 Zhi Zhao†
Zhi Zhao† Meng Li†He Zhang†Yao Yu†Lu MaWei WangYunxin FanNing HuangXinying WangKunquan Liu
Meng Li†He Zhang†Yao Yu†Lu MaWei WangYunxin FanNing HuangXinying WangKunquan Liu Shinan DongHaijuan TangJianfei WangHongsheng Zhang
Shinan DongHaijuan TangJianfei WangHongsheng Zhang Yongmei Bao*
Yongmei Bao*
- State Key Laboratory of Crop Genetics and Germplasm Enhancement, College of Agriculture, Jiangsu Collaborative Innovation Center for Modern Crop Production, Cyrus Tang Innovation Center for Crop Seed Industry, Jiangsu Province Engineering Research Center of Seed Industry Science and Technology, Nanjing Agricultural University, Nanjing, China
Rice blast, caused by Magnaporthe oryzae, is one of the most devastating diseases in rice and can affect rice production worldwide. Rice plasma membrane (PM) proteins are crucial for rapidly and precisely establishing a defense response in plant immunity when rice and blast fungi interact. However, the plant-immunity-associated vesicle trafficking network mediated by PM proteins is poorly understood. In this study, to explore changes in PM proteins during M. oryzae infection, the PM proteome was analyzed via iTRAQ in the resistant rice landrace Heikezijing. A total of 831 differentially expressed proteins (DEPs) were identified, including 434 upregulated and 397 downregulated DEPs. In functional analyses, DEPs associated with vesicle trafficking were significantly enriched, including the “transport” term in a Gene Ontology enrichment analysis, the endocytosis and phagosome pathways in a Encyclopedia of Genes and Genomes analysis, and vesicle-associated proteins identified via a protein–protein interaction network analysis. OsNPSN13, a novel plant-specific soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) 13 protein, was identified as an upregulated DEP, and transgenic plants overexpressing this gene showed enhanced blast resistance, while transgenic knockdown plants were more susceptible than wild-type plants. The changes in abundance and putative functions of 20 DEPs revealed a possible vesicle trafficking network in the M. oryzae-rice interaction. A comparative proteomic analysis of plasma membrane proteins in rice leaves revealed a plant-immunity-associated vesicle trafficking network that is provoked by blast fungi; these results provide new insights into rice resistance responses against rice blast fungi.
Introduction
Rice blast, which is caused by Magnaporthe oryzae (M. oryzae), is one of the most devastating diseases in rice growing regions worldwide (Dean et al., 2005, 2012; Skamnioti and Gurr, 2009). To resist pathogen invasion, plants have developed a complex defense system that includes enemy molecule recognition, signal transduction and defensive responses throughout the immune system (Dodds and Rathjen, 2010; Andersen et al., 2018). In the first layer of plant innate immunity, pattern recognition receptors (PRRs) on the plasma membrane (PM) can detect pathogen-associated molecular patterns (PAMPs) and activate pattern-triggered immunity (PTI), which leads to the activation of signaling cascades by mitogen-activated protein kinases (MAPKs) and calcium-dependent protein kinases (CPKs), the transcriptomic reprogramming of defense-related genes, a burst of Ca2+ and reactive oxygen species (ROS) and the deposition of callose (Chisholm et al., 2006; Jones and Dangl, 2006; Boller and Felix, 2009; Gu et al., 2017). However, adapted pathogens have evolved and release various effector proteins that cause effector-triggered susceptibility (ETS), which drives the evolution of the second layer of innate immunity in plants, termed effector-triggered immunity (ETI): ETI includes the sensing of effectors by nuclear-binding leucine-rich repeat receptors (NLRs); the hypersensitive response, especially programmed cell death (PCD); and the regulation of phytohormones (Durrant and Dong, 2004; Jones and Dangl, 2006; Dodds and Rathjen, 2010; Macho and Zipfel, 2014).
The PM acts as the primary plant-microbe interaction interface during pathogen infection. Due to its physiochemical characteristics of heterogeneity, hydrophobicity and low abundance, the PM is often underrepresented at an early stage (Marmagne et al., 2004). In recent years, diverse proteomic technologies have been widely used in plants, including one- or two-dimensional electrophoresis and liquid chromatography (LC) to separate membrane proteins, and gel-based and mass spectrometry (MS)-based (label or label-free) methods for the quantification of membrane proteins; and many proteins have been shown to be related to defense responses (Benschop et al., 2007; Komatsu, 2008; Kota and Goshe, 2011; Hopff et al., 2013; Yadeta et al., 2013; Aloui et al., 2018; Witzel et al., 2018; Wei et al., 2020). Quantitative PM proteomics have shown that in Arabidopsis, after stimulation with the flagellin peptide flg22, transporters and receptor-like kinases (RLKs), especially the flagellin receptor FLS2, change significantly, while other proteins, including H+-ATPase, Ca2+-ATPase and V-ATPase, also change, accompanied by the rapid phosphorylation of the ABC transporter PDR8/PEN3 and the NADPH oxidase RbohD as immediate responses at the early defense stage (Nühse et al., 2007; Keinath et al., 2010); after the activation of ETI, dynamic signaling events, primary and secondary metabolism, vesicle trafficking and transport change significantly (Elmore et al., 2012). In rice suspension cells expressing the disease resistance gene Xa21, H+-ATPases, phosphatases, hypersensitive-induced response protein (OsHIR1), prohibitin (OsPHB2), zinc finger proteins, universal stress proteins (USPs) and heat shock proteins are differentially regulated at the early defense stage after inoculation with bacterial blight (Xanthomonas oryzae pv. oryzae) (Chen et al., 2007). Recently, isobaric tags for relative and absolute quantification (iTRAQ) have been widely used in proteomic analysis due to their high reliability and efficiency (Karp et al., 2010; Evans et al., 2012). However, the changes in the PM proteome in rice in response to rice blast fungus are still unknown.
Notably, in plants, vesicle trafficking is a key activity at the PM in the face of pathogen attacks. The delivery of defense proteins and antimicrobial substances to the extracellular space, the deposition of callose at the cell wall and the movement of PRRs to the PM are mediated by secretory-associated membrane trafficking (Wang et al., 2015); the recycling of ligand-free and ligand-bound PRRs, the delivery of pathogen effectors and transport toward vacuoles for degradation are mediated by endocytosis-associated membrane trafficking at the location of the host-pathogen interaction (Kale and Tyler, 2011; Gu et al., 2017). Diverse proteins participate in the vesicle trafficking network at the PM, including in the process of fusion between vesicles and the PM in the secretory system and the formation of vesicles in endocytosis (Leborgne-Castel and Bouhidel, 2014). In the secretory pathway, the initial contact and recognition between vesicles and the PM are mediated by vesicle tethering factors, including the exocyst complex (SEC3, SEC5, SEC6, SEC8, SEC10, SEC15, EXO70, and EXO84) (Yu and Hughson, 2010; Žárský et al., 2013). After recognition, membrane fusion at the PM is mainly accomplished by soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) (Lipka et al., 2007).
The SNARE complex is assembled by Q-SNAREs (Qa, Qb and Qc-SNAREs) on the target membrane and an R-SNARE on the vesicle membrane and leads to membrane fusion (Fasshauer et al., 1998; Sanderfoot et al., 2000; Bassham and Blatt, 2008). Syntaxin of plants (SYP) is a Q-SNARE and participates in various defense responses. AtSYP121 in Arabidopsis belongs to the SYP1 subfamily; it forms a ternary complex with AtSNAP33 (synaptosome-associated protein of 33 kDa, a SNAP25-type protein with Qb + Qc-SNARE domains) and AtVAMP721/722 (vesicle-associated membrane protein 721/722, an R-SNARE) to deliver defense components to point of the PM where the fungus is attacking the extracellular space (Collins et al., 2003; Kwon et al., 2008). OsSYP121 in rice accumulates at the sites of rice blast fungus penetration and forms a ternary complex with the SNAP25-type proteins OsSNAP32 and OsVAMP714/724 (Bao et al., 2008b; Cao et al., 2019). SYP121/ROR2 in barley (Hordeum vulgare) forms a binary SNARE complex with HvSNAP34 and accumulates focally near the papilla to fight against powdery mildew penetration (Assaad et al., 2004; Bhat et al., 2005). SYP122 functions redundantly with its homolog SYP121 and resists penetration by facilitating cell wall deposition in Arabidopsis (Nühse et al., 2003; Assaad et al., 2004; Zhang et al., 2007). SYP123 in Arabidopsis contributes to induced systemic resistance (ISR) and the establishment of plant defense responses (Rodriguez-Furlán et al., 2016). NbSYP132 in Nicotiana benthamiana engages in the immune response to the bacterial pathogen Pseudomonas syringae by secreting vesicles containing pathogenesis-related (PR) proteins (Kalde et al., 2007). The SYP4-group proteins SYP41/42/43, located at the trans-Golgi network/early endosome (TGN/EE) in Arabidopsis, are crucial to extracellular resistance by regulating the dynamic relocation of VAMP721 to sites of plant–fungus interaction and facilitating the formation of the SYP121/VAMP721 SNARE complex (Uemura et al., 2012, 2019). The SYP7 group is a plant-specific Qc-SNARE subfamily, and transgenic plants overexpressing OsSYP71 show enhanced resistance to rice blast, while silencing SYP71 in wheat results in susceptibility to stripe rust disease (Bao et al., 2012; Liu et al., 2016). The AtBET12 Qc-SNARE interacts with the AtMEMB12 Qb-SNARE and engages in endoplasmic reticulum (ER)-to-Golgi anterograde protein trafficking, whereas it plays a repressive role in PR1 secretion (Zhang et al., 2011; Chung et al., 2018). Rab is the largest family of small GTPases, and they work with SNARE proteins to promote membrane fusion and regulate the process of secretion-dependent immune activation (Yang, 2002; Uemura et al., 2004). In plants, the Rab family can be divided into eight subfamilies, including Rab1, Rab2, Rab5, Rab6, Rab7, Rab8, Rab11, and Rab18 (Agarwal et al., 2009). RabA1b regulates the transport of the PRR Flagellin-Sensitive 2 (FLS2) from the TGN/EE to the PM (Choi et al., 2013); RabA4c contributes to the increase of callose deposition at the penetration sites of the powdery mildew fungal pathogen Golovinomyces cichoracearum (Ellinger et al., 2014); and RabE1d in Arabidopsis facilitates the secretion of PR1 and enhances resistance against Pst DC3000 (Speth et al., 2009). However, few PM proteins that participate in vesicle trafficking against M. oryzae infection in rice have been reported, and the changes that occur in the PM protein network during M. oryzae infection remain largely unknown.
In this study, the iTRAQ method was used to identify the changes in the PM proteome in the highly resistant rice landrace Heikezijing (Hei) after 24 h of M. oryzae infection. A total of 2,970 proteins were identified, 831 of which were responsive in resistance against M. oryzae. Gene Ontology (GO) enrichment analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis and a protein interaction network showed that many proteins were involved in vesicle trafficking and had obvious functional tendencies toward transport, endocytosis and phagosomes. Ten DEPs were validated at the transcript level, and a SNARE protein named NPSN (novel plant-specific SNARE) 13 actively responded to M. oryzae infection, contributed to rice blast resistance and was mainly located at the PM. Our study sheds light on some of the significant roles of PM proteins and vesicle proteins in M. oryzae–rice interactions.
Materials and Methods
Plant Materials and Growth Conditions
The rice (Oryza sativa L. japonica) landraces Hei and Su, which are resistant and susceptible, respectively, to M. oryzae strain Hoku1, along with three transgenic overexpression lines and two transgenic knockdown lines of OsNPSN13 were used in this study. Rice seeds were sown in pots containing 75% garden soil and 25% nutrient soil and grown in a greenhouse (16 h light and 8 h dark for cycle at 25°C ± 3°C) for 3 weeks for rice blast fungus inoculation.
Pathogen Inoculation and Disease Evaluation
The blast strain Hoku1 was used for blast fungus inoculation in this study. Three-week-old rice seedlings were inoculated by spraying them with a spore suspension (1 × 10–5 spores mL–1 in 0.025% [w/v] Tween 20) as previously reported (Wang et al., 2002). The inoculated seedlings were kept for 24 h in a dark incubation room with 100% relative humidity at 26°C and then moved to the greenhouse for disease development. Disease evaluation, including lesion number and lesion length, was conducted 7 days after inoculation.
Extraction and Purification of Plasma Membrane Proteins
Rice leaves that were either non-inoculated or had been inoculated for 24 h were sampled, flash frozen in liquid nitrogen and stored at –80°C for PM protein extraction. PM protein extraction and purification were performed according to the method of Cheng et al. (2009). The leaf samples were cut and ground in ice-cold homogenizing buffer containing 250 mM sucrose, 2 mM EGTA, 10% v/v glycerol, 0.5% w/v BSA, 2 mM DTT, 1 mM PMSF, 1.5% PVP, 5 mM β-mercaptoethanol, and 50 mM 1,3-bis [Tris(hydroxymethyl)-methylamino] propane (BTP) and adjusted to pH 7.8 with MES. The homogenate was filtered twice through two layers of cheesecloth and then centrifuged at 1,500 × g for 7 min and 10,000 × g for 15 min to remove debris. The supernatants were centrifuged further at 100,000 × g for 1 h. The microsomal pellets were resuspended in phase buffer (250 mM sucrose, 3 mM KCl, and 5 mM KH2PO4, pH 7.8) and then fractionated via two-phase partitioning in aqueous dextran T500 and PEG (6.3% w/v PEG 3350, 6.3% w/v dextran, 0.25 M sucrose, dissolved in 10 mM KH2PO4/K2HPO4 buffer, 35 mM NaCl, pH 7.8). The upper phase, which contained the PM vesicles, was collected and repartitioned three times against a fresh lower phase (Figure 1). The final upper phase was diluted with phase buffer and centrifuged at 160,000 × g for 40 min. The PM pellets were washed twice with resuspension buffer (250 mM sucrose, 3 mM KCl, and 5 mM BTP/MES, pH 7.8), centrifuged at 160,000 × g for 1 h, resuspended in 600 μl resuspension buffer, and stored at –80°C before use.
