 Mukesh Kumar Malviya1†Chang-Ning Li1†Prakash Lakshmanan1,2,3Manoj Kumar Solanki4Zhen Wang5Anjali Chandrol Solanki6Qian Nong1Krishan K. Verma1Rajesh Kumar Singh1Pratiksha Singh1Anjney Sharma1Dao-Jun Guo1,7Eldessoky S. Dessoky8Xiu-Peng Song1*Yang-Rui Li1,7*
Mukesh Kumar Malviya1†Chang-Ning Li1†Prakash Lakshmanan1,2,3Manoj Kumar Solanki4Zhen Wang5Anjali Chandrol Solanki6Qian Nong1Krishan K. Verma1Rajesh Kumar Singh1Pratiksha Singh1Anjney Sharma1Dao-Jun Guo1,7Eldessoky S. Dessoky8Xiu-Peng Song1*Yang-Rui Li1,7*- 1Key Laboratory of Sugarcane Biotechnology and Genetic Improvement (Guangxi), Ministry of Agriculture and Rural Affairs, Sugarcane Research Center, Chinese Academy of Agricultural Sciences, Guangxi Key Laboratory of Sugarcane Genetic Improvement, Sugarcane Research Institute, Guangxi Academy of Agricultural Sciences, Nanning, China
- 2Interdisciplinary Research Center for Agriculture Green Development in Yangtze River Basin, College of Resources and Environment, Southwest University, Chongqing, China
- 3Queensland Alliance for Agriculture and Food Innovation, University of Queensland, St. Lucia, QLD, Australia
- 4Plant Cytogenetics and Molecular Biology Group, Faculty of Natural Sciences, Institute of Biology, Biotechnology and Environmental Protection, University of Silesia in Katowice, Katowice, Poland
- 5Guangxi Key Laboratory of Agricultural Resources Chemistry and Biotechnology, College of Biology and Pharmacy, Yulin Normal University, Yulin, China
- 6Department of Agriculture Science, Mansarovar Global University, Bhopal, India
- 7College of Agriculture, Guangxi University, Nanning, China
- 8Department of Biology, College of Science, Taif University, Taif, Saudi Arabia
Considering the significant role of genetic background in plant-microbe interactions and that most crop rhizospheric microbial research was focused on cultivars, understanding the diversity of root-associated microbiomes in wild progenitors and closely related crossable species may help to breed better cultivars. This study is aimed to fill a critical knowledge gap on rhizosphere and diazotroph bacterial diversity in the wild progenitors of sugarcane, the essential sugar and the second largest bioenergy crop globally. Using a high-throughput sequencing (HTS) platform, we studied the rhizosphere and diazotroph bacterial community of Saccharum officinarum L. cv. Badila (BRS), Saccharum barberi (S. barberi) Jesw. cv Pansahi (PRS), Saccharum robustum [S. robustum; (RRS), Saccharum spontaneum (S. spontaneum); SRS], and Saccharum sinense (S. sinense) Roxb. cv Uba (URS) by sequencing their 16S rRNA and nifH genes. HTS results revealed that a total of 6,202 bacteria-specific operational taxonomic units (OTUs) were identified, that were distributed as 107 bacterial groups. Out of that, 31 rhizobacterial families are commonly spread in all five species. With respect to nifH gene, S. barberi and S. spontaneum recorded the highest and lowest number of OTUs, respectively. These results were validated by quantitative PCR analysis of both genes. A total of 1,099 OTUs were identified for diazotrophs with a core microbiome of 9 families distributed among all the sugarcane species. The core microbiomes were spread across 20 genera. The increased microbial diversity in the rhizosphere was mainly due to soil physiochemical properties. Most of the genera of rhizobacteria and diazotrophs showed a positive correlation, and few genera negatively correlated with the soil properties. The results showed that sizeable rhizospheric diversity exists across progenitors and close relatives. Still, incidentally, the rhizosphere microbial abundance of progenitors of modern sugarcane was at the lower end of the spectrum, indicating the prospect of Saccharum species introgression breeding may further improve nutrient use and disease and stress tolerance of commercial sugarcane. The considerable variation for rhizosphere microbiome seen in Saccharum species also provides a knowledge base and an experimental system for studying the evolution of rhizobacteria-host plant association during crop domestication.
Introduction
Sugarcane is an important agricultural crop grown in nearly 110 countries worldwide. China is the third-largest producer of sugarcane (a collective term for Saccharum species, but more commonly applied to cultivated Saccharum officinarum (S. officinarum) L. and Saccharum spp. inter-specific hybrids). It is a major crop in southern China, accounting for ≈90% of Chinese sugar production (Li and Yang, 2015). Over the years, sugarcane has been developed as a multi-purpose agro-industrial crop as it provides the raw material for different industries, such as food, thermal, energy/fuel, and paper (Goldemberg et al., 2008; Fischer et al., 2012). Sugarcane is mainly grown as a monoculture for extended periods resulting in yield decline, which is attributed to degraded soil, imbalanced soil biology, and build-up of pests and diseases (Shoko et al., 2007). Restoration of soil biology and soil fertility is now emerging as a priority for improving soil health, reducing the yield gap, and sustaining profitable green agriculture (Brackin et al., 2013; Schultz et al., 2017).
The rhizosphere is rich in microbial diversity and abundance. These functionally diverse microbial communities include saprophytes, epiphytes, pathogens, and also plant growth-promoting microorganisms (Avis et al., 2008). Bacteria are the most abundant rhizospheric microbiota, covering up to 15% of the total root surface (van Loon, 2007). About 2–5% of rhizobacteria are known to promote plant growth (Antoun and Prévost, 2005). Many plant growth-promoting rhizobacteria (PGPRs) are capable of nitrogen fixation, solubilization of inorganic molecules, such as phosphate, production of plant growth regulators/hormones, siderophores, and compounds that control phytopathogens (Cawoy et al., 2011; Jangu and Sindhu, 2011; Velineni and Brahmaprakash, 2011). Thus, PGPRs and other rhizosphere bacteria are now well-recognized as an essential component of sustainable agriculture systems. The most commonly found rhizospheric bacterial genera are Bacillus, Pseudomonas, Rhizobia, Arthrobacter, Agrobacterium, Micrococcus, Cellulomonas, Azotobacter, Alcaligenes, Mycobacterium, and Flavbacter (Teixeira et al., 2010; Prashar et al., 2014). There are several PGPRs and diazotrophs genera, such as Bacillus, Paenibacillus, Pseudomonas, Enterobacter, Arthrobacter, Azotobacter, Burkholderia, and Azospirillum, that are associated with sugarcane rhizosphere (Ahmad et al., 2016; Lamizadeh et al., 2016; Li et al., 2017; Malviya et al., 2019; Pereira et al., 2020; Rosa et al., 2020). The majority of PGPRs are not culturable. Thus studying those using traditional laboratory methods is challenging and time-consuming (Prashar et al., 2014; Wei et al., 2018). Rapid progress in molecular biology, especially the advent of cost-effective, high-throughput DNA sequencing technologies and the associated data analytics, has improved the understanding of rhizosphere microflora by culture-independent studies (Reuter et al., 2015; Wei et al., 2018). The next-generation sequencing approaches provide an efficient and comprehensive system to identify microbial species in the rhizosphere irrespective of microbial abundance (Uroz et al., 2013). As a result, through the sequencing of the 16S rRNA gene, remarkable progress in the taxonomic characterization of highly diversified rhizospheric bacteria has been achieved (Dong et al., 2017; Gong et al., 2019). Further, modern molecular techniques permit an in-depth analysis of soil bacterial communities' compositional and functional dynamics in changing soil environmental conditions, a recurring feature of agricultural soil (Dini-Andreote et al., 2010; Wei et al., 2018). Recently rhizosphere microbiomes community structure of three endangered Plants (Xu et al., 2020) and blueberry varieties (Wang et al., 2019) has been studied by high-throughput sequencing (HTS).
Sugarcane rhizosphere microbes need strong attention to understand their diversity and role in crop improvement. Several novel PGPRs from the sugarcane microbiome have been identified and improved crop production (Pisa et al., 2011; Schultz et al., 2017; Armanhi et al., 2018). Most of the studies on rhizosphere microbes and endophytes are limited to modern commercial sugarcane cultivars, and little is known about their occurrence and abundance in the wild sugarcane progenitor species. They are primarily involved in nitrogen fixation and plant hormone production, thus positively affecting sugarcane growth (Goldemberg et al., 2008; Mehnaz, 2011). The present study focuses on wild sugarcane progenitor species because these species are regarded as high fibrous plants with significant geographic distributions and can survive a broad range of abiotic stresses, such as droughts, saline, floods, and freezing conditions (Santchurn et al., 2019). Rhizobacteria play a significant role in nitrogen fixation in sugarcane crops (Li et al., 2017; Malviya et al., 2019). However, much remains to be learned about these diazotrophic rhizobacteria, a key driver of soil health and fertility. There are several reports of identification and characterization of PGPRs from sugarcane (Inui-Kishi et al., 2012; Lamizadeh et al., 2016) and other crops (Kumar et al., 2014; Gaikwad and Sapre, 2015; Tsegaye et al., 2019) and some of them are being used for crop productivity improvement (Kashyap et al., 2017; Swarnalakshmi et al., 2020). Not surprisingly, most sugarcane studies are conducted with modern cultivated sugarcane hybrid varieties. There is no previous attempt to understand the diversity of rhizobacteria in their wild progenitors and closely related species, such as S. officinarum, Saccharum spontaneum (S. spontaneum), Saccharum robustum (S. robustum), Saccharum barberi (S. barberi), and Saccharum sinense (S. sinense). Rhizospheric microorganisms interact with plants for their survival and nutritional requirements (Berendsen et al., 2012; Finkel et al., 2017). Many of them are beneficial to plants for nutrient uptake and to cope with pathogens and abiotic stresses (Pisa et al., 2011; Dagnaw et al., 2015; Pereira et al., 2020).
Thus, the current study is aimed to understand the role of rhizosphere bacterial communities and identify new species of nitrogen-fixing bacteria using high-throughput 16S rRNA and nifH gene sequencing using the Illumina platform. This study reports novel and valuable findings on the diversity of bacterial communities in five Saccharum progenitor species, namely, S. officinarum L. cv Badila (BRS), S. barberi Jesw. cv Pansahi (PRS), S. robustum (RRS), S. spontaneum (SRS), and S. sinense Roxb. cv Uba (URS) and provides a knowledge base to study the influence of sugarcane genotype on rhizosphere bacteria in this necessary sugar and energy crop.
Materials and Methods
Soil Sampling
Rhizospheric soil of five sugarcane species, BRS, PRS, RRS, SRS, and URS, was maintained in the sugarcane field, germplasm of Sugarcane Research Institute, Guangxi Academy of Agricultural Sciences, Nanning, Guangxi, China (22°49′ N, 108°18′ E, 800–1731 masl). Climate conditions of the site were as follows: natural night (10 h), temperature (minimum 22°C to maximum 35°C), and air humidity was 75–80%. The rhizosphere soil sample of all five sugarcane species was collected at the maturing stage of the first ratoon crop. Five soil samples were separately collected from each species by shaking roots in a sterile bag for 5 min. These soil samples were pooled to prepare a composite soil sample (≈5 g) for each species. Soil DNA from three sub-samples from the composite sample of each species were extracted separately. A schematic diagram represents the overall experimental design (Figure 1).
