 Xiaobing Wang
Xiaobing Wang Qisheng Han
Qisheng Han
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 24 February 2022
Sec. Plant Symbiotic Interactions
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.759801
This article is part of the Research Topic Plant-Microbe Interactions in Forest Ecosystems View all 7 articles
A long-standing hypothesis in biogeography predicts that a species’ abundance is highest at the center of its geographical range and decreases toward its edges. In this study, we test the abundant-center hypothesis of ectomycorrhizal (ECM) fungal communities associated with Picea crassifolia, an endemic species widely distributed in northwest China. We analyzed the taxonomic richness and the relative abundance of ECM fungi in four main distribution areas, from center to edges. In total, 234 species of ECM fungi were detected, and of these, 137 species were shared among all four sites. Inocybe, Sebacina, Tomentella, and Cortinarius were the dominant genera. ECM fungal richness and biodiversity were highest at the central and lower at peripheral sites. Our results indicated that ECM fungal species richness was consistent with the abundant-center hypothesis, while the relative abundances of individual fungal genera shifted inconsistently across the plant’s range.
Ectomycorrhiza (ECM) is an intimate symbiotic association between plant roots and ECM fungi. ECM associations benefit most terrestrial plants by enhancing nutrient uptake and tolerance to environmental stressors (Tedersoo et al., 2020). ECM fungal community structure is affected by a multitude of biotic and abiotic factors (Ishida et al., 2007). The community composition is shown to change across gradients of soil pH (Bahram et al., 2011), soil nitrogen and organic matter (Pellitier et al., 2021), elevation (Matsuoka et al., 2016), and forest age (Hui et al., 2017; Boeraeve et al., 2018). As biotic and abiotic components usually show strong interdependencies, it is methodologically difficult to assess the contributions made by each specific factor in the ECM fungal community and their response to environmental changes (Weißbecker et al., 2018).
The effect of different driving factors at different scales is variable. Physiographic factors such as altitude and soil properties including soil microorganisms and soil physicochemical properties are usually important drivers of ECM fungal communities in the local-scale sampling area, such as carbon content and pH (Vasco-Palacios et al., 2020), altitude and forest age (Matsuoka et al., 2016; Geml et al., 2017; Schön et al., 2018) and host genetic and phenotypic characteristics (Rúa, 2021). In large-scale ECM fungal studies, the role of host plants is becoming increasingly obvious. Host dispersal can affect the biogeographic patterns of the ECM fungal community (Matsuoka et al., 2019), and a significant proportion of ECM fungi exhibited host specificity in conifer broadleaf forests (Ishida et al., 2007). In addition, ECM fungal communities show a stronger correlation with host composition than soil properties, while pathogens and arbuscular mycorrhizal (AM) fungi are more strongly correlated with soil properties (Schappe et al., 2020). Hence, whether the range of host habitats is the main factor for ECM fungal community structure is worth further exploring.
The abundant-center hypothesis posits that species abundance peaks in the center of its distributional range and declines toward its edges (Hengeveld and Haeck, 1982). The high abundance in the center is because of the optimal conditions (i.e., the presence of suitable habitats), while the decline in abundance at edges is because of environmental suboptimality (Lira-Noriega and Manthey, 2014). A recent modeling study showed that the abundant-center relationships were strongly affected by populations and deterministic growth environment (Dallas and Santini, 2020). This theory provides an overview of species distributions. However, studies on abundant-center hypothesis have mainly focused on animals and plants (Pironon et al., 2017), while studies on ECM fungal communities are rare.
Unlike the animal and plant species, ECM fungi are typical symbionts and more than 90% of nutrient-absorbing roots of trees are colonized by ECM fungi (Smith and Smith, 2002). ECM fungi are estimated to comprise 20,000–25,000 species globally and form symbioses with only a tiny fraction of the terrestrial plants (2% of total plant species) (Tedersoo et al., 2012). In addition, most ECM fungi reproduce sexually and produce macroscopic fruit bodies, which differ from arbuscular and ericoid mycorrhizas (Tedersoo et al., 2010). Because these fruit bodies deposit most of their spores within in a short distance, the spatial distribution of the ECM community is probably more dependent on the host plants’ distribution than environmental factors. Hence, the distribution models between ECM fungi and host plants may be the same, or at least very similar. Most studies have been performed on the relationship between host plant species composition and ECM fungal community (Durall et al., 2006; Mucha et al., 2018), while fewer studies have addressed the effects of host plants’ abundance. The distribution of some Pinaceae species, which is certainly the oldest extant plant family symbiotic with ECM fungi (Hibbett and Matheny, 2009), have found evidence for the abundant-center hypothesis (Mimura and Aitken, 2007; Gugerli et al., 2009). Previous studies have found that ECM communities are more concentrated on species of Pinaceae and showed stronger host effects than on broadleaf species (Ishida et al., 2007; Vlk et al., 2020). However, for ECM fungal distribution modeling, it appears important to consider not only the distribution of individual species but also the ECM fungal richness and biodiversity of the whole community. Many ECM fungi may fulfill similar ecological functions and that degree of functional redundancy exists in ECM fungal communities (Dahlberg, 2001). ECM fungi are usually categorized into two major groups based on their distribution features: cosmopolitan ECM members (Cortinarius, Russula and Tomentella) and endemic ECM members (McPolin and Kranabetter, 2021). Endemic ECM fungi usually exhibit higher host specialization than cosmopolitan ECM members, which is generally driven by the host range (McPolin and Kranabetter, 2021). Picea crassifolia is one of the main tree species that belongs to the Pinaceae family in semi-arid areas of China. It forms pure stands on north-facing slopes along the northeast edge of the Qinghai-Tibetan Plateau (QTP) region and in the adjacent highlands of Helan Mountain, which has not been subject to the significant human disturbance for more than one century. The ECM fungal community associated with a single host in the pure forest of P. crassifolia was considered an excellent material for ecological studies.
In this study, we examined the ECM fungal community associated with P. crassifolia to test if the distribution of ECM fungal species fit the abundant-center hypothesis model. We set out two hypotheses: (1) Is the composition of ECM fungal communities associated with P. crassifolia generally consistent across the host’s geographical range? (2) Does the richness and diversity of these ECM fungi decrease from the center to the edge of the host’s distribution?
Picea crassifolia is mainly distributed on the north-facing slopes along the northeast edge of the QTP region and in the adjacent highlands of Helan Mountain. To show the distribution map of the P. crassifolia directly, we compiled occurrence data from the GBIF database1 and maps were generated using the tmap (Tennekes et al., 2021) and rangemap (Cobos et al., 2021) packages in R. Field sampling was conducted in four pure P. crassifolia stands in Qinghai (QH), Gansu (GS), Ningxia (NX), and Mongolia (NM) provinces as representative of its complete distribution. As shown in Figure 1, P. crassifolia has a higher abundance in QH sites (core site) and a lower abundance in NX, NM and GS sites (edge sites). In addition, the geographical distances between the core-edge QH-GS, QH-NX, and QH-NM are approximately 242, 351, and 364 km, respectively. These forests are estimated to be aged above 70 years.
