
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 24 June 2022
Sec. Plant Metabolism and Chemodiversity
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.882189
This article is part of the Research Topic Plant-derived natural compounds in drug discovery: The prism perspective between plant phylogeny, chemical composition, and medicinal efficacy View all 13 articles
 Jing Wang1,2
Jing Wang1,2 Jun Qian1
Jun Qian1 Yuan Jiang1Xiaochen Chen2
Yuan Jiang1Xiaochen Chen2 Baojiang Zheng2
Baojiang Zheng2 Shilin Chen3Fajian Yang4
Shilin Chen3Fajian Yang4 Zhichao Xu1,2*
Zhichao Xu1,2* Baozhong Duan1*
Baozhong Duan1*Members of Polygonatum are perennial herbs that have been widely used in traditional Chinese medicine to invigorate Qi, moisten the lung, and benefit the kidney and spleen among patients. However, the phylogenetic relationships and intrageneric taxonomy within Polygonatum have long been controversial because of the complexity of their morphological variations and lack of high-resolution molecular markers. The chloroplast (cp) genome is an optimal model for deciphering phylogenetic relationships in related families. In the present study, the complete cp genome of 26 species of Trib. Polygonateae were de novo assembled and characterized; all species exhibited a conserved quadripartite structure, that is, two inverted repeats (IR) containing most of the ribosomal RNA genes, and two unique regions, large single sequence (LSC) and small single sequence (SSC). A total of 8 highly variable regions (rps16-trnQ-UUG, trnS-GCU-trnG-UCC, rpl32-trnL-UAG, matK-rps16, petA-psbJ, trnT-UGU-trnL-UAA, accD-psaI, and trnC-GCA-petN) that might be useful as potential molecular markers for identifying Polygonatum species were identified. The molecular clock analysis results showed that the divergence time of Polygonatum might occur at ∼14.71 Ma, and the verticillate leaf might be the ancestral state of this genus. Moreover, phylogenetic analysis based on 88 cp genomes strongly supported the monophyly of Polygonatum. The phylogenetic analysis also suggested that Heteropolygonatum may be the sister group of the Polygonatum, but the Disporopsis, Maianthemum, and Disporum may have diverged earlier. This study provides valuable information for further species identification, evolution, and phylogenetic research of Polygonatum.
Polygonatum Mill (1754) is an essential medicinal and edible species widely distributed in warm–temperate zones of the Northern Hemisphere and Northeastern Asia (Floden, 2015). There are approximately 70 species recognized worldwide (Floden and Schilling, 2018), with 39 present in China, 20 of which are endemic to the region (Chen and Tamura, 2000). The underground rhizomes of Polygonatum have crucial medicinal value in moistening lungs, relieving thirst, replenishing the spleen, and increasing immunity (Jiao et al., 2018a). Among them, four species [Polygonatum odoratum (Mill.) Druce, Polygonatum sibiricum Red., Polygonatum cyrtonema Hua, and Polygonatum kingianum Coll. et Hemsl] were listed in the Chinese Pharmacopoeia (Chinese Pharmacopoeia Commission, 2020). Modern studies have demonstrated that some Polygonatum species were rich in nutrients and functional components and were regarded as a new enormous potential miscellaneous grain (Si and Zhu, 2021). The previous survey has revealed that Polygonati rhizoma is often contaminated with several common adulterants in herbal markets, such as Polygonatum cirrhifolium, Polygonatum humile, Polygonatum stenophyllum, Polygonatum filipes, and Polygonatum verticillatum (Yang et al., 2015; Jiao et al., 2018b; Wang Y. et al., 2019; Wang Z. W. et al., 2019). Because the morphology of these species is similar, changeable, and indistinguishable, it seriously affects the safety and effectiveness of clinical drug use (Ali et al., 2021).
In addition, the phylogenetic position of Polygonatum has been controversial for many years. Some previous taxonomy places the genus within Convallariaceae, Ruscaceae, and Asparagaceae based on morphological and molecular phylogenies (Tang, 1978; Rudall et al., 2000; Kim et al., 2012). Concerning the intraspecific relationship of Polygonatum, according to the leaf order, Baker and Esq (1875) divided it into three groups: section (sect.) Alternifolia (=sect. Polygonatum), sect. Verticillata, and sect. Oppositifolia, but Tang (1978) considered that the classification mentioned above might be inappropriate for identification. He divided the genus into eight series (ser.): including ser. Alternifolia, ser. Altelobata, ser. Bracteata, ser. Punctata, ser. Kingiana, ser. Hookeriana, ser. Verticillata, and ser. Oppositifolia. Tamura (1993) considered that Polygonatum could be divided into sect. Polygonatum and sect. Verticillata according to stamen morphology, chromosome number, karyotype, and filament. Based on the rpl16 gene and trnK gene, Wu et al. (2000) found that the opposite leaves and verticillate leaves of Polygonatum were polyphyletic. More recent molecular phylogenies based on one to several genes have suggested that the Polygonatum could be divided into three groups (Meng et al., 2014; Floden and Schilling, 2018), recommended that the petA-psbJ plastid gene region is combined with the nuclear ribosomal ITS for Polygonatum identification. However, the phylogenetic position of some species [i.e., P. franchetii Hua, P. alternicirrhosum Hand.-Mzt., P. verticillatum (L.) All., P. punctatum Royle ex Kunth] is in dispute (Meng et al., 2014; Floden, 2017; Floden and Schilling, 2018; Jiao, 2018; Zhao et al., 2019; Xia et al., 2022). Moreover, the current taxonomy of Tribe (Trib.) Polygonateae has long been debated, which has been divided into three to eight genera. For example, the genus was divided into eight genera: Polygonatum, Disporopsis, Smilacina, Maianthemum, Disporum, Clintonia, and Streptopus (Tang, 1978). Whereas Tamura et al. (1997) proposed three genera: Polygonatum, Disporopsis, and Heteropolygonatum. Thus, the boundaries and relationships of Trib. Polygonateae remain problematic.
Despite these potential issues, using the chloroplast (cp) genome for phylogenetic estimates generally shows promise for resolving deep relationships among the plant lineages (Nie et al., 2020). Compared with the traditional DNA fragments, the cp genome was relatively conserved and slightly varied (Liang et al., 2020). The method has recently been applied to many research fields, such as taxonomic revision, systematic evolution, and species identification (Chen et al., 2019; Henriquez et al., 2020). Floden and Schilling (2018) and Xia et al. (2022) used cp genome data to reconstruct the phylogeny of Polygonatum, and the results supported the three groups and their sister relationship with Heteropolygonatum. Although these studies resolved the phylogenetic relationships of some species of Polygonatum, the phylogenetic relationships among the genera of Trib. Polygonateae and some species of Polygonatum were still unclear. In addition, the reliability of some analyses still needs to be further clarified due to the limited number of samples in the previous study. Given this, it is necessary to provide further support for the intra-generic relationships, divergence times, and genomic characteristics of Trib. Polygonateae based on a larger sample size. In the present study, we de novo assembled and annotated the cp genome of 26 species, including 23 species of Asparagaceae (18 species of Polygonatum, four species of Disporopsis, and one species of Maianthemum), and three species of Disporum (Colchicaceae). Besides, comparative analysis and phylogenetic evolution of the cp genome were conducted. The present results provide a basis for species identification, phylogenetic studies, resource development, and utilization of Polygonatum medicinal plants.
The fresh and healthy leaves of Polygonatum, Disporopsis, Maianthemum, and Disporum were collected in the field or Germplasm Resource Garden (China), and then the leaf tissue was frozen fresh at –20°C. Numbers after taxa names refer to the locality, and sample information is shown in Supplementary Figure 1 and Supplementary Table 1. The specimens were identified following the taxonomic key and external morphology diagnosis proposed by related literature (Tang, 1978). The voucher specimens have been deposited at the herbarium of Dali University. Total genomic DNA was extracted from tissue samples using the Plant Genomic DNA kit (Tiangen, Beijing, China). The extracted DNA was quantified on a Nanodrop 2000 spectrophotometer (Nanodrop Technologies, Thermo Scientific, United States), and all PCR products were tested for the presence of amplified products on agarose gels. The library of each sample was prepared using 30 μl of high-quality (>100 ng/) genomic DNA. All libraries were sequenced on the Illumina NovaSeq system (Illumina, San Diego, CA, United States).
The pair-end reads were trimmed for adapter and low-quality reads (Phred score < 30) using NGS QC Toolkit v.2.3.3 software. The cp genomes of P. sibiricum (NC029485), Disporopsis fuscopicta (MW248136), Maianthemum bicolor (NC035970), and Disporum cantoniense (MW759302) were downloaded from the National Center of Biotechnology Information (NCBI). The genome above was then used as the reference sequence. The cp genome was assembled using GetOrganelle v.1.6.4, exploiting Bowtie2 v.2.4.4, SPAdes v.3.13.0, and Blast v.2.5.0 as dependencies (get_organelle_from_reads.py -1 R1.fq -2 R2.fq -o cp_output -R 15 -k 21,45,65,85,105 -F embplant) (Jin et al., 2020). All clean reads were mapped to the database, and then the mapping data were extracted based on similarity and coverage. Subsequently, the assembled contigs were visualized and removed redundant sequence by Bandage v.0.8 to generate the complete circular cp genome (Wick et al., 2015). Finally, the reads were remapped to assembled cp genome by Bowtie2, and Jellyfish v.2.2.3 was then used to determine the reverse repeat region boundary. After assembly, circular cp genomes were annotated using online tools CpGAVAS2 and GeSeq based on the reference cp genome (Michael et al., 2017; Shi et al., 2019). The Apollo was used to correct the start codons, stop codons, and intron/exon boundaries (Lewis et al., 2002). Annotated cp genome sequences were submitted to the GenBank database of the NCBI to obtain specific accession numbers (Table 1). Fully annotated cp genome circle diagrams were drawn by OrganellarGenomeDRAW (OGDRAW) online (Lohse et al., 2007).
