
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 11 February 2022
Sec. Plant Physiology
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.807249
This article is part of the Research Topic Functions of Nitric Oxide in Photosynthetic Organisms View all 8 articles
 Valérie Nicolas-Francès1†‡
Valérie Nicolas-Francès1†‡ Jordan Rossi1‡Claire Rosnoblet1†
Jordan Rossi1‡Claire Rosnoblet1† Carole Pichereaux2,3†Siham Hichami1Jeremy Astier1†Agnès Klinguer1David Wendehenne1†
Carole Pichereaux2,3†Siham Hichami1Jeremy Astier1†Agnès Klinguer1David Wendehenne1† Angélique Besson-Bard1*†
Angélique Besson-Bard1*†Tyrosine-specific protein tyrosine phosphatases (Tyr-specific PTPases) are key signaling enzymes catalyzing the removal of the phosphate group from phosphorylated tyrosine residues on target proteins. This post-translational modification notably allows the regulation of mitogen-activated protein kinase (MAPK) cascades during defense reactions. Arabidopsis thaliana protein tyrosine phosphatase 1 (AtPTP1), the only Tyr-specific PTPase present in this plant, acts as a repressor of H2O2 production and regulates the activity of MPK3/MPK6 MAPKs by direct dephosphorylation. Here, we report that recombinant histidine (His)-AtPTP1 protein activity is directly inhibited by H2O2 and nitric oxide (NO) exogenous treatments. The effects of NO are exerted by S-nitrosation, i.e., the formation of a covalent bond between NO and a reduced cysteine residue. This post-translational modification targets the catalytic cysteine C265 and could protect the AtPTP1 protein from its irreversible oxidation by H2O2. This mechanism of protection could be a conserved mechanism in plant PTPases.
Protein phosphatases and protein kinases are key enzymes that regulate many biological processes by protein dephosphorylation or phosphorylation, respectively. In humans, 197 protein phosphatases were identified. This number is quite comparable to that of Arabidopsis thaliana, as 150 protein phosphatases have already been identified (Chen et al., 2017; Bheri et al., 2021). In Eukaryotes, protein phosphatases are classified in two major categories according to the amino acid target residue(s) of the dephosphorylation process: protein tyrosine (Tyr) phosphatases (PTPases; EC 3.1.3.48) and phosphoprotein serine/threonine (Ser/Thr) phosphatases (PPases). PTPases are themselves divided into two subgroups: tyrosine-specific protein tyrosine phosphatases (Tyr-specific PTPases), which include intracellular PTPases and receptor-like PTPases, and dual-specificity PTPases that constitute a particular subgroup of PTPases since they are able to hydrolyze both phosphoserine/phosphothreonine and phosphotyrosine residues (Luan, 2003). All PTPases possess a signature motif of 11 amino acids at the active site (I/V)HCXAGXXR(S/T)G, which contains the cysteine (Cys) catalytic residue (indicated in bold) involved in the formation of a phosphoenzyme intermediate during the reaction (Guan and Dixon, 1991). Tyr phosphorylation and consequently Tyr dephosphorylation have recently emerged as important mechanisms for transmembrane signaling in plants, in particular in immunity processes (Mühlenbeck et al., 2021).
The Arabidopsis thaliana protein Tyr phosphatase 1 (AtPTP1), an intracellular PTPase, was the first PTPase characterized in higher plants. Like most of the other plant species, there is only one Tyr-specific protein phosphatase in A. thaliana (Kerk et al., 2008). Its PTPase activity was measured in vitro by Xu et al. (1998). This protein of 340 amino acids and 37.8 kDa is located in both the cytosol and the nucleus (Bartels et al., 2009). The AtPTP1 gene is preferentially expressed in roots, stems, and flowers. It is less expressed in leaves (Xu et al., 1998; data of gene expression map of Arabidopsis development from Schmid et al., 2005 using eFP Browser at bar.utoronto.ca; Winter et al., 2007). PTPases play important roles in the regulation of mitogen-activated protein kinases (MAPKs) cascades (Ichimura et al., 2002). In particular, AtPTP1 is known to dephosphorylate AtMPK6 (Gupta and Luan, 2003) and AtMPK4 (Huang et al., 2000) in vitro. In addition, it can interact in vivo with AtMPK3 and AtMPK6 (Bartels et al., 2009; Zhou et al., 2017; Zhao et al., 2020). Thus, AtPTP1 could inhibit defense reactions through MAPK inactivation by dephosphorylation and also through suppression of salicylic acid (SA) synthesis in association with AtMKP1 (MAPK Phosphatase 1), a dual-specificity PTPase whose role is partially redundant to that of AtPTP1 (Bartels et al., 2009). Recent data suggest that AtPTP1 and AtMKP1 negatively regulate H2O2 production by the NADPH oxidase AtRbohD activity and inhibit the activity levels of AtMPK3 and AtMPK6 during defense responses to Verticillium dahlia toxins (Zhao et al., 2020). Conversely, AtPTP1 is also reversibly inactivated in vitro by H2O2 treatment (Gupta and Luan, 2003).
AtPTP1 is regulated both at the transcriptional and post-translational levels. Indeed, AtPTP1 transcript expression is induced in response to salt stress (Xu et al., 1998; Zhou et al., 2017) and repressed in response to cold (Xu et al., 1998). Recently, it was reported that in poplar, the overexpression of the Tyr-specific protein phosphatase-encoding gene PdPTP1 led to an increase of the plant sensitivity to salt stress. This process was linked to increased levels of ROS and perturbations in Na+ and K+ ion homeostasis. PdPTP1 was also shown to interact with PdMPK3 and PdMPK6 in vitro and in vivo (Lu et al., 2020). Thus, it appears that the PTP1 protein has an important role in the response to salt stress in plants. In response to submergence, AtPTP1 is regulated at the post-translational level by AtSnRK1 (Sucrose non-fermenting-1-related protein kinase) through the phosphorylation of its threonine (Thr) 6, Ser 7, and Ser 8 residues. Phosphorylation of AtPTP1 would prevent its interaction with AtMPK6, which would consequently contribute to the enhancement of AtMPK6 signaling (Cho et al., 2016).
The protein most closely related to AtPTP1 in humans is the Tyr-specific protein phosphatase PTP1B. PTP1B has been extensively studied in humans because of its involvement in the insulin signaling pathway and in cell proliferation. AtPTP1 and human PTP1B share 35% identity between their amino acid sequences but the catalytic site of the two proteins, as all PTPases, is very well conserved.
