 Yu Xin Wen1
Yu Xin Wen1 Jia Yi Wang1
Jia Yi Wang1 Hui Hui Zhu1
Hui Hui Zhu1 Guang Hao Han2
Guang Hao Han2 Ru Nan Huang2
Ru Nan Huang2 Li Huang3
Li Huang3 Yi Guo Hong2
Yi Guo Hong2 Shao Jian Zheng1
Shao Jian Zheng1 Jian Li Yang1*
Jian Li Yang1* Wei Wei Chen2*
Wei Wei Chen2*- 1State Key Laboratory of Plant Physiology and Biochemistry, College of Life Sciences, Zhejiang University, Hangzhou, China
- 2Research Centre for Plant RNA Signaling and Zhejiang Provincial Key Laboratory for Genetic Improvement and Quality Control of Medicinal Plants, College of Life and Environmental Sciences, Hangzhou Normal University, Hangzhou, China
- 3Laboratory of Cell & Molecular Biology, Institute of Vegetable Science, Zhejiang University, Hangzhou, China
Deoxyribonucleic acid (DNA) methylation is an important epigenetic mark involved in diverse biological processes. Here, we report the critical function of tomato (Solanum lycopersicum) Domains Rearranged Methyltransferase7 (SlDRM7) in plant growth and development, especially in leaf interveinal chlorosis and senescence. Using a hairpin RNA-mediated RNA interference (RNAi), we generated SlDRM7-RNAi lines and observed pleiotropic developmental defects including small and interveinal chlorosis leaves. Combined analyses of whole genome bisulfite sequence (WGBS) and RNA-seq revealed that silencing of SlDRM7 caused alterations in both methylation levels and transcript levels of 289 genes, which are involved in chlorophyll synthesis, photosynthesis, and starch degradation. Furthermore, the photosynthetic capacity decreased in SlDRM7-RNAi lines, consistent with the reduced chlorophyll content and repression of genes involved in chlorophyll biosynthesis, photosystem, and photosynthesis. In contrast, starch granules were highly accumulated in chloroplasts of SlDRM7-RNAi lines and associated with lowered expression of genes in the starch degradation pathway. In addition, SlDRM7 was activated by aging- and dark-induced senescence. Collectively, these results demonstrate that SlDRM7 acts as an epi-regulator to modulate the expression of genes related to starch and chlorophyll metabolism, thereby affecting leaf chlorosis and senescence in tomatoes.
Introduction
Leaf senescence, the final stage of leaf development prior to its death, is a genetically programmed degenerative process, which is accompanied by massive macromolecular catabolism and nutrient recycling to young or storage tissues (Gan and Amasino, 1997; Guo and Gan, 2005). Characterized by leaf chlorosis due to chlorophyll loss, leaf senescence mainly results from age-dependent internal factors, and it also can be triggered by a range of other internal and external cues, including reproduction, phytohormone levels, nutritional signals, water status, light regimes, temperature change, mechanical damage and pathogen attack (Lim et al., 2007a). Although these senescence-influencing factors induce apparently similar phenotypes, the initiation and subsequent processes of senescence are controlled by different molecular modes at multiple regulatory levels (Schippers, 2015; Luo et al., 2018). At the genetic transcriptional and post-transcriptional level, well-established senescence markers include chlorophyll content (Rossi et al., 2015), photochemical efficiency (Guo and Gan, 2005), starch metabolism (Caspar et al., 1985; Zeeman et al., 1998; Zeeman and Rees, 1999), and expression of senescence-associated genes (SAGs) (Lim et al., 2007a). Various transcription factors, such as those belonging to NAC, WRKY, and MYB families (Robatzek and Somssich, 2002; Miao et al., 2004; Ay et al., 2009; Balazadeh et al., 2010; Yang et al., 2014; Lira et al., 2017; Ma et al., 2018; Ma X. et al., 2019; Woo et al., 2019; Jin et al., 2020), have been identified to modulate leaf senescence by activating the expression of downstream SAGs including chlorophyll catabolic genes (CCGs) for chlorophyll degradation (Balazadeh et al., 2010; Woo et al., 2019). In addition, histone modification and chromatin remodeling have been found to regulate certain SAGs expression at the transcriptional level (Lim et al., 2007b; Ay et al., 2009; Chen et al., 2016; Liu et al., 2019), implying a critical role of epigenetic control over leaf senescence.
The main form of conserved epigenetic modification, DNA methylation, often occurs at the 5’ carbon of cytosine base (mC) and plays roles in genome stability and gene expression (Finnegan and Kovac, 2000; Robertson, 2005). In plant, DNA methylation involves an RNA-directed DNA methylation (RdDM) pathway of mC establishment in CG, CHG, and CHH (where H is A, C, or T) contexts and mC maintenance (Zhang et al., 2006; Henderson and Jacobsen, 2007; Law and Jacobsen, 2010). The dynamics of DNA methylation are regulated by DNA methyltransferases and DNA demethylases (Finnegan et al., 1996; Penterman et al., 2007; Lei et al., 2015; Zhang et al., 2018; Liu and Lang, 2020). Plants encode DOMAINS REARRANGED METHYLTRANSFERASE (DRM), METHYLTRANSFERASE (MET), and CHROMOMETHYLASE (CMT) to establish and maintain mC by distinct pathways (Finnegan and Dennis, 1993; Henikoff and Comai, 1998; Cao and Jacobsen, 2002a). In Arabidopsis (Arabidopsis thaliana), MET1 prefers symmetric CG sites, while CMT2 chooses asymmetric CHH sites; CMT3 contributes to maintaining mCHG and, to a lesser extent, mCHH; DRM2 establishes de novo RdDM and also participates in maintaining mCHH (Chan et al., 2005; Matzke and Mosher, 2014; Zhang et al., 2018), respectively. Despite the dynamic alternations of global mC during vegetative and reproductive growth, DNA methylation plays an important role in regulating plant development, reproduction, and responses to biotic and abiotic stresses (Ronemus et al., 1996; Finnegan et al., 2000; Chinnusamy and Zhu, 2009; Slotkin et al., 2009; Dowen et al., 2012; Ay et al., 2014). Defects in RdDM and mC maintenance showed many phenotypic and developmental abnormalities, including reduced apical dominance, smaller plant size, altered leaf size with curly shape, decreased fertility, and varied flowering time (Finnegan et al., 1996; Kankel et al., 2003; Yang et al., 2019). Although neither the cmt3 mutants (Lindroth et al., 2001) nor the drm1 drm2 double mutants (Cao and Jacobsen, 2002b) show morphological differences from wild type (WT), drm1 drm2 cmt3 plants showed pleiotropic phenotypes including developmental retardation, reduced plant size, and partial sterility (Cao and Jacobsen, 2002a), whereas targeted disruption of rice OsDRM2, which caused a 13.9% decrease in genome-wide mC, displayed at vegetative and reproductive development in Oryza sativa, showing growth defects, semi-dwarfed stature, reductions in tiller number, delayed or no heading, aberrant panicle and spikelet morphology, and complete sterility (Moritoh et al., 2012).
However, limited progress has been made toward elucidating the involvement of DNA methylation during plant aging and senescence. For instance, the transcript levels of MET1, CMT3, DRM1, and DRM2 are shown to be repressed during leaf senescence (Cao and Jacobsen, 2002a; Jackson et al., 2002; Kankel et al., 2003; Law and Jacobsen, 2010). Additionally, the expression of 16 methylation-associated genes, including MET1, REPRESSOR OF SILENCING 1 (ROS1), and ARGONAUTE 10 (AGO10) were significantly downregulated in aging Arabidopsis leaves (Ay et al., 2014). Recently, Arabidopsis dml3 (DEMETER-Like DNA demethylase3) knockout (KO) mutant results in genome-wide hypermethylation, especially in the promoters of many SAGs whose expression are consequently suppressed, leading to a significant delay in leaf senescence (Yuan et al., 2020). In fact, global mC decreased dynamically during shoot aging (Ogneva et al., 2016). However, the precise relevance of such epi-modification in controlling leaf senescence, and the pertinent underlying mechanisms are still largely unknown. Here, we report that SlDRM7 (Solyc04g005250.2) impacts chloroplast development via modulating starch accumulation and senescence-related chlorophyll synthesis and imposes epi-effects on leaf senescence that affects vegetative growth in tomatoes.
Materials and Methods
Plant Materials and Growth Conditions
Wild-type tomato Solanum lycopersicum cultivar Ailsa Craig (AC) and the SlDRM7-RNAi lines (AC background) were generated and used in this study. Tomato seeds were either germinated directly in compost (Sunshine Mix 3, Sungro Horticulture Canada) or surface-sterilized and germinated on 1/2 Murashige and Skoog (MS) medium for 6 days before being transferred to 1/5 Hoagland solution (pH 5.5) for hydroponic growth. Seedlings that are 5-week-old were then transferred to composts and grown in insect-free growth rooms or greenhouses at 25°C under a 16-h-light/8-h-dark cycle with a humidity of 60 to 80% (Chen et al., 2018a).
RNA Interference Constructs, CRISPR/CAS9 Gene Editing, and Tomato Transformation
The SlDRM7 RNAi vector pRNAi-SlDRM7 was constructed as described (Chen et al., 2018b; Yao et al., 2020). A 230-bp SlDRM7 fragment was PCR-amplified using tomato cDNA as a template and cloned in the sense and antisense orientations into the pRNAi-LIC vector (Chen et al., 2018b). A pair of 20-bp sgRNA oligos targeting the exon of SlDRM7 was cloned into the plant CRISPR/Cas9-induced vector to produce the SlDRM7 gene editing constructs. Tomato transformation was performed as previously described (Yao et al., 2020). Briefly, the construct was transformed into tomato cotyledons by Agrobacterium tumefaciens strain GV3101 to induce shoots under the selection of kanamycin resistance (Dobrev and Kaminek, 2002). Regenerated shoots with 3 to 4 cm length were cut off from independent calli and transferred to the rooting medium for root development (Kit et al., 2010). To confirm the stable transformation event, putatively transformed plantlets with well-developed roots were subjected to molecular analyses through genomic PCR and RT-qPCR. Primers used for making these constructs are listed in Supplementary Table 1.
Statistical Analysis of Morphological Features
To differentiate WT and SlDRM7-RNAi tomato plants, at least 10 leaflets of each line were scanned for measurement of leaf area using ImageJ software. All images showing phenotypes were captured with a Canon digital camera.
Photosynthetic Pigment Quantification and Confocal Microscopy of Chlorophyll Auto-Fluorescence
About 0.02 g fresh leaf samples collected from the 2nd leaf of six-leaf-stage tomato seedlings were immersed in 10 ml 80% (v/v) acetone in the dark for 24 h until leaves were completely bleached. The absorbance of the supernatant was measured, respectively, at 645, 663, and 470 nm, then chlorophyll a/b and carotenoids content were calculated. The photosynthetic pigment content was calculated as described (Arnon, 1949). Three biological replicates and three technical replicates for each leaf sample were measured. To determine chlorophyll auto-fluorescence, the 2nd leaf of six-leaf-stage tomato seedlings was examined with LSM710nlo laser scanning confocal microscope (Zeiss, Germany).
Photosynthetic Measurements
The Li-6400 portable photosynthesis system (LI-COR, Lincoln, NE, United States) was used to measure the photosynthetic physiological indexes of the 2nd leaf of six-leaf-stage tomato seedlings. The reference CO2 concentration was held at 480 μmol mol–1 and leaf temperature at 25°C for all measurements. Air humidity inside the leaf chamber was equivalent to values measured inside the greenhouse (approx. 75%). The light and CO2 response curves were measured by varying light intensity from 0 to 2,500 μmol ⋅ m–2 ⋅ s–1. Net photosynthesis rate (Pn), respiration rate, transpiration rate (Tr), stomatal conductance (Gs), and intercellular CO2 concentration (Ci) were measured at 2,000 μmol ⋅ m–2 ⋅ s–1. Five replicates were measured at each light intensity. The light and CO2 response curves were simulated by a non-rectangular hyperbola model:
An(I): Pn, I: light intensity, θ: the initial slope, α: the initial photochemical efficiency, Rd: dark respiration (Thornley, 1976).
Transmission Electron Microscopy
About 1 × 3 mm2 leaf tissues were cut off from the seedlings at the six-leaf stage. Natural senescence leaf samples were collected from WT. Leaf samples were first fixed with 2.5% glutaraldehyde in phosphate buffer (0.1 M, pH 7) for more than 4 h, washed three times in the phosphate buffer for 15 min each, then post-fixed with 1% OsO4 in phosphate buffer for 1–2 h, and washed three times in the phosphate buffer. Leaf samples were first dehydrated by a graded series of ethanol and then dehydrated by pure acetone. Next, the specimen was placed in a 1:1 mixture of absolute acetone and the final Spurr resin mixture for 1 h at room temperature, then transferred to a 1:3 mixture of absolute acetone, and the final resin mixture for 3 h and transferred to final Spurr resin mixture for overnight. The specimen was placed in Eppendorf which contained Spurr resin and heated at 70°C for 9 h. The specimen was sectioned in LEICA EM UC7 ultratome and sections were stained by uranyl acetate and alkaline lead citrate for 5 to 10 min, respectively, and observed under a Hitachi Model H-7650 TEM (Hitachi, Japan).
Histological Detection of Starch and Measurements of Starch Content
Leaves were treated with 80% (v/v) ethanol to remove chlorophylls and stained with Lugol’s iodine solution to detect starch distribution (Tsai et al., 2009). Images were captured with a Canon digital camera.
Starch content was measured following the method as described by Clegg (1956). Briefly, 0.02 g of fresh leaves were ground in liquid nitrogen. Leaf powders were mixed with 80% ethanol and then incubated at 60°C for 20 min. After centrifugation at 4,000 rpm/min for 5 min, the supernatant was discarded. The samples were then resuspended in 3 ml of ddH2O and 2 ml of 9.2 M perchloric acid and incubated at 100°C for 10 min followed by centrifugation at 4,000 rpm/min for 10 min. This step was repeated three times and the supernatant was pooled. The 5 ml of anthrone reagent was added to a 0.1 ml aliquot of extract for glucose measurement. The intensity of the color formed was measured at 620 nm after heating on a boiling water bath for 10 min and rapidly cooled. The glucose concentration was estimated using a standard curve prepared from different glucose concentrations. Since 0.9 g starch yields approximately 1 g of glucose on hydrolysis, the conversion factor is 0.9 for the starch extract.