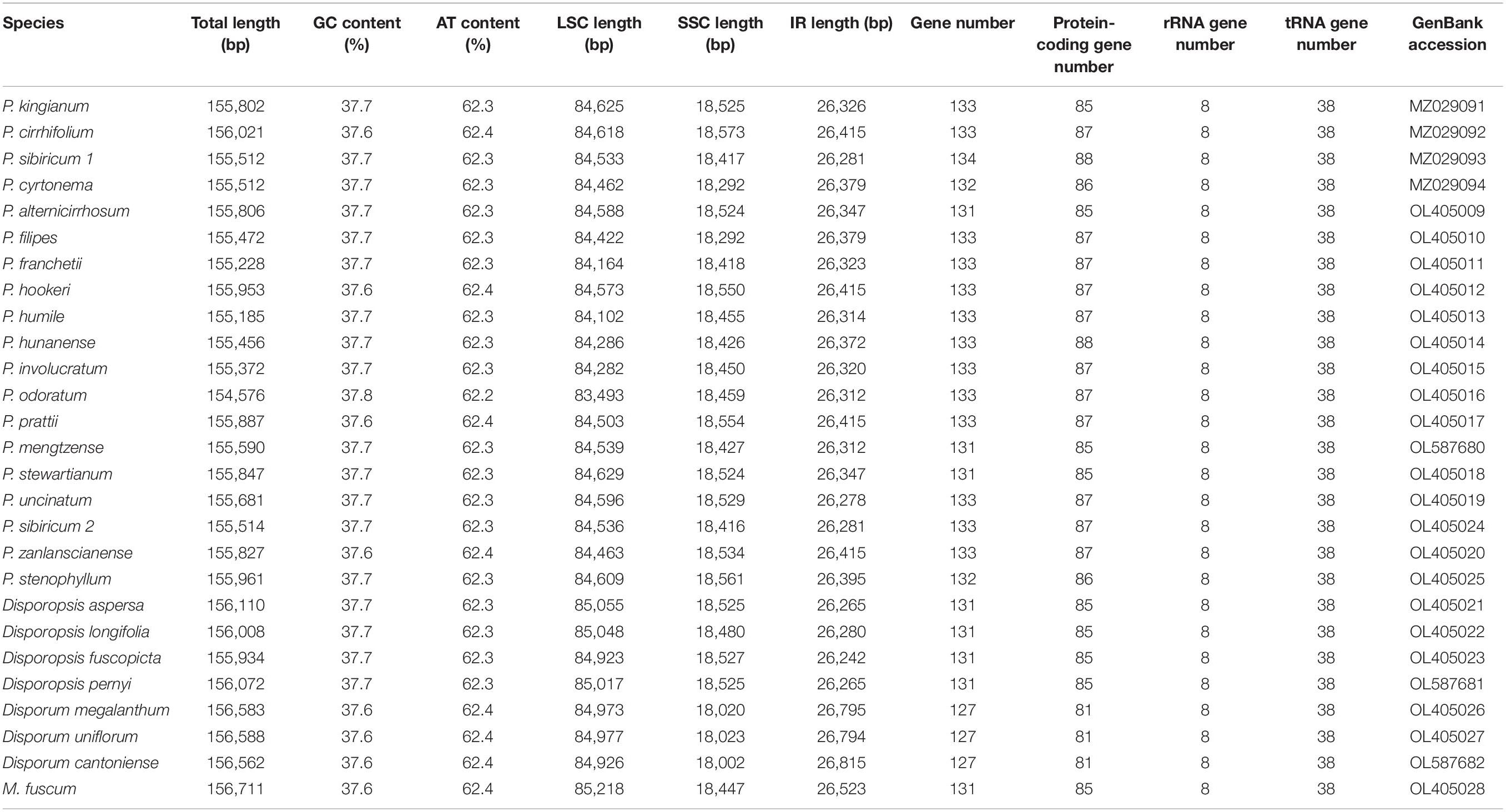
Table 1. Summary of cp genome features.
The GC content was analyzed using Geneious v.9.0. Four types of the dispersed repeat sequence, including forward (F), complementary (C), palindromic (P), and reverse (R), were detected using the REPuter program1 (Stefan et al., 2001). Tandem repeats were detected using Phobos v.3.3.122 (Mayer et al., 2010) with default parameter values. The cp genome of P. sibiricum (MZ029093) was selected as a reference for coordinate positions, and indels and SNPs were counted within the non-overlapping 150 bp window for 18 Polygonatum plastomes (Supplementary Table 7a; Liu et al., 2020). The region IRb was removed for the analyses of repeats to avoid over-representing the repeats following Abdullah et al. (2019). Spearman’s Rho correlations were calculated based on substitutions, Indels, and oligonucleotide repeats using Minitab v. 18 (Akoglu, 2018).
The criteria for repeat determination include a minimum repeat size of 20 bp with the similarity between repeat pairs of 90% by putting edit value 3. Furthermore, the simple sequence repeats (SSRs) were analyzed using MISA software3 with the parameters of “10” for mono-, “5” for di-, “4” for tri-, and “3” for tetra- and penta- nucleotide motifs (Beier et al., 2017). The cp genomes were compared with mVISTA under the Shuffle-LAGAN mode. The cp genome junctions were visualized and compared using IRscope4 online (Amiryousefi et al., 2018). The cp genomes were aligned using the MAFFT (Katoh and Standley, 2013). Additionally, the nucleotide variability across the cp genome sequences was analyzed using DnaSP v.6.12.03, with a window length of 600 sites and a step size of 200 sites.
Phylogenetic reconstruction included 27 de novo assembled sequences (Table 1), and 61 cp genomes downloaded from NCBI (Supplementary Table 2). At the same time, two species, Dioscorea esculenta (NC052854) and Dioscorea schimperian (NC039855), were used as outgroups. A total of 88 sequences were aligned using MAFFT with default parameters and trimmed using trimAl v.1.4 with option automated. Neighbor-Joining (NJ) analyses were performed using the MEGA X, applied with 1,000 bootstrap replicates at each branch node (Sudhir et al., 2018). The alignment was also evaluated using bootstrap analysis on 1,000 in a maximum likelihood (ML) by IQ-tree (Nguyen et al., 2015), with parameters: iqtree -s input -m MFP -b 1000 -nt AUTO -o NC052854, NC039855, best-fit nucleotide substitution model.
The leaf arrangement was selected to analyze the phyllotaxy evolution of Polygonatum. The phyllotaxy information was obtained from taxonomic literature and the Flora of China (Chen and Tamura, 2000; Jiao, 2018). The states of phyllotaxy were coded: alternate (A), verticillate (B), opposite (C), and the crowd (D). In the case of some species with more than two-character states, we coded the character state based on their dominant status. For example, the leaf arrangement for Polygonatum prattii was coded as alternate because it usually has alternate leaves, although there is an opposite leaf arrangement or three-verticillate leaves occasionally. For Polygonatum hunanense, its leaves were mainly verticillate, sometimes with a few alternate or opposite leaves, thus coded as verticillate in the analyses. Statistical Dispersal-Vicariance Analysis (S-DIVA) and Multistate Reconstruction BayesTraits (MRBT) method were conducted in RASP software to infer ancestral character states (Yu et al., 2015).
The divergence times of Polygonatum were calculated using the Markov chain Monte Carlo (MCMC) tree program of PAML (Puttick, 2019). IQ-tree was used to estimate the best tree topology of the data set. According to the previous study (Chen et al., 2013; Eguchi and Tamura, 2016; Wang et al., 2016; Xia et al., 2022), we used four calibration points to restrict each node: (F1) 115.9–137.4 Ma for the root node, (F2) 58.3–76.6 Ma for Asparagaceae stem age, (F3) 56.4–72.7 Ma for Asparagaceae crown age, (F4) 14.34–27.54 Ma for Polygonatum and Heteropolygonatum. The clock model uses the independent rate model (IRM), which follows a lognormal distribution. Nucleotide substitution selects the HKY model with alpha for gamma rates at sites set to 0.5. The birth–death process is used to generate uniform node age priors in the tree, using the default parameter (λ = 1, μ = 1, s = 0.1). The posterior probabilities of parameters were calculated using MCMC samples. The first 10% trees were discarded as burn-in and then sampled every 10 iterations until 20,000 samples were gathered.
The raw data of 27 individuals were filtered to remove adapters and low-quality reads; 3–5 Gb data were obtained for each species in this study. After assembly and splicing, the complete cp genomes of the circular tetrad structure were obtained (Figure 1). The annotated result suggested that the cp genome length of P. odoratum (154,576 bp) was the smallest, and the cp genome length of M. fuscum (156,711 bp) was the largest among the 27 individuals. The length of the LSC region ranged from 83,493 bp (P. odoratum) to 85,218 bp (M. fuscum). The length of the SSC region ranged from 18,002 bp (D. cantoniense) to 18,573 bp (P. cirrhifolium), and the length of the IRa and IRb regions ranged from 26,242 bp (D. fuscopicta) to 26,815 bp (D. cantoniense). The GC content of the cp genomes ranged from 37.6 to 37.8% and varied among the different regions of the cp genomes. In addition, the number of genes and introns were highly conserved (Table 1), and the same suite of rRNA genes and tRNA genes was found in all taxa. All genomes have 85–88 protein-coding genes, except for D. cantoniense, D. megalanthum, and D. uniflorum, with 83 protein-coding genes (lacking rps16 and rpl32 genes) (Supplementary Table 4). It is worth noting that 19 genes were repeated in the Polygonatum, which were involved in photosynthesis and self-replication (Supplementary Table 3).
