 Xiuyuan Wang‡Chong Teng‡Huitian Wei†‡
Xiuyuan Wang‡Chong Teng‡Huitian Wei†‡ Shuang Liu
Shuang Liu Hongzhuan XuanWentao PengQianqian LiHongyan HaoQingya Lyu
Hongzhuan XuanWentao PengQianqian LiHongyan HaoQingya Lyu Shanhua Lyu*§†
Shanhua Lyu*§† Yinglun Fan*§†
Yinglun Fan*§†- College of Agriculture, Liaocheng University, Liaocheng, China
With the advent of multiple omics and Genome-Wide Association Studies (GWAS) technology, genome-scale functional analysis of candidate genes is to be conducted in diverse plant species. Construction of plant binary expression vectors is the prerequisite for gene function analysis. Therefore, it is of significance to develop a set of plant binary expression vectors with highly efficient, inexpensive, and convenient cloning method, and easy-to-use in screening of positive recombinant in Escherichia coli. In this study, we developed a set of plant binary expression vectors, termed pBTR vectors, based on Golden Gate cloning using BsaI restriction site. Foreign DNA fragment of interest (FDI) can be cloned into the destination pBTR by one-step digestion–ligation reaction in a single tube, and even the FDI contains internal BsaI site(s). Markedly, in one digestion–ligation reaction, multiple FDIs (exemplified by cloning four soybean Glyma.02g025400, Glyma.05g201700, Glyma.06g165700, and Glyma.17g095000 genes) can be cloned into the pBTR vector to generate multiple corresponding expression constructs (each expression vector carrying an FDI). In addition, the pBTR vectors carry the visual marker, a brightness monomeric red fluorescent protein mScarlet-I, that can be observed with the unaided eye in screening of positive recombinants without the use of additional reagents/equipment. The reliability of the pBTR vectors was validated in plants by overexpression of AtMyb75/PAP1 in tomato and GUSPlus in soybean roots via Agrobacterium rhizogenes-mediated transformation, promoter activity analysis of AtGCSpro in Arabidopsis via A. tumefaciens-mediated transformation, and protein subcellular localization of the Vitis vinifera VvCEB1opt in tobacco, respectively. These results demonstrated that the pBTR vectors can be used in analysis of gene (over)expression, promoter activity, and protein subcellular localization. These vectors will contribute to speeding up gene function analysis and the process of plant molecular breeding.
Introduction
With the rapid advance of DNA sequencing technology, genomes of 800 terrestrial plants have been sequenced (Marks et al., 2021; Lu et al., 2021; Kamal et al., 2022; Peng et al., 2022; Sun et al., 2022). Concomitantly, the rapidly developing of multiple omics and Genome-Wide Association Studies (GWAS) in diverse plant species, genome-scale functional analyses of large numbers of candidate genes will have been conducted. Construction of plant binary expression vector is the prerequisite for gene function analysis. Therefore, it is important to develop a set of cost-efficient, easy-to-use, and highly efficient plant binary expression vectors. In previous studies, a series of binary expression vectors have been developed, such as pBIN19 and its derivatives (the binary pBI series) (Barton et al., 1983; Bevan, 1984; Jefferson et al., 1987), pGreen/pSoup and its derivative (pCLEAN), pPZP and its derivatives (pCAMBIA series) (Hajdukiewicz et al., 1994; van Engelen et al., 1995; Hellens et al., 2000; Zheng et al., 2001; Thole et al., 2007; Belknap et al., 2008), and Gateway binary vectors (e.g. pGWBs, pMDCs, pEarleyGate) (Curtis and Grossniklaus, 2003; Nakagawa et al., 2007; Karimi et al., 2007). In these expression vectors, to achieve the assembly of the foreign DNA fragment of interest (FDI) into the plant binary expression vectors, the main molecular cloning approaches available primarily include homologous recombination and the traditional restriction digestion–ligation methods. Homologous recombination technology that includes in vivo and in vitro assemblies depends on short homologous ends. The in vivo assembly method (Mosberg et al., 2010; Fu et al., 2012) is simple in operation and looks attractive, but it does not become popular and be widely used (Watson and García-Nafría, 2019). In vitro assembly strategies include a variety of homology-based methods, such as ligation-independent cloning (LIC) (Aslanidis and de Jong, 1990), Gateway cloning (Hartley et al., 2000; Walhout et al., 2000), SLIC (Li and Elledge, 2007), SliCE (Zhang et al., 2012; Motohashi, 2015), In-fusion cloning (Benoit et al., 2006; Sleight et al., 2010), Gibson assembly (Zhu et al., 2007; Gibson et al., 2009), and Nimble cloning (Yan et al., 2020). Homology-based cloning relies on small homologous sequences at the termini of DNA fragments generally introduced by PCR to drive the assembly of desired circular recombinants. It is simple, sequence-independent, and directional. However, it is costly to assemble genome-scale candidate FDIs into expression vectors. Compared to in vivo and in vitro homology-based cloning methods, the traditional restriction digestion–ligation method is low-cost, simple, popular, and widely used. However, it is sequence-dependent, and dozens of diverse restriction enzymes need to be stored in a lab to be able to construct different vectors. Although directional cloning can be achieved by combining two different restriction endonucleases, isocaudomers should be carefully considered to evade DNA molecular self-ligation, and some restriction enzymes show buffer incompatibility. Furthermore, digestion by restriction enzymes and ligation via ligase reactions are separately completed, and an additional DNA purification step is also required. Therefore, it is time-consuming, inefficient, and sequence-dependent. Additionally, the DNA purification step (requiring the purchase of a kit) can result in a partial loss of FDI and vector.
Engler et al. (2008) reported a Golden Gate cloning method that overcomes some shortcomings of this traditional restriction digestion–ligation method. It uses only a single restriction endonuclease, and entails a one-step digestion–ligation reaction without a DNA purification step. Based on Golden Gate cloning, Lampropoulos et al. (2013) and Emami et al. (2013) developed several plant expression vectors that can rapidly and sequentially integrate multiple pre-made modules (including promoter, FDI, terminator, selection marker, or reporter gene, etc.) into a plant expression vector. However, the expression vectors can not be directly used for cloning of FDI (requiring multiple expression-related elements integrated and arranged in a designed order). It is greatly complex in operation to clone FDI carrying one or several internal restriction enzyme site(s) into the expression vector (Engler et al., 2008; Emami et al., 2013). Furthermore, the screening of positive colonies in E. coli depends on the additional chemicals reagents. In this study, we developed a series of novel, high-efficiency plant binary expression vectors, termed pBTR vectors (representing initials of BsaI, T4 ligase, Reddish-pink colony of E. coli), based on Golden Gate cloning (Engler et al., 2008) for Agrobacterium-mediated plant transformation. Only a single BsaI restriction enzyme is used, and the FDI can be directly and conveniently cloned into the destination pBTR expression vector with almost sequence-independence and no additional DNA purification step by a one-step digestion–ligation reaction. The associated costs are substantially low. Significantly, in a single digestion–ligation reaction, multiple FDIs can be independently cloned into pBTR vector to generate multiple corresponding expression vectors (each expression vector carrying an FDI). In addition, the pBTRs allow for directional cloning, and carry directly observable visual selection marker that can be used in screening positive transformants in E. coli according to the colony color by the unaided eye. These vectors will pave the way for accelerating the speed of gene function studies and the process of plant molecular breeding.
Results
Development of the pBTRs
To achieve simple one-step cloning based on the digestion–ligation method, we developed a series of novel vectors including vectors targeted at (over)expression of genes (Figure 1), promoter analysis (Figure 2), and subcellular localization (Figure 3), based on the Golden Gate cloning method with only one restriction endonuclease BsaI (the cheapest type IIs restriction endonuclease) and T4 ligase required. For each vector type, we developed two sets, termed pBTR1 and pBTR2, respectively. pHSE401/pKSE401 (Xing et al., 2014) and pYLCRISPR/Cas9Pubi-B/H/N (Ma et al., 2014) which derived from pCAMBIA-based binary expression vectors, were used as backbones with BsaI recognition sequences eliminated. These have been widely used well in both Agrobacterium tumefaciens- and Agrobacterium rhizogenesis-mediated plant transformation. BsaI recognizes the 5’-GGTCTC(N1)/(N5)-3’ site and cuts outside of its recognition sequence at the 3’-end of DNA molecular, and thereby, producing a 5’ overhang 4 nt in length. We incorporated two tandem BsaI recognition sites arranged in reverse orientation with their neighboring sequences into the pBTR vectors. After a given vector is digested by BsaI, it lacks the original BsaI recognition site and has different 5’ non-palindromic overhangs at each terminal. For example, in the (over)expression of gene vectors, the p-35BTR1 set has CATC (5’-3’) and GGAT (5’-3’) 5’ overhangs while p-35pBTR2 has TCTA (5’-3’) and GAGC (5’-3’) 5’ overhangs (Figure 1A). Therefore, pBTR1 and pBTR2 cannot be self-religated after the single BsaI digestion (Figures 1–3). This prevents the generation of false-positive colonies of E. coli (empty vector) and ensures that FDI is integrated into the destination vector in a designed orientation.