Regarding the regulation of PTP1B activity, different mechanisms involving post-translational modifications have been characterized. PTP1B can be regulated by acetylation, phosphorylation, sulfhydration, S-nitrosation, and several levels of oxidation, which lead to reversible or irreversible modifications. PTP1B was shown to by acetylated at methionine (Met) 1 residue (Charbonneau et al., 1989) while phosphorylation occurs mostly at the Ser 50 residue (Moeslein et al., 1999; Ravichandran et al., 2001; Zhou et al., 2013) even if other Ser and some Tyr and Thr residues can be targeted (Flint et al., 1993; Liu and Chernoff, 1997; Moeslein et al., 1999; Rush et al., 2005; Zhou et al., 2013). Phosphorylation leads to activation or inhibition of PTP1B activity in vitro (Moeslein et al., 1999) and in vivo (Moeslein et al., 1999; Ravichandran et al., 2001). Both oxidation and S-nitrosation of PTP1B target the catalytic Cys residue C215. The PTPase signature motif gives to the catalytic Cys a low pKa, which increases its nucleophilicity and makes the protein particularly susceptible to inactivation by reactive oxygen species like H2O2 and superoxide (Denu and Tanner, 1998). Indeed, the catalytic Cys residue of PTPases has an unusual pKa between 4.5 and 5.5 when it is typically around 8.5. This feature allows the catalytic Cys to be under the thiolate anion form at neutral pH (Zhang and Dixon, 1993; Lohse et al., 1997). This low pKa value thus makes the Cys very sensitive to redox-dependent regulation (Salmeen and Barford, 2005). Reversible oxidation of the catalytic Cys of PTPase proteins induces a transient inhibition of their activity, which suppresses the dephosphorylation of their target proteins. The oxidation of PTP1B enzyme in solution was studied by X-ray crystallography and mass spectrometry (MS) after a treatment with H2O2. This treatment initially leads to the formation of a sulfenic acid intermediate (PTP1B-SOH), which is rapidly converted into sulphenyl-amide species. PTP1B then undergoes major conformational changes in its catalytic site that inhibit substrate binding. The formation of sulphenyl-amide is an unusual modification that protects the catalytic Cys from irreversible oxidation and promotes its reversible reduction by thiols, which allows the reactivation of PTP1B activity. This modification is reversible but after a prolonged exposure to high concentrations of H2O2, sulfenic acid is then transformed into sulfinic acid (PTP1B-SO2H) and sulfonic acid (PTP1B-SO3H), which are irreversibly oxidized forms of the enzyme (Salmeen et al., 2003; van Montfort et al., 2003). In vivo, PTP1B is reversibly inhibited by oxidation in cells following stimulation by insulin (Mahadev et al., 2001) and by the epidermal growth factor (Lee et al., 1998). Londhe et al. (2020) demonstrated the existence of a molecular interaction between the reversible oxidized form of PTP1B and the 14-3-3ζ proteins. Destabilization of this interaction prevents inactivation of PTP1B by reactive oxygen species and decreases phosphorylation of the epidermal growth factor receptor. It has been demonstrated in vitro that PTP1B is S-nitrosable by two NO donors, S-nitroso-N-acetylpenicillamine (SNAP) and diethylamine NONOate (DEA-NONOate), as well as by the trans-nitrosylating agent S-nitrosoglutathione (GSNO), on its Cys residue C215 but also C32 and C92, these last two being located at the protein surface (Chen et al., 2008). PTP1B was also found to be S-nitrosated in vivo (Li and Whorton, 2003; Hsu et al., 2016). S-nitrosation of PTP1B reduces its activity in vitro and in vivo (Li and Whorton, 2003). Interestingly, it has been shown in vitro that S-nitrosation of PTP1B may have a protective effect against protein inactivation by oxidation. Indeed, treatment of PTP1B with relatively high concentrations of H2O2 leads to a modification of Cys 215 into -SO2H/-SO3H but does not occur if PTP1B is previously treated by SNAP (Salmeen et al., 2003). In addition, Chen et al. (2008) found by mass spectrometry that pre-treatment of PTP1B with a NO donor prevents modification of the enzyme by H2O2. These data indicate that S-nitrosation would protect the enzyme from H2O2 irreversible oxidation. The prevention of irreversible oxidation and inactivation of PTP1B could also be provided by reversible sulfhydration of C215 by H2S, which was shown to inactivate the protein in vitro and in HeLa cells. This mechanism would regulate the endoplasmic reticulum (ER) stress response (Krishnan et al., 2011).
A different regulatory mechanism has been identified for the soybean GmPTP protein under oxidizing conditions (Dixon et al., 2005). GmPTP was shown to be relatively insensitive to inactivation by H2O2, unlike its mammalian counterparts such as PTP1B, but was hypersensitive to S-glutathionylation in the presence of oxidized glutathione (GSSG). More precisely, contrary to the catalytic Cys 266 (C266) residue, Cys 78 (C78) and Cys 176 (C176), which are activity regulators, are direct targets of S-glutathionylation as this modification inactivates the enzyme. The authors of this study hypothesized a chain reaction. First, C176 would undergo rapid thiolation by GSSG, which would inactivate GmPTP. Secondly, C78, which is close to the active site, could be S-glutathionylated but less easily than C176. Finally, thiolated C78 would be able to undergo a disulfide exchange with the catalytic C266, thus releasing GSH to form an intramolecular C78-C266 disulfide or a disulfide-linked dimer of the protein. This mechanism would constitute a double protection of the catalytic C266 by blocking this residue in a basal state in the form of sulfenyl-amide, as described for PTP1B (Salmeen et al., 2003; van Montfort et al., 2003), and which could be reactivated rapidly following reduction by thiols.
S-nitrosation of AtPTP1 has never been studied. Here, after producing and purifying a recombinant histidine (His)-AtPTP1 protein, we verified by mass spectrometry its S-nitrosation after application of the biotin switch method. We then tested the effects of exogenous NO or H2O2 on its activity. We found that similarly to human PTP1B, the activity of AtPTP1 is inhibited by S-nitrosation and that this post-translational modification probably would allow its protection against irreversible oxidation in vivo. The protection of the catalytic Cys residue by NO via S-nitrosation against irreversible oxidation could be a conserved mechanism in plant PTPases.
The coding sequence of AtPTP1 (At1g71860) was amplified by PCR from A. thaliana leaf cDNA using Phusion Taq Polymerase and then cloned into the PCR8 vector (Thermofisher). The coding sequence of AtPTP1 was then transferred into the pHGWA vector (Busso et al., 2005) by LR recombination, resulting in pHGWA-AtPTP1.
Arabidopsis thaliana protein tyrosine phosphatase 1 cDNA was expressed in Escherichia coli Rosetta II bacteria under the control of the T7 RNA polymerase promoter to allow synthesis of a recombinant AtPTP1 protein with a His-tag at the NH2 extremity. Bacteria were grown at 37°C in Luria-Bertani (LB) medium until the absorbance at 600 nm reached.6. Protein production was then induced by.5 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) overnight at 25°C. Bacterial cells were then lysed in lysis buffer [50 mM Tris–HCl pH 7.5, 300 mM NaCl, 1% (v/v) triton X100, Protease Inhibitor Cocktail (Roche), 2.5 mg·ml−1 lyzozyme, 21 mg·ml−1 DNAse, and 6.25 mg·ml−1 RNAse], briefly sonicated and centrifuged at 20,000g for 30 min. The soluble proteins were passed through.22 μm filters prior to purification of His-AtPTP1 by Ni2+ affinity chromatography using a His-TRAP™ column by FPLC on ÄKTA™ Purifier, according to the manufacturer’s instructions (GE Healthcare). After selection of fractions containing His-AtPTP1, these were pooled and dialyzed against 25 mM HEPES-KOH pH 7.7, 1 mM ethylenedinitrilotetraacetic acid (EDTA), and 500 mM NaCl buffer. Protein concentration was assessed by absorbance reading at 280 nm using a NanoDropTM spectrophotometer (ThermoScientific). His-AtPTP1 was stored at −20°C in 20% glycerol without loss of activity.
The biotin switch assay was performed as described by Jaffrey and Snyder (2001) with some changes described in Astier et al. (2012). Briefly, 300 μg of His-AtPTP1 were mixed with 300 μl of HEN buffer {25 mM HEPES-NaOH pH 7.7, 1 mM EDTA,.1 mM neocuproine, and.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)} containing either GSH or GSNO, which were used at a final concentration of 100 μM. The incubation was done at 25°C overnight in the dark. After removing GSH/GSNO by acetone precipitation, the pellet was resuspended in 300 μl blocking buffer [20 mM methylmethane thiosulfonate (MMTS) and 2% sodium docecyl sulfate (SDS) in HEN buffer] and was incubated 20 min at 50°C with shaking (350 rpm). MMTS was removed by acetone precipitation and the pellet was resuspended in 300 μl HENS (HEN with 1% SDS) with 1 mM N-[6-(biotinamido)hexyl]-3-(2-pyridyldithio) propionamide (biotin-HPDP; ThermoFisher Scientific) and 50 mM ascorbate. After 1 h at room temperature, the proteins were precipitated by acetone and resuspended in 100 μl of 25 mM ammonium bicarbonate (AMBIC). Proteins were digested overnight at 37°C with 6 μg of trypsin (Promega). The samples were then lyophilized and analyzed by mass spectrometry.
The peptide mixtures were analyzed by nano liquid chromatography tandem mass spectrometry (nanoLC-ESI-MS/MS) using an Orbitrap Velos mass spectrometer (ThermoFisher Scientific). Peptides were eluted on an in-house C18 reverse-phase microcolumn, 75 μm ID × 50 cm packed with Reprosil-Pur C18-AQ 3 μm resin (Dr Maisch GmbH; Ammerbuch, Germany) and analyzed by high-resolution MS, followed by tandem MS/MS of the 20 most abundant ions.
Acquired MS and MS/MS data were searched with Mascot (version 2.7.0, http://matrixscience.com) against a custom-made database containing PTP1 Arabidopsis sequence and all K12 E. coli entries from the UniProtKB database (E. coli K12_UniprotSP_20180430.fasta; 5,979 sequences; and 1,850,060 residues). The search included methionine oxidation and N-Ter acetylation. Regarding possible Cys residue modifications, we searched for biotin-HPDP addition, glutathionylation (GSH addition), or methylthiolation (-S-CH3) due to MMTS treatment. Trypsin/P was chosen as the enzyme and two missed cleavages were allowed. The mass tolerance was set to 5 ppm for the precursor ion and to.8 Da for fragment ions. Validation of identifications was performed through a false-discovery rate set to 1% at protein and peptide-sequence match level, determined by target-decoy search using the Proline software version 1.6.1 Raw MS signal extraction of identified peptides was performed with Proline across different samples. For all comparison, statistical analysis by ANOVA one way was applied to the abundance values.
