 Luyao Wu1,2†
Luyao Wu1,2† Bing Jia2†
Bing Jia2† Wenfeng Pei2Li Wang2
Wenfeng Pei2Li Wang2 Jianjiang Ma2
Jianjiang Ma2 Man Wu2
Man Wu2 Jikun Song2
Jikun Song2 Shuxian Yang2Yue Xin2
Shuxian Yang2Yue Xin2 Li Huang2Pan Feng2
Li Huang2Pan Feng2 Jinfa Zhang3*
Jinfa Zhang3* Jiwen Yu1,2*
Jiwen Yu1,2*
- 1Zhengzhou Research Base, State Key Laboratory of Cotton Biology, School of Agricultural Sciences, Zhengzhou University, Zhengzhou, China
- 2State Key Laboratory of Cotton Biology, Key Laboratory of Cotton Genetic Improvement, Ministry of Agriculture, Institute of Cotton Research of Chinese Academy of Agricultural Sciences, Anyang, China
- 3Department of Plant and Environmental Sciences, New Mexico State University, Las Cruces, NM, United States
Seed size and shape are key agronomic traits affecting seedcotton yield and seed quality in cotton (Gossypium spp.). However, the genetic mechanisms that regulate the seed physical traits in cotton are largely unknown. In this study, an interspecific backcross inbred line (BIL) population of 250 BC1F7 lines, derived from the recurrent parent Upland CRI36 (Gossypium hirsutum) and Hai7124 (Gossypium barbadense), was used to investigate the genetic basis of cotton seed physical traits via quantitative trait locus (QTL) mapping and candidate gene identification. The BILs were tested in five environments, measuring eight seed size and shape-related traits, including 100-kernel weight, kernel length width and their ratio, kernel area, kernel girth, kernel diameter, and kernel roundness. Based on 7,709 single nucleotide polymorphic (SNP) markers, a total of 49 QTLs were detected and each explained 2.91–35.01% of the phenotypic variation, including nine stable QTLs mapped in at least three environments. Based on pathway enrichment, gene annotation, genome sequence, and expression analysis, five genes encoding starch synthase 4, transcription factor PIF7 and MYC4, ubiquitin-conjugating enzyme E27, and THO complex subunit 4A were identified as candidate genes that might be associated with seed size and shape. Our research provides valuable information to improve seed physical traits in cotton breeding.
Introduction
Cotton (Gossypium spp.) is an important cash crop, grown for monetary profit from the fiber, feed, and cooking oil. Currently, research on cotton predominantly focuses on fiber yield and quality, with relatively few studies on seed quality (Deng et al., 2019; Chen et al., 2020; Liu H. et al., 2020). Seed quality is one of the most important factors considered in cotton stand establishment procedures (Sawan and Dello Ioio, 2016). Although cotton production has seen technological advances, the lack of quality cottonseed may be perceived as a pertinent issue. Good quality seeds of improved cultivars comprise one of the key inputs for attaining high cotton yield with increased economic benefits (Atique-ur-Rehman et al., 2020).
Seed size is a widely accepted measure of seed quality, and multiple earlier studies have shown that large seeds have high capacities for seedling survival, growth, and establishment (Lehtilä and Ehrlén, 2005). Compared to small-seed cultivars, large-seed cultivars exhibit increased fiber length, strength, and decreased micronaire (Main et al., 2013). Compared to small-size and mixed-size seeds, large-size and medium-size seeds achieved increased germination potential, germination rate, seed fullness, dry matter weight per plant, root-to-shoot ratio, leaf emergence rate, and leaf area (Liu et al., 1997). Seed size is the primary factor considered during harvesting and processing (Atique-ur-Rehman et al., 2020). Individual plant seed mass, in addition to total oil and protein energy content, predicts early seedling vigor (Snider et al., 2016). Additionally, oil content is largely affected by seed size (Pahlavani et al., 2008).
Quantitative trait locus (QTL) mapping uses molecular markers, based on genetic linkage maps, to determine the position of DNA segments or genes that control quantitative traits (Powder, 2020). Studies employing QTL mapping in cotton have predominantly focused on the QTL location of fiber traits (Said et al., 2013, 2015), with the related molecular mechanisms gradually revealed over time (Tian and Zhang, 2021). QTL mapping for seed quality tends to focus on oil content (Yu et al., 2012; Liu et al., 2013; Liu C. et al., 2020), protein content (Yu et al., 2012; Liu et al., 2013) and other aspects (Liu et al., 2008). Few studies have described QTL locations for seed physical traits including size and shape (Wang et al., 2019).
It is known that seed size and shape is a complex quantitative trait that is controlled by multiple genes in crops. In other crops, more QTL mapping and research on seed size and shape-related traits have been conducted, such as peanuts (Zhang et al., 2019), soybeans (Hina et al., 2020), and rice (Ying et al., 2018). Using specific site amplified fragments (SLAF) sequencing- based 7,033 single nucleotide polymorphic markers (SNPs) to construct a genetic map in a recombinant inbred line (RIL) population of 180 Upland cotton lines, Wang et al. (2019) identified 32 QTLs for four traits related to seed size, i.e., hundred seed weight (HSW), hundred kernel weight (HKW), ten kernel length (TKL), and ten kernel width (TKW). However, molecular and genomic studies on cottonseed physical traits are currently lacking. It is known that cottonseed size is associated with seedling vigor, and oil and protein content. Seed size and seed shape also affect the seed surface area, which in turn could affect the number of fiber initials and eventually lint fibers. The number of lint fibers and their length and fineness are important determinants of lint percentage, a lint yield component trait. Therefore, QTL mapping of cotton seed size and shape-related traits is of great significance for revealing the molecular mechanism of cotton seed development and improving cotton yield, seed, and fiber quality.
In a previous study, a backcross inbred line (BIL) population containing 250 BC1F7 lines, derived from an interspecific cross between recurrent parent Gossypium hirsutum L. CRI36, and Gossypium barbadense L. Hai7124, was developed and SLAF sequencing was used for SNP typing (Ma et al., 2019). The objectives of this study were to perform a QTL analysis for seed size and shape in this BIL population. Eight traits related to seed size and shape were assessed: 100-kernel weight, kernel length, kernel width, kernel length to width ratio, kernel area, kernel girth, kernel diameter, and kernel roundness. To lay a theoretical foundation for improving the quality of cotton seeds and furthering research on related genetic mechanisms, we also analyzed candidate genes for stable QTL intervals.
Materials and Methods
Plant Material and Generation of Phenotypic Data
An interspecific BIL population containing 250 BC1F7 lines was developed from a cross between G. hirsutum CRI36 (as the recurrent parent) and G. barbadense Hai7124. Ma et al. (2019) described the development details of the BILs and created a genetic linkage map composed of 7,709 SNP markers. The parents and 250 BILs were planted in five environments according to a randomized complete block design with two replications in each environment. Three field tests were conducted in the experimental farm, CRI, CAAS, Anyang (Henan Province, 36.06°N, 114.49°E) with one test in 2016 and two tests (one in the south farm and another in the east farm) in 2017. Two field tests were conducted in Shihezi (Xinjiang Uygur Autonomous Region, 44.44°N, 85.68°E) in 2016 and 2017. In each test, cotton seeds were hill-sown by hand and covered with plastic mulch applied directly by a machine in April each year. In Anyang, approximately 16 plants per 4-m-long row were retained, and the row spacing was 0.80 m. In Xinjiang, where a high-density seeding rate was used, approximately 44 plants per 5-m-long row were retained, and the row spacing was 0.38 m. Crop management practices followed the recommendations of local cotton production. The use of the two cotton production systems (i.e., normal and high plant density) allowed detection of consistent QTLs for the seed physical traits between the two production systems. The average best linear unbiased prediction (BLUP) of the five environments was also calculated and used for QTL mapping. We conducted SLAF sequencing with the G. hirsutum genetic standard TM-1 as a reference genome (Zhang et al., 2015; Hu et al., 2019) to genotype the BILs.
Twenty opened bolls were manually harvested at crop maturity. After ginning and acid-delinting of the cottonseed, a Wanshen SC-G Automatic Seed Test Analyzer was used to determine the properties of cotton kernels: the 100-kernel weight (HKW, g), kernel length (KL, mm), kernel width (KW, mm), kernel length to width ratio (KLW), kernel area (KA, mm1), kernel girth (KG, mm), kernel diameter (KD, mm), and kernel roundness (KR, mm). Analysis of variance, the frequency distribution and correlation coefficients among these traits were analyzed using SPSS (version 20.0; SPSS, Chicago, IL, United States). The lme4 package in R was used to estimate the BLUP value of the five environments, enabling its use in correlation analysis of the eight traits (Poland et al., 2011).
