 Jing Zhou
Jing Zhou Ling-Yu Yang
Ling-Yu Yang- State Key Laboratory of Tree Genetics and Breeding, Key Laboratory of Silviculture of the National Forestry and Grassland Administration, Research Institute of Forestry, Chinese Academy of Forestry, Beijing, China
Nitrate (NO3−) and ammonium (NH4+) are the primary forms of inorganic nitrogen acquired by plant roots. LncRNAs, as key regulators of gene expression, are a class of non-coding RNAs larger than 200 bp. However, knowledge about the regulatory role of lncRNAs in response to different nitrogen forms remains limited, particularly in woody plants. Here, we performed strand-specific RNA-sequencing of P. × canescens roots under three different nitrogen fertilization treatments. In total, 324 lncRNAs and 6,112 mRNAs were identified as showing significantly differential expression between the NO3− and NH4NO3 treatments. Moreover, 333 lncRNAs and 6,007 mRNAs showed significantly differential expression between the NH4+ and NH4NO3 treatments. Further analysis suggested that these lncRNAs and mRNAs have different response mechanisms for different nitrogen forms. In addition, functional annotation of cis and trans target mRNAs of differentially expressed lncRNAs indicated that 60 lncRNAs corresponding to 49 differentially expressed cis and trans target mRNAs were involved in plant nitrogen metabolism and amino acid biosynthesis and metabolism. Furthermore, 42 lncRNAs were identified as putative precursors of 63 miRNAs, and 28 differentially expressed lncRNAs were potential endogenous target mimics targeted by 96 miRNAs. Moreover, ceRNA regulation networks were constructed. MSTRG.6097.1, MSTRG.13550.1, MSTRG.2693.1, and MSTRG.12899.1, as hub lncRNAs in the ceRNA networks, are potential candidate lncRNAs for studying the regulatory mechanism in poplar roots under different nitrogen fertilization treatments. The results provide a basis for obtaining insight into the molecular mechanisms of lncRNA responses to different nitrogen forms in woody plants.
Introduction
Nitrogen is a vital nutrient for plants and has a strong influence on plant development (Oldroyd and Leyser, 2020). Plant roots play an important role in the acquisition and utilization of soil nitrogen (Wei et al., 2013; Jiao et al., 2018; Zhou et al., 2020). Plant roots mainly perceive nitrogen changes in the soil, activate the expression of key regulatory genes, such as small RNAs, transporter genes, and transcription factors, and then regulate the expression of genes related to root growth and development (Khan et al., 2011; Forde, 2014; Bellegarde et al., 2017; Massaro et al., 2019; Naulin et al., 2020; Oldroyd and Leyser, 2020). In this process, nitrate (NO3−) and ammonium (NH4+) not only serve as the main nutrients through which most plants acquire and utilize inorganic nitrogen but also play a crucial role in the plant response to nitrogen regulation as signaling molecules (O’Brien et al., 2016; Naulin et al., 2020; Wang et al., 2021). Although plants can use both ions, the physiological and molecular features of NO3− and NH4+ are different for metabolism, which leads to distinct NO3− or NH4+ preferences among plants (Patterson et al., 2010; Ruan et al., 2016). To sustain crop growth and development, obtaining a better understanding of the mechanisms of absorption and utilization of different nitrogen forms by crop roots is important.
Long non-coding RNAs (lncRNAs) are transcripts over 200 bp in length with no protein-coding capacity (Chekanova, 2015; Budak et al., 2020). LncRNAs play key regulatory roles in many important biological pathways by regulating the expression of cis and trans target mRNAs (Sun et al., 2018; Jha et al., 2020; Urquiaga et al., 2020). Moreover, lncRNAs can be used as precursors for miRNA biosynthesis (Wang et al., 2018) and as endogenous target mimics (eTMs) of miRNAs (Zhang et al., 2018). Therefore, there is a close regulatory relationship among lncRNAs, miRNAs, and mRNAs (Voshall et al., 2017). In recent years, studies on lncRNA-miRNA-mRNA regulatory networks have revealed that lncRNAs, as a type of competing endogenous RNA (ceRNA), competitively bind the same miRNA as mRNAs, which reduces the probability of miRNA-mRNA binding and increases the expression of mRNAs (Chen et al., 2021). To date, many lncRNAs involved in the nitrogen deficiency response have been identified in model plants (Chen et al., 2016; Fukuda et al., 2019, 2020; Liu et al., 2019; Lu et al., 2019; Wang et al., 2020a). For example, under low nitrogen conditions, 388 lncRNAs have been identified in poplars; among these, 126 lncRNAs responded to low nitrogen stress, 14 lncRNAs are predicted to be precursors of 25 miRNAs, and 4 lncRNAs are predicted to be target mRNAs of 29 miRNAs (Chen et al., 2016). Our preliminary research also showed that the lncRNA MSTRG.24415.1 is associated with the cis target mRNAs TIP1.1 under low nitrogen stress and affects the wood formation process in poplar trees. According to the ceRNA theory, low nitrogen treatment results in downregulated expression of the lncRNA MSTRG.4094.1, which may promote the binding of mir5021-p5 to its target mRNA TIP1.3 and thus lead to downregulated expression of TIP1:3 (Lu et al., 2019). These studies suggest that lncRNAs participate in plant responses to nitrogen deficiency. In contrast, the identification and functional resolution of lncRNAs involved in plant responses to different nitrogen forms have not been reported.
Poplars, as model woody plants, are fast-growing trees that require a large amount of supplied nitrogen (Zhang et al., 2014). Different nitrogen fertilizations lead to contrasting morphological changes in poplar roots (Rewald et al., 2016) and to the regulation of distinct genes (Luo et al., 2015; Qu et al., 2016). Nevertheless, until now, how lncRNAs affect nitrogen uptake and the assimilation of different nitrogen forms in poplar to influence physiological and biochemical characteristics has not been studied. Therefore, investigating the regulatory mechanisms of lncRNAs in the absorption and assimilation capacity for different nitrogen forms in poplar roots is important.
In this study, P. × canescens saplings were exposed to 1 mM NaNO3, 500 μM NH4NO3, and 1 mM NH4Cl for 21 days. This study aimed to perform an in-depth analysis of the regulatory mechanisms of lncRNAs in response to different nitrogen fertilization treatments in the roots of P. × canescens. To achieve this goal, we identified significant differential expression patterns of lncRNAs under treatment with different nitrogen forms, and the cis and trans target mRNAs of differentially expressed (DE)-lncRNAs were functionally annotated. Furthermore, DE-lncRNAs were identified as putative precursors of miRNAs and as potential endogenous target mimics. We also constructed lncRNA-miRNA-mRNA networks. The results provide new ideas for studying the regulatory mechanisms of woody plants in response to different nitrogen forms.
Materials and Methods
Plant Cultivation and Nitrogen Treatment
Seedlings of P. × canescens (P. tremula × P. alba, INRA 717-IB4 clone) plantlets were cultured in a tissue culture room for 4 weeks. The daily illumination time was 16 h, the photosynthetic photon flux density was 150 μmol m−2 s−1, the day/night temperature was 25/20°C, and the relative humidity was 50–55%. Subsequently, a set of 8 plants was planted in a hydroponic pot (10 pots in total) and irrigated with 8 l of Long Ashton (LA) nutrient solution every other day (Zhou et al., 2020). After 14 days in the greenhouse (under the same climatic conditions as the tissue culture chamber), plants with similar heights and ground diameters were selected, divided into 3 groups (twenty-two plants in each group), and transferred to a hydroponic system with nitrogen-free medium for 3 days (Balazadeh et al., 2014). Then, the three groups of plants were treated with different nitrogen fertilizers. The treatment conditions were as follows: one group of plants was treated with 1 mM NaNO3; the second group of plants was treated with the original LA nutrient solution as the control, which contained 500 μM NH4NO3; and the third group of plants was treated with 1 mM NH4Cl. The processing time was 21 days.
Root Measurements and Harvesting
The plant root height and dry weight were measured before harvest. Eighteen plants were examined under three nitrogen fertilization treatments, and three biological replicates of each treatment were included in the experiment. Under the three nitrogen fertilization treatments, the dry weight of another four plants was calculated after the whole root of the plant was dried in a 60°C oven for 24 h. For harvesting, the whole root of each plant was dried with absorbent paper, wrapped in tinfoil, and immediately placed into liquid nitrogen. Each whole root sample was then ground into fine powder in liquid nitrogen with a ball mill (MM400, Retsch, Haan, Germany) and stored at −80°C.
To obtain sufficient test materials, the root organization of six plants that had been subjected to the same treatment was ground in equal amounts to obtain a mixed sample. As a result, 3 mixed samples of each treatment were obtained for further analysis. The NO3− concentrations under the different nitrogen fertilization treatments were determined as described by Patterson et al. (2010), and the NH4+ concentrations under the different nitrogen fertilization treatments were analyzed spectrophotometrically according to the Berthelot reaction (Luo et al., 2013b).
RNA Extraction and Sequencing
Total RNA was isolated from poplar roots using a total RNA extraction kit (TRK1001, LianChuan (LC) Science, Hangzhou, China). An RNA 6000 Nano LabChip Kit (5067–1,511, Agilent, CA, USA) and a Bioanalyzer 2,100 (Agilent, Santa Clara, CA, USA) were used to determine the quantity of total RNA. Total RNA from the NO3−, NH4NO3, and NH4+ treatments was treated with RNase-free DNase I (E1091, Omega Bio-Tek, Norcross, GA, USA) to eliminate genomic DNA. For the sequencing of mRNAs and lncRNAs, a Ribo-Zero Gold Kit (MRZPL116, Illumina, CA, USA) was used to remove ribosomal RNA from the total RNA samples according to the kit instructions (Lu et al., 2019). Subsequently, cDNA libraries were established according to the protocol of the RNA-seq sample preparation kit (Illumina, CA, USA). Three cDNA libraries were constructed from each treatment level, and sequencing was performed according to the recommended protocol of the Illumina HiSeq 4,000 sequencer (Illumina, CA, USA) of LianChuan Science (Hangzhou, China). The raw sequence data were submitted to the Sequence Read Archive (SRA) under project ID PRJNA631840.
Identification of lncRNAs and mRNAs
The raw sequence data were purified by removing low-quality (nucleotides with quality scores lower than 20, Q < 20), adaptor contamination and undetermined base reads, and then, FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used for quality verification to obtain clean reads. Using the TopHat2 package (version: 2.0.4), high-quality reads were blasted against the P. tremula × P. alba 717-1B4 genome v1.1 sequence (http://aspendb.uga.edu/index.php/databases/spta-717-genome), and up to two mismatches were allowed during the alignment process (Trapnell et al., 2009).
LncRNAs were identified according to the method described by Wang et al. (2017). In brief, the filter ratio was less than 50% of the coverage of the transcript, and transcripts with lengths less than 200 bp were removed. The coding potential of the remaining transcripts was then assessed using Coding Potential Calculator (CPC) software (Kong et al., 2007), and Coding-Non-Coding Index (CNCI) software (Sun et al., 2013) was used for assessment. Only transcripts with CPC scores less than −1 and CNCI scores less than 0 were considered lncRNA candidates.
The fragment per kilobase of exon per million fragments mapped (FPKM) algorithm was used to quantify the expression levels of lncRNAs and mRNAs as described by Pertea et al. (2015). Based on the FPKM value, the Ballgown package was used to calculate the differential expression levels of lncRNAs and mRNAs (Frazee et al., 2015). The log2(fold change) in DE-lncRNAs and DE-mRNAs was determined using the FPKMs of the genes under NO3− or NH4+ treatments divided by those under the NH4NO3 treatments. The screening thresholds for identifying significantly DE-lncRNAs and DE-mRNAs were a p-value less than 0.05 and absolute values of log2(fold change) higher than 1.
Target mRNA Prediction and Functional Analysis of DE-lncRNAs
To further explore the functions of DE-lncRNAs, potential cis and trans target mRNAs of the DE-lncRNAs were predicted (Zhang et al., 2018). The cis targets of DE-lncRNAs were predicted using a Python script designed by LianChuan Science (Lu et al., 2019). The target mRNAs in the 100-kb region upstream or downstream of the DE-lncRNAs were considered possible cis targets (Li et al., 2020). The trans targets of DE-lncRNAs were predicted based on the complementation effect of lncRNAs on the target mRNAs and RNA duplex energy (free energy less than −50) prediction using RIsearch. Then, GO (Gene Ontology) functional classification (Wang et al., 2017) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis (Masoudi-Nejad et al., 2007) were performed for the functional analysis of the differentially expressed cis and trans target mRNAs. Functional category analysis of the differentially expressed cis and trans target mRNAs was also performed using MapMan as described by Jia et al. (2017).
Prediction of miRNA Precursors of lncRNAs
P. × canescens miRNA data were obtained using high-throughput sequencing, and 465 unique known miRNAs and 29 novel miRNAs were identified in the NO3−, NH4NO3, and NH4+ libraries (Zhou and Wu, 2022a). The raw sequence data for the small RNAs were submitted to SRA under the project ID PRJNA631845. To predict which lncRNAs can be used as potential miRNA precursors, we compared the lncRNA sequences with mature P. × canescens miRNA sequences by BLAST and selected lncRNAs that showed an alignment with 100% homology to the mature miRNA sequence and the same chromosome as the mature miRNA.