To study the effect of its S-nitrosation, the His-AtPTP1 protein was treated overnight at 25°C in HEN buffer with GSH or GSNO (100 or 500 μM), or SNAP (250 µM, 500 µM, 750 µM or 1 mM). These molecules were then removed by passing the protein samples through G50 exclusion columns (Micro Bio-Spin™ P-6 Gel Columns; Biorad), which allow the elimination of molecules whose molecular weight is lower than 6,000 Da and then the extract was treated or not with 20 mM DTT for 30 min at 25°C.
To study the effect of its oxidation, the His-AtPTP1 protein was treated for 30 min at 25°C with 50 µM, 100 µM, 250 µM, 500 µM, 750 µM, 1 mM, 2 mM or 5 mM H2O2. The H2O2 molecules were then removed by treatment with 10 μg of catalase for 30 min and then the extract was treated or not with 20 mM DTT for 30 min at 25°C.
His-AtPTP1 enzyme activity assay was performed at 25°C using p-nitrophenyl phosphate (pNPP disodium salt, Sigma Aldrich) that is hydrolyzed to p-nitrophenol (pNP). pNP production was monitored by absorbance at 405 nm. The conditions of the assay were those described by Montalibet et al. (2005). Briefly, the reaction was initiated by adding 20 ng of His-AtPTP1 to 200 μl assay buffer (50 mM BIS-TRIS pH 6.3, 2 mM EDTA, 2% glycerol,.01% triton X100, and 1 mM pNPP) in a 96-well plate and mixed vigorously for 1 min. The reaction was stopped at 15, 30, 45, or 60 min later with 20 μl of 10 M NaOH and read at 405 nm on a plate reader (Molecular Devices). To convert the absorbance value at 405 nm to pNP concentration, the constant ε.l (949 mol−1) was determined using the Beer–Lambert law from a standard range of pNP.
One-way or two-way ANOVA were performed using XLSTAT software (Addinsoft) followed by a multiple comparison procedure with the Fisher’s LSD method.
Arabidopsis thaliana Protein Tyrosine Phosphatase 1 cDNA encodes a 340 amino acid protein that shares homology with PTP1 proteins from other organisms, such as Zea mays ZmPTP1, Oryza sativa OsPTP1, Pisum sativum PsPTP1, Medicago truncatula MtPTP1, Glycine max GmPTP1, Populus nigra × (Populus deltoides × P. nigra) PdPTP1, Nicotiana tabacum NtPTP1, or Homo sapiens HsPTP1B (Figure 1). In particular, the critical region for catalysis with the Cys involved in the formation of the phosphoenzyme reaction intermediate is conserved between all of these proteins. In 1998, Xu et al. (1998) confirmed that the recombinant protein GST-AtPTP1 was a highly active Tyr-specific protein phosphatase. In regard to its key role in signaling, we investigated here whether AtPTP1 could be modified by S-nitrosation as reported for PTP1B. For this purpose, a recombinant AtPTP1 protein with a His-tag at the NH2 extremity was produced in E. coli bacteria and purified by metal-affinity chromatography. Then, 300 μg of recombinant His-AtPTP1 protein were incubated overnight either with 100 μM GSH or GSNO and subjected to the biotin switch method. This technique consists in replacing the NO groups of nitrosothiols by a biotin molecule after having protected free thiols by alkylation (Jaffrey and Snyder, 2001). After incubation with biotin-HPDP, the His-AtPTP1 protein samples were suspended in 25 mM AMBIC and digested by trypsin in the absence of a reducing agent to preserve the Cys-biotin bond. The trypsic peptides were identified by LC–MS after interrogation against the E. coli K12 library enriched with PTP1 sequences from A. thaliana. According to the sequence of the His-AtPTP1 protein, seven Cys-containing peptides can be generated by tryptic digestion of His-AtPTP1 (Figure 2A). We named the peptides generated after trypsin cleavage by their order in the protein sequence (from NH2 to COOH) and by the number of the Cys contained in the peptide (e.g., the sequence named P5-C265 is the fifth peptide generated by trypsin in the sequence and it contains cysteine C265). LC–MS analysis of GSNO- or GSH-treated His-AtPTP1 protein after application of the biotin switch method and trypsin digestion allowed the detection of the expected seven peptides. The mass difference (shift) between peptides identified the following Cys modifications: S-methylthiolation due to MMTS protection, S-biotinylation, and S-glutathionylation. The relative abundance of each Cys modification after His-AtPTP1 treatment either with GSH or GSNO was reported on Figure 2B for S-biotinylation, on Supplementary Figure S1A for S-methylthiolation and on Supplementary Figure S1B for S-glutathionylation. The GSNO-treated His-AtPTP1 control condition, without the addition of HPDP-biotin, showed that none of the peptides were found to be biotinylated under this condition, thus ensuring that the His-AtPTP1 protein was not biotinylated during its synthesis in the bacteria and that the observed biotinylation results from the application of the biotin switch method (data not shown). In contrast, methylthiolated and biotinylated forms were detected for all seven Cys-containing peptides. Nevertheless, the presence of biotinylated peptides in the absence of NO donor (in GSH condition) indicates that the protection of free Cys by MMTS was not complete as often described in the literature (Lamotte et al., 2015). GSH treatment resulted in 73%–98% methylthiolation of each Cys. A significant increase in biotinylation was only observed for the P5-C265 peptide when comparing GSH and GSNO treatments (Figure 2B). This modification was correlated to a concomitant decrease in methylthiolation (Supplementary Figure S1A). Other residues (C41 and C157 particularly) could also be targets of S-nitrosation but the data obtained between GSH and GSNO are not statistically different (Figure 2B). A significant increase in the relative percentage of glutathionylation of P4-C175, P6-C276, and P7-319 peptides in response to GSNO compared with the GSH control was also noticed (Supplementary Figure S1B). Since the C265 residue identified as S-nitrosated belongs to the active site of the enzyme, we wondered whether this post-translational modification has a positive or negative effect on the activity of the His-AtPTP1 enzyme. We sought to answer this question by measuring the enzymatic activity of His-AtPTP1 in vitro.