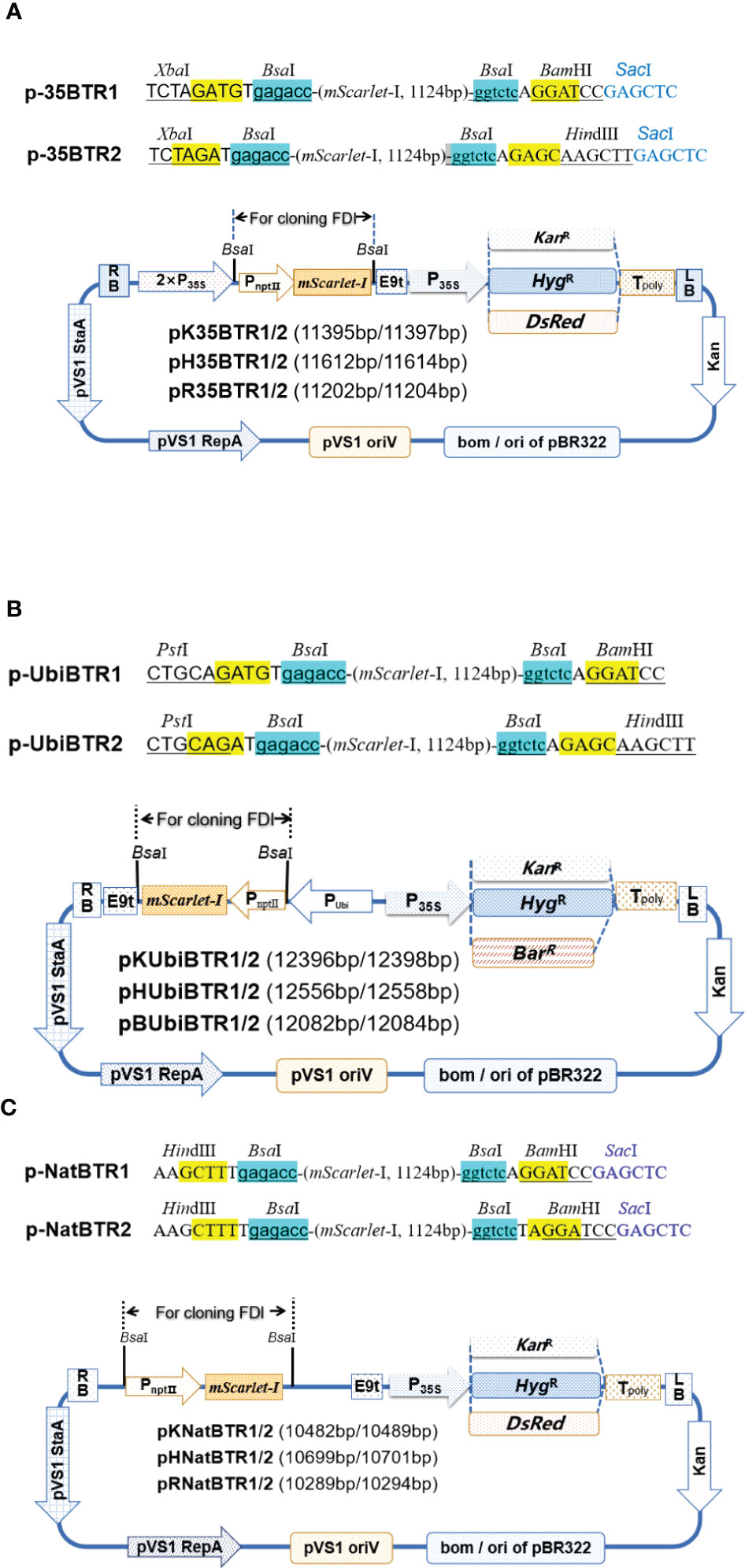
Figure 1 Diagrams of (over)expression vectors. Gene (over) expression vectors driven by CaMV 35S based on the pHSE401/pKSE401 backbone (A). Ubiquitin promoter based on the pYLCRISPR/Cas9Pubi-series of vectors backbone (B). Native promoter based on the pHSE401/pKSE401 backbone (C). KanR, kanamycin resistance gene; HygR, hygromycin resistance gene; BarR, basta resistance gene. The pNptⅡ::mScarlet-I expression cassette was used for a visual selection marker in E coli. The two BsaI recognition sites and neighbor sequences designed for cloning FDI are shown at the top of the vectors. The key restriction sites for further screening of positive recombinants are given. The KanR, HygR, DsRed, and BarR genes driven by CaMV 35S were used as selection markers/reporters in plant. 5’ overhangs (sticky end) produced after digestion by BsaI are highlighted in yellow.
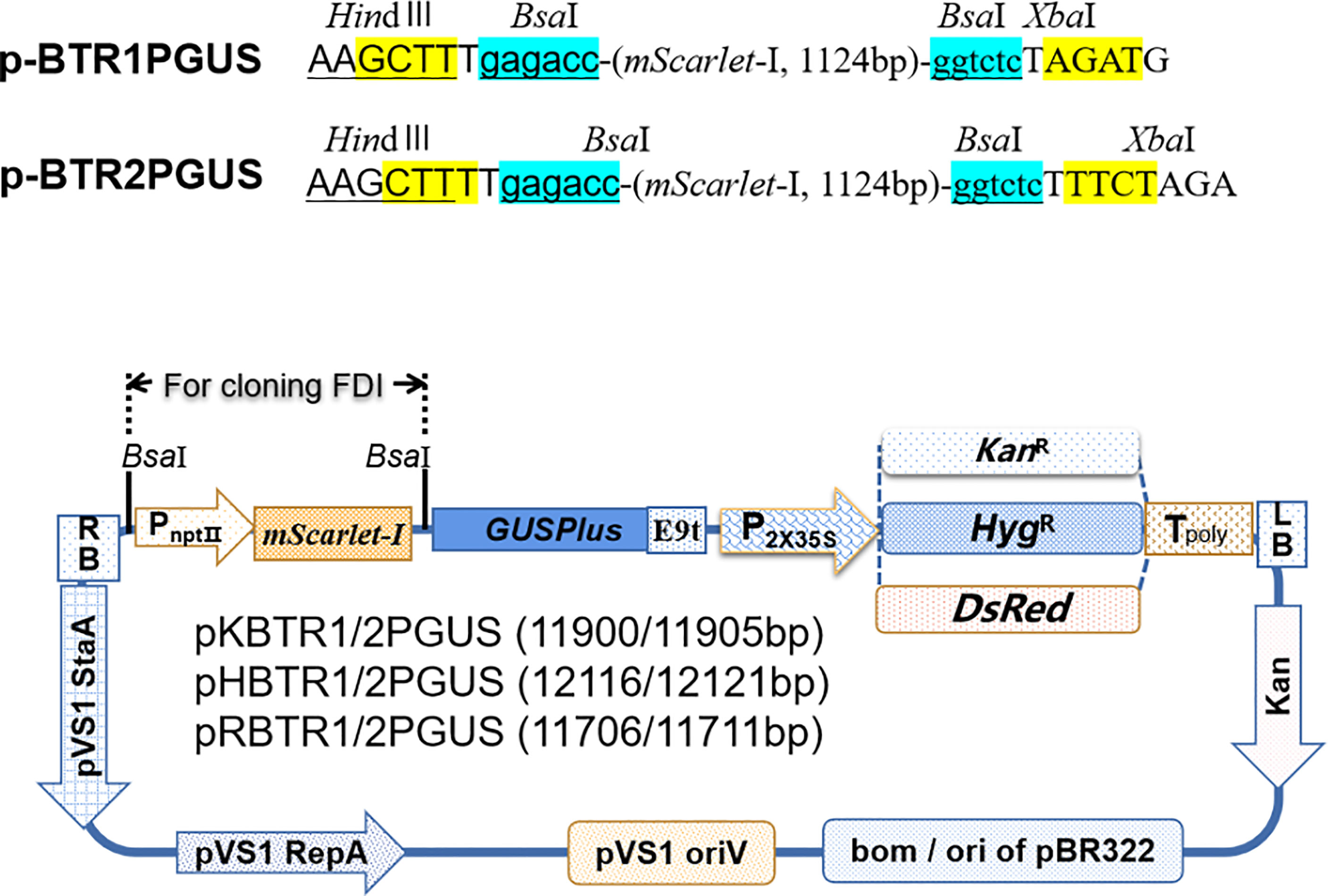
Figure 2 Diagrams of promoter analysis vectors. HindIII and XbaI restriction enzyme recognition sites for further screening of positive recombinants in E. coli are given. The vectors pKBTR1/2PGUS, pHBTR1/2PGUS, and pRBTR1/2PGUS carrying a KanR, HygR, and DsRed gene, respectively, driven by CaMV 35S (enhancer were deleted) were used as selection markers in plant. Sticky ends produced after digestion by BsaI are highlighted in yellow.