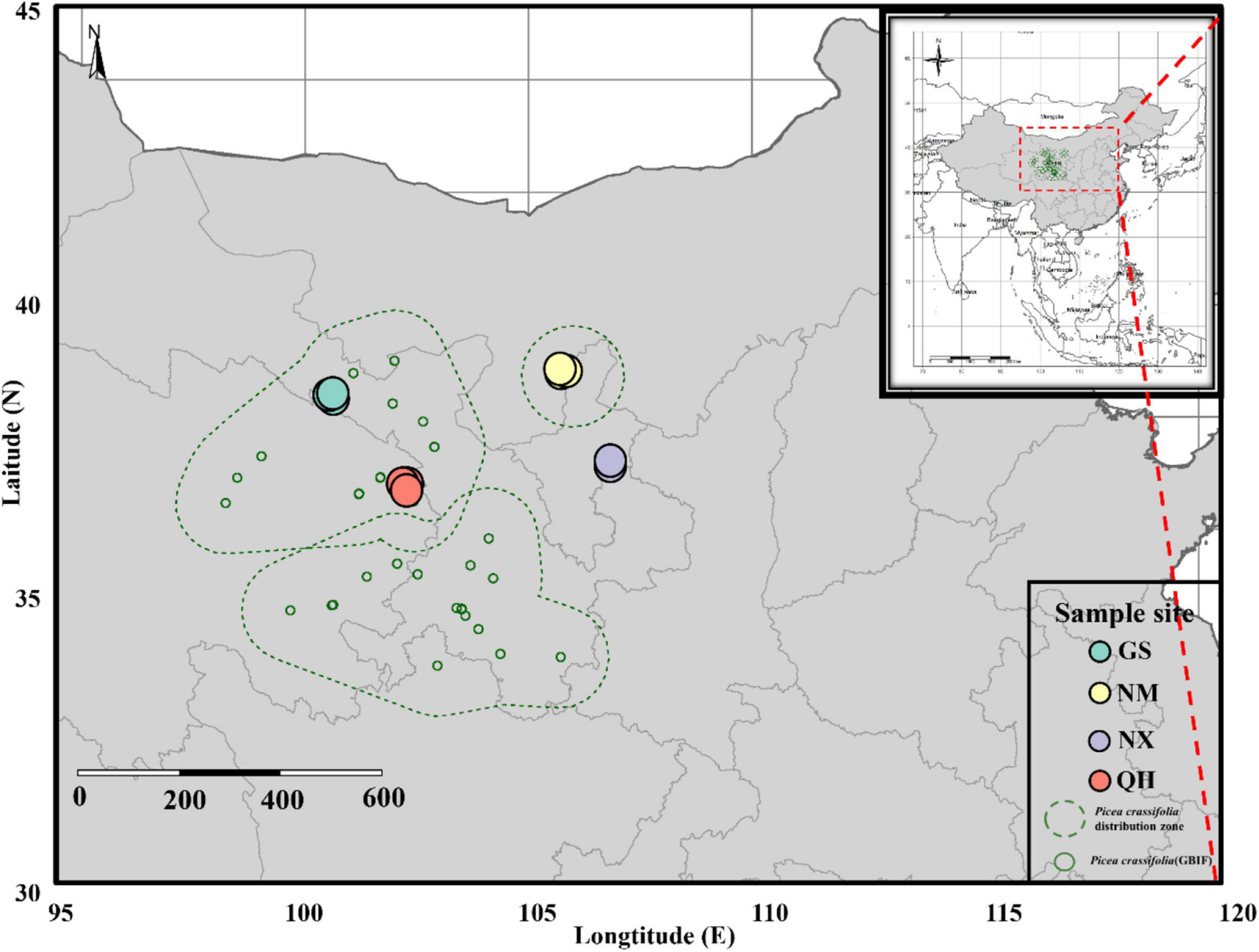
Figure 1. Map of sampling site locations in China.
Three sampling plots were randomly located in each forest stand in September 2016. At each plot, 10 spruce trees, spaced 10 m apart to avoid possible spatial autocorrelation (Lilleskov et al., 2004; Peay et al., 2010b), were selected for sampling. For each tree, three fine roots (approximately 10 cm in length) were collected within 1 m from the trunk. The collected root samples for each plot were put in a single plastic bag and stored on ice in a cooling container. Soil samples were collected from the same sampling plot. All samples were stored in the laboratory at 4°C before being processed but were not stored longer than 1 week before processing.
After removing visible plant material, parts of soil samples were dried and passed through a 2 mm sieve to determine pH and electric conductivity (EC), while other parts were further sieved through a 250 μm screen to determine soil organic carbon (SOC), total nitrogen (TN) and total phosphorus (TP). The soil pH and EC values were, respectively, measured using a Sartorius PB-10 pH meter (Sartorius Co., Ltd., Göttingen, Germany) and conductivity meter (DDS-307, Shanghai REX Instrument Factory, China) by mixing soil with distilled water (1:2). The SOC was determined using the Walkley-Black (WB) method by quantifying the amount of oxidizable soil carbon with a reaction with acidic dichromate (Cr2O72–). The TN content was determined by the semimicro Kjeldahl method (AutoAnalyzer 3, Bran + luebbe, Hamburg, Germany). The TP content was determined by the colorimetric method using the application of acid-soluble molybdenum antimony anti-determination.
Total genome DNA from the root sample was extracted using the cetyltrimethylammonium bromide (CTAB) method. The concentration and purity of DNA were monitored on 1% agarose gels. According to the concentration, DNA was diluted to 1 ng/μl using sterile water. For PCR reactions, the internal transcribed spacer (ITS) genes of distinct regions were amplified using ITS1/ITS2 primers (Sequence: TCCGTAGGTGAACCTGCGG/GCTGCGTTCTTCATCGATGC) with the barcode (Bengtsson-Palme et al., 2013). All PCR reactions were carried out using Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 2 μM of forward and reverse primers, and approximately 10 ng template DNA. Thermal cycling consisted of initial denaturation at 98°C for 1 min, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s and elongation at 72°C for 30 s; finally, 72°C for 5 min. Mix same volume of 1 × loading buffer (containing SYB green) with PCR products and operate electrophoresis on 2% agarose gel for detection. Then, mixed PCR products were purified using a Qiagen Gel Extraction Kit (Qiagen, Germany). Sequencing libraries were generated using a TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, United States), following the manufacturer’s recommendations and adding index codes. The library quality was assessed using a Qubit® 2.0 Fluorometer (Thermo Scientific, Landsmeer, Netherlands) and Agilent Bioanalyzer 2100 system. As a final step, the library was sequenced using an Illumina Hiseq 2500 platform, and 250 bp paired-end reads were generated. Paired-end reads were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Reads were then merged using FLASH (version 1.2.72) (Magoč and Salzberg, 2011), which was designed to merge paired-end reads when at least some of the reads overlap the reads generated from the opposite end of the same DNA fragment. Spliced sequences were called raw tags.
Quality filtering on the raw tags was performed under specific filtering conditions to obtain the high-quality clean tags (Bokulich et al., 2013) using the quantitative insights into microbial ecology (QIIME) (version 1.7.03) quality-controlled process (Caporaso et al., 2010). Tags were compared with a reference database (Gold database4) using the UCHIME algorithm5 to identify chimeric sequences. When detected, chimeric sequences were removed and effective tags were obtained. Sequence analyses were performed using the Uparse software (Uparse version 7.0.10016) (Edgar, 2013). Sequences with ≥97% of similarity were assigned to the same operational taxonomic unit (OTUs). OTUs represented by <10 reads were discarded as sequencing errors (Nguyen et al., 2015).
The representative sequence for each OTU was individually checked using basic local alignment search tool (BLAST) with the NCBI database7 and UNITE database8. Sequences identified as originating from non-ECM taxa (Tedersoo et al., 2010) were removed from the dataset to restrict analyses to taxa with evidence of ECM status. As mentioned earlier, we used the relative abundance of ECM fungal species instead of the absolute abundance values as the abundance index for all analyses. All the sequences reported in this study have been submitted to the DDBJ database (LC203765- LC205656).
Chao2 index, Shannon’s diversity index and Simpson’s diversity index were used to characterize OTU sample diversity at each site, and they were calculated using EsimateS (Colwell, 2021). Statistical comparisons of soil data and biodiversity indices among different samples were made with a one-way analysis of variance (ANOVA) using R (R Core Team, 2021). Analysis of similarity (ANOSIM) was used for site difference test between community groups. To statistically assess the effect of the environmental factors on the ECM composition, a multivariate analysis of variance based on dissimilarity test (ADONIS) was applied using the vegan package in R software (Oksanen, 2021). Redundancy analysis (RDA) between the relative abundance of ECM fungi and environmental factors was performed to explore the main factors determining the ECM community structure. For RDA analyses, data were log + 1 transformed. Co-linearity between environmental variables was checked using variance inflation factors (VIF), and the environmental variables with VIF > 10 were excluded from the RDA analyses. The model ran for 1,000 permutations and the significance of the explanatory variables and the first two RDA axes were assessed using ANOVA. Isolation by distance was tested through the correlation between geographical distances (calculated from GPS coordinates) and Bray–Curtis distances using a Mantel test (Smouse et al., 1986). Because of the significant relationship between species composition and distance for each sample site, we calculated a Mantel correlogram to test the strength of correlation at different distance classes by using Ecodist package (Peay et al., 2010a). To test whether ECM fungal species richness changed from the core to the edge, we modeled decrease rates with geographical distances from core to edge:
Decrease rate = (Highest observed richness in core site – the observed richness in other sites)/(Highest observed richness in core site).