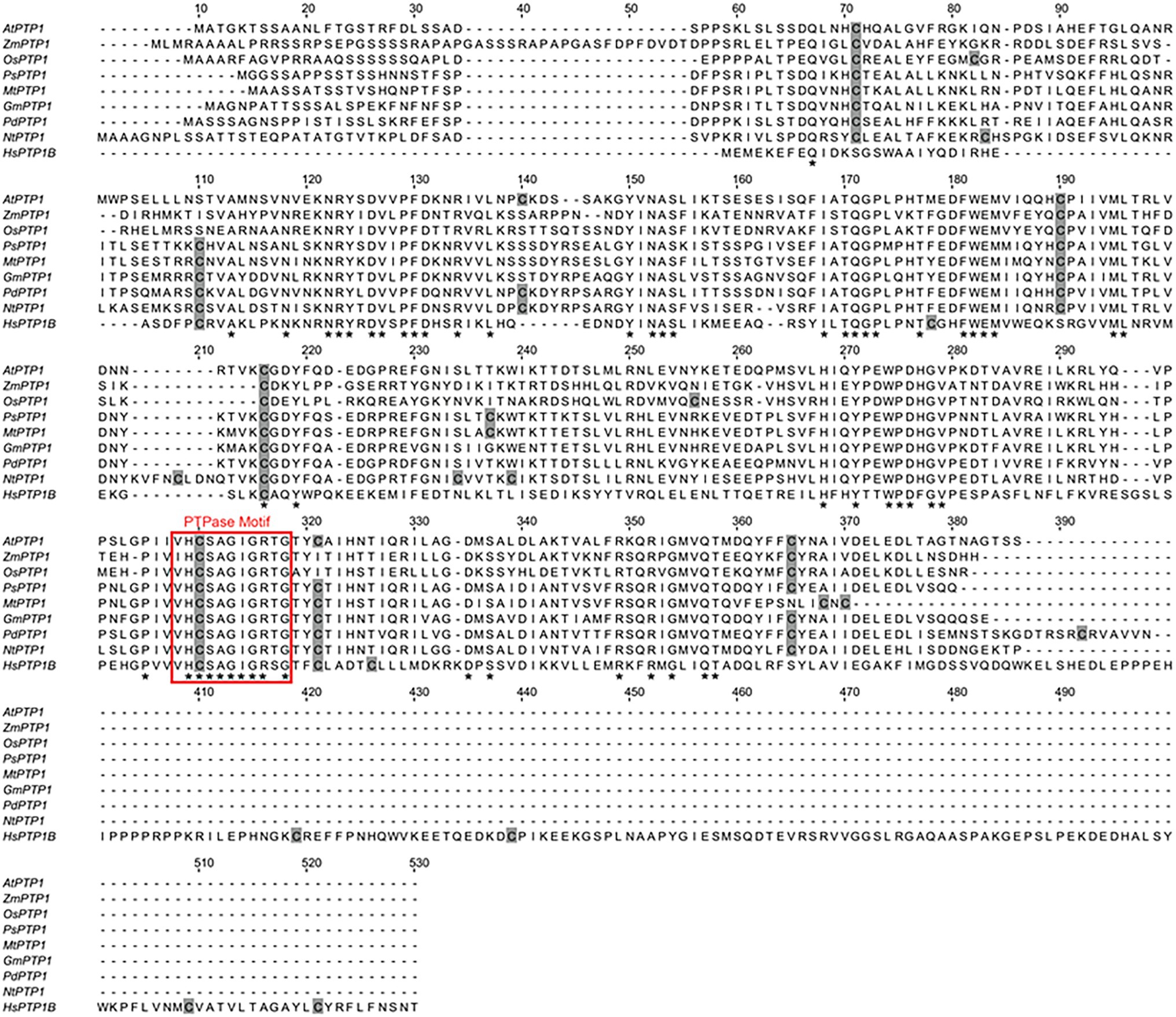
Figure 1. Sequence alignment of eight plant Protein Tyrosine Phosphatase 1 (PTP1) proteins, including Arabidopsis thaliana Protein Tyrosine Phosphatase 1 (AtPTP1), and the human PTP1B protein. Perfectly conserved amino acid residues are indicated by stars. Cys residues are indicated in gray. The typical conserved PTPase signature motif is highlighted by a red frame. The sequences compared are as follows: Arabidopsis thaliana AtPTP1, O82656; Zea mays ZmPTP1, NP_001149088; Oryza sativa OsPTP1, Q2QX07; Pisum sativum PsPTP1, O82710; Medicago truncatula MtPTP1, A0A072VQ92; Glycine max GmPTP1, O82687; Populus nigra × (Populus deltoides × P. nigra) PdPTP1, Lu et al., 2020; Nicotiana tabacum NtPTP1, A0A1S4A2F4; and Homo sapiens HsPTP1B, P18031.
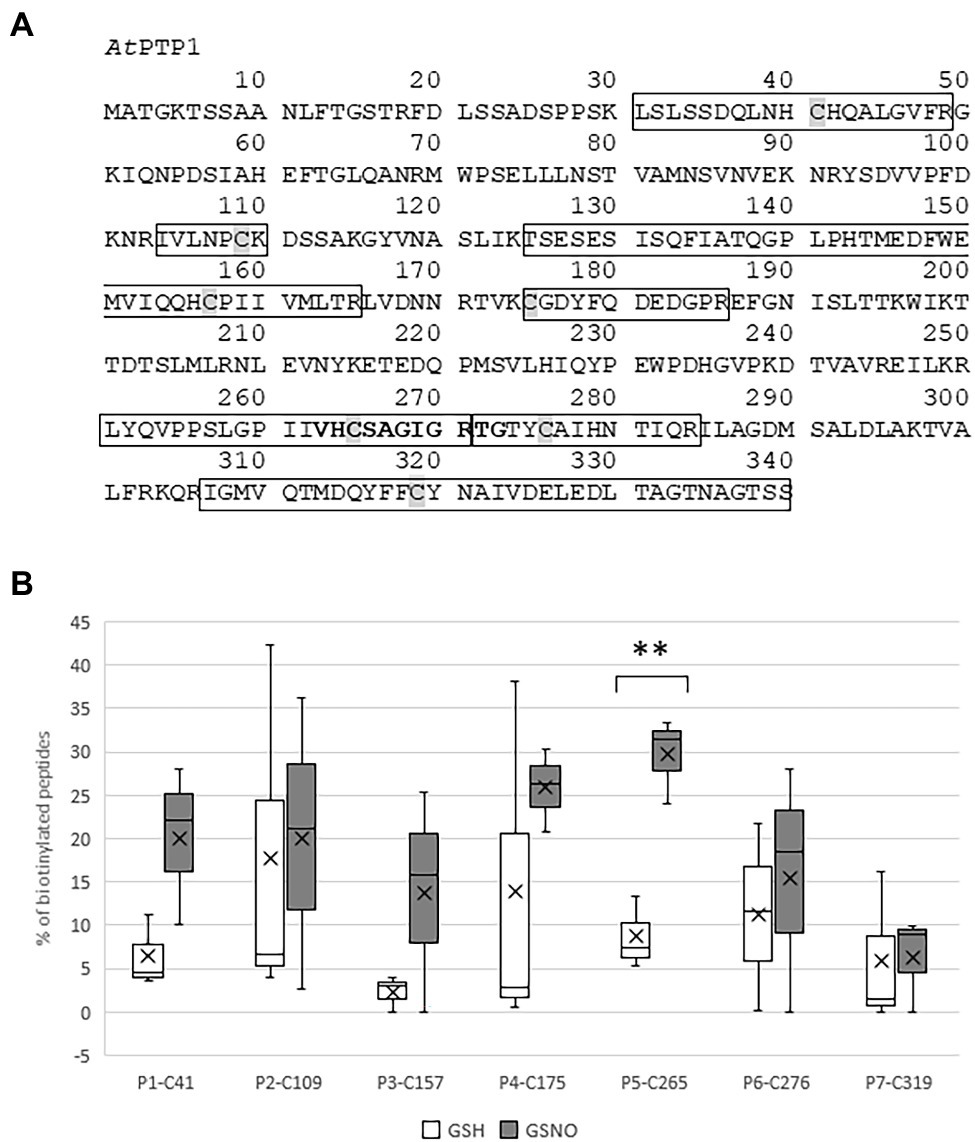
Figure 2. Identification by mass spectrometry of the Cys residues of histidine (His)-AtPTP1 targeted by S-nitrosation. (A) Primary sequence of AtPTP1 protein. The seven peptides identified by mass spectrometry are boxed, the Cys residues are shaded and the active site signature motif of PTPases is indicated in bold. (B) Relative percentage of biotinylated peptide to the total abundance of this peptide in the sample following treatment of His-AtPTP1 by 100 μM glutathione (GSH) or S-nitrosoglutathione (GSNO). Values are the results of three mass spectrometry analyses. Significant difference is denoted by an asterisk: **p ≤ .01.
The phosphatase activity of His-AtPTP1 was determined in vitro by spectrophotometric measurement of pNP (bright yellow) production from pNPP (colorless) as described by Montalibet et al. (2005). His-AtPTP1 was treated overnight with 100 or 500 μM GSH or GSNO, and the enzymatic activity was measured in the absence or in the presence of 20 mM DTT (Figure 3A). The classical protocol for measuring enzymatic activity used by Montalibet et al. (2005) requires DTT in the buffer to be optimal. However, we chose to measure the activity of His-AtPTP1 first in the absence of DTT because it breaks nitrosothiol bonds. It is therefore impossible to study the effects of S-nitrosation of the enzyme on its activity in the presence of DTT.

Figure 3. Effect of GSH or GSNO treatment on His-AtPTP1 activity in the absence or presence of DTT. (A) Experimental design. (B) Measurement of the enzymatic rate of His-AtPTP1 following treatment with 100 or 500 μM of GSNO (grey columns) or GSH (black columns) in the absence or presence of 20 mM DTT. Following the treatment, GSH or GSNO was removed through exclusion chromatography. Results are expressed relative to the enzyme activity measured without GSH/GSNO or DTT treatments. Values are means of four measurements ±SEM. The results shown are from one representative experiment among three independent experiments. Significant differences are denoted by different letters (p ≤ .05).
The left part of Figure 3B shows the rate of reaction in the absence of DTT. His-AtPTP1 protein treated with GSNO displayed about three times less activity as compared to the GSH-treated sample, thus indicating that GSNO inhibited the enzymatic activity of His-AtPTP1. Control experiments showed that no inhibition of activity was observed after GSH treatment, indicating that the strong inhibition observed after GSNO treatment was indeed dependent on NO itself.