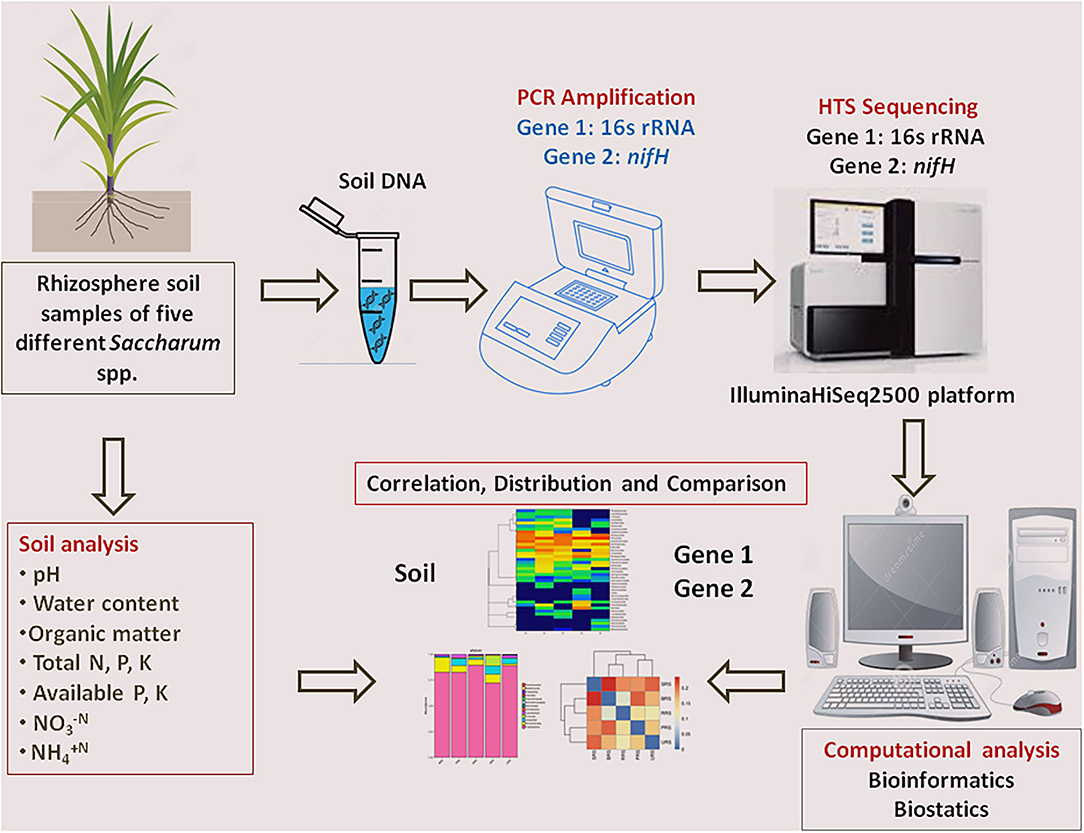
Figure 1. Schematic diagram of experimental design and analysis that used in the current study. HTS, High-throughput sequencing.
Soil Physiochemical Parameters
As described by Solanki et al. (2019), soil chemical analysis activities were carried out. Soil water content was measured by the weighing method. Soil pH was analyzed by a pH meter, and soil organic carbon was measured by the dichromate oxidation method (Walkley and Black, 1934). Total nitrogen (N) was estimated through the semi-micro-Kjeldahl method (Bremmer and Mulvaney, 1982). The FeSO4/Zn reduction method was used for the estimation of nitrate-nitrogen (NO-3N) and ammonium nitrogen (NH+4N) (Carter, 1993). Total phosphorus (P) was measured via the sodium carbonate fusion method (Carter, 1993). Available P was estimated by the sample extraction method (Bao, 2002). Total potassium (K) estimation was done by the photometry method (Bao, 2002). The ammonium acetate extraction-flame photometry method was applied to detect the available K in the soil (Bao, 2002). All analysis was performed in three biological replicates.
High-Throughput Sequencing
A culture-independent method was applied in this study to identify the bacterial composition of test species (Ranjard et al., 2000). Total microbial DNA was extracted using cetyltrimethylammonium bromide/sodium dodecyl sulfate (CTAB/SDS) isolation method with minor modifications (Barbier et al., 2019). The purity of extracted DNA was assayed with a NanoDrop One spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). Three DNA samples were pooled as one and used for DNA sequencing. Bacterial 16S rRNA was amplified with the universal primers 341F (ACTCCTACGGGAGGCAGCAG) and 806R (GGACTACHVGGGTWTCTAAT), which target the V3–V4 regions (Dong et al., 2018). The nifH gene was amplified with primers Pol-F (TGCGAYCCSAARGCBGACTC) and Pol-R (ATSGCCATCATYTCRCCGGA), as previously reported (Poly et al., 2001). PCR amplification of identified 16S rRNA and nifH genes was performed, and each PCR contains 25 μl reaction that included 12.5 μl ready to use PCR mix (Tiangen Biotech, Beijing, China), 1.0 μl of each primer (10 μM), 2.5 μl of DNA template (10 ng/ml), and 9.0 μl PCR grade water. PCR amplification consisted of a 3 min denaturation at 95°C; 25 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C; and 5 min at 72°C for 16S rRNA. For nifH gene amplification, thermal cycling consisted of initial denaturation at 98°C for 1 min, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s, and elongation at 72°C for 60 s, finally, 72°C for 5 min, following the protocols reported previously by Zhang et al. (2015). Visualization and quantification of PCR products were conducted by mixing an equal volume of 1 × loading buffer containing SYB green dye with the PCR products and electrophoresed on 2% agarose gel. The 400–450 bp DNA fragments were isolated and used for further experiments. Equimolar amounts of PCR products from all samples were pooled, and the mixture was purified using Qiagen Gel Extraction Kit (Qiagen, Germany). Sequencing libraries of purified amplicons were generated using TruSeq® DNA (Illumina, San Diego, CA, USA). PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, USA) in accordance with the manufacturer's protocol, and index codes were added. The library quality and concentration were assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific, Waltham, MA, USA) and Agilent Bioanalyzer 2100 system. To perform sequencing, the qualified libraries were fed into the IlluminaHiSeq2500 platform, and 250 bp paired-end reads were generated.
HTS Data Analysis
Based on their unique barcode, trimming of barcode, and primer sequencing were done. Reads were assembled using FLASH (V.1.2.7) (http://ccb.jhu.edu/software/FLASH/) (Magoè and Salzberg, 2011) to generate raw tags. To obtain high-quality clean tags from raw tags, we performed quality filtration using QIIME (V1.7.0) (http://qiime.org/index.html) (Caporaso et al., 2010). Removal of chimera sequences was done by comparing the tags with the reference database Unite Database (https://unite.ut.ee/) using the UCHIME algorithm (http://www.drive5.com/usearch/manual/uchime_algo.html) (Edgar et al., 2011). The above step is critical to obtain effective tags. Operational taxonomic unit (OTU) identification was done with UPARSE software (v.7.0.1001) (http://drive5.com/uparse/) (Edgar, 2013). Based on ≥95% of sequence similarity, all the effective tags were clustered into OTUs. For each OTU cluster, a representative sequence was screened to perform taxonomic annotation.
Operational Taxonomic Units were taxonomically annotated following a Basic Local Alignment Search Tool (BLAST) analysis against the Unite Database of each identified representative bacterial sequence done in QIIME software. Multiple sequence alignment was conducted with MUSCLE software (V.3.8.3) (http://www.drive5.com/muscle/) (Edgar, 2004), and phylogenetic relationship of different OTUs was established to understand the diversity of microbial species in various samples (groups). Alpha diversity analysis is carried out to find the complexity of species diversity for each sample using 6 indices, which include observed species, Chao1, Shannon, Simpson, abundance-based coverage estimator (ACE), and Good's coverage. Indices calculation for all the samples was done using QIIME and visualized in R software (V. 2.15.3). Community richness was identified with two selected indices using Chao—the Chao1 richness estimator (http://www.mothur.org/wiki/Chao). The Shannon index (http://www.mothur.org/wiki/Shannon) and Simpson index (http://www.mothur.org/wiki/Simpson) indices were used for the identification of community diversity in all the samples. To characterize the sequencing depth and coverage, the Good's coverage (http://www.mothur.org/wiki/Coverage) was used. Beta diversity analysis was performed to evaluate the differences in bacterial species among all the samples. QIIME software was used to calculate beta diversity using weighted and unweighted UniFrac distances. The raw data of 16S rRNA (accession no. PRJNA678588) and nifH gene (accession no. PRJNA681283) were submitted to the NCBI Sequence Read Archive.
Quantitative (q) PCR Analysis
Quantitative PCR was performed to determine the relative abundance of the 16S rRNA and nifH gene in each individual rhizospheric soil. The bacterial gene copy numbers were determined with the primers 341F/518R (GC-341 F 5-CCTACGGGAGGCAGCAG-3) and 518 R (5-ATTACCGCGGCTGCTGG-3) (Moore et al., 2011) and the nifH gene with the primers nifH-F (AAAGGYGGWATCGGYAARTCCACCAC) and nifH-R (TTGTTSGCSGCRTACATSGCCAT CAT) (Zhang et al., 2016). Briefly, each 20 μl PCR reaction contained 1.0 μl of DNA template (2.5 ng/μl), 1.0 μl of each primer (10 μmol/l), 7.0 μl of molecular-grade water, and 10 μl iQ™ SYBR Green SuperMix (Bio-Rad Laboratories, Hercules, CA, USA). Conditions of the qPCR for 16S rRNA are as follows: initial denaturation at 95°C for 5 min; 40 cycles of denaturation at 95°C for 15 s, and annealing at 56°C for 30 s. Condition of the qPCR for nifH is as follows: 95°C for 30 s followed by 40 cycles of 95°C for 5 s, 60°C for 40 s. The standard curve of DNA and copy number was constructed by the standard formula: y = −3.406 × 37.05 [y: Ct value; × LOG10 (copy number)]. All samples were done in three replicates.
Statistical Analysis
Using the FactoMineR and ggplot2 packages in R software, results were visualized. Later, Principal Coordinate Analysis (PCoA) was performed to obtain principal coordinates and to visualize them. To compare microbial diversity in different samples, the UniFrac method was used to generate weighted or unweighted UniFrac values among samples that were transformed to give uncorrelated/orthogonal axes. Visualization of PCoA results was done using weighted gene co-expression network analysis (WGCNA) package, stat, and ggplot2 packages in R software. QIIME software was used for hierarchical clustering by unweighted pair-group method with arithmetic mean method (UPGMA) (Sokal, 1958) to infer the distance matrix using average linkage. Heatmaps and Venn plots were generated using the package “ggplots” of software R (v3.0.3). The experiments were conducted in replicates, and data were analyzed using standard ANOVA followed by the Duncan's Multiple Range Test (DMRT) through Origin 2017SR2 software (Northampton, MA, USA). Circos plots were drawn by Circos Table Viewer v0.63-9 software (Krzywinski et al., 2009) to calculate Spearman's rank correlation coefficient between soil variables and bacterial taxa by using PAST3 software (Hammer et al., 2001), and heatmap is generated by using ClustVis online tool (Metsalu and Vilo, 2015) and Morpheus (https://software.broadinstitute.org/morpheus/).
Results
This study was aimed to understand the rhizosphere soil microbiota of five different critical ancestral sugarcane species using 16S rRNA and nifH gene sequencing to understand their bacterial community diversity, especially that of diazotrophs. This is a major knowledge gap as the significance of rhizosphere biology has a strong genetic basis. It is now increasingly recognized as a target for crop productivity improvement in all major crops, such as sugarcane.
Soil Physiochemical Parameters
The physiochemical properties of all the rhizosphere soil samples are presented in Figure 2. Higher water content values were found in S. sinense (19.1 ± 0.38%), and the low values were found in S. spontaneum (14% ± 0.33). The pH range was recorded (S. sinense) 7.1 ± 0.14 to 5.7 ± 0.11 (S. officinarum). Organic matter (OM) was observed higher in S. sinense (16.30 ± 0.33) and lowest in S. spontaneum (8.29 ± 0.17). The higher amounts of total N were recorded in S. barberi (0.78 ± 0.02), and low quantity resulted in S. spontaneum (0.48 ± 0.11). Total K, available P, and available K were recorded higher in S. officinarum and low in S. spontaneum. The higher value of available NO-3N was observed in S. officinarum (24.70 ± 0.49) and the lowest value in S. sinense (4.43 ± 0.09). A higher concentration of available NH+4N was recorded in S. sinense (22.20 ± 0.44) and low in S. spontaneum (5.79 ± 0.12).