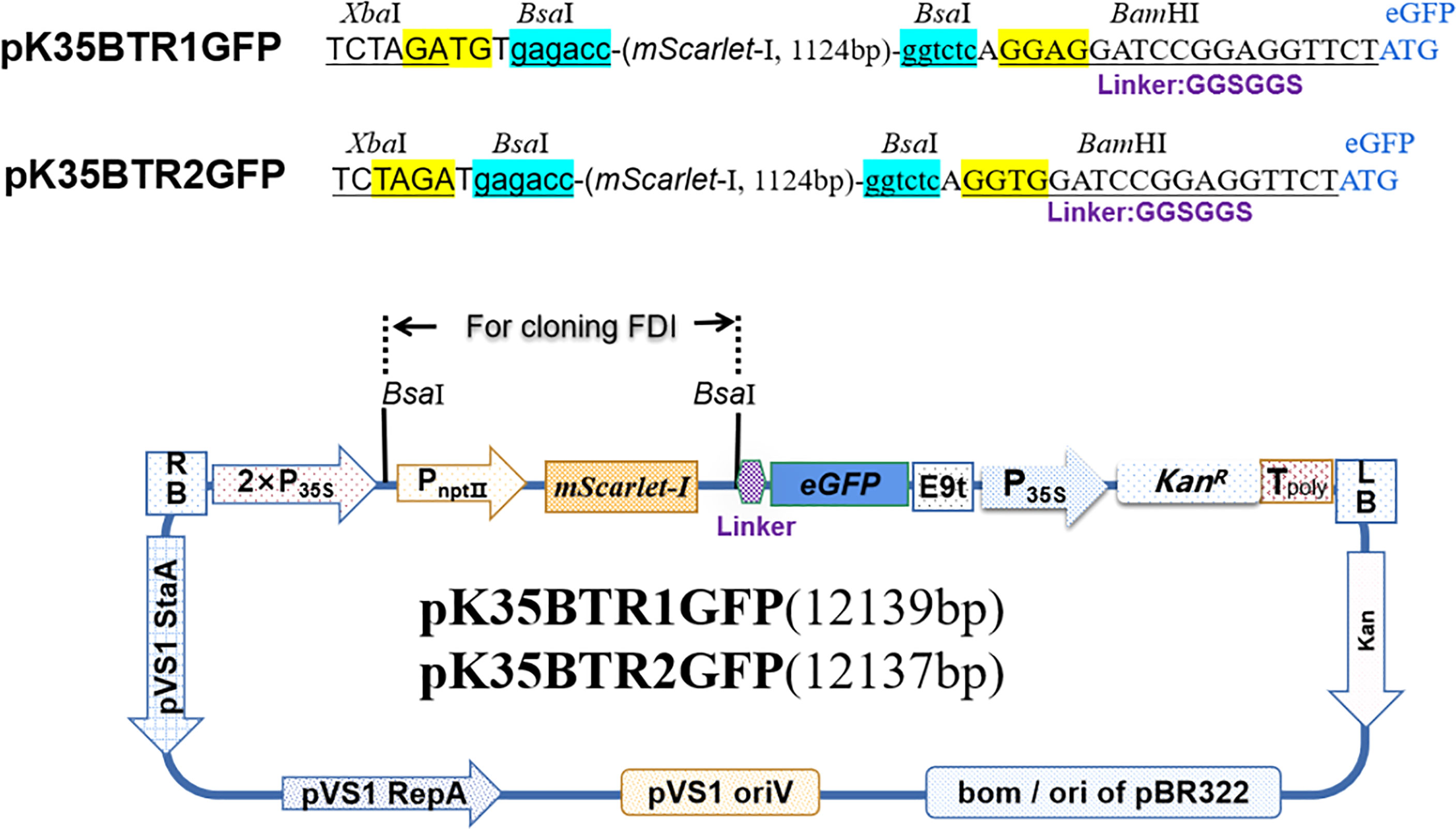
Figure 3 Diagram of subcellular localization vector.
Next, FDI was added with adapters carrying the BsaI recognition sequences at the forward and reverse primer 5’ ends introduced by PCR for cloning into the pBTR1 or pBTR2 vector. This was done to generate specific overhangs at each end of each FDI that complement those of the corresponding destination vectors in the desired orientation after digestion by BsaI. The adapter sequences applied for different expression vectors are shown in Table 1. The FDI and destination vector were concatenated together using T4 DNA ligase. The destination vector and the PCR product were digested and then ligated in a reaction mixture in one tube (Figure 4). For FDI carrying one or more internal BsaI site(s), the digestion–ligation step was performed in a thermocycler with the following program: 10 cycles at 37°C for 3 min, at 16°C for 3 min, and at 12°C for 2 min, followed by a final incubation step at 4°C for 1 h (holding ligation reaction step). For FDIs carrying no internal BsaI recognition sequences, the same program was employed, but the final incubation step was at 37°C for 10 min (holding the restriction enzyme digestion reaction step) to decrease the number of empty vectors. The resulted digestion–ligation mixtures were directly transformed into competent E. coli cells to obtain the desired clones (Figure 4).
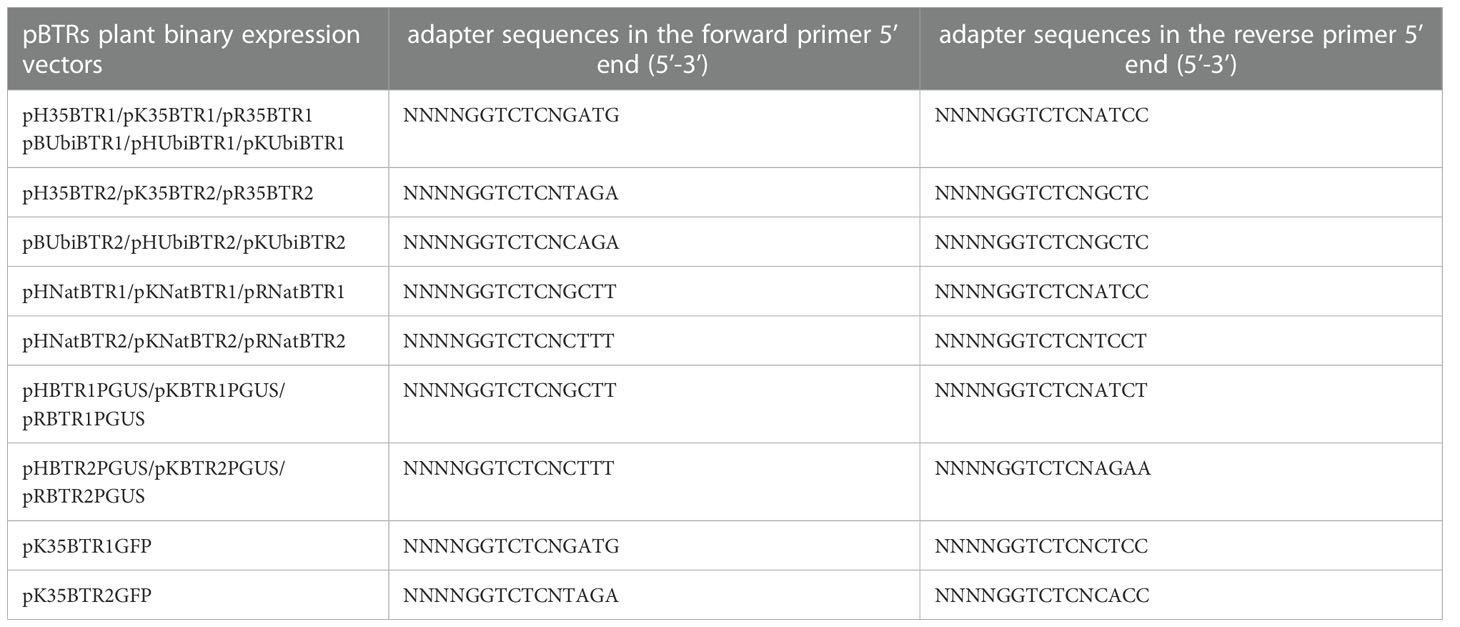
Table 1 Adapter sequences should be added in the primer 5’ end.
