
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 04 May 2022
Sec. Plant Systematics and Evolution
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.768810
 Jie Wang1,2,3†
Jie Wang1,2,3† Chao-Nan Fu1†Zhi-Qiong Mo1,3†Michael Möller4
Chao-Nan Fu1†Zhi-Qiong Mo1,3†Michael Möller4 Jun-Bo Yang2Zhi-Rong Zhang2
Jun-Bo Yang2Zhi-Rong Zhang2 De-Zhu Li2,3
De-Zhu Li2,3 Lian-Ming Gao1,5*
Lian-Ming Gao1,5*
Species of Cephalotaxus have great economic and ecological values. However, the taxonomy and interspecific phylogenetic relationships within the genus have been controversial and remained not fully resolved until now. To date, no study examined the efficiency of the complete plastome as super-barcode across Cephalotaxus species with multiple samples per taxon. In this study, we have evaluated the complete plastome in species discrimination and phylogenetic resolution in Cephalotaxus by including 32 individuals of all eight recognized species and five varieties following Farjon’s classification (2010) with multiple samples per taxon. Our results indicated that not all species recognized in recent taxonomic revisions of Cephalotaxus could be distinguished and not all were monophyletic. Based on the plastome phylogeny, a new taxonomic classification for the genus comprising nine species and two varieties, including a cryptic species, was proposed. The phylogeny also resolved all interspecific relationships. Compared to the plastome based classification, standard DNA barcodes, alone or in combination, only recognized a maximum of seven out of the nine species. Moreover, two highly variable single loci, ycf1 and rps16, each alone achieved full species discrimination. With the moderate length of 1079 bp, rps16 is proposed as a specific barcode to discriminate Cephalotaxus species. The super-barcodes and specific barcode candidates will aid in the identification of endangered Cephalotaxus species, and to help focus conservation measures.
Species delimitation is fundamental in many areas of biology (Duminil and Di Michele, 2009). Traditional species delimitation is based on the analysis of morphological variation, which is, however, affected by a large number of factors, and can result in difficulties in species identification (Hebert et al., 2003). With the development of molecular phylogenetic methods, applications of the “phylogenetic species concept” have increased, which uses monophyly to define species (de Queiroz, 1998, 2005; Zimmers et al., 2017). DNA barcoding is a technique to identify biological species, parts and products using one or several standardized DNA regions (Hebert et al., 2003; Kress et al., 2005; Hollingsworth, 2011; Hollingsworth et al., 2016). It has been widely used as a molecular tag for species identification and delimitation, and is contributing to rapid scientific progress on diverse fronts (Joly et al., 2014; Kress et al., 2015; Hollingsworth et al., 2016). Although a number of DNA markers were assessed as candidate DNA barcodes for plants, no barcode alone performed as well as COI in animals (Hollingsworth, 2011). Several different combinations of candidate DNA regions were proposed for barcoding plants (Chase et al., 2007; Kress and Erickson, 2007; Pennisi, 2007; Hollingsworth et al., 2009). However, the standard DNA barcodes suggested for flowering plants (rbcL, matK, trnL-trnF, and trnH-psbA) generally do not perform well across closely related species and recently diverged species mainly due to the lack of adequate genetic variation (Hollingsworth et al., 2009; Li et al., 2011; Yan et al., 2015; Coissac et al., 2016).
With the rapid development of next-generation sequencing (NGS) technologies and decreasing sequencing costs, a large amount of genomic data can be rapidly generated to date, thus the concept of DNA barcodes has been expanded (Coissac et al., 2016; Hollingsworth et al., 2016; Tonti-Filippini et al., 2017). The complete plastome was proposed as “ultra-barcode” (Nock et al., 2011; Kane et al., 2012) or “super-barcode” (Yang et al., 2013) for plant species delimitation, being referred to as next-generation DNA barcode (Hollingsworth et al., 2016). Compared to standard DNA barcodes, next-generation DNA barcodes may overcome the inherent limitations of traditional DNA barcodes, as they potentially possess more informative and variable sites, and therefore can enhance species discrimination power and phylogenetic resolution (Kane et al., 2012; Hollingsworth et al., 2016; Ji et al., 2019). Genome skimming is a cost-effective NGS technology to obtain the whole plastid genome (Straub et al., 2012; Ripma et al., 2014). The discriminatory efficiency of the complete plastome in plants has been assessed in several recent studies (e.g., Turner et al., 2016; Fu et al., 2019; Ji et al., 2019; Song et al., 2020). However, very few studies have been undertaken using multiple individuals per species from multiple congeneric species.
The plastid genome is uniparentally inherited in most plants (Birky, 2001; Gitzendanner et al., 2018) with an exception for most conifers such as Cephalotaxaceae, Taxaceae and Pinaceae where it is paternally inherited (Gianordoli, 1974; Mogensen, 1996), and behaves as a single non-recombining locus, providing a strong signal of phylogenetic history (Petit and Vendramin, 2007; Yu et al., 2018). With a highly conserved gene order and absence of recombination, the plastid genome is an ideal target for comparative analysis across land plants (Wicke et al., 2011; Hollingsworth et al., 2016; Gitzendanner et al., 2018). The benefits of the complete plastome have been argued for its utilization in plant species delimitation, although this may not solve delimitation failures resulting from rare biparental plastid inheritance, introgression and hybridization, such as presumed for Salix (Percy et al., 2014), and Calligonum (Song et al., 2020). However, plastome barcoding has shown a great promise for reliably distinguishing closely related species (Kane et al., 2012; Ruhsam et al., 2015; Zhang et al., 2017; Zhu et al., 2018; Fu et al., 2022). In addition, the plastid genome has a large number of evolutionarily informative variation, which has been widely used for phylogenetic reconstruction at deep to shallow levels in land plants (Moore et al., 2010; Yu et al., 2018; Ji et al., 2019; Li et al., 2019, 2021), including conifers (e.g., Fu et al., 2019; Zhang et al., 2019; Ji et al., 2021).
The genus Cephalotaxus Siebold and Zucc. ex Endl. is a small group of 7-10 species of evergreen conifers belonging to family Cephalotaxaceae in a narrow sense (Singh, 1961; Farjon, 1998, 2010) or Taxaceae in a wider sense (Hart, 1987; Price, 1990, 2003; Christenhusz et al., 2011). The plants in this genus are understory trees distributed in eastern Asia, north of the Indo-Chinese peninsula and Himalayas, mainly in China, Korea, Japan, Vietnam, Laos, Thailand, India, and Myanmar (Fu et al., 1999; Farjon, 2010; Lang et al., 2013; Yang et al., 2017; Zhang et al., 2019). The taxonomy of Cephalotaxus is primarily based on morphological characters and geographical distribution of individual taxa (Farjon, 2010; Lang et al., 2011, 2013; Yang et al., 2017). However, the morphological characters used for species delimitation are quite variable and characteristics usually overlap to a high degree between species resulting in a complex and controversial taxonomic history (Farjon, 2010; Lang et al., 2011, 2013; Zhang et al., 2019). For example, Farjon (2010) recognized eight species and three varieties in Cephalotaxus, i.e., C. fortunei Hook. (including C. f. var. fortunei and C. f. var. alpina H.L. Li), C. hainanensis H.L. Li., C. harringtonii (Knight ex J. Forbes) K. Koch (including C. h. var. harringtonii, C. h. var. nana (Nakai) Rehder and C. h. var. wilsoniana (Hayata) Kitam.), C. lanceolata K.M. Feng, C. latifolia L.K. Fu and R.R. Mill, C. mannii Hook.f., C. oliveri Mast., and C. sinensis (Rehd. and E.H. Wilson) H.L. Li. Out of these, six species were recognized in China by Fu et al. (1999), but had different taxonomic treatments. For instance, C. hainanensis was treated as a synonym of C. mannii, and C. harringtonii var. wilsoniana was treated as a variety of C. sinensis (Table 1). Whereas Lang et al. (2013), Zhang et al. (2019) only recognized seven species in recent revisions of the genus but with taxonomic differences (Table 1). Thus, the taxonomy of Cephalotaxus is controversial and has not been resolved satisfactorily.
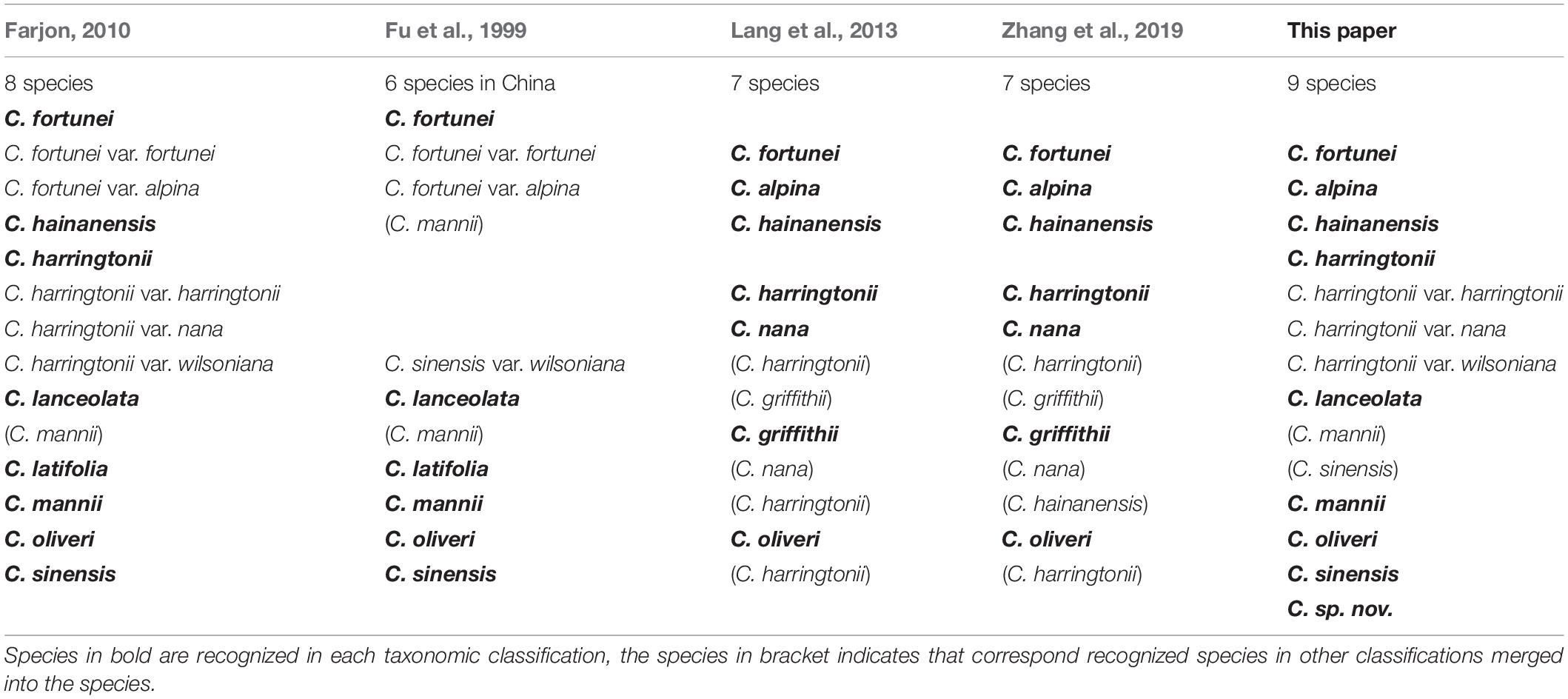
Table 1. Comparison of different taxonomic treatments of the genus Cephalotaxus.