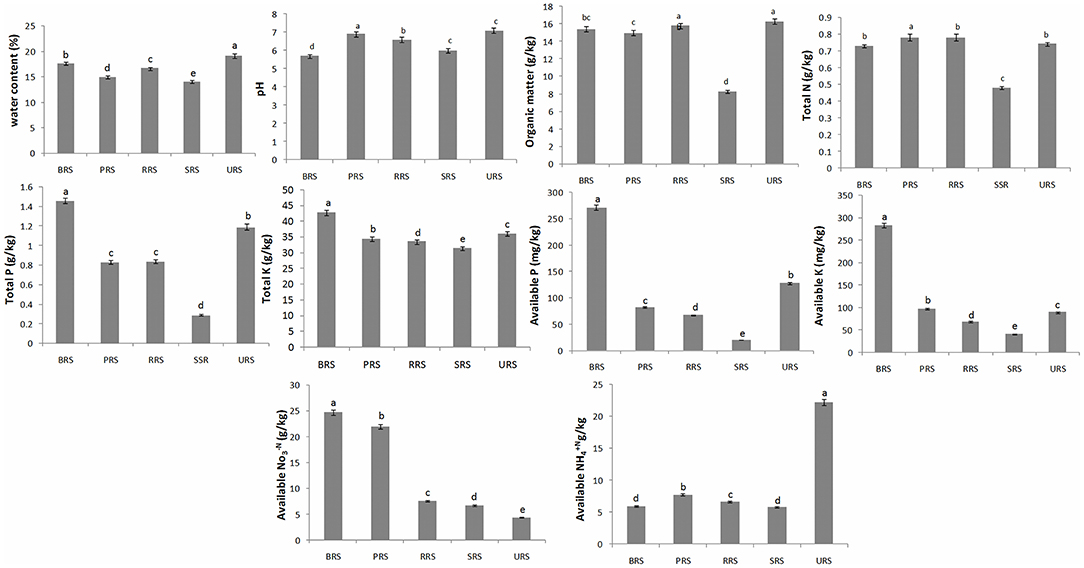
Figure 2. Soil physiochemical properties of rhizospheric soil samples of sugarcane species. Values followed by the same lowercase letters in the same bar are not significantly different according to DMRT 5%. S. officinarum L. cv Badila (BRS), S. barberi Jesw. cv Pansahi (PRS), S. robustum (RRS), S. spontaneum (SRS), and S. sinense Roxb. cv Uba (URS). DMRT, Duncan's Multiple Range test.
Sequencing Data
A total of 233,455 effective sequences with an average length of 413 bp were obtained for 16S rRNA genes from different sugarcane species samples. Sequencing of nifH genes from five different species samples produced a total of 182,185 sequences with 357 average bp length. These raw reads of 16S rRNA and nifH genes were filtered using QIIME quality filters, followed by OTU identification, clustering, and analysis, respectively (Supplementary Tables S1, S2).
Operational Taxonomic Units
The S. spontaneum sample, compared to those from all the other species, showed less species evenness at a low OTU rank. We analyzed common and unique OTUs based on 16S rRNA and nifH gene sequences for each sample (Supplementary Figures S1A,B). In the 16S rRNA sequence data, 6202 OTUs were identified from all samples collectively, of which 519 OTUs were common across all species. The relative frequency of OTUs in the studied species was as follows: S. sinense > S. robustum > S. barberi > S. officinarum > S. spontaneum (Figure 3A). A total of 1,099 OTUs were identified for nifH gene from the sequence data of species combined. Among them, 14 were common OTUs found across all the samples. The occurrence of OTUs in the plant samples was as follows: S. barberi > S. robustum > S. officinarum > S. sinense > S. spontaneum. Thus, S. barberi and S. spontaneum recorded the highest and lowest number of OTUs (Figure 3B).
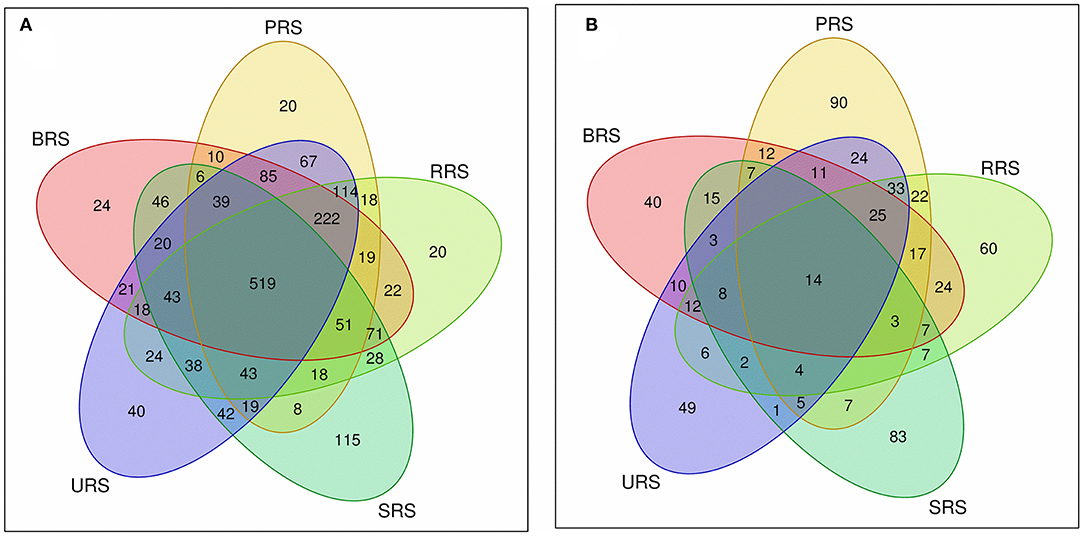
Figure 3. Venn diagram showing the OTUs distribution between the sugarcane species soil samples (A) 16S rRNA and (B) nifH gene. S. officinarum L. cv Badila (BRS), S. barberi Jesw. cv Pansahi (PRS), S. robustum (RRS), S. spontaneum (SRS), and S. sinense Roxb. cv Uba (URS). OTUs, operational taxonomic units.
Principal Component Analysis (PCA)
To understand the rhizobacterial community composition, PCA plots were generated based on 16S rRNA and nifH gene OTUs data (Supplementary Figures S2A,B). The OTUs of 16S rRNA samples show that S. officinarum and S. robustum are not identical, S. sinense and S. robustum have more similarity, and S. spontaneum is distinct from all the other species studied. PCA of nifH gene samples showed a close identity between S. barberi and S. spontaneum, whereas S. sinense, S. officinarum, and S. robustum remained distinct.
Diversity Index and Microbial Composition
Alpha diversity refers to the diversity within a particular sample individually, and it is usually represented by the microbial species (i.e., species richness) enumerated in the test samples. Alpha diversity analysis was done using Shannon, Simpson, and Chao indices Rarefaction curves for both 16S rRNA and nifH sequence data. Supplementary Figure S3A consists of plots displaying Shannon, Simpson, and Chao indices, built using 16S rRNA samples. The Shannon index was increased as both the species richness and the evenness in the community was increased. Among the 16S rRNA data of all the samples analyzed, the Shannon and Chao indices of S. sinense sample were higher than the other four samples, with a slight variation found among them. Whereas, Simpson indices was increased as the diversity was increased. In the Simpson index plot, S. officinarum samples showed the highest value, implying more species diversity than the other four samples. Shannon and Chao indices of nifH gene data of all the samples showed more species diversity in S. barberi sample than others. Like 16S rRNA data, S. officinarum sample showed the highest value in Simpson index based on nifH gene data (Supplementary Figure S3B).
The relative abundance of the microbial communalities was differed among the five sugarcane species analyzed. The abundant phyla identified in all sugarcane species using 16S rRNA data were Proteobacteria, Firmicutes, Actinobacteria, Acidobacter, Bacteroidetes, Chloroflexi, Gemmatimonadetes, Planctomycetes, and Nitrospirae (Figure 4A). Firmicutes were the highest phyla present in S. officinarum compared to other samples. Gemmatimonadetes were the highest in S. barberi, Acidobacter was more elevated in S. spontaneum, and Bacteroidetes was most increased in S. sinense. Based on nifH gene data, we found Proteobacteria and Verrucomicrobia (Figure 4B). Firmicutes were abundant in S. spontaneum samples compared to others. Many unclassified phyla were also represented abundantly in S. spontaneum followed by S. sinense and S. barberi samples.
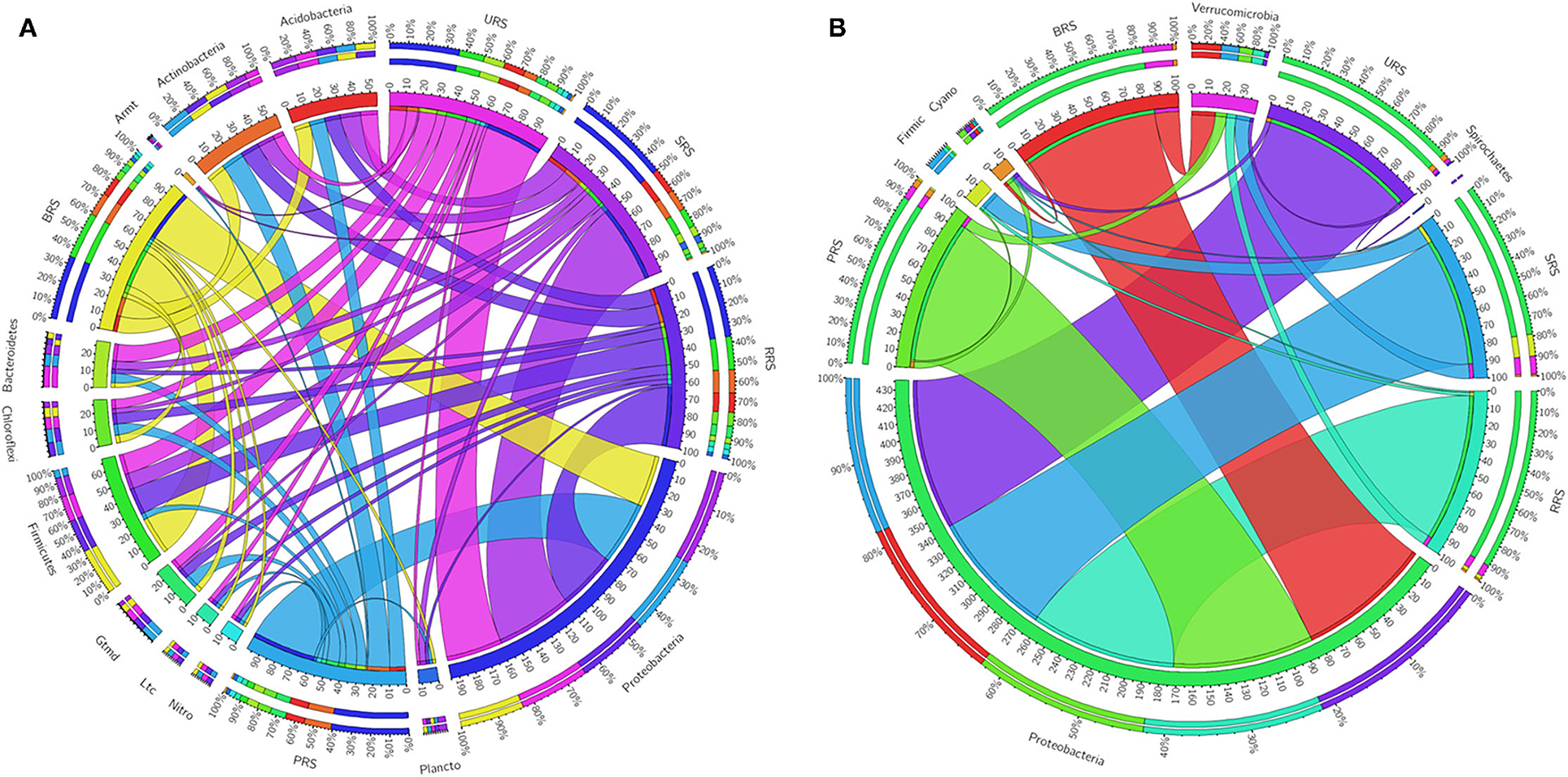
Figure 4. Circular representation of the proportional structure of bacterial communities at the phylum level based on 16s rRNA (A) and nifH gene (B) associated with the sugarcane rhizosphere of different species. Values within the inner circle indicate the number of reads of a phylum within the normalized dataset. S. officinarum L. cv Badila (BRS), S. barberi Jesw. cv Pansahi (PRS), S. robustum (RRS), S. spontaneum (SRS), and S. sinense Roxb. cv Uba (URS).