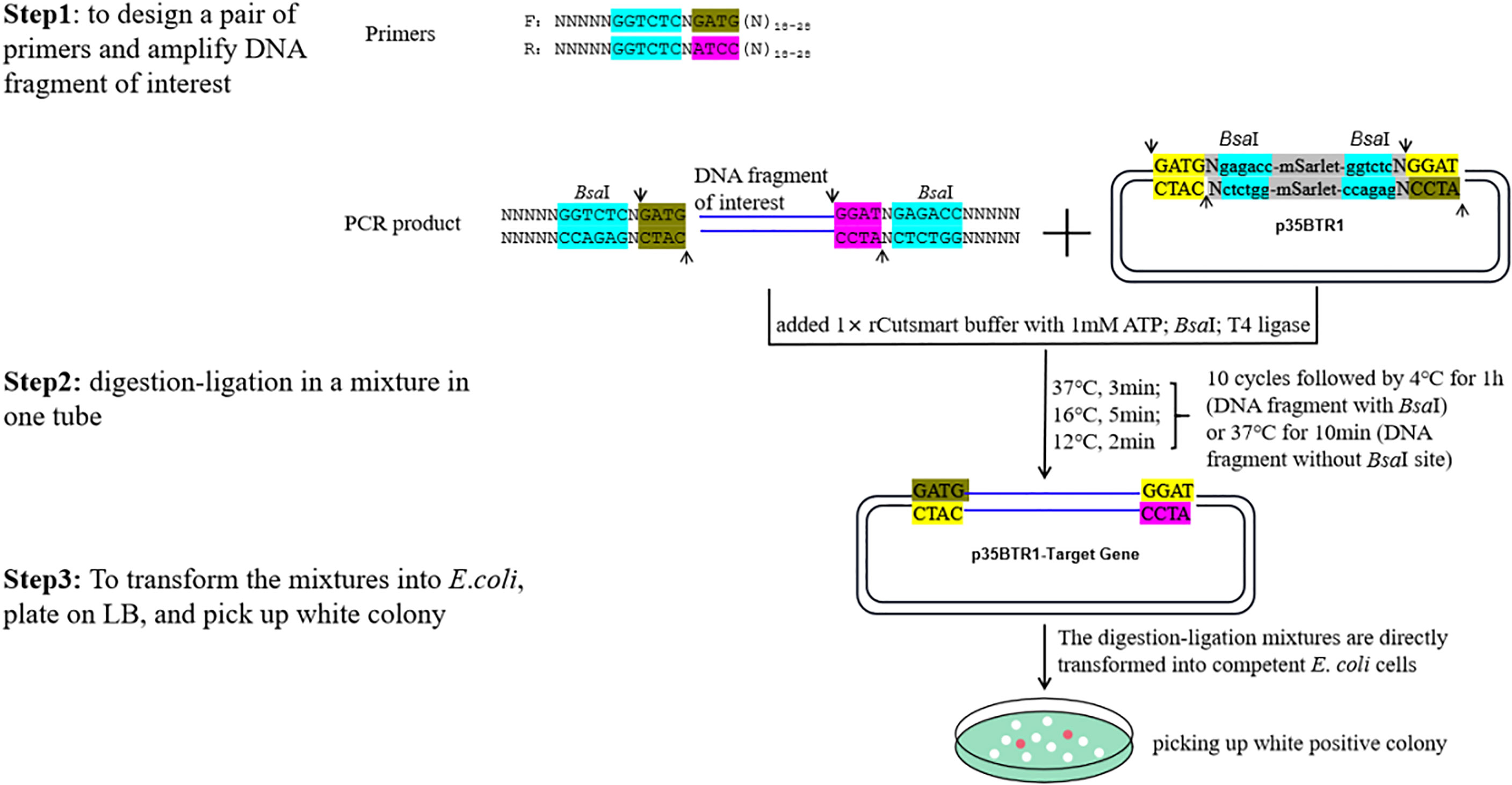
Figure 4 An example of the schematic of the construction of a pBTR vector using one-step digestion–ligation. N18-28 represents any 18–28 nt sequences (forward or reverse primers) paired with the FDI. Arrows indicate the digestion site by BsaI. The BsaI recognition sites are shown in blue. Two tandem BsaI recognition sites and neighboring sequences were incorporated into the pBTR vectors. FDI was cloned into p35BTR1, generating a recombinant product lacking the original BsaI restriction site.
In the pBTR1/2, a visual selection marker, the mScarlet-I gene (Bindels et al., 2017) driven by the neomycin phosphotransferase gene promoter (PnptⅡ) was inserted between two BsaI sites. The expression of mScarlet-I in E. coli results in a reddish-pink colony formed from negative transformants, while FDI replaced the expression cassette of PnptⅡ:: mScarlet-I from positive transformants and the positive colonies were white color (Figures 1–5). The purpose of this marker designed was to distinguish non-recombinants (reddish-pink colony) from positive recombinants (white colony) with the unaided eye without using any other reagent, substrate, or equipment. Figure 4 presents a schematic depicting vector construction (p35BTR1 used as an example).
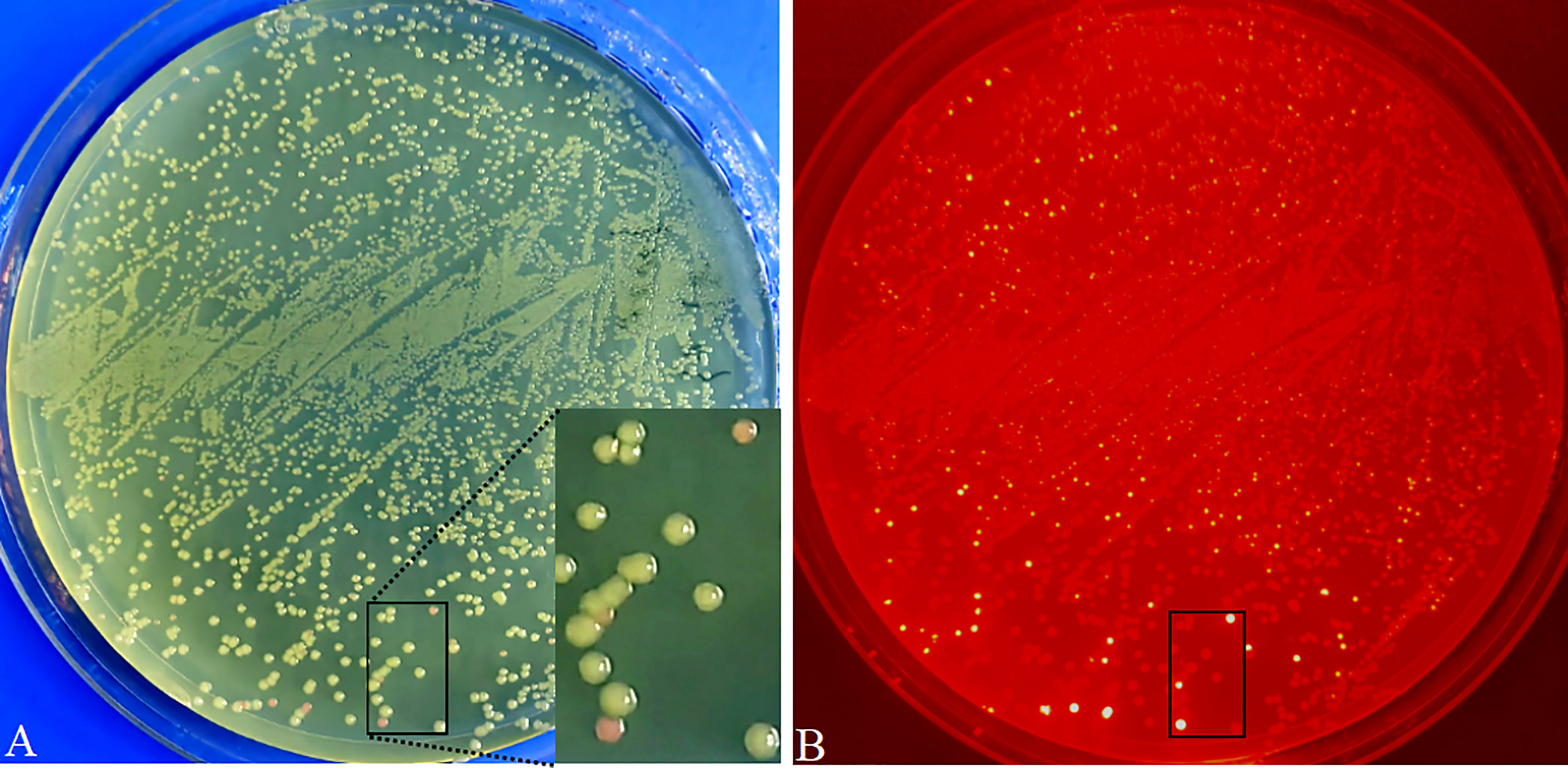
Figure 5 Visual screening of positive recombinants and cloning efficiency analysis in E coli transformed with pR35BTR2-GmbHLH293. Directly visual screening of recombinants by the naked eye under natural light. Positive recombinants are white color, while non-recombinants are reddish-pink (A). E coli transformant colonies were observed under green excitation; non-recombinants are bright red (B).