Construction of ceRNA Regulatory Networks
According to the method described by Sun et al. (2016), ceRNA regulatory networks among DE-lncRNAs, miRNAs, and DE-mRNAs were constructed. The miRNA data were based on the results of previous studies (Zhou and Wu, 2022a). The miRNA target mRNAs were predicted using Target Finder (mismatch score ≤ 2.5, penalty for mismatch and missing in strict matching zone is 1, G: U mismatch penalty is 0.5, and the penalty for non-strict matching zone is 0.5). Subsequently, target mimic prediction was performed to identify the complementary relationship between DE-lncRNAs and miRNAs. Using PsRobot, the sequences of DE-lncRNAs were entered into psRNA targets to identify miRNAs that may target DE-lncRNAs (mismatch score ≤ 2.5). Finally, the two complementary pairs were gathered to form the lncRNA-miRNA-mRNA regulatory network. The lncRNA-miRNA-mRNA regulatory network could be used as a ceRNA network if the following two conditions were met: (1) the expression patterns of interacting lncRNAs and mRNAs were up- or downregulated simultaneously because lncRNAs were reported to positively regulate the expression of mRNAs through lncRNA–miRNA–mRNA pairs (Ma et al., 2021); and (2) the interacting lncRNAs and mRNAs were significantly differentially expressed under different nitrogen fertilization treatments. The ceRNA regulatory network was imported into Cytoscape (v3.6.0) for visualization (version 3.6.0, http://chianti.ucsd.edu/cytoscape-3.6.0/).
Real-Time Quantitative PCR Validation of the Significantly DE-lncRNAs and DE-mRNAs
To validate the expression of the ceRNA regulatory network, the DE-lncRNAs, and their target mRNAs, RT–qPCR validation was performed using SYBR Green assay reagents and a LightCyclerR 480 RealTime PCR System (Roche, USA) as described by Zhou et al. (2012). Total and small RNAs were extracted using the same samples used for RNA-sequencing. For small RNA reverse transcription, universal primers (Supplementary Table S1) in the Mir-X miRNA First-Strand Synthesis (Clontech Laboratories, CA, USA) were used for reverse transcription. Mature miRNA sequences and universal primers (Supplementary Table S1) were used for RT–qPCR according to the manufacturer’s instructions for the SYBR qRT-PCR kit (Clontech Laboratories, CA, USA). For RNA reverse transcription, 2 μg of total RNA was reverse-transcribed using the PrimeScript™ RT Reagent Kit (TaKaRa BIO, Japan). The specific primers of the tested genes are shown in Supplementary Table S1. Each DE-lncRNA, DE-miRNA, and DE-mRNA was analyzed in three replicates. Relative expression levels were calculated using the 2–ΔΔCt method. Actin was used as the endogenous reference genes for DE-lncRNAs and DE-mRNAs, and 5.8S rRNA was used as the endogenous reference gene for DE-miRNAs (Supplementary Table S1).
Validation of lncRNA-miRNA-mRNA Pairs
To validate the ceRNA regulatory network, two lncRNA-miRNA-mRNA pairs were chosen, and transient coexpression analysis in N. benthamiana leaves was conducted as described by Zhou et al. (2020). Each of the eTMs (MSTRG.2693.1 and MSTRG.13550.1) and mRNAs (PcNFYA2 and PcCDL1) were individually cloned into pCAMBIA1300 vectors under the control of the 35S promoter. Similarly, fragments of two miRNA precursors (mdm-miR169b_R-1 and miR171i-3p) were inserted into the pCAMBIA2300 vectors, which also carry a 35S promoter. Through electroporation, both vectors were then individually transformed into A. tumefaciens strain GV3101 and inoculated overnight at 28°C. Before infiltration into N. benthamiana leaves, an equal amount of A. tumefaciens cell culture containing the lncRNA and its corresponding miRNA and target mRNA was mixed as described by He et al. (2008). After 2 days of incubation in the dark, N. benthamiana leaves that were infiltrated were harvested for RT–qPCR. The gene-specific primers are shown in Supplementary Table S1. The tobacco tubulin gene was used as the endogenous reference gene of mRNAs (Supplementary Table S1).
Statistical Analysis
Statgraphics software (STN, St Louis, MO, USA) was used for the statistical analyses of the data. Before statistical analyses of the data, the data were tested to determine the normality of their distribution. All the data were analyzed by one-way ANOVA using the different nitrogen treatment levels as a factor. The difference between the mean values was considered significant if the p-value from the ANOVA F test was less than 0.05.
Results
Phenotypic Responses of P. × canescens Roots to Different Nitrogen Forms
The influence of different nitrogen fertilization treatments on developing P. × canescens was monitored by measuring the root length and root dry weight. After long-term hydroponic cultivation (21 days), plants were supplied with NO3−, NH4+, or NH4NO3 (control). The plants growing under the NO3− treatment exhibited longer roots and delayed growth of lateral roots compared with those growing under the NH4NO3 (control) treatment (Figure 1). The plants growing under the NH4+ treatment exhibited shorter roots and earlier growth of lateral roots than those growing under the NH4NO3 (control) treatment (Figure 1). Moreover, the root dry weight under the NO3− and NH4+ treatments was higher than that under the NH4NO3 (control) treatment (Figure 1). However, there were no significant changes above plant height (Supplementary Figure S1). As different nitrogen forms may lead to different NO3− or NH4+ concentrations in poplar roots, the concentrations in poplar roots were analyzed. The NO3− treatment increased the NO3− concentration by 13.05% and decreased the NH4+ concentration compared with that detected under the NH4NO3 (control) treatment. Moreover, the NH4+ treatment significantly reduced the NO3− concentration by 19.65% and significantly increased the NH4+ concentration by 14.15% compared with those found under the NH4NO3 (control) treatment (Figure 1).
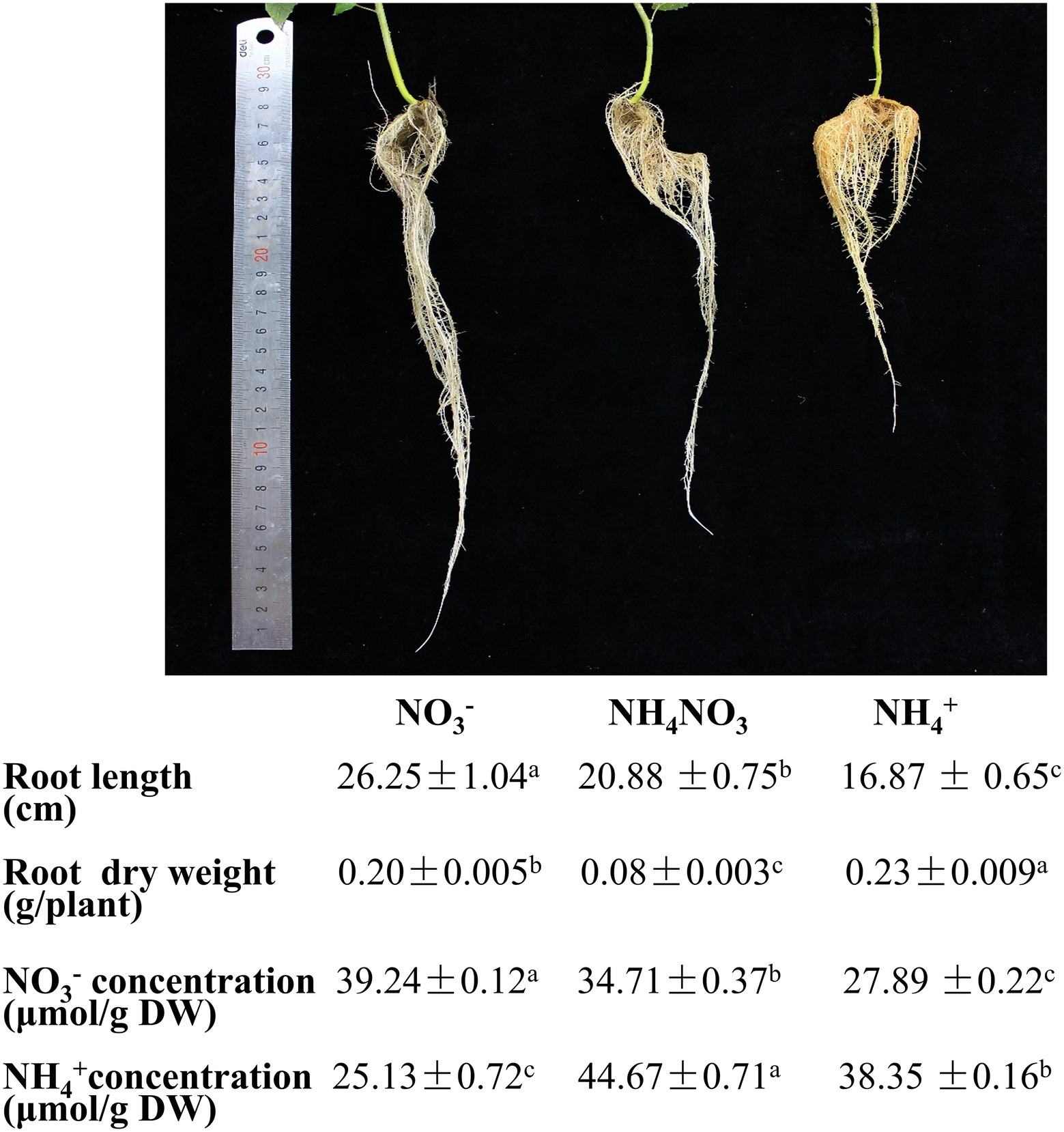
Figure 1. Morphological parameters and physiological indexes of P. x canescens roots under different nitrogen fertilization treatments for 21 days. Phenotypes of P. x canescens cultured under 1 mM NO3-, 500 μM NH4NO3 and 1 mM NH4+ for 21 days. Statistical analysis of root length, dry weight, NO3- concentration and NH4+ concentration of roots. The data indicate the mean ± SE (n = 12). a, b, and c indicate significant differences based on ANOVA and Duncan's test (P < 0.05).
Identification and Characterization of lncRNAs
cDNA libraries were constructed from P. × canescens root samples exposed to NO3−, NH4 NO3, and NH4+ for 21 days and sequenced using an Illumina HiSeq™ 4,000 platform. Three biological repeats per treatment level were used to construct the libraries. High-throughput RNA-sequencing (RNA-seq) of these nine libraries led to the generation of 755, 579, 104 clean reads and 114.52 G clean bases (Supplementary Table S2). We subsequently mapped these clean reads to the P. × canescens reference genome to identify the transcripts (Zhou et al., 2015).
A total of 4,042 novel lncRNAs were identified in P. × canescens roots under the different nitrogen fertilization treatments (Supplementary Table S3). The 4,042 lncRNAs were evenly distributed across the chromosomes of poplar, without obvious location preferences (Figure 2A). These lncRNAs were divided into 1,500 intergenic, 1,191 antisense, 632 sense, 409 intronic, and 310 bidirectional lncRNAs based on their genomic locations (Figure 2B; Supplementary Table S3). The length, exon number, and open reading frame (ORF) length of 4,042 lncRNAs were compared with 73,013 transcripts from sequencing. The length of these lncRNAs was between 201 and 4,995 nt. In contrast, the length of approximately 45% of the identified lncRNAs was less than 300 nt, whereas 76% of the mRNAs were longer than 1,000 nt (Figure 2C). Approximately 90% of the lncRNAs were composed of one or two exons, whereas the exon number of mRNAs ranged from one to nine, and approximately 22% of the mRNAs contained seven or more exons (Figure 2D). Approximately 29% of lncRNAs did not have ORFs, and nearly 50% of lncRNAs contained short-chain (<50 residues) ORFs (Figure 2E); in contrast, 79.6% of mRNAs contained 100–700 ORFs (Figure 2F).
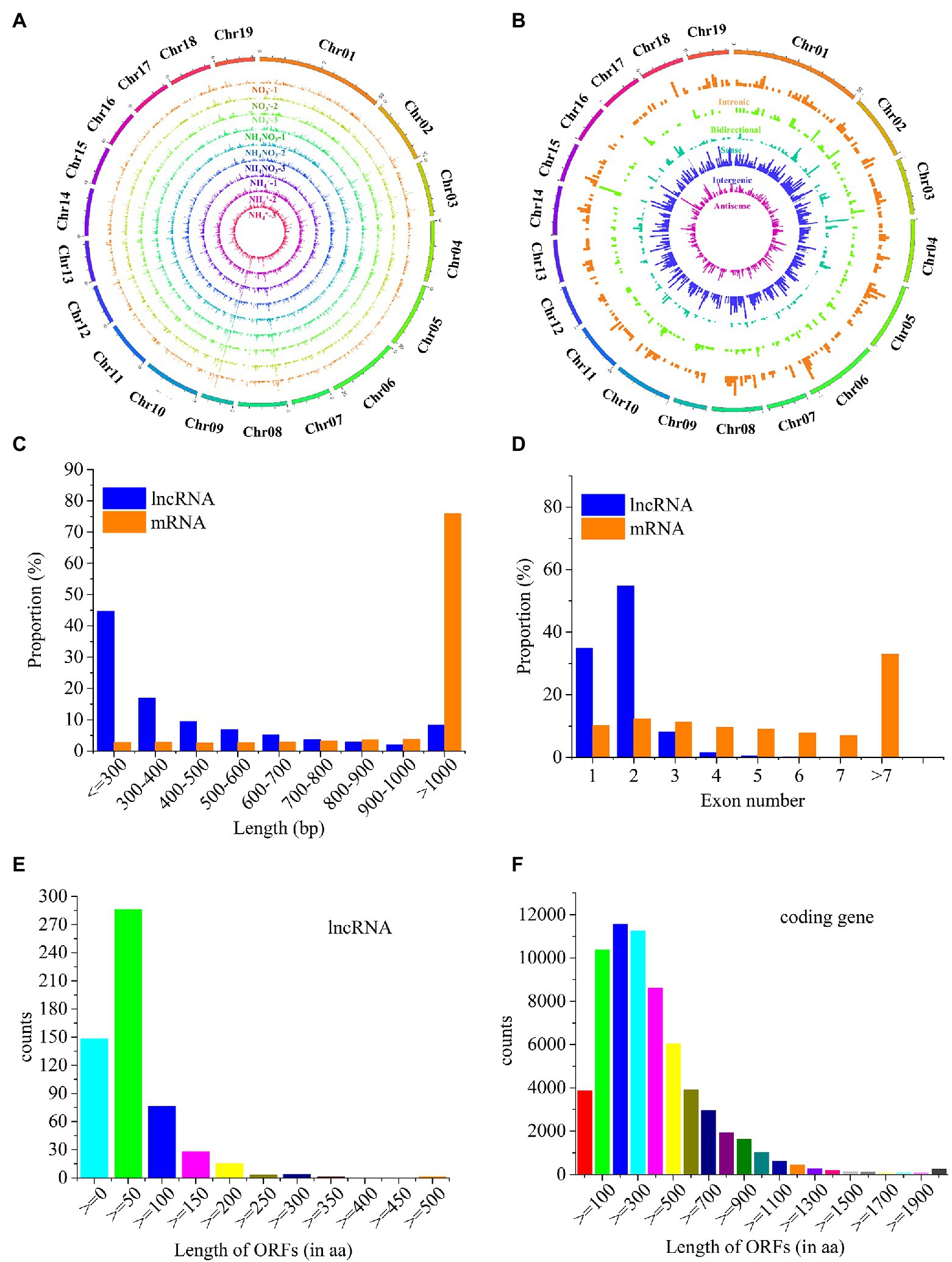
Figure 2. Comparison of structural features of lncRNAs and mRNAs. (A), The expression level of lncRNAs along the 19 chromosomes. (B), Chromosomal distribution of the five types of lncRNAs. (C), Comparison of transcript lengths of lncRNAs and mRNAs. Blue represents lncRNAs, red represents mRNAs. (D), Comparison of exon numbers of lncRNAs and mRNAs. Blue represents lncRNAs, red represents mRNAs. (E), Open reading frame (ORF) length of lncRNAs. (F), ORF length of mRNAs.