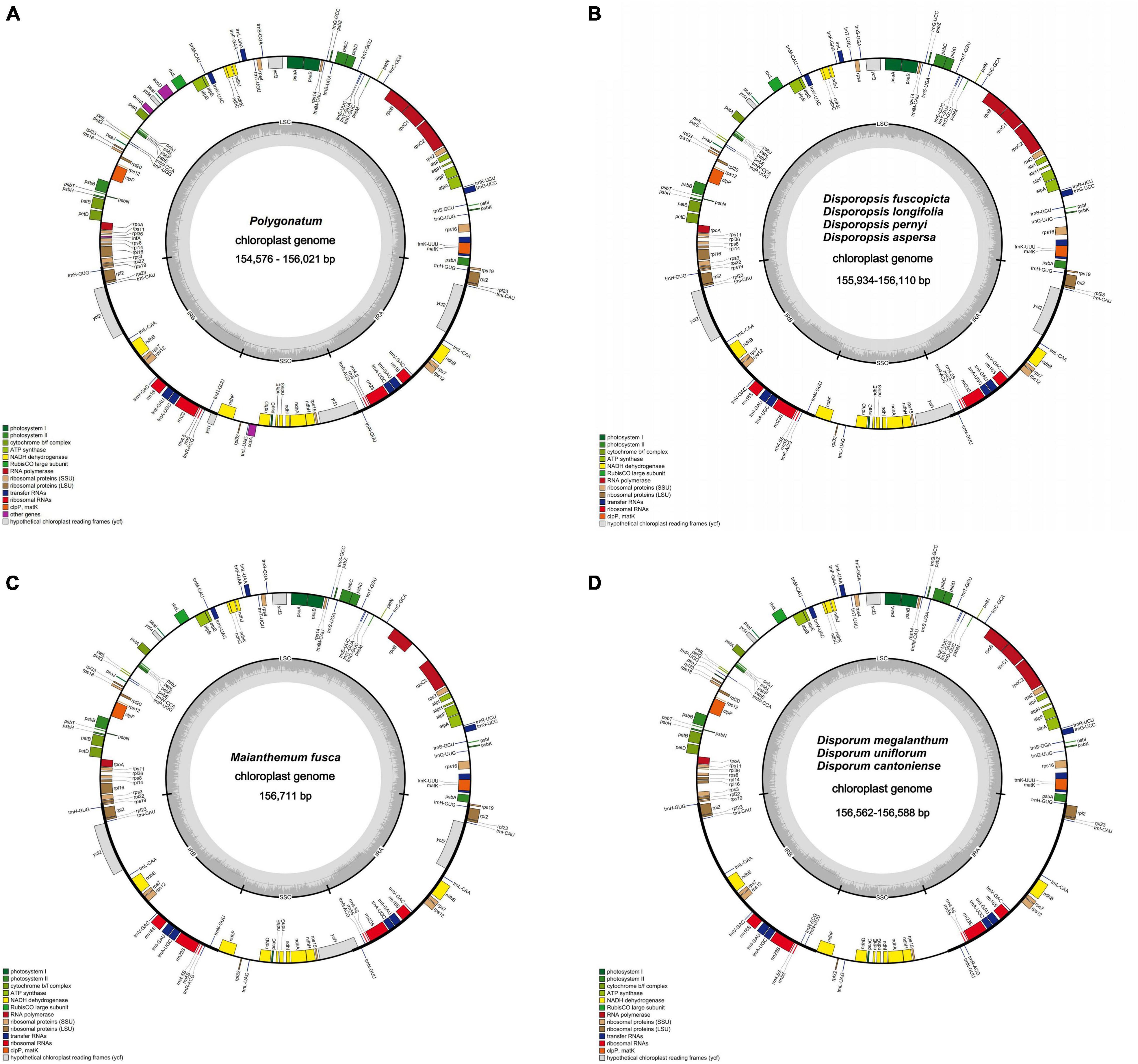
Figure 1. Chloroplast genome map: (A) Polygonatum, (B) Disporopsis, (C) Maianthemum, (D) Disporum. Genes inside and outside the circle are transcribed in a clockwise and counter-clockwise direction.
Repetitive sequences in the cp genome play a critical role in genome evolution and rearrangements. Oligonucleotide repeats analysis of four types of repeats in the cp genome, including Forward (F), Reverse (R), Palindromic (P), and Complementary (C), was performed by REPuter. The number of repeat types varied among the 26 cp genomes and presented random permutations, but most repeat sequences existed in 20–29 bp (Supplementary Figure 2). The abundance of F and P repeats was higher than that of R and C repeats (Supplementary Figure 2). The minimum number of repeats was found in Disporum uniflorum (60), whereas the maximum was found in P. cirrhifolium, P. sibiricum, and P. zanlanscianense (86). Complete details have been listed in Supplementary Tables 5, 6. Moreover, Spearman’s Rho correlation coefficients were obtained between tandem repeats, indels, and SNPs (Supplementary Table 7b). All these correlations showed a significant value (tandem repeats and indels: p < 0.001, tandem repeats and SNPs: p < 0.001, indels and SNPs: p < 0.001). The average correlation values between tandem repeats and indels, indels and SNPs, and tandem repeats in 26 Polygonatum species were 0.469, 0.351, and 0.267, respectively. Furthermore, we identified 67 (P. franchetii)–87 (M. fuscum) SSRs per cp genome consisting of mono- to hexa-nucleotide repeating units (Supplementary Table 8). Most of the SSRs were located in the intergenic areas. More than half of these SSRs (52.94–66.23%) were mononucleotide A/T motif, followed by dinucleotide (18.29–26.47%) with a predominant motif of AT/TA, tetranucleotide repeats (10.81–13.89%) with a predominant motif of AAAT/ATTT, AATC/ATTG, trinucleotide (2.94–6.94%), pentanucleotide (2.44–4.35%) with a predominant motif of AAACG/CGTTT, and hexanucleotides (0–1.35%) were absent in the cp genome of M. fuscum (Supplementary Figure 3).
The contraction and expansion of IR regions revealed variation in LSC/IR/SSC regions (Figure 2). The rpl22 gene was present in the LSC region, and rpl2 and trnH existed entirely in the IRb region. The rps19 gene was present in the junction of the IRa/IRb/LSC region in four genera (Polygonatum, Heteropolygonatum, Disporopsis, and Maianthemum), but in Disporum, the rps19 gene was absent in IRa/LSC region and existed completely in the IRb region. Notably, rps19 was started in IRa regions and integrated into the LSC by 60 base pairs in P. cyrtonema, whereas in all other species of Polygonatum, the rps19 gene exists completely in the LSC region. Additionally, the ndhF gene was observed at the junction of IRb/SSC and integrated into the SSC varied from 21 to 34 bp. Another truncated copy of the ycf1 gene was observed in all species at the IRa/SSC junction, which starts in IRa regions and integrates into the SSC. In addition, rpl2 and trnH existed in the IRa, and psbA existed in the LSC. It is worth noting that the rps19 and trnN genes existed entirely in the IRa region of four genera, whereas the two genes were missing in the Disporum at the junction of IRa and LSC (Supplementary Figure 4). Moreover, the genome alignment analysis showed that the cp genomes among the 26 species were relatively conserved, and no inversions, translocations, and genomic rearrangements were detected (Supplementary Figures 5, 6).
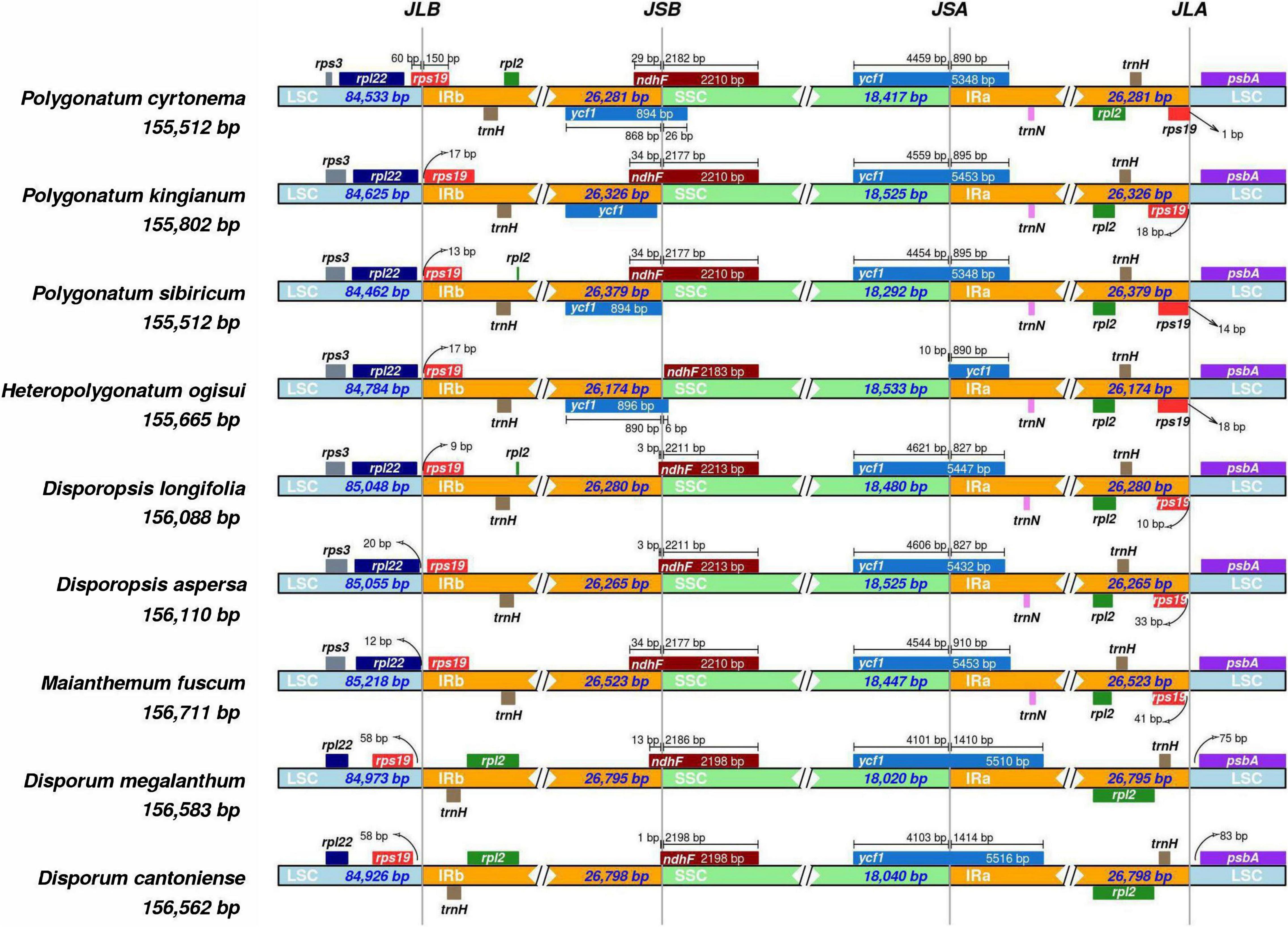
Figure 2. Comparisons of the borders of LSC, SSC, and IRa/b regions among the Polygonatum, Heteropolygonatum, Disporopsis, Maianthemum, and Disporum.