Analyses of cloning efficiency in E. coli and validation of the gene (over)expression vectors in planta
CaMV 35S and ubiquitin promoters are highly active in most transgenic plant cells and are the most widely used to drive gene overexpression in dicots and monocots. Therefore, based on pHSE401/pKSE401 and pYLCRISPR/Cas9Pubi-B/H/N backbones, we created two series of plant binary overexpression vectors, termed p-35BTR1/2 (Figure 1A) and p-UbiBTR1/2 (Figure 1B), respectively, where CaMV 35S and ubiquitin promoters were used to drive candidate gene expression. To facilitate the screening of transgenic-positive plants, we developed 12 different overexpression vectors including six the p-35BTR1/2 series and six p-UbiBTR1/2 series, respectively, with different selection markers or reporter genes. The p-35BTR1/2 series included pK35BTR1/2 (carrying the kanamycin resistance selection marker gene, KanR), pH35BTR1/2 (carrying the hygromycin resistance selection marker gene, HygR), and pR35BTR1/2 (carrying the DsRed fluorescent protein reporter gene, DsRed), respectively (Figure 1A). The p-UbiBTR1/2 series included pKUbiBTR1/2 (KanR), pHUbiBTR1/2 (HygR), and pBUbiBTR1/2 (carrying the basta resistance gene, BarR) (Figure 1B). Furthermore, we also created a series of plant binary expression vectors driven by a native promoter based on pHSE401/pKSE401 backbones (Xing et al., 2014), termed p-NatBTR1/2. This included p-NatBTR1/2 six vectors, pKNatBTR1/2 (KanR), pHNatBTR1/2 (HygR), and pRNatBTR1/2 (DsRed) (Figure 1C).
To verify the availability and cloning efficiency of these vectors, Arabidopsis AtMyb75/PAP1 (GenBank No. AY519563; no internal BsaI recognition site) (Fan et al., 2020a), GUSPlus (cloned from pCAMBIA1305.1; carrying two internal BsaI recognition sites), and soybean GmbHLH293 (Glyma.01g068600; carrying two internal BsaI sites and one BsaI site producing a palindromic sticky end) were cloned into pK35BTR1, pK35BTR2, and pR35BTR2 vectors, respectively, followed by the protocol described in Figure 4 and also described in detail in the “Material and methods” section. The digestion–ligation mixtures were directly transformed into competent E. coli cells. There were ~10, 000 colonies obtained with 100 µL E.coli chemically competent cells transformed with pK35BTR1-AtMyb75 or pK35BTR2-GUSPlus using 20 ng vector plasmid. The number of pR35BTR2-GmbHLH293 colonies was ~2, 000, less than the pK35BTR1-AtMyb75 and pK35BTR2-GUSPlus transformants, possibly because GmbHLH293 gene carrying one internal BsaI site produced a palindromic sticky end. The desired recombinants appeared as white colonies, whereas non-recombinants were reddish-pink (Figure 5). These different colonies were easily and conveniently distinguished from each other by the naked eye under natural light (Figure 5A). The red fluorescence from these reddish-pink colonies can also be observed under the control of green excitation light due to the expression of mScarlet-I (Figure 5B). We randomly selected 8 white colonies to further confirm that they were positive recombinants via restriction enzyme digestion (Figure S1A) and Sanger sequencing (Figure S1B). The results indicated that these colonies were correctly identified using our new method. All such colonies showed 100% transformants with FDI inserted (Figures S1A, B). These results also show that mScarlet-I can serve as a reliable marker for highly efficient selection of positive colonies based on colony color. More than 90% of colonies transformed with the pK35BTR1-AtMyb75 (103 white colonies/103 total colonies, 100%, Figure S1C), pK35BTR2-GUSPlus (124/124 total colonies, 100%, Figure S1D), and pR35BTR2-GmbHLH293 (236 white colonies /262 total colonies, 90%, Figure 5) recombinant vectors were positive colonies.
To validate in planta expression of the cloned gene in the pBTR1/2 vector, the pK35BTR1-AtMyb75 and pK35BTR2-GUSPlus plasmids were transformed into A. rhizogenes strain K599, and positive K599 carrying the corresponding plasmid were used to infect the hypocotyl of tomato and soybean, respectively, and to induce the generation of transgenic roots using one-step A. rhizogenes-mediated transformation (Fan et al., 2020a). Purple/red-colored anthocyanin accumulated in 35S::AtMyb75 transgenic tomato roots but not in non-transgenic roots (Figures 6A, S2A). This is consistent with previous reports that overexpression of AtMyb75/PAP1 in some plant roots can induce the accumulation of purple/red-colored anthocyanin in roots (Zuluaga et al., 2008; Fan et al., 2020b). As expected, the GUS signal was observed in overexpression-GUSPlus hairy roots of soybean (Figures 6B, S2B). These results indicate that pBTR1/2 overexpression vectors are reliable plant binary vectors for Agrobacterium-mediated plant transformation.
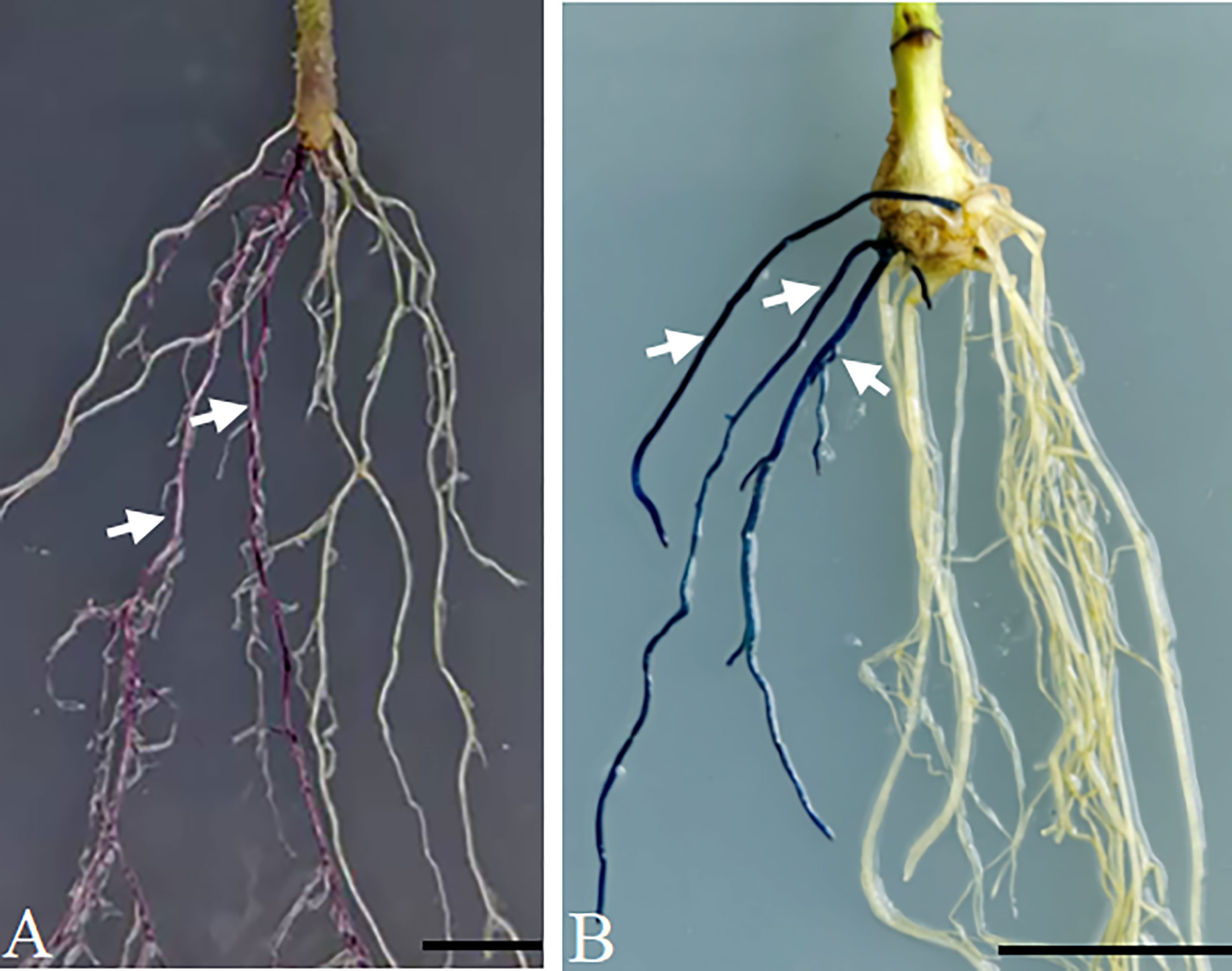
Figure 6 Verification of the overexpression vector p-35BTR1/2 in plants. Anthocyanin accumulated in the transgenic hairy roots of tomato transformed with pK35BTR1-AtMyb75 (A). The GUS signal was observed in the transgenic hairy roots of soybean transformed with pK35BTR2-GUSPlus (B). Arrows indicate the transgenic roots; bars=1 cm.