Significantly Differentially Expressed lncRNAs Under Different Nitrogen Fertilization Treatments
Among the 4,042 novel lncRNAs, 324 lncRNAs showed differential expression patterns between the NO3− and NH4NO3 treatments, with a |log2(fold change)| value greater than 1 and a p-value less than 0.05, and 333 lncRNAs showed differential expression between the NH4+ and NH4NO3 treatments (Supplementary Table S4). The heatmaps of potential DE-lncRNAs are illustrated in Figure 3. Among the identified DE-lncRNAs between the NO3− and NH4NO3 treatments, 154 lncRNAs were upregulated, and the remaining 170 were downregulated. Moreover, the analysis of the DE-lncRNAs between the NH4+ and NH4NO3 treatments revealed that 168 were upregulated, and the remaining 165 were downregulated (Supplementary Table S4). More interestingly, several DE-lncRNAs (MSTRG.5852.1, MSTRG.29402.1, MSTRG.22198.1, MSTRG.6743.1, MSTRG.24662.1, and MSTRG.5851.3) showed upregulated expression under the NO3− treatment compared with the NH4NO3 treatments but downregulated expression under the NH4+ treatment compared with the NH4NO3 treatment. In addition, MSTRG.12063.1 expression was downregulated under the NO3− treatment compared with the NH4NO3 treatments but upregulated under the NH4+ treatment compared with the NH4NO3 treatment. These results indicated that these lncRNAs have different mechanisms in response to different nitrogen forms in poplar roots. Eighteen DE-lncRNAs with a high number of reads were confirmed by RT–qPCR analysis (Supplementary Figure S2). Although the discrepancies in lncRNA expression levels did not match those obtained by sequencing in terms of magnitude, the trends of upregulation and downregulation were similar.
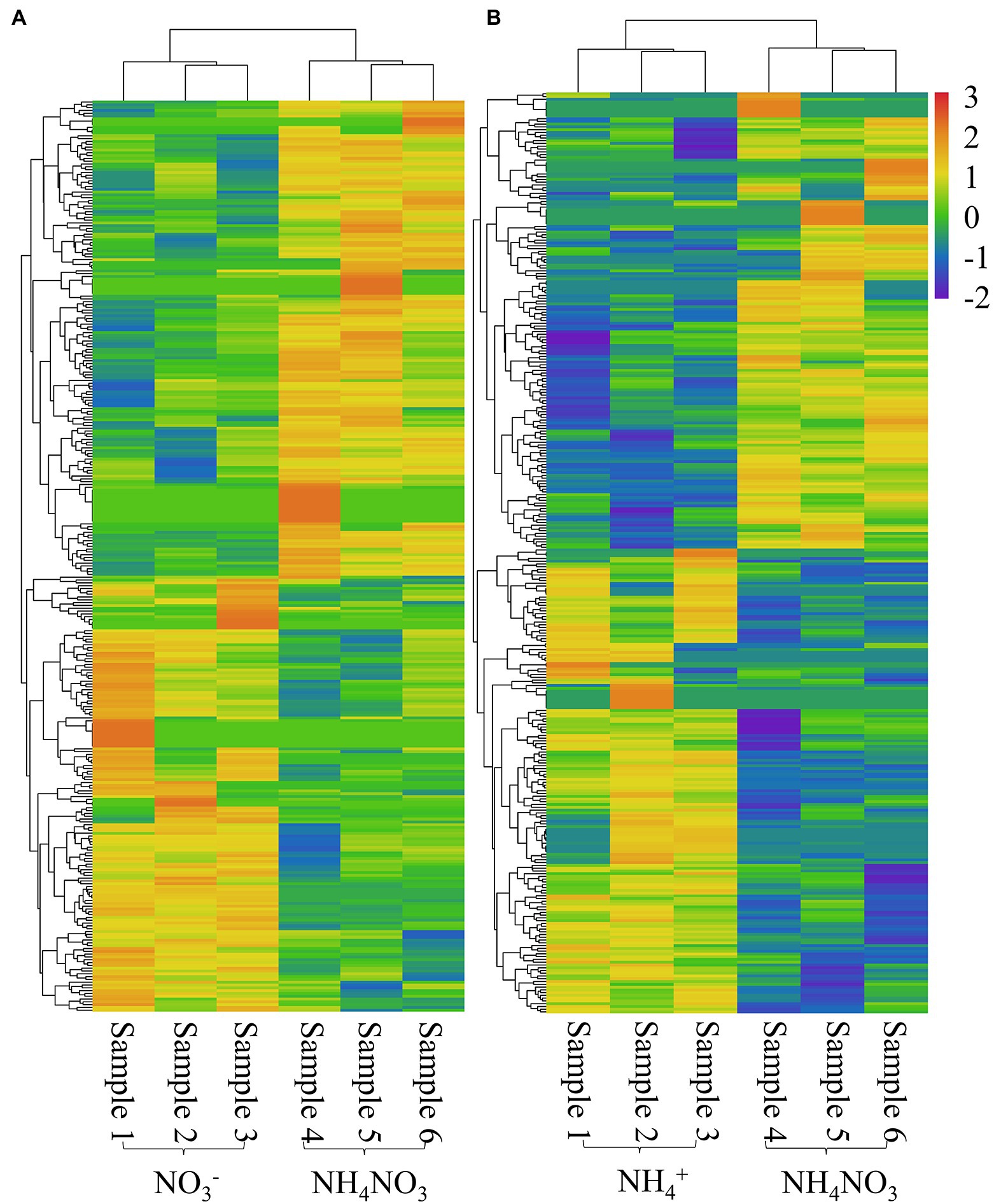
Figure 3. Heatmap of significantly differentially expressed lncRNAs with |log2(fold change)| > 1 and P value < 0.05 in the NO3- vs. NH4NO3 (A) and NH4+ vs. NH4NO3 (B) comparisons of P. x canescens.
Target Analysis and Functional Annotation of DE-lncRNAs
To globally identify mRNAs under P. × canescens exposure to NO3−, NH4NO3, and NH4+, a total of 73,013 mRNAs were identified from the nine libraries using high-throughput sequencing. Among the 73,013 mRNAs, 6,112 DE-mRNAs showing differential expression patterns, with |log2(fold change)| values higher than 1 and p-values less than 0.5, were found between NO3− and NH4NO3. Moreover, 6,007 DE-mRNAs showed differential expression patterns between NH4+ and NH4NO3 (Supplementary Table S5).
To better analyze the roles of DE-lncRNAs, we analyzed the potential cis target mRNAs of DE-lncRNAs. Among these lncRNA-mRNA pairs, 276 differentially expressed cis target mRNAs were predicted between NO3− and NH4NO3, and 265 differentially expressed cis target mRNAs were predicted between NH4+ and NH4NO3 (Supplementary Table S6). Moreover, trans targets of lncRNAs were also predicted. Among them, 561 potential trans target mRNAs, which were significantly differentially expressed, were predicted between NO3− and NH4NO3, and 567 potential trans target mRNAs were predicted between NH4+ and NH4NO3 (Supplementary Table S7). In these networks, we found that the same lncRNA could be coexpressed with multiple transcripts, and multiple lncRNAs were coexpressed with one particular transcript.
To further understand the roles of lncRNAs of potential differentially expressed cis and trans target mRNAs, a KEGG analysis was performed to gain deeper insights into the functions of DE-lncRNA targets (Figure 4). KEGG analysis showed that the target genes obtained from the NO3− vs. NH4NO3 comparison were involved in nitrogen metabolism and plant biosynthesis of amino acids, including valine, leucine, and isoleucine biosynthesis. The analysis of the target genes identified from the NH4+ vs. NH4NO3 comparison revealed that the biosynthesis of the amino acids valine, leucine, and isoleucine and metabolism of the amino acids alanine, aspartate, glutamate, D-glutamine, D-glutamate, cysteine, methionine, and phenylalanine were enriched pathways (Figure 4). These pathways are related to plant nitrogen physiological processes. A GO functional classification analysis was also conducted. The comparison of nitrogen forms at two different levels showed enriched GO terms (Supplementary Figure S3; Supplementary Tables S6, S7). Among the twenty-five biological processes, several main categories, including the regulation of transcription and response to stimulus processes, were enriched. Of the 15 cellular component categories, three important categories, namely, the nucleus, cytoplasm, and plasma membrane, were significantly enriched. Moreover, 15 molecular function categories were identified, and most of the target genes were enriched in the binding and enzymatic activity categories. This result indicated that P. × canescens DE-lncRNAs initiate broad and complex responsive processes that may play a role in binding and enzymatic activity-related functions to adapt to challenges imposed by different nitrogen form treatments (Supplementary Figure S3; Supplementary Tables S6, S7).
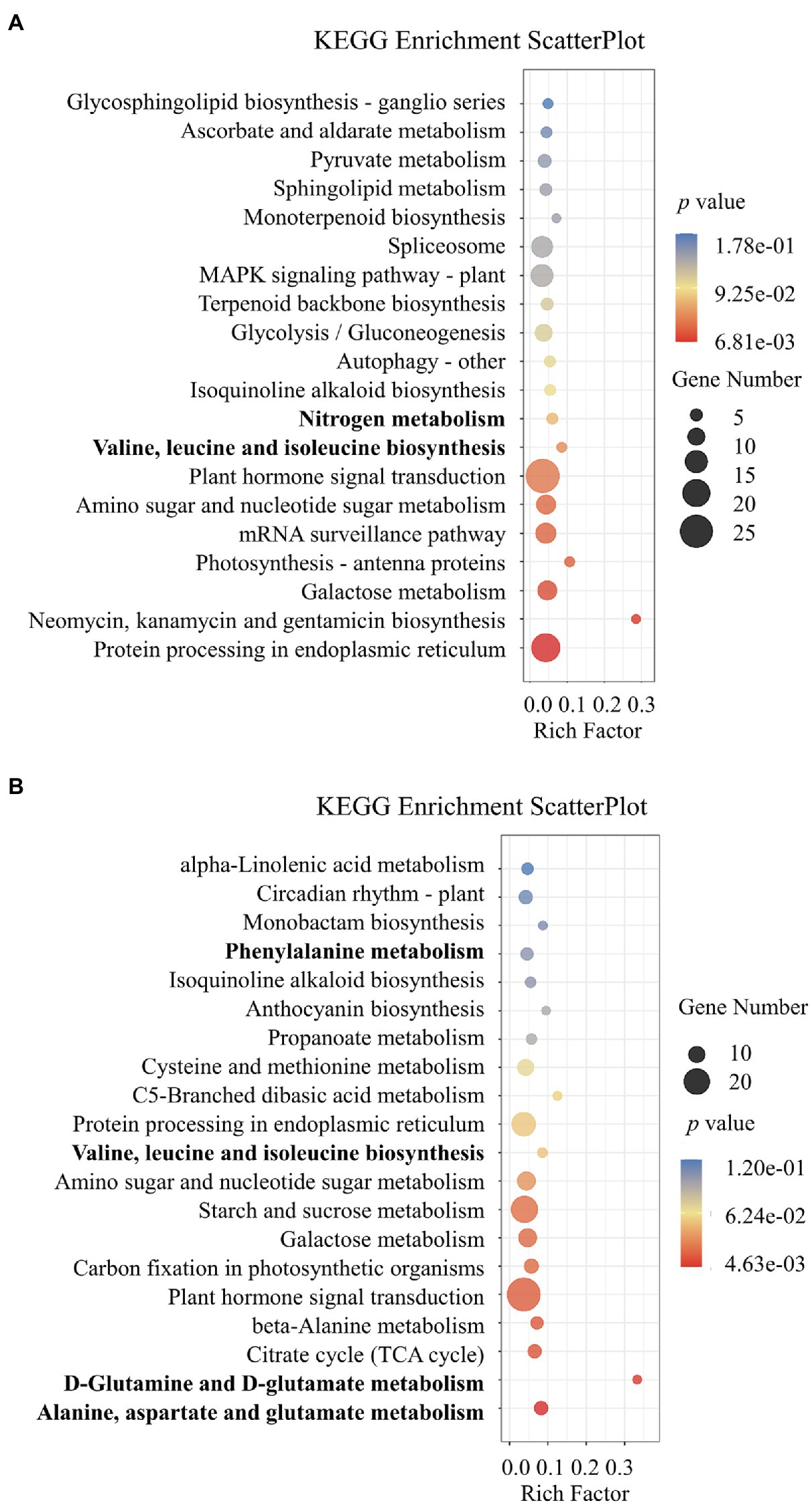
Figure 4. KEGG pathway analysis of identified differentially expressed cis and trans target genes. (A) represent NO3- vs. NH4NO3 comparsion and (B) represent NH4- vs. NH4NO3 comparsion.