Comparison of overall sequence variation showed that the cp genome within Polygonatum is highly conserved. The IR regions had lower sequence divergence than LSC and SSC regions. In addition, the coding region was more conserved than the non-coding regions. Furthermore, except for the more remarkable mutation in ndhA, ycf1, and ycf2 genes, most of the protein-coding genes of Polygonatum were pretty conserved. The highest divergence in intergenic regions was found in the rps16-trnQ-UUG, trnS-GCU-trnG-UCC, trnT-UGU-trnL-UAA, ndhC-trnV-UAC, rpl32-trnL-UAG, trnV-GAC-rps7, and accD-psaI. The most divergent in the coding region were the ycf1 and ycf2 open reading frames (Supplementary Figure 7). Moreover, the sliding window analysis demonstrated that the seven regions had greater nucleotide diversity values (>0.01), including matK-rps16, trnS-GCU-trnG-UCC, rpl32-trnL-UAG, trnC-GCA-petN, petA-psbJ, ccsA, and ycf1 (Figure 3). The polymorphism loci of these variability regions are listed in Supplementary Table 9. Among these regions, nucleotide diversity values of rps16-trnQ-UUG, trnS-GCU-trnG-UCC, rpl32-trnL-UAG, and trnC-GCA-petN were greater than 0.01, and the ycf1 gene was the lowest (0.00047). The insertions/deletions (indels) diversity of trnS-GCU-trnG-UCC and matK-rps16 were 7.076 and 5.181, respectively, with no indel events detected in the ccsA gene. Furthermore, the cp genome of Polygonatum, Heteropolygonatum, Disporopsis, and Maianthemum is similar with an average similarity of 99% but different from that of Disporum with an average similarity of 85% based on the global comparison (Supplementary Figure 8).
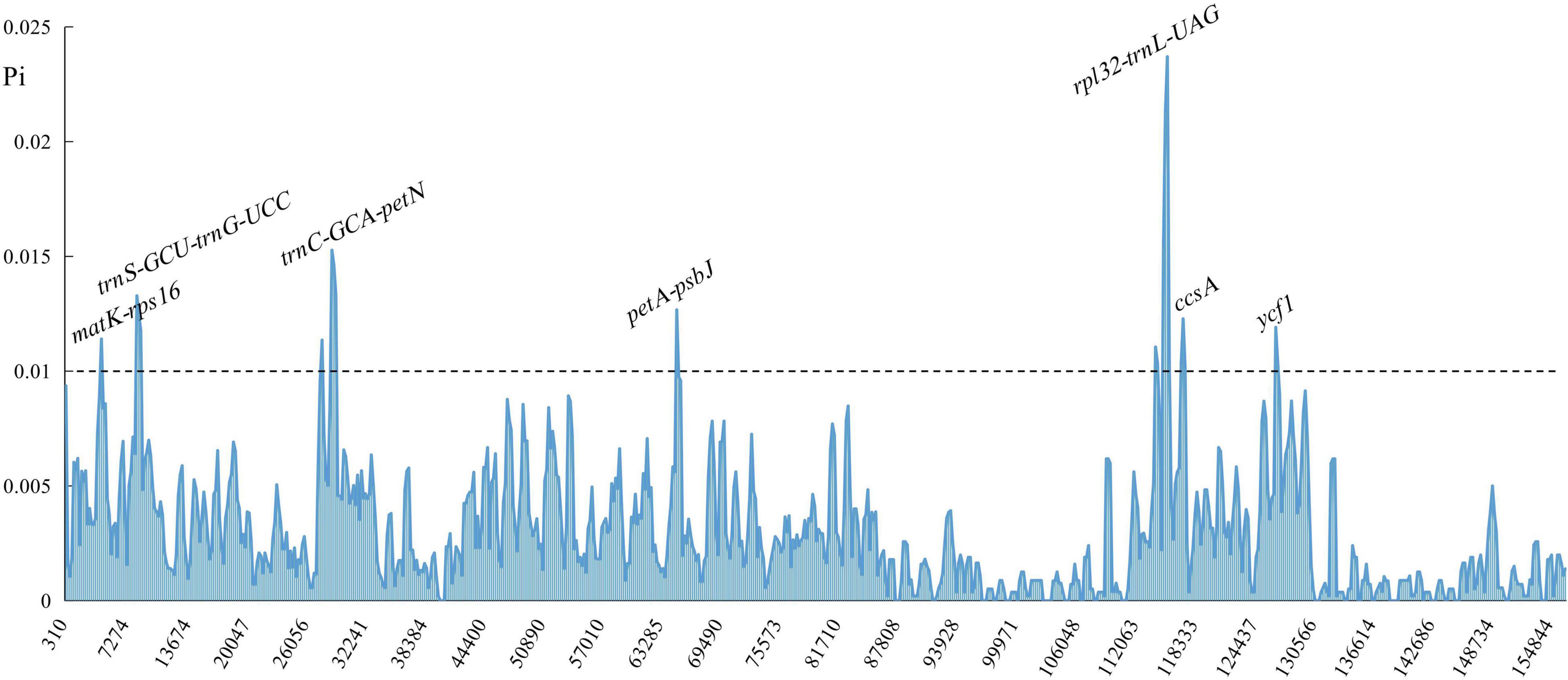
Figure 3. Sliding window analysis of Polygonatum cp genome. The X-axis represents the midpoint of the window; the Y-axis represents nucleotide diversity values.
The ML and NJ phylogenetic trees were inferred using 87 species, with the Dioscorea as the outgroup. The consensus trees obtained from the inference analyses were resolved, and most nodes were supported with maximum support (100% bootstrap support, Figure 4 and Supplementary Figure 9). The core Asparagaceae includes the subfamily of Scilloideae, Nolinoddeae, and Agavoideae, which form a monophyletic group (group I). Scilloideae and Agavoideae were sister taxa within the three subfamilies, and Nolinoddeae was a sister group to the clade of Scilloideae + Agavoideae. In addition, the results showed that most species of Trib. Polygonateae were placed in the crown of the phylogenetic tree, including Polygonatum, Heteropolygonatum, and Disporopsis, and supported the monophyly of three genera. However, within this clade, the Maianthemum and Ophiopogoneae were sisters to a clade formed by Disporopsis, Heteropolygonatum, and Polygonatum, while Disporum was polyphyletic across three separate clades and distantly related to Trib. Polygonateae. In addition, the Polygonatum is further divided into sect. Sibirica, sect. Polygonatum, and sect. Verticillata. And the sister relationship was between sect. Sibirica and sect. Polygonatum, whereas sect. Verticillata was placed as sister to sect. Polygonatum + sect. Sibirica with high support (100% B/S). It is worth mentioning that sect. Sibirica only includes a species of P. sibiricum. The NJ and ML analyses produced trees with similar topologies, although some poorly supported groups were sensitive to changes in the mode of inference. The position of several species was unresolved, including P. hunanense and P. kingianum, which varied among trees recovered using distinct phylogenetic inference methods.