Validation of promoter analysis vectors in planta
Plant growth and development involve the temporal and spatial expression of specific gene subsets in response to various physiological and environmental factors mediated by complex signal transduction pathways. Promoter functional analyses can provide evidence of the functional action of genes by appraising the spatial and temporal domains of gene expression. The GUSPlus gene is often used as a reporter gene in such analyses. Here, we developed six promoter analysis vectors, including pKBTR1/2PGUS (KanR), pHBTR1/2PGUS (HygR), and pRBTR1/2PGUS (DsRed) (Figure 2). In these vectors, the two internal BsaI recognition sites carried in the GUSPlus gene were eliminated. To analyze promoter function and validate these vectors, an Arabidopsis thaliana gamma-glutamylcysteine synthetase gene (termed AtGCS; GenBank no. AF068299.1) promoter showing highly active in broad eudicots roots (Liu et al., 2022) but not characterized in A. thaliana, was cloned into the pRBTR1PGUS vector and transformed into wild-type A. thaliana by A. tumefaciens-mediated transformation using flower dip method (Clough and Bent, 1998). The AtGCS promoter can drive GUSPlus expression of transgenes in the cotyledon, shoot apical meristem, inflorescence, and vascular areas of sepal and petal tissues, besides roots (Figures 7, S2C). This expression pattern is consistent with that of the TAIR (The Arabidopsis Information Resource, http://www.arabidopsis.org) database. The results indicated that the pBTR1/2 vector sets for promoter analysis work well in plants via Agrobacterium-mediated plant transformation.
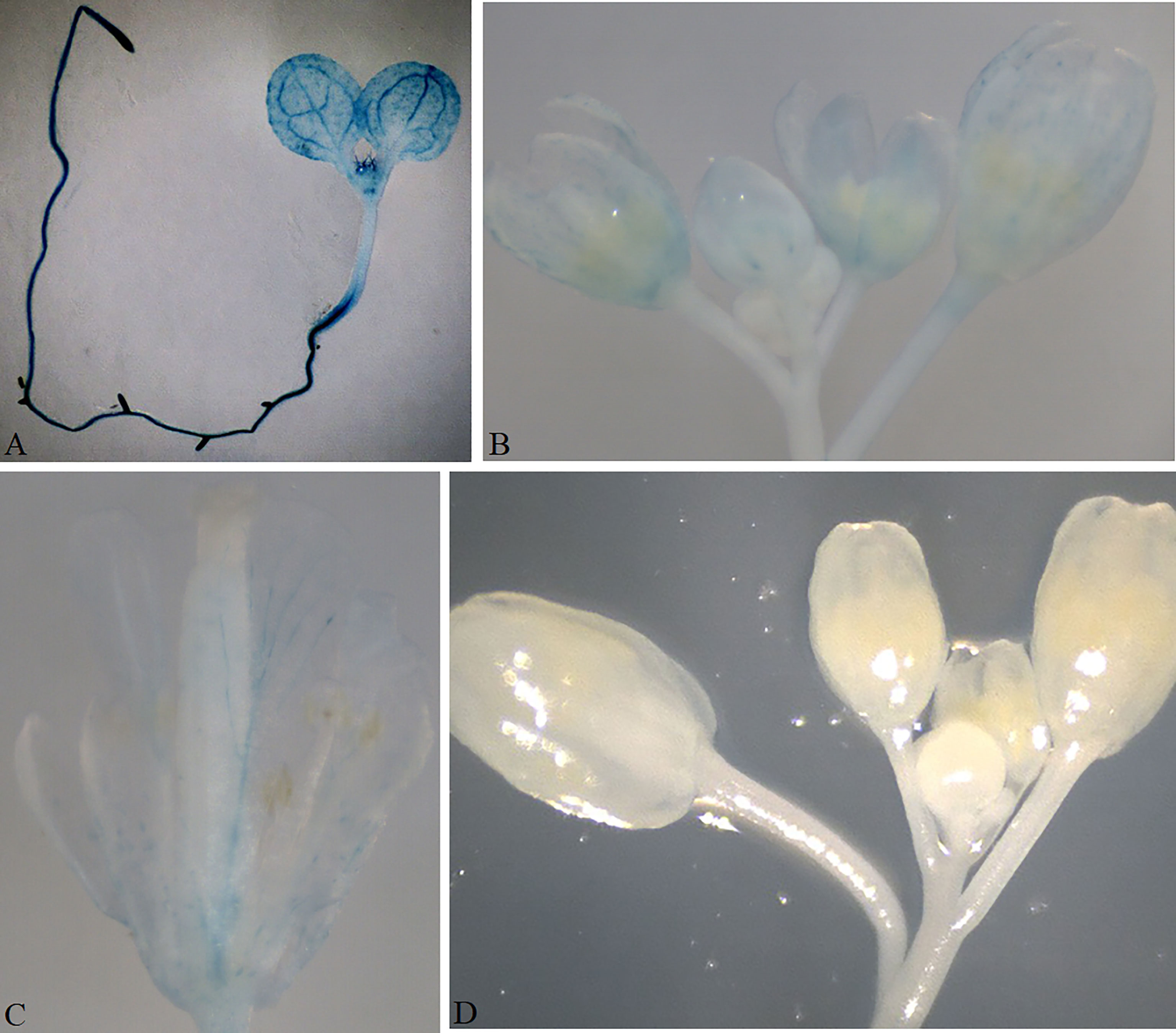
Figure 7 Validation of the promoter analysis vector in Arabidopsis. The GUS signal was observed in the cotyledon, shoot apical meristem, and the whole root of a 10-day-old transgenic seedling transformed with pRBTR1PGUS-AtGCS (A). GUS expression in an inflorescence (B); GUS expression in a single flower (C). No GUS signal was observed in the Arabidopsis transformed with an empty pRBTR1PGUS vector as a negative control (D).
Verification of protein subcellular localization vectors
Protein subcellular localization is crucial for studying plant gene function(s) and cellular biological processes. eGFP is a fluorescent marker commonly used for protein subcellular localization. We created two stable plant binary expression vectors, pK35BTR1/2GFP (KanR), that allow C-terminal protein fusion with eGFP (Figure 3). A linker sequence that codes a peptide linkage (GGSGGS), 5’-GGTGGATCCGGAGGTTCT-3’, was designed and inserted between the candidate gene and eGFP gene (Figure 3) (Hao et al., 2005). To validate that the vectors were suitable for studying protein subcellular localization, we fused the eGFP in pK35BTR2GFP with the Vitis vinifera VvCEB1opt, which encodes a transcription factor (Nicolas et al., 2013; Lim et al., 2018). The GFP fluorescent signal was detected in the nucleus when fused with VvCEB1opt (Figure 8), which is in agreement with localization of VvCEB1opt in previous reports (Nicolas et al., 2013; Lim et al., 2018). The result indicated that the protein subcellular localization vectors are reliable.

Figure 8 Analyses of the subcellular localization of VvCEB1opt in N. benthamiana leaf cells using the pK35BTR2GFP vector. Bright field (BF) (A). Fluorescent signal of VvCEB1opt-eGFP fusion protein (B). Subcellular localization of nucleus markers using mCherry fluorescent protein (mChe) (C); Merge: overlapping of the eGFP, mChe, and BF images (D). Scale bars=50 μm.