lncRNA-mRNA Pairs Participate in Nitrogen Metabolism and Amino Acid Biosynthesis and Metabolism
To identify the lncRNA-mRNA pairs that play key roles in nitrogen metabolism and amino acid metabolism, functional categories were identified using MapMan. The functional categories of the cis and trans target genes of the DE-lncRNAs indicated that 60 DE-lncRNAs corresponding to 49 cis and trans target mRNAs were involved in plant nitrogen metabolism and amino acid biosynthesis and metabolism. In the NO3− vs. NH4NO3 comparison, the MSTRG.2755.1-Potri.001G330900.3 (sulfite oxidase), MSTRG.30530.1-Potri.004G085400.2/3 (glutamine synthetase cytosolic isozyme 1), MSTRG.1921.1-Potri.004G085400.2/3 and MSTRG.7499.1-Potri.006G038400.4 (ferredoxin-dependent glutamate synthase) pairs participated in nitrogen metabolism. The MSTRG.2693.1-Potri.001G323100.1 (protein AUXIN SIGNALING F-BOX 2-like, AFB2) and MSTRG.20178.1-Potri.010G112800.6 (PIN1-like auxin transport protein) pairs participate in hormone metabolism and have been reported to participate in the NO3− level response (Vidal et al., 2010). Thirty pairs also participated in amino acid metabolism. Four transporter peptides (nitrate transmembrane transporter 1.5; NRT1.5) and two transport-related amino acids (amino acid transmembrane transporter 7; APP7) also participated in the NO3− level response (Table 1). In the NH4+ vs. NH4NO3 comparison, the MSTRG.28770.3-Potri.005G172400.1 (nitrate reductase), MSTRG.1771.1-Potri.007G047300.1 (tRNA-dihydrouridine synthase), MSTRG.441.1-Potri.012G113500.1 (glutamate dehydrogenase 2 isoform X1), MSTRG.17448.1-Potri.012G113500.1, MSTRG.1756.1-Potri.015G111000.1 (glutamate dehydrogenase 2) and MSTRG.17448.1-Potri.015G111000.1 pairs participated in nitrogen metabolism. Thirty-seven lncRNA-mRNA pairs participated in amino acid metabolism. Four transporter peptides (NRT1.5) and eighteen transport-related amino acids (APP7) also participated in the NH4+ level response (Table 1). To further analyze the relationship between DE-lncRNAs and their targets, which play key roles in nitrogen metabolism and amino acid metabolism, the expression patterns of the DE-lncRNAs and their targets under three nitrogen fertilization treatments were measured by RT–qPCR (Supplementary Figure S4).
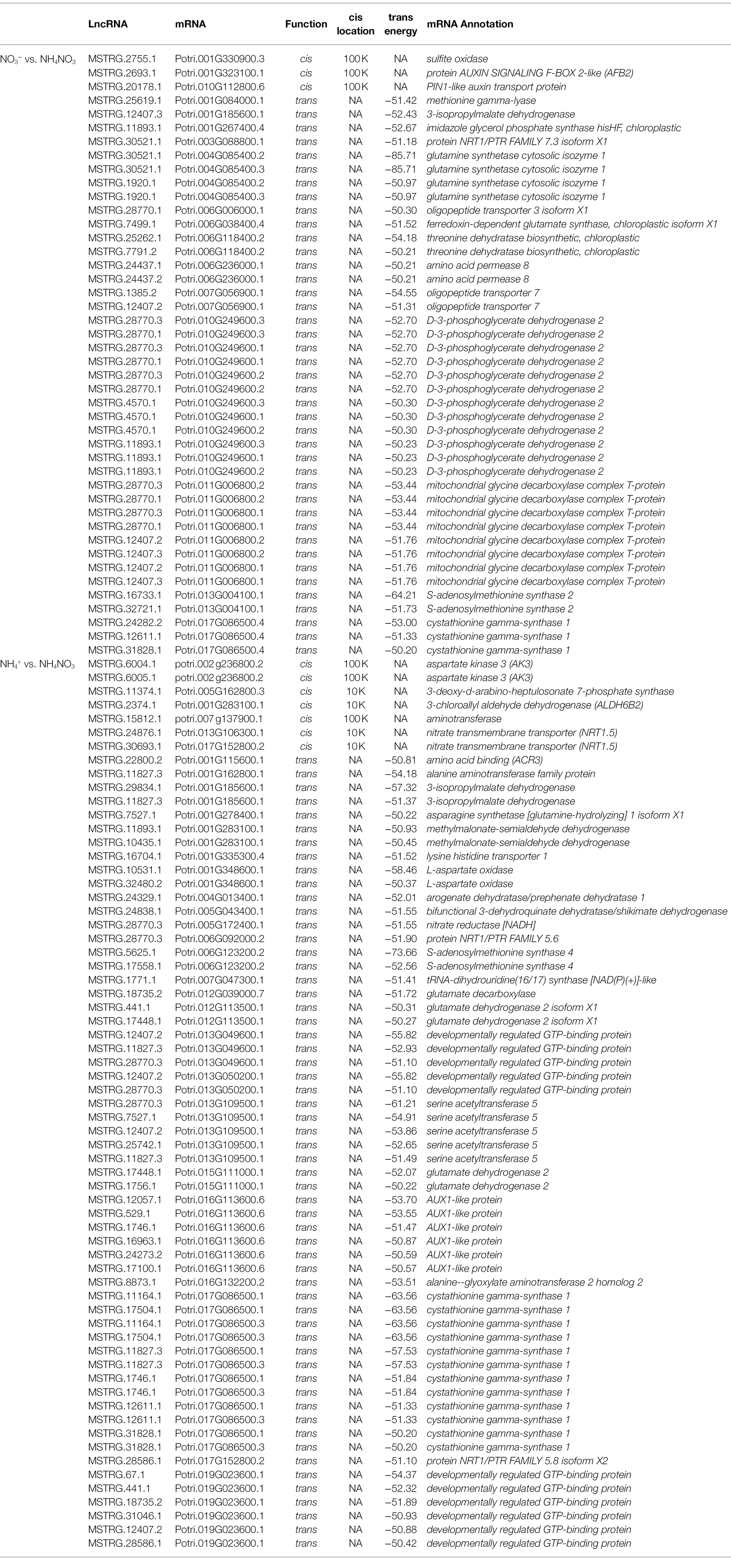
Table 1. lncRNA-mRNA pairs involved in N-metabolism.
LncRNAs as Precursors for Known and Novel miRNAs in P. × canescens
Previous studies have shown that lncRNAs can be associated with miRNAs as precursors of miRNAs (Wang et al., 2018). Using high-throughput sequencing, we identified 465 unique known miRNAs and 29 novel miRNAs in the NO3−, NH4NO3, and NH4+ libraries (Zhou and Wu, 2022a). As a result, 42 lncRNAs were identified as precursors of 60 known miRNAs and 3 novel miRNAs in poplar roots (Supplementary Table S8). Among these, 23 lncRNAs can be used as precursors of two or more miRNAs, and the remaining 19 lncRNAs can only serve as precursors of one miRNA (Figure 5A). In addition, 47 miRNAs might be produced from one lncRNA, and the remaining 15 miRNAs might be generated by two or more different lncRNAs (Figure 5B). For example, MSTRG.12806.1/2 could be aligned with 6 miR396 family member precursors. Moreover, three lncRNAs (MSTRG.10473.1, MSTRG.4365.1, and MSTRG.17270.1) were aligned with 7 miR167 family member precursors in P. × canescens. In addition, one lncRNA (MSTRG.9926.1) matched well with precursors of two novel miRNAs (PC-3p-2543_1573 and PC-5p-360552_9; Supplementary Table S8). Therefore, an in-depth analysis of lncRNAs can provide a method for identifying novel miRNAs in plants.
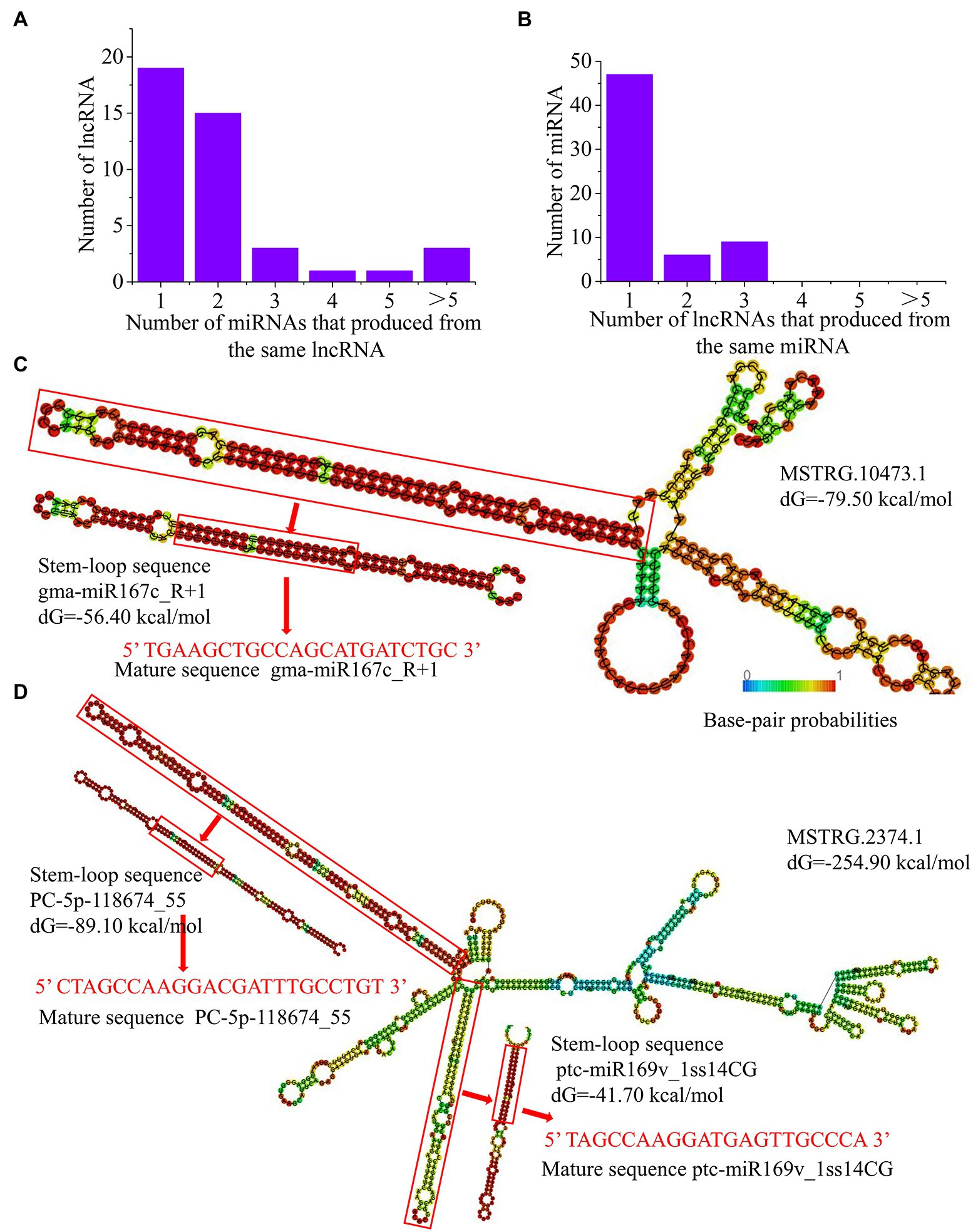
Figure 5. LncRNAs as precursors of miRNAs in P. x canescens. (A), Number of miRNAs produced by lncRNAs. (B), Number of lncRNAs that produce the same miRNA. (C), Predicted secondary structure of MSTRG.10473.1, which contains one precursor sequence of gma-miR167c R+1. (D), Predicted secondary structure of MSTRG.2374.1, which contains two precursors, namely, PC-Sp-118674_55 and ptcmiR169v lss14CG. The secondary structures were predicted using RNAfold. The color scale indicates high (red) to low (blue) probabilities of base pairing.
To further verify whether lncRNAs have the typical stem–loop structures of miRNA precursors, the secondary structures of pre-miRNAs and lncRNAs were predicted with RNAfold. For example, the long arm of the lncRNA MSTRG.10473.1 corresponds to the stem–loop structure of the precursor of gma-miR167c_R + 1, which may be cleaved by an endonuclease complex to release the precursor sequence and ultimately form mature gma-miR167c_R + 1 (Figure 5C). The other lncRNA, MSTRG.2374.1, has two long arms, which was consistent with the stem–loop structure of the precursor sequences of the novel miRNAs PC-5p-118674_55 and ptc-miR169v_1ss14CG (Figure 5D). These lncRNAs can act as miRNA precursors to regulate the expression of mature miRNAs and are involved in nitrogen metabolism and amino acid biosynthesis and metabolism.