Total RNA Extraction and Quantitative RT-PCR Analyses
Total RNA was extracted from the 2nd leaf of six-leaf-stage tomato seedlings using the RNAprep pure Plant Kit (Tiangen). Then, quantitative real-time PCR (RT-qPCR) was carried out on a LightCycler480 machine (Roche Diagnostics, Switzerland) using SYBR Premix Go Taq by CFX96™ Real-Time System (Bio-Rad, United States). The relative expression level of genes was calculated using the formula 2–ΔΔCt and normalized to the amount of Actin mRNA detected in the same samples. At least three technical replicates for each of three biological replicates for each sample were performed in this study. All primers used for real-time PCR analysis were listed in Supplementary Table 1.
Deoxyribonucleic Acid Extraction and Whole-Genome Bisulfite Sequencing
Genomic DNA was isolated from the 2nd leaf of six-leaf-stage tomato seedlings harvested from WT or SlDRM7-RNAi lines using the DNeasy Plant Mini Kit (Qiagen). Two biological replicates for each sample were performed. About 100 ng genomic DNA spiked with 0.5 ng lambda DNA were fragmented by sonication to 200–300 bp with Covaris S220. These DNA fragments were treated with bisulfite using EZ DNA Methylation-Gold™ Kit (Zymo Research). The library was constructed by Novogene Corporation (Beijing, China), and sequenced on the Illumina Novaseq platform (United States). Image analysis and base calling were performed with Illumina CASAVA pipeline and finally generated 150-bp paired-end reads. The FastQC (fastqc_v0.11.5) was used to perform basic statistics on the quality of the raw reads, which were pre-processed through fastp (fastp 0.20.0). The remaining reads that passed all the filtering steps, counted as clean reads, were mapped to the reference tomato genome1 by BSMAP. The tomato genome fasta were obtained from Ensemble Plants2. The reference genome and clean reads were transformed into bisulfite-converted version (C-to-T and G-to-A converted) and then indexed using bowtie2 (Langmead and Salzberg, 2012). Sequence reads that produce a unique best alignment were then compared to the normal genomic sequence and the methylation state of all cytosine positions was inferred. The sequencing depth and coverage were summarized using deduplicated reads.
Methylation Analysis
Results of methylation extractor were transformed into bigWig format for visualization using IGV browser. The sodium bisulfite non-conversion rate was calculated as the percentage of cytosine sequenced at cytosine reference positions in the lambda genome. Methylated sites were identified with a binomial test using the methylated counts (mC), total counts (mC+unmC), and the non-conversion rate (r). Sites with FDR-corrected P value < 0.05 were considered methylated sites. To calculate the methylation level of the sequence, Methylation Level (ML) is defined as ML(C) = reads(mC)/[reads(mC)reads(C)].
Differentially methylated regions (DMRs) were identified using the DSS, which is a new dispersion shrinkage method for estimating the dispersion parameter. According to the distribution of DMRs through the genome, we defined the genes related to DMRs as genes whose gene body region (from TSS to TES) or promoter region (2 kb upstream from the TSS) have an overlap with the DMRs. P-values less than 0.05 were considered significantly enriched by DMR-related genes.
RNA-Seq and Data Analysis
Total RNA was isolated from the same leaf materials as for the DNA extraction. Three biological replicates for each sample were performed. Five micrograms of pooled RNA were used for the RNA-seq library using the Illumina Genome Analyzer (Solexa, United States). The sequencing data was filtered with SOAPnuke (v1.5.2). Low-quality reads were removed from the raw data, and high-quality reads were aligned to the tomato genome (version SL2.50; see text footnote 1). Differential expression analysis was performed using the DESeq2 (Love et al., 2014) with a corrected P-value (q value) < 0.05. To take insight to the change of phenotype, GO3 and KEGG4 enrichment analysis of annotated different expressed genes was performed by Phyper5 based on Hypergeometric test. The significant levels of terms and pathways were corrected by q value with a rigorous threshold (q value < 0.05) by Bonferroni.
Statistical Analysis
All data in this study were presented as mean ± standard deviation (SD). Student’s t-test or One-Way ANOVA followed by multiple comparison (Tukey’s HSD, P ≤ 0.05) was performed to analyze the significant difference between genotypes.
Results
SlDRM7 Is Essential for Tomato Leaf Development and Vegetative Growth
Through a gene-specific RNAi strategy (Figure 1A), we generated two independent tomato SlDRM7-RNAi lines drm7i-1 and drm7i-2. Compared with WT, SlDRM7-RNAi resulted in leaf interveinal chlorosis and senescence in T0 and T1 to T8 seedlings of the two independent drm7i lines (Figures 1B–I). The chlorotic phenotype along with antibiotic resistance exhibited Mendelian segregation, displaying approx. 2:1 ratio of chlorotic to normal green leaves among viable seedlings in T1, and continuing to segregate in T2 to T8 generations of both lines, while seedlings without the transgene after segregation in T1 to T8 reversed to green and were sensitive to antibiotic selection (Figure 1B). Intriguingly, no homozygous transgenic plants were obtained for both SlDRM7-RNAi lines. These data indicate that the dominant leaf chlorotic phenotype is genetically linked with the presence of a single copy of the pRNAi-SlDRM7 transgene, presumably resulting from the RNAi-mediated specific suppression of SlDRM7 expression rather than non-specific off-target effect in heterozygous plants of the two SlDRM7-RNAi lines. Homozygous SlDRM7-RNAi lines may be lethal to survival (due to extreme senescence). Considering the critical role of SlDRM7 in de novo RdDM, it is possible that SlDRM7-RNAi may impose some epigenetic remodeling of its target genes that are required for proper leaf development, and such epigenetic remodeling to control leaf chlorosis is not trans-generationally heritable but relies on constant RNAi of SlDRM7.
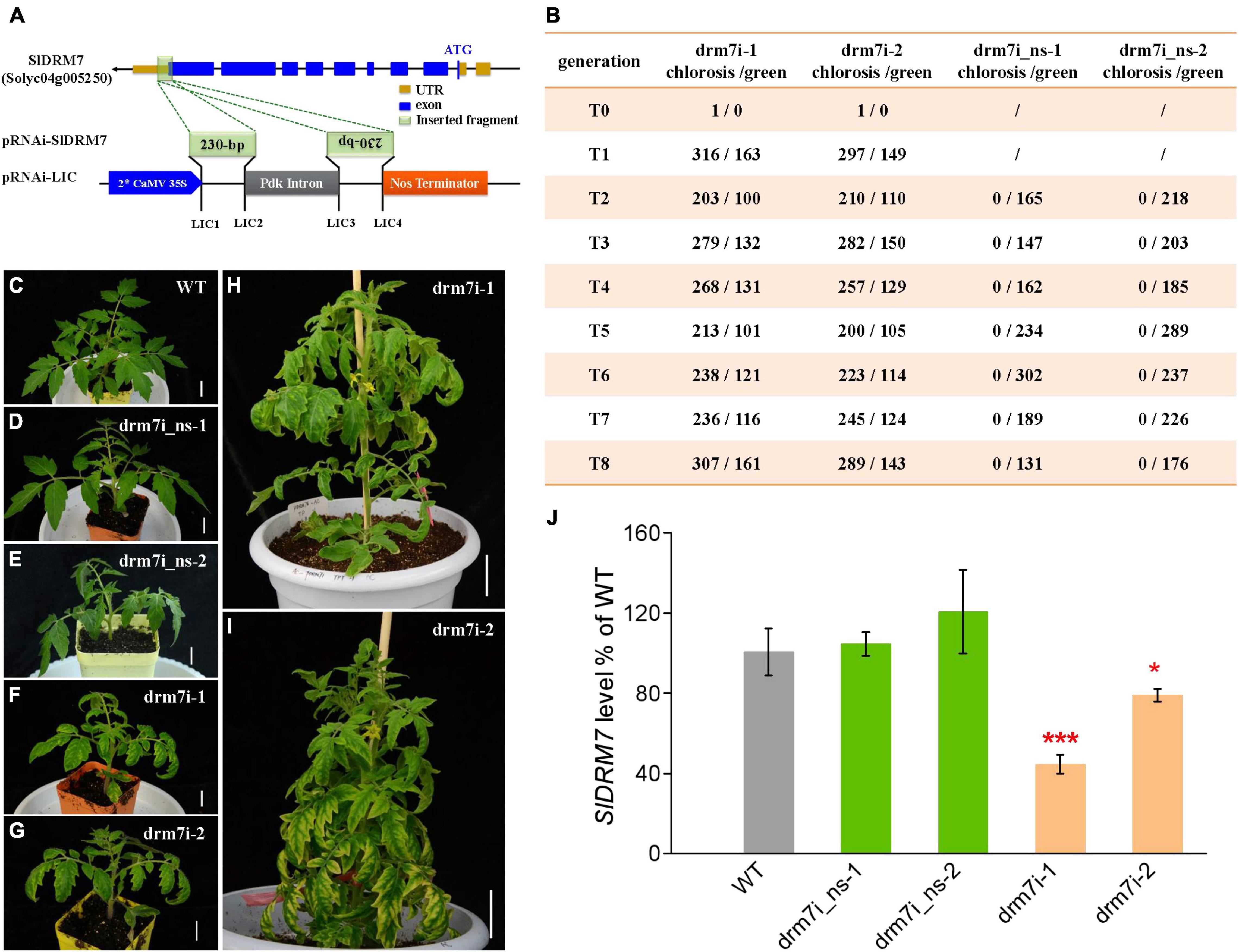
Figure 1. Generation and characterization of tomato SlDRM7-RNAi lines. (A) Schematic of the pRNAi-SlDRM7 construct. A 230-bp fragment of SlDRM7 (Solyc04g005250.2) was cloned into the pRNAi-LIC vector to generate pRNAi-SlDRM7. (B) Summary of segregation of leaf chlorosis/senescence phenotype in T0 to T8 generations of SlDRM7-RNAi lines. The numerator represents the number of progenies of SlDRM7-RNAi lines with maintained interveinal chlorosis/senescence leaves with kanamycin resistance, while the denominator represents the number of progenies with normal green leaves with kanamycin sensitivity. (C–I) Segregation of leaf chlorosis in SlDRM7-RNAi lines. wild-type (WT) seedlings display normal green leaves (C), while progenies of SlDRM7-RNAi lines display normal WT green leaves called drm7i_ns-1 (D), and drm7i_ns-2 (E), whereas progenies maintain interveinal chlorosis/senescence leaves called drm7i-1 (F,H) and drm7i-2 (G,I). Tomato seeds were spread and germinated directly in compost, and seedlings were photographed at 10 days after germination in (C–G) with bars = 1 cm, while seedlings were photographed at 6 weeks after germination in (H,I) with bars = 5 cm. (J) RT-qPCR analysis of the relative expression levels of SlDRM7 in leaves of WT and SlDRM7-RNAi lines at the six-leaf stage. Data are means ± SD of five biological replicates. Asterisks indicate the significant differences compared with WT (*P ≤ 0.05, ***P ≤ 0.001, one-way ANOVA, Tukey’s HSD). No difference with statistical significance was found for WT vs. drm7i_ns-1, and WT vs. drm7i_ns-2.
To investigate the genetic bridge between the genotype and phenotype in drm7i lines, we used drm7i_ns-1 and drm7i_ns-2 that were reverted to WT non-segregating (ns) green leaves after segregation as negative controls. Compared to WT and drm7i_ns lines, endogenous SlDMR7 expression level was dramatically reduced by approximately 30–60% in drm7i lines (Figure 1J). When the SlDRM7-RNAi affected vegetative leaf development, slight chlorosis started in the margin of the newly formed leaves of drm7i-1 during the 1st week transferred into a hydroponic culture, became more pronounced by the end of 2nd week, and gradually spread toward the interveinal regions, resulting in the characteristic phenotype of leaf interveinal chlorosis (Supplementary Figure 1A). We also generated SlDRM7-knockout (KO) lines where CRISPR/Cas9-induced gene editing resulted in nucleotide deletions of SlDRM7 (Supplementary Figure 1B). To our surprise, no defect in compound leaf development with chlorotic phenotype was observed in both KO lines vs. drm7i-1 (Supplementary Figures 1C–H). Such striking phenotypic differences between SlDRM7-RNAi and SlDRM7-KO lines are not unprecedented with genes essential for development and growth and are often associated with “transcriptional adaptation” or “genetic compensation response” (El-Brolosy et al., 2019; Ma Z.P. et al., 2019; Wang et al., 2020), which may be able to offset the effects of a completely dysfunctional SlDRM7 gene that has lost its capacity to express SlDRM7 protein in KO lines. To test this possibility, we analyzed the expression of SlDRM5, SlDRM6, and SlDRM8 in KO lines. Compared to SlDMR6 and SlDMR8, SlDRM5 was almost not expressed in tomato leaf. While the expression of SlDRM8 was not affected by the knockout of SlDRM7, that of SlDMR6 was significantly induced (Supplementary Figure 2).
In addition, SlDRM7-RNAi also affected the leaf size and plant growth at the vegetative stage (Supplementary Figure 3). At the six-leaf stage, we measured the leaf area of the first three compound leaves and found that the average lobule area of leaves was predominant in WT, followed by that in drm7i_ns lines, while the smallest in drm7i lines (Supplementary Figures 3A–C). When checking the shoot height of seedlings after 4-week hydroponic culture, we found that the average shoot height was about 16.84 (±2.18) cm in WT, 10.85 (±1.81) cm and 10.10 (±1.44) cm in drm7i_ns-1, and drm7i_ns-2, while only 7.72 (±0.99) cm and 8.63 (±0.99) cm in drm7i-1 and drm7i-2, respectively (Supplementary Figures 3D–F), consistent with the stunted plant growth exhibiting a dwarf and bushy phenotype in drm7i lines (Figures 1H,I). Phenotypically, the distinguishable differences in terms of leaf size and plant height between WT and drm7i_ns, as well as between drm7i_ns and drm7i, suggest that these phenotypic changes may result from SlDRM7-RNAi mediated epigenetic modification(s) that can be maintained in the absence of a pRNAi-SlDRM7 trigger.
Taken together, these results demonstrated that the SlDRM7-directed mechanism plays an essential role in tomato vegetative development, especially leaf development and senescence. In this work, using SlDRM7-RNAi lines drm7i-1 and drm7i-2 and the segregated lines with WT green leaves drm7i_ns-1 and drm7i_ns-2, we focus on understanding how SlDRM7 governs leaf chlorosis/senescence during vegetative growth.