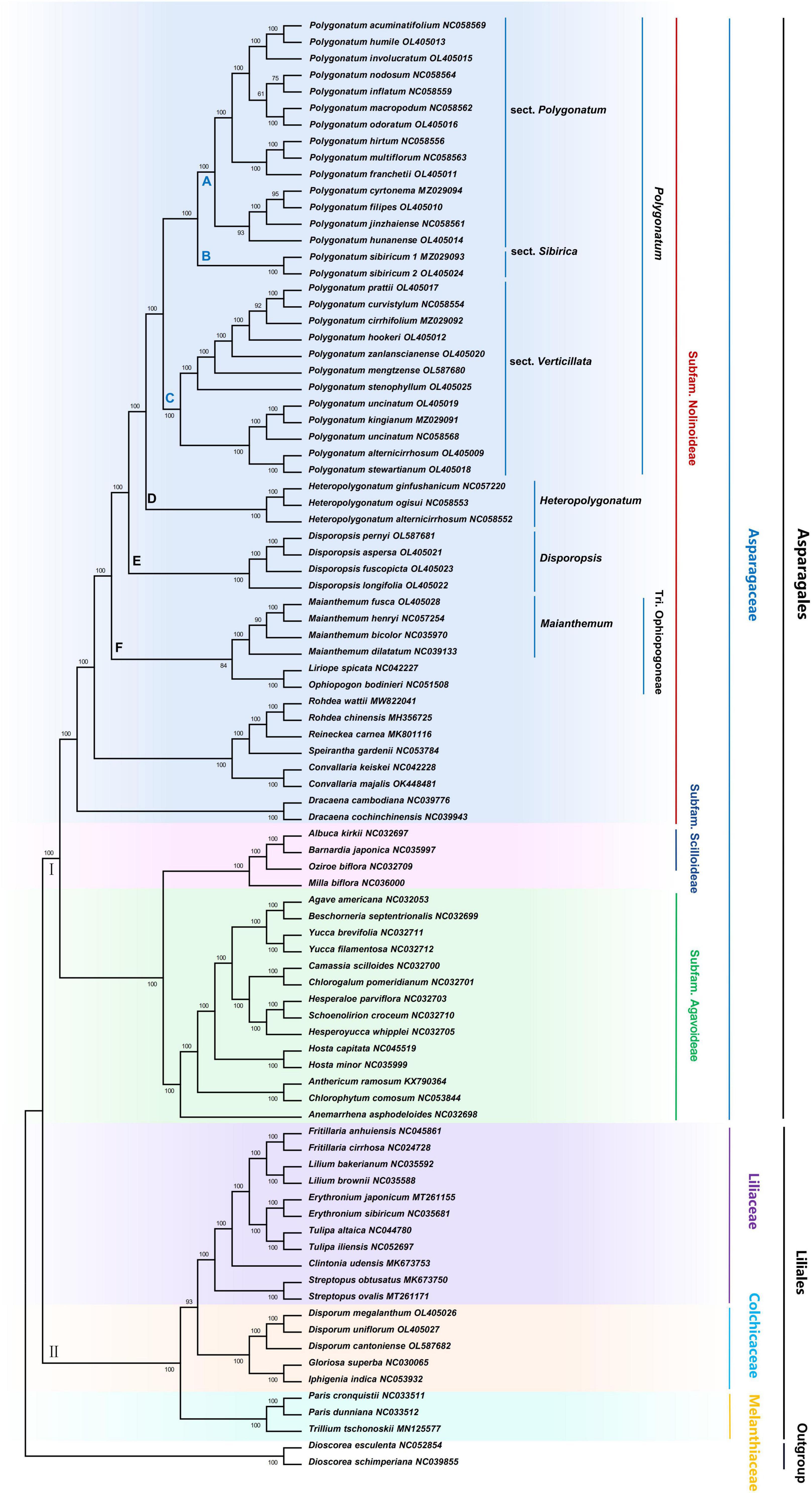
Figure 4. Maximum likelihood phylogenetic tree based on complete cp genome. Dioscorea esculenta and D. schimperiana were used as outgroups. Numbers at nodes are bootstrap support values.
Results of divergence time for the node of the 95% highest posterior density (HPD) intervals are shown in Supplementary Table 10. A complete chronogram is demonstrated in Figure 5. The extant genera of the Polygonatum and Disporopsis have shared a common ancestor at the beginning of the Eocene (41.68 Ma, 31.97–57.29, 95% HPD), while the split between Polygonatum and Heteropolygonatum is estimated to occur at 16.56 Ma (HPD = 13.57–20.56 Ma, 95%), and sect. Verticillata, sect. Polygonatum and sect. Sibirica might share a common ancestor at 14.71 Ma (1.32–18.57 Ma, 95% HPD), and the divergence times between sect. Polygonatum and sect. Sibirica was formed at approximately 11.80 Ma. Sampled specimens of Maianthemum and Ophiopogon were estimated to have originated in 52.22 Ma. Moreover, the divergence times of the Disporum occurred at 128.56 Ma, having shared a common ancestor with the Asparagaceae.
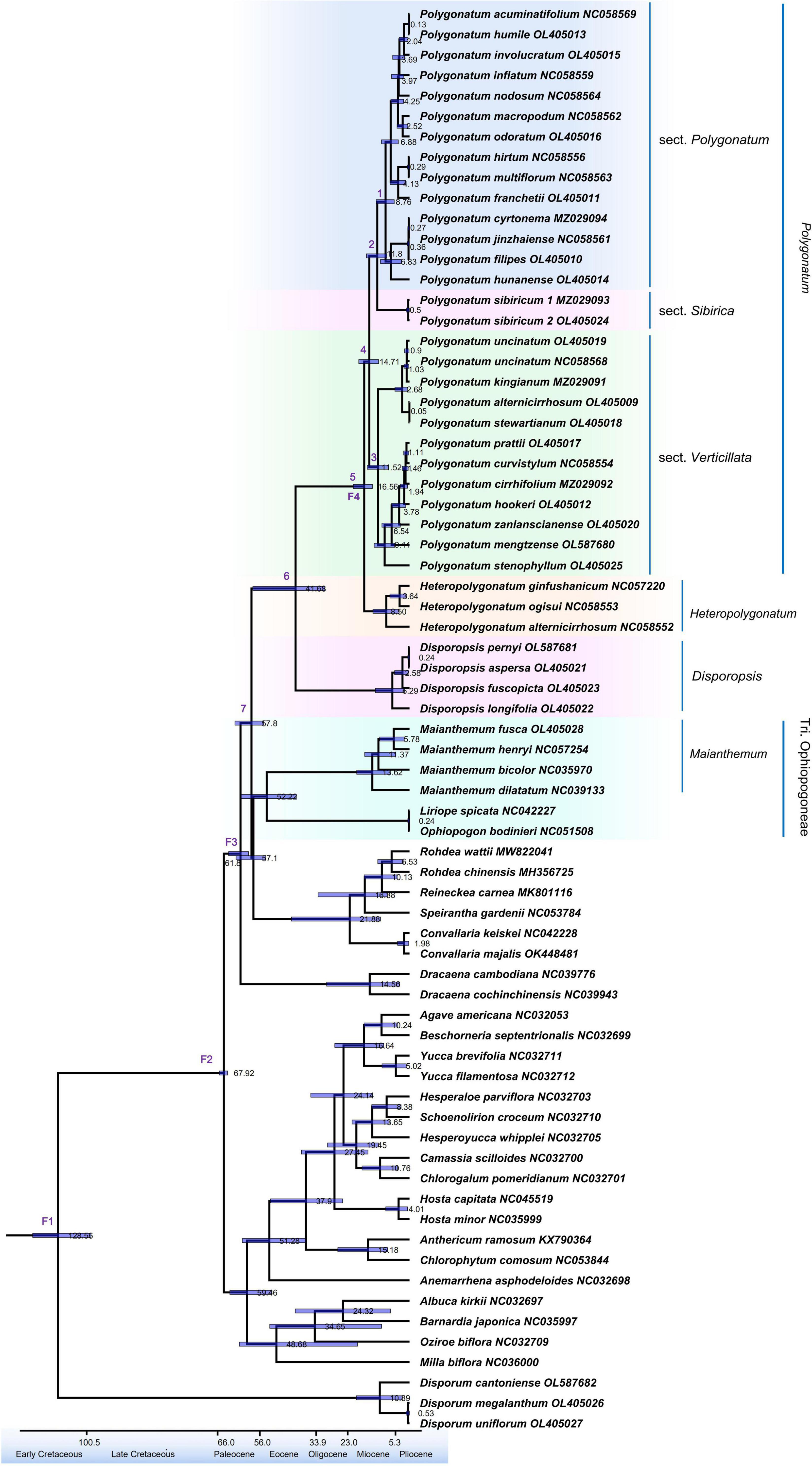
Figure 5. Divergence time estimation based on cp genome sequences. The divergence times are exhibited on each node, whereas the blue bars represent the 95% highest posterior density interval for each node age.
In the classification of Polygonatum, the arrangement of leaves was usually concerned. Phyllotaxy is one of the main characteristics of Polygonatum taxonomy, character transformation of leaf order is essential to understanding the evolution of Polygonatum. The phyllotaxy was used to reconstruct ancestral traits of Polygonatum and its relative species. As illustrated in Figure 6, the S-DIVA results showed that the verticillate leaves arrangement was the most likely ancestral state of Polygonatum, which was consistent with the MRBT method (B: p = 0.95). In addition, sect. Polygonatum is marked by phyllotaxy with an alternate leaf, except for P. hunanense. In its sister clade, sect. Sibirica is mostly a verticillate leaf arrangement. Sect. Verticillata includes species that appear to have a combination of the opposite, alternate, and verticillate leaves. In addition, alternate and verticillate leaves evolved more than once. Notably, the Heteropolygonatum and Disporopsis showed alternative phyllotaxy.