Enzyme activity was then measured in the presence of DTT 20 mM (right part of Figure 3B). In the absence of any treatment, the addition of DTT increased the measurable activity of His-AtPTP1 approximately 1.5-fold. In the presence of DTT, GSNO had no longer an inhibitory effect on His-AtPTP1 activity. This difference indicates that the modification of His-AtPTP1 by GSNO was reversible by the reductant.
In order to confirm the effect of S-nitrosation on the activity of the enzyme, we treated His-AtPTP1 with SNAP, a NO donor (Supplementary Figure S2). Treatment of His-AtPTP1 with SNAP also led to a significant decrease in its activity.
Overall, these observations suggest that the ability of GSNO or SNAP to nitrosate His-AtPTP1 protein in vitro decreased the activity of the enzyme. This inhibition seems reversible as suggested by the reduced effect of GSNO in the presence of DTT during the reaction. Indeed, it can be assumed that the presence of DTT allows a break of the Cys-NO bond, thus partially restoring the activity of the enzyme. These observations suggest that NO donors modify the enzyme in a reversible manner, which impacts the activity of the His-AtPTP1 enzyme.
The catalytic Cys residue of the human PTP1B protein can be readily oxidized in vitro and in vivo. It has been proposed that, depending on the ROS concentration, the catalytic Cys residue of PTP1B can be reversibly oxidized to sulphenic acid (Cys-SOH), but also to sulfinic acid (Cys-SO2H) or sulphonic acid (Cys-SO3H), the latter two modifications resulting in irreversible oxidation and permanent inactivation of the enzyme (Salmeen et al., 2003). His-AtPTP1 was treated in vitro for 30 min with an increasing concentration of H2O2 (50 μM to 5 mM). Residual H2O2 was removed by the addition of 10 μg of catalase prior to the measurement of enzyme activity, which was performed in the absence or presence of 20 mM DTT which eliminates the reversible modification of His-AtPTP1 (Figure 4A). A drastic reduction of His-AtPTP1 activity was observed after H2O2 treatment in the absence of DTT. This phenomenon is visible as early as the 50 μM concentration (Figure 4B, left part). This condition allows visualizing both reversible and irreversible oxidation of His-AtPTP1. In contrast, enzyme activity was partially restored after the addition of DTT (Figure 4B, right part), revealing the reversible character of some Cys oxidations. The presence of DTT eliminates the reversible modification of His-AtPTP1. This suggests that the proportion of irreversible oxidation increases with H2O2 concentration, a dose-dependent residual inactivation being observed with DTT.
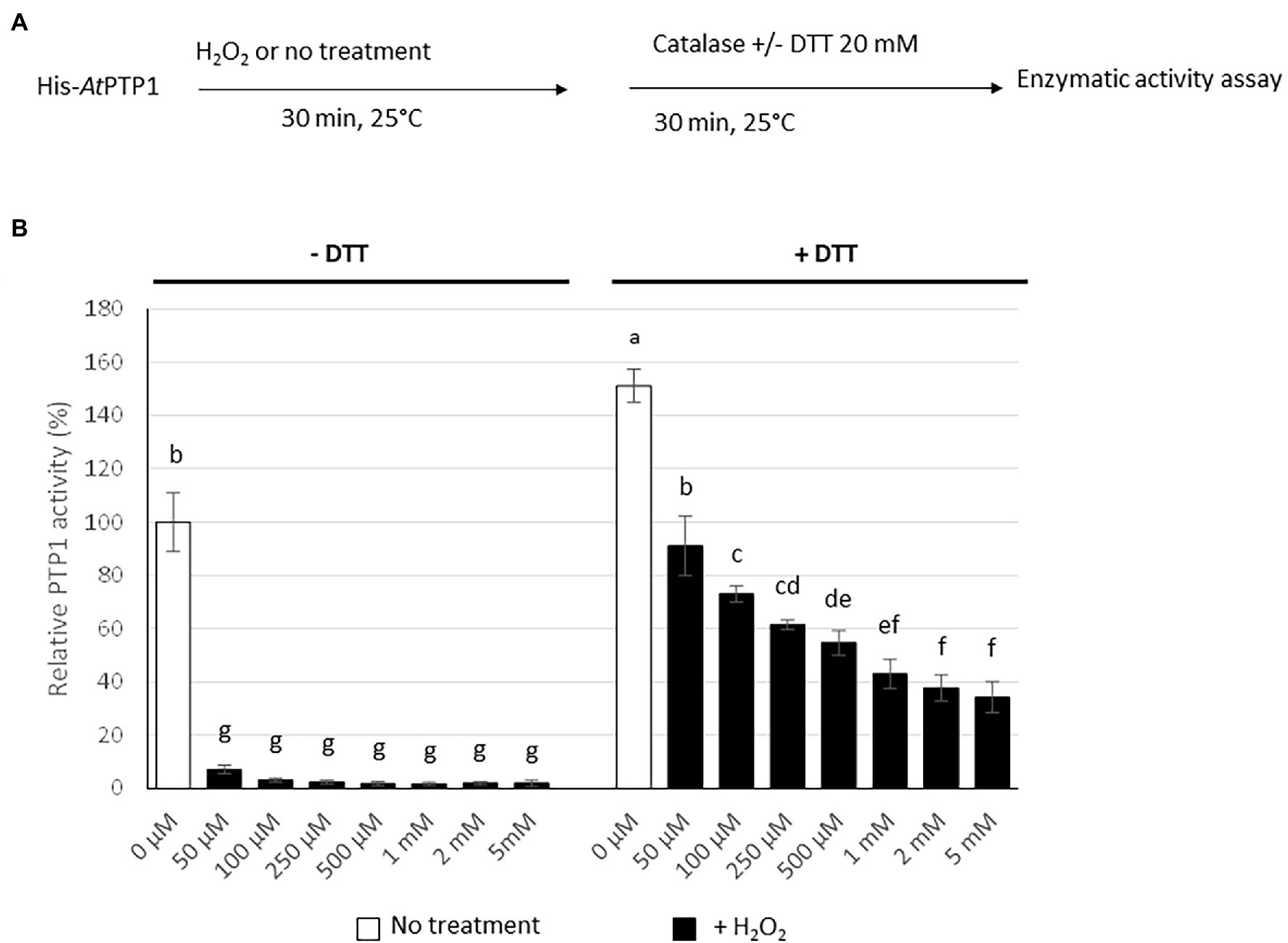
Figure 4. Effect of H2O2 treatment on His-AtPTP1 activity in the absence or presence of DTT. (A) Experimental design. (B) The enzymatic activity of His-AtPTP1 was measured following treatment with 50, 100, 250, and 500 μM or 1, 2, or 5 mM of H2O2 in the absence or presence of 20 mM DTT. Results are expressed relative to the enzyme activity measured without H2O2 or DTT treatments. Values are means of four measurements ±SEM. The results shown are from one representative experiment among three independent experiments. Significant differences are denoted by different letters (p ≤ .05).
To test the protective effect of S-nitrosation against irreversible oxidation of His-AtPTP1, we incubated it overnight with 100 or 500 μM GSH or GSNO in the absence or presence of 20 mM DTT. To avoid a potential interaction between GSH/GSNO and H2O2, we purified the protein on a G50 column to remove GSH/GSNO after the pre-treatment. His-AtPTP1 was then treated with an increasing concentration of H2O2 (between 50 μM and 1 mM) subsequently degraded by 10 μg of catalase before measuring the activity in the absence or presence of DTT (Figure 5A). We then measured the reversible and irreversible oxidation of His-AtPTP1 with or without prior S-nitrosation in the absence (Figure 5B) or in the presence of DTT (Figure 5C). As reported in Figure 4, in the absence of GSNO pre-treatment, a dose-dependent inhibitory effect of H2O2 on His-AtPTP1 activity was observed, this effect being stronger in the absence (Figure 5B) than in the presence (Figure 5C) of DTT. After pretreatment of the enzyme with 500 μM of GSNO, the effect of H2O2 treatment on its activity in the absence of DTT was reduced compared to pretreatment with GSH. Indeed, the activity measured in response to 1 mM H2O2 after pre-treatment with 500 μM GSNO was increased 7.33- and 10.8-fold following no pre-treatment or pre-treatment with 500 μM GSH, respectively (Figure 5B). The effect of GSNO was diminished following the use of DTT as it decreases to factors of 2.4 and 2.7 following no pre-treatment or pre-treatment with GSH, respectively (Figure 5C). S-nitrosation of His-AtPTP1 could therefore protect the enzyme from reversible and irreversible oxidation. As shown above, S-nitrosation by GSNO and part of the oxidation of His-AtPTP1 are reversible following DTT treatment. We assume that in the presence of DTT, we observed only the inhibitory effect due to the irreversible oxidation (Figure 5C). The decrease in His-AtPTP1 activity was less pronounced when the enzyme was pretreated with 500 μM GSNO than with GSH. Thus, the modification of His-AtPTP1 by GSNO might protect it from irreversible oxidation by H2O2. This observation agrees with the mass spectrometry results of Chen et al. (2008) showing that pre-treatment of PTP1B recombinant protein with the NO donor SNAP prevents its irreversible oxidation. To rule out the possibility that the oxidation protection effect was due to glutathionylation as in the case of the GmPTP protein, we performed the same experiment as before but use the NO donor SNAP (1 mM) instead of GSNO. The data presented in Supplementary Figure S3 show that the use of SNAP reduced the H2O2-induced inhibition of His-AtPTP1 activity by about 15.5-fold in the absence of DTT and by 4.5-fold in the presence of DTT. We confirm that S-nitrosation would protect the protein from irreversible oxidation by H2O2.