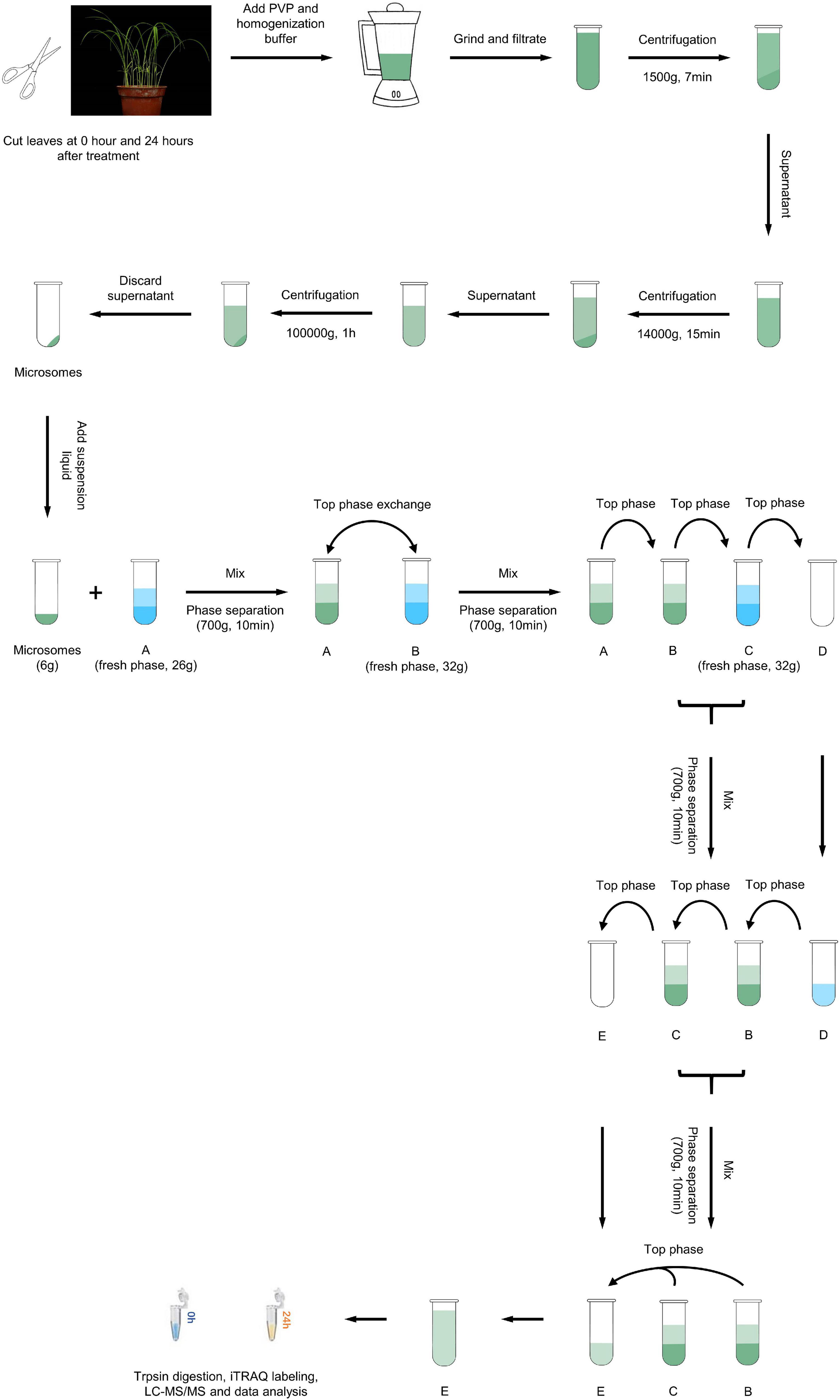
Figure 1. Overview of the experimental scheme. Schematic representation of the proteomics workflow. Leaf samples were collected at 0 and 24 h after M. oryzae infection from the resistant rice landrace Hei. Total PM protein was extracted, digested and quantified via iTRAQ labeling and LC–MS/MS analysis. Bioinformatic analyses were used to explore the constitution and function of DEPs.
Each sample was added to 30 μl of SDT buffer (4% SDS, 1 mM DTT, 150 mM Tris-HCl pH 8.0) and incubated for 5 min, then disrupted with an ultrasonic disruptor (80 W, 10 s disruption with 15 s intermission repeated 10 times), incubated for 5 min, and centrifuged at 14000 × g for 15 min; then, the supernatant was collected. Samples were qualified using a bicinchoninic acid (BCA) protein assay.
Protein Digestion and Isobaric Tags for Relative and Absolute Quantification Labeling
One mixture containing samples from all groups was used as a control and is indicated as REF. Before the iTRAQ labeling experiments, 200 μg of each sample had DTT (dithiothreitol) added to 100 mM, followed by incubation for 5 min; then, 100 μl of UA buffer (8 M urea, 150 mM Tris-HCl pH 8.0) was added, the sample was centrifuged at 14,000 × g for 15 min, and the filtrate was discarded. The cells were alkylated with 100 μl IAA (iodoacetamide) (50 mM IAA in UA) for 30 min in the dark at room temperature. For trypsin digestion, 40 μl of trypsin buffer (4 μg of trypsin in 40 μl of dissolution buffer) was added and digested at 37°C for 16–18 h. iTRAQ labeling was performed using an iTRAQ Reagent-8plex Multiplex Kit (AB SCIEX) according to the manufacturer’s protocol. iTRAQ labeling was performed as in Supplementary Table 1: REFs were labeled with iTRAQ reagent 113, the three biological replicates at 0 h were labeled with iTRAQ reagents 114, 115, and 116, and the three biological replicates at 24 h were labeled with iTRAQ reagents 117, 118, and 119.
Strong Cation Exchange Chromatography Fractionation
The peptide mixture was fractionated on an AKTA Purifier 100 (GE Healthcare, Chicago, IL, United States) equipped with a 4.6 × 100 mm polysulfoethyl column (PolyLC Inc., Columbia, MD, United States). The Strong Cation Exchange (SCX) gradient was 100% buffer A (10 mM KH2PO4 pH 3.0, 25% ACN) for 30 min, 90% buffer A and 10% buffer B (10 mM KH2PO4 pH 3.0, 500 mM KCl, 25% ACN), 80% buffer A and 20% buffer B, 55% buffer A and 45% buffer B, 100% buffer B for 10 min, and 100% buffer A for 15 min. The flow rate was 1000 μl/min. Fractions were collected every minute during the run. Subsequently, all 30 collected fractions were mixed into 10 components, lyophilized, and kept at –80°C before high-performance liquid chromatography-tandem mass spectrometry (HPLC–MS/MS) analysis.
HPLC–MS/MS Analysis
The peptides were separated on an Easy nLC system with a Thermo Scientific EASY analytical column (75 μm × 100 mm 3 μm-C18), which was interfaced with a Q-Exactive mass spectrometer (Thermo Finnigan, San Francisco, CA, United States). Mobile phase A was 0.1% formic acid, and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. Peptides were separated at a flow rate of 250 nl/min with the following gradients: 0–35% phase B, 100 min; 35–100% phase B, 8 min; and 100% phase B for 12 min. There was a full-scan mass spectrum in the Orbitrap (300–1800 m/z, 70,000 resolution) with an automatic gain control (AGC) target value of 3e6. A data-dependent acquisition method was used to collect the generated MS/MS spectra at a resolution of 17,500 with an AGC target of 1e5 and a maximum injection time (IT) of 60 ms for the top 10 ions observed in each mass spectrum. The isolation window was set at 2 m/z, the dynamic exclusion time was 40 s, and the normalized collisional energy (NCE) was set at 30 eV.
The raw data were processed using Mascot2.2 and Proteome Discoverer1.3 software (Thermo Fisher Scientific, Waltham, MA, United States). The mass spectra of the raw data were searched against the UniProt_Oryza.fasta database (144512 sequences, released February 2013). Trypsin was specified as a cleavage enzyme, allowing a maximum of 2 missing cleavages. Carbamidomethyl on cysteine was distinguished as a fixed modification. Oxidation on methionine and iTRAQ8plex (Y) were specified as variable modifications. The peptide mass tolerance was programmed to 20 ppm, and the fragment mass tolerance was programmed to 0.1 Da. The threshold score was set to 20, and the false discovery rate (FDR) threshold was adjusted to 0.01 (Sandberg et al., 2012). The quantitative analysis parameters of Proteome Discover1.3 are listed in Supplementary Table 2.
Bioinformatics Analysis
For three biological replicates, the average ratio between the protein expression level at 24 h and the protein expression level before inoculation was defined as the fold change. A 1.1-fold or 0.909-fold change as the threshold with FDR < 0.1 (p value adjusted using the Benjamini–Hochberg method for multiple comparisons) was classified as a significant change (Benedikter et al., 2019). Volcano maps and heatmaps were made using the R package ggplot2 and TBtools software (Wickham, 2016; Chen et al., 2020). The functional annotation of DEPs was mapped with GO terms1 using the UniProt database, and the enrichment was calculated using singular enrichment analysis (SEA) in agriGO with the FDR < 0.05 (Du et al., 2010). The DEPs were further functionally analyzed using the KEGG database2 and the R package clusterProfiler (Yu et al., 2012). The protein--protein interaction (PPI) network was analyzed based on the STRING database3. The PPI network was obtained by setting the confidence value at 0.4, and then Cytoscape 3.6.0 was used to adjust the PPI map (Paul et al., 2003). DEPs were mapped to a pathway enrichment in biotic stress by using MapMan software (Thimm et al., 2004).
qRT–PCR Analysis
The leaves of the resistant cultivar Hei were sampled at 0, 8, 24, 48, and 72 h after M. oryzae inoculation, frozen in liquid nitrogen and stored at –80°C. Total RNA was extracted using a universal plant total RNA extraction kit (Bioteke, Beijing, China). cDNA was synthesized using a HiScript II Q RT SuperMix kit (Vazyme, Nanjing, Jiangsu, China) for real-time quantitative PCR (qRT-PCR), which was performed using SYBR Green Master Mix (Vazyme, Nanjing, Jiangsu, China) according to the manufacturer’s protocol. The actin gene was used as an internal control, and the expression levels of genes were measured using the 2–ΔΔCT (cycle threshold) method (Pfaffl, 2001).
Generation and Identification of Transgenic Plants
Full-length and reversed full-length OsNPSN13 were inserted into the pCAMBIA1304S vector to generate the overexpression transgenic vector pCAMBIA1304S-OsNPSN13 and the antisense transgenic vector pCAMBIA1304S-rOsNPSN13, respectively. These vectors were transformed into the susceptible rice landrace Suyunuo using the Agrobacterium tumefaciens-mediated method (Toki et al., 2006). Total DNA from leaves of different transgenic lines was extracted and used to identify overexpression and antisense transgenic plants via PCR amplification. The primers used are in Supplementary Table 3.
Subcellular Localization of OsNPSN13 in Protoplasts
The full-length OsNPSN13 cDNA was amplified from Heikezijing and cloned into the pAN580 vector with the C-terminus of the fragment in frame with GFP. Protoplasts were extracted from young Heikezijing seedlings, and transient transformation with the plasmid was performed as described by Chen et al. (2006). 10 μg of plasmid DNA was added to 200 μL of suspended protoplasts (1 × 106 cells mL–1) and incubated in the dark at 28°C. OsAMT3.2-RFP was used as PM marker. After 12 h, the transformed protoplast cells were observed using a Leica SP8 laser confocal microscope.
Results
Use of Isobaric Tags for Relative and Absolute Quantification to Quantitatively Identify Plasma Membrane Proteins That Respond to Magnaporthe oryzae Infection
To investigate the roles of rice PM proteins against M. oryzae infection, we used iTRAQ to analyze the PM proteome in the resistant rice landrace Heikezijing (Hei). PM proteins were extracted from the leaves of rice seedlings and purified via two-phase partitioning in aqueous dextran T500 and PEG according to Cheng et al.’s (2009) method, then digested and labeled with iTRAQ reagents and quantitatively identified using LC–MS/MS (Figure 1). A total of 10608 unique peptides and 2,987 proteins were identified via the UniProt database (Supplementary Tables 4–6). The number of proteins in the three biological replicates of this study were 2982, 2986, and 2981 (Supplementary Figure 1), and there were 2978 common proteins in three repeats. For most proteins, fewer than 10 unique peptides were identified (Figure 2A). A total of 1,745 proteins contained at least 2 unique peptides, accounting for 58.62% of the total protein. The average protein identification coverage was 11.73%.