Genus distribution using 16S rRNA sequence data is given in Figure 5A. Bacillus was the most abundant genus in S. officinarum, S. robustum, and S. sinense, while Pseudomonas became the number one genus in S. barberi and S. spontaneum. Bacillus, Pseudomonas, Pseudarthobacter, Massilia, Lysobacter, Nitrospira, Gemmatimonas, and Streptomyces, and Rhizobium were the most abundant in S. officinarum. However, S. barberi soil sample was dominated by Bacillus, Pseudomonas, Pseudarthobacter, Lysobacter, Gemmatimonas, and Sphingomonas. S. robustum contained Bacillus, Pseudomonas, Pseudarthobacter, Massilia, Nitrospira, Gemmatimonas, Streptomyces, Paenibacillus, and Dechloromonas. S. spontaneum sample contained Pseudomonas, Pseudarthobacter, Massilia, Tumebacillus, Remibacter, Sphingomonas, and Skermanelia. S. sinense sample was dominated by Bacillus, Pseudomonas, Pseudarthobacter, Massilia, Lysobacter, Nitrospira, Faecalibacterium, and Streptococus.
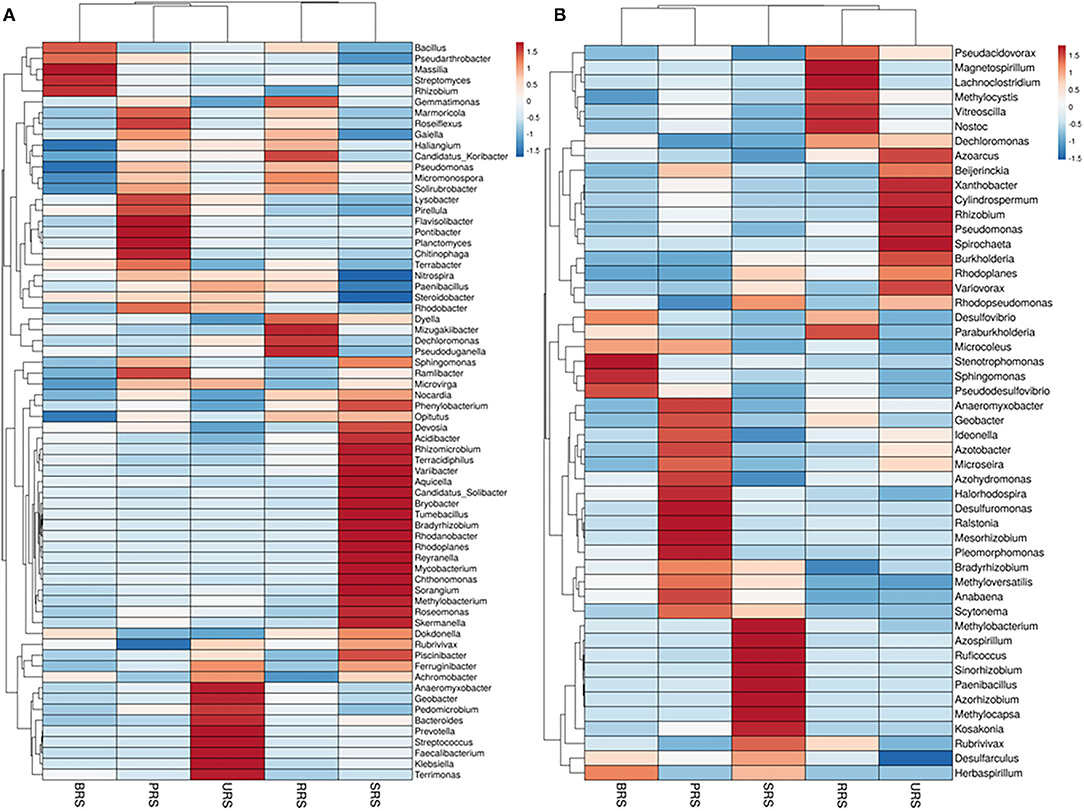
Figure 5. Heat map showing the abundances of bacterial communities at genus level in the soil samples of sugarcane species (A) 16S rRNA and (B) nifH gene. S. officinarum L. cv Badila (BRS), S. barberi Jesw. cv Pansahi (PRS), S. robustum (RRS), S. spontaneum (SRS), and S. sinense Roxb. cv Uba (URS).
Genus distribution using nifH gene data is presented in Figure 5B. Bradyrhizobium, Dechloromonas, Desulfovibrio, and Stenotrophomonas were abundant in S. officinarum, while Bradyrhizobium, Desulfovibrio, Xanthobacter, and Anaeromyxobacter were the leading genera in S. barberi. Bradyrhizobium, Dechloromonas, Desulfovibrio, and Anaeromyxobacter were the dominant groups in S. robustum. S. spontaneum contained Bradyrhizobium, Azospirillum, Methanobacterium, and Paenibacilllus species. Bradyrhizobium, Dechloromonas, Xanthobacter and Anaeromyxobacter were present in S. sinense. Genus Burkolderia was found in S. barberi, S. spontaneum, and S. sinense while Beijerinckia was recorded in S. barberi, S. spontaneum, and S. sinense. Genus Idenella was commonly present in all the samples except S. spontaneum, and Kosakonia was normally present in S. barberi and S. spontaneum.
Star analysis was conducted using the top 10 genera of each sample. The top 10 bacterial genera of 16S rRNA data-based analysis were Pseudarthobacter, Pseudomonas, Bacillus, Massilia, Gemmatimonas, Nitrospora, Haliangium, Ramibacter, Tumebacillus, and Lysobacter. From this analysis, the Bacillus genus was the leading one in S. officinarum and S. robustum samples, while Pseudomonas was the dominant one in S. barberi, S. robustum, S. spontaneum, and S. sinense. Tumebacillus genus was found only in S. spontaneum (Supplementary Figure S4A). Similarly, star analysis for nifH gene samples identified the presence of Bacillus in all the samples. Desulfovibrio was found in significant numbers in S. officinarum, S. robustum, and S. barberi. Xanthobacter was abundant in S. sinense. Anaeromyxobacter was found in all the samples except that of S. spontaneum. Pseudoacidovorax genus was found only in S. robustum, S. barberi, and S. sinense samples. Azospirillum and Methylobacterium were unique to S. spontaneum (Supplementary Figure S4B). The relative abundance of the top 20 diazotrophs at the genus level present in all sugarcane species is shown in Figure 6. Diazotrophs belonging to the genus Bradyrhizobium were present in all the samples tested.
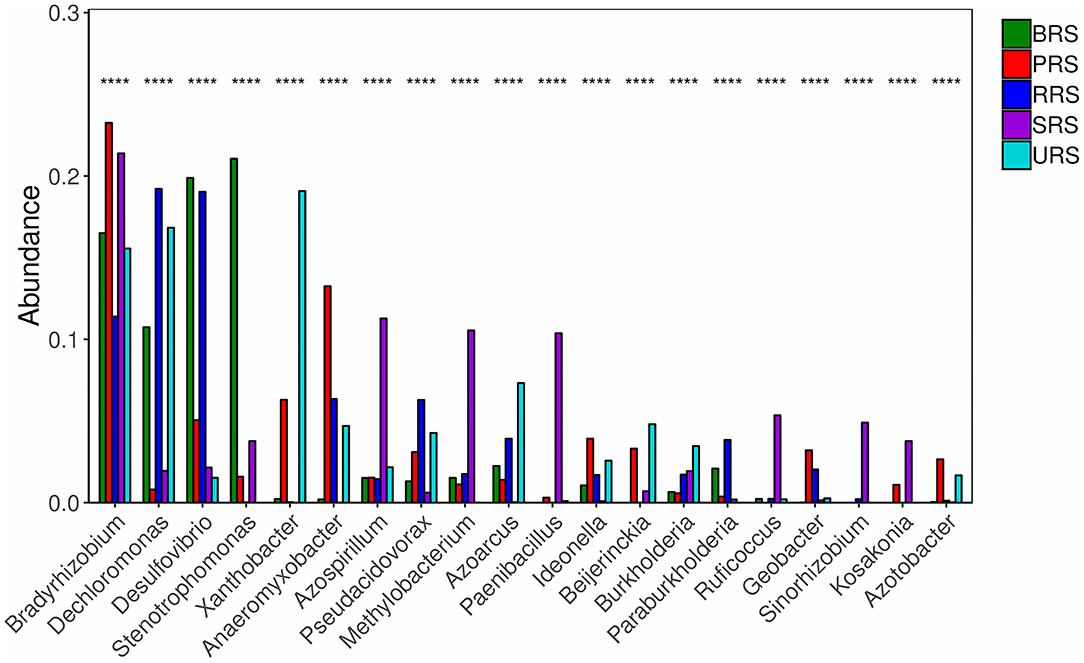
Figure 6. The relative abundances of the top 20 diazotrophs at the genus level in the soil of sugarcane species. S. officinarum L. cv Badila (BRS), S. barberi Jesw. cv Pansahi (PRS), S. robustum (RRS), S. spontaneum (SRS), and S. sinense Roxb. cv Uba (URS). Note: Select species with p < 0.05 to draw a column chart, each column is the relative abundance of different species, where **** means p < 0.0001.
Beta Diversity
Beta diversity measures species diversity among different samples collected from similar or different environments. We performed the PCoA and UniFrac-based cluster analysis to understand the beta diversity of 16S rRNA and nifH gene in the sugarcane species studied (Supplementary Figures S5, S6). Based on 16S rRNA data, S. sinense, along with S. barberi, and S. officinarum, along with S. robustum, formed independent clusters. Moreover, S. spontaneum was segregated away from all others, displaying higher beta diversity (Supplementary Figure S5A). Similarly based on nifH data, S. spontaneum was segregated away from all others samples (Supplementary Figure S6A). Hierarchical clustering based on the UniFrac cluster analysis showed similar results for both 16S rRNA and nifH gene data, containing identical sequences showing 0 (blue color) distances. S. sinense to S. spontaneum showed more distance (red color = 0.3), indicating dissimilarity in sequences of S. sinense to S. spontaneum samples (Supplementary Figures S5b, S6b).
The distribution of dominant genera based on their relative abundance performed with the Bray-Curtis algorithm showed Bacillus being the dominant genus in all the samples except those from S. spontaneum (Figure 7A). Pseudomonas was the second dominant genus, while Pseudarthobacter became the third leading one. S. spontaneum had most of the unidentified genera in our analysis. Similarly, the distribution of dominant genera in nifH samples showed the presence of Dechloromonas spp. in all samples except those from S. barberi. Xanthobacter and Bradyrhizobium were found to be prevalent in S. officinarum and S. sinense, respectively (Figure 7B).