Quantitative Trait Locus Analysis
Inclusive Composite Interval Mapping (ICIM) in the IciMapping4.2 software was used to perform QTL analyses for each seed physical trait (Meng et al., 2015). The threshold of logarithm of the odds (LOD) value was set using 1,000 permutation tests, and the detection step was set to 1 cM. Positive additive effects indicated favorable alleles derived from CRI36, while negative additive effects indicated favorable alleles from Hai7124. A QTL identified in three or more environments were considered a stable QTL (Shang et al., 2015). The naming method of QTLs followed a previous report (Gu et al., 2020). MapChart (version 2.2) was used for constructing linkage maps for mapped SNPs with QTL intervals indicated.
Candidate Gene Identification and Annotation
The physical interval of each stable QTL was determined using Basic Local Alignment Search Tool (BLAST) (Hu et al., 2019). Potential candidate genes related to seed size and shape traits were determined based on Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. GO and KEGG analyses were performed using OmicShare tools2 (Li et al., 2020). These genes were identified using CottonFGD.2 The functions of the identified genes were determined through gene annotation. The Arabidopsis thaliana homologous genes and gene function annotations of candidate genes were determined using the TM-1 genome. Based on CRI36 and Hai7124 resequencing (30×) results, candidate genes were further screened by SNP variation between the two parents. Polymorphic loci with missing or heterozygous genotypes, as well as polymorphic loci without polymorphism between parents, were filtered out. The remaining SNPs and indels were considered as effective polymorphic loci. SnpEff 4.2 software was used to predict the function of these effective polymorphic loci based on the published cotton genome sequence annotations (Cingolani et al., 2012).
Because the gene expression data for the Upland cotton parent CRI36 were not available, the expression levels of candidate genes from the sequenced TM-1 were used as a proxy to compare with these from another parent- Hai7124, both of which were based on existing RNA sequencing (RNA-seq) data (National Genomics Data Center: accession number: PRJNA490626)3 (Hu et al., 2019). Candidate genes that were poorly expressed in cotton ovules (FPKM < 2) were discarded. Screening for genes that were specifically expressed in ovules or whose expression levels were significantly different in TM-1 and Hai7124 ovules, was performed. The fold change in candidate gene expression was set to 2 as the threshold for significant differential expression between TM-1 and Hai7124 in embryos (0, 1, 3, 5, 10, and 20 days post anthesis, DPA).
Results
Phenotypic Performance in the Backcross Inbred Line Population of Gossypium hirsutum CRI36 and Gossypium barbadense Hai7124 in Five Environments
The traits of 100-kernel weight (HKW), kernel length (KL), kernel width (KW), kernel length to width ratio (KLW), kernel area (KA), kernel girth (KG), kernel diameter (KD), and kernel roundness (KR) were used to evaluate the seed size and shape of the parents, G. hirsutum CRI36 and G. barbadense Hai7124, and their interspecific BIL population in five environments. The traits of KLW, KL, KR, and KW significantly differed between the two parental lines of different species; and G. barbadense Hai7124 seeds were shorter and more rounded than the seeds of G. hirsutum CRI36. HKW, KA, KG, and KD did not significantly differ between the two parents (Table 1). However, analysis of variance (ANOVA) detected significant genetic variations for all the seed physical traits in the BIL population including these traits for which the two parents did not differ (Supplementary Table 1). The results indicate that different genes controlling the same traits with similar values between the two species, resulting in transgressive segreation.

Table 1. Comparison of the seed size and shape-related traits between two parents Gossyium hirsutum CRI36 and G. barbadense Hai7124.
The results of the descriptive statistics of phenotypic data for all traits in the five environments (except BLUP) of BIL are shown in Supplementary Table 2. Broad-sense heritability estimates were 0.74–0.86, indicating that all traits were mainly affected by the genotype. Both the skewness and kurtosis values of the eight traits in the five environments were < 1.0 except for a few cases, indicating that none of the traits deviated significantly from a normal distribution (Figures 1A–H). We further calculated correlation coefficients among the eight seed size and shape-related traits in the BIL population; there were 24 significant correlations between the eight traits (Table 2). Among them, HKW, KL, KW, KA, KG, and KD showed a positive correlation. KLW was significantly and negatively correlated with HKW, KW, and KR, and KR was significantly negatively correlated with KG, KLW, and KL. The same cottonseed physical trait was significantly correlated among different environments, suggesting the environmental stability of these traits. Taking HKW as an example, the correlation between various environments was analyzed, and it was found that there was a significant positive correlation among all environments (Supplementary Table 3).
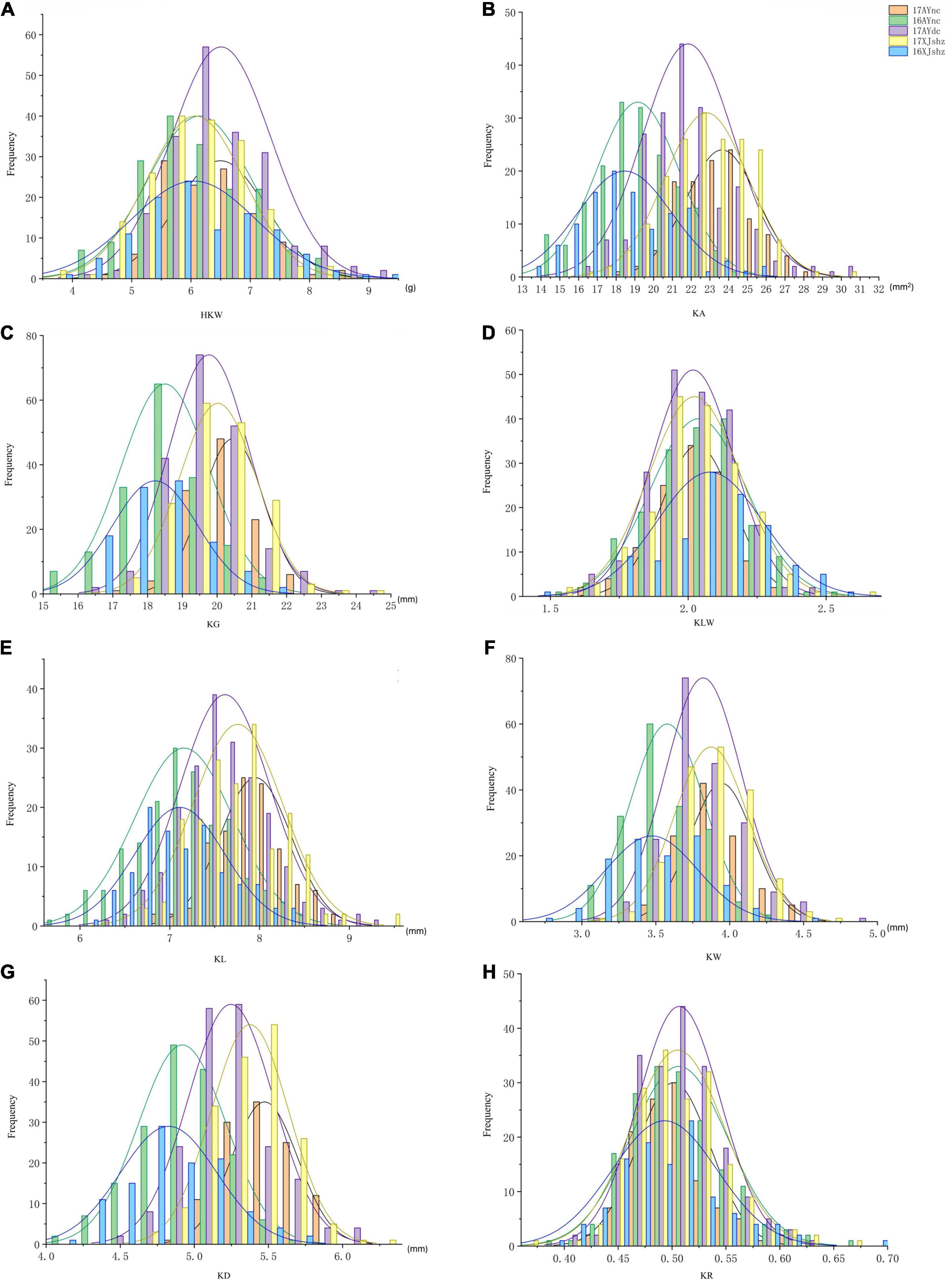
Figure 1. Frequency map of 8 traits in different environments of 250 BILs. Different colors represent different environments. (A) HKW. (B) KA. (C) KG. (D) KLW. (E) KL. (F) KW. (G) KD. (H) KR. See the footnote in Table 1 for explanations of the abbreviations.