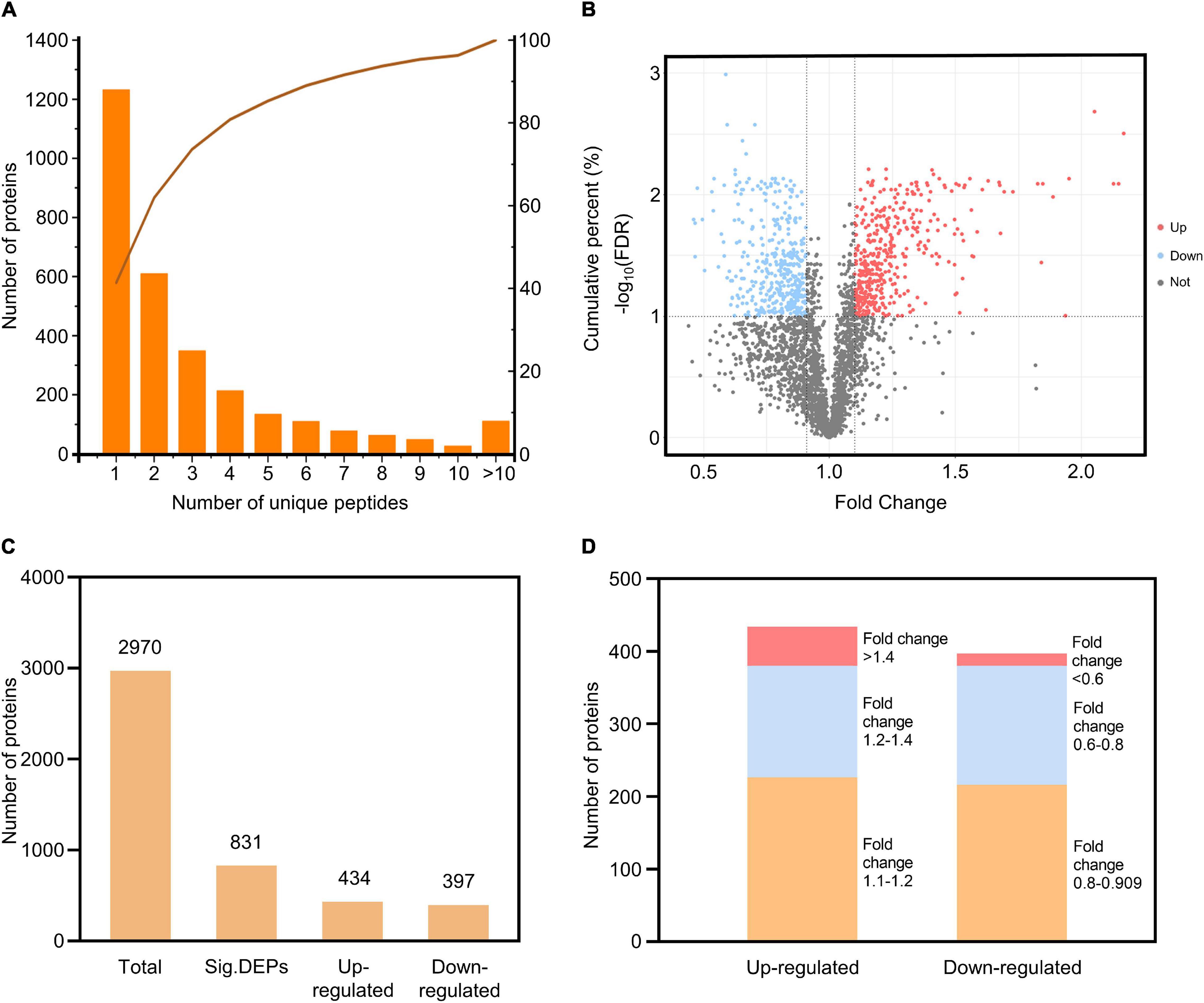
Figure 2. Quantitative identification of proteins from the iTRAQ analysis. (A) Distribution of the number of unique peptides for the identified proteins. The X-axis is the scope of the unique peptides, the left Y-axis is the number of proteins, and the right Y-axis is the cumulative percent. (B) Volcano plot of the proteins identified. The X-axis specifies the fold change between 24 h/0 h, and the Y-axis specifies the -log10 (FDR); upregulated, downregulated and non-significant proteins are shown as red, blue, and gray dots, respectively; gray vertical and horizontal lines reflect the filtering criteria (fold change > 1.1 or < 0.909 and FDR < 0.1). (C) The number of total proteins and DEPs (including upregulated and downregulated). (D) The distribution of DEP fold changes.
Based on the thresholds of FDR < 0.1 and fold change > 1.1 for upregulated DEPs and fold change < 0.909 for downregulated DEPs, 831 differentially expressed proteins (DEPs) were identified, including 434 upregulated DEPs and 397 downregulated DEPs (Figures 2B,C and Supplementary Tables 7, 8). Fifty-four upregulated DEPs had a fold change greater than 1.4, and 17 downregulated DEPs had a fold change less than 0.6 (Figure 2D). The distribution of fold changes showed that the PM proteome changed at different levels during the immune response against rice blast. The number of upregulated DEPs was approximately equal to the number of downregulated DEPs, and the three replicates for each treatment had good repeatability (Supplementary Figure 2). A total of 27.98% of all identified proteins changed during M. oryzae infection, suggesting that many PM proteins responded to and were involved in the plant defense process.
Functional Characterization of Differentially Expressed Proteins
Based on Gene Ontology (GO) annotations, 434 up-regulated DEPs and 397 down-regulated DEPs were separately categorized into three major enrichment categories: biological process, cellular component and molecular function (FDR < 0.05). The results showed that 282 up-regulated DEPs (65%) were annotated at 10 levels, including four biological processes, three cellular components, and three molecular functions; 243 down-regulated DEPs (61%) were annotated at 10 levels, including four biological processes, four cellular components, and two molecular functions (Figure 3 and Supplementary Table 9). In the biological process category, up-regulated DEPs were enriched in translation (GO:0006412), transport (GO:0006810), signal transduction (GO:0007165) and response to stimulus (GO:0050896); down-regulated DEPs were enriched in transport (GO:0006810), establishment of localization (GO:0051234), localization (GO:0051179) and cellular metabolic process (GO:0044237). Intriguingly, many DEPs were related to vesicle trafficking, including 82 DEPs (49 up-regulated and 33 down-regulated) involved in transport, which implied that the trafficking system was activated by rice blast infection. In the cellular component category, up-regulated DEPs were enriched in membrane (GO:0016020), cytoplasm (GO:0005737) and plasma membrane (GO:0005886); down-regulated DEPs were enriched in cytoplasm (GO:0005737), cell part (GO:0044464), intracellular (GO:0005622) and plasma membrane (GO:0005886). At the level of molecular function, up-regulated DEPs were associated with structural molecule activity (GO:0005198), nucleotide binding (GO:0000166) and transporter activity (GO:0005215); down-regulated DEPs were associated with catalytic activity (GO:0003824) and transporter activity (GO:0005215). Both up-regulated and down-regulated DEPs were linked with transporter activity. Overall, based on GO enrichment analysis, the functional categories of the DEPs from the three different levels all pointed to the primary conclusion that vesicle trafficking played an important role in rice resistance against M. oryzae.
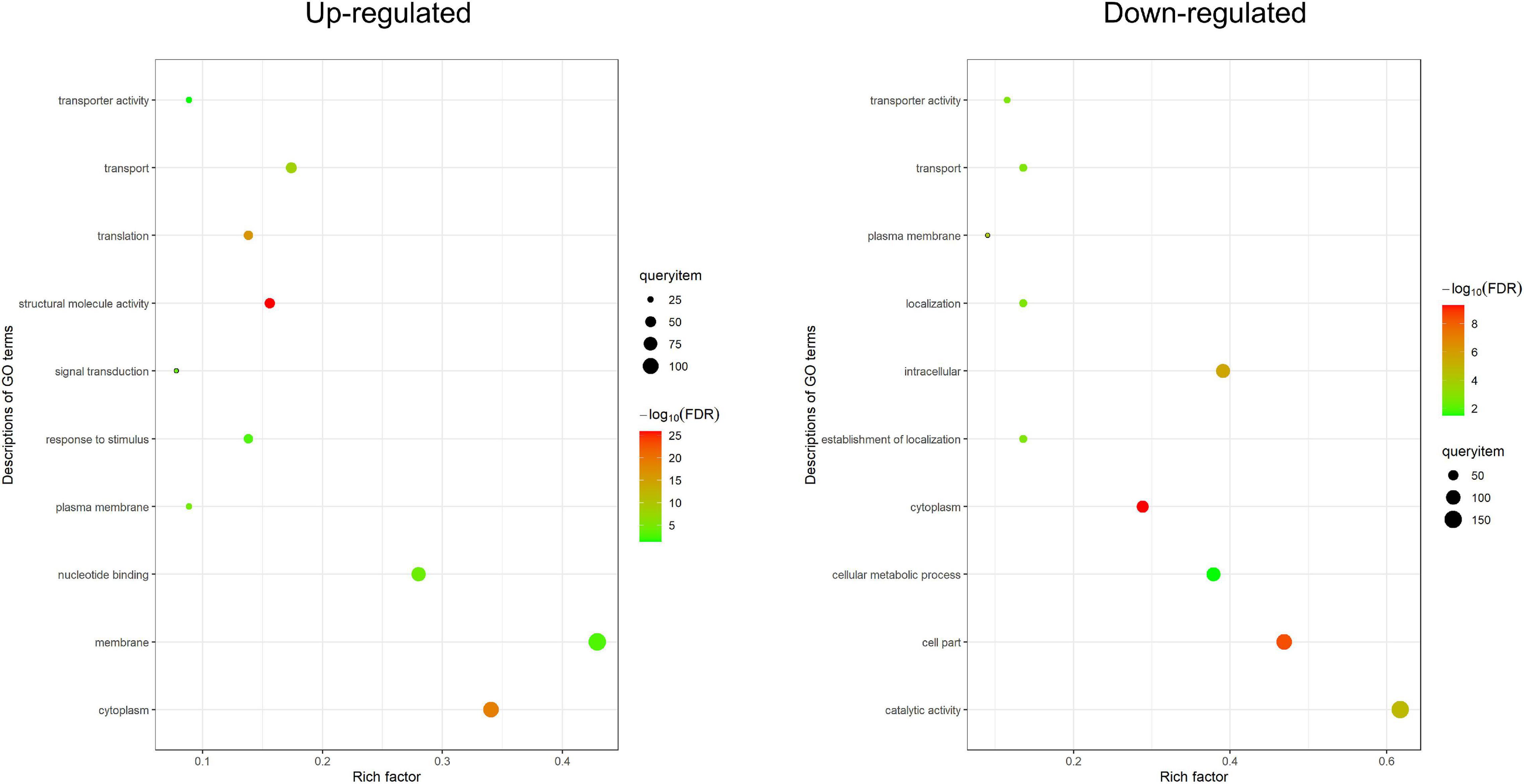
Figure 3. Gene ontology enrichment analysis. GO enrichment analysis of DEPs (FDR < 0.05). The enriched terms from three levels (biological process, molecular function, and cellular component) are shown in the figure. The X-axis is the rich factor, and the Y-axis is the description of GO term.
For further functional grouping, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed (Figure 4). Upregulated DEPs were generally assigned to the phagosome (osa04145), endocytosis (osa04144), the citrate cycle (osa00020), lysine degradation (osa00310) and valine/leucine/isoleucine degradation (osa00280); downregulated DEPs were mainly assigned to glutathione metabolism (osa00480), purine metabolism (osa00230), glycolysis (osa00010), pyruvate metabolism (osa00620) and the pentose phosphate pathway (osa00030). The top two most prominent enriched upregulated pathways were the phagosome and endocytosis, suggesting that vesicle trafficking is a crucial process in the resistance response (Table 1 and Supplementary Figure 3). The GO enrichment and KEGG pathway analyses jointly showed that multiple DEPs were engaged in vesicle trafficking, which suggested that we should analyze the detailed components and functions of this process.