To improve the normality of residuals, the geographical distance between any two points was log10 transformed. Linear discriminant analysis (LDA) effect size (LefSe) analysis was performed to investigate the differences in the ECM fungal relative abundance among all groups with the microeco statistical package (Liu et al., 2021).
Variation in soil pH among the four investigated stands was small, but the differences were significant at four sites (Table 1). The changes in the soil SOC content coincided well with those of the soil TN content; NM was the lowest (59.14 ± 5.71, 2.86 ± 0.16) and QH was the highest (129.68 ± 37.15, 5.9 ± 0.92). However, no obvious variation in the soil TP content was observed among sites (Table 1).
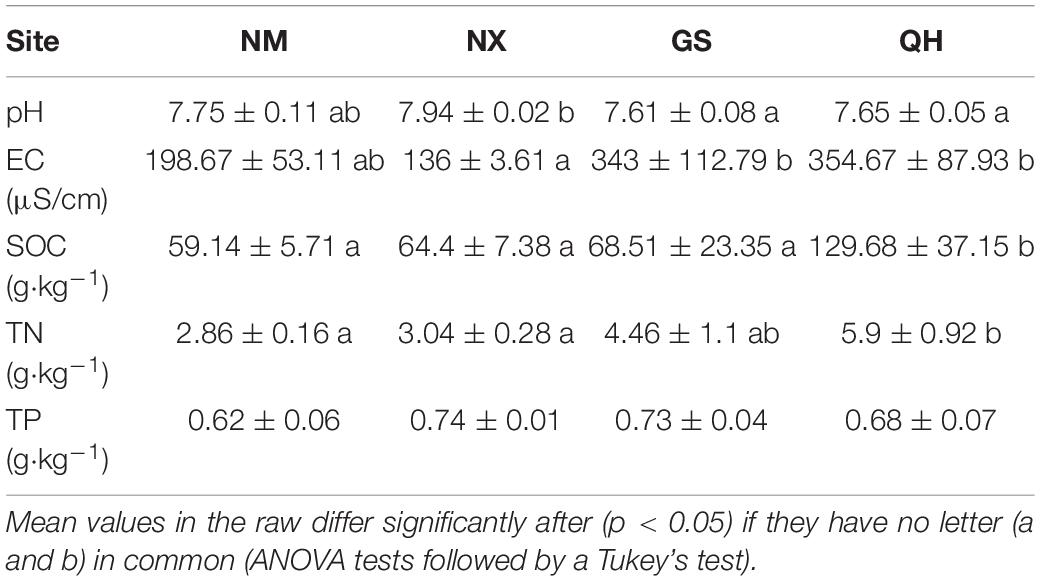
Table 1. Summary of soil characteristics recorded of the study stands.
In total, 755,738 sequence reads were generated using Illumina Hiseq 2500. The average length of the effective tags was 233.9 bq. Rarefaction curves in all sites (Figure 2A) indicate that sequencing depth can reveal fungal community composition. After removing the sequence reads that did not match ECM fungal taxa, 384,558 sequences (50.8% of total sequences) were left for analyses and interpretation. Of these, we removed 378 that possessed <10 reads. In total, 234 ECM OTUs were identified (Supplementary File 1).

Figure 2. (A) Rarefaction curves of OTU number at 97% of similarity for each site. Mean values of three replicates and the error bar are shown. (B) Alluvial diagram of the top 10 genera’s relative abundance in four sites.
Most of the ECM OTUs (93.6%) belonged to the phylum Basidiomycota, while Ascomycota only accounted for a small fraction (14 OTUs). In total, OTUs were clustered into overall 27 ECM fungal families. Among these families, Inocybaceae, Sebacinaceae, and Thelephoraceae had a higher relative abundance (43.8, 21.3, and 21.05%, respectively). Among them, 87% of the ECM OTUs could be accurately assigned at the genus level. Major ECM fungal genera in this study included the following: Inocybe (56 species), Tomentella (46 species), Cortinarius (22 species), and Sebacina (20 species). At the species level, Inocybe sp.1 was the most abundant fungal taxon, with recorded reads, followed by Inocybe flocculosa and Sebacina sp.9.
The distribution of relative abundances among these ECM genera was similar among the NX, NM, QH, and GS sites. However, Inocybe, Sebacina, Tomentella, and Cortinarius were the top four genera at the NX, QH, and GS sites, while the Russula replaced the Cortinarius as the dominant genus at the NM site. At all of the sampled edge sites (GS, NM, and NX), the fungal genus Inocybe had the highest relative abundance, while Tomentella was more dominant than Inocybe at the center site (QH) (Figure 2B).
There was a significant spatial autocorrelation between ECM fungal communities (P < 0.001), a Mantel correlogram suggested spatial autocorrelation only within ∼39 km (Figure 3A). The center site (QH) has the highest number of observed ECM richness and mean richness per site. The decrease rates of ECM fungal richness with distances followed Michaelis–Menten kinetics and the regression equation model has a good goodness of fit. The parameters for the model are shown in Figure 3B. The result showed that the richness value fell by 10% or more from core to edge. Estimated richness and diversity (Shannon’s and Simpson’s index) showed the same trends (Table 2). ECM fungal communities were dissimilar between sites (R = 0.33; P = 0.006, Anosim).

Figure 3. (A) Spatial correlogram between Mantel statistics and distance class index: significant Mantel correlation (black points) indicates spatial correlation. (B) Decrease rate of ECM fungal richness from center to edges. The data were fitted to Michaelis–Menten kinetics, and the dashed line shows a significant non-linear regression.
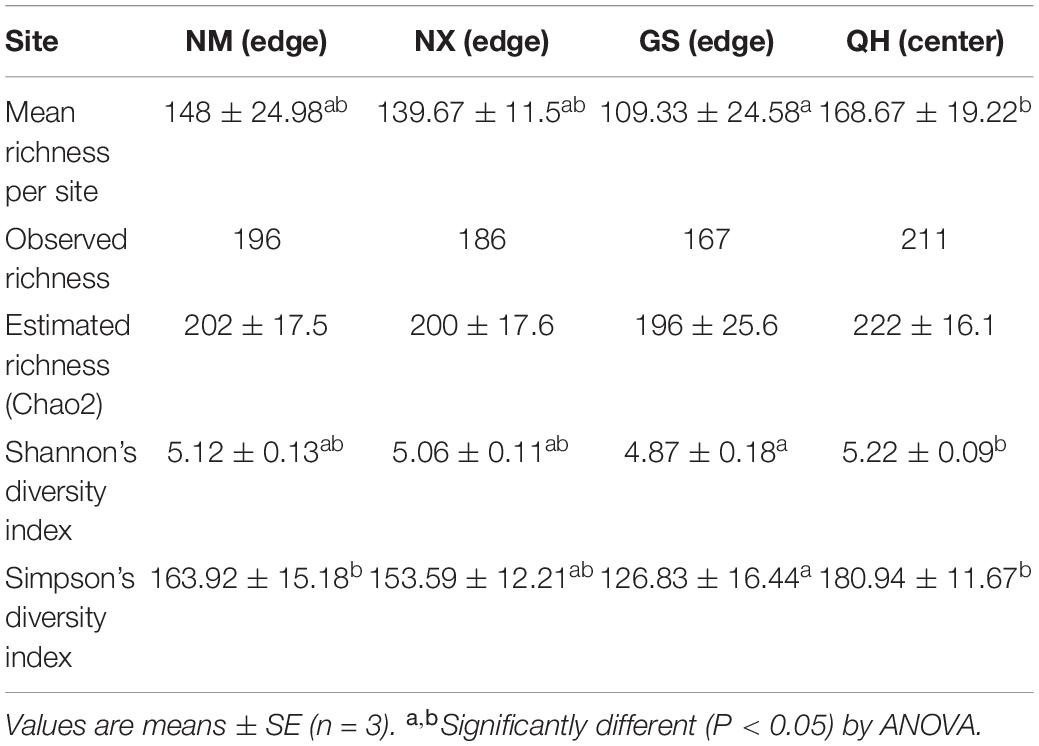
Table 2. Summary of ectomycorrhizal fungal diversity associated with P. crassifolia.