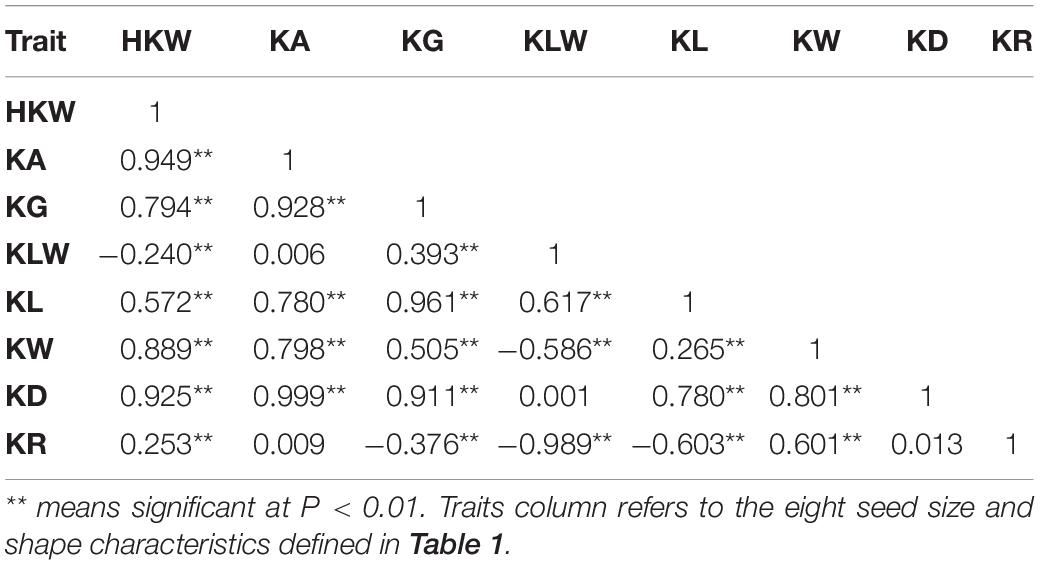
Table 2. Correlation coefficients among cotton seed size and shape-related traits in the BIL population.
Quantitative Trait Locus Analysis of Cottonseed Physical Traits
Based on the high-density genetic map and phenotypic data, a total of 49 QTLs were detected on 14 chromosomes, including 28 and 21 QTLs in the A and D subgenomes, respectively. These included five QTLs for HKW, five for KL, five for KW, five for KLW, eight for KA, two for KG, twelve for KD, and seven for KR (Supplementary Figure 1). An LOD value of 3.64–15.6 was obtained for the QTLs, with 2.91–35.01% phenotypic variation explained (PVE) by each QTL (Supplementary Table 4). The PVE of QTLs for HKW, KL, KW, KLW, KA, KG, KD and KR ranged from 5.38 to 35.01%, 5.41 to 35.01%, 13.70 to 34.10%, 6.82 to 22.80%, 5.68 to 25.10%, 8.18 to 12.91%, 2.91 to 25.28%, and 5.59 to 20.84%, respectively. A total of nine QTLs were consistently detected in at least three environments, namely qHKW-D03-1, qKW-D03-1, qKLW-D03-1, qKLW-D12-1, qKA-D03-1, qKG-D03-1, qKD-D03-1, qKR-D03-1, and qKR-D12-1 (Table 3). Among these nine stable QTLs, the additive effect of qKLW-D03-1 and qKLW-D12-1 came from CRI36, while the others came from the male parent Hai7124.
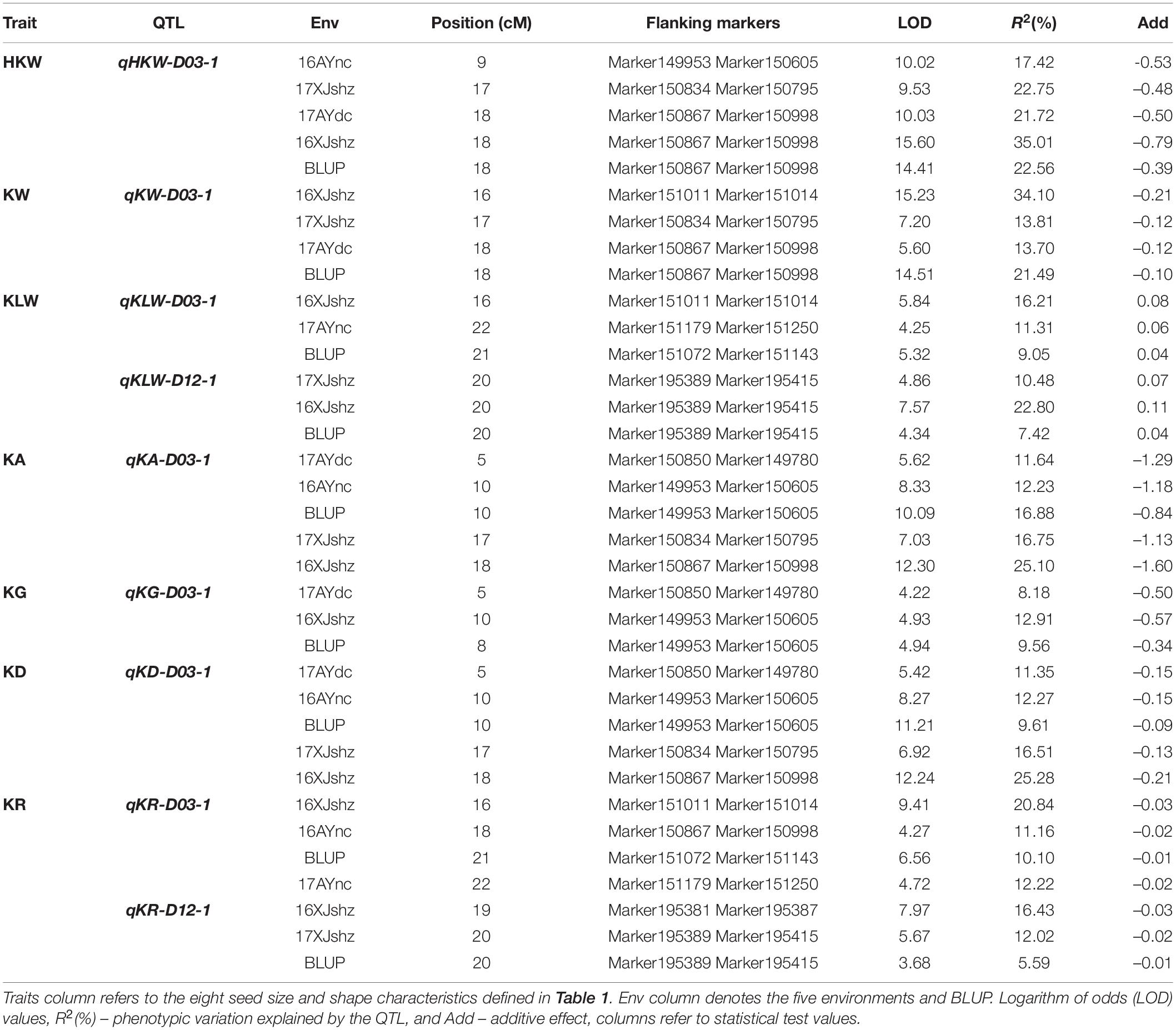
Table 3. Stable quantitative trait loci (QTLs) for cottonseed physical traits identified in five environments and BLUP.
The QTLs identified for all traits were not randomly distributed across chromosomes or chromosomal regions, some of which were closed linked in clusters. The QTLs for the same or different traits that shared an overlapping confidence interval or located in an adjacent region were estimated as the presence of a cluster (Said et al., 2013). A total of seven QTL clusters were identified in this study (Supplementary Table 5), among which four and three clusters were observed in the At and Dt subgenomes, respectively. These clusters were distributed on seven chromosomes, among which one cluster was located on each of A04, A07, A08, A13, D03, D09, and D12. The D03 QTL cluster contained the largest number of QTLs (7), followed by qClu-D09-1 (5).