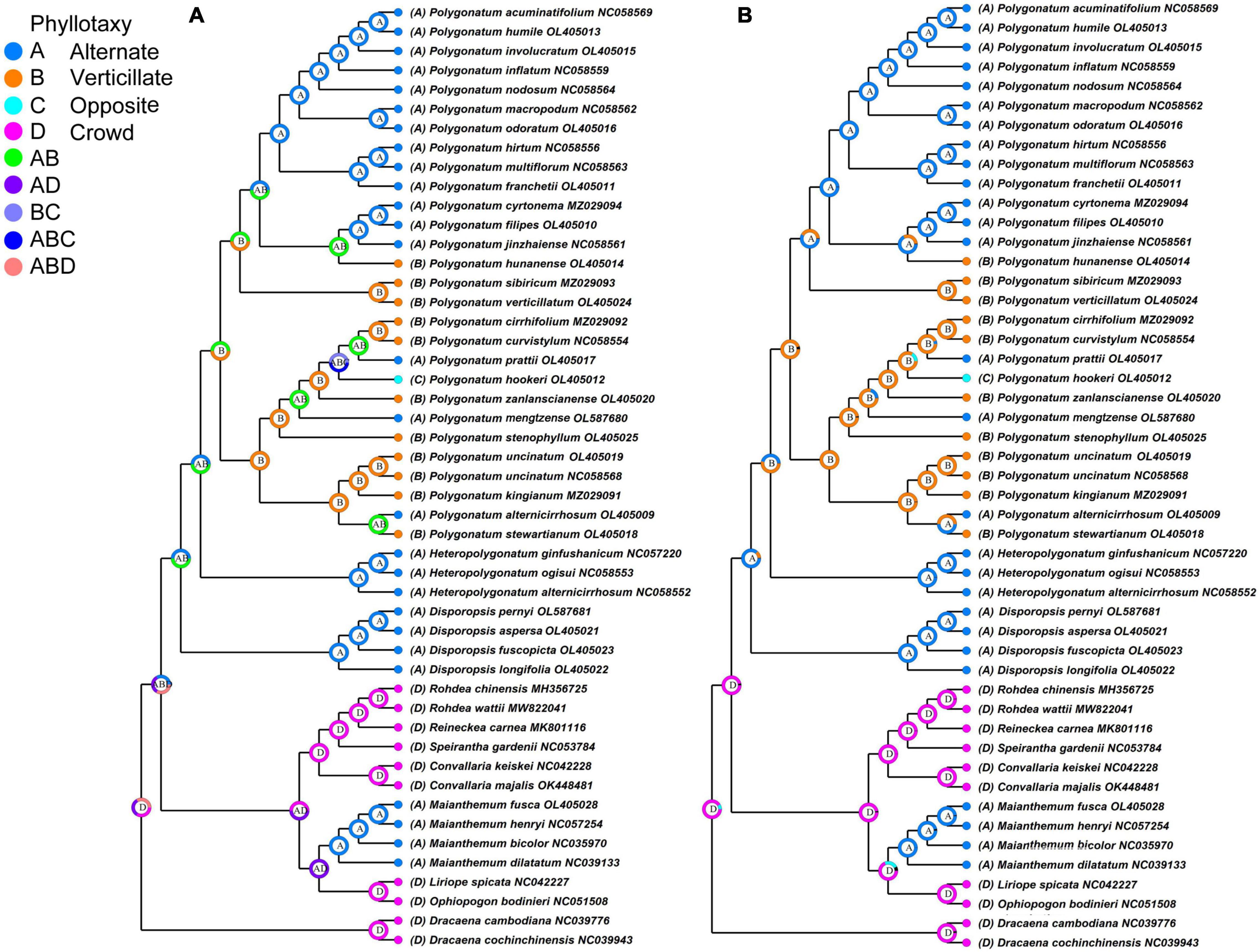
Figure 6. Ancestral state reconstructions of leaf arrangement in genus Polygonatum. (A) S-DIVA method and (B) MRBT method.
In the present study, we de novo assembled the cp genome of 26 species of Trib. Polygonateae and performed comparative analyses. The cp genome of 26 species exhibited a quadripartite structure with two IR regions separated by the LSC and SSC regions. Higher GC content was observed in the IR region compared with the LSC and SSC regions, consistent with previous reports (Liang et al., 2021). In addition, our results found that the total length, GC content, and gene composition of the cp genome were almost identical in all species. Previous studies have found that the angiosperms possessed a highly conserved nature in the cp genome at the genus level (Yu et al., 2019; Shahzadi et al., 2020), but we found that the rps16 and rpl32 genes lost in Disporum, and this variation may be specific to Disporum.
Inverted repeat contraction and expansion could cause gene duplication, the origination of pseudogenes, and length variation in the cp genome, which were considered critical evolutionary phenomena. In the present study, rps19 is present in the IR region in four genera (Polygonatum, Heteropolygonatum, Disporopsis, and Maianthemum), except for Disporum. Previous studies of monocotyledons’ cp genome revealed that the rps19 gene existed in the IR region (Ahmed et al., 2012; Nock et al., 2014; Henriquez et al., 2020). Whereas the de novo assembled genome of Disporum contrast with previous studies, revealing integration of rps19 into the LSC region. Moreover, the ycf1 gene was duplicated in the IRa and IRb regions in Polygonatum and Heteropolygonatum, while in the other three genera (Disporopsis, Maianthemum, and Disporum), this gene only exists in the junction of the SSC/IRa region, which is consistent with a previous study (Wang et al., 2011).
At the genus level, weak-to-strong correlations among tandem repeats, SNPs, and indels have been observed in the Polygonatum. We found a weak correlation between tandem repeats and SNPs, a moderate correlation between indels and SNPs, and a strong correlation between tandem repeats and indels in Polygonatum plastomes. A recent study has confirmed that the plastomes exhibited strong associations between tandem repeats, indels, and substitutions in Araceae and Malvaceae (Abdullah et al., 2020, 2021). Our results also supported prior findings that tandem repeats play an important role in generating the indels and SNPs. These results have practical implications in selecting appropriate loci for comparative analyses.
The phylogeny and classification of Polygonatum have long been debated (Feng et al., 2020). This study used 88 cp genome, including most of the basal monocot family Asparagaceae, to construct the phylogenetic tree. Results of the NJ and ML phylogenies analysis confirmed the position of Polygonatum within the Asparagaceae, which were congruent and largely concordant with recent phylogenomic studies (Zhao et al., 2019; Xia et al., 2022). There is strong support for the monophyly of many major clades of Asparagaceae, including Polygonatum, Heteropolygonatum, Disporopsis, Maianthemum, and Rohdea. In a previous study, the Polygonatum was subdivided into two sections: sect. Polygonatum and sect. Verticillata based on the trnK (Tamura et al., 1997), but in our phylogeny, the Polygonatum was recovered as monophyletic in NJ and ML analyses, which were divided into three sections: sect. Sibirica, sect. Polygonatum, and sect. Verticillata, and can be strongly supported as a sister relationship between (1) Polygonatum and Heteropolygonatum, and (2) sect. Sibirica and sect. Polygonatum, and (3) sect. Verticillata and sect. Polygonatum + sect. Sibirica, respectively, which is consistent with previous studies (Meng et al., 2014; Floden, 2017; Zhao et al., 2019; Xia et al., 2022).
Moreover, we found several interesting implications of phylogeny in this study. First, both NJ and ML analyses provided strong evidence for the monophyly of P. sibiricum in sect. Sibirica. These results were supported by the findings of other researchers based on matK, trnK, rbcL, psbA-trnH, and cp genome (Meng et al., 2014; Floden, 2017; Zhao et al., 2019; Xia et al., 2022). In contrast, the cp genome tree placed P. sibiricum nested within sect. Verticillata or sect. Sibirica in a previous study (Floden and Schilling, 2018). Therefore, additional phylogeny analysis using an enormous collection of Polygonatum should be performed in the future. Second, previous phylogenetic studies of Polygonatum based on the cp genome have shown that the Heteropolygonatum alternicirrhosum belongs to the Heteropolygonatum and strongly supported the monophyly of Heteropolygonatum (Xia et al., 2022). However, in our study, the ML and NJ tree which strongly supported P. alternicirrhosum was deeply nested within the Polygonatum and supported the monophyly of Polygonatum and Heteropolygonatum. Our analyses suggest that Polygonatum may be the sister to the Heteropolygonatum. In addition, another previous study also supports that the species belongs to Polygonatum (Chen and Tamura, 2000). Therefore, our results suggested that P. alternicirrhosum (OL405009) and H. alternicirrhosum (NC058552) should be considered as two species, in contrast with other recent estimations, mainly to the influential Floden’s (2014) study, which suggested that P. alternicirrhosum should be transferred to Heteropolygonatum based of morphological, cytological comparisons and molecular data. In addition, Zhao et al. (2019) indicated that the P. alternicirrhosum should be recovered within the Polygonatum. Another example is the Polygonatum mengtzense, which was considered most closely related to the P. punctatum (Floden, 2014). In this study, maximum likelihood analysis reveals that P. mengtzense was deeply nested within sect. Verticillata, which was supported by molecular and chromosome evidence (Floden, 2014; Xia et al., 2022). Finally, there is a significant divergence in the classification of Trib. Polygonateae. In the previous study, Kim et al. (2010) placed Disporopsis as a sister to Polygonatum + Maianthemumin in a Bayesian analysis. In the present study, the phylogenetic analyses corroborated the monophyly of Polygonatum, Heteropolygonatum, Disporopsis, Maianthemum, and Disporum. The Disporum is sister to Glorioseae and nested within the family Colchicaceae (more distantly related species), consistent with previous genetic studies (Tamura et al., 2013).