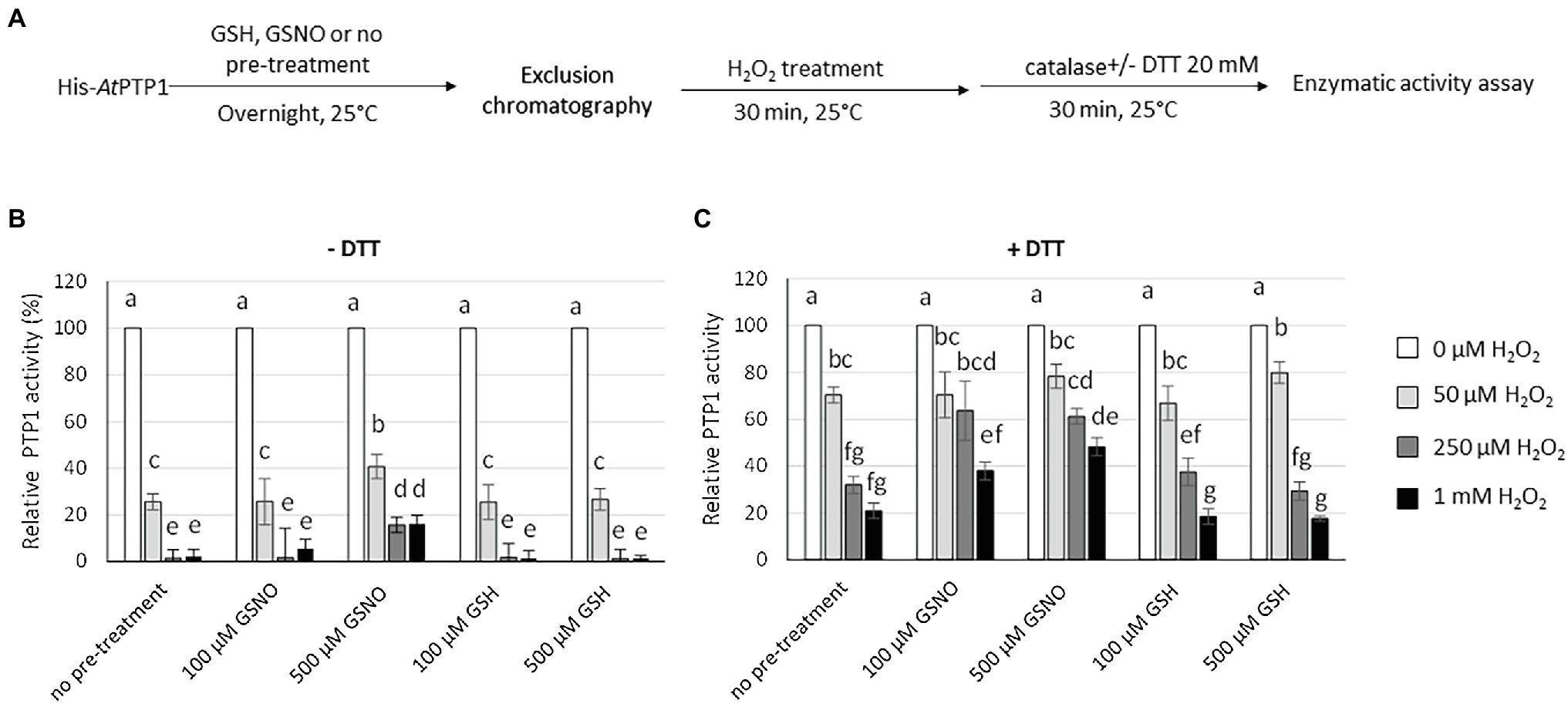
Figure 5. Effect of H2O2 treatment on His-AtPTP1 activity in the absence or presence of DTT following pre-treatment with GSH or GSNO. (A) Experimental design. (B) Measurement of the enzymatic rate of His-AtPTP1 following treatment with 50 and 250 μM or 1 mM of H2O2 in the absence of 20 mM DTT after pre-treatment with 100 or 500 μM of GSH or GSNO. (C) Measurement of the enzymatic rate of His-AtPTP1 following treatment with 50 μM, 250 μM or 1 mM of H2O2 in the presence of 20 mM DTT after pre-treatment with 100 or 500 μM of GSH or GSNO. Values are means of four measurements ±SEM. The results shown are from one representative experiment among three independent experiments. Significant differences are denoted by different letters (p ≤ .05).
After production and purification of the His-tagged AtPTP1 protein, we showed its S-nitrosation in vitro in response to the trans-nitrosylating agent GSNO. Our results suggest that the catalytic cysteine C265 is the major site of S-nitrosation on His-AtPTP1. An increase in the rate of S-nitrosation was however detected for other Cys residues but the data are not significantly different between GSH and GSNO treatments. Two studies have shown by mass spectrometry (ESI-MS) that the human PTP1B protein is a target of S-nitrosation (Chen et al., 2007, 2008). In particular, treatment of PTP1B with the NO donor SNAP led to the formation of S-nitrosothiol at C215. S-nitrosation occurs most readily at the catalytic Cys residue but two other residues, C32 and C92, were also susceptible to S-nitrosation. As these two residues are on the surface of the protein, the authors deduced that their S-nitrosation could be aspecific because of an easier access for the NO donor (Chen et al., 2008).
We next analyzed the direct effects of NO exogenous treatment on the enzymatic activity of His-AtPTP1 by measuring the colorimetric transformation of pNPP to pNP. It appears that artificially generated NO from GSNO or SNAP inhibited the activity of the protein. This process was reversible since treatment with DTT restored the activity of the enzyme. The activity of PTP1B protein on cell lysates or intact A431 or Jurkat cells was also rapidly inhibited by the addition of nitrosothiols, with the addition of DTT removing the inhibition (Li and Whorton, 2003). Moreover, using in-gel phosphatase activity assay, Chen et al. (2007) were able to prove that treatment with increasing concentrations of SNAP led to the appearance of an inactivated form of PTP1B. Thus, it appears that both AtPTP1 and PTP1B protein activities are inhibited by NO.
Oxidation of AtPTP1 by H2O2 also inhibited its activity, but unlike treatment with GSNO, this process was not completely restored following subsequent treatment with DTT. This regulatory mechanism has also been demonstrated for PTP1B (Salmeen et al., 2003; van Montfort et al., 2003). The inactivation of PTP1B was not prevented in the presence of reducing agents, but just slowed down because of the faster reaction of H2O2 with the enzyme as compared to DTT (Denu and Tanner, 1998). However, once oxidized by H2O2 to PTP1B-SOH, its activity can be reactivated by reduction with DTT, GSH, β-mercaptoethanol, or Cys (Denu and Tanner, 1998). Therefore, the activities of AtPTP1 and PTP1B undergo similar modes of regulation following H2O2 treatment.