However, the site was the strongest factor associated with compositional variation (P < 0.01, R2 = 0.37, Adonis). Direct gradient ordination of ECM fungal communities using RDA showed that the effect of site was highly significant (P < 0.001) in explaining the distribution of ECM species (Figure 4A). Shared species analysis showed that of a total of 234 OTUs, 137 were shared among all four sites (Figure 4B). We compared the values of the relative abundance at genus level and nearly half of ECM fungal genera (18) were higher at the center site than that at the edge sites (Table 3).

Figure 4. (A) Redundancy analysis (RDA) plot of ECM fungal community and environmental variables among four sites at the species level. The percentage of the X-axis and Y-axis represents the proportion of explained variance. (B) Venn diagram illustrating the number of unique and shared operational taxonomic units.
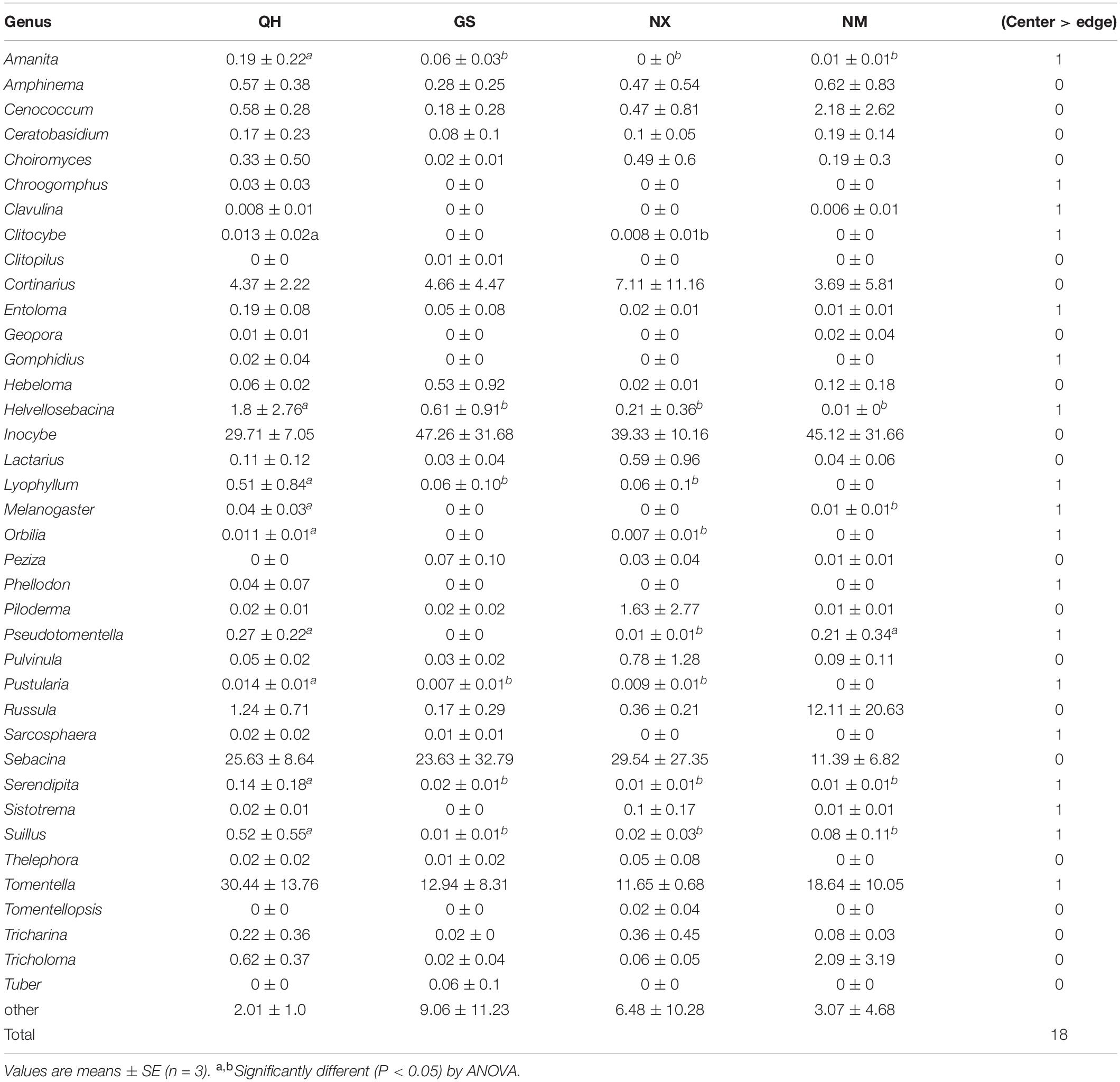
Table 3. Comparing the differences of the ECM fungal relative abundance at genus level in four sites.
We also used LEfSe to determine which taxa most likely explained differences among locations. Each OTU with an LDA value >2 was collected with higher LDA values representing greater differences. As shown in Figure 5, only the genus Amanita was significantly enriched at the GS site at the genus level. Four species (genus Inocybe), three species (genus Tomentella), one species (genus Pseudotomentella), one species (genus Sebacina), and one species (genus Amphinema) were enriched at the QH site. Two species (genus Tomentella) and one species (genus Lactarius) showed enrichment with a high LDA score at the NM site. Both of the NX (Tricholoma sp.) and GS (Cortinarius sp.8.) sites had one dominant species.
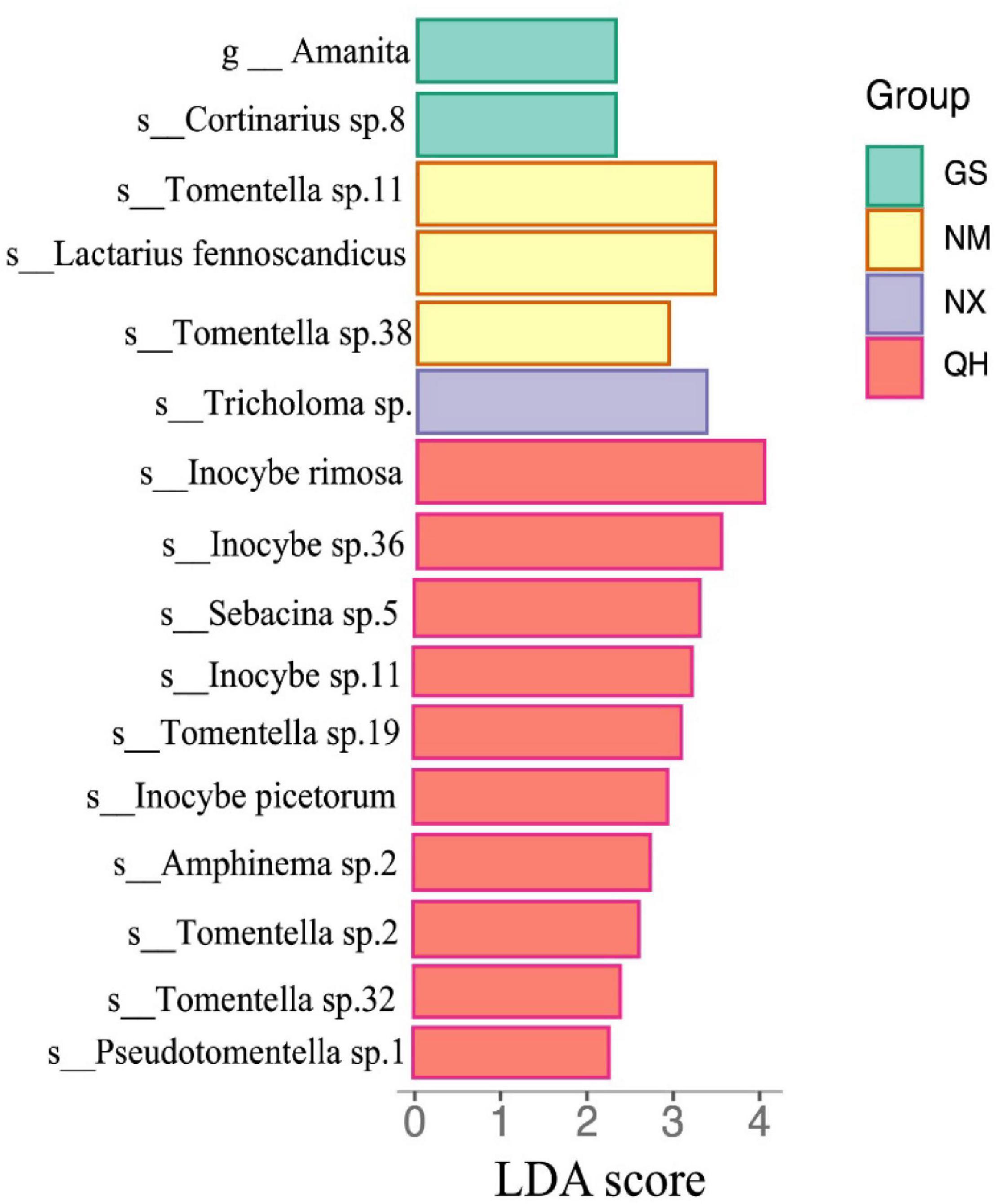
Figure 5. Histogram of LAD scores (LDA score > 2.0 and P < 0.05) for differentially abundant OTU among the four sites.