SlDRM7 Modulates Leaf Senescence and the Expression of Senescence-Associated Genes
The leaf chlorotic phenotype prompted us to investigate the function of SlDRM7 in photosynthetic capacity physiologically. At the six-leaf stage, we measured chlorophyll auto-fluorescence, photosynthetic pigment content, and photosynthetic efficiency in the 2nd compound leaves. In yellowing leaf mesophylls (YLM), but not greening leaf mesophylls (GLM) of two drm7i lines, a significant decrease in chlorophyll auto-fluorescence intensity was observed (Figure 2A). Correspondingly, the content of chlorophyll a, chlorophyll b, and carotenoid were significantly lower in these two drm7i lines vs. WT or drm7i_ns lines (Figure 2B). Since there was no obvious difference of chlorophyll auto-fluorescence and photosynthetic pigment content among WT and two drm7i_ns lines (Figures 2A,B), the influence of SlDRM7-RNAi on photosynthetic capacity was studied by comparing drm7i lines with drm7i_ns-1. Based on the light and CO2 response curves (Figure 2C), the fitted maximum photosynthetic rate (Pn) of drm7i_ns-1 was 35.53 μmol ⋅ m–2 ⋅ s–1, whereas that of drm7i-1 and drm7i-2 were reduced to only 14.34 and 8.86 μmol ⋅ m–2 ⋅ s–1, respectively (Figure 2D). Similarly, a decreasing tendency of the respiration rate, stomatal conductance (Gs), and transpiration rate (Tr) were further observed in both drm7i lines (Figure 2D). These data suggest that photosynthesis capacity was inhibited in drm7i vs. drm7i_ns.
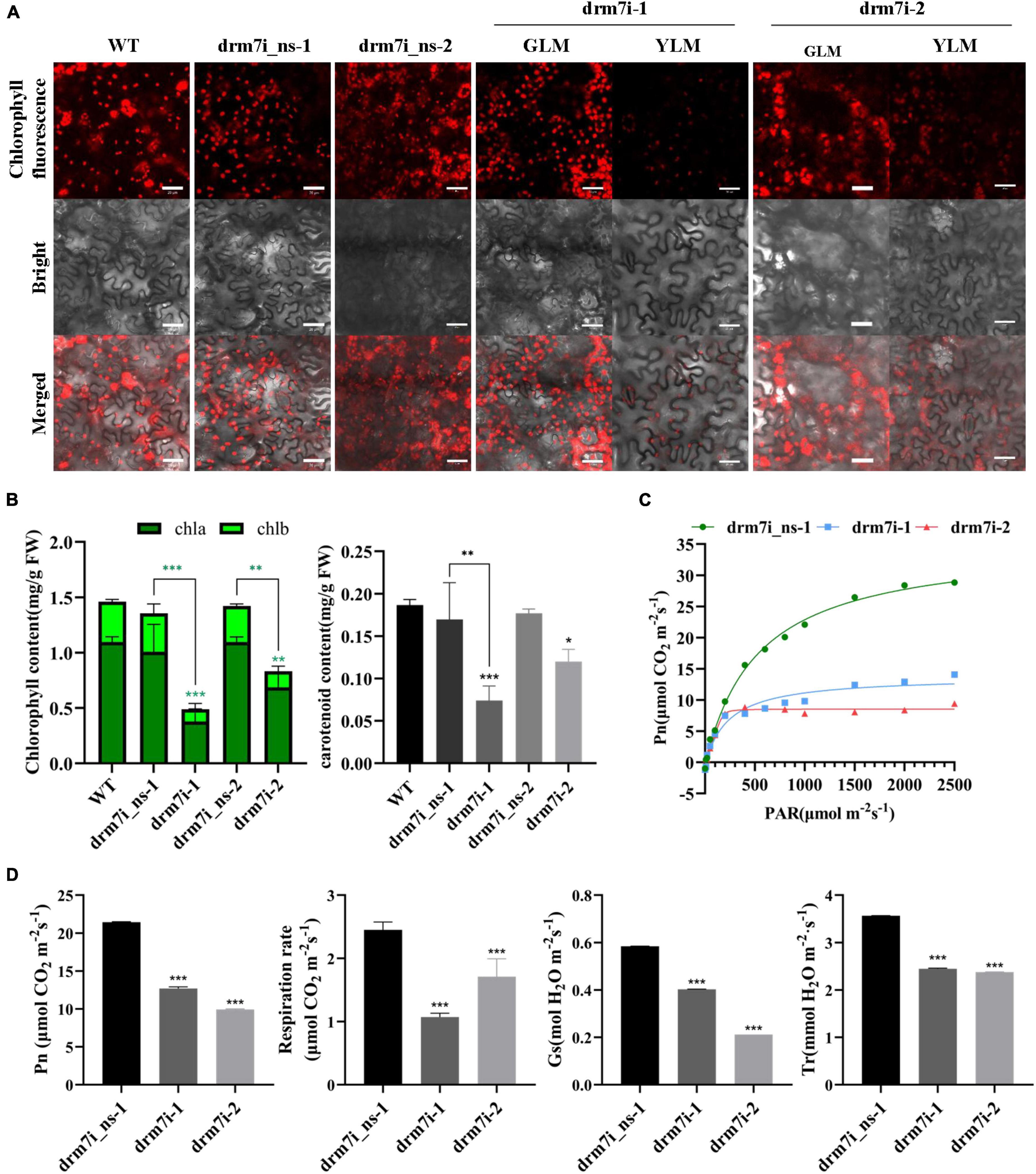
Figure 2. Measurement of photosynthetic pigment content and photosynthetic capacity. (A) The auto-fluorescence of chlorophyll in leaves of SlDRM7-RNAi lines. Strong chlorophyll auto-fluorescence intensity with almost no differences was observed in leaf Mesophyll cells from the 2nd compound leaves of wild-type AC (WT), two drm7i_ns lines, and the greening leaf Mesophyll (GLM) cells of two drm7i lines at the six-leaf stage, but much weaker in the yellowing leaf mesophyll (YLM) cells of two drm7i lines. WT, drm7i_ns-1 and drm7i_ns-2, drm7i-1 and drm7i-2 are shown. Bars = 20 μm. (B) The content of three photosynthetic pigments in leaf tissues of SlDRM7-RNAi lines. The content of chlorophyll a (chla) and chlorophyll b (chlb) (left panel), and carotenoid (right panel) was measured in the 2nd compound leaves of six-leaf-stage seedlings of WT, two drm7i_ns lines, and two drm7i lines, respectively. (C,D) Effect of SlDRM7-RNAi on photosynthesis. Light and CO2 response curves (C), as well as net photosynthetic rates (Pn), respiration rate, stomatal conductance (Gs), and transpiration rate (Tr) (D) of the 2nd compound leaves of six-leaf-stage seedlings of drm7i_ns-1 and two drm7i lines. Data are means ± SD (n = 3 in panel B, while n = 5 in panel D). Asterisks indicate the significant differences when compared with WT or drm7i_ns lines (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, one-way ANOVA, Tukey’s HSD).
The decreases in both chlorophyll content and photosynthetic efficiency led us to speculate whether the chlorosis of drm7i lines is associated with premature senescence. Therefore, we examined the expression of SAGs including SlSAG12, SlSAG13, SlSAG15, SENESCENCE-RELATED GENE1 (SlSRG1), senescence-related transcription factors (TFs) genes such as SlORE1S03, SlORE1S06, and SlNAP2, and GOLDEN2-like (SlGLK1) which is related to chlorophyll biosynthesis (Lira et al., 2017; Ma et al., 2018). Except for SlGLK1, the expression of the rest of the genes was upregulated in drm7i lines compared with WT or drm7i_ns lines (Figure 3). Furthermore, we tested the SlDRM7 expression during age-dependent and dark-induced senescence in WT plants and found that the transcript level of SlDRM7 was significantly upregulated in senescence leaves induced either naturally or darkly (Supplementary Figure 4). This finding seems contradictory to the genome-wide demethylation during plant senescence (Ogneva et al., 2016), suggesting a putative self-feedback pathway may be involved in regulating senescence by increasing SlDRM7 expression and a subsequently enhanced epi-control. Taken together, these findings reveal that SlDRM7 is required for proper vegetative growth, where SlDRM7 may work as a negative epi-regulator to repress the transcriptional expression of senescence-associated genes, leading to sustain a photosynthetic capacity and consequently inhibit the initiation of leaf senescence in tomatoes.
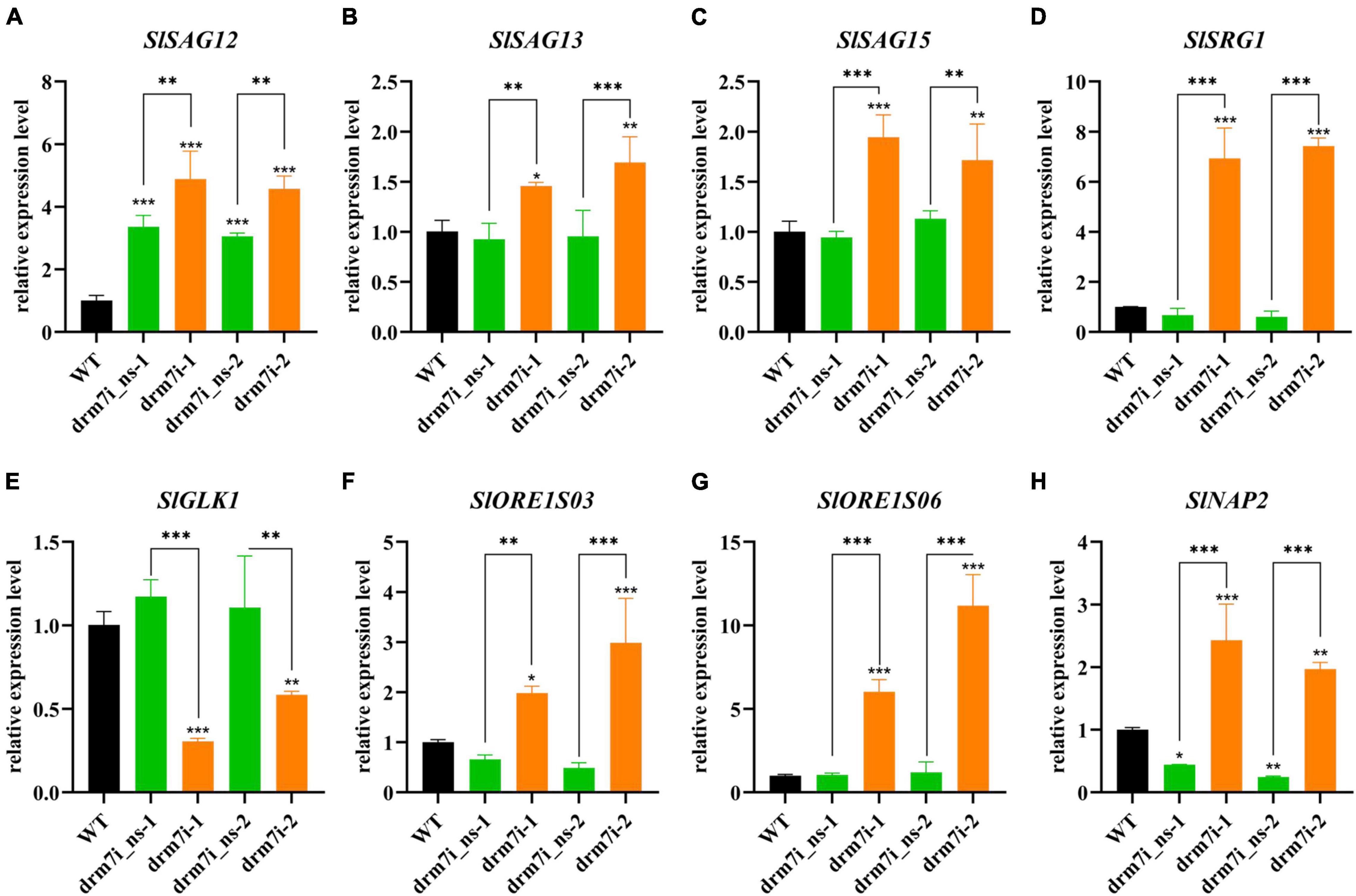
Figure 3. Expression analysis of senescence-associated genes in WT and SlDRM7-RNAi leaf tissues. (A–H) RT-qPCR analysis of the relative expression levels of SlSAG12 (A), SlSAG13 (B), SlSAG15 (C), SlSRG1 (D), SlGLK1 (E), SlORE1S03 (F), SlORE1S06 (G), SlNAP2 (H) in mature leaves of the WT, two drm7i_ns lines, and two drm7i lines, respectively. Data are means ± SD of three biological duplicates. Asterisks (above error bar represent the difference with WT; above the short line represent the difference with drm7i_ns lines) indicate the significant differences (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, one-way ANOVA, Tukey’s HSD).
Effect of SlDRM7 on Genome-Wide Methylome and Transcriptional Profiling
To elucidate how SlDRM7-RNAi regulates the leaf interveinal chlorosis/senescence, whole-genome bisulfite sequencing (WGBS) was performed on the 2nd leaf collected from WT, drm7i_ns-1, and drm7i-1 seedlings at the six-leaf stage. We observed that SlDRM7-RNAi did not cause any significant alterations in genome-wide mC (Figure 4A; Supplementary Tables 2, 3). However, upon evaluating methylation levels within gene body or promoter regions, we found genome-wide hypermethylation at CHG, CG, and CHH sites of drm7i-1 compared to WT and drm7i_ns-1 (Figure 4B), resulting in the production of a number of differentially methylated regions (DMRs) and differentially methylated genes (DMGs) in drm7i-1. A similar scenario where the increased whole-genome methylation caused by SlMET1 RNAi has been reported previously (Yao et al., 2020). Generally, most hypermethylated DMRs (hyper-DMRs) occurred at CHH sites, where mCHH-type hyper-DMRs/DMGs accounted for more than half (Figure 4C). Although mCHH-type DMRs were much more abundant than mCG-type or mCHG-type DRMs, their methylation levels were lowest and less influenced by SlDRM7 silencing (Figures 4D,E). In addition, when assessing the DMRs associated with different gene features of DMGs, there were 12 DMGs possessing DMRs in all three contexts located at gene body, and 4 DMGs located at promoters, respectively (Figure 4F).