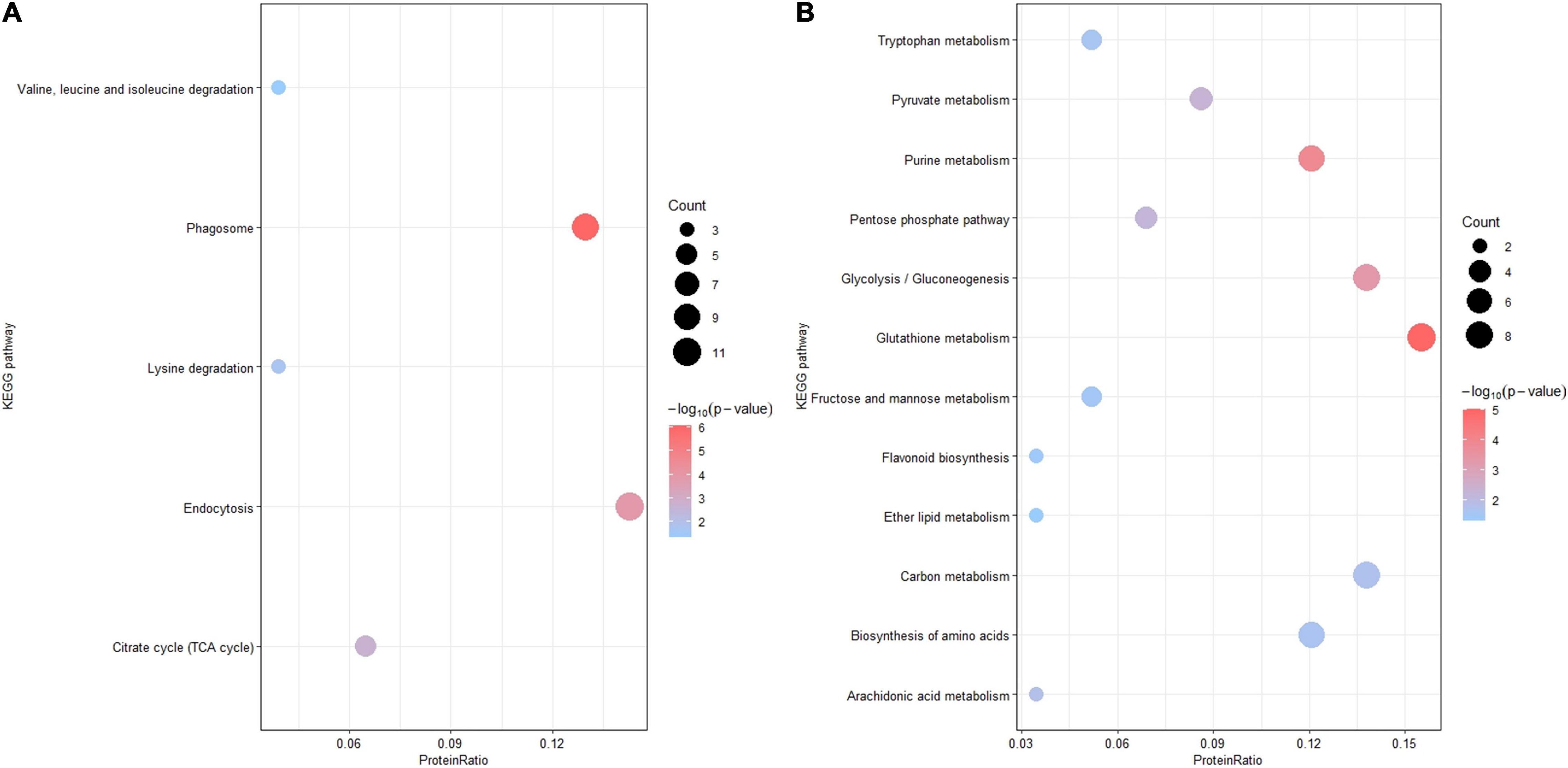
Figure 4. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis. KEGG pathway enrichment analysis of DEPs (p-value < 0.05). (A) KEGG pathway enrichment analysis of upregulated DEPs. (B) KEGG pathway analysis of downregulated DEPs. The X-axis is the protein ratio (the number of DEPs annotated with this pathway term/the total number of DEPs), the Y-axis is the KEGG pathway, the size of the bubble represents the protein count, and the color of the bubble represents –log10 (p-value).
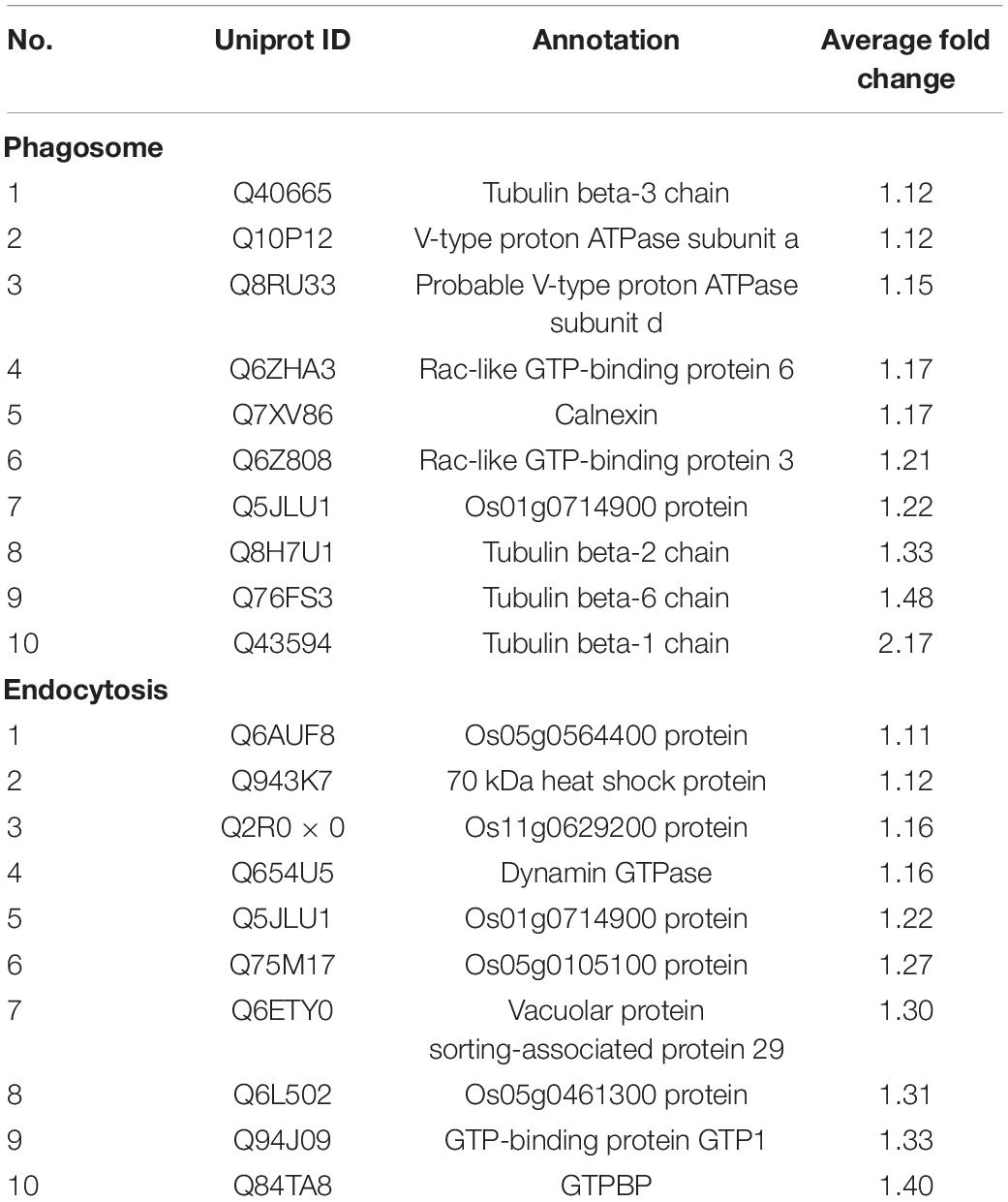
Table 1. The list of up-regulated DEPs involved in phagosome and endocytosis according to KEGG enrichment analysis.
Of the 831 DEPs, 496 could be mapped in MapMan, and 136 of those were associated with biotic stress (Supplementary Figure 4). Forty-seven DEPs associated with signaling were the most enriched group, including 34 upregulated DEPs and 13 downregulated DEPs. Other physiological activities, including respiratory burst, hormone signaling and proteolysis, were also significantly changed. Diverse types of proteins were identified, including peroxidases, glutathione-S-transferases, transcription factors, beta-glucanases, heat shock proteins and secondary metabolites. The various DEPs engaged in rice blast resistance at the PM suggest that there must be an intricate and dynamic network linked with various biological processes at the PM during the plant immune response and during plant–pathogen interactions.
A Protein–Protein Interaction Network of Selected Differentially Expressed Proteins Based on the STRING Database
To decipher the functional networks, we used the STRING database to construct a PPI network of DEPs and obtain a putative association network. The results of the functional analysis showed that many DEPs participated in vesicle trafficking, so we focused on these DEPs, which encompassed one major GO enrichment term (transport) and two major pathways from the KEGG analysis (phagosome and endocytosis), to further understand their functions. The maximum confidence (score) cutoff was adjusted to 0.40, and the result was visualized using Cytoscape (Figure 5). The results showed that the most closely interacting DEPs (>9 interaction degrees) were found between multiple small GTPase subfamily proteins, including the Ras protein OsRIC1 (P40392) and unclassified Ras-related proteins (Q5JLU1, Q8RZ83, Q75M17, Q6AUF8, Q6L502), the Rab proteins OsRab6 (Q8H4Q9) and OsRab8a (Q84TA8), and the GTP-binding protein GTP1 (Q94J09); other types of proteins, including the vacuolar protein sorting-associated proteins VPS18 (Q6ZKF1), VPS26 (Q2R0X0) and VPS29 (Q6ETY0), a serine/threonine phosphatase (Q6K3D4), tubulin beta-1 chain protein TuBB1 (Q43594) and tubulin beta-6 chain protein TuBB6 (Q76FS3), also had close interactions. Thirteen of these 15 proteins were upregulated, which meant that active transportation was essential in rice blast resistance. Nine proteins were small GTPase-related transporting and vacuolar protein sorting-associated proteins, indicating that different types of vesicles and organelles participated in plant immunity.

Figure 5. Protein–protein interaction network of DEPs. A protein–protein interaction network of DEPs associated with vesicle trafficking (including the transport terms from the GO analysis and the phagosome and endocytosis terms from the KEGG analysis) was built using the STRING database. Identified interactions with confidence scores ≥ 0.4 were visualized using Cytoscape software. The red nodes are upregulated DEPs, and the green nodes are downregulated DEPs; the size of the nodes represents the number of related interactions in the network; and the colors of the edges indicate the strength of the interaction: orange represents a strong interaction, and blue represents a weak interaction.
Examination of Differentially Expressed Proteins at the Transcriptional Level Using qRT–PCR
We examined the transcriptional pattern of ten selected DEPs using qRT–PCR. Except for OsRem4.1, the 10 DEPs were selected because they are typically related to vesicle trafficking; OsRem4.1 was chosen because it is typically located at the PM, and it has been reported that in Arabidopsis, Rem1.2 is related to plant immunity (Huang et al., 2019). The expression pattern analysis included seven genes that encoded upregulated proteins, including a Qc-SNARE protein (OsSYP71), a novel plant-specific SNARE protein (OsNPSN13), a vesicle-associated membrane protein (VAMP), an exocyst complex component protein (Sec6), a Sec3-PIP2 bind domain-containing protein (Sec3), a secretory carrier-associated membrane protein (SCAMP) and an exocyst subunit Exo70 family protein (Exo70), as well as three genes that encoded downregulated proteins, including Remorin (OsRem4.1), Sec1 and Sec7. The transcriptional levels of OsSYP71, OsNPSN13, VAMP, Sec6, Sec7, OsRem4.1, Sec3, SCAMP, and Exo70 were upregulated at 8 h, 24 h and 72 h after M. oryzae infection, while Sec1 was downregulated at 8 h (Figure 6). OsSYP71 and OsNPSN13 were upregulated at 8 h, Sec3 was upregulated at 24 h, and SCAMP and Exo70 were upregulated at 72 h. According to the iTRAQ results, the transcriptional trend of OsRem4.1 and Sec7 was reversed, and some factors, such as mRNA splicing, translation and posttranslational regulation, as well as protein degradation, all influenced the correlation between the transcriptome and proteome data (Wei et al., 2009; Chen et al., 2015; Su et al., 2016). Additionally, to test the accuracy of the proteomic data, we identified DEP transcriptional patterns that changed slightly (fold change between 0.8 and 0.909 or 1.1 and 1.2), and the results showed that 9 of 16 proteins had comparatively good consistency (Supplementary Figure 5). Based on the iTRAQ and qRT–PCR analysis, the upregulated protein OsNPSN13 was selected for further functional analysis in resistance to rice blast.