Intrageneric phylogenetic relationships of Cephalotaxus also remained largely unresolved or disputable in previous studies which did not include all extant lineages or only used a few gene sequences. For example, Zhang et al. (2000) employed AFLP markers to discriminate only four species and four varieties of Cephalotaxus. Hao et al. (2008) investigated the interspecific relationships of Taxaceae and Cephalotaxaceae based on four cpDNA regions and the nrDNA ITS region with a complete sampling of 12 species and two varieties of Cephalotaxus. In their studies, Cephalotaxus latifolia fell on the basal branch sister to the rest of the species resolved as polytomy, with sister relationships of C. griffithii and C. oliveri, C. mannii and C. hainanensis, C. lanceolata and C. fortunei, and a clade C. harringtonia (C. wilsoniana, C. koreana, C. harringtonia cv. fastigiata). Ji et al. (2021) reconstructed a phylogeny of Cephalotaxus based on 81 plastid protein-coding genes and ten sampled species. Their interspecific relationships were fully resolved with strong support, but differed with that in Hao et al. (2008) despite both being based on plastid DNA loci. The conflicting relationships of Cephalotaxus in previous studies need to be verified using more robust molecular markers and more species sampled. While misidentification of samples will also lead to erroneous phylogenetic relationships, a correct species identification is fundamental for phylogeny reconstruction.
The species of Cephalotaxus have important economic values and are used as ornamental plants, and also as medicinal plants due to their alkaloids (Abdelkafi and Nay, 2012). A few species are listed as endangered and vulnerable by IUCN1. Thus, a correct taxonomy and accurate species delimitation are crucial to the utilization and conservation of extant Cephalotaxus species. In this study, we performed genome skimming and assembled the complete plastome of 32 samples, including multiple samples for all Cephalotaxus species recognized in past treatments, and covering the genus’ main distribution range. We aimed to: (1) delimit and discriminate the species of Cephalotaxus using the entire plastome and a phylogenetic approach using the taxonomic classification of Farjon (2010) as baseline; (2) to compare these results to those obtained using standard DNA barcodes (matK, rbcL, trnH-psbA, and trnL-trnF); (3) investigate candidates for specific barcodes; (4) based on the plastome tree propose a phylogeny-based species classification; and (5) discuss the importance of correct species identification and classification on conservation assessments for the genus.
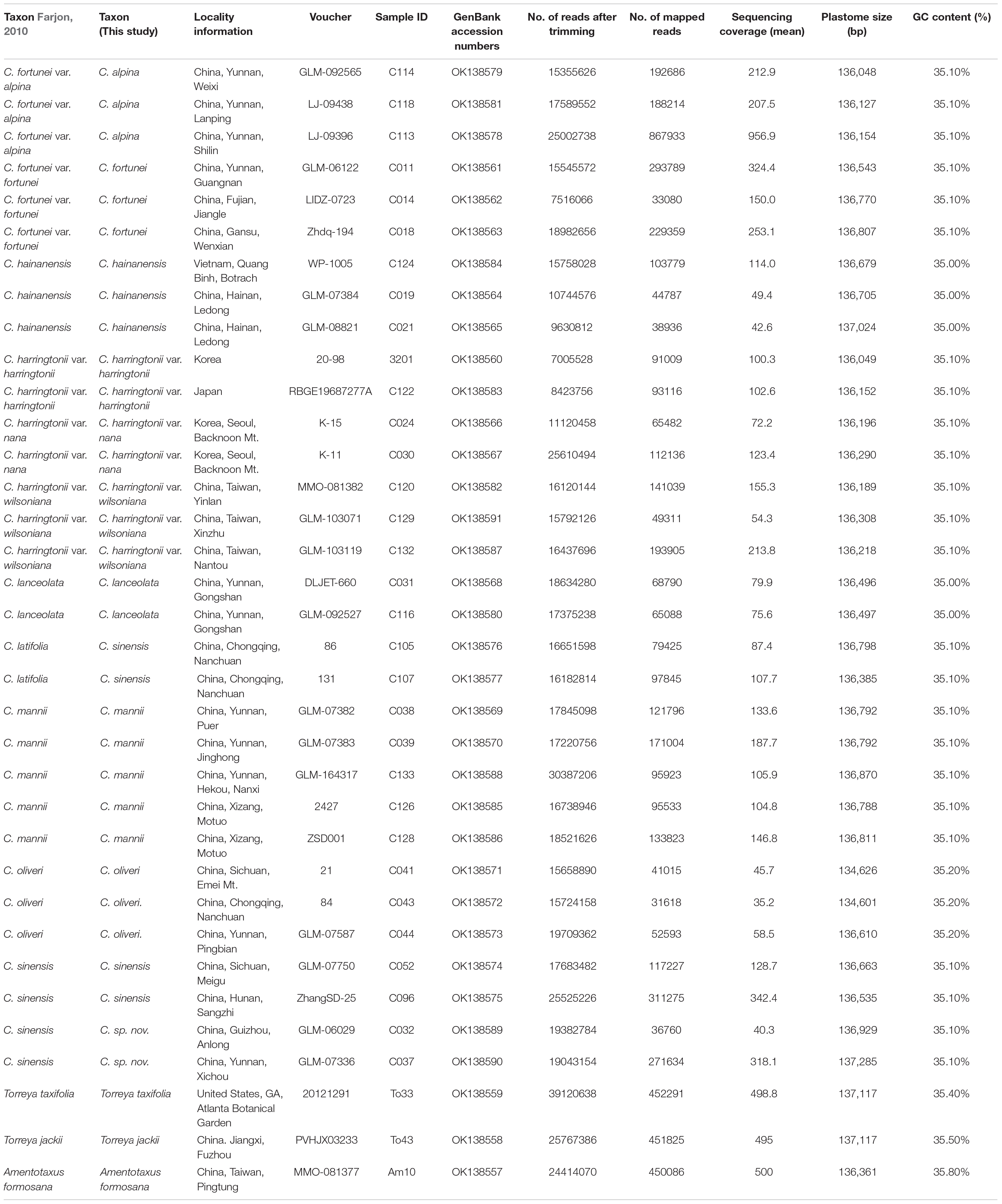
Table 2. Taxa included in this study with locality, voucher, GenBank accession numbers, and NGS performance for the 32 sequenced samples of Cephalotaxus (according to Farjon, 2010) and outgroups.
To cover the maximum diversity within species, 2-5 individuals from different populations per species and varieties were sampled in the field for this study. A total of 32 individuals of 8 species and 3 varieties in Cephalotaxus following Farjon’s latest classification (Farjon, 2010) were sampled, which also covered all the taxa raised in other recent taxonomic revisions of this genus (e.g., Fu et al., 1999; Lang et al., 2013; Zhang et al., 2019). These samples covered most of the geographic distribution range of Cephalotaxus. We selected Torreya jackii Chun, T. taxifolia Arn and Amentotaxus formosana H.L. Li as outgroups (Hao et al., 2008; Ran et al., 2018; Ji et al., 2021). More detailed samples information can be found in Table 2. Healthy leaves were collected and dried immediately in silica-gel for DNA extraction. A few samples were collected from herbarium specimens. Voucher specimens of most sampled taxa were deposited at the Herbarium of Kunming Institute of Botany (KUN), Chinese Academy of Sciences.
Total genomic DNA was extracted from ∼20 mg of leaf samples by a modified CTAB method (Doyle and Doyle, 1987). Approximately 5 μg of purified genomic DNA was used to construct shotgun libraries with a TruSeq DNA Sample Prep Kit following the manufacturer’s instructions (NEBNext® Ultra IITMDNA Library Prep Kit for Illumina®). Paired-end sequencing from both ends of 150 bp fragments was performed on an Illumina HiSeq X Ten platform (Illumina, San Diego, CA, United States) at BGI (Wuhan, China) to generate approximately 3 Gb data for each individual sample. Raw reads were filtered to remove adaptors and low quality reads using the NGS QC Toolkit (Patel and Jain, 2012) with default parameters.
The plastomes were de novo assembled from the genome skimming data using the GetOrganelle toolkit (Jin et al., 2020). The complete plastome of C. hainanensis (NC_042392) was used as the reference. Plastid genes were annotated using PGA (Qu et al., 2019) and coupled with manual adjustment in Geneious v8.0.2 (Kearse et al., 2012). The tRNAs were checked with tRNAscan-SE v2.0.3 (Lowe and Chan, 2016). Final plastid genome map was drawn using OGDRAW (Lohse et al., 2013).
We employed the complete plastome sequences for species delimitation using tree-based and genetic distance methods. To compare the discriminatory power of standard DNA barcode and super-DNA barcode (the whole plastome), the standard DNA barcodes of matK, rbcL, trnH-psbA and trnL-trnF sequences were extracted from the complete plastomes. Besides, DnaSP v6.12 (Rozas et al., 2017) was used to perform a sliding window analysis with a step size of 200 bp and a window length of 800 bp on the alignment of all Cephalotaxus species plastomes, to detect the rapidly evolving candidate molecular markers for species delimitation and discrimination. Nucleotide diversity (Pi) was computed for constructing a DNAsp graph.