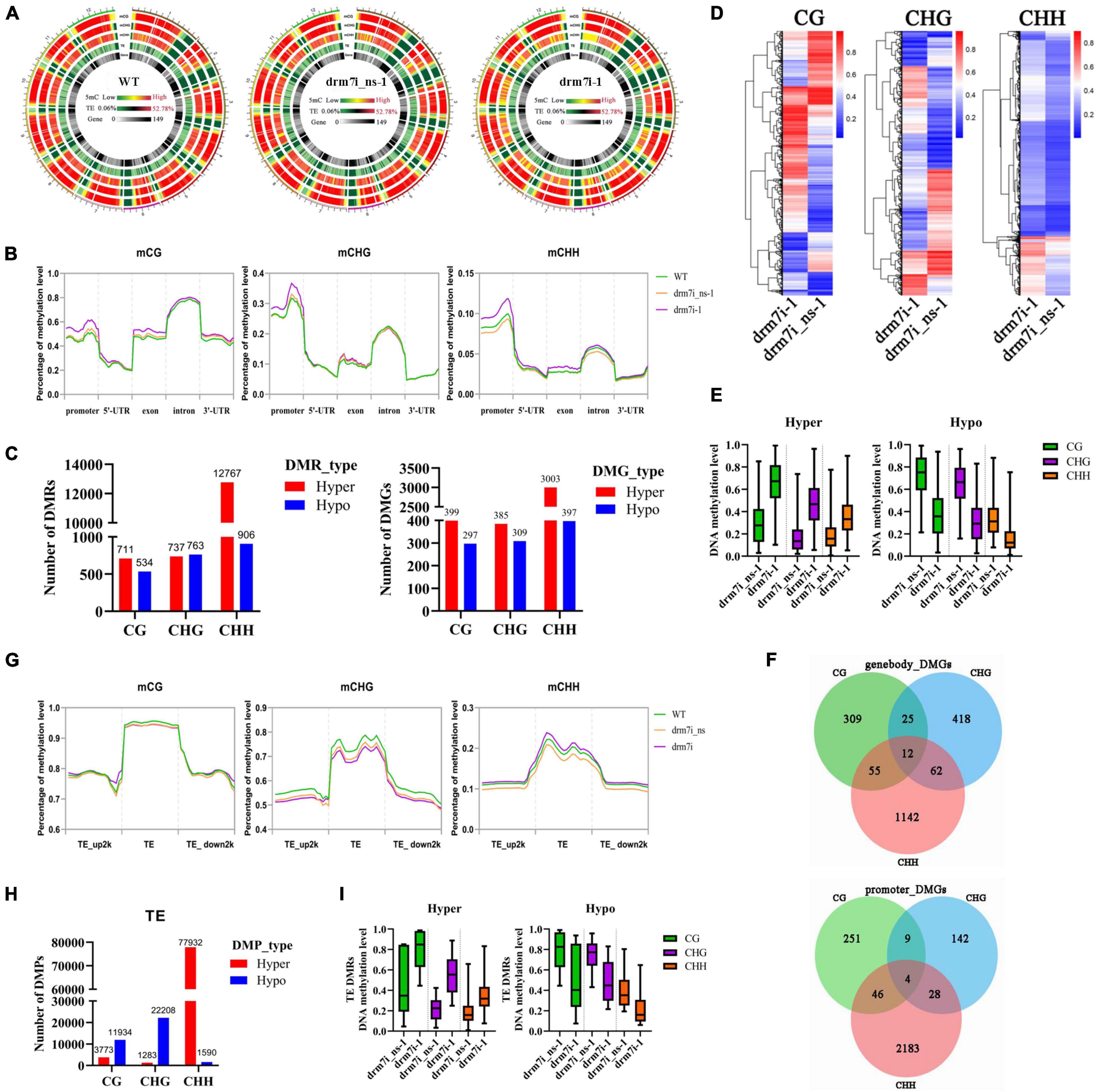
Figure 4. Effect of SlDRM7-RNAi on whole-genome DNA methylation. (A) Whole-genome bisulfite sequencing (WGBS) reveals DNA methylation profiles globally. Genome-wide methylations of 12 tomato chromosomes from WT, drm7i_ns-1, and drm7i-1 samples are illustrated in the mCG, mCHG, and mCHH contexts (where H is A, T, or C), and mC for TEs and genes. (B) Density plots of differential mCG, mCHG, and mCHH at the regions of the promoter, 5’-UTR, exon, intron, and 3’-UTR. Green, orange, and purple lines represent WT, drm7i_ns-1, and drm7i-1, respectively. (C) Numbers of differentially methylated regions (DMRs) and differentially methylated genes (DMGs) at CG, CHG, and CHH sites in drm7i vs. drm7i_ns. (D) Heat maps of the methylation level of DMRs at CG, CHG, and CHH sites in drm7i vs. drm7i_ns. (E) Overall DNA methylation level of hyper- and hypo-DMRs at CG, CHG, and CHH sites identified in drm7i_ns vs. drm7i. (F) Venn diagrams of DMGs associated with gene body (left) and promoter regions (right) in drm7i. (G) Density plots of differential mCG, mCHG, and mCHH at TEs and their 2-kb upstream and downstream regions. WT, drm7i_ns-1, and drm7i-1 are indicated. (H) Numbers of differentially methylated probes (DMPs) of TEs at CG, CHG, and CHH sites in drm7i-1 vs. drm7i_ns-1. (I) Methylation levels of hyper- and hypo-TEs in CG, CHG, and CHH sites.
Previous studies have suggested that hypermethylation of transposable elements (TEs) is probably responsible for the suppression of active transposons (Nuthikattu et al., 2013; Yang et al., 2019). We further analyzed methylation levels within Tes and their flanking regions. In general, mCG, mCHG, and mCHH all show higher levels in TEs than in both 2-kb upstream and downstream regions in the tomato genome (Figure 4G), consistent with the methylation patterns in Arabidopsis and rice genomes (Le et al., 2014; Zhang et al., 2015). However, by contrast with WT and drm7i_ns-1, mCHG and mCG decreased while mCHH increased within TEs of drm7i-1 (Figure 4G). Moreover, 118,720 differentially methylated probes (DMPs) were identified, of which the majority were preferentially hypermethylated in the CHH context, although greater differences of methylation levels were observed in mCG and mCHG rather than that in mCHH (Figures 4H,I).
To understand the biological functions of DMGs, Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. The GO enrichment showed that the hyper-DMGs were significantly enriched in terms of “extracellular region” and “catalytic activity,” which contained 37 and 565 DMGs, respectively (Supplementary Figure 5A). The KEGG pathway enrichment revealed that abundant hyper-DMGs were mainly assigned into five pathways related to “metabolic pathways,” “biosynthesis of secondary metabolites”, “starch and sucrose metabolism,” “linoleic acid metabolism,” and “phenylpropanoid biosynthesis,” containing 166, 94, 26, 22, 6, and 22 DMGs, respectively (Supplementary Figure 5B). For hypo-DMGs, neither GO nor KEGG enriched any terms significantly (Supplementary Figures 5C,D).
To further investigate whether SlDRM7-mediated DMRs affect gene expression, RNA-seq was performed using the same leaf samples for WGBS. From the comparative “drm7i-1 vs. drm7i_ns-1” transcriptomes, 709 and 968 differentially expressed genes (DEGs) were identified to be up- and downregulated, respectively (Supplementary Figure 6A). However, there were only 289 unique DEGs with either mCG, mCHG, or mCHH-type DMRs (Figure 5A), which were designed as meth-DEGs thereafter. Among those meth-DEGs possessing both hyper- and hypo-DMRs, 22 were upregulated, while the other 3 are downregulated (Figure 5B). Most DMRs of meth-DEGs occurred in the CHH context, except that hypo-DMRs of upregulated meth-DEGs dominated in the CG context (Figure 5C). Indeed, hyper-DMRs of upregulated meth-DEGs preferentially occurred in promoters, while others were predominantly in the gene body (Figure 5D). Apart from mCHH with a moderate increase in drm7i-1, the methylation levels of upregulated meth-DEGs in CG and CHG contexts reduced 22 and 31%, respectively (Figure 5E). These results establish that different meth-DEGs may involve in different epi-regulatory modes mediated by silencing of SlDRM7. It is worthwhile noting that our comparative transcriptional profiling does not reveal any off-target effect on the expression of genes that may be related to SlDRM7 in these RNAi lines.
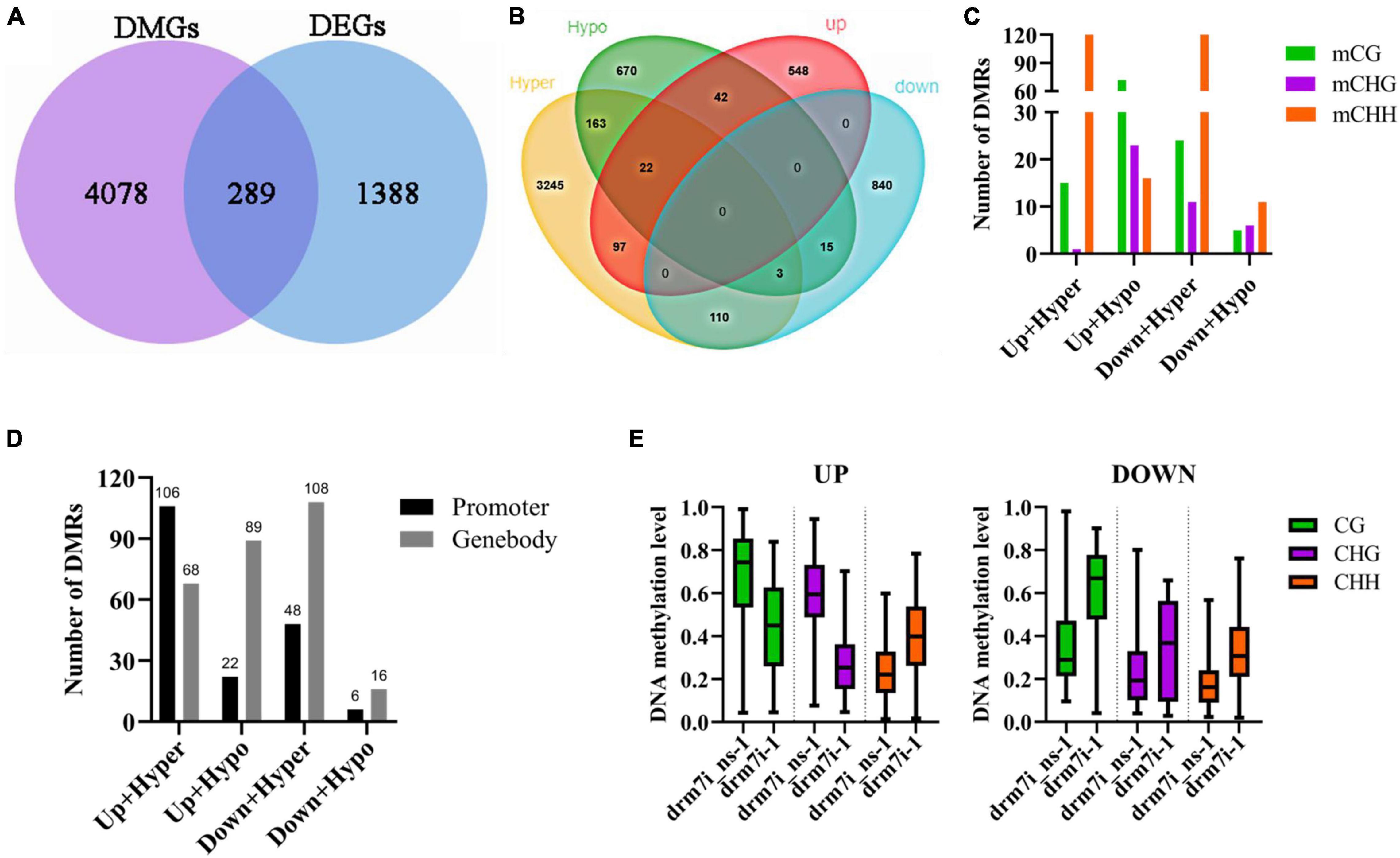
Figure 5. Combinational analysis of differential DNA methylated genes (DMGs) and differentially expressed genes (DEGs). (A) Venn diagrams of DMGs and DEGs (fold change > 2, FDR < 0.05, and drm7i-1 or drm7i_ns-1 FPKM > 1). (B) Venn diagrams of hypermethylated/hypomethylated genes and up-/downregulated genes. (C) Numbers of meth-DMGs in CG, CHG, and CHH contexts. (D) Numbers of meth-DMGs with DMRs located at promoters or gene bodies. (E) Overall DNA methylation levels of meth-DEGs in drm7i-1 vs. drm7i_ns-1.
SlDRM7 Epi-Controls Gene Expression Related to Photosynthesis and Chlorophyll Metabolism
To explore which biological processes the above meth-DEGs participate in, using GO enrichment analysis, the above 289 meth-DEGs were implied in “beta-glucosidase activity,” “photosystem II,” “glucosidase activity,” and “oxidoreductase activity” (Supplementary Figure 6B; Supplementary Table 4). Exhilaratingly, 128 downregulated meth-DEGs were significantly enriched in 20 putative pathways, most of which were linked to “chloroplast-related cellular component metabolism or photosynthesis” (Figure 6A; Supplementary Table 5), and 16 candidates out of them were further screened for RT-qPCR analysis. Consistent with RNA-seq data (Supplementary Table 5), the transcript levels of these genes were significantly depressed in drm7i-1 compared with drm7i_ns-1 or WT (Figure 6B). Apart from Solyc03g096850 that encodes a mitochondrial out membrane transporter protein presenting hypomethylation at CHH sites within its promoter, the rest of 15 meth-DEGs possessed hypermethylation at either CG, CHG, or CHH sites within promotors or gene body regions (Figure 6C).