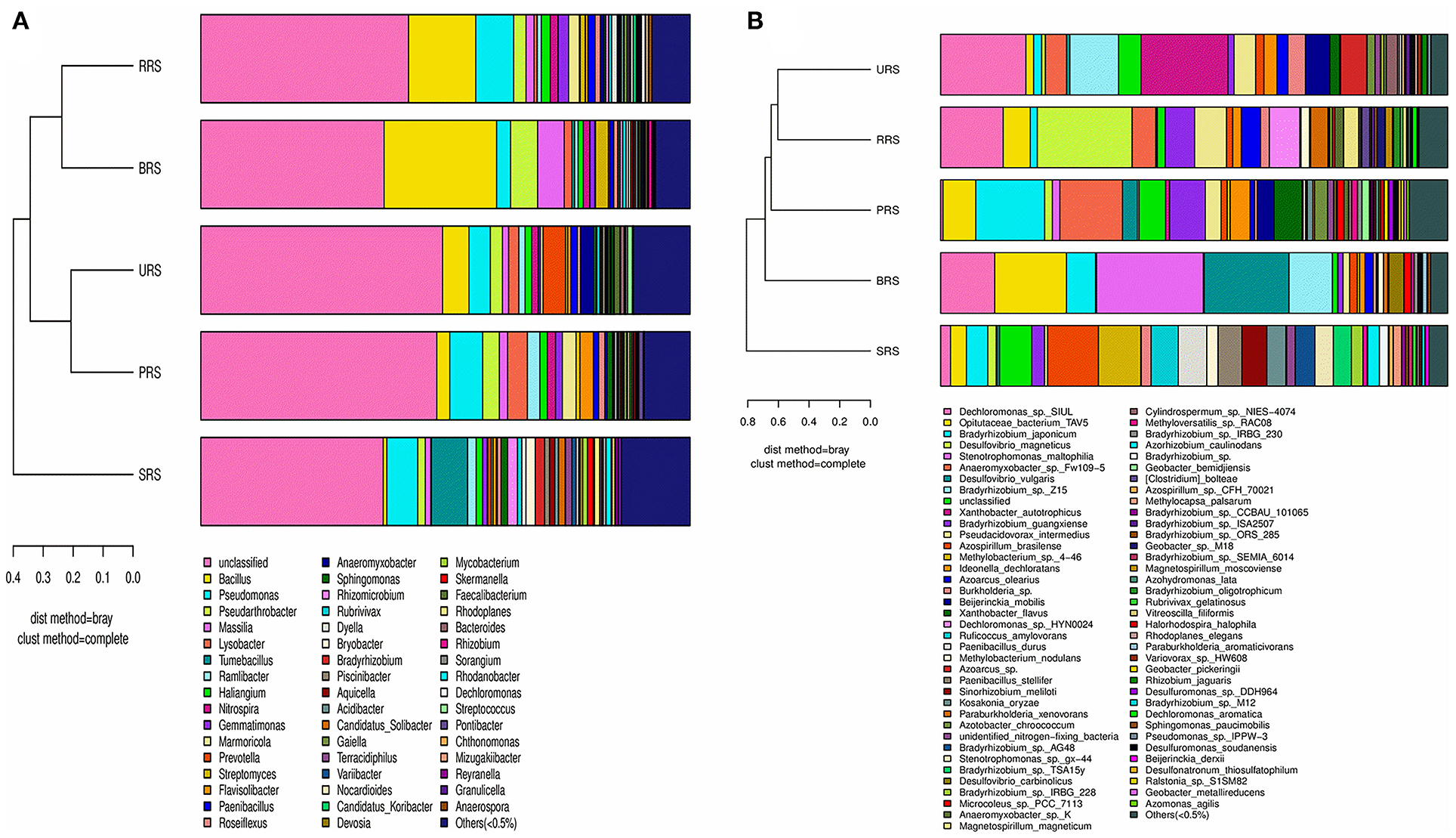
Figure 7. A combination of horizontal multi-sample similarity trees and histograms 16S rRNA (A) and nifH genes (B). On the left is hierarchical clustering between samples based on community composition (Bray-Curtis algorithm), and on the right is a column chart of the sample's community structure. S. officinarum L. cv Badila (BRS), S. barberi Jesw. cv Pansahi (PRS), S. robustum (RRS), S. spontaneum (SRS), and S. sinense Roxb. cv Uba (URS).
qPCR
To quantify bacterial abundance in all five species, we performed qPCR analysis using the 16S rRNA and nifH gene-specific primer (Figure 8). Occurrences of gene copy number, indicating bacterial abundance, in all species were as follows: S. sinense > S. robustum > S. barberi > S. officinarum > S. spontaneum (Figure 8A). The nifH gene copy number results showed that N-fixing bacterial population was highest in S. barberi followed by S. robustum > S. officinarum > S. sinense with the least in S. spontaneum (Figure 8B). Thus, S. sinense had the most considerable rhizospheric bacterial abundance, while N-fixing bacteria were most prolific in the rhizosphere of S. barberi.
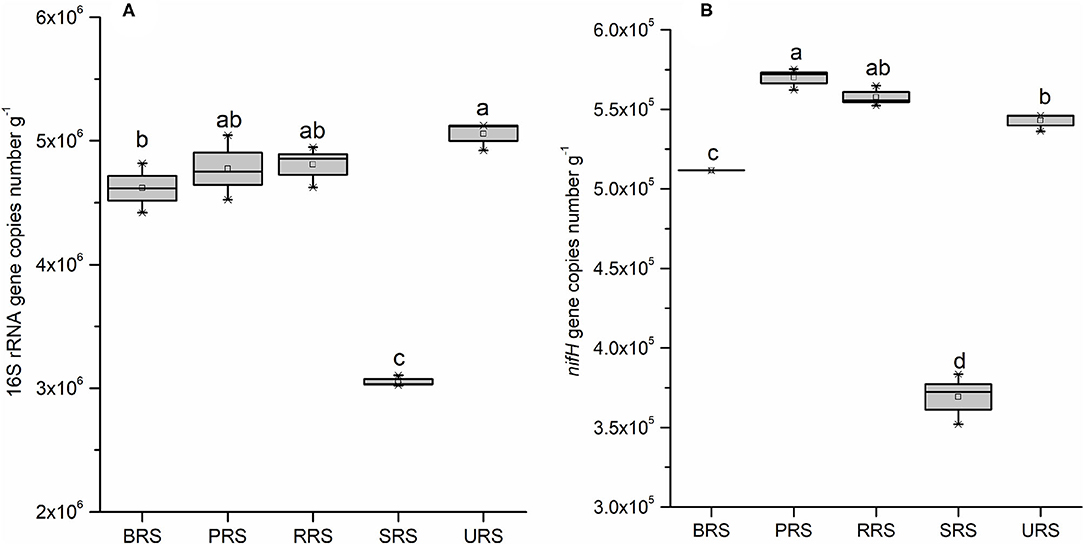
Figure 8. Box plot estimated values of gene copies of 16S rRNA (A), and nifH (B) in the soil samples of sugarcane species. Within each box, Small squares denote mean values; boxes extend from the 10th to the 90th percentile of each group's distribution of values. Values followed by the same lowercase letters in the same column are not significantly different according to DMRT 5%. DMRT, Duncan's Multiple Range Test.
Spearman's Rank Correlation
Spearman rank correlation analysis was calculated based on 16S rRNA and nifH gene abundance (top 50), diversity indices, gene copy, and soil variables; and values were illuminated in a heat map (Figure 9). Spearman's rank correlation coefficients were calculated to assess the association between soil physiochemical properties and dominant (16S rRNA and nifH) bacterial genera. The relationships were estimated between these taxa and physiochemical properties to understand the role of the microbial community shift. Related heat maps show that soil physiochemical factors significantly affected the relative abundance of bacterial taxa (phyla and genera). 16S rRNA bacterial abundance shows a strong positive correlation (r = 0.8, p < 0.1) between pH and abundance of bacteria, such as Nitrospira, Flavisolibacter, and Paenibacillus. OM had a positive correlation (p < 0.05) with Paenibacillus, Anaeromyxobactor, and Microvirga. Total N had a positive correlation (r = 0.9, p < 0.05) with Marmoricola and Gaiella. Total P had positive correlation (p < 0.05) with Bacillus (r = 0.9) and Streptomyces (r = 0.6). Total K had positive correlation (r = 0.9, p < 0.01) with Pseudarthobacter and Streptomyces. Available P had positive correlation with Pseudarthobacter (r = 0.9, p < 0.05). While, available K had positive correlation with Pseudarthobacter (r = 1, p < 0.05) and Massilia (r = 0.9, p < 0.05). Moreover, available NO-3N showed a positive correlation with Massilia (r = 0.9, p < 0.05), Terrabacter (r = 0.9, p < 0.5), and Streptomyces (r = 0.8, p < 0.1). Available NH+4N had a positive correlation with Lysobacter (r = 0.8, p < 0.1), Nitrospira (r = 0.9, p < 0.05), Paenibacillus (r = 0.9, p < 0.05), Anaeromyxobacter (r = 0.9, p < 0.05), and Steroidobacter (r = 0.9, p < 0.05). Paenibacillus and Anaeromyxobacter strongly correlated with obs, Chao, PD_whole tree, and OM. However, Nocardia had a significant negative correlation with total K, while Tumebacillus showed a negative correlation with pH. In the case of nifH gene, diversity indices (observed species, Ace, Chao) had a strong positive correlation with Anaeromyxobacter (r = 0.9, p < 0.05), Lachnoclostridium (r = 0.9, p < 0.05), and Vitreoscilla (r = 0.8, p < 0.1). The qRT-PCR data (nifH gene copy number) had a strong positive correlation (p < 0.5) with Anaeromyxobacter, Ideonella, Geobacter, Azotobacter, and Vitreoscilla. WC had positive correlation with Azoarcus (r = 0.9, p < 0.05), and soil pH showed a strong positive correlation with Ideonella (r = 0.8, p < 0.1), Azotobacter (r = 0.8, p < 0.1), Rhizobium (r = 0.9, p < 0.05), and Pseudomonas (r = 0.8, p < 0.1). Similarly, soil OM had a strong positive correlation with Azoarus (r = 1, p < 0.05), Pseudomonas (r = 0.9, p < 0.05), and Nostoc (r = 0.8, p < 0.1). Soil total N revealed a strong positive correlation with Vitreoscilla (r = 0.9, p < 0.05), and available K showed with Pseudosulfovibrio (r = 0.9, p < 0.05). While, soil available NO-3N was correlated positively to Desulfovibrio (r = 0.9, p < 0.05), Microcoleus (r = 0.97, p < 0.05), Halorhodospira (r = 0.8, p < 0.1), Sphingnomonas (r = 0.9, p < 0.05), and Pseudosulfovibrio (r = 0.9, p < 0.05). Moreover, soil available NH+4N showed strong correlation with Xanthobacter (r = 0.9, p < 0.05), Ideonella (r = 0.9, p < 0.05), Azotobacter (r = 0.9, p < 0.05), Pseudomonas (r = 0.9, p < 0.05), and Azohydromonas (r = 0.08, p < 0.1). However, pH was correlated significantly negative with the Stenotrophomonas (r = −0.9, p < 0.05), and OM was correlated strongly negative with Desulfarculus (r = −0.9, p < 0.05) and Anabaena (r = −0.9, p < 0.1). Similarly, soil available NH+4N showed a negative correlation with Methylobacter (r = −0.9, p < 0.05) and Desulfarculus (r = −0.9, p < 0.05).
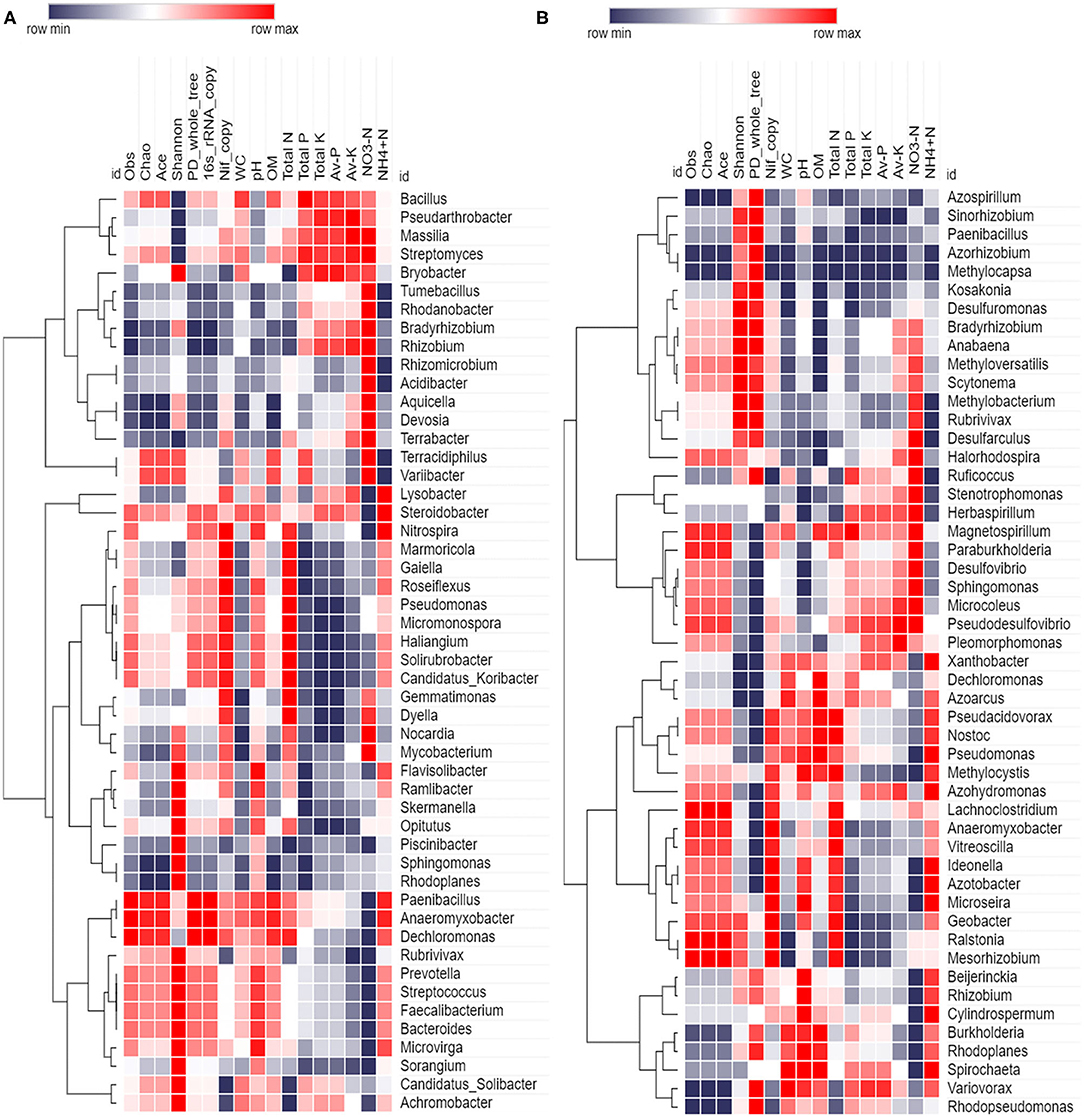
Figure 9. Spearman correlation heatmap based on abundance (top 50 genera), diversity indices, gene copy, and soil variables (A) 16S rRNA (B) nifH gene.