In tree-based analyses, since some inversions were found in the plastomes of the outgroups (Torreya jackii, T. taxifolia and Amentotaxus formosana) compared with Cephalotaxus, we adjusted the plastid genome structure of the outgroups using Draft in the MULAN web server2 (Ovcharenko et al., 2005) with Cephalotaxus fortunei (C011) as the reference sequence to make the gene order and direction collinear across the genome for dataset alignment. Sixteen datasets were constructed in this study: (a) the complete plastomes, (b) six based on the standard DNA barcodes and some of their combinations: matK, rbcL, trnH-psbA, trnL-trnF, matK + rbcL, and matK + rbcL + trnH-psbA + trnL-trnF, (c) eight from selected high variation loci: trnI-rrn16, ycf1, chlN-ycf1, clpP-accD, rps16, accD, ycf2, ndhF-trnR, and (d) added the 10 sampled species of Ji et al. (2021) to our dataset and used only the 81 plastid protein-coding genes. The complete plastomes and the DNA loci in each datasets were aligned separately using the program MAFFT v7.221 (Katoh and Standley, 2013) with manual adjustments where necessary and concatenated into data matrices. Phylogenetic analyses for each dataset using maximum likelihood (ML) were performed in RAxML v8.2 (Stamatakis, 2014) under the GTRGAMMA model. The best-scoring ML tree for each dataset was produced with 1,000 bootstrap replicates to provide support values for each node. Taxon with multiple individuals resolved as monophyletic with bootstrap support (BS) over 50% were treated as successfully discriminated against Farjon’s (2010) classification.
To assess the barcoding gap, inter- and intraspecific distances were calculated using the Kimura 2-parameter (K2P) distance in MEGA v10.1.8 (Kumar et al., 2018) for each of the 16 data sets. A discrete distribution difference between the average intraspecific and the average interspecific genetic distance is indicative of the existence of a barcoding gap (Hebert et al., 2004).
High-quality complete plastid genomes of all 32 sampled individuals of Cephalotaxus were obtained and assembled into circular molecules (Figure 1 and Table 2). For the 32 individuals, 31,618 (C. oliveri C043) to 867,933 (C. fortunei var. alpina C113) reads were mapped to the newly assembled plastomes with an average sequencing depth ranging from 35.2× to 956.9×. The length of the 32 de novo assembled Cephalotaxus plastomes ranged from 134,601 to 137,285 bp with very similar GC contents (35.1–35.2%) (Table 2). The plastomes of all Cephalotaxus samples included 113 unique genes in identical order, comprising 4 rRNA genes, 27 tRNA genes and 82 protein-coding genes (Table 3). Identical to the published Cephalotaxus plastomes, an inverted repeat region was lost, resulting in difficulties in defining the boundary between the large and small single-copy regions (Figure 1).

Table 3. Gene content of the Cephalotaxus plastomes.
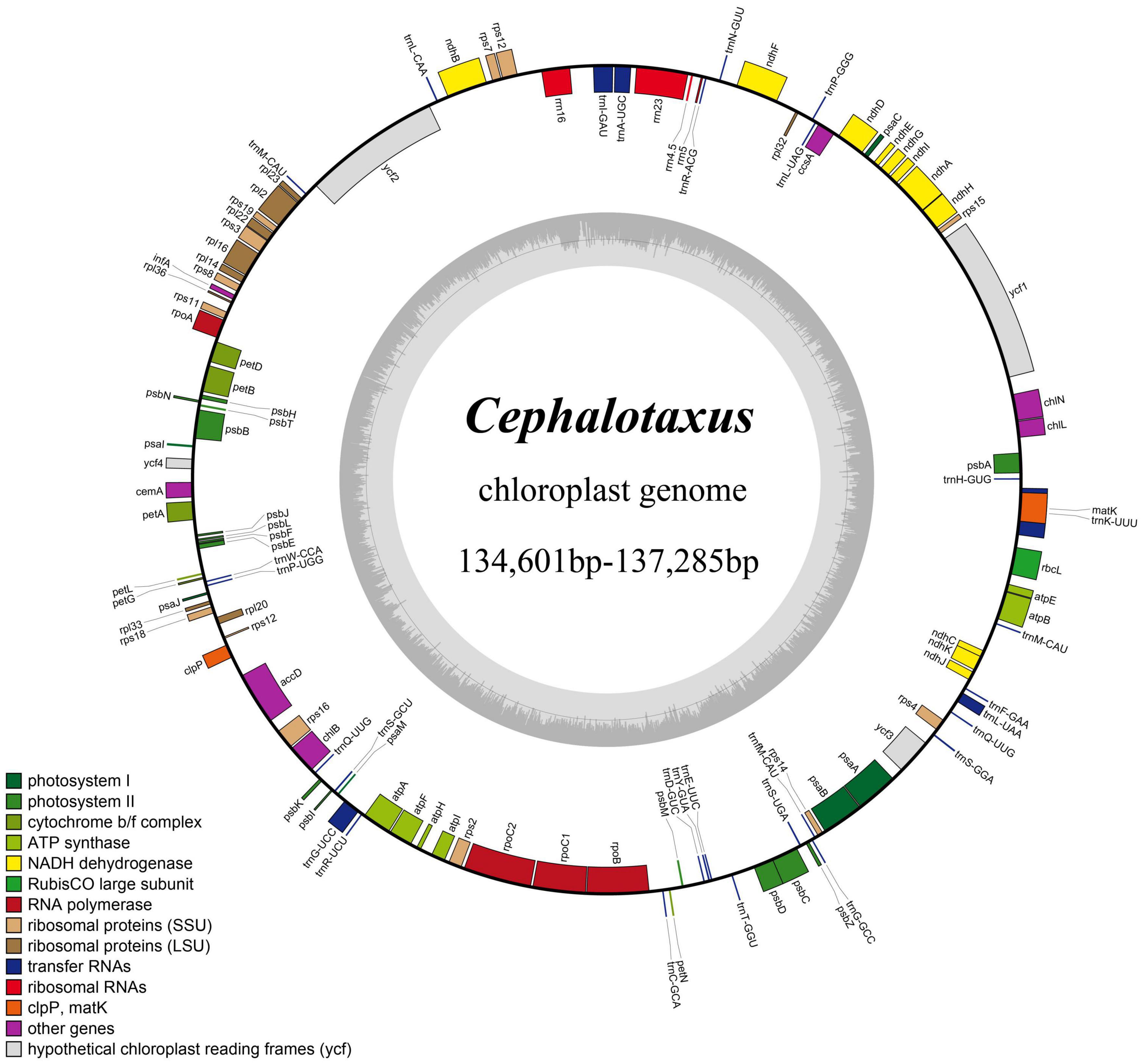
Figure 1. Map of the Cephalotaxus plastomes. Genes shown outside the circle are transcribed clockwise, and genes inside are transcribed counterclockwise. The dark gray area in the inner circle indicates the GC content of the plastomes.
We obtained an alignment matrix of 142,194 bp, which had 3,795 (2.67%) variable sites including 3,467 (2.44%) parsimony informative (PI) sites (Table 4). The ML tree showed that all the taxon nodes (the node grouping individuals from the same species/variety) had maximum support values (BP = 100%), except for C. harringtonii var. harringtonii (BP = 73%) (Figure 2). Based on the classification of Farjon (2010), five species (C. hainanensis, C. harringtonii, C. lanceolata, C. mannii, and C. oliveri) out of eight of his recognized species (62.5%) were successfully discriminated with highest support values, while the remaining three, C. latifolia, C. fortunei, and C. sinensis, were non-monophyletic. Within C. harringtonii, samples of each variety formed a clade respectively, and C. harringtonii var. nana was sister to the other two conspecific varieties. Samples of C. sinensis (C052 and C096) and C. latifolia (C105 and C107) were recovered in a clade (BP = 100%), but neither species was monophyletic. Interestingly, two individuals (C032 and C037) of C. sinensis formed a distinct clade as sister to a clade (BP = 100%) that included samples of C. fortunei, C. sinensis (C052 and C096) and C. latifolia (Figure 2). Although samples of the two varieties of C. fortunei (C. fortunei var. fortunei, C. fortunei var. alpina) each formed a separate clade, they were not monophyletic (Figure 2). The phylogenetic tree including additionally 10 Cephalotaxus species used in Ji et al. (2021) based on 81 protein-coding genes recovered the same topology with strong support as the complete plastome tree. In this tree, six out of the ten species grouped in their respective species clades. Two species, C. griffithii (= C. lanceolata) and C. nana (= C. latifolia), grouped into our C. fortunei var. alpina clade, and the other two species, C. fortunei and C. alpina, fell in the clade of C. mannii (Supplementary Figure S1).