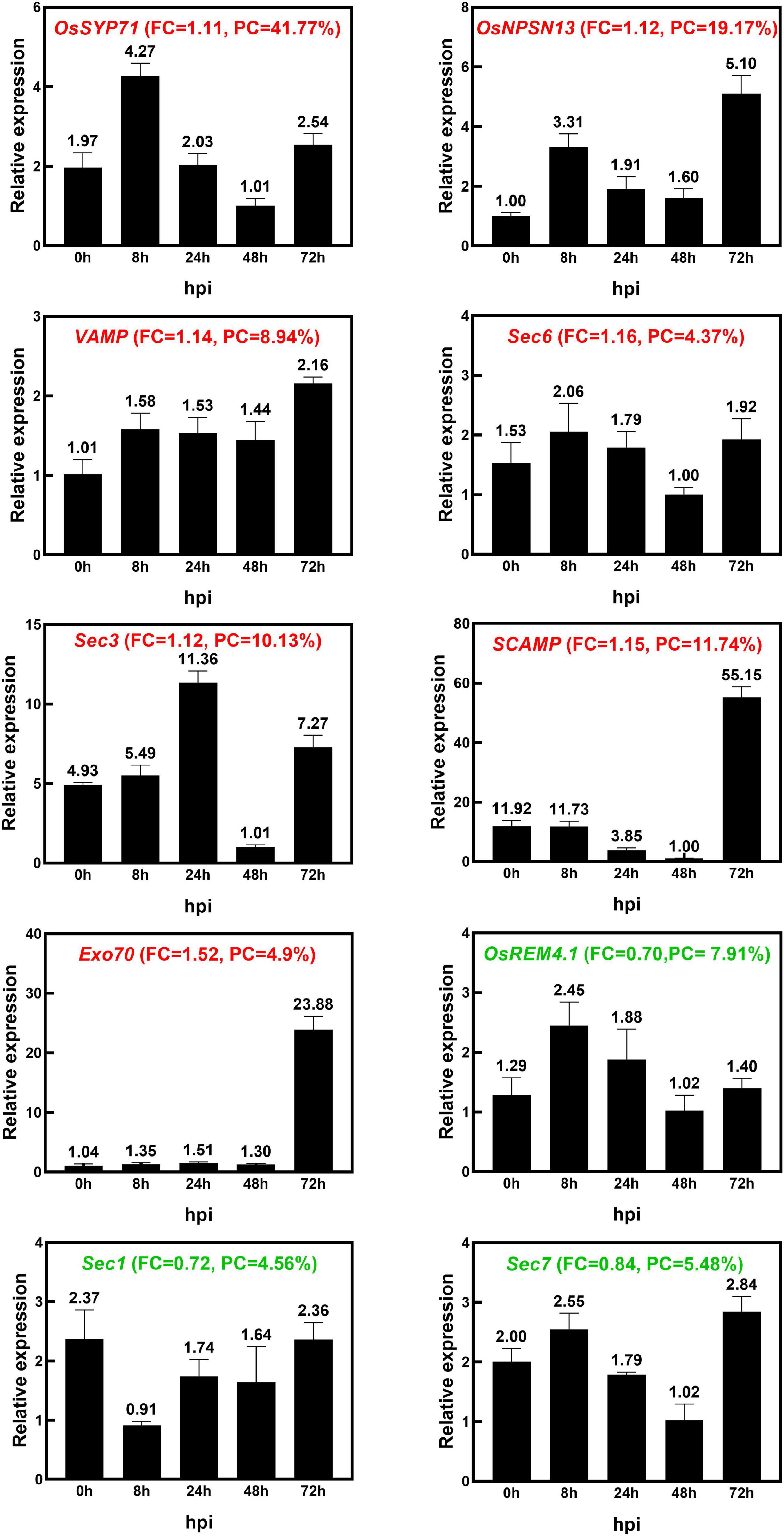
Figure 6. The identification of transcriptional expression patterns of 10 DEPs after M. oryzae infection. The expression patterns of 10 selected DEPs in Heikezijing (Hei) inoculated with the M. oryzae strain Hoku1 were investigated via qRT–PCR. Hei seedlings were collected 0, 8, 24, 48, and 72 h after inoculation. The rice actin gene was used as an internal control. The bar chart shows the average ± standard deviation (n = 3). The information of Fold Change (FC) and Peptide Coverage (PC) in proteomic data was also shown in the figure. Red and green colored names represent upregulated and downregulated, respectively, in the proteomic analysis.
OsNPSN13 Is Involved in the Resistance Response Against Magnaporthe oryzae
Three OsNPSN13-overexpressing transgenic lines (S13-7, S13-11, and S13-12) and two knockdown transgenic lines (A13-3 and A13-4) in Su were obtained using the Agrobacterium tumefaciens-mediated method (driven by the 35S promoter). The characterization of the transgenic lines is shown in Supplementary Figure 6. After inoculation with rice blast fungus (strain Hoku1) at the seedling stage, three transgenic overexpression lines showed significantly enhanced resistance compared with wild-type Su, including fewer and shorter lesions, whereas the two knockdown transgenic lines were more susceptible to rice blast than was wild-type Su (Figures 7A–C). These results showed that OsNPSN13 was associated with resistance to rice blast fungus. The subcellular localization in protoplasts showed that OsNPSN13-GFP was mainly localized to the PM (Figure 7D).
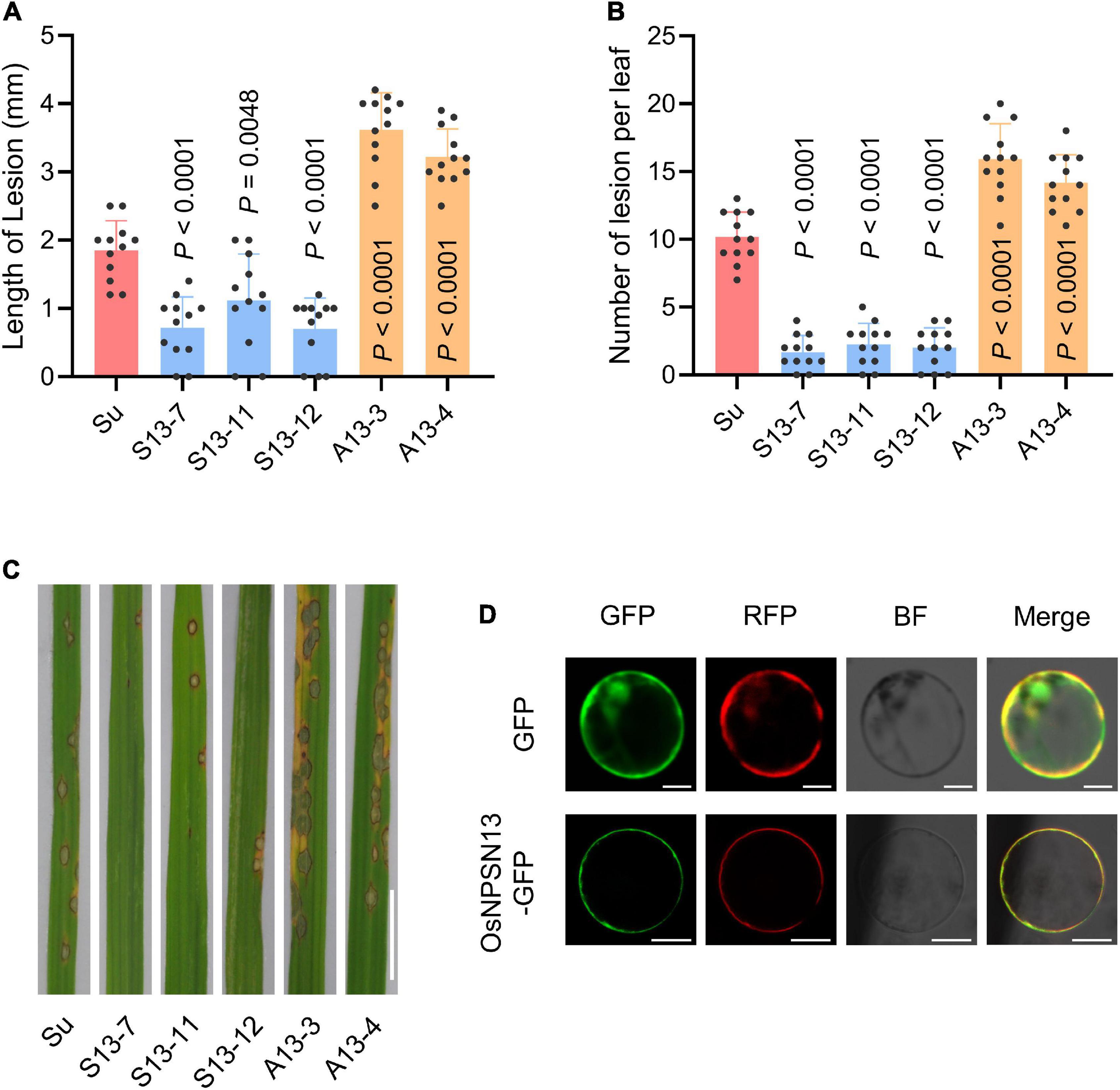
Figure 7. Resistance phenotype of representative transgenic plants expressing OsNPSN13 and the subcellular localization of OsNPSN13. (A) Lesion length in wild-type Su and transgenic plants expressing OsNPSN13 at 7 dpi with M. oryzae. (B) Lesion number per leaf in wild-type Su and transgenic plants expressing OsNPSN13 inoculated with M. oryzae at 7 dpi. S13, transgenic overexpression lines; A13, transgenic knockdown lines. The bar charts show the average, standard deviation (n = 12) and p-value. (C) Rice blast resistance phenotypes of transgenic plants and wild-type Su inoculated with M. oryzae strain Hoku1. The leaves with lesions are shown. Bar = 1 cm. (D) The subcellular localization of OsNPSN13 transiently expressed in rice protoplasts. OsAMT3.2-RFP was used as PM marker. Bar = 5 μm.
Discussion
Multiple proteins associated with blast resistance in rice have been extensively studied. However, proteomic analyses of the PM protein network that participates in rice resistance against M. oryzae infection remain limited. In this study, a comparative iTRAQ-based proteomic analysis was performed to elucidate the changes in PM proteins during M. oryzae infection. A total of 831 DEPs were identified, including 434 upregulated and 397 downregulated proteins. GO enrichment analysis showed that the biological process of transport and the molecular functions associated with transporter activity were related to vesicle trafficking. KEGG analysis indicated that endocytosis and the phagosome were highly enriched pathways in the upregulated DEPs, and glutathione metabolism and glycolysis/gluconeogenesis were highly enriched pathways in the downregulated DEPs. DEPs associated with vesicle trafficking based on the GO and KEGG analyses were used to construct a protein interaction network, which showed that there were tight interactions between small GTPases and VPS proteins. A functional analysis of the DEPs yielded 20 essential proteins related to vesicle trafficking that could be mapped to a potential PM trafficking network against M. oryzae (Figure 8).
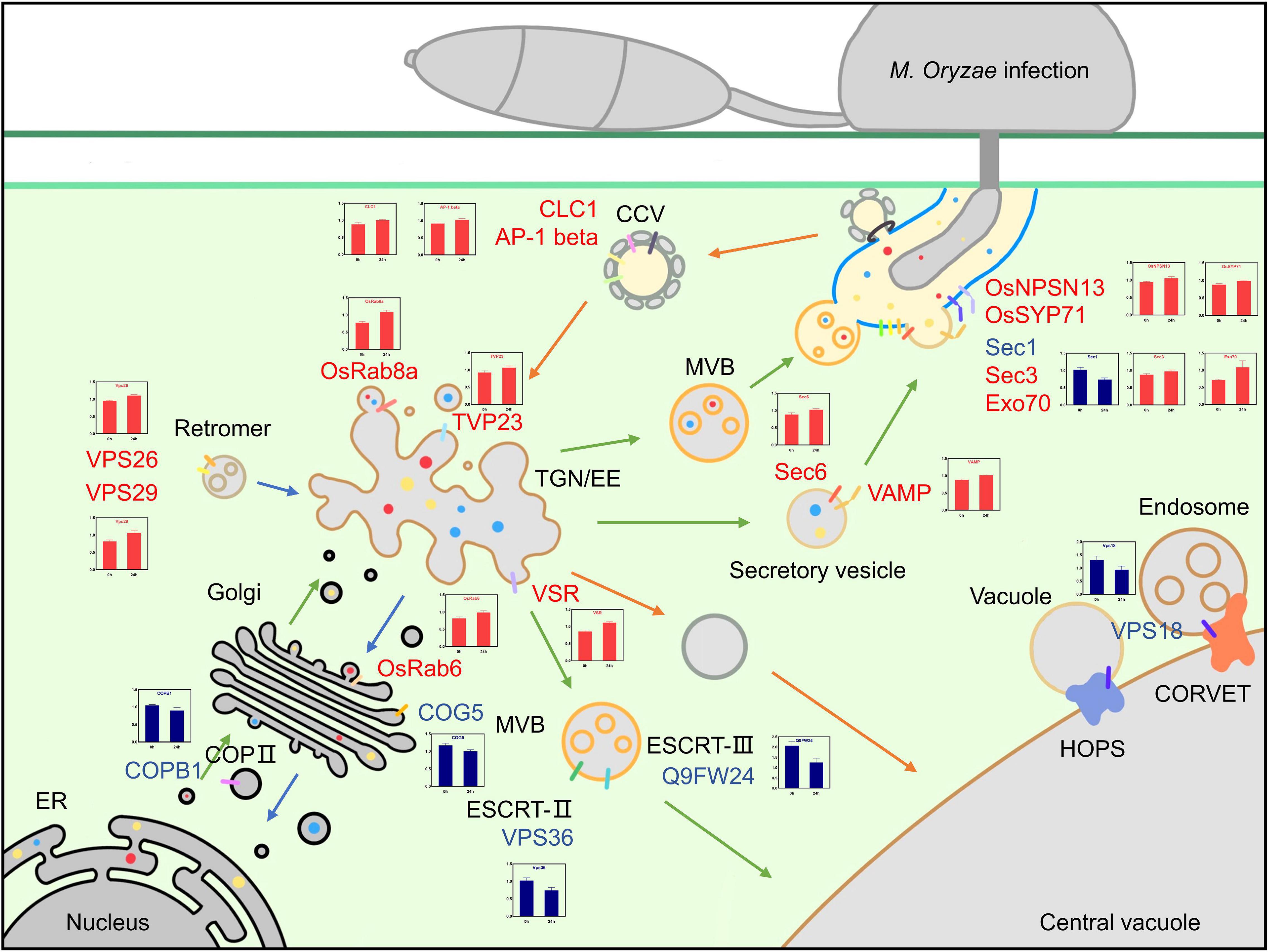
Figure 8. Overview of the possible vesicle trafficking network of DEPs. Possible model for the roles of DEPs in vesicle trafficking during the rice-blast fungus interaction. Upregulated DEPs are shown in red, downregulated DEPs are shown in blue, green arrows indicate exocytosis, orange arrows indicate endocytosis, and the translational patterns of DEPs are shown in the charts.