Next-generation sequencing technologies have enabled large-scale analyses of the complex and normally species-rich ECM communities, showing high consistency with the clone-based Sanger sequencing (Kauserud et al., 2012). High-throughput sequencing technology has greatly facilitated the acquisition of genome data (Li et al., 2020), which allows us to investigate the ecophysiology of uncultured microbes (Wagner, 2009). In our study, the number of ECM species (196 OTUs) detected that were associated with P. crassifolia from the region of Inner Mongolia (NM group) is much higher than 11 OTUs at the same site (Fan et al., 2016). A total of 10 ECM species (all except 1) were found using both methods (Table 4).
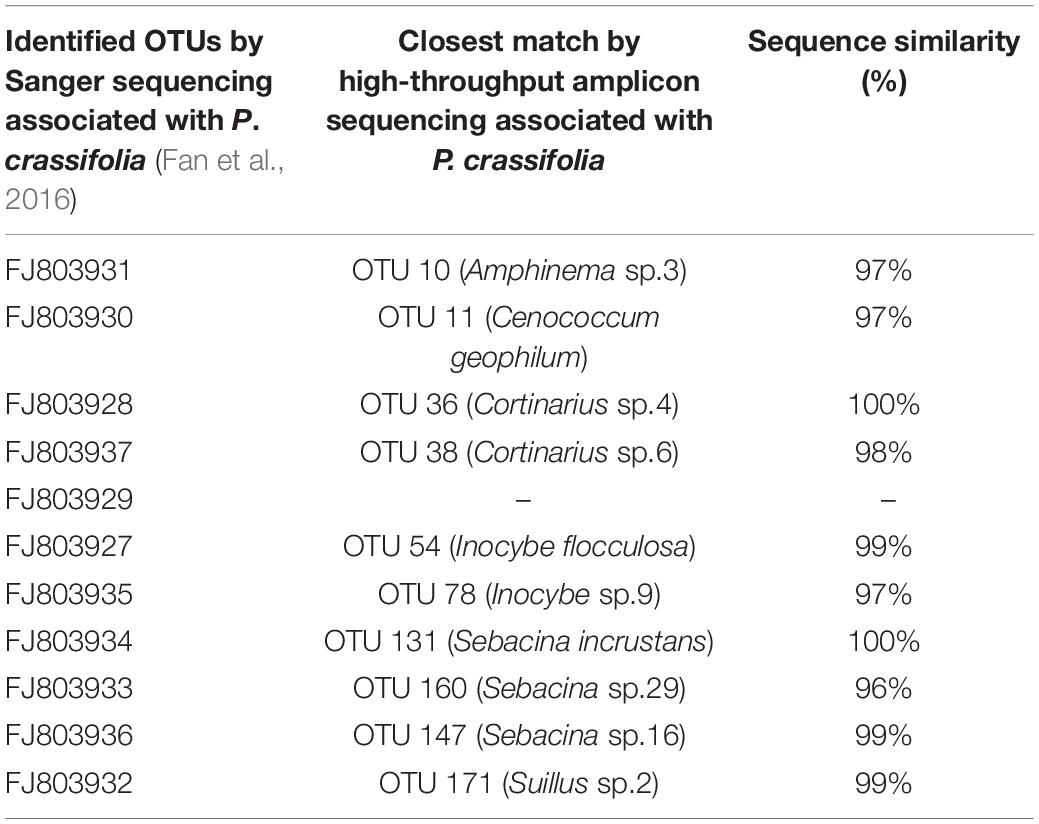
Table 4. The similarity of OTU sequences between the Sanger sequencing and high-throughput amplicon sequencing associated with P. crassifolia.
Our results clearly showed Tomentella was the most species-rich genus ahead of Cortinarius, Inocybe, and Sebacina. However, none of Tomentella species was detected using Sanger sequencing (Fan et al., 2016). Thus, relative to Sanger sequencing, high-throughput sequencing can capture a greater proportion of species, which is considered to be more effective in the field of community ecology (Paul et al., 2018). We found that the most species-rich ECM fungal genera were still the most common one in many other kinds of forest trees (Tedersoo et al., 2003; Richard et al., 2005; Bergemann and Garbelotto, 2006). These genera are often considered to be widespread, forming symbiotic relationships with more host plants than other ECM genera (Halling, 1987; Frøslev et al., 2005; Jakucs and Eros-Honti, 2008).
A number of studies have reported that the ECM fungal community was significantly affected by the host, soil, atmospheric deposition, geographical distance and climate (Ishida et al., 2007; Thompson et al., 2017; van der Linde et al., 2018; Wang et al., 2019). In addition to soil characteristics or even the disturbance regime of the system, the ECM fungal community structure and diversity could be determined by host trees (Johnson et al., 2005). The host age, photosynthetic activity, distributions, forest management and species composition affected the diversity or community structure of the ECM fungi (Goldmann et al., 2015; Erlandson et al., 2016; Hui et al., 2017; Unuk et al., 2019; Matsuoka et al., 2020). To explore the effects of abiotic factors, the most efficient way to eliminate the host effect is to focus on only one distributed worldwide host species such as Pinus tabulaeformis, Quercus liaotungensis, P. crassifolia, and Populus simonii. Consistent with our previous work (Long et al., 2016; Han et al., 2017; Wang et al., 2017), we found that site was the overriding factor affecting ECM fungal community structure compared with other factors at large scales (R2 = 0.37, P = 0.009, PERMANOVA). Most species of ECM fungi are thought to have limited distributions, which are commonly shaped by those of their hosts (Tedersoo et al., 2020). In general, the dominant ECM fungal genus associated with any specific host species is relatively consistent across the plant’s range. For instance, the dominant ECM fungal genus associated with Coccoloba uvifera was Scleroderma on a global scale (Séne et al., 2018). Tomentella, Inocybe, Clavulina, and Russula were the dominant ECM fungal genera in the Alnus trees (Roy et al., 2013). Consistent with our study, we found that Inocybe, Sebacian, and Tomentella were the dominant ECM fungal genera across the whole distribution area, accounting for more than 80% of the total relative abundance (Figure 2).
