementary Table S10). In agreement with the BUSCO analysis result, orthologous gene clustering further revealed that as many as 91.2% (70117) of C. dactylon genes are members of 17,632 multiple-copy gene families, which is much higher than that of other ten species (Figure 2A; Supplementary Table S8). A phylogenetic tree was constructed based on the 112 shared single-copy orthologous genes (Figure 2A). The result indicated that O. thomaeum was closest to C. dactylon and the estimated divergence time of the two species was between 17.85 to 29.19 (midvalue of 23.52) million years ago (MYA). In line with phylogenic relationships, C. dactylon shared more orthologous gene families with members of the PACMAD (acronym for Panicoideae, Aristidoideae, Chloridoideae, Micrairoideae, Arundinoideae, and Danthonioideae) clade of grasses, including O. thomaeum, S. bicolor, and S. viridis, compared with O. sativa belonging to the BEP (acronym for Bambusoideae, Ehrhartoideae, and Pooideae) clade of grasses (Supplementary Figure S8).
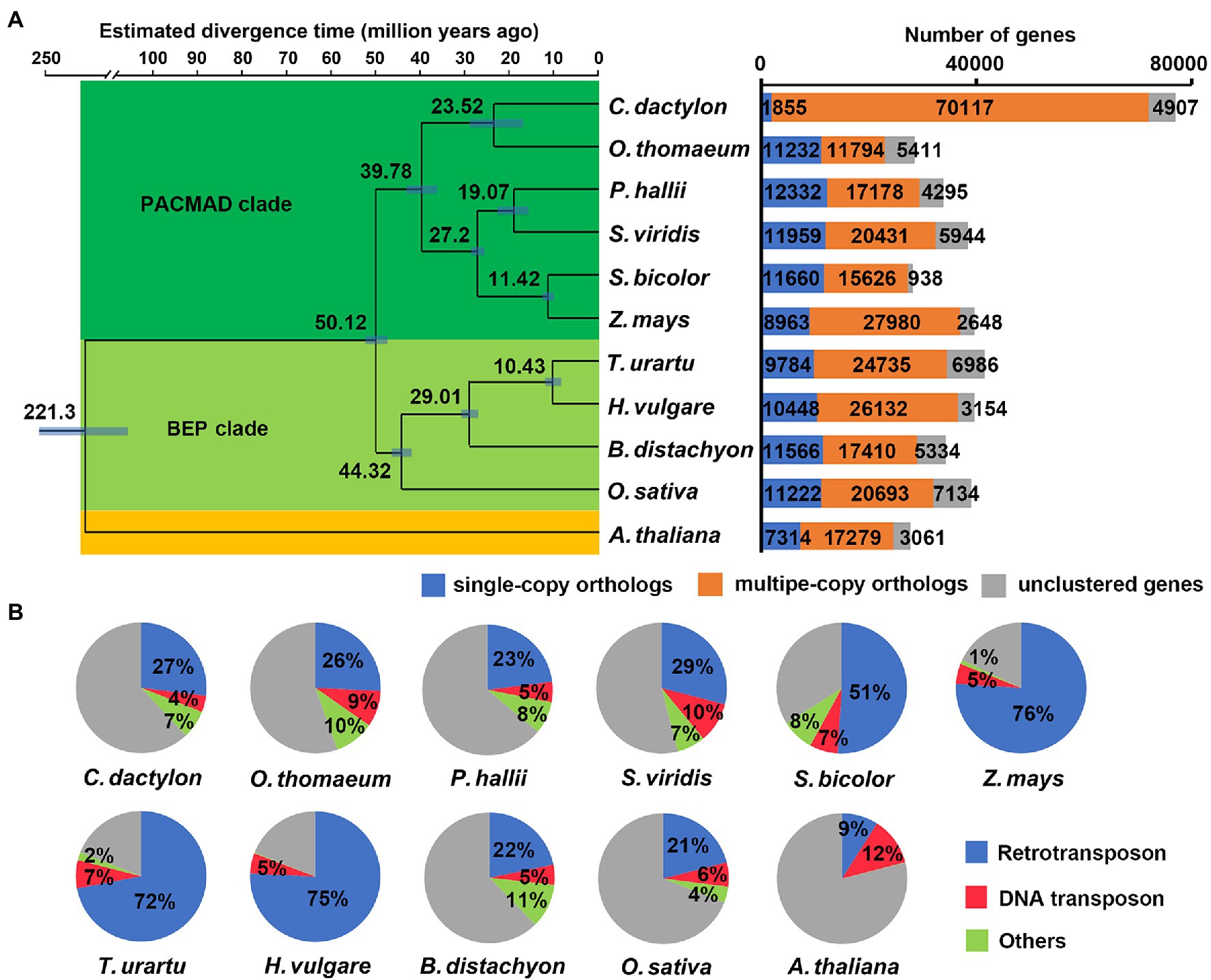
Figure 2. Comparative genomic analysis among C. dactylon and other plant species. (A) Phylogenetic relationship, divergence time, and gene family clustering of C. dactylon and other ten plant species. Left panel, Maximum parsimony (MP) species tree was constructed using protein sequences of 112 shared single-copy orthologous genes. The numbers in the brackets indicate the estimated divergence time of each node, and the blue bars show the 95% confidence interval of divergence time. All the nodes are 100% bootstrap support. Right panel, Orthologous gene families of C. dactylon and other ten plant species. (B) Comparison of repetitive sequences in C. dactylon and other ten plant species.
A total of 381.3 Mb of repetitive sequences were also annotated in the assembled C. dactylon genome (Table 2 and Supplementary Table S11). The most abundant repetitive sequences are retrotransposons (70.95% of repetitive sequences and 26.9% of genome assembly), among which LTR-RTs and non-LTR-RT represent 84.73 and 15.27%, respectively. DNA transposons make up 11.29% of the repetitive sequences (4.28% of genome assembly), whereas tandem repeats and unclassified repeat sequences account for 2.79 and 3.75% of the assembled genome, respectively. Interestingly, the total repetitive sequence content and retrotransposon content in C. dactylon (37.91 and 26.9%, respectively) were similar to those of closely related species, including O. thomaeum (45 and 26%, respectively), P. hallii (36 and 23%, respectively), and S. viridis (46 and 29%, respectively), but much lower than those of distantly related species, including Z. mays (82 and 76%, respectively), T. urartu (81 and 72%, respectively), and H. vulgare (80 and 75%, respectively; Figure 2B). It is also noteworthy that genes are unevenly distributed in different chromosomes (39.87 to 104.80 Mb−1 in density), whereas similar distributions of repetitive sequences were found on all chromosomes of C. dactylon (482.23 to 559.93 Mb−1 in density; Supplementary Table S12). The annotated LTR-RTs were further used to calculate the LAI of the assembled genome. The total LAI score of C. dactylon genome is 13.63, implying that the current assembly of C. dactylon genome reached the reference genome level (Supplementary Table S13; Ou et al., 2018).