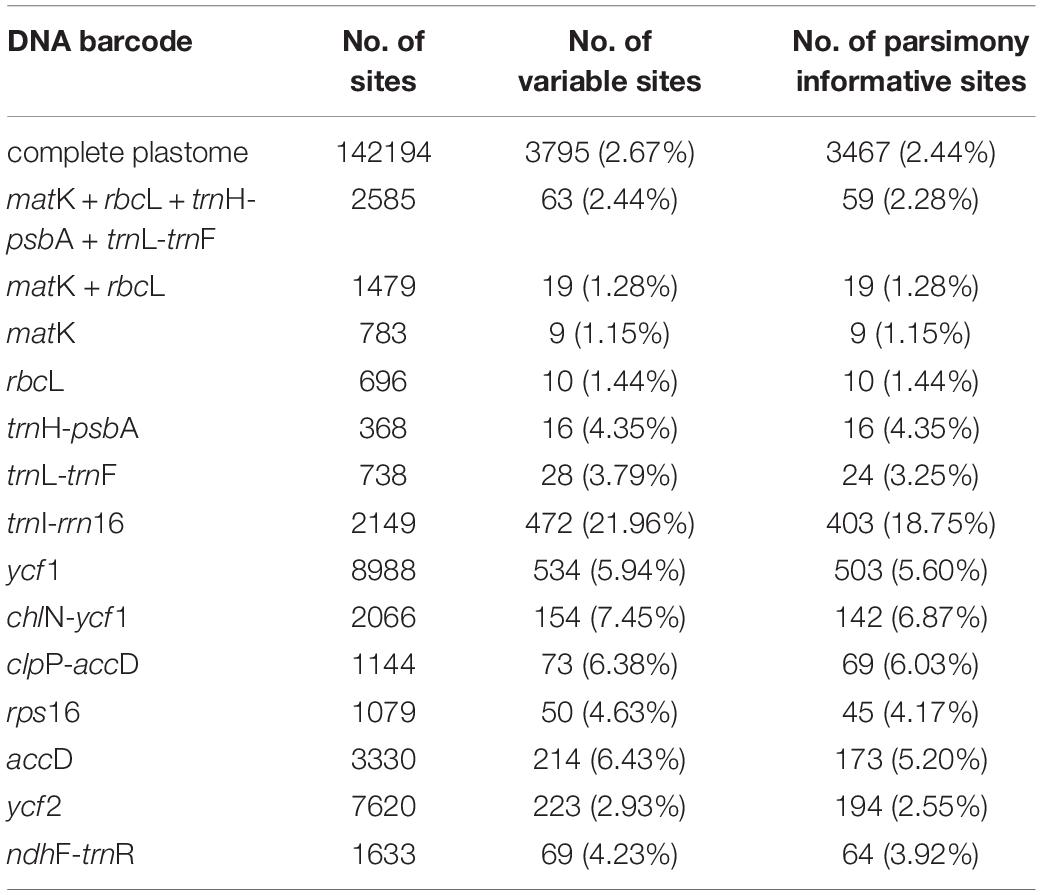
Table 4. Sequence characteristics of the complete plastome, different DNA loci and the combinations for Cephalotaxus.
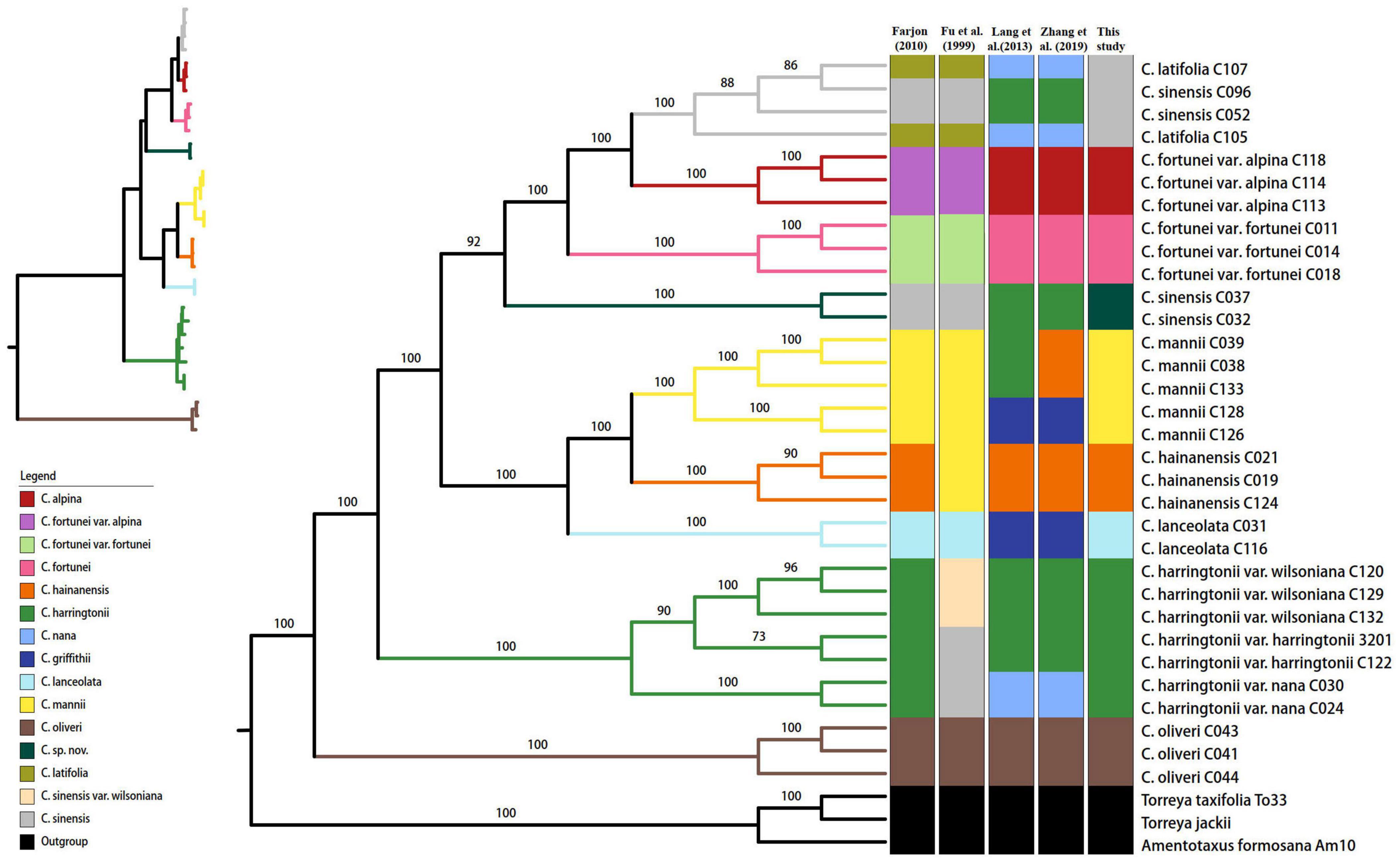
Figure 2. Phylogenetic tree reconstructed via maximum-likelihood (ML) analysis of the complete plastome data. Numbers above branches indicate the bootstrap support values. In the top left is an ML phylogenetic tree with branch length excluding outgroups. The branch colors correspond with the species recognized in this study.
Four species recognized by Lang et al. (2013), Zhang et al. (2019), i.e., C. griffithii (C031, C 116, C126, and C128), C. hainanensis (C19, C21, C124, C038, C039, and C133), C. harringtonii (C120, C122, C129, C132, 3201, C032, C037, C052, and C096), and C. nana (C024, C030, C105, and C107) that differed in their taxonomic treatment with Farjon (2010), were not resolved as monophyletic by the complete plastome data; only three species (C. fortunei, C. alpina, and C. oliveri) recognized by Lang et al. (2013), Zhang et al. (2019) were identified successfully. Based on the phylogenetic tree of the entire plastid genome, nine species including a cryptic lineage (C. sp. nov. C032/C037) for Cephalotaxus were successfully identified and received high support here (Figure 2), which corresponded well with their geographic distribution and morphology. From here on, we used these species names proposed in this study (Table 1).
All the interspecific relationships within Cephalotaxus were fully resolved with strong support values (>92%) based on the phylogenetic tree using complete plastome data (Figure 2). Cephalotaxus oliveri was on the earliest diverging branch, and C. harringtonii formed the subsequent clade which was sister to the rest of the species. The remaining species formed two clades, i.e., C. lanceolata (C. hainanensis + C. mannii), and C. sp. nov. (C. fortunei (C. alpina + C. sinensis)).
Among the four single standard DNA barcodes (rbcL, matK, trnH-psbA, trnL-trnF), trnL-trnF had the highest number of PI sites of 24 (3.25%, the percentage of informative sites in relation to the length of the sequenced fragment), followed by trnH-psbA of 16 (4.35%) and rbcL of 10 (1.44%), with matK showing the lowest number with 9 (1.15%) (Table 4). The combination of matK + rbcL resulted in a matrix of 1479 bp in length, with 19 (1.28%) PI sites, while the combination of the four standard plastid DNA barcodes (matK + rbcL + trnH-psbA + trnL-trnF) had 2,585 bp and 59 (2.28%) PI sites. Based on the tree-based analysis of the six datasets of the standard DNA barcodes, the single barcodes and combination of matK + rbcL distinguished a maximum of four (50%) and five (55.6%) species (trnL-trnF) for the classification of Farjon (2010) and the one proposed in this study respectively. However, the four-barcode combination (matK + rbcL + trnH-psbA + trnL-trnF) resolved five (62.5%) and seven (77.8%) species, for the respective classifications (Figure 3 and Supplementary Figure S2).
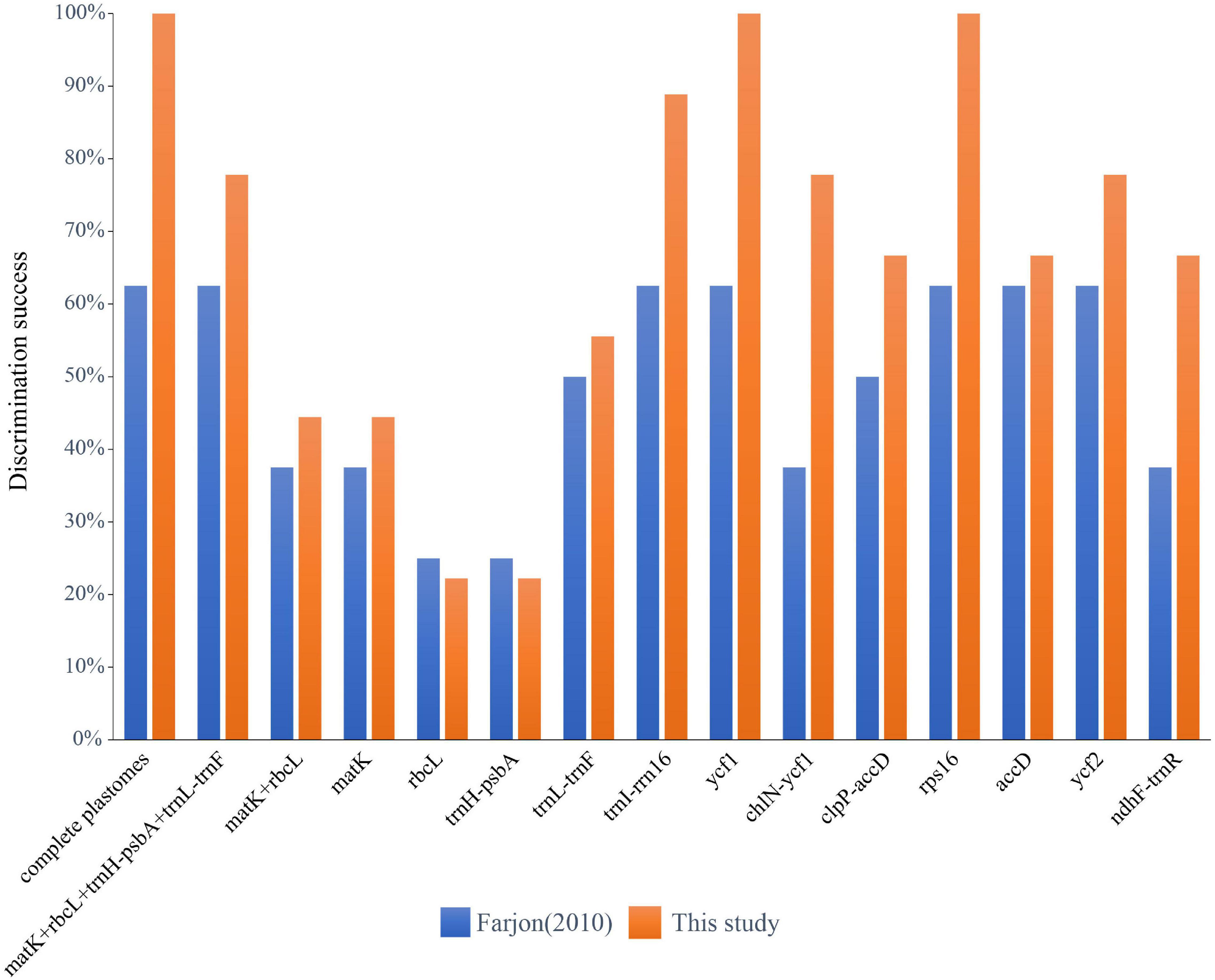
Figure 3. Species discrimination rate of the complete plastome, all single fragments and two DNA barcoding combinations based on the tree-building method.