Prediction of Candidate Genes in Stable Quantitative Trait Locus
There were 641 candidate genes within the nine stable QTL regions. First, GO enrichment and KEGG analyses were performed on these candidate genes. In the GO analysis results (Figures 2A,B and Supplementary Table 6), the number of genes related to the metabolic process of the biological process category was 200. The number of genes identified as being related to cellular processes in the biological process category was 184, and the number of genes identified as being related to the combination of molecular functional categories was 199. Among the top 20 GO enrichment results, cellular component organization, or biogenesis, and carbohydrate metabolic process had the most enriched genes. In the KEGG analysis results (Figures 2C,D and Supplementary Table 7), the number of genes enriched in the metabolic pathway was 33, and 13 genes were enriched in the biosynthesis of secondary metabolites. In the top 20 KEGG pathways, several genes from the spliceosome (Jiang et al., 2011), protein processing in the endoplasmic reticulum (Yi et al., 2021), starch and sucrose metabolism (Yin et al., 2020), and purine metabolism (Qi and Xiong, 2013) were found to be related to seed size. In addition, in the remaining KEGG pathways, ABC transporters are also related to seed size (Do et al., 2018). Plant hormone signal transduction (Li et al., 2011; Liu et al., 2015; Ji et al., 2019), mitogen-activated protein kinase (MAPK) signaling (Li and Li, 2015) and ubiquitin-mediated proteolysis (Li et al., 2008; Xia et al., 2013) pathways have been studied extensively in the context of seed size. A total of 22 candidate genes were enriched in these pathways related to seed size. We infer that these genes may play a key role in the development of cotton seed size and shape.
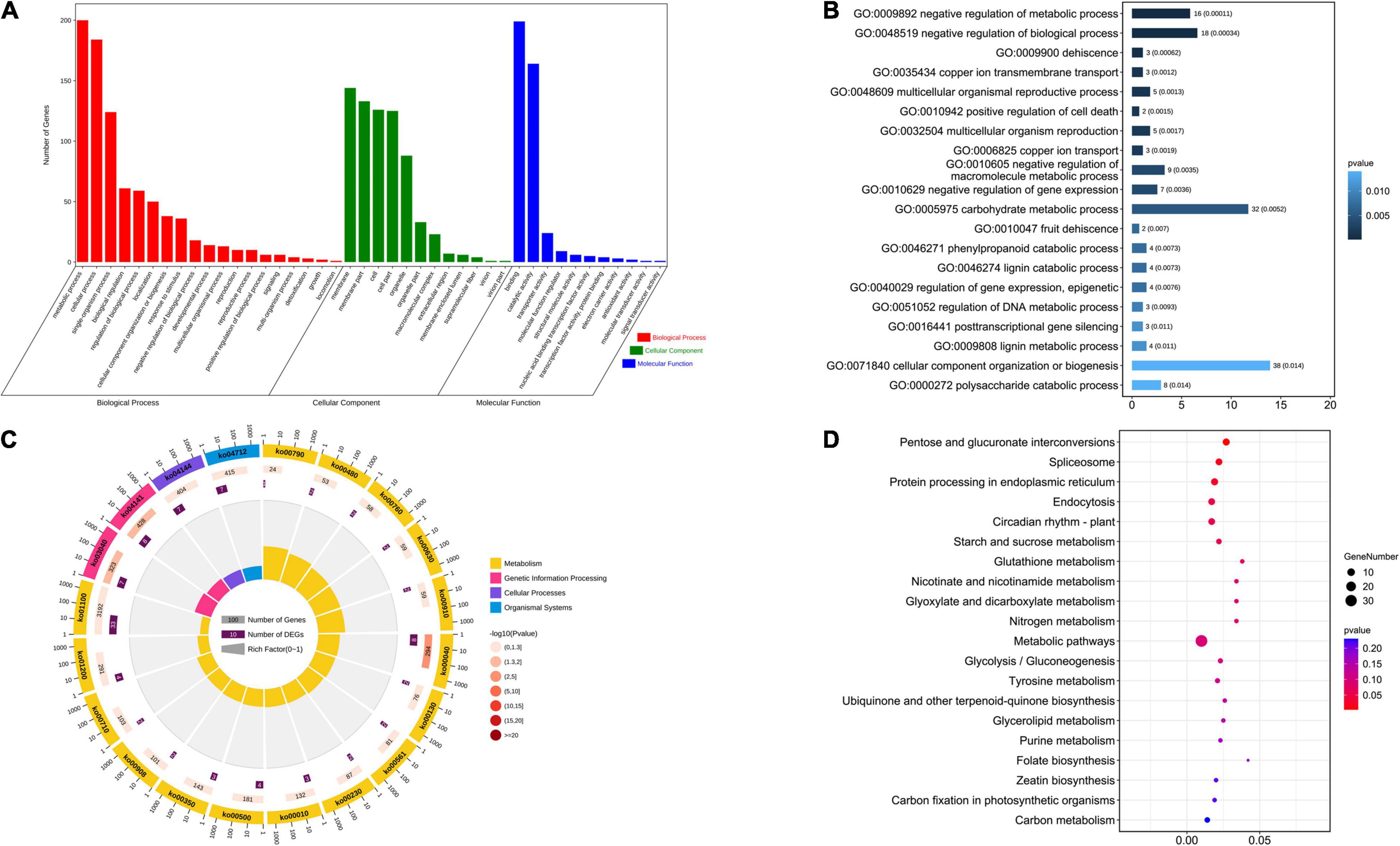
Figure 2. Analysis of the GO enrichment and KEGG of stable QTLs related to seed size and shape. (A) Analysis of the GO enrichment of stable QTLs. (B) Top 20 GO terms enrichment in the molecular function category. (C) Analysis of the KEGG of stable QTLs. (D) Top 20 of KEGG enrichment.
Based on the functional annotation of orthologs in Arabidopsis spp. of 22 candidate genes, 10 candidate genes within the stable QTLs were further identified that may be involved in cotton seed development (Supplementary Table 8). We analyzed the SNP variation of these 10 candidate genes between the two parents, and it contained a total of 1,334 effective SNPs (including intergenic regions). According to the annotations, non-synonymous mutations (9), start gained (1), synonymous variant (7), stop gained (2), splice acceptor variant, and intron variant (1) exist in eight candidate genes, which may affect the biological function of these genes (Table 4).
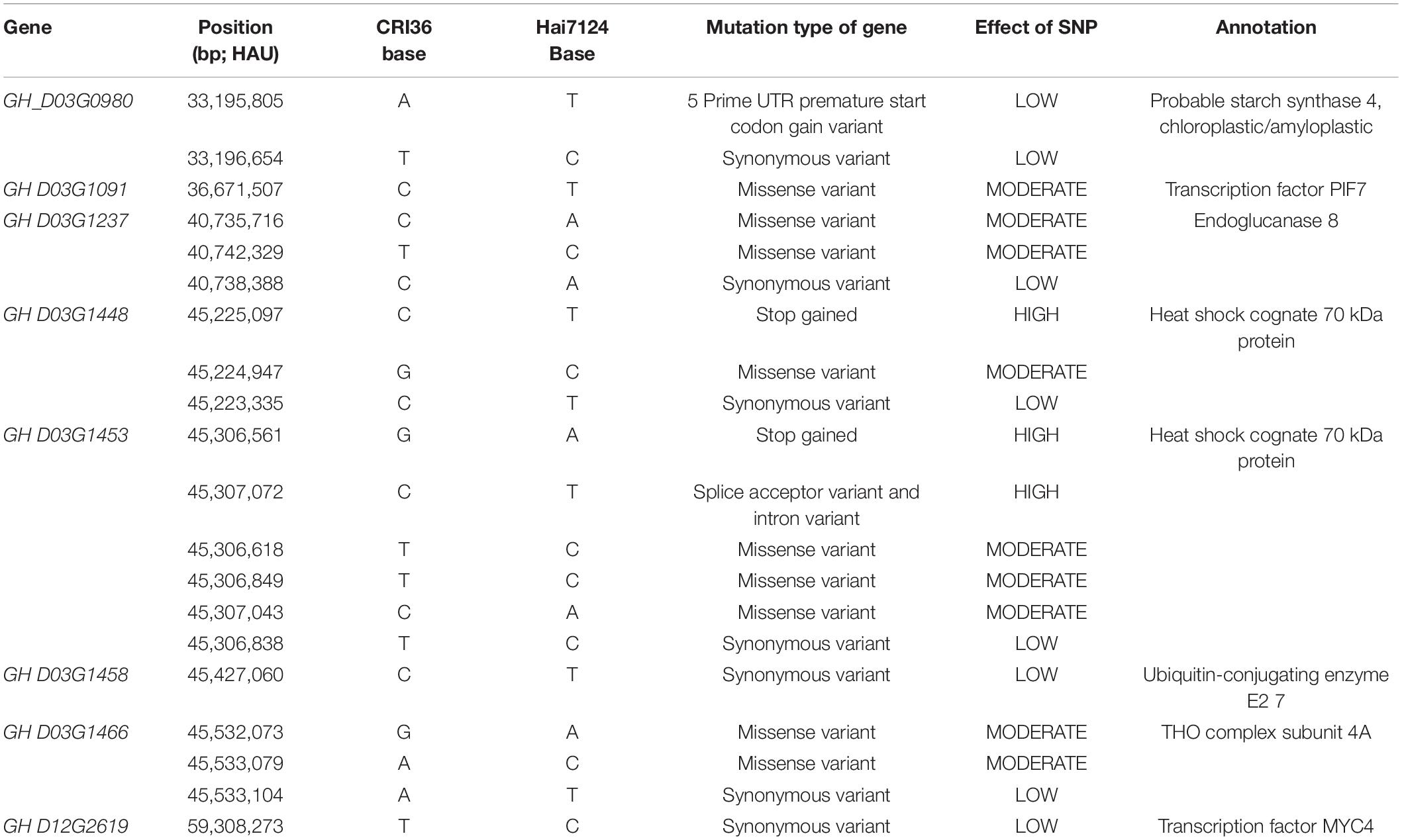
Table 4. SNPs in candidate genes between the two parents.