Intra-genomic syntenic analysis totally detected 845 syntenic blocks containing 84,649 pairs of homoeologous genes in the C. dactylon genome, whereas 643 syntenic blocks containing 52,590 pairs of homoeologous genes were found between C. dactylon and O. thomaeum through inter-genomics syntenic analysis (Figure 3A and Supplementary Figure S9). Interestingly, the syntenic depth ratios of C. dactylon versus O. thomaeum and C. dactylon itself were 4:1 and 4:4, respectively, implying that C. dactylon genome is composed of four haplotypes containing the same number of chromosomes (Supplementary Figure S9). To distinguish homoeologous chromosomes from the four haplotypes of C. dactylon, putative centromeric array tandem repeat sequences were identified from the 36 chromosomes and were used to construct a maximum likelihood phylogenetic tree as previously described (Supplementary Table S14; VanBuren et al., 2020). The result indicated that the 36 centromeric array sequences showed distinguishing polymorphisms and could be clustered in four clades (Supplementary Figure S10). Based on this classification result and chromosome length variance, the four haplotypes, which were named as A1, A2, B1 and B2, respectively, were successfully resolved in the C. dactylon genome (Supplementary Table S15). In addition, syntenic analysis also revealed that chromosome 2, 3, and 10 of O. thomaeum are split and merged into chromosome 2 and 7 in four haplotypes of C. dactylon, whereas other chromosomes all have one-to-one correspondence (Figure 3A and Supplementary Figure S9).
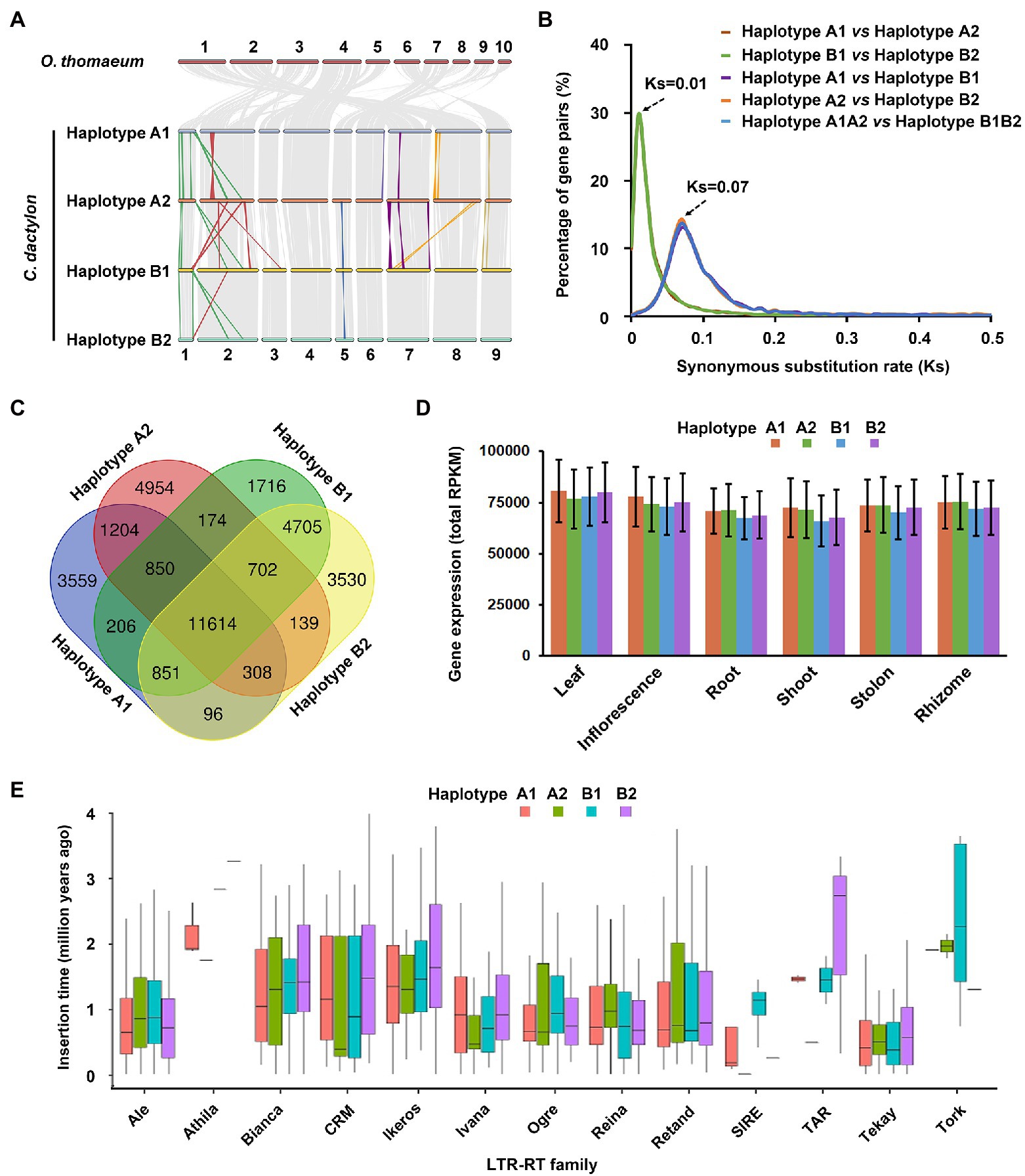
Figure 3. Subgenomic organization and variation of C. dactylon. (A) Schematic representation of syntenic genes among O. thomaeum and four haplotypes of C. dactylon. Gray lines depict homologous genome blocks. Color lines indicate inversion and translocation on the homologous chromosomes. (B) Distribution of synonymous nucleotide substitution levels (Ks) of syntenic gene pairs between different haploptypes of C. dactylon. (C) Venn diagram of alleles and orphan genes in the four haploptypes of C. dactylon. (D) Total gene expression level of the 11,614 four-copy alleles based on their relative expression level in six organs of C. dactylon. Error bars represent SE of the three sequencing replicates. (E) Box plots showing the insertion dynamics of 13 LTR-RT families in four haploptypes of C. dactylon. Box boundaries indicate the 25th and 75th percentiles of the insertion time and whiskers extend to 1.5 times the interquartile range.
Calculation of Ks of homologous gene pairs in the inter-genomic and intra-genomic synteny blocks not only confirmed the phylogenic analysis result that C. dactylon and O. thomaeum diverged at approximately 21.54 MYA (Ks = 0.28), but also indicated that two rounds of WGD events occurred in the evolutionary history of C. dactylon (Supplementary Figure S11). Specifically, the first WGD event occurred at approximately 5.38 MYA (Ks = 0.07), whereas the second WGD event occurred lately at about 0.77 MYA (Ks = 0.01; Supplementary Figure S11). Interestingly, the two WGD time points equivalent exactly to the divergence time of haplotypes A1/A2 with haplotypes B1/B2 and haplotype A1 with haplotype A2 (the same as haplotype B1 with haplotype B2), respectively (Figure 3B).