Although ECM fungal communities were dissimilar between sites (R = 0.33; P = 0.006, Anosim), the composition of the ECM fungal community was generally consistent among the four study sites. The number of overlapping species (137) far exceeds the number of unique species in each area (Figure 4B). We suggest that host identity should be the major factor that determines ECM fungal community composition. However, this finding is different from the study of the host plant Castanopsis sieboldii (Matsuoka et al., 2019), which suggested the explanatory power of the host was less than for climatic filtering and/or fungal dispersal. The cause of this difference could be that seawater blocked the transmission of ECM fungal spores among the whole hosts’ distribution. Furthermore, the LEfSe analysis identified 16 indicator taxa for differences among ECM fungal communities associated with P. crassifolia trees (Figure 5). The differences in ECM fungal community structure seem to be largely responsible for the shifts in the abundances of rare ECM fungal genera (Hui et al., 2017). Weißbecker et al. (2018) found that ECM fungal communities showed the greatest overlap of two sampled communities in a fungal functional group (approximately 80% of ECM fungal community similarity), although ECM fungal communities showed the highest pairwise community dissimilarities.
The distribution models consider the factors beyond consumable resources, such as space, to play a significant role in determining community structure (VanDerWal et al., 2009; McPolin and Kranabetter, 2021). The biogeographic position of each population on the mainland is typically classified as edge, sub edge or core (De Kort et al., 2021). QH is the core distribution of P. crassifolia, while other sites are at the edge in this study. ECM fungal richness decreases with the distances increasing from the core to the edge (Figure 3B), which agrees with the abundant-center hypothesis. Our model is similar to the non-linear model for endemic ECM species about ECM fungal community dissimilarity (based on species incidence) and interplot distance (McPolin and Kranabetter, 2021). However, unlike the pattern of ECM fungal richness distribution, the relative abundances of individual ECM fungal genera are difficult to link directly to the abundant-center hypothesis (Table 3). Mixed support for the abundant-center hypothesis has also been found in population genetic studies of other plants (e.g., Dixon et al., 2013). Furthermore, the center site (QH) has the most endemic ECM fungi (Figure 4B) and different biomarkers (Figure 5). The reason for this could be the changes in the relative abundance of dominant ECM fungi might be influenced by a specific environmental factor (Wakelin et al., 2007). For instance, nitrogen enrichment can lead to an increase in the biomass and abundance of the genus Tomentella (Brearley et al., 2005). In contrast, species in the genus Sebacina were usually dominant in the high-phosphorus forest (Zavišić et al., 2016). In addition, the dominant genera usually belong to the cosmopolitan group, which displayed lower species turnover across the host distribution (McPolin and Kranabetter, 2021). However, most of the low relative abundance of ECM fungi, belonging to the endemic ECM group, was generally driven by environmental history as well as biological traits (Hobohm and Tucker, 2014). At a continental scale, the distributions of these ECM fungal genera may reflect dispersal and colonization limitations, as well as greater species turnover at a local scale (McPolin and Kranabetter, 2021). These forces could produce ECM fungal distributions that conform to the predictions of the abundant-center hypothesis.
A species would achieve higher growth rates or greater stability at the core of a host species’ fundamental ecological niche than at sites farther from these optimal conditions. Thus, at core sites, hosts may have the highest diversity of ECM fungi as host effects play a critical role in the ECM fungal community assemblage and taxonomic diversity. Our results show that ECM fungal species richness was greatest at the center of the host plant’s range, consistent with the abundant-center hypothesis, while the relative abundances of individual fungal genera shifted inconsistently across the plant’s range. We encourage other researchers to test this hypothesis on other ECM fungal host species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ddbj.nig.ac.jp, LC203765-LC205656.
QH and XW carried out the experiments. QH wrote the manuscript. Both authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 51809269), the Co-ordination Foundation of the Chinese Academy of Agricultural Sciences (No. FIRI 2019-03-01), and the National Cotton Industrial Technology System (CARS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.759801/full#supplementary-material
Bahram, M., Põlme, S., Kõljalg, U., and Tedersoo, L. (2011). A single European aspen (Populus tremula) tree individual may potentially harbour dozens of Cenococcum geophilum ITS genotypes and hundreds of species of ectomycorrhizal fungi. FEMS Microbiol. Ecol. 75, 313–320. doi: 10.1111/j.1574-6941.2010.01000.x
Bengtsson-Palme, J., Ryberg, M., Hartmann, M., Branco, S., Wang, Z., Godhe, A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evolut. 4, 914–919. doi: 10.1111/2041-210X.12073
Bergemann, S., and Garbelotto, M. (2006). High diversity of fungi recovered from the roots of mature tanoak (Lithocarpus densiflorus) in northern California. Can. J. Bot. 84, 1380–1394. doi: 10.1139/b06-097
Boeraeve, M., Honnay, O., and Jacquemyn, H. (2018). Effects of host species, environmental filtering and forest age on community assembly of ectomycorrhizal fungi in fragmented forests. Fungal Ecol. 36, 89–98. doi: 10.1016/j.funeco.2018.08.003
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Brearley, F. Q., Scholes, J. D., and Su See, L. (2005). Nitrogen nutrition and isotopic discrimination in tropical ectomycorrhizal fungi. Res. Microbiol. 156, 184–190. doi: 10.1016/j.resmic.2004.09.003
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cobos, M. E., Barve, V., Barve, N., Jimenez-Valverde, A., and Nuñez-Penichet, C. (2021). rangemap: Simple Tools for Defining Species Ranges. Available online at: https://CRAN.R-project.org/package=rangemap (Accessed September 23, 2021).
Colwell, R. K. (2021). EstimateS: Biodiversity Estimation. Available online at: http://viceroy.eeb.uconn.edu/estimates/ (Accessed June 3, 2021)
Dahlberg, A. (2001). Community ecology of ectomycorrhizal fungi: an advancing interdisciplinary field. New Phytol. 150, 555–562. doi: 10.1046/j.1469-8137.2001.00142.x
Dallas, T. A., and Santini, L. (2020). The influence of stochasticity, landscape structure and species traits on abundant–centre relationships. Ecography 43, 1341–1351. doi: 10.1111/ecog.05164
De Kort, H., Prunier, J. G., Ducatez, S., Honnay, O., Baguette, M., Stevens, V. M., et al. (2021). Life history, climate and biogeography interactively affect worldwide genetic diversity of plant and animal populations. Nat. Commun. 12:516. doi: 10.1038/s41467-021-20958-2
Dixon, A. L., Herlihy, C. R., and Busch, J. W. (2013). Demographic and population-genetic tests provide mixed support for the abundant centre hypothesis in the endemic plant Leavenworthia stylosa. Mol. Ecol. 22, 1777–1791. doi: 10.1111/mec.12207
Durall, D. M., Gamiet, S., Simard, S. W., Kudrna, L., and Sakakibara, S. M. (2006). Effects of clearcut logging and tree species composition on the diversity and community composition of epigeous fruit bodies formed by ectomycorrhizal fungi. Botany 84, 966–980.