Furthermore, we analyzed the expression levels of these eight candidate genes in the ovules of TM-1 (as a proxy to the Upland cotton parent, CRI36) and Hai7124 using previously published RNA-seq data (Hu et al., 2019). Among them, three genes on chromosome D03, GH_D03G1237, GH_D03G1448, and GH_D03G1453, were not expressed during the developmental stages of cottonseed ovules (Supplementary Table 9), and were therefore not further analyzed. Among the remaining five genes, four were on the D03 chromosome including GH_D03G0980 encoding probable starch synthase 4 (within the region of QTLs- qKLW-D03-1 and qKR-D03-1) and GH_D03G1091 encoding transcription factor PIF7 (with the region of QTLs- qHKW-D03-1, qKA-D03-1, qKG-D03-1, and qKD-D03-1) in a close proximity, and GH_D03G1458 encoding ubiquitin-conjugating enzyme E27 (within the region of QTLs- qKLW-D03-1 and qKR-D03-1) and GH_D03G1466 encoding THO complex subunit 4A (within the region of QTLs- qKLW-D03-1 and qKR-D03-1) in a close proximity, and GH_D12G2619 encoding transcription factor MYC4 (within the region of QTLs- qKLW-D12-1 and qKR-D12-1) on D12. The expression levels of these genes were different during different ovule developmental stages between the two parents (Figure 3A). However, because developing ovules were not harvested for RNA extraction from representative BILs with differing seed physical traits, a comparative quantitative RT-PCR analysis was not performed in this study.
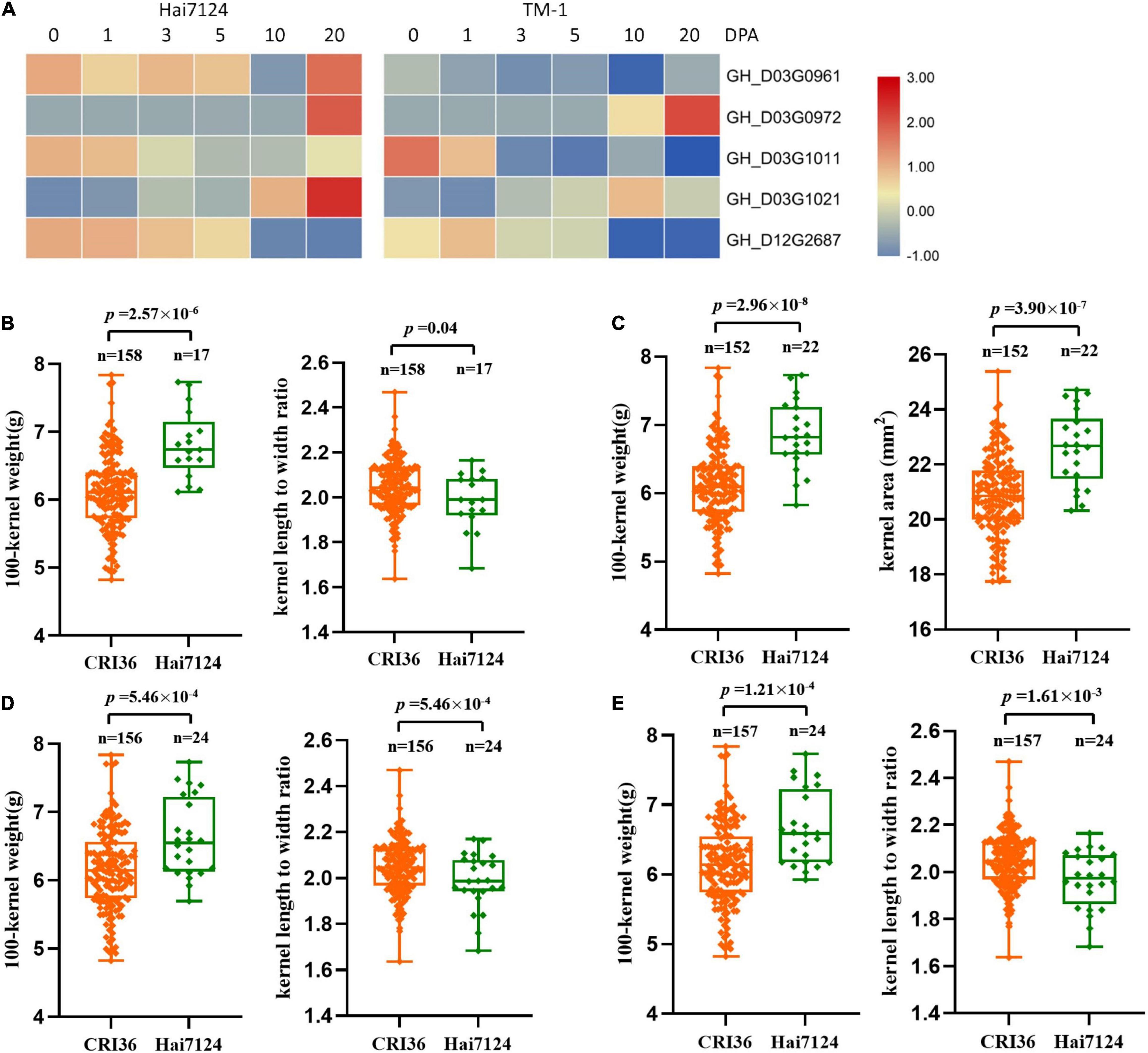
Figure 3. The expression levels and genotypic evaluation of five candidate genes in ovules of each development stage (0, 1, 3, 5, 10, and 20 days post anthesis) of G. hirsutum TM-1 and G. barbadense Hai7124. (A) The expression levels of five candidate genes in ovules of each development stage (0, 1, 3, 5, 10, and 20 days post anthesis) of G. hirsutum TM-1 and G. barbadense Hai7124. (B–E) The distribution and means of seed size and shape traits in the BIL population based on SNP alleles from two parents for GH_D03G0980, GH_D03G1091, GH_D03G1458, and GH_D03G1466, respectively.
To further determine the effective allelic variation of the candidate genes, we analyzed SNPs of these candidate genes and their contributions to variation in seed size and shape traits. The results showed that SNPs in four candidate genes were significantly associated with changes in seed size and shape (Figures 3B–E). The possible roles of these candidate genes in relation to cottonseed size and shape will be discussed in Discussion.
Discussion
Crop germplasm, including crop varieties, strains, types, wild species, and relatives, is the source of genes for genetically improving crops (Wang et al., 2005). Cotton, like other crops, has heterosis to varying degrees between species and varieties. Using cotton heterosis is an effective way to increase cotton yield (Xing et al., 2007). Since G. barbadense and Upland cotton belong to two different species under the genus Gossypium, different genetic loci are involved in seed development. The interspecific hybrids exhibit heterosis, which is reflected in many aspects, such as fiber quality and yield (Zhang et al., 1994, 2014; Wang and Zhe, 2013; Lu et al., 2017). The phenotypic data of all size and shape-related traits between BILs of G. barbadense Hai7124 and G. hirsutum CRI36 showed rich variation. For example, the minimum value of HKW is 3.63 and the maximum is 9.36, which has obvious transgressive segregation, even though there were no significant differences between the two parents. QTLs mapped by BILs will be the choice for MAS to improve the quality of cotton seeds by transferring favorable alleles to cotton.