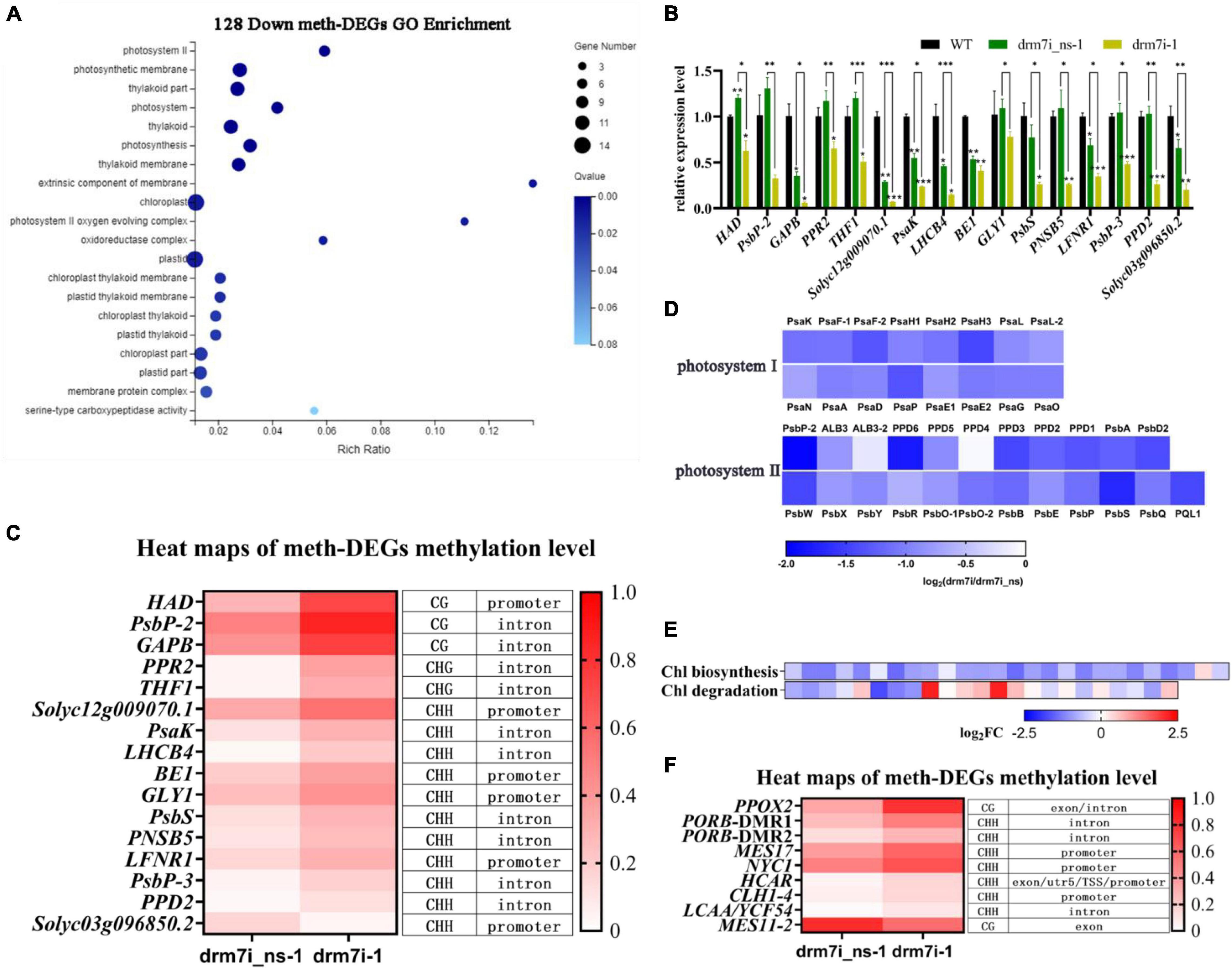
Figure 6. Analysis of down-regulated meth-DEGs. (A) Gene ontology enrichment analysis of 128 downregulated meth-DEGs. The size of the circle represents gene numbers, and the color represents the FDR. (B) Validation of RNA-seq using RT-qPCR for meth-DEGs involved in photosynthetic or chloroplast-related terms in (A). Data are shown as means ± SD of three biological duplicates. Asterisks (above error bar represent the difference with WT; above the short line represent the difference with drm7i_ns) indicate the significant differences (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, one-way ANOVA, Tukey’s HSD). (C) Heat map illustration of the methylation level of meth-DEGs shown in (B). (D) Heat map display of gene expression in the photosynthesis light reactions. Gene expression was determined by RNA-seq. (E) Heat map of expression of genes related to chlorophyll biosynthesis and chlorophyll degradation. Gene expression was determined by RNA-seq. (F) Heat map of the methylation levels of meth-DEGs in (E).
It was known that SlPSKA, SlLHCB4, SlPsbP, SlPsbP-2, SlPsbP-3, and Solyc10g047410.1 encode photosystem family proteins to form core complexes as well as light-harvesting complexes (LHCI and LHCII). The light reaction is catalyzed by two photosystems, photosystem I (PS I), and photosystem II (PS II), where the light energy is harvested to drive the transfer of electrons from water, via a series of electron donors and acceptors, to the final acceptor NADP+, which is finally reduced to NADPH (Nelson and Yocum, 2006; Jarvi et al., 2015). By multiple sequences alignment with those encoding core complexes of light reactions in Arabidopsis, a total of 88 tomato orthologous were identified, of which 42 genes were expressed in leaves (WT FPKM > 1) (Supplementary Table 6). Compared with drm7i_ns-1, the transcriptional level of 19 genes in drm7i-1 was significantly inhibited (Figure 6D; Supplementary Table 6). Since a set of LHCI and LHCII subunits binds to the core complex to capture the light and to bound chlorophylls (Li et al., 2000; Scheller et al., 2001; Jarvi et al., 2015), we also found 32 genes encoding LHC proteins in tomato. Compared with WT, the expression of these LHC genes were significantly repressed in drm7i-1, except for Solyc05g056080.2 and Solyc05g056060.2 (Supplementary Figure 6C). Moreover, 3 out of 32 genes, i.e., Solyc08g067320.1, Solyc07g063600.2, and Solyc09g014520.2, were meth-DEGs with mCHH-type hyper-DMRs located at promotor or intron regions (Supplementary Figure 6D).
Considering that chlorophyll content decreased in drm7i leaves (Figure 2B, left panel), we further investigated whether candidates involved in chlorophyll biosynthesis and/or degradation could be identified as a meth-DEG. By screening a tomato orthologous that is linked to chlorophyll metabolism as previously indicated in Arabidopsis (Kobayashi and Masuda, 2016; Lin et al., 2016) (Supplementary Table 7), expression levels of most genes involved in chlorophyll biosynthesis were found to be significantly decreased in drm7i-1 (Figure 6E; Supplementary Figure 6E), where SlPPOX2 and SlPORB were meth-DEGs (Figure 6F). On the other hand, another 6 meth-DEGs were observed to be related to chlorophyll degradation (Figure 6F). Nevertheless, unlike DEGs associated with chlorophyll biosynthesis, no obvious variation trend was observed in the expression of DEGs linked to chlorophyll degradation, among which some were unchanged, some were downregulated, and some were upregulated (Figure 6E; Supplementary Table 7). These findings imply that the inhibition of chlorophyll biosynthesis, rather than acceleration of chlorophyll degradation, might be important to induce chlorophyll loss, leading to the subsequent formation of chlorosis in drm7i leaves.
Silencing of SlDRM7 Blocked Starch Degradation and Caused Chloroplast Dysfunction
The comparative transcriptomes revealed that 59 unique DEGs in drm7i lines were linked to terms of “starch and sucrose metabolism,” the most significant category in the KEGG enrichment pathway (Supplementary Figure 7A). Starch is the transient storage photosynthate in many higher plants. During rapid growth, starch accumulates in the chloroplast in the daytime and degrades at night, thereby providing a steady supply of carbohydrates for organs to sustain metabolism and growth (Zeeman et al., 2002). Defects in starch turnover have been shown to impact plant growth in various species (Eimert et al., 1995; Corbesier et al., 1998; Harrison et al., 2000; Yu et al., 2001; Nashilevitz et al., 2009; Vriet et al., 2010). Therefore, we tracked diurnal variation of starch in leaves evidenced by Lugol’s staining. Notably, after growing in daylight for 12 h, WT, drm7i_ns-1, and dim7i-1 exhibited bright blue-purple color (Figure 7A, upper panel); however, after undergoing in dark for 12 h, the staining color of starch almost completely disappeared in WT and drm7i_ns-1, while remained substantially in drm7i-1, especially in leaf lamina where chlorosis developed (Figure 7A, bottom panel). This finding suggested that, in drm7i-1 leaves, starch degradation occurred in green regions near the vein but not in interveinal yellowing regions. Consistently, after 12-h treatment of daylight or dark, starch content in drm7i was significantly higher than that in WT or drm7i_ns, indicating that the starch degradation was inhibited in drm7i leaves (Figure 7B).
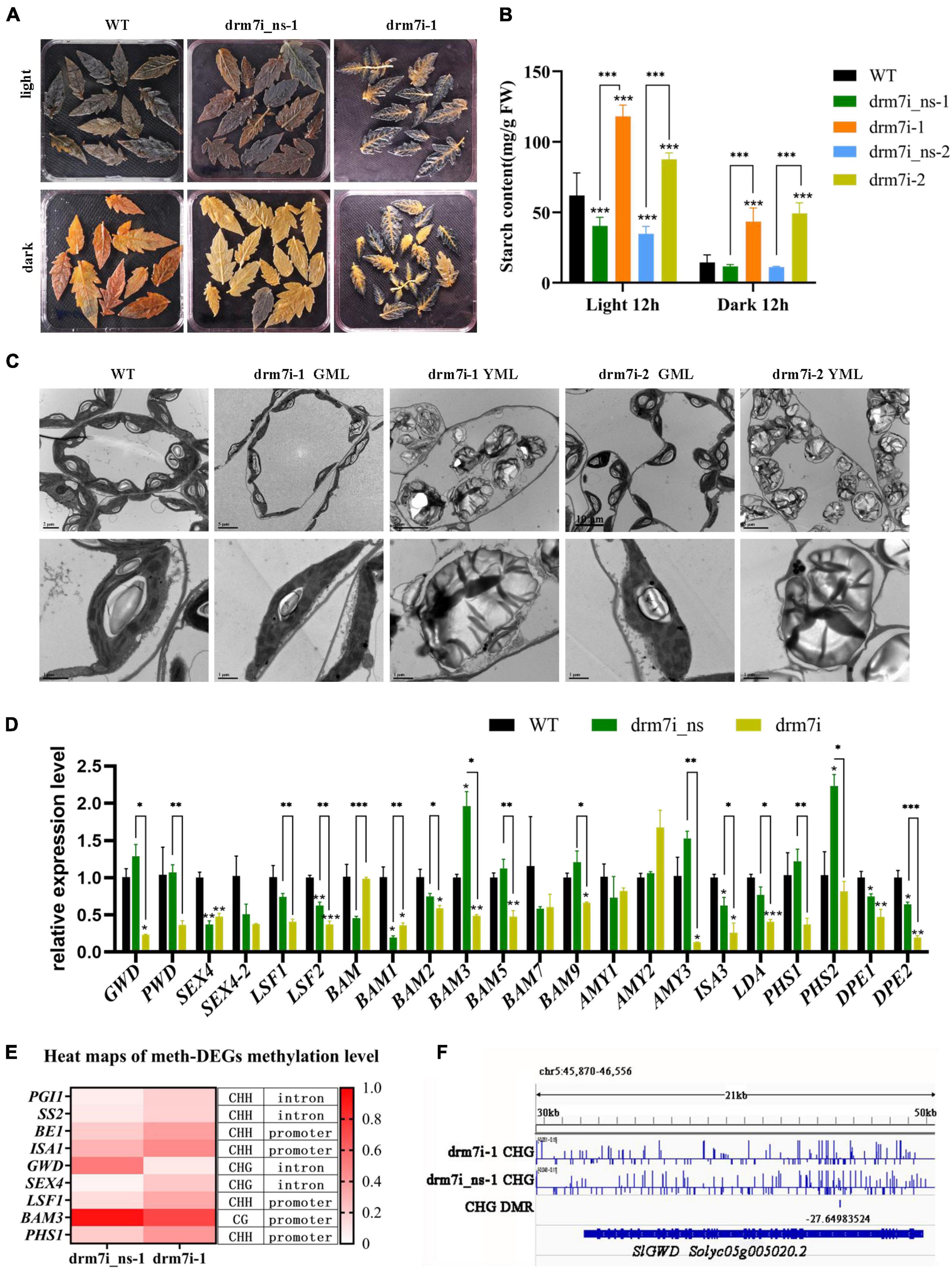
Figure 7. SlDRM7 functions in starch metabolism. (A,B) Iodine-iodide kalium staining (A) and measurement (B) of starch content of the leaves in a 12-h photoperiod. By harvesting at the end of daylight (light 12 h) and the end of the night (dark 12 h), leaves at the same position of the drm7i_ns-1 and drm7i-1 lines at the six-leaf stage were stained with Lugol’s solution to visualize starch, and then the starch content was measured physiologically. (C) TEM examination of chloroplast structures. Leaf mesophyll cells were collected from the 2nd leaves of AC, two SlDRM7-RNAi lines drm7i-1 and drm7i-2 at the six-leaf stage. The Upper and lower panels show a group of or individual chloroplasts. Bars are inserted as indicated. S, starch granule; OG, osmiophilic plastoglobuli; GL, Grana lamella; SL, stroma lamella. (D) Relative expression of genes related to starch degradation in WT, drm7i_ns-1, and drm7i-1. Data are represented as means ± SD of three biological duplicates. Asterisks (above error bar represent the difference with WT; above the short line represent the difference with drm7i_ns) indicate the significant differences (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, one-way ANOVA, Tukey’s HSD). (E) Heat map of the methylation levels of meth-DEGs involved in starch metabolism. (F) Detailed DNA methylation profile at the SlGWD (Solyc05g005020.2) loci. Differential DNA methylation is seen in the drm7i vs. drm7i_ns. A screenshot from the Integrative Genomics Viewer (IGV) display of whole-genome bisulfite sequencing data is shown. Vertical bars on each track indicate DNA methylation levels. Red boxes represent the DMRs.
In severe starch excess (sex) mutant, whole-chloroplast degradation is due to the highly accumulated starch caused by an imbalance between the daytime excessive starch synthesis and night-time limited breakdown (Stettler et al., 2009), which prompted us to examine the cellular ultrastructure of mesophyll cells in drm7i-1 leaves using transmission electron microscopy (TEM). As shown in Figure 7C, when compared with the chloroplasts in the leaves of WT, chloroplasts in GLM of drm7i lines resembled WT, but often contained grana lamella with a slightly disorganized appearance. However, it was noteworthy that in YLM of drm7i lines, highly misshapen chloroplasts were observed, in which grana and stroma thylakoids were completely disrupted, leaving an aberrant accretion of starch granules that appeared as swollen. Interestingly, the chloroplast degradation that occurred in YLM of drm7i lines was distinguishably different from that occurred in naturally senescent WT leaves, where thylakoid membrane system and starch granules were normally degraded (Supplementary Figure 7B), suggesting a linkage between starch accumulation and chloroplast homeostasis in SlDRM7-mediated leaf chlorosis/senescence. Taken together, our findings demonstrate that silencing of SlDRM7 led to an excessive accumulation of starch in chloroplasts that causes chloroplast dysfunction, resulting in tomato leaf chlorosis/senescence, a similar phenotype in sex mutants.