It is interesting to note that the Maianthemum was traditionally included with Trib. Polygonateae based on morphology (Chen and Tamura, 2000), and other research based on multiple plastid markers (atpB, ndhF, rbcL, matK, psbA-trnH, trnC-petN, atpB-rbcL, and rps16) also supports its placement in Trib. Polygonateae (Chen et al., 2013; Meng et al., 2014; Zhao et al., 2019). However, ML analysis in our study reveals that the Maianthemum was deeply nested within Trib. Ophiopogoneae rather than Trib. Polygonateae, which agrees with former studies based on multiple loci (e.g., petA-psbJ, ETS, ITS, and rps10; or trnL-F, rps16, rpl16, psbA-trnH, rbcL, trnK, trnC-petN, and ITS) (Floden, 2017; Floden and Schilling, 2018; Meng et al., 2021). Our results suggested that the Trib. Polygonateae should include only three genera (Disporopsis, Heteropolygonatum, and Polygonatum). Therefore, the results revealed that the convergent evolution of some traits may have misled previous relationships. Further phylogenetic analysis is needed within the Maianthemum.
Results of divergence time estimates suggest that the elevated diversification rates of Polygonatum occurred from approximately 15–0.1 Ma during the late Miocene and early Pliocene. The two main lineages, sect. Verticillata and sect. Sibirica + sect. Polygonatum seem to have radiated since the mid-Miocene (sect. Verticillata: 11.52 Ma; sect. Sibirica + sect. Polygonatum: 11.80 Ma; Figure 5; Supplementary Table 10). Notably, the diversification rates of Polygonatum slowly increased during this period, attributed to uplifts of the Qinghai–Tibetan Plateau in the early Miocene (Xue et al., 2021). In addition to the aforementioned tectonic rearrangements and mountain formation in East Asia, the global climatic fluctuations and aridification that occurred in the Mid-Miocene Climatic Optimum (MMCO, 15–17 Ma; Zachos et al., 2001) also accelerated the diversification rates of Polygonatum. Global warming occurred at approximately 15 Ma (MMCO), followed by a gradual decrease in temperature (Zachos et al., 2001). These climatic changes might have influenced the plant diversification and promoted radiation of Polygonatum species.
Variation in phyllotaxy morphology represents an important character source for species delimitation. The phyllotaxy diversity (alternate, opposite, and verticillate) caused some confusion in classifying the Polygonatum. Our study showed the evolutionary trend of Polygonatum from verticillate leaves to alternate leaves, and this suggests that verticillate leaf is the ancestral state and agrees with the previous molecular studies of Polygonatum (Xia et al., 2022). It is noteworthy that the phyllotaxy of Polygonatum is an unstable character even for the same species. Therefore, phyllotaxy cannot be used as the unique taxonomic feature for classifying Polygonatum.
In this study, the complete cp genome of 26 species of Trib. Polygonateae was de novo assembled from Illumina reads. In all of our analyses, these cp genomes were generally conserved and exhibited similar gene content and genomic structure. A total of 8 highly variable loci were identified across the Polygonatum cp genome, which could serve as potential markers for phylogenetic and population genetics studies. The monophyly of Polygonatum was confirmed, and phylogenetic analysis indicated that the genus consists of three sections (sect. Sibirica, sect. Polygonatum, and sect. Verticillata). Meanwhile, the phylogenetic analysis suggested that Heteropolygonatum may be the sister group of the Polygonatum, but the Disporopsis, Maianthemum, and Disporum may have diverged earlier. In conclusion, our results enhanced the genomic information for Polygonatum and provided valuable insight into the phylogenetic relationships among the genera involved in Trib. Polygonateae. The results also contribute to the bioprospecting and conservation of the Polygonatum.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
JW, ZX, SC, and BD participated in the conception and design of the research. JW, JQ, FY, YJ, and BZ collected and identified the species. JW, JQ, and XC were responsible for analyzing and processing data. JW wrote the manuscript. ZX and BD revised the manuscript. All authors agreed to the submitted version of the manuscript.
This work was supported by the Yunnan Academician Expert Workstation (202205AF150026 and 202105AF150053), the National Natural Science Foundation of China (Grant No. 31860080), the key technology projects in Yunnan Province of China (202002AA100007), the National Key R&D Program of China (Grant No. 2019YFC1711100), Heilongjiang Touyan Innovation Team Program (Tree Genetics and Breeding Innovation Team), and the Yunnan Xingdian Talent Support Plan (YNWR-QNBJ-2020251).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Zhongyu Zhou, Jie Liu, and Qingshu Yang for their assistance in obtaining specimens for this study. We thank Yaping Wang at Jinan University for her assistance with language polishing. We are also thankful to Northeast Forestry University and the China Academy of Chinese Medical Sciences for technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.882189/full#supplementary-material
Abdullah, Henrique, C. L., Croat, T. B., Poczai, P., and Ahmed, I. (2020). Mutational dynamics of Aroid chloroplast genomes II. Front. Genet. 11:610838. doi: 10.3389/fgene.2020.610838
Abdullah Mehmood, F., Shahzadi, I., Ali, Z., Islam, M., Naeem, M., et al. (2021). Correlations among oligonucleotide repeats, nucleotide substitutions, and insertion-deletion mutations in chloroplast genomes of plant family Malvaceae. J. Syst. Evol. 59, 388–402. doi: 10.1111/jse.12585
Abdullah Shahzadi, I., Mehmood, F., Ali, Z., Malik, M. S., Waseem, S., et al. (2019). Comparative analyses of chloroplast genomes among three Firmiana species: identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Genet. 19:100199. doi: 10.1016/j.plgene.2019.100199
Ahmed, I., Bigg, P. J., Matthews, P. J., Collins, L. J., Hendy, M. D., Lockhart, P. J., et al. (2012). Mutational dynamics of Aroid chloroplast genomes. Genom. Biol. Evol. 4, 1316–1323. doi: 10.1093/gbe/evs110
Akoglu, H. (2018). User’s guide to correlation coefficients. Turk. J. Emerg. Med. 3, 91–93. doi: 10.1016/j.tjem.2018.08.001
Ali, M., Liu, Y. J., Xia, Q. P., Bahadur, S., and Shuaib, M. (2021). Pollen micromorphology of eastern Chinese Polygonatum and its role in taxonomy by using scanning electron microscopy. Microsc. Res. Techniq. 84, 1451–1461. doi: 10.1002/jemt.23701
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Baker, J. G., and Esq, F. L. S. (1875). Revision of the genera and species of Asparagaceae. Bot. J. Linn. Soc. 80, 547–632. doi: 10.1111/j.1095-8339.1875.tb00349.x
Beier, S., Thie, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Chen, Q., Wu, X. B., and Zhang, D. Q. (2019). Phylogenetic analysis of Fritillaria cirrhosa D. Don and its closely related species based on complete chloroplast genomes. Peer J. 7:e7480. doi: 10.7717/peerj.7480
Chen, S., Kim, D. K., Chase, M. W., and Kim, J. H. (2013). Networks in a large-scale phylogenetic analysis: reconstructing evolutionary history of Asparagales (Lilianae) based on four plastid genes. PLoS One 8:e59472. doi: 10.1371/journal.pone.0059472
Chen, X. Q., and Tamura, M. N. (2000). Flora of China. Science Press. Beijing: Beijing and Missouri Botanical Garden Press, St. Louis.
Chinese Pharmacopoeia Commission (2020). Pharmacopoeia of the People’s Republic of China. Beijing: China medicine science and technology press.
Eguchi, S., and Tamura, M. N. (2016). Evolutionary timescale of monocots determined by the fossilized birth-death model using a large number of fossil records. Evolution 70, 1136–1144. doi: 10.1111/evo.12911
Feng, T. H., Jia, Q. J., Meng, X., Chen, X. L., and Liang, Z. S. (2020). Evaluation of genetic diversity and construction of DNA fingerprinting in Polygonatum Mill. based on EST-SSR and SRAP molecular markers. 3 Biotech 10:322. doi: 10.1007/s13205-020-02316-z
Floden, A. J. (2014). New names in Heteropolygonatum (Asparagaceae). Phytotaxa 188, 218–226. doi: 10.11646/phytotaxa.188.4.4
Floden, A. J. (2015). A new Polygonatum (Asparagaceae) endemic to the Truong Son of southern Vietnam. Phytotaxa 197, 125–131. doi: 10.11646/phytotaxa.197.2.5
Floden, A. J. (2017). Molecular phylogenetic Studies of the Genera of Tribe Polygonateae (Asparagaceae: Nolinoideae): Disporopsis, Heteropolygonatum, and Polygonatum. PhD diss., University of Tennessee. Knoxville: University of Tennessee
Floden, A., and Schilling, E. E. (2018). Using phylogenomics to reconstruct phylogenetic relationships within tribe Polygonateae (Asparagaceae), with a special focus on Polygonatum. Mol. Phylogenet. Evol. 129, 202–213. doi: 10.1016/j.ympev.2018.08.017
Henriquez, C. L., Abdullah Ahmed, I., Carlsen, M. M., and Mckain, M. R. (2020). Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 112, 2349–2360. doi: 10.1016/j.ygeno.2020.01.006
Jiao, J. (2018). A Study on Germplasm Resources of Polygonati Rhizoma. Doctoral thesis, Northwest A&F University, Xianyang, 1–274.