In this study, we detected 49 QTLs for cotton seed size and shape-related traits that were distributed in seven QTL clusters, representing one of the first such a comprehensive study in cotton. Nine QTLs were stably detected in multiple environments and were located on chromosomes D03 and D12. Previously, a QTL for plant height (Ma et al., 2019) and a QTL for micronaire (Pei et al., 2021) were detected in this BIL population. Interestingly, the physical interval of the QTL mapping of the seed size and shape overlapped with the QTLs for these two traits. There were also other QTL studies on seed index, with QTLs mapped on the D03 chromosome (Shang et al., 2016; Liu et al., 2018). In a previous QTL mapping study for the four traits (HSW, HKW, TKL, TKW) of cottonseed (Wang et al., 2019), QTLs for the three traits (HSW, HKW, and TKW) were also detected on the D03 chromosome. In addition to two QTL clusters on D03 for the cotton physical traits, our current study detected two new QTLs- qKLW-D12-1 and qKR-D12-1 for cotton seed size and shape. These common QTLs and new stable QTLs will be the first choice for MAS to improve cottonseed quality by transferring favorable alleles to cotton cultivars.
Among the 641 genes within nine stable QTLs, we further identified five candidate genes for their possible involvement of regulating seed size and shape based on differential gene expression and sequence variation. The exact roles of these five genes in relation to cottonseed size and shape are currently unknown and should be further studied. The following discussion was solely based on relevant studies in other plants.
GH_D03G0980 encodes starch synthase 4. Starch synthase 4 (SS4) is required for proper starch granule initiation in Arabidopsis thaliana; and ss4 mutants grow poorly even under long-day conditions (Ragel et al., 2013). In rice, four starch synthase I (SSI)-deficient mutant lines did not alter seed morphology (Fujita et al., 2006). Fujita et al. (2011) further showed that rice endosperm requires the presence of either SS I or IIIa for starch biosynthesis, whose mutations led to reduced dehulled seed weight. In wheat, all three SSII genes on A, B and D subgenomes had to be missing or inactive for a change in seed weight and other traits (Konik-Rose et al., 2007). Therefore, seed weight may be affected by SS.
GH_D03G1091 encodes the transcription factor PIF7, which is a basic helix-loop-helix (bHLH)-type transcription factor. GH_D12G2619 encodes another bHLH transcription factor MYC4, which is homologous to AT4G00870 in Arabidopsis. This bHLH transcription factor family in plants is widely involved in biological processes, including the response to hormone signals (Friedrichsen et al., 2002; Yin et al., 2005), and flower and fruit development (Rajani and Sundaresan, 2001; Liljegren et al., 2004; Szecsi et al., 2006). It was found in Arabidopsis that the bHLH subgroup IIID transcription factors (bHLH 3, bHLH 13, bHLH 14, and bHLH 17) have a negative regulatory effect on the jasmonate (JA) response, and can act as a transcription inhibitor to coordinate the JA response, thereby regulating the defense and development of plants (Song et al., 2013). bHLH transcription factors may be also involved in determining seed size and shape. In rice, two bHLH proteins- POSITIVE REGULATOR OF GRAIN LENGTH 1 (PGL1) and its antagonistic partner ANTAGONIST OF PGL1 (APG) were involved in determining rice grain length by controlling cell length in the lemma/palea. Heang and Sassa (2012) showed that overexpression of PGL1 and silencing of APG each increased grain length and weight in transgenic rice, suggesting that APG was a negative regulator whose function was inhibited by PGL1. Other transcription factors can also affect seed size and shape. For example, most recently, Sun et al. (2021) showed that three SNPs related to ZmBES1/BZR1-5 were significantly correlated with kernel width and four SNPs in the gene were related to 100-kernel weight. They further confirmed that transgenic Arabidopsis and rice with ZmBES1/BZR1-5 displayed significantly increased seed size and weight, while Mu transposon insertion and EMS maize mutants in the gene possessed smaller kernels.
GH_D03G1458 encodes the ubiquitin-conjugating enzyme E2 7. The ubiquitin-26S proteasome pathway (UPP) is a crucial regulatory mechanism for selective protein degradation in a wide variety of plant developmental processes. Ubiquitin-binding (UBC) E2 enzyme, an important part of it, also plays a vital role in plant growth and development (Wang, 2010; Gao et al., 2017). Xu (2014) showed that the null UBC 22 mutants produced larger plants and larger and heavier seeds that stored a higher amount of protein and fatty acids in Arabidopsis. However, Mao et al. (2020) showed that overexpression of a soybean UBC gene (GmUBC1) in Arabidopsis significantly increased the 1,000-grain weight and total amino acid content. Similar to genes for bHLH transcription factors, different gene family members may have an opposite effect on seed weight and shape.
GH_D03G1466 encodes THO complex subunit 4A. THO is a multi-protein complex promoting coupling between transcription and mRNA processing. It is demonstrated the THO complex is involved in regulating female germline specification and disease resistance in Arabidopsis (Pan et al., 2012; Su et al., 2017). The destruction of ALY1, ALY2, ALY3, and ALY4 (orthologs of genes involved in the THO complex) in Arabidopsis caused nutritional and reproductive defects, including severe growth slowdowns, changes in flower morphology, and abnormal ovules and female gametophytes, resulting in reduced seed yield (Pfaff et al., 2018). However, the role of the complex in relation to seed size and shape is current unknown.
In cotton, the roles of those five candidate genes in relation to seed physical traits are not understood. However, we showed that these genes may be target genes for the genetic improvement of cotton seed size and shape. Among them, SNPs in four candidate genes were significantly associated with changes in seed size and shape traits such as HKW and KLW. The results provided important alleles for molecular breeding to improve cotton physical traits.
Conclusion
In summary, 49 QTLs for eight seed size and shape-related traits were identified by QTL mapping using an interspecific BIL population derived from G. barbadense Hai7124 and G. hirsutum CRI36 as the recurrent parent. Nine stable QTLs and 641 putative genes were identified within these QTL intervals. After further analysis, five genes encoding enzymes and transcription factors were identified as possible candidate genes that may be associated with cotton seed size and shape for further studies. These results represent the first study on the genetic basis for most seed physical traits in cotton. Their relationships with lint percentage and yield and fiber quality should be studied, which will facilitate breeding for high-quality and high-yield cotton.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
LWu analyzed, summed all the data, and wrote the manuscript. BJ performed SNP analysis and wrote the introduction part of the manuscript. WP, JM, JS, SY, and MW conducted instrument debugging, managed, and collected phenotypic data. LWa participated in the discussion of the manuscript. YX, LH, and PF participated in the analysis of SNP markers. JY and JZ guided the experiment and manuscript revision. All authors read and approved the final manuscript.
Funding
This present study was funded by the Natural Science Foundation of Xinjiang Uygur Autonomous Region of China (grant nos. 2021D01B113 and 2020D01A135), the National Natural Science Foundation of China (grant no. 31621005), and the Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.837984/full#supplementary-material
Supplementary Figure 1 | QTLs for seed size and shape-related traits under five environments and BLUP. The lines on the right side of the linkage groups indicate the QTL likelihood intervals. Map distances in centiMorgans are indicated on the left side of the linkage groups.