To elucidate the possible cause(s) underlying the sex-like phenotype of drm7i lines, genes related to starch metabolism and sugar transporter were chosen from the tomato genome (Supplementary Table 8). The RT-qPCR analysis showed that, compared with WT and drm7i_ns-1, some starch biosynthesis-associated genes were downregulated, but none was upregulated in drm7i-1 (Supplementary Figure 7C), despite 4 of them were hyper-DMGs at CHH sites (Figure 7E), indicating that the sex-like phenotype of drm7i lines did not result from the acceleration of starch biosynthesis. Besides, expression of most starch degradation-associated genes was repressed in drm7i-1 (Figure 7D), including 4 meth-DEGs, i.e., SlGWD, SlLSF1, SlBAM3, and SlPHS1, which encode glucan water dikinase, phosphoglucan phosphatase, beta-amylase-3, and alpha-1, 4 glucan phosphorylase isozyme, respectively. The methylation levels of SlGWD and SlBAM3 were decreased by 42.6% in the CHG context located at intron regions, and 20.4% in CG context located at promoter regions, respectively (Figure 7F). Both SlLSF1 and SlPHS1 had hypermethylation at CHH sites (approximately 20%) within promoter regions (Figure 7E). It is worth noting that SlGWD is a key enzyme in controlling the phosphate content of starch and that Arabidopsis gwd mutant exhibits the most severe sex phenotype (Yu et al., 2001). These data suggested that the suppression of starch degradation is a key factor for starch accumulation and is closely linked with leaf chlorosis mediated by SlDRM7-silencing-induced epi-regulation.
In addition, we found a decreased expression of three plastidic phosphate translocator genes and two sugar transporter genes in drm7i-1 vs. WT or drm7i_ns-1, including glucose-6-phosphate/phosphate translocator (SlGPT1), phosphoenolpyruvate/phosphate translocator (SlPPT2), triose-phosphate/phosphate translocator (SlTPT), maltose transporter (SlMEX1), and glucose transporter (SlGLT1). These data suggested that the translocation between phosphate and photosynthates was repressed, resulting in growth retardation of drm7i lines (Supplementary Figure 7D). However, none of these genes possesses obvious DMRs, thereby, their expression may be unlikely affected directly by SlDRM7-mediated epigenetic control.
Discussion
SlDRM7 Impacts Tomato Methylome
Plant DRMs are not only required for de novo methylation but also act with CMT3 to maintain non-CG methylation (Cao and Jacobsen, 2002a; Cao et al., 2003). For instance, rice OsDRM2, Arabidopsis DRM2, and tobacco (Nicotiana tabacum) NtDRM1 have de novo methylation activity and a preference for non-CG methylation (Cao et al., 2003; Wada et al., 2003; Moritoh et al., 2012; Gouil and Baulcombe, 2016). Surprisingly, although no obvious changes in overall genome-wide methylation level were caused by SlDRM7-RNAi (Figure 4A), methylation levels of different gene features in drm7i lines, especially at promoter and exon regions, were different from that in WT and drm7i_ns lines (Figure 4B). In addition, the hypermethylation and hypomethylation levels of DMRs were equivalent (Figures 4E,I), which might be accountable for no significant alterations in whole-genome mC. While the majority of DMRs were preferentially hypermethylated in the CHH context, the magnitude of differential methylation was much greater for CG and CHG contexts (Figure 4D). Similarly, increased CHH methylation was observed in the Arabidopsis met1 mutant (Mathieu et al., 2007). One possible explanation is that CHH methylation might be a result of changes in DMLs expression that was affected by SlDRM7 (Supplementary Figure 8). It has been reported that CHH hypermethylation occurred when DNA demethylase (DMLs) was repressed (Liu and Lang, 2020).
SlDRM7 Affects Plant Growth and Development
We reveal that SlDRM7 is required for proper plant growth and development and, more importantly, for preventing leaf chlorosis and premature senescence (Figure 1; Supplementary Figures 1, 3). Although the function of genes involved in de novo DNA methylation and maintenance in plant growth and development have been reported previously, the interveinal chlorosis exhibited in tomato drm7i lines was never observed in other plant species. For example, rice osdrm2 disruption displayed pleiotropic developmental defects at both vegetative and reproductive stages including semi-dwarfed stature, reductions in tiller number, abnormal panicle, and spikelet morphology (Moritoh et al., 2012). Maize ortholog of DRM2 loss-of-function lines, dmt103, had developmental defects in the reproductive stage but no morphological phenotypes (Garcia-Aguilar et al., 2010). However, Arabidopsis drm1 drm2 mutant had WT phenotypes (Cao and Jacobsen, 2002b). The different effects of DRMs on plant development in different species may be ascribed to the distribution of repeats and TEs and different genome sizes or the abundance (He et al., 2011). We observed a higher degree of methylation within the TEs enrichment region than a gene body in the tomato genome (Figure 4G). Compared to Arabidopsis, crop genomes such as tomato, rice, and maize have not only more abundant TEs but also more genes with highly methylated TEs (Lang et al., 2017). In addition, given the more complex genome (∼900 Mb) of tomato in comparison with Arabidopsis genome (∼125 Mb) or rice (∼370 Mb), it is possible that the tomato SlDRMs play more intricate functions in growth and development.
SlDRM7-Mediated Epi-Control Influences Starch Metabolic Pathways
First, RNA-seq showed that a considerable proportion of genes related to starch degradation were downregulated in drm7i lines compared to drm7i_ns and WT plants (Figure 7D). Second, several genes were identified as meth-DEGs including SlGWD, suggesting that these genes could be regulated through SlDRM7-induced epi-control. In Arabidopsis, GWD was reported to be involved in the initiation of starch degradation, since gwd mutants had more severe sex phenotypes than mutations affecting steps downstream of GWD (Critchley et al., 2001; Lu and Sharkey, 2004). In the absence of GWD activity, starch degradation was impaired leading to a severe sex phenotype not only in Arabidopsis but also in potato (Solanum tuberosum) and Lotus japonicas (Lorberth et al., 1998; Yu et al., 2001; Nashilevitz et al., 2009; Garcia-Aguilar et al., 2010). Here, TEM analysis demonstrated that silencing of SlDRM7 resulted in excessive accumulation of starch granules in chloroplasts, which triggered severe sex phenotype in tomato leaves as well (Figure 7C). Additionally, pollen in tomato gwd mutant with a sex phenotype was associated with a reduction in pollen germination and caused a male gametophytic lethality (Nashilevitz et al., 2009), indicating that the deficiency of SlGWD affects both the vegetative and reproductive growth in tomatoes. Moreover, SlPWD and other genes acting downstream of SlGWD contributing to starch degradation were also suppressed in drm7i (Figure 7D), reflecting the key position of SlGWD to initiate the biodegradation of starch.
We further found that starch accumulation was accompanied by chloroplasts dysfunction in drm7i lines, indicating that starch metabolism is critical for chloroplast homeostasis (Figure 7C). In agreement with our supposition, Arabidopsis maltose excess 1 (mex1) mutant accumulated high levels of starch in chloroplasts and displays autophagy-like chloroplast degradation (Stettler et al., 2009). However, leaf senescence and senescence-related chlorophyll catabolism are not induced in mex1 (Stettler et al., 2009), whereas our results showed silencing of SlDRM7 reduced chlorophyll content and repressed expression of genes involved in the chlorophyll biosynthesis and subsequently induced leaf senescence (Figures 2, 7B). These findings indicated that, apart from starch metabolism, SlDRM7-induced epi-control has a wide range of effects on plant growth and development in tomatoes.
SlDRM7 Modulates Leaf Chlorosis and Senescence
Leaf chlorosis and senescence involve complex genetic programming and less understood epigenetic re-programming (Gan, 2003; Guo and Gan, 2005; Ay et al., 2014; Woo et al., 2019). In comparison to drm7i_ns-1, we found 289 genes, such as SlGWD, in the whole genome were meth-DEGs in drm7i-1 (Figure 5A). Whether SlDRM7 directly affects methylation levels at these specific sites or indirectly through other processes remains unknown. Also, it is unclear whether DNA methylation changes are directly related to gene expression regulation. In some cases, DNA methylation changes appear to be associated closely with the transcriptional control of specific loci in plants (Kinoshita and Seki, 2014; Yong-Villalobos et al., 2015; Feng et al., 2016). Here, we found that intron hypomethylation of SlGWD is related to the expression repression in SlDRM7i lines (Figure 7E). Gene body methylation can be related to transcriptional upregulation and has been suggested to protect genes from aberrant transcription caused by cryptic promoters (Zhang et al., 2006; Feng et al., 2016). On the other hand, a poor correlation between DNA methylation and gene expression has also been reported in Arabidopsis (Meng et al., 2016; Yen et al., 2017; Chen et al., 2018c; Fan et al., 2020). Indeed, there are a large number of DEGs including SAGs, TFs related to senescence, and genes involved in the photosynthetic system, chlorophyll metabolism, and Calvin cycle, whose methylation levels remain unchanged in SlDRM7i lines (Figure 3; Supplementary Tables 6–9).
NAC family proteins are found to act as regulators during leaf senescence (Woo et al., 2019), and the orthologs of Arabidopsis ORESARA1 (AtORE1), namely SlORE1S02, SlORE1S03, and SlORE1S06, have received special attention for encoding a master regulator of senescence initiation, which can induce senescence-related genes SlSAG12 and interact physically with and inactivates the chloroplast maintenance-related TF SlGLK1 (Garapati et al., 2015; Lira et al., 2017). The SlNAP2 has a complex role in establishing ABA homeostasis during leaf senescence (Ma et al., 2018; Ma X. et al., 2019). The decrease of Calvin cycle activity in chloroplasts leads to an increased generation of reactive oxygen species (ROS) in the respective organelle, which worked as signaling molecules involved in the regulation of senescence (Navabpour et al., 2003). Therefore, the pleiotropic phenotype in drm7i lines may be affected by multiple pathways, and there might be the indirect effects of SlDRM7-mediated epi-control on regulating the expression of many development-related genes.
It is found that global DNA demethylation occurs during Arabidopsis leaf senescence (Ogneva et al., 2016). However, SlDRM7 expression was induced in both natural senescence and dark-induced aging (Supplementary Figure 4). Therefore, tomatoes might have a feedback regulation mechanism to inhibit leaf senescence by increasing SlDRM7 expression and consequently affecting genome-wide DNA methylation levels, and SlDRM7 seems to be a necessary anti-senescence factor during the growth and development in tomatoes. Based on our results, we propose a regulatory model to illustrate the relationship between SlDRM7-mediated epi-control and leaf senescence (Figure 8). The SlDRM7 functions as an epi-regulator to modulate the expression of meth-DEGs to affect leaf development and senescence. Silencing of SlDRM7 leads to hypermethylation or hypomethylation within promoter or intron regions, which directly or indirectly suppresses a set of meth-DEGs involved in photosynthesis, chlorophyll biosynthesis, photosystem, and starch degradation. Subsequently, the inhibition of these genes reduces chlorophyll content and photosynthetic capacity and triggers chloroplast breakdown as well, which resulted in leaf chlorosis and senescence. Meanwhile, an unknown self-feedback regulatory pathway is established by activating SlDRM7 expression to make a balance between vegetative growth and senescence.
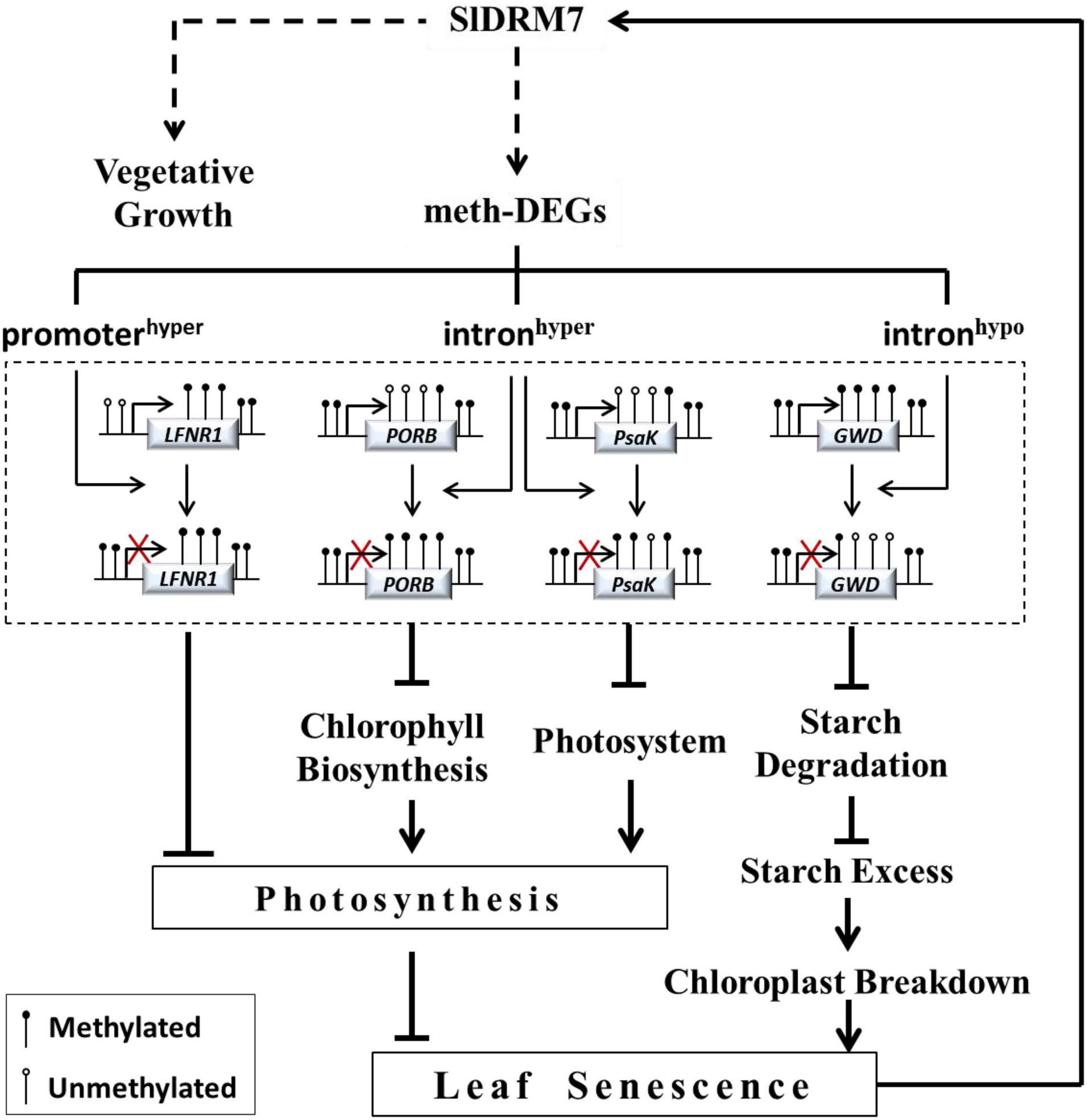
Figure 8. A model for SlDRM7 epi-controls leaf chlorosis and senescence. SlDRM7-mediated DMRs affect gene expression, which is designed as meth-DEGs. Silencing of SlDRM7 influences DNA methylation in promoter and gene body, and leads to transcriptional inhibition of genes directly or indirectly as exemplified by SlLFNR1, SlPORB, SlPsaK, and SlGWD. We hypothesized that these changes including promoter hypermethylation of SlLFNR1, intron hypermethylation of SlPORB and SlPsaK, and intron hypomethylation of SlGWD resulted in their expression repression, which inhibited photosynthesis and starch degradation, eventually leading to leaf chlorosis and senescence. Conversely, leaf senescence can induce SlDRM7, forming a feedback regulatory loop, to balance vegetative growth and senescence.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: All tomato materials, including wild-type Solanum lycopersicum cv. Ailsa Craig (AC), the SlDRM7-RNAi lines and SlDRM7-KO lines (AC background), are deposited in our lab. Sequence data can be found in NCBI (https://www.ncbi.nlm.nih.gov/) and Phytozome (https://phytozome-next.jgi.doe.gov/), two comparative platform for green plant genomics. Gene ID was shown in Supplementary Tables 1, 4–8. WGBS and RNA-seq data sets have been deposited in NCBI under Bioproject ID: PRJNA773102 and PRJNA772527, respectively.