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Erlandson, S. R., Savage, J. A., Cavender-Bares, J. M., and Peay, K. G. (2016). Soil moisture and chemistry influence diversity of ectomycorrhizal fungal communities associating with willow along an hydrologic gradient. FEMS Microbiol. Ecol. 92:fiv148. doi: 10.1093/femsec/fiv148
Fan, Y. J., Grebenc, T., Wei, J., Zhao, Y. L., Yan, W., and Wang, L. B. (2016). Association of ectomycorrhizal fungi with Picea crassifolia (Pinaceae, Piceoidae) from high-altitude stands in Mount Helan Nature Reserve, China. Genet. Mol. Res. 15, 1–14. doi: 10.4238/gmr.15038604
Frøslev, T. G., Matheny, P. B., and Hibbett, D. S. (2005). Lower level relationships in the mushroom genus Cortinarius (Basidiomycota, Agaricales): a comparison of RPB1, RPB2, and ITS phylogenies. Mol. Phylogenet. Evol. 37, 602–618. doi: 10.1016/j.ympev.2005.06.016
Geml, J., Morgado, L. N., Semenova-Nelsen, T. A., and Schilthuizen, M. (2017). Changes in richness and community composition of ectomycorrhizal fungi among altitudinal vegetation types on Mount Kinabalu in Borneo. New Phytol. 215, 454–468. doi: 10.1111/nph.14566
Goldmann, K., Schöning, I., Buscot, F., and Wubet, T. (2015). Forest management type influences diversity and community composition of soil fungi across temperate forest ecosystems. Front. Microbiol. 6:1300. doi: 10.3389/fmicb.2015.01300
Gugerli, F., Rüegg, M., and Vendramin, G. G. (2009). Gradual decline in genetic diversity in Swiss stone pine populations (Pinus cembra) across Switzerland suggests postglacial re-colonization into the Alps from a common eastern glacial refugium. Bot. Helvetica 119, 13–22. doi: 10.1007/s00035-009-0052-6
Halling, R. E. (1987). A revision of the genus Inocybe in Europe. I. Subgenus Inosperma and the smooth-spored species of subgenus Inocybe. Brittonia 39:406. doi: 10.2307/2807145
Han, Q., Huang, J., Long, D., Wang, X., and Liu, J. (2017). Diversity and community structure of ectomycorrhizal fungi associated with Larix chinensis across the alpine treeline ecotone of Taibai Mountain. Mycorrhiza 27, 487–497. doi: 10.1007/s00572-017-0766-z
Hengeveld, R., and Haeck, J. (1982). The distribution of abundance. I. measurements. J. Biogeogr. 9, 303–316. doi: 10.2307/2844717
Hibbett, D. S., and Matheny, P. B. (2009). The relative ages of ectomycorrhizal mushrooms and their plant hosts estimated using Bayesian relaxed molecular clock analyses. BMC Biol. 7:13. doi: 10.1186/1741-7007-7-13
Hobohm, C., and Tucker, C. M. (2014). “The increasing importance of endemism: responsibility, the media and education,” in Plant and Vegetation. Endemism in Vascular Plants, ed. C. Hobohm (Dordrecht: Springer), 3–9.
Hui, N., Liu, X., Kotze, D. J., Jumpponen, A., Francini, G., and Setälä, H. (2017). Ectomycorrhizal fungal communities in urban parks are similar to those in natural forests but shaped by vegetation and park age. Appl. Environ. Microbiol. 83, e01797-17. doi: 10.1128/AEM.01797-17
Ishida, T. A., Nara, K., and Hogetsu, T. (2007). Host effects on ectomycorrhizal fungal communities: insight from eight host species in mixed conifer-broadleaf forests. New Phytol. 174, 430–440. doi: 10.1111/j.1469-8137.2007.02016.x
Jakucs, E., and Eros-Honti, Z. (2008). Morphological-anatomical characterization and identification of Tomentella ectomycorrhizas. Mycorrhiza 18, 277–285. doi: 10.1007/s00572-008-0183-4
Johnson, D., IJdo, M., Genney, D. R., Anderson, I. C., and Alexander, I. J. (2005). How do plants regulate the function, community structure, and diversity of mycorrhizal fungi? J. Exp. Bot. 56, 1751–1760. doi: 10.1093/jxb/eri192
Kauserud, H., Kumar, S., Brysting, A. K., Nordén, J., and Carlsen, T. (2012). High consistency between replicate 454 pyrosequencing analyses of ectomycorrhizal plant root samples. Mycorrhiza 22, 309–315. doi: 10.1007/s00572-011-0403-1
Li, J., Xie, D.-F., Guo, X.-L., Zheng, Z.-Y., He, X.-J., and Zhou, S.-D. (2020). Comparative analysis of the complete plastid genome of five bupleurum species and new insights into DNA barcoding and phylogenetic relationship. Plants 9:543. doi: 10.3390/plants9040543
Lilleskov, E. A., Bruns, T. D., Horton, T. R., Lee Taylor, D., and Grogan, P. (2004). Detection of forest stand-level spatial structure in ectomycorrhizal fungal communities. FEMS Microbiol. Ecol. 49, 319–332. doi: 10.1016/j.femsec.2004.04.004
Lira-Noriega, A., and Manthey, J. D. (2014). Relationship of Genetic diversity and niche centrality: a survey and analysis. Evolution 68, 1082–1093. doi: 10.1111/evo.12343
Liu, C., Cui, Y., Li, X., and Yao, M. (2021). microeco: an R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 97:fiaa255. doi: 10.1093/femsec/fiaa255
Long, D., Liu, J., Han, Q., Wang, X., and Huang, J. (2016). Ectomycorrhizal fungal communities associated with Populus simonii and Pinus tabuliformis in the hilly-gully region of the Loess Plateau, China. Sci. Rep. 6:24336. doi: 10.1038/srep24336
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Matsuoka, S., Iwasaki, T., Sugiyama, Y., Kawaguchi, E., Doi, H., and Osono, T. (2019). Biogeographic patterns of ectomycorrhizal fungal communities associated with Castanopsis sieboldii across the japanese archipelago. Front. Microbiol. 10:2656. doi: 10.3389/fmicb.2019.02656
Matsuoka, S., Mori, A. S., Kawaguchi, E., Hobara, S., and Osono, T. (2016). Disentangling the relative importance of host tree community, abiotic environment and spatial factors on ectomycorrhizal fungal assemblages along an elevation gradient. FEMS Microbiol. Ecol. 92:fiw044. doi: 10.1093/femsec/fiw044
Matsuoka, S., Sugiyama, Y., Tateno, R., Imamura, S., Kawaguchi, E., and Osono, T. (2020). Evaluation of host effects on ectomycorrhizal fungal community compositions in a forested landscape in northern Japan. R. Soc. Open Sci. 7:191952. doi: 10.1098/rsos.191952
McPolin, M. C., and Kranabetter, J. M. (2021). Influence of endemic versus cosmopolitan species on the local assembly of ectomycorrhizal fungal communities. New Phytol. 229, 2395–2399. doi: 10.1111/nph.17015
Mimura, M., and Aitken, S. N. (2007). Increased selfing and decreased effective pollen donor number in peripheral relative to central populations in Picea sitchensis (Pinaceae). Am. J. Bot. 94, 991–998. doi: 10.3732/ajb.94.6.991
Mucha, J., Peay, K. G., Smith, D. P., Reich, P. B., Stefański, A., and Hobbie, S. E. (2018). Effect of simulated climate warming on the ectomycorrhizal fungal community of boreal and temperate host species growing near their shared ecotonal range limits. Microb. Ecol. 75, 348–363. doi: 10.1007/s00248-017-1044-5
Nguyen, N. H., Smith, D., Peay, K., and Kennedy, P. (2015). Parsing ecological signal from noise in next generation amplicon sequencing. New Phytol. 205, 1389–1393. doi: 10.1111/nph.12923
Oksanen, J. (2021). vegan: Community Ecology Package. Available Online at: https://CRAN.R-project.org/package=vegan (Accessed June 4, 2021).