Footnotes
References
Atique-ur-Rehman, Kamran, M., and Afzal, I. (2020). “Production and processing of quality cotton seed,” in Cotton Production and Uses, eds S Ahmad and M Hasanuzzaman. (Singapore: Springer). doi: 10.1007/978-981-15-1472-2_27
Chen, F., Guo, Y., Chen, L., Gan, X., and Xu, W. (2020). Global identification of genes associated with xylan biosynthesis in cotton fiber. J. Cotton Res. 3:15. doi: 10.1186/s42397-020-00063-3
Cingolani, P., Platts, A., Wang Le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Deng, X., Gong, J., Liu, A., Shi, Y., Gong, W., Ge, Q., et al. (2019). QTL mapping for fiber quality and yield-related traits across multiple generations in segregating population of CCRI 70. J. Cotton Res. 2:13. doi: 10.1186/s42397-019-0029-y
Do, T. H. T., Martinoia, E., and Lee, Y. (2018). Functions of ABC transporters in plant growth and development. Curr. Opin. Plant Biol. 41, 32–38. doi: 10.1016/j.pbi.2017.08.003
Friedrichsen, D. M., Nemhauser, J., Muramitsu, T., Maloof, J. N., Alonso, J., Ecker, J. R., et al. (2002). Three redundant brassinosteroid early response genes encode putative bHLH transcription factors required for normal growth. Genetics 162, 1445–1456. doi: 10.1093/genetics/162.3.1445
Fujita, N., Satoh, R., Hayashi, A., Kodama, M., Itoh, R., Aihara, S., et al. (2011). Starch biosynthesis in rice endosperm requires the presence of either starch synthase I or IIIa. J. Exp. Bot. 62, 4819–4831. doi: 10.1093/jxb/err125
Fujita, N., Yoshida, M., Asakura, N., Ohdan, T., Miyao, A., Hirochika, H., et al. (2006). Function and characterization of starch synthase I using mutants in rice. Plant Physiol. 140, 1070–1084. doi: 10.1104/pp.105.071845
Gao, Y., Yi, W., Xin, H., Li, S., and Liang, Z. (2017). Involvement of ubiquitin-conjugating enzyme (E2 Gene Family) in ripening process and response to cold and heat stress of. Sci. Rep. 7:13290. doi: 10.1038/s41598-017-13513-x
Gu, Q., Ke, H., Liu, Z., Lv, X., Sun, Z., Zhang, M., et al. (2020). A high-density genetic map and multiple environmental tests reveal novel quantitative trait loci and candidate genes for fibre quality and yield in cotton. Theor. Appl. Genet. 133, 3395–3408. doi: 10.1007/s00122-020-03676-z
Heang, D., and Sassa, H. (2012). Antagonistic actions of HLH/bHLH proteins are involved in grain length and weight in rice. PLoS One 7:e31325. doi: 10.1371/journal.pone.0031325
Hina, A., Cao, Y., Song, S., Li, S., Sharmin, R. A., Elattar, M. A., et al. (2020). High-esolution mapping in two ril populations refines major “QTL hotspot” regions for seed size and shape in soybean (Glycine max L.). Int. J. Mol. Sci. 21:1040. doi: 10.3390/ijms21031040
Hu, Y., Chen, J., Fang, L., Zhang, Z., Ma, W., Niu, Y., et al. (2019). Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748. doi: 10.1038/s41588-019-0371-5
Ji, X., Du, Y., Li, F., Sun, H., and Zhao, Q. (2019). The basic helix-loop-helix transcription factor, OsPIL15, regulates grain size via directly targeting a purine permease gene OsPUP7 in rice. Plant Biotechnol. J. 17, 1527–1537. doi: 10.1111/pbi.13075
Jiang, Q. T., Liu, T., Ma, J., Wei, Y. M., Lu, Z. X., Lan, X. J., et al. (2011). Characterization of barley Prp1 gene and its expression during seed development and under abiotic stress. Genetica 139, 1283–1292. doi: 10.1007/s10709-012-9630-4
Konik-Rose, C., Thistleton, J., Chanvrier, H., Tan, I., Halley, P., Gidley, M., et al. (2007). Effects of starch synthase IIa gene dosage on grain, protein and starch in endosperm of wheat. Theor. Appl. Genet. 115, 1053–1065. doi: 10.1007/s00122-007-0631-0
Lehtilä, K., and Ehrlén, J. (2005). Seed size as an indicator of seed quality: a case study of Primula veris. Acta Oecol. 28, 207–212. doi: 10.1016/j.actao.2005.04.004
Li, N., and Li, Y. (2015). Maternal control of seed size in plants. J. Exp. Bot. 4:1087. doi: 10.1093/jxb/eru549
Li, X., Xing, B., Liu, X., Jiang, X. W., and Zhao, Q. C. (2020). Network pharmacology-based research uncovers cold resistance and thermogenesis mechanism of Cinnamomum cassia. Fitoterapia 149:104824. doi: 10.1016/j.fitote.2020.104824
Li, Y., Fan, C., Xing, Y., Jiang, Y., Luo, L., Sun, L., et al. (2011). Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 43, 1266–1269. doi: 10.1038/ng.977
Li, Y., Zheng, L., Corke, F., Smith, C., and Bevan, M. W. (2008). Control of final seed and organ size by the DA1 gene family in Arabidopsis thaliana. Genes Dev. 22, 1331–1336. doi: 10.1101/gad.463608
Liljegren, S. J., Roeder, A., Kempin, S. A., and Gremski, K. (2004). Control of fruit patterning in Arabidopsis by indehiscent. Cell 116, 843–853. doi: 10.1016/s0092-8674(04)00217-x
Liu, C., Li, Z., Dou, L., Yuan, Y., and Xiao, G. (2020). A genome-wide identification of the BLH gene family reveals BLH1 involved in cotton fiber development. J. Cotton Res. 3:26. doi: 10.1186/s42397-020-00068-y
Liu, H., Zhang, L., Mei, L., Quampah, A., and Zhu, S. (2020). QOil-3, a major QTL identification for oil content in cottonseed across genomes and its candidate gene analysis. Ind. Crops Prod. 145:112070. doi: 10.1016/j.indcrop.2019.112070
Liu, L., Tong, H., Xiao, Y., Che, R., and Chu, A. C. (2015). Activation of big grain1 significantly improves grain size by regulating auxin transport in rice. Proc. Natl. Acad. Sci. U.S.A. 112:11102. doi: 10.1073/pnas.1512748112
Liu, R., Gong, J., Xiao, X., Zhang, Z., Li, J., Liu, A., et al. (2018). GWAS analysis and QTL identification of fiber quality traits and yield components in upland cotton using enriched high-density SNP markers. Front. Plant Sci. 9:1067. doi: 10.3389/fpls.2018.01067
Liu, X., Li, J., Yu, X., Shi, Y., Fei, J., Sun, F., et al. (2013). Identification of QTL for cottonseed oil and protein content in upland cotton(Gossypium hirsutum L.) based on a RIL population. Mol. Plant Breed. 11, 520–528.
Liu, X., Yuan, J., Li, Y., and Pan, Z. (1997). Influences of seed quality on growth and yield in cotton. J. Shanxi Agric. Sci. 4, 40–43.
Liu, Z. H., Zhao, G. H., Jing-Shan, L. U., Shui, Y., and Gang, W. U. (2008). Studies on content of cotton gossypol and characteristics of pest resistance. Xinjiang Agric. Sci. 45, 409–413. doi: 10.2967/jnmt.107.044081
Lu, Q., Shi, Y., Xiao, X., Li, P., Gong, J., Gong, W., et al. (2017). Transcriptome analysis suggests that chromosome introgression fragments from sea island cotton (Gossypium barbadense) increase fiber strength in upland cotton (Gossypium hirsutum). G3 7, 3469–3479. doi: 10.1534/g3.117.300108
Ma, J., Pei, W., Ma, Q., Geng, Y., Liu, G., Liu, J., et al. (2019). QTL analysis and candidate gene identification for plant height in cotton based on an interspecific backcross inbred line population of Gossypium hirsutum x Gossypium barbadense. Theor. Appl. Genet. 132, 2663–2676. doi: 10.1007/s00122-019-03380-7
Main, C. L., Barber, L. T., Boman, R. K., Chapman, K., Dodds, D. M., Duncan, S., et al. (2013). Effects of nitrogen and planting seed size on cotton growth, development, and yield. Agron. J. 105, 1853–1859. doi: 10.2134/agronj2013.0154
Mao, Z. Z., Gong, Y., Shi, G. X., Li, Y. L., Yu, Y., and Huang, F. (2020). Cloning of the soybean E2 ubiquitin-conjugating enzyme GmUBC1 and its expression in Arabidopsis thaliana. Hereditas 42, 788–798. doi: 10.16288/j.yczz.20-141
Meng, L., Li, H., Zhang, L., and Wang, J. (2015). QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations - ScienceDirect. Crop J. 3, 269–283.