LncRNA Transcripts as eTMs of miRNAs in P. × canescens
Recent studies have shown that lncRNAs can act as eTMs for miRNAs and work together to regulate mRNA expression in plants (Zhang et al., 2018). In our investigation, 14 eTMs were targeted by 38 miRNAs between NO3− and NH4NO3, and 14 eTMs were targeted by 58 miRNAs between NH4+ and NH4NO3 (Supplementary Table S9). Among them, one eTM could be targeted by 1–3 miRNAs, whereas one miRNA could pair with several eTMs. For example, in the NO3− vs. NH4NO3 comparison, MSTRG.6920.1 was targeted by 2 miRNAs belonging to the miR11607 and miR7486 family members. Specifically, MSTRG.2693.1 was targeted by a novel miRNA (PC-5p-118674_55) and miR169 family members (Supplementary Table S9). miR171 family members were identified in 3 eTMs (MSTRG.6097.1, MSTRG.27072.1 and MSTRG.13550.1). In the NH4+ vs. NH4NO3 comparison, miR396 family members were identified in 5 eTMs (MSTRG.12806.2, MSTRG.17795.3, MSTRG.33125.2, MSTRG.32317.2 and MSTRG.34633.2) (Supplementary Table S9). Specifically, MSTRG.3753.1 and MSTRG.8235.2 were targeted by 2 novel miRNAs, namely, PC-3p-256804_17 and PC-3p-134932_46, respectively (Supplementary Table S9).
Construction of ceRNA Regulatory Networks
The development of regulatory network research has revealed that lncRNAs, as ceRNAs, participate in the regulation of target miRNA expression. Moreover, miRNAs can target mRNAs, inhibit target translation or degrade mRNAs (Qiu et al., 2012). To further explore the potential regulatory function of DE-lncRNAs, we examined the possible regulation of ceRNA networks in P. × canescens under different nitrogen fertilization treatments (Supplementary Table S10). The miRNA data were based on the results from previous studies (Zhou and Wu, 2022a). The network included 20 DE-lncRNAs, 47 miRNAs, and 143 DE-mRNAs (Figure 6; Supplementary Table S10). DE-mRNAs in the ceRNA regulatory network were assigned to functional categories using MapMan (Supplementary Table S10). Some functional categories, including amino acid metabolism and development, were associated with the regulation of nitrogen metabolism (Supplementary Table S10). Among the identified DE-mRNAs, ATMS1 (methionine synthase), CYSC1 (CYSTEINE SYNTHASE C1), and C2 domain-containing protein, which are targeted by MSTRG.8235.2, MSTRG.3196.1, and MSTRG.33125.2, are responsible for amino acid metabolism. Several targets, including the NFYA1/2/6/7/11 (nuclear transcription factor Y subunit A), ARF1/4 (auxin response factors), MYB116 (myb domain protein 116), and NAC90 (NAC transcription factor) transcription factors, belong to the miR169, miR171, miR166, and miR396 families, respectively. These transcription factors may participate in nitrogen metabolism and plant growth and development (Supplementary Table S10). To further confirm the expression pattern of DE-lncRNAs and DE-mRNAs in ceRNA regulatory networks, we detected several lncRNA-miRNA-mRNA pairs under different nitrogen fertilization treatments by RT–qPCR and found that the DE-lncRNA expression patterns were consistent with those of their corresponding DE-mRNAs (Figure 7).
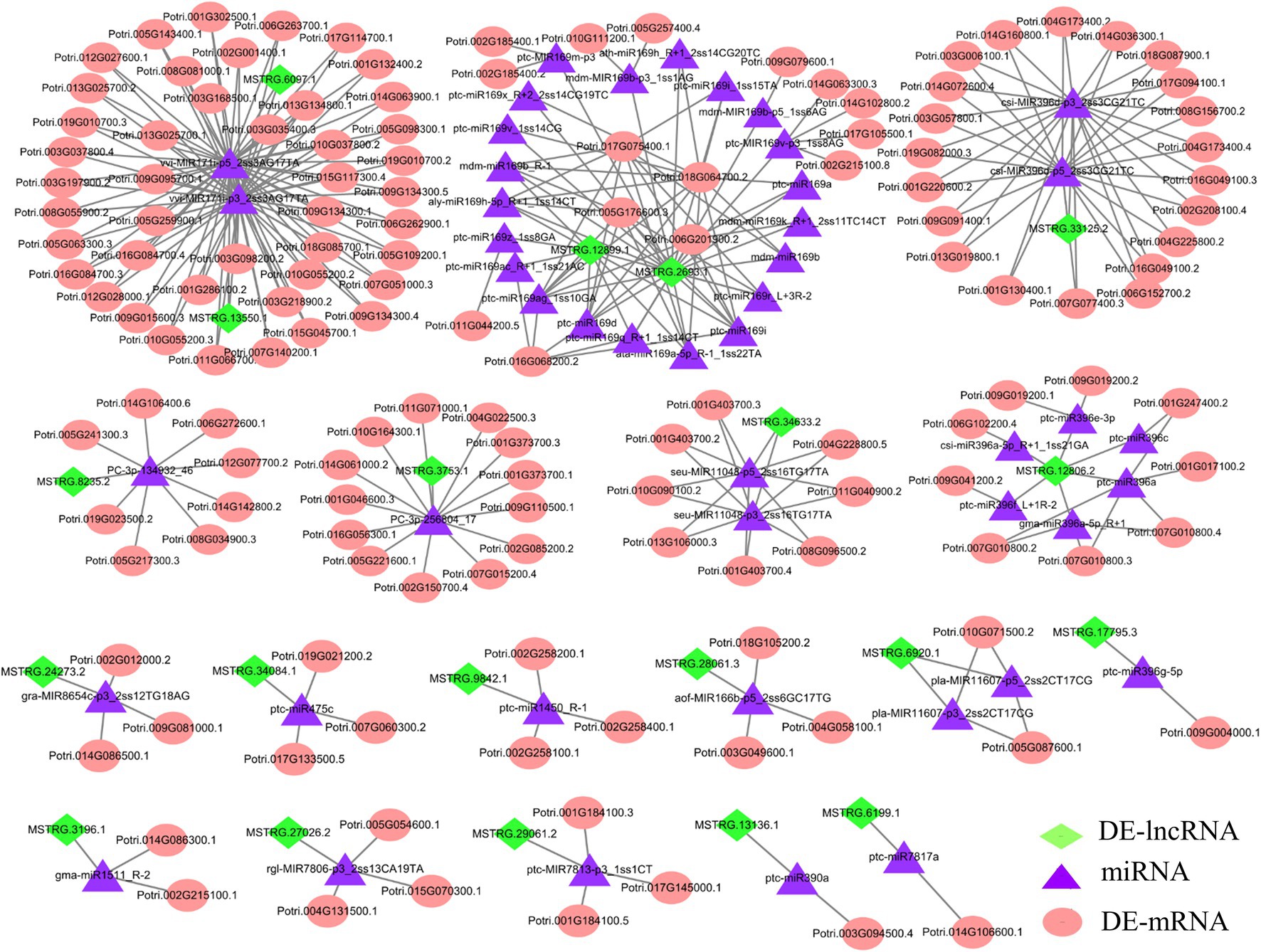
Figure 6. CeRNA networks in P. x canescens roots in response to different nitrogen forms. The green diamond-shaped, purple triangular, and orange round nodes represent DE-lncRNAs, miRNAs, and DE-mRNAs, respectively. Details regarding the abbreviations of the genes are listed in Supplementary Table S10.
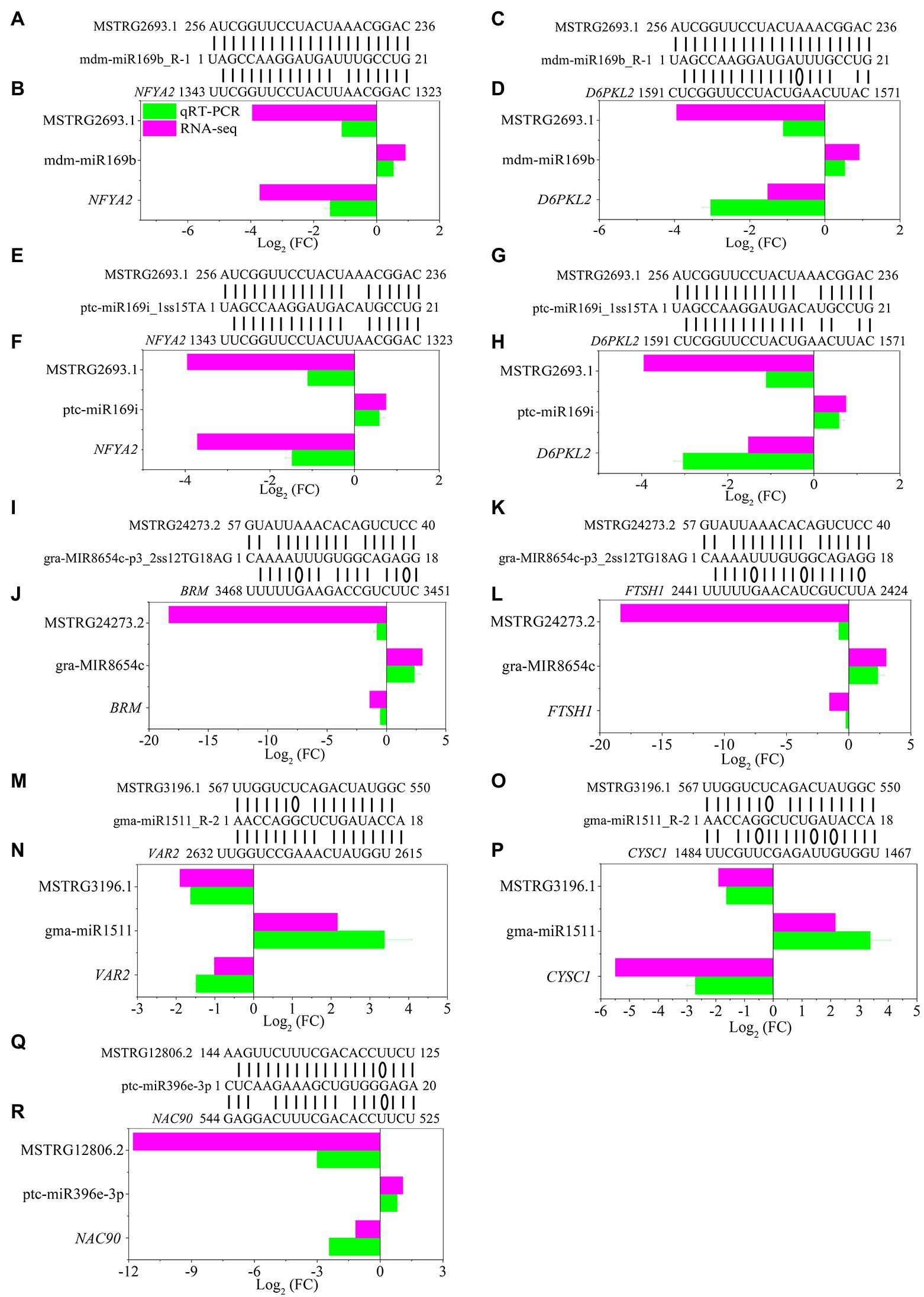
Figure 7. Validation of significantly differentially expressed lncRNAs, miRNAs and mRNAs under different nitrogen fertilization treatments in P. x canescens determined by sRNA-seq and RT—qPCR. (B,D,F,H,J,I) represent NO3- vs. NH4NO3 comparsion and (N,P,R) represent NH4+ vs. NH4NO3 comparsion. The bars show the mean ± SE (n = 4). Sequence complementary positions of predicted lncRNA-miRNA-mRNA pairs (A,C,E,G,I,K,M,O,Q). Details regarding the abbreviations of mRNAs are listed in Supplementary Table S10.
In the ceRNA network, we found that most nodes were connected to MSTRG.13550.1, MSTRG.6097.1, MSTRG.2693.1 and MSTRG.12899.1 (Figure 6; Supplementary Table S10). MSTRG.6097.1 was significantly upregulated in the NO3− vs. NH4NO3 comparison and targeted two miR171 family members (vvi-MIR171i-p3_2ss3AG17TA and vvi-MIR171i-p5_2ss3AG17TA). Because MSTRG.6097.1 targets vvi-MIR171i-p3_2ss3AG17TA and vvi-MIR171i-p5_2ss3AG17TA as ceRNAs, 21 target mRNAs corresponding to two miR171 family members were significantly upregulated. Moreover, in the NH4+ vs. NH4NO3 comparison, MSTRG.13550.1 was significantly downregulated and targeted two miR171 family members (vvi-MIR171i-p3_2ss3AG17TA and vvi-MIR171i-p5_2ss3AG17TA), and 23 target mRNAs corresponding to two miR171 family members were also significantly downregulated. More interestingly, we found that among the 21 and 23 target mRNAs, two target mRNAs (Potri.013G025700.2 and Potri.013G025700.1), which encode serine/threonine-protein kinase CDL1 (CDL1), coexisted under both treatments. However, the pattern of expression was indeed the opposite (Supplementary Table S10). Moreover, in the NO3− vs. NH4NO3 comparison, 19 miR169 family members were targeted to the same lncRNA (MSTRG.2693.1), which was significantly downregulated, resulting in significantly downregulated expression of NFYA3/10. In the NH4+ vs. NH4NO3 comparison, 9 miR169 family members were targeted to the same lncRNA (MSTRG.12899.1), which was significantly upregulated, resulting in the significantly upregulated expression of NFYA2 (Figure 6; Supplementary Table S10).