In combination with the orthologous gene clustering result, syntenic analysis totally identified 20,849 alleles in C. dactylon (Supplementary Table S16). Among these alleles, 11,614 have four allelic copies in all haplotypes, 2,711 have three allelic copies in three of four haplotypes, and 6,524 have two allelic copies in two of four haplotypes (Figure 3C and Supplementary Figure S12; Supplementary Table S17). Meanwhile, 3559, 4954, 1716, and 3530 orphan genes that exist as single-copy genes were also identified from haplotype A1, A2, B1, and B2, respectively (Figure 3C and Supplementary Figure S12; Supplementary Table S17). KOBAS enrichment analyses indicated that alleles were enriched in valine/leucine/isoleucine degradation, proteasome, brassinosteroid biosynthesis, and other eight pathways, whereas orphan genes were enriched in plant-pathogen interaction, base excision repair, DNA replication, and other nine pathways (q value <0.05; Supplementary Table S18). The expression abundance of alleles and orphan genes were further analyzed using the organ-specific transcriptome sequencing data of C. dactylon cultivar Yangjiang (Chen et al., 2021; Supplementary Table S19). The result indicated that similar portions of alleles and orphan genes in the four haplotypes were significantly expressed in six organs of bermudagrass; however, gene numbers of alleles and orphan genes enhance- or enrich-expressed in different organs, especially the three types of stems, varied greatly in the four haplotypes (Supplementary Figure S13; Supplementary Tables S20 and S21). Accordingly, the 11,614 four-copy alleles of the four haplotypes showed similar total expression abundance in the six organs (Figure 3D).
The distribution of repeat sequences in C. dactylon was also analyzed at the haplotype level. Among the four haplotypes, haplotype A2 and B1 has the minimum and maximum number of RTs, respectively (Supplementary Figure S14). By contrast, maximum number of four types of DNA transposons, including Tcl/mariner, EnSpm/CACTA, hAT, and muDR, was observed in haplotype B2, while haplotype A1 has the fewest muDR- and Helitron-type of DNA transposons (Supplementary Figure S14). Notably, total sequence length of Ty3-Gypsy LTR-RTs in haplotype B1 was 2.2 Mb larger than that of haplotype B2, which contributed approximately 40% of size variance between the two haplotypes, whereas another type of LTR-RTs, Ty1-Copia, showed similar sequence length in the two haplotypes (Supplementary Figure S14; Supplementary Tables S22 and S23). Moreover, 5,066 intact LTR-RTs were further used to estimate the insertion time of different families of LTR-RTs in C. dactylon genome (Supplementary Table S24). The results indicated that four families of LTR-RTs, including Athila, SIRE, TAR, and Tork, inserted into the four haplotypes of C. dactylon genome at different time, whereas other nine families showed similar insertion time range in the four haplotypes (Figure 3E). Interestingly, among the 244 active LTR-RTs with an insertion time of zero, 153 were located in three chromosomes of haplotype B1, 47 were located in two chromosomes of haplotype A1, whereas only 29 and 16 were located in single chromosome of haplotype A2 and B1, respectively (Supplementary Table S24).
As a polyploid plant species with four sets of chromosomes, C. dactylon might develop a mechanism to control proper pairing and segregation of chromosomes during meiosis thus maintain its genome stability (Svačina et al., 2020). ZMM proteins, which stabilize the D-loop crossover intermediate of synapsis, are important homologous recombination regulators in all eukaryotes (Pyatnitskaya et al., 2019; Figure 4A). Previous studies have illustrated that gene copy number reduction of a ZMM protein, MSH4, could prevent meiotic crossovers between non-homologous chromosomes and stabilize the genome in allotetraploid Brassica napus (Gonzalo et al., 2019). Similar gene copy number reduction of MSH4 was also observed in other two polyploidy plants, allotetraploid Gossypium hirsutum and hexaploid Triticum aestivum (Figure 4B; Supplementary Table S25). However, MSH4 and other four ZMM genes, including ZYP1, MER3, SHOC1, and MSH5, all existed as four-copy alleles in C. dactylon genome. By contrast, two ZMM genes, PTD and HEL10, existed as two-copy alleles, and another ZMM gene, ZIP4, existed as single-copy orphan gene in haplotype B1 of C. dactylon genome (Figure 4B; Supplementary Tables S25 and S26). Syntenic analysis further indicated that different sizes of chromosomal fragments containing ZIP4 were lost in other three haplotypes (Figure 4C). These results collectively implied that C. dactylon also evolved a ZMM-dependent regulatory mechanism to maintain its genome stability as other polyploidy plants did; however, the key regulator might be ZIP4 rather than MSH4.

Figure 4. Evolution of homologous recombination-regulating genes in C. dactylon. (A) Diagram depicting the essential roles of ZMM proteins in regulating homologous recombination. (B) Comparison of the gene copy number of eight ZMM proteins in C. dactylon, O. thomaeum, and other nine species with different ploidy levels. (C) Enlarged chromosomal gene location map showing the loss of ZIP4 gene and other contiguous genes in haplotype A1, A2, and B2 of C. dactylon. The ZIP4 gene in haplotype B1 was shown in red color.
As a widely used turfgrass species with two types of specialized stems, stolon and rhizome, C. dactylon exhibits a prostrate plant architecture owing to increased tiller angles of the two specialized stems (Dong and de Kroon, 1994). Previous studies have successfully identified several tiller-angle-regulating genes, including PROG1, TAC1, and LA1, in rice and other plants (Li et al., 2007; Yu et al., 2007; Jin et al., 2008; Tan et al., 2008; Figure 5A). Eight PROG1-like genes, four TAC1-like genes, and two LA1-like genes were also identified in C. dactylon (Figure 5B). Similar to semi-prostrate and prostrate growing Oryza genus plants, the family/genome gene number ratio of two prostrate growth-promoting genes, PROG1-like and TAC1-like, were higher than that of erect-growth-promoting LA1-like gene in C. dactylon (Figure 5B). Syntenic and phylogenic analysis revealed that six of the eight PROG1-like genes existed as three-copy alleles and the remaining two genes existed as two-copy alleles, the four TAC1-like genes existed as four-copy alleles, whereas two LA1-like genes existed as two-copy alleles (Figure 5C and Supplementary Figures S15, S16). Pfam domain analysis further indicated that all eight PROG1-like proteins have the conserved C2H2-type zinc finger domain identified in the functional OsPROG1 and OgPROG7 proteins, however, both two LA1-like proteins of C. dactylon lack the functional C-terminal conserved region V (Figure 5D and Supplementary Figure S15). Moreover, the LA1-like protein encoded by the allele of haplotype A2 further lack the functional N-terminal conserved region I and two other conserved regions II and III (Yoshihara and Spalding, 2020; Figure 5D). In combination with the observation that large chromosome fragments containing the LA1-like gene locus were lost in other two haplotypes (Supplementary Figure S16), sequence variation of LA1-like genes in the two residual alleles suggested that LA1 protein activity was inhibited in C. dactylon. In addition, both two LA1-like genes were weakly expressed in stolon and rhizome, whereas three of four TAC1-like genes were preferentially expressed in the two specialized stems (Supplementary Figure S16; Supplementary Table S27). These results collectively implied that different tiller-angle-regulating genes were synergistically evolved to promote a prostrate plant architecture in C. dactylon.