The exocytosis pathway is an integral component of the plant immune mechanism, and it contributes to basal resistance by transporting defense-related proteins, including PRRs; delivering antimicrobial metabolites; and mediating callose deposition (Gu et al., 2017). Five DEPs that were part of the vesicle-mediated transport term in the GO enrichment analysis, COPB1, OsRab6, COG5, OsRab8a and TVP23, were associated with protein transport from the ER to the Golgi, TGN and PM. Coatomer subunit beta COPB1 (B8BJD1) reversibly associates with Golgi non-clathrin-coated vesicles and further mediates biosynthetic protein transport from the ER via the Golgi to the TGN; OsRab6 (Q8H4Q9), conserved oligomeric Golgi complex subunit 5 COG5 (A2Y424) and TGN vesicle protein 23 TVP23 (Q5W723) localize to the Golgi apparatus and are involved in intra-Golgi vesicle-mediated transport and constitutive secretion (Osterrieder et al., 2009; Bottanelli et al., 2011); OsRab8a (Q84TA8) participates in vesicle trafficking between the TGN and PM to ensure the release of secretory vesicles (Huber et al., 1993; Nielsen et al., 2008). In tomato, Rab8 was identified as a potential protein that interacts with avrPto from the pathogen Pseudomonas sp., and this interaction possibly blocks the extracellular release of plant antimicrobial peptides (Bogdanove and Martin, 2000).
Eight DEPs, OsNPSN13, OsSYP71, VAMP, SCAMP, Sec1, Sec3, Sec6, and Exo70, were enriched in the transport term and were related to vesicle recognition and PM fusion. The Qb-SNARE protein OsNPSN13 (Q5XQQ5) localizes to the PM and is related to abiotic stress, and its expression is significantly activated in rice seedlings treated with H2O2 but is downregulated under NaCl and PEG 6000 stresses, suggesting that it might be involved in different aspects of signal transduction cascades (Bao et al., 2008a). NPSN11, a homolog of NPSN13, has been demonstrated to play an important role in disease resistance. In wheat, TaNPSN11 interacted with TaSYP132 and was distributed in vesicle structures near the cell membrane toward Puccinia striiformis f. sp. tritici (Pst) hypha; this behavior may play a role in vesicle-mediated resistance to stripe rust (Wang et al., 2014). ShNPSN11 in tomato is located at the PM and plays an important role in defense reactions to O. neolycopersici (Lian et al., 2020). In Arabidopsis, AtSYP71 belongs to the Qc-SNARE family and localizes to the PM and the ER (Suwastika et al., 2008). In rice, OsSYP71 (Q6I633) has been reported to positively contribute to rice blast resistance, and OsSYP71 overexpression obviously enhanced resistance, including lower lesion scores and higher survival rates after M. oryzae inoculation (Bao et al., 2012). TaSYP71 in wheat also plays a positive role in wheat defense against Puccinia striiformis f. sp. tritici (Pst), and it can elevate tolerance to H2O2 and influence the expression of pathogenesis-related genes (Liu et al., 2016). However, SYP71 can also be a negative factor since it mediates the fusion of turnip mosaic potyvirus (TuMV) 6K2 vesicles with host chloroplasts during TuMV infection, which contributes to the robust replication of the viral genome. Silencing the SYP71 gene inhibits TuMV infection and arrests the formation of the chloroplast-bound 6K2 complex (Wei et al., 2013). The SYP7 family has not been identified in yeast or mammalian SNAREs, so they probably have unique functions in the plant vesicle system (Ebine et al., 2008). Although the VAMP family protein (Q2QLJ6) of the SNARE complex and secretory carrier membrane protein (SCAMP) family protein (B8AZR2) have not been precisely classified, it is clear that these two families play an important role in vesicle trafficking. Sec1 (Q6K7L5) is involved in vesicle docking in exocytosis and contributes to membrane fusion by interacting with SNAREs or nascent trans-SNARE complexes (Südhof and Rothman, 2009; Karnahl et al., 2018). The exocyst complex (including SEC3, SEC5, SEC6, SEC8, SEC10, SEC15, EXO70 and EXO84) tethers secretory vesicles to specific sites on the PM and facilitates exocytosis (He and Guo, 2009). The exocyst subunit Sec3 (B8AMN4) belongs to the SEC3 family and plays a pivotal role in exocytosis and pollen tube growth (Bloch et al., 2016); it directly interacts with Sso1/2, facilitates the assembly of the Sso-Sec9 t-SNARE complex and stimulates membrane fusion in yeast (Yue et al., 2017). Exocyst subunit Sec6 (A2X9M7) belongs to the SEC6 family and plays a role in polar auxin transport in Arabidopsis (Tan et al., 2016). Exocyst subunit Exo70 (B9FW54) functions together with Sec3 in targeting the exocyst to the PM (He et al., 2007). Exocyst complexes, including Sec3, Sec6, and Exo70, may mediate precise recognition and tethering between vesicles and PM. Subsequently, Sec1, OsNPSN13, OsSYP71, VAMP, and SCAMP contribute to membrane fusion and release antimicrobial compounds and defense-related proteins to fight M. oryzae infection.
The endocytosis of PM-localized PRRs and LRR-receptor-like proteins (LRR-RLPs) is required for the proper dynamic localization of activated and inactivated PRRs to regulate the immune signaling response (Wang et al., 2015). Endocytic trafficking is initiated by vesicle formation and fusion and subsequently routes to endosomes and lytic vacuoles for degradation or recycling back to the PM. The major route of endocytosis is clathrin-mediated endocytosis (CME), and the cargo and vesicle coats are recruited into clathrin-coated vesicles (CCVs) and transported from the PM to early endosomes. Clathrin-coated vesicles contain two layers outside of the membrane, including adaptor proteins (APs) as the inner layer and clathrin as the outer layer. Eight DEPs, including AP-1 beta, CLC1, VPS18, VPS26, VPS29, VPS36, VSR, and Q9FW24, were included in the endocytosis and transport terms in the KEGG and GO enrichment analyses, respectively. AP-1 complex subunit beta (Q0DRT9) mediates TGN-endosome and Golgi-vacuole intracellular protein transport (Zwiewka et al., 2011; Feng et al., 2017); Clathrin light chain 1 CLC1 (Q7XKE9) combines with clathrin heavy chain (CHC) and forms the outer layer of clathrin-coated vesicles (CCVs). We hypothesize that the activated PRRs at the PM are essential for the continuation of immune signaling, so the downregulated proteins can lead to fewer activated PRRs being recycled and degraded in the vacuole.
After transport from the PM to the TGN/EE by clathrin-coated vesicles, the recycled proteins are sorted into different locations, including to the vacuole for degradation or recruitment to the PM. Vacuolar protein sorting-associated proteins (VPSs) play an essential role in this process. Two tethering complexes, homotypic fusion and protein sorting (HOPS) and class C core vacuole/endosome tethering (CORVET), mediate transport from endosomes to vacuoles and share mutual core proteins, including VPS11, VPS18 (Q6ZKF1), and VPS16 (Vukašinovic and Žárský, 2016; Takemoto et al., 2018). VPS36 (B8AB94) is a component of endosomal sorting complexes required for transport (ESCRT)-II, which has ubiquitin-binding activity and is critical for MVB formation and vacuolar biogenesis to degrade proteins (Teo et al., 2004; Hsua and Jauh, 2017; Wang et al., 2017). VPS36 has been reported to be associated with the transport of PIN1, AUX1, and PIP1 family members and with root gratitropism in Arabidopsis, but its function in plant immunity remains unknown. The SNF7 domain-containing protein (Q9FW24) is the core component of ESCRT-III that mediates MVB biogenesis by facilitating membrane bending, scission and fusion (Ibl et al., 2012). Vacuolar-sorting receptor VSR (Q0JA85) mediates Golgi-to-vacuole transport. VPS18, VPS36, and Q9FW24 were downregulated, and this result suggested that the degradation of some specific proteins may be restricted during M. oryzae infection, but the details are still elusive and require further investigation.
In addition to being degraded by vacuoles, some proteins may be recruited back to the PM by retromers to perform their function. It has been reported that flg22-activated FLS2 is recruited to the extrahaustorial membrane (EHM) induced by Phytophthora infestans (Bozkurt et al., 2015). The retromer is a conserved complex located on the cytosolic face of endosomes and VPS26 (Q2R0X0), VPS29 (Q6ETY0) and VPS35; it regulates the retrieval of cargo from the endocytic system to the TGN or PM (Zelazny et al., 2013; Priya et al., 2015). VPS26 and VPS29 were upregulated, suggesting that retrograde transport was enhanced and potentially included the recruitment of defense-associated proteins to the PM. The roles of receptor-mediated endocytosis may be complicated in plants; however, our current knowledge about its roles in regulating the activity of immunity-related proteins remains limited. Many DEPs associated with endocytosis were found in this study, which provides a direction for future research efforts.
An overview of the potential vesicle trafficking response to M. oryzae infection is shown in Figure 8. In exocytosis, COPB1, COG5, OsRab6, OsRab8a, and TVP23 mediate the transport of synthesized proteins from the ER to the Golgi and TGN/EE, assembly in vesicles and movement toward the PM; Sec1, Sec3, Sec6, and Exo70 ensure recognition and tethering between vesicles and the PM; and membrane fusion is completed by VAMP, OsNPSN13, and OsSYP71. In endocytosis, the formation and assembly of clathrin-coated vesicles require AP-1 beta and CLC1; the sorting of recycled proteins and vacuole-targeted degradation are mediated by VPS18, VPS36, VSR and Q9FW24; and the recruitment of proteins to the Golgi or PM is completed by retromers, including VPS26 and VPS29.
After thoroughly exploring the iTRAQ data via the functional analysis of DEPs and identification via qRT–PCR, we ultimately focused on the significantly changed SNARE protein OsNPSN13. Overexpressing OsNPSN13 in rice led to enhanced resistance, while knocking down OsNPSN13 expression led to serious susceptibility. SNARE proteins are related to the secretory transport of PRRs and PR proteins to the PM (Beck et al., 2012). It has been reported that in wheat, knocking down TaNPSN13 reduced ROS-mediated plant defense at stripe rust infection sites, indicating that it might be involved in the delivery of ROS-related materials; in addition, TaNPSN11 and its structural homologs mediated the vesicle trafficking of plant defense-related materials between the Golgi apparatus and the PM based on the bubbly layered structures in immunolocalization assays (Wang et al., 2014). In rice, OsNPSN13 has been related to H2O2 stress, and in this study, it was mainly located at the PM, with a small portion in the cytoplasm, and it showed resistance against M. oryzae. We hypothesize that OsNPSN13 may have a dual function in transporting both ROS and defense-related proteins to the PM by forming different SNARE complexes in different vesicles. Our study provides initial evidence that OsNPSN13 is involved in vesicle-mediated plant immunity, but the detailed molecular mechanism requires further investigation.
The PM proteomic analysis presented an overview of the protein expression profile in resistant rice in response to M. oryzae infection at 24 h. The iTRAQ data presented here will help us to further understand the molecular mechanisms of resistance to rice blast. Many DEPs were involved in transport, phagosome and endocytosis. The protein interaction network showed higher degrees of interaction among multiple Ras-related proteins, including OsRab6, OsRab8a, GTP1, OsRIC1, VPS18, VPS26, VPS29, TuBB1, TuBB6 and serine/threonine phosphatases. qRT–PCR proved the accuracy of the iTRAQ data at the transcriptional level. OsNPSN13 was upregulated during M. oryzae infection, and the identification of overexpression and knockdown mutants showed that it indeed played a role in the process of rice blast resistance. Our study uncovered a complex network that associates the PM (especially membrane trafficking) with rice blast resistance. In the future, studies on the functions of the specific proteins found in this study will help to explore the mechanisms of resistance against M. oryzae infection.
Data Availability Statement
The datasets presented in this study can be found in online repository. The name of the repository is PRIDE proteomics repository, and the accession number is PXD028545.