We finally studied the effect of H2O2 on AtPTP1 activity and the impact of prior S-nitrosation on this process. It appeared that a pre-treatment with GSNO or SNAP prevented the irreversible oxidation of AtPTP1 by H2O2. Although we cannot totally exclude the protection of C165 from oxidation by glutathionylation as shown for GmPTP, our results show that this process would be rather provided by S-nitrosation. In addition, the mechanism would anyway be different from the one described for GmPTP as the C78 residue involved in the disulfide bridge formation with the catalytic Cys residue is not conserved in A. thaliana. However, the protective effect of NO against AtPTP1 oxidation that we measured in vitro needs to be verified in vivo. On the one hand, the effects of exogenous treatments with NO donors or H2O2, or endogenously in response to stress, could perhaps be detected by mass spectrometry at the catalytic Cys residue. On the other hand, it is complicated to consider measuring an effect on protein activity given the number of proteins capable of hydrolyzing pNPP to pNP. Previously, Chen et al. (2008) highlighted an increased cellular level of NO in COS7 cells, due to pre-treatment with a NO donor or to ectopically activated NOS expression. Such NO increase inhibited ROS-induced irreversible oxidation of the endogenous PTP1B protein. The authors of this study indicated that the data obtained in vitro could be verified in vivo to analyze the redox status of endogenous PTPases in response to a stimulus that induces NO and/or ROS production in cells. They suggested to immunoprecipitate the PTPase of interest from a cell lysate thanks to specific antibodies and then to identify the S-nitrosated residues by nanoLC-nano-ESI-MS/MS analyses. This method could be used to demonstrate that AtPTP1 is regulated by S-nitrosation in vivo. However, such specific antibodies are not yet available.
Protection from irreversible oxidation by S-nitrosation is not unique to PTPases as other proteins such as galectin-2 are regulated in this way. Galectin-2 are soluble animal lectins defined by their conserved carbohydrate recognition domain and their affinity for β-galactosides. Mouse galectin-2 (mGal-2) which is mainly expressed in the gastrointestinal tract is regulated by oxidation and S-nitrosation. Oxidation of Cys 57 of mGal-2 by H2O2 results in the loss of its sugar-binding capacity and thus its inactivation. In contrast, pretreatment of mGal-2 with S-nitrosocysteine, an S-nitrosating agent, has no effect on its sugar-binding capacity but prevents H2O2-induced inactivation (Tamura et al., 2015, 2016). It appears that S-nitrosation slows down the H2O2-induced aggregation of mGal-2 by stabilization of the hydrophobic pocket around Cys 57 (Sakakura et al., 2018). Understanding the molecular mechanisms of protection of PTP1B or AtPTP1 by S-nitrosation against oxidative inactivation may provide a better understanding of the protective function of PTPases in response to oxidative stress.
Under standard culture conditions, Bartels et al. (2009) did not identify any macroscopic effects of the mutation in the AtPTP1 gene when measuring leaf mass of 21-day-old null mutant plants, which would indicate that it is primarily AtMKP1 that has a role in Arabidopsis. In contrast, the mkp1ptp1 double mutant has a stronger phenotype than the mkp1 mutant. AtPTP1 could therefore have a complementary role to that of AtMPK1, especially since AtPTP1 and AtMKP1 display different cellular localizations. The nuclear localization of AtPTP1 could especially give it a specific regulatory role. Only few data are available regarding the regulation of AtMKP1. It can be phosphorylated at Ser and Thr residues by AtMPK6 but its sensitivity to oxidation or S-nitrosation has not been studied. The possibility of regulating AtPTP1 activity by oxidation and/or S-nitrosation could allow a finer adjustment in response to abiotic stress. Again, if a specific antibody would be available to immunoprecipitate AtPTP1, it would be relevant to study post-translational modifications of the enzyme in response to (a)biotic stresses.
Overall, our data showed that the Arabidopsis thaliana PTP1 protein has a similar regulation of its activity as other PTPases in response to oxidative stress. S-nitrosation of PTPase proteins is likely to be a general mechanism for preventing the permanent inactivation of these enzymes caused by oxidative stress.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: ProteomeXchange, PXD030071.
AB-B, DW, and VN-F supervised the research and wrote the manuscript. JR and VN-F designed the experiments. AK, CR, JR, and VN-F carried out His-AtPTP1 production and purification. JR, VN-F, and SH measured the enzymatic activities of His-AtPTP1. CP analyzed the samples by mass spectrometry. JA performed sequence alignments of the different PTP1s. All authors have read and approved the final manuscript.
Work in our lab is supported by the Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation, Investissements d’Avenir program, project ISITE-BFC NOISELESS (contract ANR-15-IDEX-0003; grant NOISELESS-RA18041.AEC.IS), the Agence Nationale de la Recherche, project ALGAE-NOS (grant ANR-18-CE20-0022-02), and the Région de Bourgogne, project RHINO (grant 2018Y-07113).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.807249/full#supplementary-material
Astier, J., Besson-Bard, A., Lamotte, O., Bertoldo, J., Bourque, S., Terenzi, H., et al. (2012). Nitric oxide inhibits the ATPase activity of the chaperone-like AAA+ ATPase CDC48, a target for S-nitrosylation in cryptogein signalling in tobacco cells. Biochem. J. 447, 249–260. doi: 10.1042/BJ20120257
Bartels, S., Anderson, J. C., González Besteiro, M. A., Carreri, A., Hirt, H., Buchala, A., et al. (2009). MAP KINASE PHOSPHATASE1 and PROTEIN TYROSINE PHOSPHATASE1 are repressors of salicylic acid synthesis and snc1-mediated responses in Arabidopsis. Plant Cell 21, 2884–2897. doi: 10.1105/tpc.109.067678
Bheri, M., Mahiwal, S., Sanyal, S. K., and Pandey, G. K. (2021). Plant protein phosphatases: what do we know about their mechanism of action? FEBS J. 288, 756–785. doi: 10.1111/febs.15454
Busso, D., Delagoutte-Busso, B., and Moras, D. (2005). Construction of a set gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal. Biochem. 343, 313–321. doi: 10.1016/j.ab.2005.05.015
Charbonneau, H., Tonks, N. K., Kumar, S., Diltz, C. D., Harrylock, M., Cool, D. E., et al. (1989). Human placenta protein-tyrosine-phosphatase: amino acid sequence and relationship to a family of receptor-like proteins. Proc. Natl. Acad. Sci. U. S. A. 86, 5252–5256. doi: 10.1073/pnas.86.14.5252
Chen, Y. Y., Chu, H. M., Pan, K. T., Teng, C. H., Wang, D. L., Wang, A. H., et al. (2008). Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J. Biol. Chem. 283, 35265–35272. doi: 10.1074/jbc.M805287200
Chen, M. J., Dixon, J. E., and Manning, G. (2017). Genomics and evolution of protein phosphatases. Sci. Signal. 10:eaag1796. doi: 10.1126/scisignal.aag1796
Chen, Y. Y., Huang, Y. F., Khoo, K. H., and Meng, T. C. (2007). Mass spectrometry-based analyses for identifing and characterizing S-nitrosylation of protein tyrosine phosphatases. Methods 42, 243–249. doi: 10.1016/j.ymeth.2007.03.002
Cho, H. Y., Wen, T. N., Wang, Y. T., and Shih, M. C. (2016). Quantitative phosphoproteomics of protein kinase SnRK1 regulated protein phosphorylation in Arabidopsis under submergence. J. Exp. Bot. 67, 2745–2760. doi: 10.1093/jxb/erw107
Denu, J. M., and Tanner, K. G. (1998). Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic intermediate and implications for redox regulation. Biochemistry 37, 5633–5642. doi: 10.1021/bi973035t
Dixon, D. P., Fordham-Skelton, A. P., and Edwards, R. (2005). Redox regulation of a soybean tyrosine-specific protein phosphatase. Biochemistry 44, 7696–7703. doi: 10.1021/bi047324a
Flint, A. J., Gebbink, M. F. G. B., Franza, B. R. Jr., Hill, D. E., and Tonks, N. K. (1993). Multi-site phosphorylation of the protein tyrosine phosphatase, PTP1B: identification of cell cycle regulated and phorbol ester stimulated sites of phosphorylation. EMBO J. 12, 1937–1946. doi: 10.1002/j.1460-2075.1993.tb05843.x