Experimental Validation of Two lncRNA-miRNA-mRNA Pairs
To validate the role of lncRNAs as eTMs with miRNAs, we carried out transient coexpression in Nicotiana benthamiana leaves. Two lncRNA-miRNA-mRNA pairs were selected for the transient coexpression assays (MSTRG.13550.1-miR171i-PcCDL1.1 and MSTRG.2693.1-miR169b-PcNFYA2). After 2 days of coexpression in N. benthamiana leaves, the upregulation of miR171i-p3 and miR169b_R-1 significantly decreased the transcript levels of the target mRNAs PcCDL1.1 and PcNFYA2, respectively (Figure 8). Coexpression of miR171i-p3 with MSTRG.13550.1 restored the expression level of PcCDL1.1 (Figures 8A,B). Similarly, MSTRG.2693.1 antagonized the inhibitory effects of mdm-miR169b_R-1 on the mRNA levels of PcNFYA2 in poplars, and coexpression of mdm-miR169b_R-1 with MSTRG.2693.1 restored the expression level of PcNFYA2 (Figures 8C,D). These results indicate that MSTRG.13550.1 and MSTRG.2693.1 likely antagonize the inhibitory effects of miR171i-p3 and mdm-miR169b_R-1 on the transcript levels of the target mRNAs PcCDL1.1 and PcNFYA2 in poplar roots, respectively.
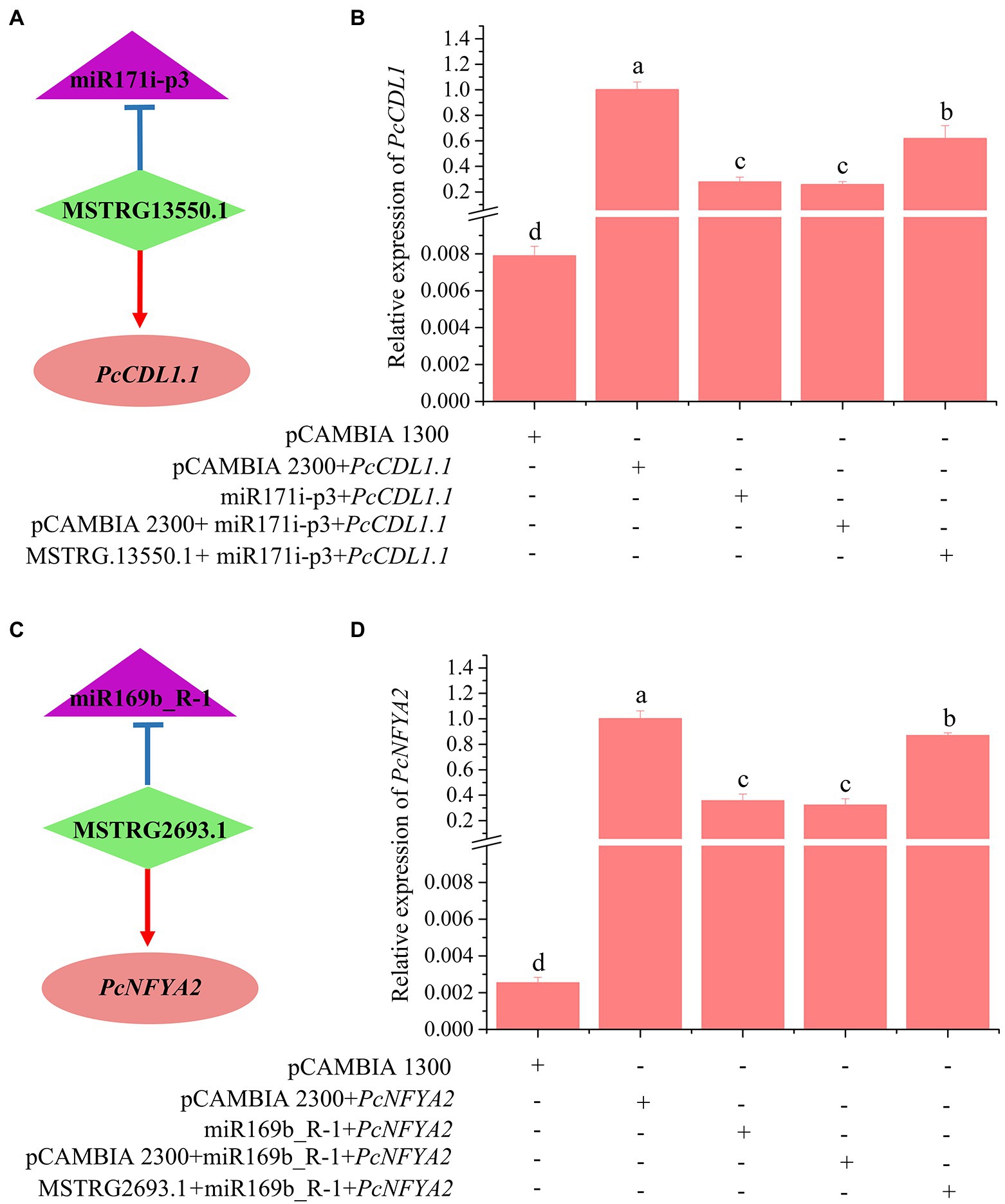
Figure 8. Validation of 1ncRNA-miRNA-mRNA pairs using Nicotiana benthamiana leaves transient coexpression system by RT—qPCR (B,D). The bars show the mean ± SE (n = 4). Different letters on the error bars indicate significant differences. (A), MSTRG.13550.1 targets miR171i-p3 and acts as an eTM for miR171i-p3; (C), MSTRG2693.1 targets miR169b R-1 and acts as an eTM for miR169b R1.
Discussion
Poplar Roots Exhibit Adaptability and Plasticity Under Different Nitrogen Fertilization Treatments
Nitrogen is one of the essential mineral nutrients for plants and plays a very important role in regulating plant growth and development (Kung et al., 2013). NO3− and NH4+, as the main inorganic nitrogen sources in soil, can be absorbed and utilized by plant roots (Oldroyd and Leyser, 2020). Studies have shown that different nitrogen fertilizations can alter the root morphological configuration of plants (Qu et al., 2016) and that the roles of NO3− and NH4+ in root morphogenesis are different (Dai and Zhao, 2011; Luo et al., 2013a), which is due to the dynamic adaptation of the plant root morphological configuration to the dynamic adjustment of nitrogen forms supplied in soil (Hu et al., 2015). In P. simonii × P. nigra, after 21 days of treatment with different nitrogen fertilizations, the root length of poplar when NH4+ served as the only nitrogen source was lower than that obtained under the NO3− and NH4NO3 treatments (Qu et al., 2016), and this finding is consistent with our results. In our study, after 21 days of being supplied with different nitrogen fertilizations, the roots under the NO3− and NH4NO3 treatments were longer than those under the NH4+ treatment. Moreover, the root dry weight under NO3− treatment was higher than that under NH4NO3 treatment, which was also consistent with the findings reported by Rewald et al. (2016). These results indicated that the root system of P. × canescens exhibited strong adaptability and plasticity to changes in the availability of different nitrogen forms.
Many DE-lncRNAs in Poplar Roots Are Involved in the Response to Different Nitrogen Forms
In addition to physiological regulation, molecular regulation, particularly small RNA-mediated regulation, plays a key role in plant nitrogen absorption and assimilation, especially small RNA-mediated regulation (Raheel et al., 2018; Zhou et al., 2020; Zhou and Wu, 2022b). To date, research on small RNAs has focused on lncRNAs and miRNAs with regulatory functions (Qiao et al., 2018; Dos Santos et al., 2019; Lu et al., 2019). For example, in poplar wood, 91 DE-lncRNAs have been found under low nitrogen treatment. These lncRNAs participate in the regulation of wood properties and physiological processes of poplar under low nitrogen stress (Lu et al., 2019). However, the mechanism by which lncRNAs respond to different nitrogen forms in poplar roots has not been studied. In this study, genome-wide identification of lncRNAs was conducted, and a functional analysis of the DE-lncRNAs in poplar root responses to different nitrogen forms was performed. As a result, 324 and 333 lncRNAs showed differential expression patterns in the NO3− vs. NH4NO3 and NH4+ vs. NH4NO3 comparisons, respectively. More interestingly, the same lncRNAs exhibited different expression patterns under different nitrogen fertilization treatments, which suggests that these lncRNAs have different response mechanisms for different nitrogen forms. More attention should thus be given to these lncRNAs, and their functions should be further studied.
The Regulation of cis and trans Target mRNAs by lncRNAs Is Key for the Poplar Root Response to Different Nitrogen Forms
Previous studies have shown that lncRNAs can regulate the expression of their target mRNAs in both cis and trans manners depending on their neighboring gene and complementary pairing of bases (Li et al., 2020). To further resolve the biological functions of the DE-lncRNAs, the differentially expressed cis and trans target mRNAs of DE-lncRNAs were predicted, and the functions of these target genes were annotated. Both KEGG and MapMan analyses indicated that the cis and trans target genes of DE-lncRNAs play an important role in nitrogen metabolism, biosynthesis of amino acids, and plant amino acid metabolism. For example, MSTRG.2755.1 was downregulated in the roots of P. × canescens treated with NO3−, and the transcription of its potential cis target mRNA, Potri.001G330900.3, which encodes sulfite oxidase, was downregulated under NO3− treatment (Supplementary Figure S4A). Potri.001G330900.3 is homologous to Arabidopsis sulfite oxidase (AT3G01910) and is reportedly involved in nitrate metabolism (Qiu et al., 2012). Potri.001G323100.1, a cis target mRNA of downregulated MSTRG.2693.1, was downregulated in the roots of NO3−-treated P. × canescens (Supplementary Figure S4B). This cis target mRNA is homologous to the Arabidopsis protein AUXIN SIGNALING F-BOX 2-like (AFB2). It has been reported that AFB2 expression is induced by NO3− treatment in Arabidopsis roots and affects their growth and development (Vidal et al., 2010). In addition, the lncRNAs MSTRG.6004.1 and MSTRG.6005.1 share the same cis target mRNA, potri.002 g236800.2, which is homologous to an Arabidopsis aspartate kinase (AK3) (AT3G02020) and is reportedly involved in amino acid biosynthesis and metabolism (Less and Galili, 2008; Jander and Joshi, 2009). Moreover, thirty and thirty-seven lncRNA-mRNA pairs identified from the NO3− vs. NH4NO3 and NH4+ vs. NH4NO3 comparisons, respectively, participated in amino acid metabolism. Taken together, the results suggest that these lncRNA-mRNA pairs, as hub genes, might be essential for the response of P. × canescens roots to different nitrogen forms. Moreover, the expression of several hub lncRNA-mRNA pairs was verified by RT–qPCR (Supplementary Figure S4), which further confirmed the accuracy of the sequencing results.
The Function of lncRNAs as miRNA Precursors and eTMs Is Crucial for the Poplar Root Response to Different Nitrogen Forms
Studies have shown that lncRNAs interact with miRNAs as miRNA precursors, target mimics, or targets to affect plant growth and development or in response to abiotic stress (Wu et al., 2013; Lu et al., 2019; Wang et al., 2020b; Feng et al., 2021). Interestingly, we found that the same lncRNA would correspond to multiple miRNA precursors or become an eTM of multiple miRNAs. Similarly, several lncRNAs may correspond to the same miRNA precursor or eTMs becoming the same miRNA (Wang et al., 2020b). This result demonstrated the existence of a complex regulatory interaction between lncRNAs and miRNAs. miR167 is the first reported miRNA involved in the nitrogen response (Gifford et al., 2008). In Arabidopsis, 5 mM NO3− treatment for 12 h inhibits the expression of pericyclic cell miR167 and promotes the expression of its target ARF8 (AUXIN RESPONSE FACTOR 8), which results in an effect on the root growth and development process (Gifford et al., 2008; Gutierrez, 2012). In this study, three lncRNAs (MSTRG.10473.1, MSTRG.4365.1, and MSTRG.17270.1) were aligned with 7 miR167 family member precursors involved in the nitrogen response in P. × canescens roots. This study further suggested that miR167 family members play a key role in regulating plant growth and development by participating in the response to different nitrogen forms. Moreover, this study provides a new idea for the regulatory mechanisms of miR167 family members involved in the response to different nitrogen forms and lays the foundation for studying the regulatory mechanisms of lncRNA-miR167 family members in response to different nitrogen forms.
Competing Endogenous RNA Networks Are Crucial for the Poplar Root Response to Different Nitrogen Forms
Recent studies have shown that lncRNAs mainly interact with miRNAs as ceRNAs of miRNAs, which prevents the interaction between miRNAs and their target mRNAs and thereby enhances the function of encoding transcripts by inhibiting the translation of miRNAs to their target mRNAs (Qiu et al., 2012). The ceRNA network plays a key role in the response of plants to nitrogen deficiency (Chen et al., 2016; Borah et al., 2018; Shin et al., 2018; Lu et al., 2019). For example, in poplar trees, low nitrogen stress leads to downregulated expression of MSTRG.4094.1, which may promote the binding of mir5021-p5 to TIP1.3 and thus lead to a reduction in TIP1:3 transcription and a reduction in the vessel elements and lumina of fibers (Lu et al., 2019). However, the regulatory mechanism of ceRNA networks in poplar roots under different nitrogen forms has not been reported.