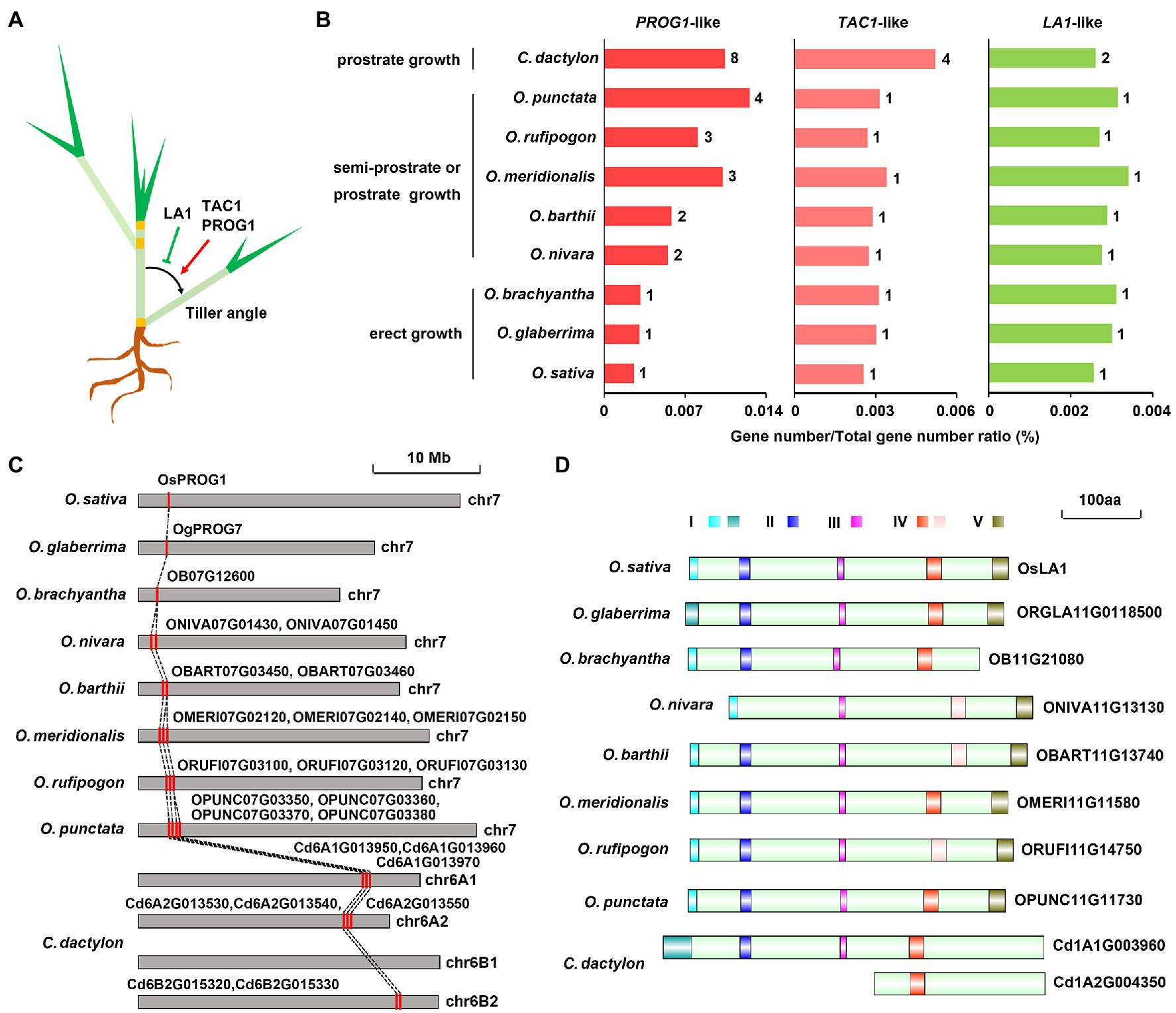
Figure 5. Evolution of tiller angle-regulating genes in C. dactylon. (A) Diagram depicting the positive/negative regulatory roles of PROG1, LAZY1, and TAC1 genes in tiller angle control of grasses. (B) Comparison of the gene number of PROG1-, LAZY1-, and TAC1-like genes in C. dactylon and eight species of Oryza genus with different growth habits. (C) Syntenic relationship of PROG1-like genes in C. dactylon and eight species of Oryza genus with different growth habits. (D) Diagram showing the deficiency of key functional motifs in two LAZY1-like proteins of C. dactylon compared with those of eight species of Oryza genus.
Turfgrasses are important groups of grass species serving essential functions, including soil stabilization, water conservation, filtration of air, and water borne pollutants, in urban and suburban landscapes (Huang, 2021). In the past several years, genome sequences of many turfgrass species, including zoysiagrasses (Zoysia japonica and Zoysia matrella), perennial ryegrass (Lolium perenne), centipedegrass (Eremochloa ophiuroides), and African bermudagrass (C. transvaalensis), were successfully sequenced and assembled using different techniques (Tanaka et al., 2016; Cui et al., 2021; Frei et al., 2021; Wang et al., 2021). In this study, we reported a high-quality haplotype-resolved genome of another important turfgrass species, common bermudagrass (C. dactylon), consisting of 36 pseudo chromosomes with a contig N50 of 2.65 Mb and a LAI score of 13.63 (Figure 1; Table 1 and Supplementary Table S13). The assembled nome of C. dactylon not only offers a solid foundation to study the molecular basis of valuable agronomic traits as well as molecular breeding of this important turfgrass species, but also provides an essential resource for comparative genomic analysis among different turfgrasses and other grasses.