Generation of multiple expression vectors cloned with different FDIs by a single step digestion–ligation reaction
Genome-scale functional annotation of large numbers of candidate genes will be conducted in the post-genome era. A fast and highly efficient cloning method is the prerequisite for the functional annotation of such genes. Next, we tested whether several FDIs could be independently cloned into the pBTR vectors to generate multiple corresponding expression vectors carrying different FDIs by a single step digestion–ligation reaction (each expression vector cloned an FDI and no different FDIs stacked in one vector). For this purpose, four genes, Glyma.02g025400 (3200 bp), Glyma.05g201700 (2616 bp), Glyma.06g165700 (2068 bp), and Glyma.17g095000 (2852 bp), were cloned into pR35BTR2. Among these four genes, Glyma.06g165700 carries a BsaI recognition site. Ten white colonies were randomly selected after the digestion–ligation mixtures were transformed into competent E. coli cells. The plasmids were extracted and subjected to digesting by HindIII to identify the inserts (Figure S3A). The inserted fragments were further determined by Sanger sequencing (Figure S3B). All ten white colonies were positive transformants and carried the expected inserts. The recombinant frequencies of the four FDIs were as follows: Glyma.02g025400 (2/10, 20%), Glyma.05g201700 (3/10, 30%), Glyma.06g165700 (2/10, 20%), and Glyma.17g095000 (3/10, 30%). The result shows that the pBTR1/2 vectors can be used for multiple FDIs cloning to construct expression vectors by one-step digestion–ligation reaction in one tube by a single transformation.
Discussion
pBTR vectors: highly efficient, cost-efficient, and convenient plant binary expression vectors
Gene (over)expression analysis, promoter activity analysis, and protein subcellular localization are crucial for studying plant gene function(s) and cellular processes. In these analyses, the first step is the construction of expression vectors. To this end, Golden Gate cloning provides a rapid, inexpensive assembly of multi-component DNA fragments that can be inserted into a destination cloning vector (Engler et al., 2008). Lampropoulos et al. (2013) used this method to develop a GreenGate system that can rapidly and sequentially integrate multiple pre-made modules into a plant expression vector (added two BsaI recognition sequences arranged in reverse orientation in multiple cloning sites) by one-step digestion–ligation reaction completed in one tube. Emami et al. (2013) developed three plant expression vectors based on Golden Gate cloning. However, only two plant selection markers/reporters can be used for plant transformation. In addition, expression-related cassettes, such as promoters and terminators, have not been pre-recombined into the expression vector. Multiple DNA fragments including the FDI need to be sequentially recombined into the expression vector. However, cloning efficiency significantly decreases with an increasing number of cloned modules.
In this study, based on the Golden Gate cloning method (Engler et al., 2008), we developed a series of novel pBTR vectors in which expression-related modules and several types of plant selection markers or reporter genes (e.g., KanR, HygR, BarR, and DsRed) have been previously cloned into the expression vectors (Figures 1–3). The diverse types of plant selection markers or reporter gene in the different pBTR vectors provide a convenient selection applied for different plant transformation events. The pBTR expression vectors require only a single FDI cloned into them, which is greatly convenient and highly efficient.
One limitation of Golden Gate cloning is to clone FDI carrying one or several internal restriction enzyme site(s), such as BsaI used, into the expression vector. Engler et al. (2008) suggested three possible methods to bypass this problem. The first strategy is to run the digestion–ligation program, inactivate the BsaI by heating, and then append new T4 ligase to the mixtures sequentially. This is efficiently validated (Engler et al., 2008; Emami et al., 2013), but it is a complex operation. In addition, the gel-purify step needs to be added in GreenGate system (Emami et al., 2013). The second strategy requires another set of expression cloning vectors that are different from the BsaI enzyme used, which is not recommended by Engler et al. (2008). The third strategy is to eliminate any internal BsaI restriction site(s) carried in the FDI, which is also time-consuming, complex, and laborious. In this study, using pBTR vectors and our modified digestion–ligation program, even FDI carries two internal BsaI recognition sites (exemplified by cloning of GUSPlus and GmbHLH293), which can also be highly efficiently cloned into the pBTR vector using our one-step digestion–ligation program (Figure 4). In our modified digestion–ligation program, inactivation of BsaI via heating and re-supplement of fresh ligase to the digestion–ligation mixtures are not required. FDI carrying one, or more recognition site(s) is to be cloned into the pBTR vector, in that case, after running the program of cycles, the final incubation temperature is set at 4°C for 1 h in order to religate the linearized FDI by BsaI. Hence, it is a simple operation to clone a single FDI carrying internal BsaI site(s) into the expression vector.
A DNA molecule carrying BsaI site(s) would produce a 4-nt overhang after digestion by BsaI; thus, theoretically, 256 (44) different sticky ends can be produced. BsaI recognizes six base pair recognition sequences and cuts the DNA molecule outside its recognition sequences at the 3’ terminus. Restriction sites are expected to be present on average every 4, 096 bp (46) in genomes. It is greatly low frequency to present the same sticky ends produced between an FDI (internal BsaI recognition site) and the pBTR vectors (410=1,048,576) because only a single FDI was cloned. In contrast, in expression vectors based on the GreenGate system, internal BsaI sites carried by FDIs can complicate cloning because digestion at such sites might generate the same sticky end sequences, thereby greatly decreasing the correctness and integrity of multi-component DNA fragments into the destination vector. In this study, to avoid this issue, we developed two sets of pBTR vector, each of which carried a different neighbor sequence flanked by BsaI recognition sites and would thus generate different sticky ends. If a particular FDI with a BsaI recognition sequence has the same sticky end as that of the pBTR1 set, in such case, the pBTR2 set can be used. That is, any two sets of pBTR vector will be suitable for almost all FDIs and are also sequence-independent.
We modified the digestion–ligation procedure of Engler et al. (2008) as more time cycles replacing a single digestion–ligation reaction. In our procedure, in each cycle, the FDI and original pBTR vector are digested by BsaI at 37°C for 3 min, then the completely digested FDI and pBTR vector are recombined as a circular DNA at 16°C for 5 min; at which point another gradient ligation at 12°C for 2 min is run. In the subsequent cycles, if the FDI is cloned into the original destination pBTR vector, the BsaI recognition sequences in that vector would be eliminated in the novel recombinant DNAs. Therefore, it would not be possible to re-cut these DNAs in subsequent cycles. The left non-recombinant original pBTR vector would continue to be cut over and over again by BsaI in the following digestion steps. It doesn’t be re-digested in the digestion step after FDI is recombined into the pBTR vector. This creates a one-way digestion–ligation reaction in one tube. Even FDI carries internal BsaI recognition site(s), the digested FDI caused in the digestion step can be re-ligated into an integrated DNA molecular again in the final ligation step at 4°C for 1 h. Therefore, the FDI with internal BsaI recognition site(s) can also efficiently be cloned into the pBTR vector (different overhangs at 5’-end and 3’-end with the vector). Furthermore, these vectors can be used to clone multiple FDIs to construct corresponding multiple expression vectors in a single-step digestion–ligation and transformation event. Hence, the pBTR vectors are cost-efficient, easy-to-use, and highly efficient.
A marker or reporter gene is often used to facilitate the screening of positive recombinants in E. coli. To this end, antibiotic genes such as lac-Z or ccdb are commonly used (Xing et al., 2014; Ma et al., 2015). However, this requires additional chemical reagents. Emami et al. (2013) developed three plant expression vectors based on Golden Gate cloning using lac-Z as a selective marker in E. coli, which requires the chemical reagent X-gal. The fluorescent proteins, such as GFP, YFP, RFP, CFP, etc., are often used as a reporter in screening of transgenic plants in various plant species (Fan et al., 2020b). However, to date, no such expression vector carrying fluorescent protein has been found and be used as an indicator in E. coli to select recombinant colonies by the unaided eye. Recently, it was reported that mScarlet-I is a monomeric red fluorescent protein with fast maturity time and high brightness (Bindels et al., 2017; Mccullock et al., 2020). In this study, we developed pBTR vectors carrying mScarlet-Ⅰ to enable to visualize recombinant colonies by the naked eye without a need for additional substrate/reagents or special E. coli strains. This is greatly convenient for screening positive colonies. Additionally, to facilitate the user to recombine expression-related modules or special tags with flexibility due to special destination or designation in need, we also developed a set of p-SBTR1/2 vectors with a visual mScarlet-I selection marker that can meet the requirements (Figure S4).
Some crucial steps to consider when using the pBTR vectors
To achieve directional cloning, special adapters added at the 5’-end of primers for different destination vectors should be selected carefully (listed in Table 1). To clone an FDI into the pBTR vectors, for one thing, as for no internal BsaI recognition site(s) presented in the FDI, both pBTR1 and pBTR2 sets can be used. For another, one or more internal BsaI site(s) presented in the FDI, the same sticky ends produced by BsaI between FDI and vector should be avoided. In the digestion–ligation reaction program (Figure 4), the final incubation temperature is held at 4°C for 1 h. This is done with the aim to religate the linearized FDI (with internal BsaI sites) by BsaI. Generally, the molecular molar ratio of FDI to vector should be 1:1 when no internal BsaI site(s) is carried by the FDI or when different non-palindromic sticky ends are produced with internal BsaI site(s). We recommend that the molar ratio of foreign DNA to vector should be 3–5:1 for palindrome sticky end(s) generated from FDI. To reduce costs without sacrificing BsaI digestion efficiency, we recommend adding four protective bases to the 5’ terminus of primers.