Pahlavani, M. H., Miri, A. A., and Kazemi, G. (2008). Response of oil and protein content to seed size in cotton. Int. J. Agric. Biol. 10, 643–647. doi: 10.2478/v10129-009-0004-8
Pan, H., Liu, S., and Tang, D. (2012). The THO/TREX complex functions in disease resistance in Arabidopsis. Plant Signal. Behav. 7, 422–424. doi: 10.4161/psb.18991
Pei, W., Song, J., Wang, W., Ma, J., Jia, B., Wu, L., et al. (2021). Quantitative trait locus analysis and identification of candidate genes for micronaire in an interspecific backcross inbred line population of Gossypium hirsutum × Gossypium barbadense. Front. Plant Sci. 12:2308. doi: 10.3389/fpls.2021.763016
Pfaff, C., Ehrnsberger, H. F., Flores-Tornero, M., Sorensen, B. B., Schubert, T., Langst, G., et al. (2018). ALY RNA-Binding proteins are required for nucleocytosolic mRNA transport and modulate plant growth and development. Plant Physiol. 177, 226–240. doi: 10.1104/pp.18.00173
Poland, J. A., Bradbury, P. J., Buckler, E. S., and Nelson, R. J. (2011). Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc. Natl. Acad. Sci. U.S.A. 108, 6893–6898. doi: 10.1073/pnas.1010894108
Powder, K. E. (2020). Quantitative trait loci (QTL) mapping. Methods Mol. Biol. 2082, 211–229. doi: 10.1007/978-1-0716-0026-9_15
Qi, Z., and Xiong, L. (2013). Characterization of a purine permease family gene OsPUP7 involved in growth and development control in rice. J. Integr. Plant Biol. 55, 1119–1135. doi: 10.1111/jipb.12101
Ragel, P., Streb, S., Feil, R., Sahrawy, M., Annunziata, M. G., Lunn, J. E., et al. (2013). Loss of starch granule initiation has a deleterious effect on the growth of arabidopsis plants due to an accumulation of ADP-glucose. Plant Physiol. 163, 75–85. doi: 10.1104/pp.113.223420
Rajani, S., and Sundaresan, V. (2001). The Arabidopsis myc/bHLH gene ALCATRAZ enables cell separation in fruit dehiscence. Curr. Biol. 11, 1914–1922. doi: 10.1016/S0960-9822(01)00593-0
Said, J. I., Knapka, J. A., Song, M., and Zhang, J. (2015). Cotton QTLdb: a cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 290, 1615–1625. doi: 10.1007/s00438-015-1021-y
Said, J. I., Lin, Z., Zhang, X., Song, M., and Zhang, J. (2013). A comprehensive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genomics 14:776. doi: 10.1186/1471-2164-14-776
Sawan, Z. M., and Dello Ioio, R. (2016). Cottonseed yield and its quality as affected by mineral nutrients and plant growth retardants. Cogent Biol. 2:1. doi: 10.1080/23312025.2016.1245938
Shang, L., Abduweli, A., Wang, Y., and Hua, J. P. (2016). Genetic analysis and QTL mapping of oil content and seed index using two recombinant inbred lines and two backcross populations in Upland cotton. Plant Breed. 135, 224–231. doi: 10.1111/pbr.12352
Shang, L., Liang, Q., Wang, Y., Wang, X., Wang, K., Abduweli, A., et al. (2015). Identification of stable QTLs controlling fiber traits properties in multi-environment using recombinant inbred lines in Upland cotton (Gossypium hirsutum L.). Euphytica 205, 877–888. doi: 10.1007/s10681-015-1434-z
Snider, J. L., Collins, G. D., Whitaker, J., Chapman, K. D., and Horn, P. (2016). The impact of seed size and chemical composition on seedling vigor, yield, and fiber quality of cotton in five production environments. Field Crops Res. 193, 186–195. doi: 10.1016/j.fcr.2016.05.002
Song, S., Qi, T., Fan, M., Zhang, X., Gao, H., Huang, H., et al. (2013). The bHLH subgroup IIId factors negatively regulate jasmonate-mediated plant defense and development. PLoS Genetics 9:e1003653. doi: 10.1371/journal.pgen.1003653
Su, Z., Zhao, L., Zhao, Y., Li, S., Won, S., Cai, H., et al. (2017). The THO complex non-cell-autonomously represses female germline specification through the TAS3-ARF3 module. Curr. Biol. 27, 1597–1609. doi: 10.1016/j.cub.2017.05.021
Sun, F., Ding, L., Feng, W., Cao, Y., Lu, F., Yang, Q., et al. (2021). Maize transcription factor ZmBES1/BZR1-5 positively regulates kernel size. J. Exp. Bot. 72, 1714–1726. doi: 10.1093/jxb/eraa544
Szecsi, J., Joly, C., Bordji, K., Varaud, E., Cock, J. M., Dumas, C., et al. (2006). BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J. 25, 3912–3920. doi: 10.1038/sj.emboj.7601270
Tian, Y., and Zhang, T. (2021). MIXTAs and phytohormones orchestrate cotton fiber development. Curr. Opin. Plant Biol. 59:101975. doi: 10.1016/j.pbi.2020.10.007
Wang, F. R., Zhang, J., Liu, R. Z., Liu, Q. H., Zhang, C. Y., and Liu, G. D. (2005). Genetic analysis of upland cotton germplasm obtained from introduced DNA from island cotton. Sci. Agric. Sin. 38, 1528–1533.
Wang, J. S. (2010). Progress on functions of ubiquitin-conjugating enzyme(E2) in plants. Biotechnol. Bull. 4, 7–10. doi: 10.13560/j.cnki.biotech.bull.1985.2010.04.002
Wang, Q., and Zhe, L. (2013). Heterosis and combining ability analysis of fiber quality traits of sea-land hybrid F_1 generation. J. Henan Sci. Technol. 41, 12–18.
Wang, W., Sun, Y., Yang, P., Cai, X., Yang, L., Ma, J., et al. (2019). A high density SLAF-seq SNP genetic map and QTL for seed size, oil and protein content in upland cotton. BMC Genomics 20:599. doi: 10.1186/s12864-019-5819-6
Xia, T., Li, N., Dumenil, J., Li, J., Kamenski, A., Bevan, M. W., et al. (2013). The ubiquitin receptor DA1 interacts with the E3 ubiquitin ligase DA2 to regulate seed and organ size in Arabidopsis. Plant Cell 25, 3347–3359. doi: 10.1105/tpc.113.115063
Xing, C. Z., Jing, S. R., and Xing, Y. H. (2007). Review and prospect on cotton heterosis utilization and study in China. Cotton Sci. 5, 337–345.
Xu, Y. (2014). Characterization Of The Peroxisomal Ubiquitin-Conjugating Enzyme 22 Protein In Arabidopsis Thaliana. M.S. thesis, Michigan State University, Michigan, United States.
Yi, F., Gu, W., Li, J., Chen, J., Hu, L., Cui, Y., et al. (2021). Miniature Seed6, encoding an endoplasmic reticulum signal peptidase, is critical in seed development. Plant Physiol. 185, 985–1001. doi: 10.1093/plphys/kiaa060
Yin, S., Li, P., Xu, Y., Liu, J., Yang, T., Wei, J., et al. (2020). Genetic and genomic analysis of the seed-filling process in maize based on a logistic model. Heredity 124, 122–134. doi: 10.1038/s41437-019-0251-x
Yin, Y., Vafeados, D., Tao, Y., Yoshida, S., and Chory, J. (2005). A new class of transcription factors mediates brassinosteroid-regulated gene expression in Arabidopsis. Cell 120, 249–259. doi: 10.1016/j.cell.2004.11.044
Ying, J. Z., Ma, M., Bai, C., Huang, X. H., Liu, J. L., Fan, Y. Y., et al. (2018). TGW3, a major QTL that negatively modulates grain length and weight in rice. Mol. Plant 11, 750–753. doi: 10.1016/j.molp.2018.03.007
Yu, J., Yu, S., Fan, S., Song, M., Zhai, H., Li, X., et al. (2012). Mapping quantitative trait loci for cottonseed oil, protein and gossypol content in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Euphytica 187, 191–201. doi: 10.1007/s10681-012-0630-3
Zhang, J. F., Gong, Z. P., Sun, J. Z., and Liu, J. L. (1994). Heterosis of yield and fiber performance in interspecific crosses be-tween Gossypium hirsutum and G. barbadense. Mian Hua Xue Bao. 3, 140–145.
Zhang, J. F., Percy, R. G., and McCarty, J. C. Jr. (2014). Introgression genetics and breeding between upland and pima cotton- a review. Euphytica 198, 1–12. doi: 10.1007/s10681-014-1094-4
Zhang, S., Hu, X., Miao, H., Chu, Y., Cui, F., Yang, W., et al. (2019). QTL identification for seed weight and size based on a high-density SLAF-seq genetic map in peanut (Arachis hypogaea L.). BMC Plant Biol. 19:537. doi: 10.1186/s12870-019-2164-5
Keywords: Gossypium hirsutum, Gossypium barbadense, backcross inbred lines, seed size and shape, quantitative trait locus, candidate genes
Citation: Wu L, Jia B, Pei W, Wang L, Ma J, Wu M, Song J, Yang S, Xin Y, Huang L, Feng P, Zhang J and Yu J (2022) Quantitative Trait Locus Analysis and Identification of Candidate Genes Affecting Seed Size and Shape in an Interspecific Backcross Inbred Line Population of Gossypium hirsutum × Gossypium barbadense. Front. Plant Sci. 13:837984. doi: 10.3389/fpls.2022.837984
Received: 17 December 2021; Accepted: 31 January 2022;
Published: 22 March 2022.
Edited by:
Linghe Zeng, United States Department of Agriculture (USDA), United StatesReviewed by:
Narayanan Manikanda Boopathi, Tamil Nadu Agricultural University, IndiaDevendra Pandeya, Texas A&M University, United States
Copyright © 2022 Wu, Jia, Pei, Wang, Ma, Wu, Song, Yang, Xin, Huang, Feng, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinfa Zhang, amluemhhbmdAbm1zdS5lZHU=; Jiwen Yu, eXVqdzY2NkBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work