The most prominent characteristics of C. dactylon genome are the presence of four haplotypes, named as A1, A2, B1, and B2, respectively. As an allotetraploid plants with high heterozygosity (1.92%), C. dactylon has four sets of chromosomes with significant differences that could be discriminated as different haplotypes using the newest haplotype-resolving Hifiasm algorithm; thus, an A1A2B1B2 genome assembly with 36 chromosomes, rather than an AB genome assembly with 18 chromosomes, was finally obtained (Kyriakidou et al., 2018). Similar result was also observed in the haplotype-phased genome assembly of tetraploid blueberry (2n = 4x = 48), which also reported a four haplotype-resolved genome containing 48 pseudo chromosomes (Colle et al., 2019). Notably, all the four haplotypes of C. dactylon have nine chromosomes; however, the total chromosome size showed variance among different haplotypes (Figure 3A; Supplementary Table S15). Specifically, haplotype A1 and A2 have a similar size (236.96 Mb and 231.36 Mb), whereas haplotype B1 and B2 have another similar size (271.19 Mb and 266.17 Mb; Supplementary Table S15). Accordingly, genes and repeat sequences in haplotype A1 and A2 are fewer than those in haplotype B1 and B2 (Supplementary Table S15). Interestingly, the size of four haplotypes are similar to the genome size of O. thomaeum (10 chromosomes, 243 Mb) and the monoploid genome size of Eragrostis tef (10 chromosomes, 288 Mb), two grass species belonging to the Chloridoideae subfamily of PACMAD clade of grasses as C. dactylon does, but much smaller than the genome size of African bermudagrass C. transvaalensis (nine chromosomes, 444 Mb), which is classified along with C. dactylon in the same Cynodon genus (VanBuren et al., 2015, 2020; Cui et al., 2021). Similar chromosome size variation was also observed between Morus notabilis and M. alba, both of which belongs to the same Morus genus (Xuan et al., 2022). These findings collectively suggested that genome size variation among different plant species might not be simply correlated with their phylogenic relationships.
Whole-genome duplication is an extreme mechanism of gene duplication that leads to a sudden increase in both genome size and the entire gene set thus plays important roles in plant genome evolution (Clark and Donoghue, 2018). Ks analysis revealed that two rounds of WGD events occurred in C. dactylon, which is in correspondence to the divergence time of haplotypes A1/A2 with haplotypes B1/B2 at 5.38 MYA and haplotype A1 with haplotype A2 at 0.77 MYA (the same as haplotype B1 with haplotype B2), respectively (Figure 3B and Supplementary Figure S11). These results collectively implied a complex evolutionary history of C. dactylon. At approximately 5.38 MYA, the ancestor of haplotype A1 and A2, named as A, might hybridized with B, the ancestor of haplotype B1 and B2, to form an AB hybrid species. At about 0.77 MYA, either an autopolyploidization event occurred in the AB hybrid species that doubled the genome to AABB or a secondary hybridization event occurred between two AB hybrid species to form an ABAB hybrid species through allopolyploidization, both of which could finally evolved into the present A1A2B1B2 genome of C. dactylon. The latter allopolyplodization mechanism seems more possible because the ratio of coupling to repulsion linkage phase of nondistorted mapped loci was approximately 1: 1 in an SSR-maker based linkage mapping of the first-generation selfed population of C. dactylon (Guo et al., 2017). Similar two rounds of WGD events were also observed in the formation of the polyploidy genome of Miscanthus floridulus and Saccharum spontaneum, suggesting a conserved evolution mechanism might exist in different genus of polyploid grasses (Zhang et al., 2018b, 2021).
A dominant subgenome often emerges immediately following the WGD event in the genome of allopolyploids (Liang and Schnable, 2018). However, some recent allopolyploids, including the above-mentioned M. floridulus and S. spontaneum, display indistinguishable or slight subgenome dominance (Zhang et al., 2018b, 2021). Orthologous gene clustering analysis indicated that four haplotypes of C. dactylon shared similar number of gene families with O. thomaeum (Supplementary Figure S8). Syntenic analysis further revealed that the four haplotypes have 12,197 (68.50% of 17,805), 12,039 (68.40% of 17,600), 14,406 (69.20% of 20,818), and 14,347 (69.46% of 20,656) syntenic orthologs to O. thomaeum, respectively (Supplementary Figure S9). These results suggested that four subgenomes of C. dactylon did not experience biased gene loss during evolution. Moreover, although a few genes from different haplotypes showed biased expression in different organs, overall gene expression levels showed high similarity among the four haplotypes (Figure 3D and Supplementary Figure S13). In addition, similar distribution and insertion time of LTRs were also observed in the four haplotypes (Figure 3E and Supplementary Figure S14). Taken together, these analyses collectively implied subgenome dominance is also unobvious in C. dactylon.
Polyploidy brings many advantages to polyploid plants. Heterosis could foster a greater biomass and accelerated development, whereas gene redundancy could mask deleterious mutations and diversify the functions of extra gene copies (Comai, 2005). As a worldwidely distributed grass species inhabiting diverse and harsh environments, allotetraploid C. dactylon undoubtedly benefits from these advantages. However, long-term survival of polyploid plants also require a mechanism to withstand the extensive genomic instability that accompanies with the presence of multiple pairing chromosomes in meiosis (Mason and Mason and Wendel, 2020). As a clonal plant with stolons and rhizomes, C. dactylon reproduces asexually through regenerating new plants from axillary buds of stolon and rhizome node (Dong and de Kroon, 1994), thus bypasses meiosis and recombination in gamete generation process. On the other hand, a ZMM-dependent regulatory mechanism to maintain genome stability during meiosis was also identified in C. dactylon (Figure 4). Owing to these belt and braces strategies, four unbiased haplotypes of subgenome are stably maintained in C. dactylon genome.
Tiller angle (branch angle in eudicot plants) is an important plant architectural trait affecting the density of growing plants (Wang et al., 2022). Cereal grasses often have compact and erect plant architecture characteristics with small tiller angles, which is essential for high yields. Specifically, successful domestication of cultivated rice from wild rice ancestors depended on the transition from prostrate growth to erect growth, in which process the tiller angle was greatly reduced (Li et al., 2007; Yu et al., 2007; Jin et al., 2008; Tan et al., 2008). However, for turfgrasses including C. dactylon, prostrate growth mode with large tiller angle is more preferable because it could accelerate turf formation, increase soil coverage, and diminish mowing frequency (Wang et al., 2021). Blast searches indicated that key tiller-angle-regulating genes reported in rice and other plants, including PROG1, LA1, and TAC1, were highly conserved in C. dactylon (Figure 5B). Similar to prostrate growing wild rice species, clustering of PROG1-like C2H2 transcription factor genes in adjacent positions of chromosomes were observed in C. dactylon (Wu et al., 2018; Huang et al., 2020; Figure 5C and Supplementary Figure S15). By contrast, LA1-like genes that promote erect growth not only experienced gene copy lost due to large chromosomal fragment deletions but also mutated to form truncated proteins (Figure 5D and Supplementary Figure S16). These results strongly suggested that similar selection pressure might also exist in C. dactylon to form the prostrate plant architecture characteristics as the domestication of rice from wild rice; however, the selection target might be LA1 rather than PROG1.
The genome of a widely used warm-season turfgrass species, C. dactylon, was sequenced and annotated in this study. The assembled genome contains 36 pseudo chromosomes, includes 37.91% genome size of repeat sequences, and encodes 76,879 protein-coding genes. The polyploid C. dactylon genome is consists of four haplotypes derived from two rounds of WGD events. Although a few haplotype-specific genes and transposons were identified, no global subgenome dominance was detected among the four haplotypes. A ZMM-dependent regulatory mechanism to maintain the genome stability was successfully identified. Furthermore, synergistic evolution of tiller-angle-regulating genes was also observed. In summary, the extensive datasets and analyses presented in this study not only offer an essential resource for basic studies and breeding researches of turfgrasses, but also provide new insights into regulation mechanisms underlying polyploid genome stability and prostrate growth.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: National Center for Biotechnology Information (NCBI) BioProject database under accession numbers PRJNA430136, PRJNA685207, and PRJNA805105.