Sliding window analyses for all the newly assembled Cephalotaxus plastid genomes identified eight highly variable regions (mutational hotspots) (trnI-rrn16, ycf1, chlN-ycf1, clpP-accD, rps16, accD, ycf2, ndhF-trnR) (Figure 4). All eight hypervariable specific loci provided higher (maximally 62.5%) or the same (37.5%: clpP-accD and ndhF-trnR showed the same discriminative power as matK, and matK + rbcL) species resolution than any single standard DNA barcode or the combination of matK + rbcL for the classification of Farjon (2010) (Figure 3). Discrimination success was always higher for the specific loci for the classification proposed in the present study. For this classification, out of these eight specific loci, two, ycf1 and rps16, yielded the maximum species discriminatory rate, identical to that of the complete plastome, followed by trnI-rrn16 which identified eight out of nine (89%) species of Cephalotaxus (Figure 3 and Supplementary Figure S2).
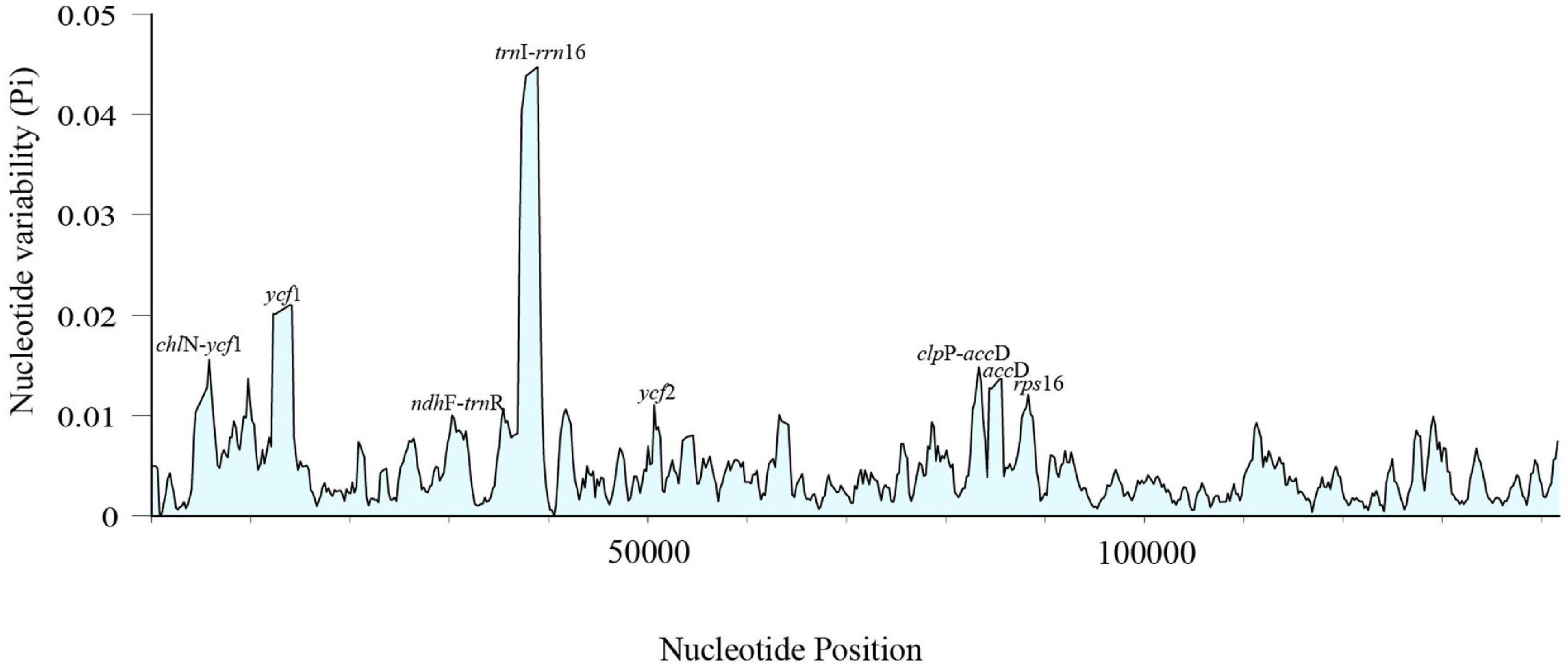
Figure 4. Sliding window analysis of 32 Cephalotaxus plastomes (window length: 800 bp, step size: 200 bp). X-axis: position of the midpoint of a window; Y-axis: nucleotide diversity of each window.
The intraspecific and interspecific K2P distance varied among the 16 datasets. Out of these, the four standard DNA barcodes and their combination, plus ycf2, had an interspecific distance of zero, and chlN-ycf1 the highest intraspecific distance with 2.41%. Although there was some degree of overlapping between the minimum interspecific distance and the maximum intraspecific distance of these loci/combinations (Supplementary Table S1), relatively distinct barcoding gaps were identified for the complete plastome, plastid protein-coding genes and rps16, ndhF-trnR and ycf2 (Supplementary Figure S3). Out of these, four datasets, i.e., the complete plastome, plastid protein-coding genes, ycf1, and rps16, showed maximum species resolution.
With the ever more cost-effective NGS technology, the complete plastid genome assembled from genome skimming data has been used several times as super-barcode with promising potential for plant species delimitation (Kane et al., 2012; Turner et al., 2016; Tonti-Filippini et al., 2017; Fu et al., 2019; Ji et al., 2019; Song et al., 2020). In the present study, compared to the standard barcodes, the complete plastome showed much higher species discrimination rates, and distinguished five out of the eight (62.5%) species of Cephalotaxus recognized by Farjon (2010). Only two (25%) to four (50.0%) species were resolved by any single barcode and the combination of matK + rbcL, while the combination of all four standard barcodes recovered five species (62.5%), the same as the whole plastome. With view to the nine species proposed here, that all species were highly supported in the phylogeny based on the entire plastomes and the plastid coding genes, the standard DNA barcodes (single barcode and their combinations) only discriminated a maximum of seven (77.8%) of those species (with the combination of matK + rbcL + trnH-psbA + trnL-trnF) and with relatively low support values (Figure 3 and Supplementary Figure S2).
Our species discrimination results indicated that the complete plastome can provide more variation to discriminate Cephalotaxus species compared to the standard barcodes, which has also been demonstrated in other studies (e.g., Ji et al., 2020; Song et al., 2020; Fu et al., 2022). In addition, the complete plastome approach based on genome skimming can circumvent issues of locus choice, and low PCR and sequencing recovery rate, which are sometimes encountered in standard DNA barcoding studies (Ruhsam et al., 2015; Curci et al., 2016). Therefore, the whole plastome as super-barcode illustrated the benefits of moving beyond standard barcodes in Cephalotaxus (Tonti-Filippini et al., 2017; Nevill et al., 2020).
Although the super-barcode effectively distinguished species beyond the standard barcodes, its use may sometimes be limited due to the relatively higher costs and complex analyses of genome skimming data compared to the standard PCR and Sanger sequencing barcode approach. A “specific” barcode is a fragment of DNA sequence that has a sufficiently high mutation rate to enable high species identification within a given taxonomic group beyond the standard DNA barcodes, and a more wider use of plant group-specific barcodes for discriminating closely related species was predicted (Li et al., 2015). In this study, we identified eight highly variable loci from the plastid genomes of Cephalotaxus. Among them, although trnI-rrn16 was the most divergent locus, it achieved only a discriminatory rate of 8 out of 9 species (88.9%) (Figure 3), which implied that the selection of barcoding loci based on maximum sequence divergence may not be in line with maximum species discrimination ability. The two loci ycf1 and rps16 showed the maximum species discrimination ability, the same as the complete plastome (Figure 3). Considering the length of ycf1 (8988 bp), it is not suitable as a hypervariable specific barcode. With a PCR-manageable length of 1079 bp, rps16 can be proposed here as a specific barcode for discriminating Cephalotaxus species. In addition, these identified highly variable loci represent valuable genetic resources for future population genetics and phylogeographic studies.