Author Contributions
YW, WC, and JY designed and performed the experiments, analyzed the data, and wrote the manuscript. YW and LH performed the bioinformatics analyses. HZ, JW, GH, and RH cultured the plants and performed the experiments. SZ was involved in data analysis and helped writing the manuscript. YH, JY, and WC initiated the project, conceived the experiments, analyzed the data, and wrote the manuscript. All authors edited and finalized the manuscript.
Funding
This work was financially supported by grants from Zhejiang Provincial Natural Science Foundation (Z22C158590; 2021C02065; and LY19C150006), National Natural Science Foundation of China (NSFC 31601765 and 31872636), Ministry of Agriculture of the People’s Republic of China (National Transgenic Program of China 2016ZX08009001-004), Ministry of Science and Technology of China (National Key R&D Program 2017YFE0110900), China Postdoctoral Science Foundation (2019M652064), China Scholarship Council [(2016)3035], and Hangzhou Innovation Project for Returned Chinese Scholars.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Yuele Liu (Tsinghua University, China) for his kind gift of the pRNAi-LIC vector, and Qijun Chen (China Agricultural University, China) for his kind gift of gene editing vector induced by CRISPR-CAS9. We are deeply indebted to Junhua Kong (Chinese Academy of Sciences in Beijing, China) for her initial participation in this project. We are also grateful to Chaoqun Wu and Hui Wang for their technical supports.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.836015/full#supplementary-material
Footnotes
- ^ https://solgenomics.net
- ^ http://ftp.ensemblgenomes.org/pub/plants/current/fasta/solanum_lycopersicum/dna/
- ^ http://www.geneontology.org/
- ^ https://www.kegg.jp/
- ^ https://en.wikipedia.org/wiki/Hypergeometric_distribution
References
Arnon, D. I. (1949). Copper enzymes in isolated chloroplasts. Polyphenoloxidase in Beta vulgaris. Plant Physiol. 24, 1–15. doi: 10.1104/pp.24.1.1
Ay, N., Irmler, K., Fischer, A., Uhlemann, R., Reuter, G., and Humbeck, K. (2009). Epigenetic programming via histone methylation at WRKY53 controls leaf senescence in Arabidopsis thaliana. Plant J. 58, 333–346. doi: 10.1111/j.0960-7412.2009.03782.x
Ay, N., Janack, B., and Humbeck, K. (2014). Epigenetic control of plant senescence and linked processes. J. Exp. Bot. 65, 3875–3887. doi: 10.1093/jxb/eru132
Balazadeh, S., Siddiqui, H., Allu, A. D., Matallana-Ramirez, L. P., Caldana, C., Mehrnia, M., et al. (2010). A gene regulatory network controlled by the NAC transcription factor ANAC092/AtNAC2/ORE1 during salt-promoted senescence. Plant J. 62, 250–264. doi: 10.1111/j.1365-313X.2010.04151.x
Cao, X. F., Aufsatz, W., Zilberman, D., Mette, M. F., Huang, M. S., Matzke, M., et al. (2003). Role of the DRM and CMT3 Methyltransferases in RNA-directed DNA methylation. Curr. Biol. 13, 2212–2217. doi: 10.1016/s0960-9822(03)00908-4
Cao, X. F., and Jacobsen, S. E. (2002a). Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. U.S.A. 99, 16491–16498. doi: 10.1073/pnas.162371599
Cao, X. F., and Jacobsen, S. E. (2002b). Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 12, 1138–1144. doi: 10.1016/s0960-9822(02)00925-9
Caspar, T., Huber, S. C., and Somerville, C. (1985). Alterations in Growth, Photosynthesis, and Respiration in a Starchless Mutant of Arabidopsis thaliana (L.) Deficient in Chloroplast Phosphoglucomutase Activity. Plant Physiol. 79, 11–17. doi: 10.1104/pp.79.1.11
Chan, S. W. L., Henderson, I. R., and Jacobsen, S. E. (2005). Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 6, 351–360. doi: 10.1038/nrg1601
Chen, W. W., Jin, J. F., Lou, H. Q., Liu, L., Kochian, L. V., and Yang, J. L. (2018a). LeSPL-CNR negatively regulates Cd acquisition through repressing nitrate reductase-mediated nitric oxide production in tomato. Planta 248, 893–907. doi: 10.1007/s00425-018-2949-z
Chen, W. W., Zhang, X., Fan, Y. Y., Li, B., Ryabov, E., Shi, N. N., et al. (2018b). A genetic network for systemic RNA silencing in plants. Plant Physiol. 176, 2700–2719. doi: 10.1104/pp.17.01828
Chen, X. C., Schoenberger, B. S., Menz, J., and Ludewig, U. (2018c). Plasticity of DNA methylation and gene expression under zinc deficiency in Arabidopsis roots. Plant Cell Physiol. 59, 1790–1802. doi: 10.1093/pcp/pcy100
Chen, X. S., Lu, L., Mayer, K. S., Scalf, M., Qian, S. M., Lomax, A., et al. (2016). POWERDRESS interacts with HISTONE DEACETYLASE 9 to promote aging in Arabidopsis. eLife 5:e17214. doi: 10.7554/eLife.17214
Chinnusamy, V., and Zhu, J. K. (2009). Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 12, 133–139. doi: 10.1016/j.pbi.2008.12.006
Clegg, K. M. (1956). The application of the anthrone reagent to the estimation of starch in cereals. J. Sci. Food Agric. 7, 40–44. doi: 10.1002/jsfa.2740070108
Corbesier, L., Lejeune, P., and Bernier, G. (1998). The role of carbohydrates in the induction of flowering in Arabidopsis thaliana: comparison between the wild type and a starchless mutant. Planta 206, 131–137. doi: 10.1007/s004250050383
Critchley, J. H., Zeeman, S. C., Takaha, T., Smith, A. M., and Smith, S. M. (2001). A critical role for disproportionating enzyme in starch breakdown is revealed by a knock-out mutation in Arabidopsis. Plant J. 26, 89–100. doi: 10.1046/j.1365-313x.2001.01012.x
Dobrev, P. I., and Kaminek, M. (2002). Fast and efficient separation of cytokinins from auxin and abscisic acid and their purification using mixed-mode solid-phase extraction. J. Chromatogr. A. 950, 21–29. doi: 10.1016/s0021-9673(02)00024-9
Dowen, R. H., Pelizzola, M., Schmitz, R. J., Lister, R., Dowen, J. M., Nery, J. R., et al. (2012). Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. U.S.A. 109, E2183–E2191. doi: 10.1073/pnas.1209329109
Eimert, K., Wang, S. M., Lue, W. L., and Chen, J. C. (1995). Monogenic recessive mutations causing both late floral initiation and excess starch accumulation in Arabidopsis. Plant Cell 7, 1703–1712. doi: 10.2307/3870031
El-Brolosy, M. A., Kontarakis, Z., Rossi, A., Kuenne, C., Gunther, S., Fukuda, N., et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. doi: 10.1038/s41586-019-1064-z
Fan, S. K., Ye, J. Y., Zhang, L. L., Chen, H. S., Zhang, H. H., Zhu, Y. X., et al. (2020). Inhibition of DNA demethylation enhances plant tolerance to cadmium toxicity by improving iron nutrition. Plant Cell Environ. 43, 275–291.
Feng, S. J., Liu, X. S., Tao, H., Tan, S. K., Chu, S. S., Oono, Y., et al. (2016). Variation of DNA methylation patterns associated with gene expression in rice (Oryza sativa) exposed to cadmium. Plant Cell Environ. 39, 2629–2649.
Finnegan, E. J., and Dennis, E. S. (1993). Isolation and identification by sequence homology of a putative cytosine methyltransferase from Arabidopsis thaliana. Nucleic Acids Res. 21, 2383–2388. doi: 10.1093/nar/21.10.2383
Finnegan, E. J., and Kovac, K. A. (2000). Plant DNA methyltransferases. Plant Mol. Biol. 43, 189–201. doi: 10.1023/a:1006427226972
Finnegan, E. J., Peacock, W. J., and Dennis, E. S. (1996). Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. U.S.A. 93, 8449–8454. doi: 10.1073/pnas.93.16.8449
Finnegan, E. J., Peacock, W. J., and Dennis, E. S. (2000). DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev 10, 217–223.
Gan, S. S. (2003). Mitotic and postmitotic senescence in plants. Sci. Aging Knowledge Environ. 38:RE7. doi: 10.1126/sageke.2003.38.re7
Gan, S. S., and Amasino, R. M. (1997). Making sense of senescence - Molecular genetic regulation and manipulation of leaf senescence. Plant Physiol. 113, 313–319. doi: 10.1104/pp.113.2.313
Garapati, P., Xue, G. P., Munne-Bosch, S., and Balazadeh, S. (2015). Transcription Factor ATAF1 in Arabidopsis promotes senescence by direct regulation of key chloroplast maintenance and senescence transcriptional cascades. Plant Physiol. 168, 1122–1139. doi: 10.1104/pp.15.00567
Garcia-Aguilar, M., Michaud, C., Leblanc, O., and Grimanelli, D. (2010). Inactivation of a DNA methylation pathway in maize reproductive organs results in apomixis-like phenotypes. Plant Cell 22, 3249–3267. doi: 10.1105/tpc.109.072181
Gouil, Q., and Baulcombe, D. C. (2016). DNA methylation signatures of the plant chromomethyltransferases. PLoS Genet. 12:e1006526. doi: 10.1371/journal.pgen.1006526
Guo, Y. F., and Gan, S. S. (2005). Leaf senescence: signals, execution, and regulation. Curr. Top. Dev. Biol. 71, 83–112. doi: 10.1016/s0070-2153(05)71003-6
Harrison, C. J., Mould, R. M., Leech, M. J., Johnson, S. A., Turner, L., Schreck, S. L., et al. (2000). The rug3 locus of pea encodes plastidial phosphoglucomutase. Plant Physiol. 122, 1187–1192. doi: 10.1104/pp.122.4.1187
He, X. J., Chen, T. P., and Zhu, J. K. (2011). Regulation and function of DNA methylation in plants and animals. Cell Res. 21, 442–465. doi: 10.1038/cr.2011.23
Henderson, I. R., and Jacobsen, S. E. (2007). Epigenetic inheritance in plants. Nature 447, 418–424. doi: 10.1038/nature05917
Henikoff, S., and Comai, L. (1998). A DNA methyltransferase homolog with a chromodomain exists in multiple polymorphic forms in Arabidopsis. Genetics 149, 307–318. doi: 10.1093/genetics/149.1.307
Jackson, J. P., Lindroth, A. M., Cao, X. F., and Jacobsen, S. E. (2002). Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416, 556–560. doi: 10.1038/nature731
Jarvi, S., Suorsa, M., and Aro, E. M. (2015). Photosystem II repair in plant chloroplasts - Regulation, assisting proteins and shared components with photosystem II biogenesis. Biochim. Biophys. Acta 1847, 900–909. doi: 10.1016/j.bbabio.2015.01.006
Jin, J. F., Wang, Z. Q., He, Q. Y., Wang, J. Y., Li, P. F., Xu, J. M., et al. (2020). Genome-wide identification and expression analysis of the NAC transcription factor family in tomato (Solanum lycopersicum) during aluminum stress. BMC Genomics 21:14. doi: 10.1186/s12864-020-6689-7
Kankel, M. W., Ramsey, D. E., Stokes, T. L., Flowers, S. K., Haag, J. R., Jeddeloh, J. A., et al. (2003). Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163, 1109–1122. doi: 10.1093/genetics/163.3.1109
Kinoshita, T., and Seki, M. (2014). Epigenetic memory for stress response and adaptation in plants. Plant Cell Physiol. 55, 1859–1863.