BZ and J-YL planned and managed the project and wrote the manuscript. J-YL provided the research fund. BZ, SC, JC, and DL conducted the research and analyzed the data. JL provided the plant material and helped to write the manuscript. Y-BY helped to analyze the data and write the manuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported by the Science and Technology Development Foundation of Tsinghua University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors want to appreciate Professor Hong-Wei Wang at Tsinghua University for his invaluable suggestions and Qiang Gao and Zhaoyang Wang at BGI-Shenzhen for their helps in accomplishment of this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.890980/full#supplementary-material
1. ^https://github.com/BGI-flexlab/SOAPnuke
2. ^https://github.com/gmarcais/Jellyfish
3. ^http://qb.cshl.edu/genomescope
4. ^https://github.com/chhylp123/hifiasm
5. ^https://github.com/isovic/racon
6. ^https://github.com/bwa-mem2/bwa-mem2
7. ^https://github.com/broadinstitute/pilon
8. ^https://bionanogenomics.com/support/software-downloads
9. ^https://github.com/aidenlab/juicer
10. ^https://github.com/aidenlab/3d-dna
11. ^https://www.girinst.org/server/RepBase
12. ^http://www.repeatmasker.org
13. ^http://www.repeatmasker.org/RepeatModeler
14. ^http://www.drive5.com/piler
15. ^http://bix.ucsd.edu/repeatscout
16. ^http://tlife.fudan.edu.cn/tlife/ltr_finder
17. ^https://tandem.bu.edu/trf/trf.html
18. ^https://github.com/oushujun/LTR_retriever
19. ^https://phytozome-next.jgi.doe.gov
20. ^https://www.ebi.ac.uk/Tools/psa/genewise
21. ^https://github.com/Gaius-Augustus/Augustus
22. ^https://genome.crg.es/software/geneid/
24. ^http://ccb.jhu.edu/software/glimmerhmm
25. ^http://argonaute.mit.edu/GENSCAN.html
26. ^http://research-pub.gene.com/gmap
27. ^https://anaconda.org/bioconda/pasa
28. ^http://ccb.jhu.edu/software/tophat
29. ^http://cole-trapnell-lab.github.io/cufflinks
30. ^http://evidencemodeler.github.io
32. ^http://eggnog-mapper.embl.de
33. ^http://www.geneontology.org
34. ^http://www.genome.jp/kegg
36. ^http://bioinfo.bti.cornell.edu/tool/itak
37. ^http://planttfdb.gao-lab.org
38. ^https://github.com/tseemann/barrnap
39. ^http://trna.ucsc.edu/tRNAscan-SE
40. ^http://eddylab.org/infernal
42. ^https://github.com/davidemms/OrthoFinder
43. ^https://www.ebi.ac.uk/Tools/msa/muscle
44. ^http://abacus.gene.ucl.ac.uk/software/
46. ^https://ftp.ncbi.nlm.nih.gov/blast/executables/
47. ^https://github.com/wyp1125/MCScanX
48. ^http://daehwankimlab.github.io/hisat2
Birney, E., Clamp, M., and Durbin, R. (2004). GENEWISE and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chaves, A. L. A., Chiavegatto, R. B., Benites, F. R. G., and Techio, V. H. (2019). Comparative karyotype analysis among cytotypes of Cynodon dactylon (L.) Pers. (Poaceae). Mol. Biol. Rep. 46, 4873–4881. doi: 10.1007/s11033-019-04935-z
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6. doi: 10.1093/gigascience/gix120
Chen, S., Xu, X., Ma, Z., Liu, J., and Zhang, B. (2021). Organ-specific transcriptome analysis identifies candidate genes involved in the stem specialization of bermudagrass (Cynodon dactylon L.). Front. Genet. 12:678673. doi: 10.3389/fgene.2021.678673
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Clark, J. W., and Donoghue, P. C. J. (2018). Whole-genome duplication and plant macroevolution. Trends Plant Sci. 23, 933–945. doi: 10.1016/j.tplants.2018.07.006
Colle, M., Leisner, C. P., Wai, C. M., Ou, S., Bird, K. A., Wang, J., et al. (2019). Haplotype-phased genome and evolution of phytonutrient pathways of tetraploid blueberry. Gigascience 8:giz012. doi: 10.1093/gigascience/giz012
Comai, L. (2005). The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6, 836–846. doi: 10.1038/nrg1711
Cui, F., Taier, G., Li, M., Dai, X., Hang, N., Zhang, X., et al. (2021). The genome of the warm-season turfgrass African bermudagrass (Cynodon transvaalensis). Horticult. Res. 8:93. doi: 10.1038/s41438-021-00519-w
Dong, M., and de Kroon, H. (1994). Plasticity in morphology and biomass allocation in Cynodon dactylon, a grass species forming stolons and rhizomes. Oikos 70, 99–106. doi: 10.2307/3545704
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S., et al. (2016). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Farsani, T. M., Etemadi, N., Sayed-Tabatabaei, B. E., and Talebi, M. (2012). Assessment of genetic diversity of bermudagrass (Cynodon dactylon) using ISSR markers. Int. J. Mol. Sci. 13, 383–392. doi: 10.3390/ijms13010383
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U. S. A. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Frei, D., Veekman, E., Grogg, D., Stoffel-Studer, I., Morishima, A., Shimizu-Inatsugi, R., et al. (2021). Ultralong Oxford Nanopore reads enable the development of a reference-grade perennial ryegrass genome assembly. Genome Biol. Evol. 13, evab159. doi: 10.1093/gbe/evab159
Gaut, B. S., Morton, B. R., McCaig, B. C., and Clegg, M. T. (1996). Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. U. S. A. 93, 10274–10279. doi: 10.1073/pnas.93.19.10274
Ghosh, S., and Chan, C. K. (2016). Analysis of RNA-seq data using TopHat and Cufflinks. Methods Mol. Biol. 1374, 339–361. doi: 10.1007/978-1-4939-3167-5_18
Glover, J. D., Reganold, J. P., Bell, L. W., Borevitz, J., Brummer, E. C., Buckler, E. S., et al. (2010). Increased food and ecosystem security via perennial grains. Science 328, 1638–1639. doi: 10.1126/science.1188761
Gonzalo, A., Lucas, M. O., Charpentier, C., Sandmann, G., Lloyd, A., and Jenczewski, E. (2019). Reducing MSH4 copy number prevents meiotic crossovers between non-homologous chromosomes in Brassica napus. Nat. Commun. 10:2354. doi: 10.1038/s41467-019-10010-9