In this study, the regulatory mechanism of ceRNA networks in poplar roots under different nitrogen fertilization treatments included 20 DE-lncRNAs, 47 miRNAs, and 143 DE-mRNAs (Figure 6; Supplementary Table S10). In the ceRNA regulation networks, most nodes were connected to MSTRG.6097.1 and MSTRG.13550.1. MSTRG.6097.1 and MSTRG.13550.1, as ceRNA targets for two miR171 family members, warrant attention. Two target mRNAs (Potri.013G025700.2 and Potri.013G025700.1), which encode serine/threonine-protein kinase CDL1 (CDL1), coexisted in both treatment comparisons (NO3− vs. NH4NO3 and NH4+ vs. NH4NO3), but their pattern of expression was indeed the opposite (Figure 9; Supplementary Table S10). The Potri.013G025700 gene is homologous to Arabidopsis serine/threonine-protein kinase (AT1G54820) and is reportedly involved in the nitrate response (Jander and Joshi, 2009).
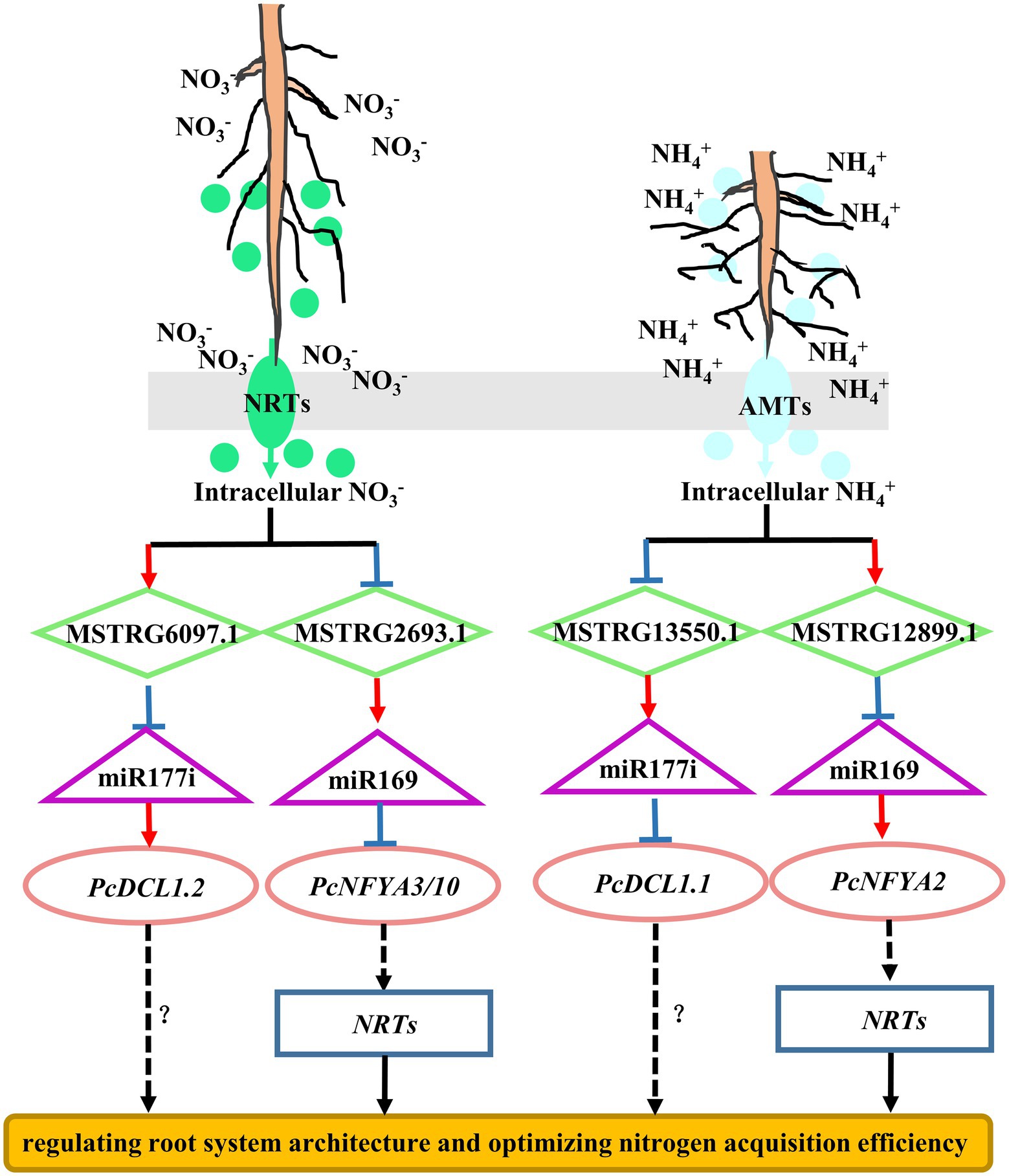
Figure 9. Simple model illustrating MSTRG.6097.1, MSTRG.13550.1, MSTRG.2693.1 and MSTRG.12899.1 as hub lncRNAs in the ceRNA networks. The target genes CDL1 and NFYAs may enable woody plants to optimize nitrogen acquisition efficiency by regulating root system architecture and nitrogen uptake acticity under different nitrogen fertilization treatments. The arrow indicates positive regulation; the T-line represents negative regulation. The solid line represents the experimentally determined pathway, and the dotted line represents the predicted pathway.
Several studies have indicated that NFYA family members play a key role in the response to nitrogen by regulating nitrate transporter (NRT) gene family members in A. thaliana (Fukuda et al., 2019), wheat (Qu et al., 2015), rice (Yu et al., 2018) and poplar (Lu et al., 2019). In wheat, TaNFYA-B1, TANRT1.1, and TANRT2.1 overexpression is induced, which increases NO3− influx in wheat roots and promotes lateral root growth (Qu et al., 2015). In previous studies, we found that the NFYA transcription factor, as the target mRNA of miR169 family members, participates in the alteration of poplar root morphology in response to different nitrogen forms (Zhou and Wu, 2022a). The network obtained in this study also identified several members of the miR169 family (Figure 6; Supplementary Table S10). In the NO3− vs. NH4NO3 comparison, 19 miR169 family members were targeted to the same lncRNA (MSTRG.2693.1), resulting in significantly downregulated expression of NFYA3/10. In the NH4+ vs. NH4NO3 comparison, nine miR169 family members were targeted to the same lncRNA (MSTRG.12899.1), resulting in significantly upregulated expression of NFYA2. These results suggest that different lncRNAs can target different members of the same miRNA family and simultaneously act on different targets under different nitrogen fertilization treatments. Moreover, different members of the same gene family responded to different nitrogen form treatments, which further indicates that the response mechanisms of woody plant roots to different nitrogen forms are different. Therefore, a complex mechanism may exist for ceRNA regulation networks to regulate the expression profile of lncRNAs, miRNAs, and their targets.
In conclusion, MSTRG.6097.1, MSTRG.13550.1, MSTRG. 2693.1, and MSTRG.12899.1, as hub lncRNAs in ceRNA regulation networks, are potential candidate genes for studying the regulatory mechanism in poplar roots under different nitrogen form treatments. Moreover, in the ceRNA network formed by the four candidate hub lncRNAs, the target mRNAs CDL1.1/2 and NFYAs may enable woody plants to optimize nitrogen acquisition efficiency by regulating the root system architecture and nitrogen uptake activity under different nitrogen form treatments (Figure 9). Verification of this hypothesis will require further functional analysis of these candidate lncRNAs through experimental investigation. The results of this study provide clues to comprehensively elucidate the physiological and molecular mechanisms of poplar root responses to different nitrogen forms.
Data Availability Statement
The data presented in the study are deposited in the NCBI repository, accession numbers PRJNA631840, https://dataview.ncbi.nlm.nih.gov/object/PRJNA631840 and PRJNA631845, https://dataview.ncbi.nlm.nih.gov/object/PRJNA631845.
Author Contributions
JZ conceived the experiment and performed most of the experimental work. JZ, L-YY, and XC performed the experiments and data analyses. JZ, W-GS, S-RD, and Z-BL interpreted the experimental data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 32171739 and 31500507) and the Fundamental Research Funds for the Central Nonprofit Research Institution of CAF (grant no. CAFYBB2016QB005).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful for bioinformatics support from Zhang-Yan Xu (LC-Bio Technology Co., Ltd). The platform of the facilities from the State Key Laboratory of Tree Genetics and Breeding is acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.890453/full#supplementary-material
References
Balazadeh, S., Schildhauer, J., Araujo, W. L., Munne-Bosch, S., Fernie, A. R., Proost, S., et al. (2014). Reversal of senescence by N resupply to N-starved Arabidopsis thaliana: transcriptomic and metabolomic consequences. J. Exp. Bot. 65, 3975–3992. doi: 10.1093/jxb/eru119
Bellegarde, F., Gojon, A., and Martin, A. (2017). Signals and players in the transcriptional regulation of root responses by local and systemic N signaling in Arabidopsis thaliana. J. Exp. Bot. 68, 2553–2565. doi: 10.1093/jxb/erx062
Borah, P., Das, A., Milner, M. J., Ali, A., Bentley, A. R., and Pandey, R. (2018). Long non-coding RNAs as endogenous target mimics and exploration of their role in low nutrient stress tolerance in plants. Genes (Basel) 9:459. doi: 10.3390/genes9090459
Budak, H., Kaya, S. B., and Cagirici, H. B. (2020). Long non-coding RNA in plants in the era of reference sequences. Front. Plant Sci. 11:276. doi: 10.3389/fpls.2020.00276
Chekanova, J. A. (2015). Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 27, 207–216. doi: 10.1016/j.pbi.2015.08.003
Chen, Q., Liu, K., Yu, R., Zhou, B., Huang, P., Cao, Z., et al. (2021). From "dark matter" to "star": insight Into the regulation mechanisms of plant functional long non-coding RNAs. Front. Plant Sci. 12:650926. doi: 10.3389/fpls.2021.650926
Chen, M., Wang, C., Bao, H., Chen, H., and Wang, Y. (2016). Genome-wide identification and characterization of novel lncRNAs in Populus under nitrogen deficiency. Mol. Gen. Genomics. 291, 1663–1680. doi: 10.1007/s00438-016-1210-3
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39, W155–W159. doi: 10.1093/nar/gkr319
Dos Santos, T. B., Soares, J. D. M., Lima, J. E., Silva, J. C., Ivamoto, S. T., Baba, V. Y., et al. (2019). An integrated analysis of mRNA and sRNA transcriptional profiles in Coffea arabica L. roots: insights on nitrogen starvation responses. Funct. Integr. Genomics 19, 151–169. doi: 10.1007/s10142-018-0634-8
Feng, S., Fang, H., Liu, X., Dong, Y., Wang, Q., and Yang, K. Q. (2021). Genome-wide identification and characterization of long non-coding RNAs conferring resistance to Colletotrichum gloeosporioides in walnut (Juglans regia). BMC Genomics 22:15. doi: 10.1186/s12864-020-07310-6
Forde, B. G. (2014). Nitrogen signalling pathways shaping root system architecture: an update. Curr. Opin. Plant Biol. 21, 30–36. doi: 10.1016/j.pbi.2014.06.004
Frazee, A. C., Pertea, G., and Jaffe, A. E. (2015). Ballgown bridges the gap between transcriptome assembly and expression analysis. Nature Biotechnol. 33, 243–246. doi: 10.1038/nbt.3172
Fukuda, M., Fujiwara, T., and Nishida, S. (2020). Roles of non-coding RNAs in response to nitrogen availability in plants. Int. J. Mol. Sci. 21:8508. doi: 10.3390/ijms21228508
Fukuda, M., Nishida, S., Kakei, Y., Shimada, Y., and Fujiwara, T. (2019). Genome-wide analysis of long intergenic noncoding RNAs responding to low-nutrient conditions in Arabidopsis thaliana - possible involvement of trans-acting siRNA3 in response to low nitrogen. Plant Cell Physiol. 60, 1961–1973. doi: 10.1093/pcp/pcz048/5406927
Gifford, M. L., Dean, A., Gutierrez, R. A., Coruzzi, G. M., and Birnbaum, K. D. (2008). Cell-specific nitrogen responses mediate developmental plasticity. PNAS 105, 803–808. doi: 10.1073/pnas.0709559105
Gutierrez, R. A. (2012). Systems biology for enhanced plant nitrogen nutrition. Science 336, 1673–1675. doi: 10.1126/science.1217620
He, X. F., Fang, Y. Y., Feng, L., and Guo, H. S. (2008). Characterization of conserved and novel microRNAs and their targets, including a TuMV-induced TIR-NBS-LRR class R gene-derived novel miRNA in brassica. FEBS Lett. 582, 2445–2452. doi: 10.1016/j.febslet.2008.06.011
Hu, B., Wang, W., Ou, S., Tang, J., Li, H., Che, R., et al. (2015). Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 47, 834–838. doi: 10.1038/ng.3337
Jander, G., and Joshi, V. (2009). Aspartate-derived amino acid biosynthesis in Arabidopsis thaliana. Arabidopsis Book 7:e0121. doi: 10.1199/tab.0121
Jha, U. C., Nayyar, H., Jha, R., Khurshid, M., Zhou, M., Mantri, N., et al. (2020). Long non-coding RNAs: emerging players regulating plant abiotic stress response and adaptation. BMC Plant Biol. 20:466, 1–20. doi: 10.1186/s12870-020-02595-x
Jia, J., Zhou, J., Shi, W., Cao, X., Luo, J., Polle, A., et al. (2017). Comparative transcriptomic analysis reveals the roles of overlapping heat−/drought-responsive genes in poplars exposed to high temperature and drought. Sci. Rep. 7:43215. doi: 10.1038/srep43215
Jiao, Y., Chen, Y., Ma, C., Qin, J., Nguyen, T. H. N., Liu, D., et al. (2018). Phenylalanine as a nitrogen source induces root growth and nitrogen-use efficiency in Populus × canescens. Tree Physiol. 38, 66–82. doi: 10.1093/treephys/tpx109
Khan, G. A., Declerck, M., Sorin, C., Hartmann, C., Crespi, M., and Lelandais-Brière, C. (2011). MicroRNAs as regulators of root development and architecture. Plant Mol. Biol. 77, 47–58. doi: 10.1007/s11103-011-9793-x
Kong, L., Zhang, Y., Ye, Z. Q., Liu, X. Q., Zhao, S. Q., Wei, L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35, W345–W349. doi: 10.1093/nar/gkm391
Kung, J. T., Colognori, D., and Lee, J. T. (2013). Long noncoding RNAs: past, present, and future. Genetics 193, 651–669. doi: 10.1534/genetics.112.146704
Less, H., and Galili, G. (2008). Principal transcriptional programs regulating plant amino acid metabolism in response to abiotic stresses. Plant Physiol. 147, 316–330. doi: 10.1104/pp.108.115733