Author Contributions
YB and ZZ conceived of the presented idea and drafted the manuscript with input from all authors. YB, ZZ, ML, HeZ, YY, LM, WW, YF, NH, XW, KL, SD, and HT implemented, collected, and analyzed the data. YB, ZZ, LM, HeZ, and YY conducted most experiments. YB was involved in the planning and supervision of the work. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by grants from the National Key Project for Jiangsu Agriculture Science and Technology Innovation Fund [CX(2019)1008], the National Key Research and Development Program of China (2020YFE0202900), the Natural Science Foundation of China (32072033, 31871602, 31171516), the Natural Science Foundation of Jiangsu Province (BK20211212), the Fundamental Research Funds for the Central Universities (Y0202100126) and the project was supported by the Guidance Foundation, the Sanya Institute of Nanjing Agricultural University (NAUSY-MS06).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Wei Zhang for their technical help and constructive criticism.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.853195/full#supplementary-material
Footnotes
References
Agarwal, P., Reddy, M. K., Sopory, S. K., and Agarwal, P. K. (2009). Plant rabs: characterization, functional diversity, and role in stress tolerance. Plant Mol. Biol. Rep. 27, 417–430. doi: 10.1007/s11105-009-0100-9
Aloui, A., Recorbet, G., Lemaître-Guillier, C., Mounier, A., Balliau, T., Zivy, M., et al. (2018). The plasma membrane proteome of Medicago truncatula roots as modified by arbuscular mycorrhizal symbiosis. Mycorrhiza 28, 1–16. doi: 10.1007/s00572-017-0789-5
Andersen, E. J., Ali, S., Byamukama, E., Yen, Y., and Nepal, M. P. (2018). Disease resistance mechanisms in plants. Genes 9:339. doi: 10.3390/genes9070339
Assaad, F. F., Qiu, J.-L., Youngs, H., Ehrhardt, D., Zimmerli, L., Kalde, M., et al. (2004). The PEN1 syntaxin defines a novel cellular compartment upon fungal attack and is required for the timely assembly of papillae. Mol. Biol. Cell 15, 5118–5129. doi: 10.1091/mbc.e04-02-0140
Bao, Y. M., Sun, S. J., Li, M., Li, L., Cao, W. L., Luo, J., et al. (2012). Overexpression of the Qc-SNARE gene OsSYP71 enhances tolerance to oxidative stress and resistance to rice blast in rice (Oryza sativa L.). Gene 504, 238–244. doi: 10.1016/j.gene.2012.05.011
Bao, Y. M., Wang, J. F., Huang, J., and Zhang, H. S. (2008a). Cloning and characterization of three genes encoding Qb-SNARE proteins in rice. Mol. Genet. Genomics 279, 291–301. doi: 10.1007/s00438-007-0313-2
Bao, Y. M., Wang, J. F., Huang, J., and Zhang, H. S. (2008b). Molecular cloning and characterization of a novel SNAP25-type protein gene OsSNAP32 in rice (Oryza sativa L.). Mol. Biol. Rep. 35, 145–152. doi: 10.1007/s11033-007-9064-8
Bassham, D. C., and Blatt, M. R. (2008). SNAREs: cogs and coordinators in signaling and development. Plant Physiol. 147, 1504–1515. doi: 10.1104/pp.108.121129
Beck, M., Heard, W., Mbengue, M., and Robatzek, S. (2012). The INs and OUTs of pattern recognition receptors at the cell surface. Curr. Opin. Plant Biol. 15, 367–374. doi: 10.1016/j.pbi.2012.05.004
Benedikter, B. J., Bouwman, F. G., Heinzmann, A. C. A., Vajen, T., Mariman, E. C., Wouters, E. F. M., et al. (2019). Proteomic analysis reveals procoagulant properties of cigarette smoke-induced extracellular vesicles. J. Extracell. Vesicles 8:1585163. doi: 10.1080/20013078.2019.1585163
Benschop, J. J., Mohammed, S., O’Flaherty, M., Heck, A. J. R., Slijper, M., and Menke, F. L. H. (2007). Quantitative phosphoproteomics of early elicitor signaling in Arabidopsis. Mol. Cell. Proteomics 6, 1198–1214. doi: 10.1074/mcp.M600429-MCP200
Bhat, R. A., Miklis, M., Schmelzer, E., Schulze-Lefert, P., and Panstruga, R. (2005). Recruitment and interaction dynamics of plant penetration resistance components in a plasma membrane microdomain. Proc. Natl. Acad. Sci. U.S.A. 102, 3135–3140. doi: 10.1073/pnas.0500012102
Bloch, D., Pleskot, R., Pejchar, P., Potocký, M., Trpkošová, P., Cwiklik, L., et al. (2016). Exocyst SEC3 and phosphoinositides define sites of exocytosis in pollen tube initiation and growth. Plant Physiol. 172, 980–1002. doi: 10.1104/pp.16.00690
Bogdanove, A. J., and Martin, G. B. (2000). AvrPto-dependent Pto-interacting proteins and AvrPto-interacting proteins in tomato. Proc. Natl. Acad. Sci. U.S.A. 97, 8836–8840. doi: 10.1073/pnas.97.16.8836
Boller, T., and Felix, G. (2009). A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 60, 379–407. doi: 10.1146/annurev.arplant.57.032905.105346
Bottanelli, F., Foresti, O., Hanton, S., and Denecke, J. (2011). Vacuolar transport in Tobacco leaf epidermis cells involves a single route for soluble cargo and multiple routes for membrane cargo. Plant Cell 23, 3007–3025. doi: 10.1105/tpc.111.085480
Bozkurt, T. O., Belhaj, K., Dagdas, Y. F., Chaparro-Garcia, A., Wu, C.-H., Cano, L. M., et al. (2015). Rerouting of plant late endocytic trafficking toward a pathogen interface. Traffic 16, 204–226. doi: 10.1111/tra.12245
Cao, W. L., Yu, Y., Li, M. Y., Luo, J., Wang, R. S., Tang, H. J., et al. (2019). OsSYP121 accumulates at fungal penetration sites and mediates host resistance to rice blast. Plant Physiol. 179, 1330–1342. doi: 10.1104/pp.18.01013
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, F., Yuan, Y., Li, Q., and He, Z. (2007). Proteomic analysis of rice plasma membrane reveals proteins involved in early defense response to bacterial blight. Proteomics 7, 1529–1539. doi: 10.1002/pmic.200500765
Chen, Q., Guo, W., Feng, L., Ye, X., Xie, W., Huang, X., et al. (2015). Transcriptome and proteome analysis of Eucalyptus infected with Calonectria pseudoreteaudii. J. Proteomics 115, 117–131. doi: 10.1016/j.jprot.2014.12.008
Chen, S., Tao, L., Zeng, L., Vega-Sanchez, M. E., Umemura, K., and Wang, G. L. (2006). A highly efficient transient protoplast system for analyzing defence gene expression and protein-protein interactions in rice. Mol. Plant Pathol. 7, 417–427. doi: 10.1111/j.1364-3703.2006.00346.x
Cheng, Y., Qi, Y., Zhu, Q., Chen, X., Wang, N., Zhao, X., et al. (2009). New changes in the plasma-membrane-associated proteome of rice roots under salt stress. Proteomics 9, 3100–3114. doi: 10.1002/pmic.200800340
Chisholm, S. T., Coaker, G., Day, B., and Staskawicz, B. J. (2006). Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124, 803–814. doi: 10.1016/j.cell.2006.02.008
Choi, S., Tamaki, T., Ebine, K., Uemura, T., Ueda, T., and Nakano, A. (2013). RABA members act in distinct steps of subcellular trafficking of the FLAGELLIN SENSING2 receptor. Plant Cell 25, 1174–1187. doi: 10.1105/tpc.112.108803
Chung, K. P., Zeng, Y., Li, Y., Ji, C., Xia, Y., and Jiang, L. (2018). Signal motif-dependent ER export of the Qc-SNARE BET12 interacts with MEMB12 and affects PR1 trafficking in Arabidopsis. J. Cell Sci. 131:2. doi: 10.1242/jcs.202838
Collins, N. C., Thordal-Christensen, H., Lipka, V., Bau, S., Kombrink, E., Qiu, J. L., et al. (2003). SNARE-protein-mediated disease resistance at the plant cell wall. Nature 425, 973–977. doi: 10.1038/nature02076
Dean, R., Van Kan, J. A. L., Pretorius, Z. A., Hammond-Kosack, K. E., Di Pietro, A., Spanu, P. D., et al. (2012). The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 414–430. doi: 10.1111/j.1364-3703.2011.00783.x
Dean, R. A., Talbot, N. J., Ebbole, D. J., Farman, M. L., Mitchell, T. K., Orbach, M. J., et al. (2005). The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434, 980–986. doi: 10.1038/nature03449
Dodds, P. N., and Rathjen, J. P. (2010). Plant immunity: towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 11, 539–548. doi: 10.1038/nrg2812
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, 64–70. doi: 10.1093/nar/gkq310
Durrant, W. E., and Dong, X. (2004). Systemic acquired resistance. Annu. Rev. Phytopathol. 42, 185–209. doi: 10.1146/annurev.phyto.42.040803.140421
Ebine, K., Okatani, Y., Uemura, T., Goh, T., Shoda, K., Niihama, M., et al. (2008). A SNARE complex unique to seed plants is required for protein storage vacuole biogenesis and seed development of Arabidopsis thaliana. Plant Cell 20, 3006–3021. doi: 10.1105/tpc.107.057711
Ellinger, D., Glöckner, A., Koch, J., Naumann, M., Stürtz, V., Schütt, K., et al. (2014). Interaction of the Arabidopsis GTPase RabA4c with its effector PMR4 results in complete penetration resistance to powdery mildew. Plant Cell 26, 3185–3200. doi: 10.1105/tpc.114.127779
Elmore, J. M., Liu, J., Smith, B., Phinney, B., and Coaker, G. (2012). Quantitative proteomics reveals dynamic changes in the plasma membrane during Arabidopsis immune signaling. Mol. Cell. Proteomics 11:e014555. doi: 10.1074/mcp.M111.014555
Evans, C., Noirel, J., Ow, S. Y., Salim, M., Pereira-Medrano, A. G., Couto, N., et al. (2012). An insight into iTRAQ: where do we stand now? Anal. Bioanal. Chem. 404, 1011–1027. doi: 10.1007/s00216-012-5918-6
Fasshauer, D., Sutton, R. B., Brunger, A. T., and Jahn, R. (1998). Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. U.S.A. 95, 15781–15786. doi: 10.1073/pnas.95.26.15781
Feng, Q. N., Li, S., and Zhang, Y. (2017). Update on adaptor protein-3 in Arabidopsis. Plant Signal. Behav. 12:e1356969. doi: 10.1080/15592324.2017.1356969
Gu, Y., Zavaliev, R., and Dong, X. (2017). Membrane trafficking in plant immunity. Mol. Plant 10, 1026–1034. doi: 10.1016/j.molp.2017.07.001
He, B., and Guo, W. (2009). The exocyst complex in polarized exocytosis. Curr. Opin. Cell Biol. 21, 537–542. doi: 10.1016/j.ceb.2009.04.007
He, B., Xi, F., Zhang, X., Zhang, J., and Guo, W. (2007). Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 26, 4053–4065. doi: 10.1038/sj.emboj.7601834
Hopff, D., Wienkoop, S., and Lüthje, S. (2013). The plasma membrane proteome of maize roots grown under low and high iron conditions. J. Proteomics 91, 605–618. doi: 10.1016/j.jprot.2013.01.006
Hsua, Y. W., and Jauh, G. Y. (2017). VPS36-mediated plasma membrane protein turnover is critical for Arabidopsis root gravitropism. Plant Signal. Behav. 12:e1307495. doi: 10.1080/15592324.2017.1307495
Huang, D., Sun, Y., Ma, Z., Ke, M., Cui, Y., Chen, Z., et al. (2019). Salicylic acid-mediated plasmodesmal closure via Remorin-dependent lipid organization. Proc. Natl. Acad. Sci. U.S.A. 116, 21274–21284. doi: 10.1073/pnas.1911892116
Huber, L. A., Pimplikar, S., Parton, R. G., Virta, H., Zerial, M., and Simons, K. (1993). Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J. Cell Biol. 123, 35–45. doi: 10.1083/jcb.123.1.35
Ibl, V., Csaszar, E., Schlager, N., Neubert, S., Spitzer, C., and Hauser, M. T. (2012). Interactome of the plant-specific ESCRT-III component AtVPS2.2 in Arabidopsis thaliana. J. Proteome Res. 11, 397–411. doi: 10.1021/pr200845n
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kalde, M., Nühse, T. S., Findlay, K., and Peck, S. C. (2007). The syntaxin SYP132 contributes to plant resistance against bacteria and secretion of pathogenesis-related protein 1. Proc. Natl. Acad. Sci. U.S.A. 104, 11850–11855. doi: 10.1073/pnas.0701083104
Kale, S. D., and Tyler, B. M. (2011). Entry of oomycete and fungal effectors into plant and animal host cells. Cell Microbiol. 13, 1839–1848. doi: 10.1111/j.1462-5822.2011.01659.x
Karnahl, M., Park, M., Krausea, C., Hiller, U., Mayer, U., Stierhof, Y. D., et al. (2018). Functional diversification of Arabidopsis SEC1-related SM proteins in cytokinetic and secretory membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 115, 6309–6314. doi: 10.1073/pnas.1722611115
Karp, N. A., Huber, W., Sadowski, P. G., Charles, P. D., Hester, S. V., and Lilley, K. S. (2010). Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 9, 1885–1897. doi: 10.1074/mcp.M900628-MCP200
Keinath, N. F., Kierszniowska, S., Lorek, J., Bourdais, G., Kessler, S. A., Shimosato-Asano, H., et al. (2010). PAMP (pathogen-associated molecular pattern)-induced changes in plasma membrane compartmentalization reveal novel components of plant immunity. J. Biol. Chem. 285, 39140–39149. doi: 10.1074/jbc.M110.160531