The inclusion of multiple individuals from a single species sampled is highly important to confirm species delimitation successfully and accurately (Liu et al., 2012; Fu et al., 2022). Therefore, in this study, all species/varieties of Cephalotaxus had more than one individual included (two to five individuals per taxon). Samples of five species and all varieties in Farjon’s (2010) taxonomic classification were resolved as monophyletic with strong support in the complete plastome phylogeny, while another three species, C. latifolia, C. sinensis and C. fortunei, were not individually resolved as monophyletic. Samples of C. latifolia and C. sinensis together formed a highly supported clade, but were not resolved in their respective species monophyly. This indicated that they belonged to the same genetic lineage, and suggested a merging of C. latifolia into C. sinensis. This is supported by their sympatric distribution and overlapping of some morphological traits. Notably, the two samples (C032 and C037) collected from the karst region of southwest Guizhou and southeast Yunnan, China, identified as C. sinensis under the classification of Farjon (2010), were recovered in a highly supported isolated clade, suggesting a cryptic species of Cephalotaxus (Figure 2). In a study including more individuals collected from this karst region, in adjacent areas in southwest Guizhou, southeast Yunnan and north Vietnam, standard DNA barcodes already indicated the existence of this cryptic species, though it was not distinguished due to a lack of sufficient sequence variations in the nrDNA ITS region (Gao et al., 2015). The new taxon may be adapted to the special karst habitats and has become a distinct species over time, as also found in Taxus (Liu et al., 2011; Möller et al., 2013, Möller et al., 2020) and Amentotaxus (Gao et al., 2017, 2019) in this region. More detailed morphological study for this group is needed to describe the new species. Cephalotaxus fortunei var. fortunei and C. fortunei var. alpina were each resolved as monophyletic respectively, but the species was not monophyletic. This supports the view that the two varieties are best represented at species rank (Lang et al., 2011, 2013; Zhang et al., 2019).
The 81 protein-coding gene phylogenetic tree that included all ten species of Cephalotaxus used in Ji et al. (2021) showed that only six of the ten species (C. oliveri C. harringtonii, C. wilsoniana, C. hainanensis, C. mannii, and C. sinensis) grouped into the respective species clade. However, four species (C. fortunei, C. alpina, C. griffithii, C. nana) fell into the clade of other species. For instance, two species, C. fortunei (MT555080) and C. alpina (MT555079) grouped together with C. mannii (MT555084) with very short branch length within C. mannii clade, indicating that the samples identified as C. fortunei and C. alpina in Ji et al. (2021) are misidentified and are likely C. mannii. Similarly, C. griffithii (MT555081) and C. nana (MT555085) were placed in the C. fortunei var. alpina clade, hinting they were also misidentified and likely represent C. alpina (Supplementary Figure S1). According to the geographic distribution of the four species, their identification is sound. For example, C. griffithii (MT555081) treated as C. lanceolata in Farjon (2010) was collected from Dulongjiang, Gongshan, Yunnan, its type locality. Cephalotaxus fortunei was from Nanchuan, Chongqing, and C. nana from west Hubei treated as C. latifolia in Fu et al. (1999), Farjon (2010). The unusual placement of the four species might be the result from experimental and/or identification errors.
Cephalotaxus oliveri has a distinct morphology and no taxonomic controversy in all classifications (Farjon, 1998, 2010; Lang et al., 2011, 2013; Zhang et al., 2019), and the species can be discriminated in all barcoding datasets. Our results did not support the merging of C. lanceolata into C. griffithii, C. sinensis into C. harringtonii (Lang et al., 2011, 2013; Zhang et al., 2019), and C. mannii into C. harringtonii (Lang et al., 2013) or C. hainanensis (Zhang et al., 2019), nor supported C. harringtonii var. wilsoniana as a variety of C. sinensis (Fu et al., 1999). Cephalotaxus lanceolata formed a distinct clade separate from C. griffithii (C126 and C128), and morphologically differed from C. griffithii by lanceolate leaves and rounded base. Within C. harringtonii, each of the three varieties and the species itself were resolved as monophyletic. The variety C. harringtonii var. wilsoniana is endemic to Taiwan, China, the other two taxa mainly occur in Japan and Korea. We observed a low sequence variation within the species and samples were missing of C. harringtonii var. nana from Japan. Therefore, we refrained from a decision of whether to raise the varieties to species level until more samples of this species particularly from Japan are studied.
Phylogenetic relationships of Cephalotaxus in previous studies were not fully resolved likely due to a low number of DNA loci used, incomplete sampling, or both (e.g., Cheng et al., 2000; Hao et al., 2008). In this study, the interspecific phylogenetic relationships of Cephalotaxus were fully resolved (Figure 2), and the topology is basically consistent with the phylogeny based on 81 plastid protein-coding genes (Ji et al., 2021), but with some differences in interspecific relationships possibly due to experimental error or misidentification (see above). In the present study, C. oliveri was on the first diverging branch. C. harringtonii formed the subsequent grade and sister to the rest of the species, which was also recovered in Ji et al. (2021), but such relationship was not recovered by the combination of four plastid loci (matK, rbcL, trnL-F, and psbA-trnH) and nrITS region (Hao et al., 2008). The remaining species formed two clades in this study, i.e., C. lanceolata (C. hainanensis + C. mannii), and C. sp. nov. (C. fortunei (C. alpina + C. sinensis)). The sister relationship of C. hainanensis and C. mannii, and monophyly of C. harringtonii was supported in Hao et al. (2008), but other interspecific relationships were not recovered. While in Ji et al. (2021), C. alpina and C. latifolia (representing C. nana in Ji et al., 2021) were resolved as sister and then as sister to C. sinensis. Another difference is represented by C. fortunei; this species grouped in the clade of C. hainanensis (C. mannii (C. lanceolata + C. fortunei)) being sister to C. lanceolata (with low branch support 75%) with very short branch length (Ji et al., 2021). Such inconsistent relationships were likely either due to species misidentification or experimental errors (see above). Therefore, the use of the entire plastome and multiple samples per species from different populations resulted in a fully resolved and accurate phylogeny of Cephalotaxus.
Accurate species delimitation and identification are key to proper species management and conservation (Trias-Blasi and Vorontsova, 2015). Because of the morphological similarities between Cephalotaxus taxa, it is often difficult to unambiguously identify species. In this study, the whole plastome as super-barcode in combination with a revised classification enabled an accurate delimitation of taxa, including disputed ones in Cephalotaxus, and suggested the presence of a cryptic species. In this genus, C. lanceolata and C. hainanensis are listed as endangered, C. mannii and C. oliveri are vulnerable in the global IUCN Red List (see text footnote 1). Of these, only C. oliveri is recognized in all classifications (Table 1). In recent taxonomic revisions of the genus (Lang et al., 2013; Zhang et al., 2019), C. lanceolata and C. mannii were merged into C. griffithii and C. hainanensis respectively, and their conservation rank changed consequently. However, these taxonomic treatments were not supported by our results, and we propose to retain these species. This has conservation consequences. For example, C. lanceolata is a species with extremely small populations which are confined to a small area in northwest Yunnan and the adjacent region of north Myanmar, and it is highly endangered. If C. lanceolata would be treated as a synonym of C. griffithii (Lang et al., 2011, 2013; Zhang et al., 2019), the populations in Yunnan and Myanmar would no longer receive attention for conservation since C. griffithii is not listed as endangered species. Our study also discovered a new cryptic species (C. sp. nov.). Based on IUCN criteria v3.1 (IUCN, 2001), this cryptic species would be assessed as threatened species due to small population size fewer than 2,500 mature individuals, ongoing habitat degradation from surrounding agriculture and forest clearances in this region. While plants of this taxon had always been identified as C. sinensis, a species categorized as Least Concern (LC), and never considered endangered.
In this study, we have evaluated the species discrimination efficiency of a super-barcode (the complete plastome), standard DNA and specific DNA barcode using 32 plastomes from all recognized species/varieties in different taxonomic classifications of Cephalotaxus with multiple samples per taxon. Our phylogenetic results based on the complete plastome sequences indicated that nine species, including a cryptic taxon, could be discriminated phylogenetically and the interspecific relationships of Cephalotaxus were fully resolved with strong support. The standard barcodes, alone or in combinations, were unable to discriminate all nine species. Of the eight selected hypervariable specific barcode loci, we found that only ycf1 and rps16 alone were able to distinguish all nine species, and, because of its PCR-convenient length, propose rps16 as a specific barcode. In addition, our result indicates that the inclusion of multiple samples per species from different population is necessary for both plant DNA barcoding and phylogeny studies. The new insights into the species delineation of Cephalotaxus in this study will facilitate their identification and would allow targeted conservation management of the endangered Cephalotaxus species in the future.
The original contributions presented in the study are publicly available. This data can be found here: National Center for Biotechnology Information (NCBI) BioProject database under accession numbers BankIt2499120 AM10 OK138557, BankIt2499138 To43 OK138558, BankIt2499138 To33 OK138559, BankIt2499502 3201 OK138560, BankIt2499502 C011 OK138561, BankIt2499502 C014 OK138562, BankIt2499502 C018 OK138563, BankIt2499502 C019 OK138564, BankIt2499502 C021 OK138565, BankIt2499502 C024 OK138566, BankIt2499502 C030 OK138567, BankIt2499502 C031 OK138568, BankIt2499502 C038 OK138569, BankIt2499502 C039 OK138570, BankIt2499502 C041 OK138571, BankIt2499502 C043 OK138572, BankIt2499502 C044 OK138573, BankIt2499502 C052 OK138574, BankIt2499502 C096 OK138575, BankIt2499502 C105 OK138576, BankIt2499502 C107 OK138577, BankIt2499502 C113 OK138578, BankIt2499502 C114 OK138579, BankIt2499502 C116 OK138580, BankIt2499502 C118 OK138581, BankIt2499502 C120 OK138582, BankIt2499502 C122 OK138583, BankIt2499502 C124 OK138584, BankIt2499502 C126 OK138585, BankIt2499502 C128 OK138586, BankIt2499502 C132 OK138587, BankIt2499502 C133 OK138588, BankIt2499536 C032 OK138589, BankIt2499536 C037 OK138590, and BankIt2500849 C129 OK138591.
L-MG and D-ZL designed the experiments. L-MG and MM collected the plant samples. JW, C-NF, and Z-QM performed the experiments and analyzed the data. JW, L-MG, and MM wrote the manuscript. All authors read and approved the final manuscript.