Li, X., Shahid, M. Q., Wen, M., Chen, S., Yu, H., Jiao, Y., et al. (2020). Global identification and analysis revealed differentially expressed lncRNAs associated with meiosis and low fertility in autotetraploid rice. BMC Plant Biol. 20:82. doi: 10.1186/s12870-020-2290-0
Liu, F., Xu, Y., Chang, K., Li, S., Liu, Z., Qi, S., et al. (2019). The long noncoding RNA T5120 regulates nitrate response and assimilation in Arabidopsis. New Phytol. 224, 117–131. doi: 10.1111/nph.16038
Lu, Y., Deng, S., Li, Z., Wu, J., Liu, Q., Liu, W., et al. (2019). Competing endogenous RNA networks underlying anatomical and physiological characteristics of poplar wood in acclimation to low nitrogen availability. Plant Cell Physiol. 60, 2478–2495. doi: 10.1093/pcp/pcz146
Luo, J., Qin, J., He, F., Li, H., Liu, T., Polle, A., et al. (2013a). Net fluxes of ammonium and nitrate in association with H+ fluxes in fine roots of Populus popularis. Planta 237, 919–931. doi: 10.1007/s00425-012-1807-7
Luo, J., Qin, J., He, F., Li, H., Liu, T., Polle, A., et al. (2013b). Net fluxes of ammonium and nitrate in association with H+ fluxes in fine roots of Populus popularis. Planta 237, 919–931. doi: 10.1007/s00425-012-1807-7
Luo, J., Zhou, J., Li, H., Shi, W., Polle, A., Lu, M., et al. (2015). Global poplar root and leaf transcriptomes reveal links between growth and stress responses under nitrogen starvation and excess. Tree Physioloy 35, 1283–1302. doi: 10.1093/treephys/tpv091
Ma, P., Zhang, X., Luo, B., Chen, Z., He, X., Zhang, H., et al. (2021). Transcriptomic and genome-wide association study reveal long noncoding RNAs responding to nitrogen deficiency in maize. BMC Plant Biol. 21:93. doi: 10.1186/s12870-021-02847-4
Masoudi-Nejad, A., Goto, S., Endo, T. R., and Kanehisa, M. (2007). KEGG bioinformatics resource for plant genomics research. Methods Mol. Biol. 406, 437–458. doi: 10.1007/978-1-59745-535-0_21
Massaro, M., De Paoli, E., Tomasi, N., Morgante, M., Pinton, R., and Zanin, L. (2019). Transgenerational response to nitrogen deprivation in Arabidopsis thaliana. Int. J. Mol. Sci. 20:5587. doi: 10.3390/ijms20225587
Naulin, P. A., Armijo, G. I., Vega, A. S., Tamayo, K. P., Gras, D. E., de la Cruz, J., et al. (2020). Nitrate induction of primary root growth requires Cytokinin signaling in Arabidopsis thaliana. Plant Cell Physiol. 61, 342–352. doi: 10.1093/pcp/pcz199
O'Brien, J. A., Vega, A., Bouguyon, E., Krouk, G., Gojon, A., Coruzzi, G., et al. (2016). Nitrate transport, sensing, and responses in plants. Mol. Plant 9, 837–856. doi: 10.1016/j.molp.2016.05.004
Oldroyd, G. E. D., and Leyser, O. (2020). A plant’s diet, surviving in a variable nutrient environment. Science 368:eaba0196. doi: 10.1126/science.aba0196
Patterson, K., Cakmak, T., Cooper, A., Lager, I., Rasmusson, A. G., and Escobar, M. A. (2010). Distinct signalling pathways and transcriptome response signatures differentiate ammonium- and nitrate-supplied plants. Plant Cell Environ. 33, 1486–1501. doi: 10.1111/j.1365-3040.2010.02158.x
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Qiao, Q., Wang, X., Yang, M., Zhao, Y., Gu, J., and Xiao, K. (2018). Wheat miRNA member TaMIR2275 involves plant nitrogen starvation adaptation via enhancement of the N acquisition-associated process. Acta Physiol. Plant. 40, 1–13. doi: 10.1007/s11738-018-2758-9
Qiu, J. A., Wilson, H. L., and Rajagopalan, K. V. (2012). Structure-based alteration of substrate specificity and catalytic activity of sulfite oxidase from sulfite oxidation to nitrate reduction. Biochemistry 51, 1134–1147. doi: 10.1021/bi201206v
Qu, B., He, X., Wang, J., Zhao, Y., Teng, W., Shao, A., et al. (2015). A wheat CCAAT box-binding transcription factor increases the grain yield of wheat with less fertilizer input. Plant Physiol. 167, 411–423. doi: 10.1104/pp.114.246959
Qu, C. P., Xu, Z. R., Hu, Y. B., Lu, Y., Yang, C. J., Sun, G. Y., et al. (2016). RNA-SEQ reveals transcriptional level changes of poplar roots in different forms of nitrogen treatments. Front. Plant Sci. 7:51. doi: 10.3389/fpls.2016.00051
Raheel, S., Widyanti, H. P., Mohammed, A., Mohamed, E., Elsayed, N., Shah, F., et al. (2018). Dynamic roles of microRNAs in nutrient acquisition and plant adaptation under nutrient stress: a review. Plant Omics 11, 58–79. doi: 10.21475/poj.11.01.18.pne1014
Rewald, B., Kunze, M. E., and Godbold, D. L. (2016). NH4: NO3 nutrition influence on biomass productivity and root respiration of poplar and willow clones. GCB Bioenergy 8, 51–58. doi: 10.1111/gcbb.12224
Ruan, L., Wei, K., Wang, L., Cheng, H., Zhang, F., Wu, L., et al. (2016). Characteristics of NH4(+) and NO3(−) fluxes in tea (Camellia sinensis) roots measured by scanning ion-selective electrode technique. Sci. Rep. 6:38370. doi: 10.1038/srep38370
Shin, S. Y., Jeong, J. S., Lim, J. Y., Kim, T., Park, J. H., Kim, J. K., et al. (2018). Transcriptomic analyses of rice (Oryza sativa) genes and non-coding RNAs under nitrogen starvation using multiple omics technologies. BMC Genomics 19:532. doi: 10.1186/s12864-018-4897-1
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41:e166. doi: 10.1093/nar/gkt646
Sun, J., Yan, J., Yuan, X., Yang, R., Dan, T., Wang, X., et al. (2016). A computationally constructed ceRNA interaction network based on a comparison of the SHEE and SHEEC cell lines. Cell. Mol. Biol. Lett. 21:21. doi: 10.1186/s11658-016-0022-0
Sun, X., Zheng, H., and Sui, N. (2018). Regulation mechanism of long non-coding RNA in plant response to stress. Biochem. Biophys. Res. Commun. 503, 402–407. doi: 10.1016/j.bbrc.2018.07.072
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Urquiaga, M. C. O., Thiebaut, F., Hemerly, A. S., and Ferreira, P. C. G. (2020). From trash to luxury: the potential role of plant LncRNA in DNA methylation during abiotic stress. Front. Plant Sci. 11:603246. doi: 10.3389/fpls.2020.603246
Vidal, E. A., Araus, V., Lu, C., Parry, G., Green, P. J., Coruzzi, G. M., et al. (2010). Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. PNAS 107, 4477–4482. doi: 10.1073/pnas.0909571107
Voshall, A., Kim, E. J., Ma, X., Yamasaki, T., Moriyama, E. N., and Cerutti, H. (2017). miRNAs in the alga Chlamydomonas reinhardtii are not phylogenetically conserved and play a limited role in responses to nutrient deprivation. Sci. Rep. 7:5462. doi: 10.1038/s41598-017-05561-0
Wang, J., Chen, Q., Wu, W., Chen, Y., Zhou, Y., Guo, G., et al. (2020a). Genome-wide analysis of long non-coding RNAs responsive to multiple nutrient stresses in Arabidopsis thaliana. Funct. Integr. Genomics 21, 17–30. doi: 10.1007/s10142-020-00758-5
Wang, X., Feng, C., Tian, L., Hou, C., Tian, W., Hu, B., et al. (2021). A transceptor-channel complex couples nitrate sensing to calcium signaling in Arabidopsis. Molecular Plant online 14, 774–786. doi: 10.1016/j.molp.2021.02.005
Wang, Y., Zhang, H., Li, Q., Jin, J., Chen, H., Zou, Y., et al. (2020b). Genome-wide identification of lncRNAs involved in fertility transition in the photo-Thermosensitive genic male sterile Rice line Wuxiang S. Front. Plant Sci. 11:580050. doi: 10.3389/fpls.2020.580050
Wang, T., Zhao, M., Zhang, X., Liu, M., Yang, C., Chen, Y., et al. (2017). Novel phosphate deficiency-responsive long non-coding RNAs in the legume model plant Medicago truncatula. J. Exp. Bot. 68, 5937–5948. doi: 10.1093/jxb/erx384
Wang, Z., Zhu, T., Ma, W., Wang, N., Qu, G., Zhang, S., et al. (2018). Genome-wide analysis of long non-coding RNAs in Catalpa bungei and their potential function in floral transition using high-throughput sequencing. BMC Genet. 19:86. doi: 10.1186/s12863-018-0671-2
Wei, H., Yordanov, Y. S., Georgieva, T., Li, X., and Busov, V. (2013). Nitrogen deprivation promotes Populus root growth through global transcriptome reprogramming and activation of hierarchical genetic networks. New Phytol. 200, 483–497. doi: 10.1111/nph.12375
Wu, H. J., Wang, Z. M., Wang, M., and Wang, X. J. (2013). Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 161, 1875–1884. doi: 10.1104/pp.113.215962
Yu, C., Chen, Y., Cao, Y., Chen, H., Wang, J., Bi, Y. M., et al. (2018). Overexpression of miR169o, an overlapping MicroRNA in response to both nitrogen limitation and bacterial infection, promotes nitrogen use efficiency and susceptibility to bacterial blight in Rice. Plant Cell Physiol. 59, 1234–1247. doi: 10.1093/pcp/pcy060
Zhang, G., Chen, D., Zhang, T., Duan, A., Zhang, J., and He, C. (2018). Transcriptomic and functional analyses unveil the role of long non-coding RNAs in anthocyanin biosynthesis during sea buckthorn fruit ripening. DNA Res. 25, 465–476. doi: 10.1093/dnares/dsy017
Zhang, C., Meng, S., Li, Y., and Zhao, Z. (2014). Net NH4+ and NO3− fluxes, and expression of NH4+ and NO3− transporter genes in roots of Populus simonii after acclimation to moderate salinity. Trees 28, 1813–1821. doi: 10.1007/s00468-014-1088-9
Zhou, X., Jacobs, T. B., Xue, L. J., Harding, S. A., and Tsai, C. J. (2015). Exploiting SNPs for biallelic CRISPR mutations in the outcrossing woody perennial Populus reveals 4-coumarate:CoA ligase specificity and redundancy. New Phytol. 208, 298–301. doi: 10.1111/nph.13470
Zhou, J., Liu, M., Jiang, J., Qiao, G., Lin, S., Li, H., et al. (2012). Expression profile of miRNAs in Populus cathayana L. and Salix matsudana Koidz under salt stress. Mol. Biol. Rep. 39, 8645–8654. doi: 10.1007/s11033-012-1719-4
Zhou, J., Lu, Y., Shi, W. G., Deng, S. R., and Luo, Z. B. (2020). Physiological characteristics and RNA sequencing in two root zones with contrasting nitrate assimilation of Populus × canescens. Tree Physiol. 40, 1392–1404. doi: 10.1093/treephys/tpaa071
Zhou, J., and Wu, J. T. (2022a). Nitrate/ammonium-responsive microRNA-mRNA regulatory networks affect root system architecture in Populus × canescens. BMC Plant Biol. 22, 96. doi: 10.1186/s12870-022-03482-3
Keywords: nitrate, ammonium, Populus × canescens, roots, lncRNAs, ceRNAs
Citation: Zhou J, Yang L-Y, Chen X, Shi W-G, Deng S-R and Luo Z-B (2022) Genome-Wide Identification and Characterization of Long Noncoding RNAs in Populus × canescens Roots Treated With Different Nitrogen Fertilizers. Front. Plant Sci. 13:890453. doi: 10.3389/fpls.2022.890453
Edited by:
Francisco M. Cánovas, University of Malaga, SpainReviewed by:
Rafael Antonio Cañas, University of Malaga, SpainYuepeng Song, Beijing Forestry University, China
Copyright © 2022 Zhou, Yang, Chen, Shi, Deng and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Zhou, Z2FoYTIwMDhAMTI2LmNvbQ==