Komatsu, S. (2008). Plasma membrane proteome in Arabidopsis and rice. Proteomics 8, 4137–4145. doi: 10.1002/pmic.200800088
Kota, U., and Goshe, M. B. (2011). Advances in qualitative and quantitative plant membrane proteomics. Phytochemistry 72, 1040–1060. doi: 10.1016/j.phytochem.2011.01.027
Kwon, C., Neu, C., Pajonk, S., Yun, H. S., Lipka, U., Humphry, M., et al. (2008). Co-option of a default secretory pathway for plant immune responses. Nature 451, 835–840. doi: 10.1038/nature06545
Leborgne-Castel, N., and Bouhidel, K. (2014). Plasma membrane protein trafficking in plant-microbe interactions: a plant cell point of view. Front. Plant Sci. 5:735. doi: 10.3389/fpls.2014.00735
Lian, Q., Meng, Y., Zhao, X., Xu, Y., Wang, Y., Day, B., et al. (2020). ShNPSN11, a vesicle-transport-related gene, confers disease resistance in tomato to Oidium neolycopersici. Biochem. J. 477, 3851–3866. doi: 10.1042/BCJ20190776
Lipka, V., Kwon, C., and Panstruga, R. (2007). SNARE-ware: the role of SNARE-domain proteins in plant biology. Annu. Rev. Cell Dev. Biol. 23, 147–174. doi: 10.1146/annurev.cellbio.23.090506.123529
Liu, M., Peng, Y., Li, H., Deng, L., Wang, X., and Kang, Z. (2016). TaSYP71, a Qc-SNARE, contributes to wheat resistance against Puccinia striiformis f. sp. tritici. Front. Plant Sci. 7:544. doi: 10.3389/fpls.2016.00544
Macho, A. P., and Zipfel, C. (2014). Plant PRRs and the activation of innate immune signaling. Mol. Cell 54, 263–272. doi: 10.1016/j.molcel.2014.03.028
Marmagne, A., Roue, M. A., Ferro, M., Rolland, N., Alcon, C., Joyard, J., et al. (2004). Identification of new intrinsic proteins in Arabidopsis plasma membrane proteome. Mol. Cell. Proteomics 3, 675–691. doi: 10.1074/mcp.M400001-MCP200
Nielsen, E., Cheung, A. Y., and Ueda, T. (2008). The regulatory RAB and ARF GTPases for vesicular trafficking. Plant Physiol. 147, 1516–1526. doi: 10.1104/pp.108.121798
Nühse, T. S., Boller, T., and Peck, S. C. (2003). A plasma membrane syntaxin is phosphorylated in response to the bacterial elicitor flagellin. J. Biol. Chem. 278, 45248–45254. doi: 10.1074/jbc.M307443200
Nühse, T. S., Bottrill, A. R., Jones, A. M. E., and Peck, S. C. (2007). Quantitative phosphoproteomic analysis of plasma membrane proteins reveals regulatory mechanisms of plant innate immune responses. Plant J. 51, 931–940. doi: 10.1111/j.1365-313X.2007.03192.x
Osterrieder, A., Carvalho, C. M., Latijnhouwers, M., Johansen, J. N., Stubbs, C., Botchway, S., et al. (2009). Fluorescence lifetime imaging of interactions between Golgi tethering factors and Small GTPases in plants. Traffic 10, 1034–1046. doi: 10.1111/j.1600-0854.2009.00930.x
Paul, S., Andrew, M., Owen, O., Nitin, S. B., Jonathan, T. W., Daniel, R., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303.metabolite
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. doi: 10.1093/nar/29.9.e45
Priya, A., Kalaidzidis, I. V., Kalaidzidis, Y., Lambright, D., and Datta, S. (2015). Molecular insights into Rab7-mediated endosomal recruitment of core retromer: deciphering the role of Vps26 and Vps35. Traffic 16, 68–84. doi: 10.1111/tra.12237
Rodriguez-Furlán, C., Salinas-Grenet, H., Sandoval, O., Recabarren, C., Arraño-Salinas, P., Soto-Alvear, S., et al. (2016). The root hair specific SYP123 regulates the localization of cell wall components and contributes to rizhobacterial priming of induced systemic resistance. Front. Plant Sci. 7:1081. doi: 10.3389/fpls.2016.01081
Sandberg, A., Lindell, G., Källström, B. N., Branca, R. M., Danielsson, K. G., Dahlberg, M., et al. (2012). Tumor proteomics by multivariate analysis on individual pathway data for characterization of vulvar cancer phenotypes. Mol. Cell. Proteomics 11, e016998. doi: 10.1074/mcp.M112.016998
Sanderfoot, A. A., Assaad, F. F., and Raikhel, N. V. (2000). The Arabidopsis genome. An abundance of soluble N-ethylmaleimide-sensitive factor adaptor protein receptors. Plant Physiol. 124, 1558–1569. doi: 10.1104/pp.124.4.1558
Skamnioti, P., and Gurr, S. J. (2009). Against the grain: safeguarding rice from rice blast disease. Trends Biotechnol. 27, 141–150. doi: 10.1016/j.tibtech.2008.12.002
Speth, E. B., Imboden, L., Hauck, P., and He, S. Y. (2009). Subcellular localization and functional analysis of the Arabidopsis GTPase RabE 1. Plant Physiol. 149, 1824–1837. doi: 10.1104/pp.108.132092
Su, Y., Xu, L., Wang, Z., Peng, Q., Yang, Y., Chen, Y., et al. (2016). Comparative proteomics reveals that central metabolism changes are associated with resistance against Sporisorium scitamineum in sugarcane. BMC Genomics 17:800. doi: 10.1186/s12864-016-3146-8
Südhof, T. C., and Rothman, J. E. (2009). Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477. doi: 10.1126/science.1161748
Suwastika, I. N., Uemura, T., Shiina, T., Sato, M. H., and Takeyasu, K. (2008). SYP71, a plant-specific Qc-SNARE protein, reveals dual localization to the plasma membrane and the endoplasmic reticulum in Arabidopsis. Cell Struct. Funct. 33, 185–192. doi: 10.1247/csf.08024
Takemoto, K., Ebine, K., Askani, J. C., Krüger, F., Gonzalez, Z. A., Ito, E., et al. (2018). Distinct sets of tethering complexes, SNARE complexes, and Rab GTPases mediate membrane fusion at the vacuole in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 115, E2457–E2466. doi: 10.1073/pnas.1717839115
Tan, X., Feng, Y., Liu, Y., and Bao, Y. (2016). Mutations in exocyst complex subunit SEC6 gene impaired polar auxin transport and PIN protein recycling in Arabidopsis primary root. Plant Sci. 250, 97–104. doi: 10.1016/j.plantsci.2016.06.001
Teo, H., Perisic, O., González, B., and Williams, R. L. (2004). ESCRT-II, an endosome-associated complex required for protein sorting: crystal structure and interactions with ESCRT-III and membranes. Dev. Cell 7, 559–569. doi: 10.1016/j.devcel.2004.09.003
Thimm, O., Bläsing, O., Gibon, Y., Nagel, A., Meyer, S., Krüger, P., et al. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939. doi: 10.1111/j.1365-313X.2004.02016.x
Toki, S., Hara, N., Ono, K., Onodera, H., Tagiri, A., Oka, S., et al. (2006). Early infection of scutellum tissue with Agrobacterium allows high-speed transformation of rice. Plant J. 47, 969–976. doi: 10.1111/j.1365-313X.2006.02836.x
Uemura, T., Kim, H., Saito, C., Ebine, K., Ueda, T., Schulze-Lefert, P., et al. (2012). Qa-SNAREs localized to the trans-Golgi network regulate multiple transport pathways and extracellular disease resistance in plants. Proc. Natl. Acad. Sci. U.S.A. 109, 1784–1789. doi: 10.1073/pnas.1115146109
Uemura, T., Nakano, R. T., Takagi, J., Wang, Y., Kramer, K., Finkemeier, I., et al. (2019). A golgi-released subpopulation of the trans-golgi network mediates protein secretion in Arabidopsis 1. Plant Physiol. 179, 519–532. doi: 10.1104/pp.18.01228
Uemura, T., Ueda, T., Ohniwa, R. L., Nakano, A., and Takeyasu, K. (2004). Systematic analysis of SNARE molecules in Arabidopsis : dissection of the post-Golgi network in plant cells. Cell Struct. Funct. 65, 49–65. doi: 10.1247/csf.29.49
Vukašinovic, N., and Žárský, V. (2016). Tethering complexes in the Arabidopsis endomembrane system. Front. Cell Dev. Biol. 4:46. doi: 10.3389/fcell.2016.00046
Wang, H. J., Hsu, Y. W., Guo, C. L., Jane, W. N., Wang, H., Jiang, L., et al. (2017). VPS36-dependent multivesicular bodies are critical for plasmamembrane protein turnover and vacuolar biogenesis. Plant Physiol. 173, 566–581. doi: 10.1104/pp.16.01356
Wang, J. F., He, X. J., Zhang, H. S., and Chen, Z. Y. (2002). Genetic analysis of blast resistance in japonica rice landrace heikezijing from Taihu region. Yi Chuan Xue Bao 29, 803–807.
Wang, W., Liu, P., Xu, Y., Xiao, S., and Wang, W. (2015). Protein trafficking during plant innate immunity. J. Integr. Plant Biol. 58, 284–298. doi: 10.1111/jipb.12426
Wang, X., Wang, X., Deng, L., Chang, H., Dubcovsky, J., Feng, H., et al. (2014). Wheat TaNPSN SNARE homologues are involved in vesicle-mediated resistance to stripe rust (Puccinia striiformis f. sp. tritici). J. Exp. Bot. 65, 4807–4820. doi: 10.1093/jxb/eru241
Wei, T., Zhang, C., Hou, X., Sanfaçon, H., and Wang, A. (2013). The SNARE protein SYP71 is essential for turnip mosaic virus infection by mediating fusion of virus-induced vesicles with chloroplasts. PLoS Pathog. 9:e1003378. doi: 10.1371/journal.ppat.1003378
Wei, Y., Jiang, C., Han, R., Xie, Y., Liu, L., and Yu, Y. (2020). Plasma membrane proteomic analysis by TMT-PRM provides insight into mechanisms of aluminum resistance in tamba black soybean roots tips. PeerJ 8:e9312. doi: 10.7717/peerj.9312
Wei, Z., Hu, W., Lin, Q., Cheng, X., Tong, M., Zhu, L., et al. (2009). Understanding rice plant resistance to the brown planthopper (Nilaparvata lugens): a proteomic approach. Proteomics 9, 2798–2808. doi: 10.1002/pmic.200800840
Witzel, K., Matros, A., Møller, A. L. B., Ramireddy, E., Finnie, C., Peukert, M., et al. (2018). Plasma membrane proteome analysis identifies a role of barley membrane steroid binding protein in root architecture response to salinity. Plant Cell Environ. 41, 1311–1330. doi: 10.1111/pce.13154
Yadeta, K. A., Mitch Elmore, J., and Coaker, G. (2013). Advancements in the analysis of the Arabidopsis plasma membrane proteome. Front. Plant Sci. 4:86. doi: 10.3389/fpls.2013.00086
Yang, Z. (2002). Small GTPases: versatile signaling switches in plants. Plant Cell 14, 375–389. doi: 10.1105/tpc.001065
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. doi: 10.1089/omi.2011.0118
Yu, I., and Hughson, F. M. (2010). Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 26, 137–156. doi: 10.1146/annurev.cellbio.042308.113327
Yue, P., Zhang, Y., Mei, K., Wang, S., Lesigang, J., Zhu, Y., et al. (2017). Sec3 promotes the initial binary t-SNARE complex assembly and membrane fusion. Nat. Commun. 8:14236. doi: 10.1038/ncomms14236
Žárský, V., Kulich, I., Fendrych, M., and Pečenková, T. (2013). Exocyst complexes multiple functions in plant cells secretory pathways. Curr. Opin. Plant Biol. 16, 726–733. doi: 10.1016/j.pbi.2013.10.013
Zelazny, E., Santambrogio, M., Pourcher, M., Chambrier, P., Berne-Dedieu, A., Fobis-Loisy, I., et al. (2013). Mechanisms governing the endosomal membrane recruitment of the core retromer in Arabidopsis. J. Biol. Chem. 288, 8815–8825. doi: 10.1074/jbc.M112.440503
Zhang, X., Zhao, H., Gao, S., Wang, W. C., Katiyar-Agarwal, S., Huang, H. D., et al. (2011). Arabidopsis argonaute 2 regulates innate immunity via miRNA393-mediated silencing of a Golgi-localized SNARE gene, MEMB12. Mol. Cell 42, 356–366. doi: 10.1016/j.molcel.2011.04.010
Zhang, Z., Feechan, A., Pedersen, C., Newman, M. A., Qiu, J. L., Olesen, K. L., et al. (2007). A SNARE-protein has opposing functions in penetration resistance and defence signalling pathways. Plant J. 49, 302–312. doi: 10.1111/j.1365-313X.2006.02961.x
Keywords: Oryza sativa L., rice blast, plasma membrane, iTRAQ, proteomics, vesicle trafficking
Citation: Zhao Z, Li M, Zhang H, Yu Y, Ma L, Wang W, Fan Y, Huang N, Wang X, Liu K, Dong S, Tang H, Wang J, Zhang H and Bao Y (2022) Comparative Proteomic Analysis of Plasma Membrane Proteins in Rice Leaves Reveals a Vesicle Trafficking Network in Plant Immunity That Is Provoked by Blast Fungi. Front. Plant Sci. 13:853195. doi: 10.3389/fpls.2022.853195
Received: 12 January 2022; Accepted: 16 March 2022;
Published: 25 April 2022.
Edited by:
Ian S. Wallace, University of Nevada, Reno, United StatesReviewed by:
Caiji Gao, South China Normal University, ChinaWilliam Underwood, Agricultural Research Service (USDA), United States
Copyright © 2022 Zhao, Li, Zhang, Yu, Ma, Wang, Fan, Huang, Wang, Liu, Dong, Tang, Wang, Zhang and Bao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongmei Bao, eW9uZ21laWJhb0BuamF1LmVkdS5jbg==
†These authors have contributed equally to this work