Guan, K. L., and Dixon, J. E. (1991). Evidence of protein tyrosine phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem. 266, 17026–17030. doi: 10.1016/S0021-9258(19)47335-3
Gupta, R., and Luan, S. (2003). Redox control of protein tyrosine phosphatases and mitogen-activated protein kinases in plants. Plant Physiol. 132, 1149–1152. doi: 10.1104/pp.103.020792
Hsu, M. F., Pan, K. T., Chang, F. Y., Khoo, K. H., Urlaub, H., Cheng, C. F., et al. (2016). S-nitrosylation of endogenous protein tyrosine phosphatases in endothelial insulin signaling. Free Radic. Biol. Med. 99, 199–213. doi: 10.1016/j.freeradbiomed.2016.08.012
Huang, Y., Li, H., Gupta, R., Morris, P. C., Luan, S., and Kieber, J. J. (2000). ATMPK4, an Arabidopsis homolog of mitogen-activated protein kinase, is activated in vitro by AtMEK1 through threonine phosphorylation. Plant Physiol. 122, 1301–1310. doi: 10.1104/pp.122.4.1301
Ichimura, K., Shinozaki, K., Tena, G., Sheen, J., Henry, Y., Champion, A., et al. (2002). Mitogen-activated protein kinase cascades in plants: a new nomenclature. Trends Plant Sci. 7, 301–308. doi: 10.1016/S1360-1385(02)02302-6
Jaffrey, S. R., and Snyder, S. H. (2001). The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001:pl1. doi: 10.1126/stke.2001.86.pl1
Kerk, D., Templeton, G., and Moorhead, G. B. (2008). Evolutionary radiation pattern of novel protein phosphatases revealed by analysis of protein data from the completely sequenced genomes of humans, green algae, and higher plants. Plant Physiol. 146, 351–367. doi: 10.1104/pp.107.111393
Krishnan, N., Fu, C., Pappin, D. J., and Tonks, N. K. (2011). H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 4:ra86. doi: 10.1126/scisignal.2002329
Lamotte, O., Bertoldo, J. B., Besson-Bard, A., Rosnoblet, C., Aimé, S., Hichami, S., et al. (2015). Protein S-nitrosylation: specificity and identification strategies in plants. Front. Chem. 2:114. doi: 10.3389/fchem.2014.00114
Lee, S. R., Kwon, K. S., Kim, S. R., and Rhee, S. G. (1998). Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372. doi: 10.1074/jbc.273.25.15366
Li, S., and Whorton, A. R. (2003). Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch. Biochem. Biophys. 410, 269–279. doi: 10.1016/s0003-9861(02)00696-3
Liu, F., and Chernoff, J. (1997). Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor. Biochem. J. 327, 139–145. doi: 10.1042/bj3270139
Lohse, D. L., Denu, J. M., Santoro, N., and Dixon, J. E. (1997). Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1. Biochemistry 36, 4568–4575. doi: 10.1021/bi963094r
Londhe, A. D., Bergeron, A., Curley, S. M., Zhang, F., Rivera, K. D., Kannan, A., et al. (2020). Regulation of PTP1B activation through disruption of redox-complex formation. Nat. Chem. Biol. 16, 122–125. doi: 10.1038/s41589-019-0433-0
Lu, Y., Su, W., Bao, Y., Wang, S., He, F., Wang, D., et al. (2020). Poplar PdPTP1 gene negatively regulates salt tolerance by affecting ion and ROS homeostasis in Populus. Int. J. Mol. Sci. 21:1065. doi: 10.3390/ijms21031065
Luan, S. (2003). Protein phosphatases in plants. Annu. Rev. Plant Biol. 54, 63–92. doi: 10.1146/annurev.arplant.54.031902.134743
Mahadev, K., Zilbering, A., Zhu, L., and Goldstein, B. J. (2001). Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J. Biol. Chem. 276, 21938–21942. doi: 10.1074/jbc.C100109200
Moeslein, F. M., Myers, M. P., and Landreth, G. E. (1999). The CLK family kinases, CLK1 and CLK2, phosphorylate and activate the tyrosine phosphatase, PTP-1B. J. Biol. Chem. 274, 26697–26704. doi: 10.1074/jbc.274.38.26697
Montalibet, J., Skorey, K. I., and Kennedy, B. P. (2005). Protein tyrosine phosphatase: enzymatic assays. Methods 35, 2–8. doi: 10.1016/j.ymeth.2004.07.002
Mühlenbeck, H., Bender, K. W., and Zipfel, C. (2021). Importance of tyrosine phosphorylation for transmembrane signaling in plants. Biochem. J. 478, 2759–2774. doi: 10.1042/BCJ20210202
Ravichandran, L. V., Chen, H., Li, Y., and Quon, M. J. (2001). Phosphorylation of PTP1B at Ser50 by Akt impairs its ability to dephosphorylate the insulin receptor. Mol. Endocrinol. 15, 1768–1780. doi: 10.1210/mend.15.10.0711
Rush, J., Moritz, A., Lee, K. A., Guo, A., Goss, V. L., Spek, E. J., et al. (2005). Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101. doi: 10.1038/nbt1046
Sakakura, M., Tamura, M., Fujii, N., Takeuchi, T., Hatanaka, T., Kishimoto, S., et al. (2018). Structural mechanisms for the S-nitrosylation-derived protection of mouse galectin-2 from oxidation-induced inactivation revealed by NMR. FEBS J. 285, 1129–1145. doi: 10.1111/febs.14397
Salmeen, A., Andersen, J. N., Myers, M. P., Meng, T. C., Hinks, J. A., Tonks, N. K., et al. (2003). Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773. doi: 10.1038/nature01680
Salmeen, A., and Barford, D. (2005). Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal. 7, 560–577. doi: 10.1089/ars.2005.7.560
Schmid, M., Davison, T. S., Henz, S. R., Pape, U. J., Demar, M., Vingron, M., et al. (2005). A gene expression map of Arabidopsis thaliana development. Nat. Genet. 37, 501–506. doi: 10.1038/ng1543
Tamura, M., Saito, M., Yamamoto, K., Takeuchi, T., Ohtake, K., Tateno, H., et al. (2015). S-nitrosylation of mouse galectin-2 prevents oxidative inactivation by hydrogen peroxide. Biochem. Biophys. Res. Commun. 457, 712–717. doi: 10.1016/j.bbrc.2015.01.055
Tamura, M., Sasai, A., Ozawa, R., Saito, M., Yamamoto, K., Takeuchi, T., et al. (2016). Identification of the cysteine residue responsible for oxidative inactivation of mouse galectin-2. J. Biochem. 160, 233–241. doi: 10.1093/jb/mvw029
van Montfort, R. L., Congreve, M., Tisi, D., Carr, R., and Jhoti, H. (2003). Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423, 773–777. doi: 10.1038/nature01681
Winter, D., Vinegar, B., Nahal, H., Ammar, R., Wilson, G. V., and Provart, N. J. (2007). An “electronic fluorescent pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS One 2:e718. doi: 10.1371/journal.pone.0000718
Xu, Q., Fu, H. H., Gupta, R., and Luan, S. (1998). Molecular characterization of a tyrosine-specific protein phosphatase encoded by a stress-responsive gene in Arabidopsis. Plant Cell 10, 849–857. doi: 10.1105/tpc.10.5.849
Zhang, Z. Y., and Dixon, J. E. (1993). Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 32, 9340–9345. doi: 10.1021/bi00087a012
Zhao, J., Chen, Q., Zhou, S., Sun, Y., Li, X., and Li, Y. (2020). H2Bub1 regulates RbohD-dependent hydrogen peroxide signal pathway in the defense responses to Verticillium dahliae toxins. Plant Physiol. 182, 640–657. doi: 10.1104/pp.19.00913
Zhou, S., Chen, Q., Sun, Y., and Li, Y. (2017). Histone H2B monoubiquitination regulates salt stress-induced microtubule depolymerization in Arabidopsis. Plant Cell Environ. 40, 1512–1530. doi: 10.1111/pce.12950
Keywords: Arabidopsis thaliana, protein tyrosine phosphatase 1, nitric oxide, S-nitrosation, H2O2, oxidation, post-translational modifications, mitogen-activated protein kinases
Citation: Nicolas-Francès V, Rossi J, Rosnoblet C, Pichereaux C, Hichami S, Astier J, Klinguer A, Wendehenne D and Besson-Bard A (2022) S-Nitrosation of Arabidopsis thaliana Protein Tyrosine Phosphatase 1 Prevents Its Irreversible Oxidation by Hydrogen Peroxide. Front. Plant Sci. 13:807249. doi: 10.3389/fpls.2022.807249
Edited by:
Noelia Foresi, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) Mar del Plata, ArgentinaReviewed by:
Mirko Zaffagnini, University of Bologna, ItalyCopyright © 2022 Nicolas-Francès, Rossi, Rosnoblet, Pichereaux, Hichami, Astier, Klinguer, Wendehenne and Besson-Bard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angélique Besson-Bard, YW5nZWxpcXVlLmJlc3Nvbi1iYXJkQHUtYm91cmdvZ25lLmZy
†ORCID: Valérie Nicolas-Francès, orcid.org/0000-0002-1883-4228
Claire Rosnoblet, orcid.org/0000-0002-1906-0922
Carole Pichereaux, orcid.org/0000-0002-7559-5358
Jeremy Astier, orcid.org/0000-0002-3713-6230
David Wendehenne, orcid.org/0000-0002-1088-102X
Angélique Besson-Bard, orcid.org/0000-0003-4832-738X
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.