Discussion
The present study focuses on wild sugarcane progenitors because these species are distributed in different geographic distributions and service in various biotic and abiotic stresses (Santchurn et al., 2019). To understand the PGPRs and nitrogen-fixing diazotrophs, we need to study the sugarcane rhizosphere microbiome community. In most crops, such as sugarcane, a large proportion of PGPRs are yet to be identified. Bacteria are the most abundant of all the rhizospheric microbiota, and many are known to promote plant growth (Antoun and Prévost, 2005; van Loon, 2007; Dagnaw et al., 2015). The present study made an effort to understand the diverse and complex bacterial communities present in the rhizospheric soils of progenitors and closely related species of modern sugarcane hybrids as they play critical roles in plant nutrition, biotic and abiotic stress tolerance, and growth and development (Baldani et al., 2002; Li et al., 2017).
16S rRNA sequence data revealed 6,202 OTUs assigned to different bacterial species colonizing the rhizosphere of different sugarcane species studied. Analysis of these OTUs showed that S. sinense rhizosphere has the most significant number of rhizobacterial communities compared to other related species studied here. These results were also confirmed by qPCR analysis. qPCR analysis results demonstrated that 16S rRNA gene copy numbers were observed highest in S. sinense and lowest in S. spontaneum. The highest nifH gene numbers were observed in S. robustum and lowest in S. spontaneum. Besides this, alpha diversity analysis also predicted the highest bacterial diversity in S. sinense samples. However, other sugarcane species also showed considerable species diversity. Phylum level distribution studies identified the dominance of Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, Chloroflexi, Gemmatimonadetes, Planctomycetes, and Nitrospirae. Many of them were reported to be present in the sugarcane rhizosphere previously (Gao et al., 2019). Out of the total OTUs identified, 1,099 were from diazotrophs based on nifH gene data. The nifH gene sequence analysis helped to identify the top twenty genera (Bradyrhizobium, Dechloromonas, Desulfovibrio, Stenotrophomonas, Xanthobacter Anthobacter, Anaeromyxobacter, Azospirillum, Pseudoacidovorax, Methylobacterium, Azoarcus, Paenibacillus, Ideonella, Beijerinckia, Paraburkholderia, Burkholderia, Ruficoccus, Geobacter, Sinorhizobium, Kosakonia, and Azotobacter). Out of these 20 genera, most of the genera were found to fix nitrogen in sugarcane and other plants. Genus Bradyrhizobium is known to fix nitrogen in sorghum and sugarcane (de Matos et al., 2019; Hara et al., 2019). Azotobacter genus consists of seven different species and they are involved in atmospheric nitrogen fixation in different crops (Jiménez et al., 2011). Genera Azotobacter and Beijerinckia were studied for diazotrophic attributes in the early 1960s in rice and other cereal crops (Dobereiner, 1961; Pankievicz et al., 2019). Contrary to Azotobacter, the Beijerinckia genus is restricted mainly to the tropics, and its nitrogen fixation ability has been reported in various plants (Nassar et al., 2020). Kosakonia spp. fixes nitrogen on cucumber roots (Sun et al., 2018). Roots of switchgrass are inhabited with nitrogen-fixing bacteria that belong to the genera Dechloromonas, Desulfovibrio, Azoarcus, Ideonella, Paraburkholderia, and Burkholderia (Bahulikar et al., 2019). It is hard to identify and classify most bacteria in culture because of their morphological similarities. But, culture-independent methods, such as 16S rRNA sequencing, are highly efficient, cost-effective and provide accurate identification and classification of rhizobacteria. Overall, the dominant genera identified in this study are known to fix atmospheric nitrogen, facilitating plant growth.
In the present study, we observed a few taxa, such as Proteobacteria, Acidobacteria, Actinobacteria, Firmicute, Sphingomonas, Bradyrhizobium, and Gemmatimonas, were dominant with Bacillus, Pseudomonas, Bradyrhizobium, Burkholderia, and Rhizobium as leading genera. In our previous studies, we observed the occurrence of Proteobacteria, Acidobacteria, Actinobacteria, Firmicute, Bacteroidetes, Sphingomonas, Bradyrhizobium, Bryobacter, and Gemmatimonas in the sugarcane rhizosphere (Solanki et al., 2018, 2020). Some genera, such as Bacillus, Pseudomonas, and Burkholderia, known for their plant-growth-promoting and nitrogen-fixing properties, were found to be enriched in the sugarcane rhizosphere (Li et al., 2017; Malviya et al., 2019; Singh et al., 2020). Community composition analysis of 16S rRNA sequence data helped to track phylum and genus level distribution of rhizobacteria among different sugarcane species studied. Interestingly, we observed the presence of a few genera, namely Streptococcus, Rhodanobacter, Anaeromyxobacter, and Prevotella, shared among all the species studied here. The members of these genera were found commonly in soil and can colonize crop plants. From the previous reports, it appears that Rhodanobacter, Anaeromyxobacter, and Prevotella species were isolated from different soil and plant sources, and they were found to have nifH gene and nitrogen fixation abilities (Igai et al., 2016; Espenberg et al., 2018). Thus, we believe that more characterization of these bacteria colonizing the sugarcane rhizosphere will be beneficial for developing bio-based crop products to improve sugarcane crop productivity. Diversity among these nitrogen-fixing bacteria was revealed by alpha and beta diversity analyses. The top genera with the highest abundance were found to be Bacillus, Desulfovibrio, Xanthobacter, Anaeromyxobacter, Pseudoacidovorax, Azospirillum, and Methylobacterium. Among these, Bacillus is a common bacterial diazotroph in sugarcane (Reis et al., 2007). Desulfovibrio, Anaeromyxobacter, Azospirillum, and Xanthobacter are nitrogen fixers in rice (Sessitsch et al., 2012; Bao et al., 2014; Yoneyam et al., 2017; Rosenblueth et al., 2018). Correlation results showed that among all bacteria, genera Bacillus, Pseudomonas, Massilia, Pseudarthobacter, and Streptomyces correlate positively with various soil parameters. Genera Bacillus, Pseudomonas, and Streptomyces are well-known for plant growth and plant disease suppression activities in sugarcane and other crops (Wang et al., 2019; Amna et al., 2020; Chandra et al., 2020; Jiao et al., 2021). Genus Massilia is a significant group of rhizosphere and root colonizing bacteria of many plant species (Ofek et al., 2012). Genus Pseudarthobacter plays an essential role in biodegradation (Gupta et al., 2020; Chen et al., 2021). Genera Azotobacter and Ideonella revealed a positive correlation with NH+4N, and several previous reports also demonstrated that these bacteria could fix atmospheric nitrogen efficiently and enhance the crop yield (Bhromsiri and Bhromsiri, 2010; Aasfar et al., 2021). Diazotroph Anaeromyxobacter correlated positively with NH+4N and recently reported that Anaeromyxobacter enhances the nitrogen-fixing activity of paddy soils (Masuda et al., 2020). Desulfovibrio had a positive correlation with available NO-3N. Desulfovibrio is known for its sulfur-reducing property and plays a particular and significant role in the fixation of biological nitrogen (Mistry et al., 2022). On the contrary, soil physiochemical, such as OM, showed a negative correlation with genera Desulfarculus and Anabaena, while NH+4N showed a negative correlation with genera Methylobacter and Desulfarculus. It may be due to a decrease of soil nutrients in the sugarcane rhizosphere that increases microbial completion (Jones et al., 2018; Solanki et al., 2020).
Conclusions
The sugarcane species studied here showed a significant number of N-fixing rhizobacteria, which strengthens the contention that exploring rhizosphere bacteria may help to develop a sustainable low resource input sugarcane crop production system, particularly, for meeting its N requirement from atmospheric nitrogen fixation. Substantial genetic variation for rhizobacteria, such as diazotroph communities, exists among different progenitors and closely related species of modern cultivated sugarcane hybrids. However, considering the vast natural habitats of these wild species spanning tropics and subtropics, similar studies using accessions sourced from different locations and environmental conditions will significantly advance our understanding of sugarcane rhizobiome. Future research should also focus on Saccharum species introgression breeding and isolation and practical application of beneficial PGPRs, especially diazotrophs. Filling the significant knowledge gap on microbiota and sugarcane interactions is critical for exploiting these beneficial microbes for sustainable sugarcane agriculture.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
MM and C-NL conceived and designed the experiments and performed the experiments. MM, C-NL, and MS analyzed the data. ZW, ACS, KV, AS, RS, PS, and ED contributed to reagents/materials/analysis tools. Y-RL contributed to project administration. C-NL, QN, X-PS, and Y-RL contributed to funding acquisition. MM, MS, and PL contributed in writing-original draft and revision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Guangxi Innovation Teams of Modern Agriculture Technology (No. gjnytxgxcxtd-2021-03-01 to Y-RL), National Natural Science Foundation of China (Nos. 31701489 to QN, 31801288 to C-NL, and 31901594 to X-PS), the Natural Science Foundation of Guangxi Province (Nos. 2019GXNSFDA185004 and 2021GXNSFAA196041 to C-NL), and Guangxi Academy of Agricultural Sciences Fund (Nos. Guinongke 2021YT09 and Guinongke 2021JM01 to C-NL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors extend their appreciation to National Natural Science Foundation of China, China Postdoctoral Foundation, the Youth program of the National Natural Science Foundation of China, Guangxi Innovation Term of Modern Agriculture Technology, and Guangxi Academy of Agricultural Sciences, Nanning, Guangxi, China, for providing the necessary funding and facilities for this study. The authors also extend their appreciation to Taif University Researchers Supporting Project number (TURSP-2020/85).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.829337/full#supplementary-material
References
Aasfar, A., Bargaz, A., Yaakoubi, K., Hilali, A., Bennis, I., Zeroual, Y., et al. (2021). Nitrogen fixing azotobacter species as potential soil biological enhancers for crop nutrition and yield stability. Front. Microbiol. 12, 628379. doi: 10.3389/fmicb.2021.628379
Ahmad, F., Ahmad, I., Aqil, F., Ahmed Wani, A., and Sousche, Y. S. (2016). Plant growth promoting potential of free-living diazotrophs and other rhizobacteria isolated from Northern Indian soil. Biotechnol. J. 1, 1112–1123. doi: 10.1002/biot.200600132
Amna, Xia, Y., Farooq, M. A., Javed, M. T., Kamran, M. A., Mukhtar, T., Ali, J., et al. (2020). Multi-stress tolerant PGPR Bacillus xiamenensis PM14 activating sugarcane (Saccharum officinarum L.) red rot disease resistance. Plant Physiol. Biochem. 151, 640–649. doi: 10.1016/j.plaphy.2020.04.016
Antoun, H., and Prévost, D. (2005). “Ecology of plant growth promoting rhizobacteria,” in PGPR: Biocontrol and Biofertilization, ed Z. A. Siddiqui (Netherlands: Springer), 1–38. doi: 10.1007/1-4020-4152-7_1
Armanhi, J. S., de Souza, R. S., Damasceno, N. D., de Araújo, L. M., Imperial, J., and Arruda, P. A. (2018). Community-based culture collection for targeting novel plant growth-promoting bacteria from the sugarcane microbiome. Front. Plant Sci. 8, 2191. doi: 10.3389/fpls.2017.02191
Avis, T. J., Gravel, V., Antoun, H., and Tweddell, R. J. (2008). Multifaceted beneficial effects of rhizosphere microorganisms on plant health and productivity. Soil Biol. Biochem. 40, 1733–1740. doi: 10.1016/j.soilbio.2008.02.013
Bahulikar, R. A., Chaluvadi, S. R., Torres-Jerez, I., Mosali, J., Bennetzen, J. L., and Udvardi, M. (2019). Nitrogen fertilization reduces nitrogen fixation activity of diverse diazotrophs in switchgrass roots. Phytobiomes J. 5, 80–87. doi: 10.1094/PBIOMES-09-19-0050-FI
Baldani, J. I., Reism, V. M., Baldani, V. L., and Döbereiner, J. (2002). A brief story of nitrogen fixation in sugarcane-reasons for success in Brazil. Funct. Plant Biol. 29, 417–423. doi: 10.1071/PP01083
Bao, S. D.. (2002). Soil Agricultural Chemical Analysis, 3rd Edn. Beijing: China Agricultural Press.