Grossman, A. Y., Andrade, M. H. M. L., Chaves, A. L. A., Mendes Ferreira, M. T., Techio, V. H., Lopez, Y., et al. (2021). Ploidy level and genetic parameters for phenotypic traits in bermudagrass (Cynodon spp.) germplasm. Agronomy 11:912. doi: 10.3390/agronomy11050912
Guo, L., Plunkert, M., Luo, X., and Liu, Z. C. (2021). Developmental regulation of stolon and rhizome. Curr. Opin. Plant Biol. 59:101970. doi: 10.1016/j.pbi.2020.10.003
Guo, Y., Wu, Y., Anderson, J. A., Moss, J. Q., Zhu, L., and Fu, J. (2017). SSR marker development, linkage mapping, and QTL analysis for establishment rate in common bermudagrass. Plant Genome 10:1. doi: 10.3835/plantgenome2016.07.0074
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Harlan, J. R., and de Wet, J. M. J. (1969). Sources of variation in Cynodon dactylon (L). Pers. Crop Sci 9, 774–778. doi: 10.2135/cropsci1969.0011183X000900060031x
Hill, G. M., Gates, R. N., and West, J. W. (2001). Advances in bermudagrass research involving new cultivars for beef and dairy production. J. Anim. Sci. 79, E48–E58. doi: 10.2527/jas2001.79E-SupplE48x
Huang, B. R. (2021). Grass research for a productive, healthy and sustainable society. Grass Res. 1, 1–2. doi: 10.48130/GR-2021-0001
Huang, L., Liu, H., Wu, J., Zhao, R., Li, Y., Melaku, G., et al. (2020). Evolution of plant architecture in Oryza driven by the PROG1 locus. Front. Plant Sci. 11:876. doi: 10.3389/fpls.2020.00876
Jin, J., Huang, W., Gao, J. P., Yang, J., Shi, M., Zhu, M. Z., et al. (2008). Genetic control of rice plant architecture under domestication. Nat. Genet. 40, 1365–1369. doi: 10.1038/ng.247
Khanal, S., Kim, C., Auckland, S. A., Rainville, L. K., Adhikari, J., Schwartz, B. M., et al. (2017). SSR-enriched genetic linkage maps of bermudagrass (Cynodon dactylon × transvaalensis), and their comparison with allied plant genomes. Theor. Appl. Genet. 130, 819–839. doi: 10.1007/s00122-017-2854-z
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kneebone, W. R. (1966). Bermuda grass-worldly, wily, wonderful weed. Econ. Bot. 20, 94–97. doi: 10.1007/BF02861931
Kronenberg, Z. N., Rhie, A., Koren, S., Concepcion, G. T., Peluso, P., Munson, K. M., et al. (2021). Extended haplotype-phasing of long-read de novo genome assemblies using Hi-C. Nat. Commun. 12:1935. doi: 10.1038/s41467-020-20536-y
Kyriakidou, M., Tai, H. H., Anglin, N. L., Ellis, D., and Strömvik, M. V. (2018). Current strategies of polyploid plant genome sequence assembly. Front. Plant Sci. 9:1660. doi: 10.3389/fpls.2018.01660
Li, P., Wang, Y., Qian, Q., Fu, Z., Wang, M., Zeng, D., et al. (2007). LAZY1 controls rice shoot gravitropism through regulating polar auxin transport. Cell Res. 17, 402–410. doi: 10.1038/cr.2007.38
Liang, Z., and Schnable, J. C. (2018). Functional divergence between subgenomes and gene pairs after whole genome duplications. Mol. Plant 11, 388–397. doi: 10.1016/j.molp.2017.12.010
Ma, Z., Chen, S., Wang, Z., Liu, J., and Zhang, B. (2021). Proteome analysis of bermudagrass stolons and rhizomes provides new insights into the adaptation of plant stems to aboveground and underground growth. J. Proteome 241:104245. doi: 10.1016/j.jprot.2021.104245
Ma, J., Devos, K. M., and Bennetzen, J. L. (2004). Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869. doi: 10.1101/gr.1466204
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Mason, A. S., and Wendel, J. F. (2020). Homoeologous exchanges, segmental allopolyploidy, and polyploid genome evolution. Front. Genet. 11:1014. doi: 10.3389/fgene.2020.01014
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Nachtweide, S., and Stanke, M. (2019). Multi-genome annotation with AUGUSTUS. Methods Mol. Biol. 1962, 139–160. doi: 10.1007/978-1-4939-9173-0_8
Nagori, B. P., and Solanki, R. (2011). Cynodon dactylon (L.) Pers.: a valuable medicinal plant. Res. J. Med. Plant 5, 508–514. doi: 10.3923/rjmp.2011.508.514
Nautiyal, A. K., Gani, U., Sharma, P., Kundan, M., Fayaz, M., Lattoo, S. K., et al. (2020). Comprehensive transcriptome analysis provides insights into metabolic and gene regulatory networks in trichomes of Nicotiana tabacum. Plant Mol. Biol. 102, 625–644. doi: 10.1007/s11103-020-00968-2
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Ou, S., Chen, J., and Jiang, N. (2018). Assessing genome assembly quality using the LTR assembly index (LAI). Nucleic Acids Res. 46:e126. doi: 10.1093/nar/gky730
Ozias-Akins, P., and Conner, J. A. (2020). Clonal reproduction through seeds in sight for crops. Trends Genet. 36, 215–226. doi: 10.1016/j.tig.2019.12.006
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Pyatnitskaya, A., Borde, V., and De Muyt, A. (2019). Crossing and zipping: molecular duties of the ZMM proteins in meiosis. Chromosoma 128, 181–198. doi: 10.1007/s00412-019-00714-8
Satorre, E. H., Rizzo, F. A., and Arias, S. P. (1996). The effect of temperature on sprouting and early establishment of Cynodon dactylon. Weed Res. 36, 431–440. doi: 10.1111/j.1365-3180.1996.tb01672.x
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Svačina, R., Sourdille, P., Kopecký, D., and Bartoš, J. (2020). Chromosome pairing in polyploid grasses. Front. Plant Sci. 11:1056. doi: 10.3389/fpls.2020.01056
Tan, L., Li, X., Liu, F., Sun, X., Li, C., Zhu, Z., et al. (2008). Control of a key transition from prostrate to erect growth in rice domestication. Nat. Genet. 40, 1360–1364. doi: 10.1038/ng.197
Tan, C., Wu, Y., Taliaferro, C. M., Bell, G. E., Martin, D. L., Smith, M. W., et al. (2014). Selfing and outcrossing fertility in common bermudagrass under open-pollinating conditions examined by SSR markers. Crop Sci. 54, 1832–1837. doi: 10.2135/cropsci2013.12.0816
Tanaka, H., Hirakawa, H., Kosugi, S., Nakayama, S., Ono, A., Watanabe, A., et al. (2016). Sequencing and comparative analyses of the genomes of zoysiagrasses. DNA Res. 23, 171–180. doi: 10.1093/dnares/dsw006
Tempel, S. (2012). Using and understanding RepeatMasker. Methods Mol. Biol. 859, 29–51. doi: 10.1007/978-1-61779-603-6_2