This study was supported by the Large-scale Scientific Facilities of the Chinese Academy of Sciences (2017-LSFGBOWS-02 to L-MG), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB31000000 to L-MG), the Key Basic Research program of Yunnan Province, China (Grant No. 202101BC070003 to L-MG), and the Biodiversity Survey and Assessment Project of the Ministry of Ecology and Environment, China (2019HJ2096001006 to L-MG). Molecular experiments and data analysis were performed at the Laboratory of Molecular Biology and iFlora High Performance Computing Center of Germplasm Bank of Wild Species in Southwest China, Kunming Institute of Botany. The Royal Botanic Garden Edinburgh is supported by the Scottish Government’s Rural and Environment Science and Analytical Services division.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Drs. Jie Liu, Dequan Zhang, Yumin Shui, Xiaohua Jin, Shudong Zhang, Cunxia Zeng, Jianjun Jin, Mr. Bo Wang, and Profs. Lisong Wang, Kevin Burgess for their help with plant materials or technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.768810/full#supplementary-material
Supplementary Figure S1 | Maximum Likelihood phylogenetic tree of 42 Cephalotaxus samples including the 10 species used in Ji et al. (2021) based on 81 plastid protein-coding genes with proportional branch lengths. Numbers to the right of nodes indicate bootstrap support values.
Supplementary Figure S2 | Maximum Likelihood phylogenetic trees of 32 Cephalotaxus samples based on the eight highly variable plastid loci, the four single standard DNA barcode and two combinations of standard DNA barcodes (a. trnI-rrn16, b. ycf1, c. chlN-ycf1, d. clpP-accD, e. rps16, f. accD, g. ycf2, h. ndhF-trnR, i. matK + rbcL + trnH-psbA + trnL-trnF, j. matK, k. matK + rbcL, l. rbcL, m. trnH-psbA, n. trnL-trnF). Numbers to the right of nodes indicate bootstrap support values.
Supplementary Figure S3 | Relative distribution of intraspecific and interspecific distances for the datasets of the complete plastome, 81 plastid protein-coding genes, combination of the four standard barcodes, and eight highly variable regions (trnI-rrn16, ycf1, chlN-ycf1, clpP-accD, rps16, accD, ycf2, ndhF-trnR). x-axes relate to K2P distances arranged in intervals, and the y-axes correspond to the percentage of occurrences.
Supplementary Table S1 | Examination of the 15 DNA loci used for discriminating the recognized species of Cephalotaxus in this study.
Abdelkafi, H., and Nay, B. (2012). Natural products from Cephalotaxus sp.: chemical diversity and synthetic aspects. Nat. Prod. Rep. 29, 845–869. doi: 10.1039/D1NP00035G
Birky, C. W. Jr. (2001). The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Annu. Rev. Genet. 35, 125–148. doi: 10.1146/annurev.genet.35.102401.090231
Chase, M. W., Cowan, R. S., Hollingsworth, P. M., Van Den Berg, C., Madriñán, S., Petersen, G., et al. (2007). A proposal for a standardised protocol to barcode all land plants. Taxon 56, 295–299. doi: 10.1002/tax.562004
Cheng, Y., Nicolson, R. G., Tripp, K., and Chaw, S. M. (2000). Phylogeny of Taxaceae and Cephalotaxaceae genera inferred from chloroplast matK gene and nuclear rDNA ITS region. Mol. Phylogenet. Evol. 14, 353–365. doi: 10.1006/mpev.1999.0710
Christenhusz, M. J., Reveal, J. L., Farjon, A., Gardner, M. F., Mill, R. R., and Chase, M. W. (2011). A new classification and linear sequence of extant gymnosperms. Phytotaxa 19, 55–70. doi: 10.11646/PHYTOTAXA.19.1.3
Coissac, E., Hollingsworth, P. M., Lavergne, S., and Taberlet, P. (2016). From barcodes to genomes: extending the concept of DNA barcoding. Mol. Ecol. 25, 1423–1428. doi: 10.1111/mec.13549
Curci, P. L., De Paola, D., and Sonnante, G. (2016). Development of chloroplast genomic resources for Cynara. Mol. Ecol. Resour. 16, 562–573. doi: 10.1111/1755-0998.12457
de Queiroz, K. (1998). “The general lineage concept of species, species criteria, and the process of speciation,” in Endless Forms: Species and Speciation, eds D. J. Howard and S. H. Berlocher (Oxford: Oxford University Press), 57–75.
de Queiroz, K. (2005). A unified concept of species and its onsequences for the future of taxonomy. Proc. Calif. Acad. Sci. 56, 196–215.
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Duminil, J., and Di Michele, M. (2009). Plant species delimitation: a comparison of morphological and molecular markers. Plant Biosyst. 143, 528–542. doi: 10.1080/11263500902722964
Farjon, A. (1998). World Checklist and Bibliography of Conifers. Richmond: Royal Botanical Gardens at Kew.
Fu, C. N., Mo, Z. Q., Yang, J. B., Cai, J., Ye, L. J., Zou, J. Y., et al. (2022). Testing genome skimming for species discrimination in the large and taxonomically difficult genus Rhododendron. Mol. Ecol. Resour. 22, 404–414. doi: 10.1111/1755-0998.13479
Fu, C. N., Wu, C. S., Ye, L. J., Mo, Z. Q., Liu, J., Chang, Y. W., et al. (2019). Prevalence of isomeric plastomes and effectiveness of plastome super-barcodes in yews (Taxus) worldwide. Sci. Rep. 9, 1–11. doi: 10.1038/s41598-019-39161-x
Fu, L. G., Li, N., and Mill, R. (1999). Cephalotaxaceae. Flora of China (Volume 4). Beijing: Science Press.
Gao, L. M., Li, D. Z., and Liu, J. (2015). DNA barcoding for identification of Cephalotaxus and the discovery of new species. Genome 58, 219–219.
Gao, L. M., Li, Y., Phan, L. K., Yan, L. J., Thomas, P., Phan, L. K., et al. (2017). DNA barcoding of East Asian Amentotaxus (Taxaceae): potential new species and implications for conservation. J. Syst. Evol. 55, 16–24. doi: 10.1111/jse.12207
Gao, L. M., Tan, S. L., Zhang, G. L., and Thomas, P. (2019). A new species of Amentotaxus (Taxaceae) from China, Vietnam, and Laos. PhytoKeys 130, 25–32. doi: 10.3897/phytokeys.130.33956
Gianordoli, M. (1974). “A cytological investigation on gametes and fecundation among Cephalotaxus drupacea,” in Fertilization in Higher Plants, ed. H. F. Linskens (Amsterdam: North-Holland), 221–232.
Gitzendanner, M. A., Soltis, P. S., Wong, G. K. S., Ruhfel, B. R., and Soltis, D. E. (2018). Plastid phylogenomic analysis of green plants: a billion years of evolutionary history. Am. J. Bot. 105, 291–301. doi: 10.1002/ajb2.1048
Hao, D. C., Xiao, P. G., Huang, B., Ge, G. B., and Yang, L. (2008). Interspecific relationships and origins of Taxaceae and Cephalotaxaceae revealed by partitioned Bayesian analyses of chloroplast and nuclear DNA sequences. Plant Syst. Evol. 276, 89–104. doi: 10.1007/s00606-008-0069-0
Hart, J. A. (1987). A cladistic analysis of conifers: preliminary results. J. Arnold. Arbor. 26, 9–307.
Hebert, P. D., Cywinska, A., Ball, S. L., and Dewaard, J. R. (2003). Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B 270, 313–321. doi: 10.1098/rspb.2002.2218
Hebert, P. D., Stoeckle, M. Y., Zemlak, T. S., and Francis, C. M. (2004). Identification of birds through DNA barcodes. PLoS. Biol. 2:e312. doi: 10.1371/journal.pbio.0020312
Hollingsworth, M. L., Andra, C. A., Forrest, L. L., Richardson, J., Pennington, R. T., Long, D. G., et al. (2009). Selecting barcoding loci for plants: evaluation of seven candidate loci with species-level sampling in three divergent groups of land plants. Mol. Ecol. Resour. 9, 439–457. doi: 10.1111/j.1755-0998.2008.02439.x
Hollingsworth, P. M. (2011). Refining the DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 108, 19451–19452. doi: 10.1073/pnas.1116812108
Hollingsworth, P. M., Li, D. Z., Van Der Bank, M., and Twyford, A. D. (2016). Telling plant species apart with DNA: from barcodes to genomes. Philos. Trans. R. Soc. B 371:20150338. doi: 10.1098/rstb.2015.0338
IUCN (2001). IUCN Red List Categories and Criteria: Version 3.1. Gland: IUCN Species Survival Commission, IUCN, ii+30.
Ji, Y., Liu, C., Landis, J. B., Deng, M., and Chen, J. (2021). Plastome phylogenomics of Cephalotaxus (Cephalotaxaceae) and allied genera. Ann. Bot. 127, 697–708. doi: 10.1093/aob/mcaa201
Ji, Y. H., Liu, C. K., Yang, J., Jin, L., Yang, Z. Y., and Yang, J. B. (2020). Ultra-barcoding discovers a cryptic species in Paris yunnanensis (Melanthiaceae), a medicinally important plant. Front. Plant Sci. 11:411. doi: 10.3389/fpls.2020.00411
Ji, Y. H., Liu, C. K., Yang, Z. Y., Yang, L. F., He, Z. S., Wang, H. C., et al. (2019). Testing and using complete plastomes and ribosomal DNA sequences as the next-generation DNA barcodes in Panax (Araliaceae). Mol. Ecol. Resour. 19, 1333–1345. doi: 10.1111/1755-0998.13050
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Depamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Joly, S., Davies, T. J., Archambault, A., Bruneau, A., Derry, A., Kembel, S. W., et al. (2014). Ecology in the age of DNA barcoding: the resource, the promise and the challenges ahead. Mol. Ecol. Resour. 14, 221–232. doi: 10.1111/1755-0998.12173
Kane, N., Sveinsson, S., Dempewolf, H., Yang, J. Y., Zhang, D., Engels, J. M., et al. (2012). Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 99, 320–329. doi: 10.3732/ajb.1100570
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kress, W. J., and Erickson, D. L. (2007). A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 2:e508. doi: 10.1371/journal.pone.0000508
Kress, W. J., García-Robledo, C., Uriarte, M., and Erickson, D. L. (2015). DNA barcodes for ecology, evolution, and conservation. Trends Ecol. Evol. 30, 25–35. doi: 10.1016/j.tree.2014.10.008
Kress, W. J., Wurdack, K. J., Zimmer, E. A., Weigt, L. A., and Janzen, D. H. (2005). Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. U.S.A. 102, 8369–8374. doi: 10.1073/pnas.0503123102
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lang, X. D., Su, J. R., Lu, S. G., and Zhang, Z. J. (2013). A taxonomic revision of the genus Cephalotaxus (Taxaceae). Phytotaxa 84, 1–24.