Bao, Z., Okubo, T., Kubota, K., Kasahara, Y., Tsurumaru, H., Anda, M., et al. (2014). Metaproteomic identification of diazotrophic methanotrophs and their localization in root tissues of field-grown rice plants. Appl. Environ. Microbiol. 80, 5043–5052. doi: 10.1128/AEM.00969-14
Barbier, F. F., Chabikwa, T. G., Ahsan, M. U., Cook, S. E., Powell, R., Tanurdzic, M., et al. (2019). A phenol/chloroform-free method to extract nucleic acids from recalcitrant, woody tropical species for gene expression and sequencing. Plant Methods 15, 62. doi: 10.1186/s13007-019-0447-3
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bhromsiri, C., and Bhromsiri, A. (2010). Isolation, screening of growth-promoting activities and diversity of rhizobacteria from vetiver grass and rice plants. Thai J. Agric. Sci. 43, 217–230.
Brackin, R., Robinson, N., Lakshmanan, P., and Schmidt, S. (2013). Microbial function in adjacent subtropical forest and agricultural soil. Soil Biol. Biochem. 57, 68–77. doi: 10.1016/j.soilbio.2012.07.015
Bremmer, J. M., and Mulvaney, C. S. (1982). “Total nitrogen,” in methods of soil analysis—Part 2,” in Chemical and Microbiological Properties, ed. C. A. Bluck (Madison: American Society of Agronomy), 595–624.
Caporaso, J., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cawoy, H., Bettiol, W., Fickers, P., and Ongena, M. (2011). Bacillus-based biological control of plant diseases. Pesticides Modern World-Pesticides Use Manage. 21, 273–302. doi: 10.5772/17184
Chandra, H., Kumari, P., Bisht, R., Prasad, R., and Yadav, S. (2020). Plant growth promoting Pseudomonas aeruginosa from Valeriana wallichii displays antagonistic potential against three phytopathogenic fungi. Mol. Biol. Rep. 47, 6015–6026. doi: 10.1007/s11033-020-05676-0
Chen, F. Y., Chen, Y., Chen, C., Feng, L., Dong, Y., Chen, J., et al. (2021). High-efficiency degradation of phthalic acid esters (PAEs) by Pseudarthro bacter defluvii E5: performance, degradative pathway, and key genes. Sci. Total Environ. 794, 148719. doi: 10.1016/j.scitotenv.2021.148719
Dagnaw, F., Assefa, F., Gebrekidan, H., and Argaw, A. (2015). Characterization of plant growth promoting bacteria from sugarcane (Saccharum officinarum L.) rhizosphere of Wonji-shoa sugar estate and farmers landraces of Ethiopia. Biotechnol. 14, 58. doi: 10.3923/biotech.2015.58.64
de Matos, G. F., Zilli, J. E., de Araújo, J. L. S., Parma, M. M., Melo, I. S., Radl, V., et al. (2019). Bradyrhizobium sacchari sp. nov., a legume nodulating bacterium isolated from sugarcane roots. Arch. Microbiol. 199, 1251–1258. doi 10.1007/s00203-017-1398-6
Dini-Andreote, F., Andreote, F. D., Costa, R., Taketani, R. G., van Elsas, J. D., and Araújo, W. L. (2010). Bacterial soil community in a Brazilian sugarcane field. Plant Soil 336, 337–349. doi: 10.1007/s11104-010-0486-z
Dobereiner, J.. (1961). Nitrogen-fixing bacteria of the genus Beijerinckia Derx in the rhizosphere of sugar cane. Plant Soil. 15, 211–216. doi: 10.1007/BF01400455
Dong, L., Xu, J., Zhang, L., Yang, J., Liao, B., Li, X., et al. (2017). High-throughput sequencing technology reveals that continuous cropping of American ginseng results in changes in the microbial community in arable soil. Chinese Med. 12, 18. doi: 10.1186/s13020-017-0139-8
Dong, M., Yang, Z., Cheng, G., Peng, L., Xu, Q., and Xu, J. (2018). Diversity of the bacterial microbiome in the roots of four saccharum species: S. spontaneum, S. robustum, S. barberi, and S. officinarum. Front. Microbiol. 9, 267. doi: 10.3389/fmicb.2018.00267
Edgar, R. C.. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5, 113. doi: 10.1186/1471-2105-5-113
Edgar, R. C.. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinform 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Espenberg, M., Truu, M., Mander, Ü., Kasak, K., Nõlvak, H., Ligi, T., et al. (2018). Differences in microbial community structure and nitrogen cycling in natural and drained tropical peat land soils. Sci. Rep. 8, 4742. doi: 10.1038/s41598-018-23032-y
Finkel, O. M., Castrillo, G., Herrera Paredes, S., Salas González, I., and Dangl, J. L. (2017). Understanding and exploiting plant beneficial microbes. Curr. Opin. Plant Biol. 38, 155–163. doi: 10.1016/j.pbi.2017.04.018
Fischer, D., Pfitzner, B., Schmid, M., Simoes-Araujo, J. L., Reis, V. M., Pereira, W., et al. (2012). Molecular characterization of the diazotrophic bacterial community in uninoculated and inoculated field-grown sugarcane (Saccharum sp.). Plant Soil 356, 83–99. doi: 10.1007/s11104-011-0812-0
Gaikwad, S., and Sapre, V. (2015). Structural and functional diversity of rhizobacterial strains isolated from rhizospheric zone of different plants of Sholapur- Maharashtra Region, India. Int. J. Curr. Microbiol. Appl. Sci. 4, 263–273.
Gao, X., Wu, Z., Liu, R., Wu, J., Zeng, Q., and Qi, Y. (2019). Rhizosphere bacterial community characteristics over different years of sugarcane ratooning in consecutive monoculture. BioMed. Res. Int. 2019:4943150. doi: 10.1155/2019/4943150
Goldemberg, J., Coelho, S. T., and Guardabassi, P. (2008). The sustainability of ethanol production from sugarcane. Energy Policy 36, 2086–2097. doi: 10.1016/j.enpol.2008.02.028
Gong, B., Cao, H., Peng, C., Perčulija, V., Tong, G., and Fang, H. (2019). High-throughput sequencing and analysis of microbial communities in the mangrove swamps along the coast of Beibu Gulf in Guangxi, China. Sci. Rep. 9, 9377. doi: 10.1038/s41598-019-45804-w
Gupta, N., Skinner, K. A., Summers, Z. M., Edirisinghe, J. N., Weisenhorn, P. B., Faria, J. P., et al. (2020). Draft genome sequence of Pseudarthrobacter sp. strain ATCC 49442 (Formerly Micrococcus luteus), a pyridine-degrading bacterium. Microbiol. Resour. Announc. 9, e00299-20 doi: 10.1128/MRA.00299-20
Hammer, Ø., Harper, D. A. T., and Ryan, P. D. (2001). Past: paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4, 178. Available online at: http://palaeo-electronica.org/2001_1/past/issue1_01.htm
Hara, S., Morikawa, T., Wasai, S., Kasahara, Y., Koshiba, T., Yamazaki, K., et al. (2019). Identification of nitrogen-fixing bradyrhizobium associated with roots of field-grown sorghum by metagenome and proteome analyses. Front. Microbiol. 10, 407. doi: 10.3389/fmicb.2019.00407
Igai, K., Itakura, M., Nishijima, S., Tsurumaru, H., Suda, W., Tsutaya, T., et al. (2016). Nitrogen fixation and nifH diversity in human gut microbiota. Sci. Rep. 6, 31942. doi: 10.1038/srep31942
Inui-Kishi, R. N., Kishi, L. K., Picchi, S. C., Barbosa, J. C., Lemos, M. T. O., Marcondes, J., et al. (2012). Phosphorus solubilizing and IAA production activities in plant growth promoting rhizobacteria from Brazilian soils under sugarcane cultivation. J. Eng. Appl. Sci. 27, 1819–6608.