Uhlén, M., Hallström, B. M., Lindskog, C., Mardinoglu, A., Pontén, F., and Nielsen, J. (2016). Transcriptomics resources of human tissues and organs. Mol. Syst. Biol. 12:862. doi: 10.15252/msb.20155865
VanBuren, R., Bryant, D., Edger, P. P., Tang, H., Burgess, D., Challabathula, D., et al. (2015). Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527, 508–511. doi: 10.1038/nature15714
VanBuren, R., Ching, M. W., Wang, X., Pardo, J., Yocca, A. E., Wang, H., et al. (2020). Exceptional subgenome stability and functional divergence in the allotetraploid Ethiopian cereal teff. Nat. Commun. 11:884. doi: 10.1038/s41467-020-14724-z
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, W., Gao, H., Liang, Y., Li, J., and Wang, Y. (2022). Molecular basis underlying rice tiller angle: current progress and future perspectives. Mol. Plant 15, 125–137. doi: 10.1016/j.molp.2021.12.002
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wang, J., Zi, H., Wang, R., Liu, J., Wang, H., Chen, R., et al. (2021). A high-quality chromosome-scale assembly of the centipedegrass [Eremochloa ophiuroides (Munro) Hack.] genome provides insights into chromosomal structural evolution and prostrate growth habit. Horticult. Res. 8:201. doi: 10.1038/s41438-021-00636-6
Wu, Y. Q., Taliaferro, C. M., Bai, G. H., and Anderson, M. P. (2004). AFLP analysis of Cynodon dactylon (L.) Pers. var. dactylon genetic variation. Genome 47, 689–696. doi: 10.1139/g04-032
Wu, Y. Q., Taliaferro, C. M., Bai, G. H., Martin, D. L., Anderson, J. A., Anderson, M. P., et al. (2006). Genetic analyses of Chinese Cynodon accessions by flow cytometry and AFLP markers. Crop Sci. 46, 917–926. doi: 10.2135/cropsci2005.08.0256
Wu, Y. Q., Taliaferro, C. M., Martin, D. L., Anderson, J. A., and Anderson, M. P. (2007). Genetic variability and relationships for adaptive, morphological, and biomass traits in Chinese bermudagrass accessions. Crop Sci. 47, 1985–1994. doi: 10.2135/cropsci2007.01.0047
Wu, T. D., and Watanabe, C. K. (2005). GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. doi: 10.1093/bioinformatics/bti310
Wu, Y., Zhao, S., Li, X., Zhang, B., Jiang, L., Tang, Y., et al. (2018). Deletions linked to PROG1 gene participate in plant architecture domestication in Asian and African rice. Nat. Commun. 9:4157. doi: 10.1038/s41467-018-06509-2
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Xu, J., Wang, Z., and Cheng, J. J. (2011). Bermuda grass as feedstock for biofuel production: a review. Bioresour. Technol. 102, 7613–7620. doi: 10.1016/j.biortech.2011.05.070
Xuan, Y., Ma, B., Li, D., Tian, Y., Zeng, Q., and He, N. (2022). Chromosome restructuring and number change during the evolution of Morus notabilis and Morus alba. Horticult. Res 9:uhab030. doi: 10.1093/hr/uhab030
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, L., Wu, Y. Q., Moss, J. Q., Zhong, S., and Yang, B. (2018). Molecular identification and characterization of seeded turf bermudagrass cultivars using simple sequence repeat markers. Agron. J. 110, 2142–2150. doi: 10.2134/agronj2018.01.0068
Yao, H., Guo, L., Fu, Y., Borsuk, L. A., Wen, T. J., Skibbe, D. S., et al. (2005). Evaluation of five ab initio gene prediction programs for the discovery of maize genes. Plant Mol. Biol. 57, 445–460. doi: 10.1007/s11103-005-0271-1
Yoshihara, T., and Spalding, E. P. (2020). Switching the direction of stem gravitropism by altering two amino acids in AtLAZY1. Plant Physiol. 182, 1039–1051. doi: 10.1104/pp.19.01144
Yu, B., Lin, Z., Li, H., Li, X., Li, J., Wang, Y., et al. (2007). TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. 52, 891–898. doi: 10.1111/j.1365-313X.2007.03284.x
Zhang, B., Fan, J., and Liu, J. (2019). Comparative proteomic analysis provides new insights into the specialization of shoots and stolons in bermudagrass (Cynodon dactylon L.). BMC Genomics 20:708. doi: 10.1186/s12864-019-6077-3
Zhang, G., Ge, C., Xu, P., Wang, S., Cheng, S., Han, Y., et al. (2021). The reference genome of Miscanthus floridulus illuminates the evolution of Saccharinae. Nat. Plants 7, 608–618. doi: 10.1038/s41477-021-00908-y
Zhang, B., and Liu, J. (2018). Molecular cloning and sequence variance analysis of the TEOSINTE BRANCHED1 (TB1) gene in bermudagrass [Cynodon dactylon (L.) Pers]. J. Plant Physiol. 229, 142–150. doi: 10.1016/j.jplph.2018.07.008
Zhang, B., Liu, J., Wang, X., and Wei, Z. (2018a). Full-length RNA sequencing reveals unique transcriptome composition in bermudagrass. Plant Physiol. Bioch. 132, 95–103. doi: 10.1016/j.plaphy.2018.08.039
Zhang, W., Liu, J., Zhang, Y., Qiu, J., Li, Y., Zheng, B., et al. (2020). A high-quality genome sequence of alkaligrass provides insights into halophyte stress tolerance. Sci. China Life Sci. 63, 1269–1282. doi: 10.1007/s11427-020-1662-x
Zhang, J., Zhang, X., Tang, H., Zhang, Q., Hua, X., Ma, X., et al. (2018b). Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565–1573. doi: 10.1038/s41588-018-0237-2
Zheng, Y., Jiao, C., Sun, H., Rosli, H. G., Pombo, M. A., Zhang, P., et al. (2016). iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 9, 1667–1670. doi: 10.1016/j.molp.2016.09.014
Keywords: Cynodon dactylon, common bermudagrass, genome, haplotype, tiller angle
Citation: Zhang B, Chen S, Liu J, Yan Y-B, Chen J, Li D and Liu J-Y (2022) A High-Quality Haplotype-Resolved Genome of Common Bermudagrass (Cynodon dactylon L.) Provides Insights Into Polyploid Genome Stability and Prostrate Growth. Front. Plant Sci. 13:890980. doi: 10.3389/fpls.2022.890980
Edited by:
Surya Saha, Boyce Thompson Institute (BTI), United StatesReviewed by:
Liangsheng Zhang, Zhejiang University, ChinaCopyright © 2022 Zhang, Chen, Liu, Yan, Chen, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin-Yuan Liu, bGl1anlAbWFpbC50c2luZ2h1YS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.