Lang, X. D., Su, J. R., Zhang, Z. J., and Lu, S. G. (2011). The taxonomic history and original literatures arrangements of living Cephalotaxus (Cephalotaxaceae). Am. J. Plant Sci. 2, 496–506. doi: 10.4236/ajps.2011.23058
Li, D. Z., Gao, L. M., Li, H. T., Wang, H., Ge, X. J., Liu, J. Q., et al. (2011). Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. U.S.A. 108, 19641–19646. doi: 10.1073/pnas.1104551108
Li, H. T., Luo, Y., Gan, L., Ma, P. F., Gao, L. M., Yang, J. B., et al. (2021). Plastid phylogenomic insights intorelationships of all flowering plant families. BMC Biol. 19:232. doi: 10.1186/s12915-021-01166-2
Li, H. T., Yi, T. S., Gao, L. M., Ma, P. F., Zhang, T., Yang, J. B., et al. (2019). Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 5, 461–470. doi: 10.1038/s41477-019-0421-0
Li, X., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y., and Chen, S. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. 90, 157–166. doi: 10.1111/brv.12104
Liu, J., Moeller, M., Gao, L. M., Zhang, D. Q., and Li, D. Z. (2011). DNA barcoding for the discrimination of Eurasian yews (Taxus L., Taxaceae) and the discovery of cryptic species. Mol. Ecol. Resour. 11, 89–100. doi: 10.1111/j.1755-0998.2010.02907.x
Liu, J., Provan, J., Gao, L. M., and Li, D. Z. (2012). Sampling strategy and potential utility of indels for DNA barcoding of closely related plant species: a case study in Taxus. Int. J. Mol. Sci. 13, 8740–8751. doi: 10.3390/ijms13078740
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Mogensen, H. L. (1996). INVITED SPECIAL PAPER: the hows and whys of cytoplasmic inheritance in seed plants. Am. J. Bot. 83, 383–404. doi: 10.1002/j.1537-2197.1996.tb12718.x
Möller, M., Gao, L. M., Mill, R. R., Liu, L., Zhang, D. Q., Poudel, R. C., et al. (2013). A multidisciplinary approach reveals hidden taxonomic diversity in the morphologically challenging Taxus wallichiana complex. Taxon 62, 1161–1177. doi: 10.12705/626.9
Möller, M., Liu, J., Li, Y., Li, J. H., Ye, L. J., Mill, R., et al. (2020). Repeated intercontinental migrations and recurring hybridizations characterise the evolutionary history of yew (Taxus L.). Mol. Phylogenet. Evol. 153:106952. doi: 10.1016/j.ympev.2020.106952
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Nevill, P. G., Zhong, X., Tonti-Filippini, J., Byrne, M., Hislop, M., Thiele, K., et al. (2020). Large scale genome skimming from herbarium material for accurate plant identification and phylogenomics. Plant Methods 16:1. doi: 10.1186/s13007-019-0534-5
Nock, C. J., Waters, D. L., Edwards, M. A., Bowen, S. G., Rice, N., Cordeiro, G. M., et al. (2011). Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9, 328–333. doi: 10.1111/j.1467-7652.2010.00558.x
Ovcharenko, I., Loots, G. G., Giardine, B. M., Hou, M., Ma, J., Hardison, R. C., et al. (2005). Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res. 15, 184–194. doi: 10.1101/gr.3007205
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next-generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Pennisi, E. (2007). Wanted: a barcode for plants. Science 318, 190–191. doi: 10.1126/science.318.5848.190
Percy, D. M., Argus, G. W., Cronk, Q. C., Fazekas, A. J., Kesanakurti, P. R., Burgess, K. S., et al. (2014). Understanding the spectacular failure of DNA barcoding in willows (Salix): does this result from a trans-specific selective sweep? Mol. Ecol. 23, 4737–4756. doi: 10.1111/mec.12837
Petit, R. J., and Vendramin, G. G. (2007). “Plant phylogeography based on organelle genes: an introduction,” in Phylogeography of Southern European Refugia, eds S. Weiss and N. Ferrand (Dordrecht: Springer), 23–97.
Price, R. A. (1990). The genera of Taxaceae in the southeastern United States. J. Arnold Arbor. 71, 69–91.
Price, R. A. (2003). Generic and familial relationships of the Taxaceae from rbcL and matK sequence comparisons. Acta Hortic. 615, 235–237. doi: 10.17660/ActaHortic.2003.615.23
Qu, X. J., Moore, M. J., Li, D. Z., and Yi, T. S. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 1–12. doi: 10.1186/s13007-019-0435-7
Ran, J. H., Shen, T. T., Wang, M. M., and Wang, X. Q. (2018). Phylogenomics resolves the deep phylogeny of seed plants and indicates partial convergent or homoplastic evolution between Gnetales and angiosperms. Proc. R. Soc. B Biol. Sci. 285:20181012. doi: 10.1098/rspb.2018.1012
Ripma, L. A., Simpson, M. G., and Hasenstab-Lehman, K. (2014). Geneious! Simplified genome skimming methods for phylogenetic systematic studies: a case study in Oreocarya (Boraginaceae). Appl. Plant Sci. 2:1400062. doi: 10.3732/apps.1400062
Rozas, J., Ferrer-Mata, A., Sánchez-Delbarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Ruhsam, M., Rai, H. S., Mathews, S., Ross, T. G., Graham, S. W., Raubeson, L. A., et al. (2015). Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Resour. 15, 1067–1078. doi: 10.1111/1755-0998.12375
Singh, H. (1961). The life history and systematic position of Cephalotaxus drupacea Sieb. and Zicc. Phytomorphology 11, 153–197. doi: 10.1007/BF03052338
Song, F., Li, T., Burgess, K. S., Feng, Y., and Ge, X. J. (2020). Complete plastome sequencing resolves taxonomic relationships among species of Calligonum L. (Polygonaceae) in China. BMC Plant Biol. 20:5. doi: 10.1186/s12870-020-02466-5
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Straub, S. C., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., and Liston, A. (2012). Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364. doi: 10.3732/ajb.1100335
Tonti-Filippini, J., Nevill, P. G., Dixon, K., and Small, I. (2017). What can we do with 1000 plastid genomes? Plant J. 90, 808–818. doi: 10.1111/tpj.13491
Trias-Blasi, A., and Vorontsova, M. (2015). Plant identification is key to conservation. Nature 521, 161–161. doi: 10.1038/521161c
Turner, B., Paun, O., Munzinger, J., Chase, M. W., and Samuel, R. (2016). Sequencing of whole plastid genomes and nuclear ribosomal DNA of Diospyros species (Ebenaceae) endemic to New Caledonia: many species, little divergence. Ann. Bot. 117, 1175–1185. doi: 10.1093/aob/mcw060
Wicke, S., Schneeweiss, G. M., Depamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Yan, L. J., Liu, J., Moeller, M., Zhang, L., Zhang, X. M., Li, D. Z., et al. (2015). DNA barcoding of Rhododendron (Ericaceae), the largest Chinese plant genus in biodiversity hotspots of the Himalaya-Hengduan Mountains. Mol. Ecol. Resour. 15, 932–944. doi: 10.1111/1755-0998.12353
Yang, J. B., Tang, M., Li, H. T., Zhang, Z. R., and Li, D. Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Yang, Y., Wang, Z., and Xu, X. (2017). Taxonomy and Distribution of Global Gymnosperms. China: Scientific Books, 1045–1056.
Yu, X., Yang, D., Guo, C., and Gao, L. (2018). Plant phylogenomics based on genome-partitioning strategies: progress and prospects. Plant Divers. 40, 158–164. doi: 10.1016/j.pld.2018.06.005
Zhang, D., Dirr, M. A., and Price, R. A. (2000). Discrimination and genetic diversity of Cephalotaxus accessions using AFLP markers. J. Am. Soc. Hort. Sci. 125, 404–412. doi: 10.21273/JASHS.125.4.404
Zhang, J. W., D’rozario, A., Liang, X. Q., and Zhou, Z. K. (2019). Middle Miocene Cephalotaxus (Taxaceae) from Yunnan, Southwest China, and its implications to taxonomy and evolution of the genus. Palaeoworld 28, 381–402. doi: 10.1016/j.palwor.2019.01.002
Zhang, S. D., Jin, J. J., Chen, S. Y., Chase, M. W., Soltis, D. E., Li, H. T., et al. (2017). Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 214, 1355–1367. doi: 10.1111/nph.14461
Zhu, S., Niu, Z., Xue, Q., Wang, H., Xie, X., and Ding, X. (2018). Accurate authentication of Dendrobium officinale and its closely related species by comparative analysis of complete plastomes. Acta Pharm. Sin. B 8, 969–980. doi: 10.1016/j.apsb.2018.05.009
Keywords: Cephalotaxus, complete plastome, species discrimination, cryptic species, phylogenetic relationships, standard DNA barcodes
Citation: Wang J, Fu C-N, Mo Z-Q, Möller M, Yang J-B, Zhang Z-R, Li D-Z and Gao L-M (2022) Testing the Complete Plastome for Species Discrimination, Cryptic Species Discovery and Phylogenetic Resolution in Cephalotaxus (Cephalotaxaceae). Front. Plant Sci. 13:768810. doi: 10.3389/fpls.2022.768810
Received: 01 September 2021; Accepted: 07 March 2022;
Published: 04 May 2022.
Edited by:
Antonio Gonzalez-Rodriguez, Universidad Nacional Autónoma de México, MexicoReviewed by:
Sofia Solorzano, National Autonomous University of Mexico, MexicoCopyright © 2022 Wang, Fu, Mo, Möller, Yang, Zhang, Li and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lian-Ming Gao, Z2FvbG1AbWFpbC5raWIuYWMuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.