Jiao, J., Huang, W. L., Bai, Z. Q., Liu, F., Ma, C. D., Liang, Z., et al. (2018a). DNA barcoding for the efficient and accurate identification of medicinal Polygonati Rhizoma in China. PLoS One 13:e0201015. doi: 10.1371/journal.pone.0201015
Jiao, J., Jia, X. R., Liu, P., Zhang, Q. M., Liu, F., Pengzhou, Xi, et al. (2018b). Species identification of Polygonati Rhizoma in China by both morphological and molecular marker methods. C. R. Biol. 341, 102–110. doi: 10.1016/j.crvi.2017.10.004
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., DePamphilis, C. W., Yi, T.-S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genom. Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, D. K., Kim, J. S., and Kim, J. H. (2012). The phylogenetic relationships of Asparagales in Korea based on five plastid DNA regions. J. Plant Biol. 55, 325–341. doi: 10.1007/s12374-011-0016-4
Kim, J. H., Kim, D. K., Forest, F., Fay, M. F., and Chase, M. W. (2010). Molecular phylogenetics of Ruscaceae sensu lato and related families (Asparagales) based on plastid and nuclear DNA sequences. Ann. Bot. 106, 775–790. doi: 10.1093/aob/mcq167
Lewis, S. E., Searle, S., Harris, N., Gibson, M., and Clamp, M. E. (2002). Apollo: a sequence annotation editor. Genome Biol. 3, 1–82.14. doi: 10.1186/gb-2002-3-12-research0082
Liang, C. L., Wang, L., Ma, W. S., and Xu, J. (2021). A comparative study of complete chloroplast genome for the genus Salvia. J. Plant Biochem. Biot. 30, 117–125. doi: 10.1007/s13562-020-00575-8
Liang, H., Zhang, Y., Deng, J. B., Gao, G., and Yang, R. W. (2020). The complete chloroplast genome sequences of 14 Curcuma species: insights into genome evolution and phylogenetic relationships within Zingiberales. Front. Genet. 11:802. doi: 10.3389/fgene.2020.0080
Liu, Q., Li, X. Y., Li, M. Z., Xu, W. K., Schwarzacher, T., Heslop-Harrison, J. S., et al. (2020). Comparative chloroplast genome analyses of Avena: insights into evolutionary dynamics and phylogeny. BMC Plant Biol. 20:406. doi: 10.1186/s12870-020-02621-y
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Mayer, C., Leese, F., and Tollrian, R. (2010). Genome-wide analysis of tandem repeats in Daphnia pulex - a comparative approach. BMC Genom. 11:277. doi: 10.1186/1471-2164-11-277
Meng, R., Meng, Y., Yang, Y. P., and Nie, Z. L. (2021). Phylogeny and biogeography of Maianthemum (Asparagaceae: Nolinoideae) revisited with emphasis on its divergence pattern in SW China. Plant Divers. 43, 93–101. doi: 10.1016/j.pld.2021.02.001
Meng, Y., Nie, Z. L., Deng, T., Wen, J., and Yang, Y. P. (2014). Phylogenetics and evolution of phyllotaxy in the Solomon’s seal genus Polygonatum (Asparagaceae: Polygonateae). Bot. J. Linn. Soc. 176, 435–451. doi: 10.1111/boj.12218
Michael, T., Pascal, L., Tommaso, P., Elena, S. U. E., Axel, F., Bock, R., et al. (2017). GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 47, W6–W11. doi: 10.1093/nar/gkx391
Nguyen, L., Schmidt, H. A., Haeseler, A. V., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nie, L. P., Cui, Y. X., Chen, X. L., Xu, Z. C., Sun, W., Song, J., et al. (2020). Complete chloroplast genome sequence of the medicinal plant Arctium lappa (Burdock). Genome 63, 53–60. doi: 10.1139/gen-2019-0070
Nock, C. J., Baten, A., and King, G. J. (2014). Complete chloroplast genome of Macadamia integrifolia confirms the position of the Gondwanan early-diverging eudicot family Proteaceae. BMC Genom. 9:S13. doi: 10.1186/1471-2164-15-S9-S13
Puttick, M. N. (2019). MCMCtreeR: functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics 35, 5321–5322. doi: 10.1093/bioinformatics/btz554
Rudall, P. J., Rudall, J. G., and Chase, M. W. (2000). Systematics of Ruscaceae/Convallariaceae: a combined morphological and molecular investigation. Bot. J. Linn. Soc. 134, 73–92. doi: 10.1006/bojl.2000.0365
Shahzadi, I., Abdullah Mehmood, F., Ali, Z., Ahmed, I., Mirza, B., et al. (2020). Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 112, 1454–1463. doi: 10.1016/j.ygeno.2019.08.016
Shi, L. C., Chen, H. M., Jiang, M., Wang, L. Q., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Si, J. P., and Zhu, A. G. (2021). Polygonati Rhizoma-a new high quality crop with great potential and not occupying farmland. Sci. Sin. Vitae 51, 1477–1484.
Stefan, K., Choudhuri, J. V., Enno, O., Chris, S., Jens, S., Giegerich, R., et al. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Sudhir, K., Glen, S., Li, M., Christina, K., and Koichiro, T. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Tamura, M. N. (1993). Biosystematic studies on the genus Polygonatum (Liliaceae) III. Morphology of staminal filaments and karyology of eleven Eurasian species. Bot. Jahrb. Syst. 115, 1–26.
Tamura, M. N., Schwarzbach, A. E., Kruse, S., and Reski, R. (1997). Biosystematic studies on the genus Polygonatum (Convallariaceae) IV. molecular phylogenetic analysis based on restriction site mapping of the chloroplast gene trnK. Feddes Rep. 108, 159–168. doi: 10.1002/fedr.19971080306
Tamura, M. N., Yamada, M., Fuse, S., and Hotta, M. (2013). Molecular phylogeny and axonomy of the genus Disporum (Colchicaceae). Acta Phytotaxon. Geobot. 64, 137–147. doi: 10.18942/apg.KJ00008918923
Tang, Y. C. (1978). “Polygonatum Mill,” in Flora Reipublicae Popularis Sinicae, eds F. T. Wang and T. Tang (Beijing China: Science Press).
Wang, J. J., Yang, Y. P., Hang, S., Wen, J., Tao, D., Nie, Z. L., et al. (2016). The biogeographic South-North divide of Polygonatum (Asparagaceae Tribe Polygonateae) within Eastern Asia and its recent dispersals in the Northern Hemisphere. PLoS One. 11:e166134. doi: 10.1371/journal.pone.0166134
Wang, W. Q., Messing, J., and Badger, J. H. (2011). High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS One. 6:e24670. doi: 10.1371/journal.pone.0024670
Wang, Y., Liu, X. Q., Su, H., Yin, S. K., Han, C. X., Hao, D., et al. (2019). The regulatory mechanism of chilling-induced dormancy transition from endo-dormancy to non-dormancy in Polygonatum kingianum Coll. et Hemsl rhizome bud. Plant Mol. Biol. 99, 205–217. doi: 10.1007/s11103-018-0812-z
Wang, Z. W., Du, F. Q., Zhang, L., Wang, S. M., and Wei, S. H. (2019). Advances in classification and identification of the plants of Polygonati Rhizoma and its adulterants. North Hortic. 24, 130–136. doi: 10.11937/bfyy.20191881
Wick, R. R., Schultz, M. B., Justin, Z., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, S. A., Lu, H. L., Yang, J., Rao, G. Y., You, R. L., and Zhong, Y. (2000). Molecular systematic studies on the tribe Polygonateae (s.l.) in China based on RFLPs data of PCR-amplified chloroplast DNA fragments. Acta Phytotaxon. Sin. 38, 97–110.
Xia, M. Q., Liu, Y., Liu, J. J., Chen, D. H., Shi, Y., Chen, Z., et al. (2022). Out of the Himalaya-Hengduan Mountains: phylogenomics, biogeography and diversification of Polygonatum Mill. (Asparagaceae) in the Northern Hemisphere. Mol. Phylogenet. Evol. 169:107431. doi: 10.1016/j.ympev.2022.107431
Xue, T., Gadagkar, S. R., Albright, T. P., Yang, X., Li, J., Xia, C., et al. (2021). Prioritizing conservation of biodiversity in an alpine region: distribution pattern and conservation status of seed plants in the Qinghai-Tibetan plateau. Glob. Ecol. Conserv. 32:e01885. doi: 10.1016/j.gecco.2021.e01885
Yang, P., Zhou, H., Xin, T. Y., Ma, S. J., Duan, B. Z., Yao, H., et al. (2015). Identification study of DNA Barcode sequences in the medicinal plants of Polygonatum. World Chin. Med. 10, 1173–1176.
Yu, X. Y., Zuo, L. H., Lu, D. D., Lu, B., Yang, M. S., Wang, J., et al. (2019). Comparative analysis of chloroplast genomes of five Robinia species: genome comparative and evolution analysis. Gene 689, 141–151. doi: 10.1016/j.gene.2018.12.023
Yu, Y., Harris, A. J., Blair, C., and He, X. (2015). RASP (Reconstruct Ancestral State in Phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49. doi: 10.1016/j.ympev.2015.03.008
Zachos, J., Pagani, M., Sloan, L., Thomas, E., and Billups, K. (2001). Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 292, 686–693. doi: 10.1126/science.1059412
Keywords: Polygonatum, chloroplast genome, phylogenetics, divergence time, Trib. Polygonateae
Citation: Wang J, Qian J, Jiang Y, Chen X, Zheng B, Chen S, Yang F, Xu Z and Duan B (2022) Comparative Analysis of Chloroplast Genome and New Insights Into Phylogenetic Relationships of Polygonatum and Tribe Polygonateae. Front. Plant Sci. 13:882189. doi: 10.3389/fpls.2022.882189
Received: 23 February 2022; Accepted: 03 June 2022;
Published: 24 June 2022.
Edited by:
Da-Cheng Hao, Dalian Jiaotong University, ChinaReviewed by:
Aaron Floden, Missouri Botanical Garden, United StatesCopyright © 2022 Wang, Qian, Jiang, Chen, Zheng, Chen, Yang, Xu and Duan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhichao Xu, emN4dUBuZWZ1LmVkdS5jbg==; Baozhong Duan, YnpkdWFuQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.