Jangu, O. P., and Sindhu, S. S. (2011). Differential response of inoculation with indole acetic acid producing Pseudomonas sp. in green gram (Vignaradiata L.) and black gram (Vignamungo L.). Microbiol. J. 1, 159–173. doi: 10.3923/mj.2011.159.173
Jiao, X., Takishita, Y., Zhou, G., and Smith, D. L. (2021). Plant associated rhizobacteria for biocontrol and plant growth enhancement. Front. Plant Sci. 17, 634796. doi: 10.3389/fpls.2021.634796
Jiménez, D. J., Montaña, J. S., and Martínez, M. M. (2011). Characterization of free nitrogen fixing bacteria of the genus azotobacterin organic vegetable-grown colombian soils. Braz. J. Microbiol. 42, 1517–8382. doi: 10.1590/S1517-83822011000300003
Jones, D. L., Magthab, E. A., Gleeson, D. B., Hill, P. W., Sánchez-Rodríguez, A. R., Roberts, P., et al. (2018). Microbial competition for nitrogen and carbon is as intense in the subsoil as in the topsoil. Soil Biol. Biochem. 117, 72–82. doi: 10.1016/j.soilbio.2017.10.024
Kashyap, A. S., Pandey, V. K., Kannojia, P., Singh, U., and Sharma, P. K. (2017). “Role of plant growth-promoting rhizobacteria for improving crop productivity in sustainable agriculture,” in Plant-Microbe Interactions in Agro-Ecological Perspectives, eds D. Singh, H. Singh, and R. Prabha (Singapore: Springer), pp. 673–693. doi: 10.1007/978-981-10-6593-4_28
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, A., Maurya, B. R., and Raghuwanshi, R. (2014). Isolation and characterization of PGPR and their effect on growth, yield and nutrient content in wheat (Triticum aestivum L.). Biocatal. Agric. Biotechnol. 3, 121–128. doi: 10.1016/j.bcab.2014.08.003
Lamizadeh, E., Enayatizamir, N., and Motamedi, H. (2016). Isolation and identification of plant growth-promoting rhizobacteria (pgpr) from the rhizosphere of sugarcane in saline and non-saline soil. Int. J. Curr. Microbiol. Appl. Sci. 5, 1072–1083. doi: 10.20546/ijcmas.2016.510.113
Li, H. B., Singh, R. K., Singh, P., Song, Q. Q., Xing, Y. X., Yang, L. T., et al. (2017). Genetic diversity of nitrogen-fixing and plant growth promoting Pseudomonas species isolated from sugarcane rhizosphere. Front. Microbiol. 8, 1268. doi: 10.3389/fmicb.2017.01268
Li, Y. R., and Yang, L. T. (2015). Sugarcane agriculture and sugar industry in China. Sugar Tech. 17, 1–8. doi: 10.1007/s12355-014-0342-1
Magoè, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinform 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Malviya, M. K., Solanki, M. K., Li, C. N., Htun, R., Singh, R. K., Singh, P., et al. (2019). Beneficial linkages of endophytic Burkholderia anthina MYSP113 towards sugarcane growth promotion. Sugar Tech. 21, 737–748. doi: 10.1007/s12355-019-00703-2
Masuda, Y., Yamanaka, H., Xu, Z. X., Shiratori, Y., Aono, T., Amachi, S., et al. (2020). Diazotrophic anaeromyxobacter isolates from soils. Appl. Environ. Microbiol. 86, e00956-20. doi: 10.1128/AEM.00956-20
Mehnaz, S.. (2011). Plant growth-promoting bacteria associated with sugarcane. in bacteria in agrobiology. Crop Ecosyst. 11, 165–187. doi: 10.1007/978-3-642-18357-7_7
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, 566–570. doi: 10.1093/nar/gkv468
Mistry, H., Thakor, R., and Bariy, H. (2022). “Isolation and identification of associative symbiotic n2 fixing microbes: Desulfovibrio,” in Practical Handbook on Agricultural Microbiology, eds N. Amaresan, P. Patel, and D. Amin (New York, NY: Springer Protocols Handbooks), 77–83. doi: 10.1007/978-1-0716-1724-3_10
Moore, T. A., Xing, Y., Lazenby, B., Lynch, M. D. J., Schiff, S., Robertson, W. D., et al. (2011). Prevalence of anaerobic ammonium-oxidizing bacteria in contaminated groundwater. Environ. Sci. Technol. 45, 7217–7225. doi: 10.1021/es201243t
Nassar, R., Seleem, E. A., Caruso, G., Sekara, A., and Abdelhamid, M. T. (2020). The Nitrogen-fixing bacteria—effective enhancers of growth and chemical composition of Egyptian henbane under varied mineral n nutrition. Agronomy 10, 921. doi: 10.3390/agronomy10070921
Ofek, M., Hadar, Y., and Minz, D. (2012). Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS One 7, e40117. doi: 10.1371/journal.pone.0040117
Pankievicz, V. C. S., Irving, T. B., Maia, L. G. S., and An,é, J. M. (2019). Are we there yet? The long walk towards the development of efficient symbiotic associations between nitrogen-fixing bacteria and non-leguminous crops. BMC Biol. 17, 99. doi: 10.1186/s12915-019-0710-0
Pereira, L. B., Andrade, G. S., Meneghin, S. P., Vicentini, R., and Ottoboni, L. M. M. (2020). Prospecting plant growth promoting bacteria isolated from the rhizosphere of sugarcane under drought stress. Curr. Microbiol. 76, 1345–1354. doi: 10.1007/s00284-019-01749-x
Pisa, G., Magnani, G. S., Weber, H., Souza, E. M., Faoro, H., Monteiro, R. A., et al. (2011). Diversity of 16S rRNA genes from bacteria of sugarcane rhizosphere soil. Braz. J. Med. Biol. Res. 4, 1215–1221. doi: 10.1590/S0100-879X2011007500148
Poly, F., Monrozier, L. J., and Bally, R. (2001). Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res. Microbiol. 152, 95–103. doi: 10.1016/S0923-2508(00)01172-4
Prashar, P., Kapoor, N., and Sachdeva, S. (2014). Rhizosphere: its structure, bacterial diversity and significance. Rev. Environ. Sci. Biotechnol. 13, 63–77. doi: 10.1007/s11157-013-9317-z
Ranjard, L., Poly, F., and Nazaret, S. (2000). Monitoring complex bacterial communities using culture-independent molecular techniques: application to soil environment. Res. Microbiol. 151, 167–177. doi: 10.1016/S0923-2508(00)00136-4
Reis, V., Lee, S., and Kennedy, C. (2007). “Biological nitrogen fixation in sugarcane,” in Associative and Endophytic Nitrogen-fixing Bacteria and Cyanobacterial Associations, vol 5, eds C. Elmerich, and W. E. Newton (Dordrecht: Springer), pp. 213–232.
Reuter, J. A., Spacek, D. V., and Snyder, M. P. (2015). High-throughput sequencing technologies. Mol. Cell. 58, 586–597. doi: 10.1016/j.molcel.2015.05.004
Rosa, P. A. L., Mortinho, E. S., Jalal, A., Galindo, F. S., Buzetti, S., Fernandes, G. C., et al. (2020). Inoculation with growth-promoting bacteria associated with the reduction of phosphate fertilization in sugarcane. Front. Environ. Sci. 8, 32. doi: 10.3389/fenvs.2020.00032
Rosenblueth, M., Ormeño-Orrillo, E., López-López, A., Rogel, M. A., Reyes-Hernández, B. J., Martínez-Romero, J. C., et al. (2018). Nitrogen fixation in cereals. Front. Microbiol. 9, 1794. doi: 10.3389/fmicb.2018.01794
Santchurn, G., Badaloo, M. G. H., Zhou, M., and Labuschagne, M. T. (2019). Contribution of sugarcane crop wild relatives in the creation of improved varieties in Mauritius. Plant Genet. Resour. 17, 151–163. doi: 10.1017/S1479262118000552
Schultz, N., Pereira, W., Silva, P., d, A., Baldani, J. I., Boddey, R. M., et al. (2017). Yield of sugarcane varieties and their sugar quality grown in different soil types and inoculated with a diazotrophic bacteria consortium. Plant Produc. Sci. 20, 366–374. doi: 10.1080/1343943X.2017.1374869
Sessitsch, A., Hardoim, P., Döring, J., Weilharter, A., Krause, A., and Woyke, T. (2012). Functional characteristics of an endophyte community colonizing rice roots as revealed by metagenomic analysis. Mol. Plant-Microbe Interact. 25, 28–36. doi: 10.1094/MPMI-08-11-0204
Shoko, M. D., Tagwira, F., and Zhou, M. (2007). The potential of reducing nitrogen fertilizers in a soybean-sugarcane production system in Zimbabwe. Afr. J. Agric. Res. 2, 475–480. Available online at: https://academicjournals.org/journal/AJAR/article-full-text-pdf/D6D871A33883 (accessed July 26, 2007).
Singh, R. K., Singh, P., Li, H. B., Song, Q. Q., Guo, D. J., Solanki, M. K., et al. (2020). Diversity of nitrogen-fixing rhizobacteria associated with sugarcane: a comprehensive study of plant-microbe interactions for growth enhancement in Saccharum spp. BMC plant Biol. 20, 220. doi: 10.1186/s12870-020-02400-9
Sokal, M. A.. (1958). Statistical method for evaluating systematic relationships. Univ. Kansas Sci. Bull. 38, 1409–1438.
Solanki, M. K., Wang, F. Y., Li, C. N., Wang, Z., Lan, T. J., Singh, R. K., et al. (2019). Impact of sugarcane–legume intercropping on diazotrophic microbiome. Sugar Tech. 22, 52–64. doi: 10.1007/s12355-019-00755-4
Solanki, M. K., Wang, F. Y., Wang, Z., Li, C. N., Lan, T. J., Singh, R. K., et al. (2018). Rhizospheric and endospheric diazotrophs mediated soil fertility intensification in sugarcane-legume intercropping systems. J. Soil Sedi. 19, 1911–1927. doi: 10.1007/s11368-018-2156-3
Solanki, M. K., Wang, Z., Wang, F. Y., Li, C. N., Lal, C. G., Singh, R. K., et al. (2020). Assessment of diazotrophic proteobacteria in sugarcane rhizosphere when intercropped with legumes (Peanut and Soybean) in the field. Front. Microbiol. 11, 1814. doi: 10.3389/fmicb.2020.01814
Sun, S., Chen, Y., Cheng, J., Li, Q., Zhang, Z., and Lan, Z. (2018). Isolation, characterization, genomic sequencing, and GFP-marked insertional mutagenesis of a high-performance nitrogen-fixing bacterium, Kosakonia radicincitans GXGL-4A and visualization of bacterial colonization on cucumber roots. Folia Microbiol. 63, 789–802. doi: 10.1007/s12223-018-0608-1
Swarnalakshmi, K., Yadav, V., Tyagi, D., Dhar, D. W., Kannepalli, A., and Kumar, S. (2020). Significance of plant growth promoting rhizobacteria in grain legumes: growth promotion and crop production. Plants 9, 1596. doi: 10.3390/plants9111596
Teixeira, L. C., Peixoto, R. S., Cury, J. C., Sul, W. J., Pellizari, V. H., Tiedje, J., et al. (2010). Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J. 4, 989–1001. doi: 10.1038/ismej.2010.35
Tsegaye, Z., Gizaw, B., Tefera, G., Feleke, A., Chaniyalew, S., Alemu, T., et al. (2019). Isolation and biochemical characterization of Plant Growth Promoting (PGP) bacteria colonizing the rhizosphere of Tef crop during the seedling stage. J. Plant Sci. Phytopathol. 3, 013–027. doi: 10.29328/journal.jpsp.1001027
Uroz, S., Ioannidis, P., Lengelle, J., Cébron, A., Morin, E., Buée, M., et al. (2013). Functional assays and metagenomic analyses reveals differences between the microbial communities inhabiting the soil horizons of a Norway spruce plantation. PLoS One 8:e55929. doi: 10.1371/journal.pone.0055929
van Loon, L. C.. (2007). Plant responses to plant growth promoting bacteria. Euro. J. Plant Pathol. 119, 243–254. doi: 10.1007/s10658-007-9165-1
Velineni, S., and Brahmaprakash, G. P. (2011). Survival and phosphate solubilizing ability of Bacillus megaterium in liquid inoculants under high temperature and desiccation stress. J. Agric. Sci. Technol. 13, 795–802.
Walkley, A., and Black, I. A. (1934). An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 37, 29–38. doi: 10.1097/00010694-193401000-00003
Wang, Z., Solanki, M. K., Yu, Z. X., Yang, L. T., An, Q. L., Dong, D. F., et al. (2019). Draft genome analysis offers insights into the mechanism by which Streptomyces chartreusis wzs021 increases drought tolerance in sugarcane. Front. Microbiol. 9, 3262. doi: 10.3389/fmicb.2018.03262
Wei, Y. J., Wu, Y., Yan, Y. Z., Zou, W., Xue, J., Ma, W. R., et al. (2018). High-throughput sequencing of microbial community diversity in soil, grapes, leaves, grape juice and wine of grapevine from China. PloS one 13:e0193097. doi: 10.1371/journal.pone.0193097
Xu, D., Yu, X., Yang, J., Zhao, X., and Bao, Y. (2020). High-throughput sequencing reveals the diversity and community structure in rhizosphere soils of three endangered plants in western ordos, China. Curr. Microbiol. 77, 2713–2723. doi: 10.1007/s00284-020-02054-8
Yoneyam, T., Terakado-Tonooka, J., and Minamisawa, K. (2017). Exploration of bacterial N2-fixation systems in association with soil-grown sugarcane, sweet potato, and paddy rice: a review and synthesis. Soil Sci. Plant Nutri. 63, 578–590. doi: 10.1080/00380768.2017.1407625
Zhang, B., Penton, C. R., Xue, C., Wang, Q., Zheng, T., and Tiedje, J. M. (2015). Evaluation of the Ion Torrent Personal Genome Machine for gene-targeted studies using amplicons of the nitrogenase gene nifH. Appl. Environ. Microbiol. 81, 4536–4545. doi: 10.1128/AEM.00111-15
Keywords: rhizosphere soil, sugarcane, nifH, 16S rRNA, diazotroph diversity
Citation: Malviya MK, Li C-N, Lakshmanan P, Solanki MK, Wang Z, Solanki AC, Nong Q, Verma KK, Singh RK, Singh P, Sharma A, Guo D-J, Dessoky ES, Song X-P and Li Y-R (2022) High-Throughput Sequencing-Based Analysis of Rhizosphere and Diazotrophic Bacterial Diversity Among Wild Progenitor and Closely Related Species of Sugarcane (Saccharum spp. Inter-Specific Hybrids). Front. Plant Sci. 13:829337. doi: 10.3389/fpls.2022.829337
Received: 05 December 2021; Accepted: 04 January 2022;
Published: 24 February 2022.
Edited by:
Freddy Mora-Poblete, University of Talca, ChileReviewed by:
Peifang Zhao, Yunnan Academy of Agricultural Sciences, ChinaJay Prakash Verma, Banaras Hindu University, India
Copyright © 2022 Malviya, Li, Lakshmanan, Solanki, Wang, Solanki, Nong, Verma, Singh, Singh, Sharma, Guo, Dessoky, Song and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiu-Peng Song, xiupengsong@gxaas.net; Yang-Rui Li, liyr@gxaas.net; orcid.org/0000-0002-7559-9244
†These authors have contributed equally to this work