Kit, A. H., Boureau, L. Stammitti-Bert, L., Rolin, D., Teyssier, E., and Gallusci, P. (2010). Functional analysis of SlEZ1 a tomato Enhancer of zeste (E(z)) gene demonstrates a role in flower development. Plant Mol. Biol. 74, 201–213. doi: 10.1007/s11103-010-9657-9
Kobayashi, K., and Masuda, T. (2016). Transcriptional Regulation of Tetrapyrrole Biosynthesis in Arabidopsis thaliana. Front. Plant Sci. 7:1811. doi: 10.3389/fpls.2016.01811
Lang, Z. B., Wang, Y. H., Tang, K., Tang, D. G., Datsenk, A. T., Cheng, J. F., et al. (2017). Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. U.S.A. 114, E4511–E4519. doi: 10.1073/pnas.1705233114
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Le, T. N., Schumann, U., Smith, N. A., Tiwari, S., Au, P. C. K., Zhu, Q. H., et al. (2014). DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 15:458. doi: 10.1186/s13059-014-0458-3
Lei, M. G., Zhang, H. M., Julian, R., Tang, K., Xie, S. J., and Zhu, J. K. (2015). Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 112, 3553–3557. doi: 10.1073/pnas.1502279112
Li, X. P., Bjorkman, O., Shih, C., Grossman, A. R., Rosenquist, M., Jansson, S., et al. (2000). A pigment-binding protein essential for regulation of photosynthetic light harvesting. Nature 403, 391–395. doi: 10.1038/35000131
Lim, P. O., Kim, H. J., and Nam, H. G. (2007a). Leaf senescence. Annu. Rev. Plant Biol. 58, 115–136. doi: 10.1146/annurev.arplant.57.032905.105316
Lim, P. O., Kim, Y., Breeze, E., Koo, J. C., Woo, H. R., Ryu, J. S., et al. (2007b). Overexpression of a chromatin architecture-controlling AT-hook protein extends leaf longevity and increases the post-harvest storage life of plants. Plant J. 52, 1140–1153. doi: 10.1111/j.1365-313X.2007.03317.x
Lin, Y. P., Wu, M. C., and Charng, Y. Y. (2016). Identification of a chlorophyll dephytylase involved in chlorophyll turnover in Arabidopsis. Plant Cell 28, 2974–2990. doi: 10.1105/tpc.16.00478
Lindroth, A. M., Cao, X. F., Jackson, J. P., Zilberman, D., McCallum, C. M., Henikoff, S., et al. (2001). Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292, 2077–2080. doi: 10.1126/science.1059745
Lira, B. S., Gramegna, G., Trench, B. A., Alves, F. R. R., Silva, E. M., Silva, G. F. F., et al. (2017). Manipulation of a senescence-associated gene improves fleshy fruit yield. Plant Physiol. 175, 77–91. doi: 10.1104/pp.17.00452
Liu, P., Zhang, S., Zhou, B., Luo, X., Zhou, X. F., Cai, B., et al. (2019). The Histone H3K4 Demethylase JMJ16 represses leaf senescence in Arabidopsis. Plant Cell 31, 430–443. doi: 10.1105/tpc.18.00693
Liu, R. E., and Lang, Z. B. (2020). The mechanism and function of active DNA demethylation in plants. J. Integr. Plant Biol. 62, 148–159. doi: 10.1111/jipb.12879
Lorberth, R., Ritte, G., Willmitzer, L., and Kossmann, J. (1998). Inhibition of a starch-granule-bound protein leads to modified starch and repression of cold sweetening. Nat. Biotechnol. 16, 473–477. doi: 10.1038/nbt0598-473
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, Y., and Sharkey, T. D. (2004). The role of amylomaltase in maltose metabolism in the cytosol of photosynthetic cells. Planta 218, 466–473. doi: 10.1007/s00425-003-1127-z
Luo, C., Hajkova, P., and Ecker, J. R. (2018). Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340. doi: 10.1126/science.aat6806
Ma, X., Balazadeh, S., and Mueller-Roeber, B. (2019). Tomato fruit ripening factor NOR controls leaf senescence. J. Exp. Bot. 70, 2727–2740. doi: 10.1093/jxb/erz098
Ma, X., Youjun, Z., Tureckova, V., Gang-Ping, X., Fernie, A. R., Mueller-Roeber, B., et al. (2018). The NAC transcription factor SlNAP2 regulates leaf senescence and fruit yield in tomato. Plant Physiol. 177, 1286–1302. doi: 10.1104/pp.18.00292
Ma, Z. P., Zhu, P. P., Shi, H., Guo, L. W., Zhang, Q. H., Chen, Y. N., et al. (2019). PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263. doi: 10.1038/s41586-019-1057-y
Mathieu, O., Reinders, J., Caikovski, M., Smathajitt, C., and Paszkowski, J. (2007). Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 130, 851–862. doi: 10.1016/j.cell.2007.07.007
Matzke, M. A., and Mosher, R. A. (2014). RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
Meng, D. Z., Dubin, M., Zhang, P., Osborne, E. J., Stegle, O., Clark, R. M., et al. (2016). Limited contribution of DNA methylation variation to expression regulation in Arabidopsis thaliana. PLoS Genet. 12:e1006141. doi: 10.1371/journal.pgen.1006141
Miao, Y., Laun, T., Zimmermann, P., and Zentgraf, U. (2004). Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol. Biol. 55, 853–867. doi: 10.1007/s11103-004-2142-6
Moritoh, S., Eun, C. H., Ono, A., Asao, H., Okano, Y., Yamaguchi, K., et al. (2012). Targeted disruption of an orthologue of DOMAINS REARRANGED METHYLASE 2, OsDRM2, impairs the growth of rice plants by abnormal DNA methylation. Plant J. 71, 85–98. doi: 10.1111/j.1365-313X.2012.04974.x
Nashilevitz, S., Melamed-Bessudo, C., Aharoni, A., Kossmann, J., Wolf, S., and Levy, A. A. (2009). The legwd mutant uncovers the role of starch phosphorylation in pollen development and germination in tomato. Plant J. 57, 1–13. doi: 10.1111/j.1365-313X.2008.03664.x
Navabpour, S., Morris, K., Allen, R., Harrison, E., A-H-Mackerness, S., and Buchanan-Wollaston, V. (2003). Expression of senescence-enhanced genes in response to oxidative stress. J. Exp. Bot. 54, 2285–2292. doi: 10.1093/jxb/erg267
Nelson, N., and Yocum, C. F. (2006). Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 57, 521–565. doi: 10.1146/annurev.arplant.57.032905.105350
Nuthikattu, S., McCue, A. D., Panda, K., Fultz, D., DeFraia, C., Thomas, E. N., et al. (2013). The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21-22 Nucleotide Small Interfering RNAs. Plant Physiol. 162, 116–131. doi: 10.1104/pp.113.216481
Ogneva, Z., Dubrovina, A., and Kiselev, K. (2016). Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in Arabidopsis thaliana. Biol. Plant. 60, 628–634. doi: 10.1007/s10535-016-0638-y
Penterman, J., Zilberman, D., Huh, J. H., Ballinger, T., Henikoff, S., and Fischer, R. L. (2007). DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. U.S.A. 104, 6752–6757. doi: 10.1073/pnas.0701861104
Robatzek, S., and Somssich, I. E. (2002). Targets of AtWRKY6 regulation during plant senescence and pathogen defense. Genes. Dev. 16, 1139–1149. doi: 10.1101/gad.222702
Robertson, K. D. (2005). DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610. doi: 10.1038/nrg1655
Ronemus, M. J., Galbiati, M., Ticknor, C., Chen, J. C., and Dellaporta, S. L. (1996). Demethylation-induced developmental pleiotropy in Arabidopsis. Science 273, 654–657. doi: 10.1126/science.273.5275.654
Rossi, M., Bermudez, L., and Carrari, F. (2015). Crop yield: challenges from a metabolic perspective. Curr. Opin. Plant Biol. 25, 79–89. doi: 10.1016/j.pbi.2015.05.004
Scheller, H. V., Jensen, P. E., Haldrup, A., Lunde, C., and Knoetzel, J. (2001). Role of subunits in eukaryotic Photosystem I. Biochim. Biophys. Acta 1507, 41–60. doi: 10.1016/s0005-2728(01)00196-7
Schippers, J. H. M. (2015). Transcriptional networks in leaf senescence. Curr. Opin. Plant Biol. 27, 77–83. doi: 10.1016/j.pbi.2015.06.018
Slotkin, R. K., Vaughn, M., Borges, F., Tanurdzic, M., Becker, J. D., Feijo, J. A., et al. (2009). Epigenetic Reprogramming and Small RNA silencing of transposable elements in pollen. Cell 136, 461–472. doi: 10.1016/j.cell.2008.12.038
Stettler, M., Eicke, S., Mettler, T., Messerli, G., Hortensteiner, S., and Zeeman, S. C. (2009). Blocking the metabolism of starch breakdown products in Arabidopsis Leaves Triggers Chloroplast Degradation. Mol. Plant 2, 1233–1246. doi: 10.1093/mp/ssp093
Thornley, J. H. M. (1976). Mathematical Models in Plant Physiology. (London: Academic Press), 86–110.
Tsai, H. L., Lue, W. L., Lu, K. J., Hsieh, M. H., Wang, S. M., and Chen, J. (2009). Starch synthesis in arabidopsis is achieved by spatial cotranscription of core starch metabolism genes. Plant Physiol. 151, 1582–1595. doi: 10.1104/pp.109.144196
Vriet, C., Welham, T., Brachmann, A., Pike, M., Pike, J., Perry, J., et al. (2010). A Suite of Lotus japonicus starch mutants reveals both conserved and novel features of starch metabolism. Plant Physiol. 154, 643–655. doi: 10.1104/pp.110.161844
Wada, Y., Ohya, H., Yamaguchi, Y., Koizumi, N., and Sano, H. (2003). Preferential de Novo methylation of cytosine residues in non-CpG sequences by a domains rearranged DNA methyltransferase from tobacco plants. J. Biol. Chem. 278, 42386–42393. doi: 10.1074/jbc.M303892200
Wang, R. F., Angenent, G. C., Seymour, G., and de Maagd, R. A. (2020). Revisiting the role of master regulators in tomato ripening. Trends Plant Sci. 25, 291–301. doi: 10.1016/j.tplants.2019.11.005
Woo, H. R., Kim, H. J., Lim, P. O., and Nam, H. G. (2019). Leaf Senescence: systems and dynamics aspects. Annu. Rev. Plant Biol. 70, 347–376. doi: 10.1146/annurev-arplant-050718-095859
Yang, J. D., Worley, E., and Udvardi, M. (2014). A NAP-AAO3 regulatory module promotes chlorophyll degradation via ABA Biosynthesis in Arabidopsis Leaves. Plant Cell 26, 4862–4874. doi: 10.1105/tpc.114.133769
Yang, Y., Tang, K., Datsenka, T. U., Liu, W. S., Lv, S. H., Lang, Z. B., et al. (2019). Critical function of DNA methyltransferase 1 in tomato development and regulation of the DNA methylome and transcriptome. J. Integr. Plant Biol. 61, 1224–1242. doi: 10.1111/jipb.12778
Yao, M. Q., Chen, W. W., Kong, J. H., Zhang, X., Shi, N. N., Zhong, S. L., et al. (2020). METHYLTRANSFERASE1 and ripening modulate vivipary during tomato fruit development. Plant Physiol. 183, 1883–1897. doi: 10.1104/pp.20.00499
Yen, M. R., Suen, D. F., Hsu, F. M., Tsai, Y. H., Fu, H. Y., Schmidt, W., et al. (2017). Deubiquitinating enzyme OTU5 contributes to DNA methylation patterns and is critical for phosphate nutrition signals. Plant Physiol. 175, 1826– 1838.
Yong-Villalobos, L., González-Morales, S. I., Wrobel, K., Gutiérrez-Alanis, D., Cervantes-Peréz, A. S., Hayano-Kanashiro, C., et al. (2015). Methylome analysis reveals an important role for epigenetic changes in the regulation of the Arabidopsis response to phosphate starvation. Proc. Natl. Acad. Sci. U.S.A. 112, E7293–E7302. doi: 10.1073/pnas.1522301112
Yu, T. S., Kofler, H., Hausler, R. E., Hille, D., Flugge, U. I., Zeeman, S. C., et al. (2001). The Arabidopsis sex1 mutant is defective in the R1 protein, a general regulator of starch degradation in plants, and not in the chloroplast hexose transporter. Plant Cell 13, 1907–1918. doi: 10.1105/tpc.13.8.1907
Yuan, L., Wang, D., Cao, L. W., Yu, N. N., Liu, K., Guo, Y. F., et al. (2020). Regulation of Leaf Longevity by DML3-Mediated DNA Demethylation. Mol. Plant 13, 1149–1161. doi: 10.1016/j.molp.2020.06.006
Zeeman, S. C., Northrop, F., Smith, A. M., and Rees, T. (1998). A starch-accumulating mutant of Arabidopsis thaliana deficient in a chloroplastic starch-hydrolysing enzyme. Plant J. 15, 357–365. doi: 10.1046/j.1365-313X.1998.00213.x
Zeeman, S. C., and Rees, T. (1999). Changes in carbohydrate metabolism and assimilate export in starch-excess mutants of Arabidopsis. Plant Cell Environ. 22, 1445–1453. doi: 10.1046/j.1365-3040.1999.00503.x
Zeeman, S. C., Tiessen, A., Pilling, E., Kato, K. L., Donald, A. M., and Smith, A. M. (2002). Starch synthesis in Arabidopsis, Granule synthesis, composition, and structure. Plant Physiol. 129, 516–529. doi: 10.1104/pp.003756
Zhang, H. M., Lang, Z. B., and Zhu, J. K. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506. doi: 10.1038/s41580-018-0016-z
Zhang, J., Liu, Y., Xia, E. H., Yao, Q. Y., Liu, X. D., and Gao, L. Z. (2015). Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. U.S.A. 112, E7022–E7029. doi: 10.1073/pnas.1515170112
Keywords: SlDRM7, DNA methylation, leaf chlorosis and senescence, starch excess, chlorophyll metabolism
Citation: Wen YX, Wang JY, Zhu HH, Han GH, Huang RN, Huang L, Hong YG, Zheng SJ, Yang JL and Chen WW (2022) Potential Role of Domains Rearranged Methyltransferase7 in Starch and Chlorophyll Metabolism to Regulate Leaf Senescence in Tomato. Front. Plant Sci. 13:836015. doi: 10.3389/fpls.2022.836015
Received: 15 December 2021; Accepted: 10 January 2022;
Published: 08 February 2022.
Edited by:
Cécile Raynaud, UMR 9213 Institut des Sciences des Plantes de Paris Saclay (IPS2), FranceReviewed by:
Yingfang Zhu, Henan University, ChinaDaisuke Miki, Shanghai Center for Plant Stress Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences (CAS), China
Copyright © 2022 Wen, Wang, Zhu, Han, Huang, Huang, Hong, Zheng, Yang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Li Yang, eWFuZ2ppYW5saUB6anUuZWR1LmNu; Wei Wei Chen, MTU4NTgyMjM4MDdAMTYzLmNvbQ==