Paul, F., Otte, J., Schmitt, I., and Dal Grande, F. (2018). Comparing sanger sequencing and high-throughput metabarcoding for inferring photobiont diversity in lichens. Sci. Rep. 8:8624. doi: 10.1038/s41598-018-26947-8
Peay, K. G., Kennedy, P. G., Davies, S. J., Tan, S., and Bruns, T. D. (2010b). Potential link between plant and fungal distributions in a dipterocarp rainforest: community and phylogenetic structure of tropical ectomycorrhizal fungi across a plant and soil ecotone. New Phytol. 185, 529–542. doi: 10.1111/j.1469-8137.2009.03075.x
Peay, K. G., Garbelotto, M., and Bruns, T. D. (2010a). Evidence of dispersal limitation in soil microorganisms: Isolation reduces species richness on mycorrhizal tree islands. Ecology 91, 3631–3640. doi: 10.1890/09-2237.1
Pellitier, P. T., Zak, D. R., Argiroff, W. A., and Upchurch, R. A. (2021). Coupled shifts in ectomycorrhizal communities and plant uptake of organic nitrogen along a soil gradient: an isotopic perspective. Ecosystems 24, 1–15. doi: 10.1007/s10021-021-00628-6
Pironon, S., Papuga, G., Villellas, J., Angert, A. L., García, M. B., and Thompson, J. D. (2017). Geographic variation in genetic and demographic performance: new insights from an old biogeographical paradigm. Biol. Rev. 92, 1877–1909. doi: 10.1111/brv.12313
R Core Team (2021). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Richard, F., Millot, S., Gardes, M., and Selosse, M.-A. (2005). Diversity and specificity of ectomycorrhizal fungi retrieved from an old-growth Mediterranean forest dominated by Quercus ilex. New Phytol. 166, 1011–1023. doi: 10.1111/j.1469-8137.2005.01382.x
Roy, M., Rochet, J., Manzi, S., Jargeat, P., Gryta, H., Moreau, P.-A., et al. (2013). What determines Alnus-associated ectomycorrhizal community diversity and specificity? A comparison of host and habitat effects at a regional scale. New Phytol. 198, 1228–1238. doi: 10.1111/nph.12212
Rúa, M. A. (2021). Characterizing ectomycorrhizal fungal community structure and function of two varieties of pinus clausa that differ in disturbance history. Forests 12:219. doi: 10.3390/f12020219
Schappe, T., Albornoz, F. E., Turner, B. L., and Jones, F. A. (2020). Co-occurring fungal functional groups respond differently to tree neighborhoods and soil properties across three tropical rainforests in Panama. Microb. Ecol. 79, 675–685. doi: 10.1007/s00248-019-01446-z
Schön, M. E., Nieselt, K., and Garnica, S. (2018). Belowground fungal community diversity and composition associated with Norway spruce along an altitudinal gradient. PLoS One 13:e0208493. doi: 10.1371/journal.pone.0208493
Séne, S., Selosse, M.-A., Forget, M., Lambourdière, J., Cissé, K., Diédhiou, A. G., et al. (2018). A pantropically introduced tree is followed by specific ectomycorrhizal symbionts due to pseudo-vertical transmission. ISME J. 12, 1806–1816. doi: 10.1038/s41396-018-0088-y
Smith, S. E., and Smith, F. A. (2002). Diversity and Integration in Mycorrhizas: Proceedings of the 3rd International Conference on Mycorrhizas (ICOM3) Adelaide, Australia, 8–13 July 2001. Dordrecht: Springer. doi: 10.1007/978-94-017-1284-2
Smouse, P. E., Long, J. C., and Sokal, R. R. (1986). Multiple regression and correlation extensions of the mantel test of matrix correspondence. Syst. Biol. 35, 627–632. doi: 10.2307/2413122
Tedersoo, L., Bahram, M., and Zobel, M. (2020). How mycorrhizal associations drive plant population and community biology. Science 367:eaba1223. doi: 10.1126/science.aba1223
Tedersoo, L., Bahram, M., Toots, M., Diédhiou, A. G., Henkel, T. W., Kjøller, R., et al. (2012). Towards global patterns in the diversity and community structure of ectomycorrhizal fungi. Mol. Ecol. 21, 4160–4170. doi: 10.1111/j.1365-294X.2012.05602.x
Tedersoo, L., Kõljalg, U., Hallenberg, N., and Larsson, K.-H. (2003). Fine scale distribution of ectomycorrhizal fungi and roots across substrate layers including coarse woody debris in a mixed forest. New Phytol. 159, 153–165. doi: 10.1046/j.1469-8137.2003.00792.x
Tedersoo, L., May, T. W., and Smith, M. E. (2010). Ectomycorrhizal lifestyle in fungi: global diversity, distribution, and evolution of phylogenetic lineages. Mycorrhiza 20, 217–263. doi: 10.1007/s00572-009-0274-x
Tennekes, M., Nowosad, J., Gombin, J., Jeworutzki, S., Russell, K., Zijdeman, R., et al. (2021). tmap: Thematic Maps. Available online at: https://CRAN.R-project.org/package=tmap (Accessed September 23, 2021).
Thompson, L. R., Sanders, J. G., McDonald, D., Amir, A., Ladau, J., Locey, K. J., et al. (2017). A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463. doi: 10.1038/nature24621
Unuk, T., Martinović, T., Finžgar, D., Šibanc, N., Grebenc, T., and Kraigher, H. (2019). Root-associated fungal communities from two phenologically contrasting silver fir (Abies alba Mill.) groups of trees. Front. Plant Sci. 10:214. doi: 10.3389/fpls.2019.00214
van der Linde, S., Suz, L. M., Orme, C. D. L., Cox, F., Andreae, H., Asi, E., et al. (2018). Environment and host as large-scale controls of ectomycorrhizal fungi. Nature 558, 243–248. doi: 10.1038/s41586-018-0189-9
VanDerWal, J., Shoo, L. P., Johnson, C. N., and Williams, S. E. (2009). Abundance and the environmental niche: environmental suitability estimated from niche models predicts the upper limit of local abundance. Am. Nat. 174, 282–291. doi: 10.1086/600087
Vasco-Palacios, A. M., Bahram, M., Boekhout, T., and Tedersoo, L. (2020). Carbon content and pH as important drivers of fungal community structure in three Amazon forests. Plant Soil 450, 111–131. doi: 10.1007/s11104-019-04218-3
Vlk, L., Tedersoo, L., Antl, T., Vìtrovskě, T., Abarenkov, K., Pergl, J., et al. (2020). Alien ectomycorrhizal plants differ in their ability to interact with co-introduced and native ectomycorrhizal fungi in novel sites. ISME J. 14, 2336–2346. doi: 10.1038/s41396-020-0692-5
Wagner, M. (2009). Single-cell ecophysiology of microbes as revealed by Raman microspectroscopy or secondary ion mass spectrometry imaging. Annu. Rev. Microbiol. 63, 411–429. doi: 10.1146/annurev.micro.091208.073233
Wakelin, S. A., Colloff, M. J., Harvey, P. R., Marschner, P., Gregg, A. L., and Rogers, S. L. (2007). The effects of stubble retention and nitrogen application on soil microbial community structure and functional gene abundance under irrigated maize. FEMS Microbiol. Ecol. 59, 661–670. doi: 10.1111/j.1574-6941.2006.00235.x
Wang, X., Liu, J., Long, D., Han, Q., and Huang, J. (2017). The ectomycorrhizal fungal communities associated with Quercus liaotungensis in different habitats across northern China. Mycorrhiza 27, 441–449. doi: 10.1007/s00572-017-0762-3
Wang, Y.-L., Gao, C., Chen, L., Ji, N.-N., Wu, B.-W., Li, X.-C., et al. (2019). Host plant phylogeny and geographic distance strongly structure Betulaceae-associated ectomycorrhizal fungal communities in Chinese secondary forest ecosystems. FEMS Microbiol. Ecol. 95:fiz037. doi: 10.1093/femsec/fiz037
Weißbecker, C., Wubet, T., Lentendu, G., Kühn, P., Scholten, T., Bruelheide, H., et al. (2018). Experimental evidence of functional group-dependent effects of tree diversity on soil fungi in subtropical forests. Front. Microbiol. 9:2312. doi: 10.3389/fmicb.2018.02312
Keywords: ectomycorrhizal fungi, abundance-centre hypothesis, Picea crassifolia, richness, community structure
Citation: Wang X and Han Q (2022) A Closer Examination of the ‘Abundant-Center’ for Ectomycorrhizal Fungal Community Associated With Picea crassifolia in China. Front. Plant Sci. 13:759801. doi: 10.3389/fpls.2022.759801
Received: 17 August 2021; Accepted: 27 January 2022;
Published: 24 February 2022.
Edited by:
Luciano Kayser Vargas, Department of Agricultural Research and Diagnosis, State Secretariat for Agriculture, Livestock and Irrigation, BrazilReviewed by:
Priscila Pauly Ribas, Samsung R&D Institute Brazil (SRBR), BrazilCopyright © 2022 Wang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qisheng Han, aGFucWlzaGVuZ0BjYWFzLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.