Conclusion
A novel series of plant binary expression vectors, termed pBTR vectors, were developed. The pBTR vectors were used with a highly simplified cloning step, single BsaI restriction enzyme required, DNA purification-free, almost sequence-independent, directional cloning, multiple target genes cloning, cost-effective, and directly visual selection marker for screening positive recombinants colonies with the unaided eye. We anticipate that these vectors will be widely used for analyses of gene (over)expression, promoter activity, and protein subcellular localization. These vectors will promote functional genomics research in plants and are free for users by non-profit researchers.
Methods
Plant materials and growth conditions
Seeds of soybean (Glycine max cv. Williams82), tomato (Solanum lycopersicum, local variety Maofen802) , Arabidopsis thaliana Columbia (Col-0), and transgenic Nicotiana benthamiana were kept in our lab. The N. benthamiana was transformed with a mCherry fluorescent protein marker fused with nuclear localization peptide signal of SV40 (Simian virus 40) large T antigen (van der Krol and Chua, 1991). All plants were cultivated in a growth chamber (24–26°C, 16 h light/ 8 h dark cycle).
Construction of gene (over)expression vectors p-35BTR1/2, p-UbiBTR1/2, and p-NatBTR1/2
To construct p-35BTR1/2 and p-UbiBTR1/2, the CRISPR/Cas9 vectors pHSE401/pKSE401 and pYLCRISPR/Cas9Pubi-H/B/N (Ma et al., 2015) were used. To remove the sequences between the two HindIII sites (including two BsaI recognition sequences, the SpR-gRNA-scaffold, and the expression cassette of SmR), the pHSE401/pKSE401 vectors were digested with HindIII at 37°C for 30 min in 1×T4 ligation buffer followed by 65°C for 15 min (HindIII inactivated), and then 10 U T4 ligase was added to the reaction mixtures and held at 16°C for 1 h to religate the vectors. The digestion–ligation mixtures were transformed into chemically competent TreliefTM5α (>1010cfu/ug, Tsingke Biotech Co., Ltd., Qingdao, China) E. coli cells via heat shock at 42°C for 60 s. The two generated vectors, in which 1919 bp sequences between the two HindIII sites were deleted (primary regions 268–2187 bp in pHSE401 [Addgene Plasmid #62201], and 268–2187 bp in pKSE401 [Addgene Plasmid #62202]; Xing et al., 2014), were termed pH35Cas9 and pK35Cas9, respectively. To construct the pH35BTR1/pK35BTR1 and pH35BTR2/pK35BTR2 vectors (Figure 1A), the Cas9 gene was replaced with the cassette of PnptⅡ:: mScarlet-I based on pH35Cas9 and pK35Cas9. The nptII promoter (PnptⅡ) and mScarlet-I coding sequences were amplified via PCR with two primer sets, mSXb1F/mSSa1R and mSXb2F/mSSa2R, respectively, using pMRE-Tn5-155 (Addgene Plasmid #118537) (Schlechter et al., 2018) as the template. The PCR products were digested using restriction enzymes XbaI and SacI for cloning into pH35Cas9 and pK35Cas9 that were previously digested by XbaI and SacI, thereby producing pH35BTR1/pK35BTR1 and pH35BTR2/pK35BTR2, respectively (Figure 1A).
To construct vectors p-35BTR1/2 with various selected markers, a DsRed gene was used to replace the hygromycin-resistance gene in the pH35BTR1 and pH35BTR2 vectors. DsRed (from pRed1305 plasmid; Fan et al., 2020c) was cloned into pH35BTR1 and pH35BTR2 between the EcoRI and SacII restriction sites using EcoRI and SacII digestion, respectively, thereby generating pR35BTR1 and pR35BTR2 (Figure 1A).
To construct p-UbiBTR1/2 (Figure 1B), pYLCRISPR/Cas9Pubi-H/B/N backbones were used (Ma et al., 2014). The mScarlet gene and E9 terminator cassettes were amplified via PCR with primer sets mSYL1/2 and mSYL2/3 using plasmids pR35BTR1 and pR35BTR2 as the templates with the High-Fidelity DNA Polymerase Enzyme KOD-plus kit (TOYOBO Biotech, Osaka, Japan), respectively. The two PCR products were digested with PstI for cloning into pYLCRISPR/Cas9Pubi-H/B/N that was previously digested by PstI and PmeI. These recombinant vectors were termed as pHUbiBTR1/2, pBUbiBTR1/2, and pKUbiBTR1/2, respectively (Figure 1B).
To construct expression vectors p-NatBTR1/2 (Figure 1C) driven by the native promoter, the mScarlet-I expression cassette was amplified by PCR with two primer sets, mSHin3/mSSa1R and mSHi32/mSSa4R, respectively, using pMRE-Tn5-155 as the template. The PCR products were digested with HindIII and SacI for cloning into pH35Cas9, pK35Cas9, and pR35BTR1 that were previously digested with HindIII and SacI, respectively, creating the expression vectors pHNatBTR1/2, pKNatBTR1/2, and pRNatBTR1/2 (Figure 1C).
To construct the pK35BTR1-AtMyb75, pK35BTR2-GUSPlus, and pR35BTR2-GmbHLH293 vectors, AtMyb75, GUSPlus, and GmbHLH293 were amplified via PCR with the primers At75BF/At75BR (for AtMyb75), GUS1F/GUS1R (for GUSPlus), and Gmb293F/Gmb293R (for GmbHLH293), using p35SAt75 plasmid (Fan et al., 2020b), pCAMBIA1305.1 plasmid, and Williams82 genomic DNA as templates, respectively. The PCR products were digested with BsaI for cloning into pK35BTR1, pK35BTR2, and pR35BTR2, thereby generating pK35BTR1-AtMyb75, pK35BTR2-GUSPlus, and pR35BTR2-GmbHLH293 vectors, respectively. The one-step restriction digestion–ligation reaction was performed as follows. Vector p-35BTR1/2 (~20 ng), PCR products (~20 ng), BsaI (10U, New England Biolabs, Ipswich, MA, USA, #R3535L), T4 ligase (10U, New England Biolabs, Ipswich, MA, USA, #M0202L), and 1×NEB rCutsmart buffer were mixed with 1mM ATP in one tube. The one-step restriction digestion–ligation reaction was performed in a thermo-cycler as follows: 10 cycles at 37°C for 3 min, at 16°C for 3 min, and at 12°C for 2 min, followed by a final incubation step at 4°C for 1 h (to clone GUSPlus and GmbHLH293 into pK35BTR2 and pR35BTR2) or at 37°C for 10 min (to clone AtMyb75 into pK35BTR1). Subsequently, the digestion–ligation mixtures were transformed into competent TreliefTM5α. The white colonies were selected to identify the positive recombinants. The recombinant plasmids were further tested using HindIII and XbaI (for pK35BTR1-AtMyb75) and HindIII (for pK35BTR2-GUSPlus and pR35BTR2-GmbHLH293), and Sanger sequencing, respectively.
Construction of promoter analysis vectors pKBTR1/2PGUS, pHBTR1/2PGUS, and pRBTR1/2PGUS
To construct pKBTR1/2PGUS, pHBTR1/2PGUS, and pRBTR1/2PGUS (Figure 2), GUSPlus gene (without internal BsaI recognition site) was used to replace the expression cassette PnptⅡ::mScarlet-I in the pH35BTR1 vector, following the protocol described in Figure 4. GUSPlus was amplified via PCR using the primers GUS2F and GUS2R with pGSE401 plasmid (Fan et al., 2020a) as a template, generating the intermediate vectors pH35GUS. The expression cassette of PnptⅡ::mScarlet-I was amplified with primers mSHin3 and mSXba1(to produce pHBTR1G), and primers mSHi32 and mSXb12 (to produce pHBTR2G), using pR35BTR2 as a template. The PCR products were digested with HindIII and XbaI and directly ligated into pH35GUS, which were previously digested with HindIII and XbaI, and produced two mediate vectors, pHBTR1G and pHBTR2G, respectively.
Because the 35S enhancer sequences presented in pHBTR1G and pHBTR2G vectors may influence the expression activity of nearby promoters, we used the double 35S to drive the expression of selection reporter gene/marker (HygR, KanR, and DsRed) and replaced the enhanced CaMV35S promoter sequences in the pHBTR1G and pHBTR2G. Two oligos DNA, MCS41 and MCS42, were chemically synthesized and annealed at 65°C for 5 min. Then the annealed DNA fragment was inserted into the site of HindIII of pH35BTR1, and thus produced the new intermediate vector pH35BTR1MCS, which was introduced a restriction endonuclease site of EcoRI. Three primer sets, HygS1 and HygS2, KanS1 and KanS2, RedS1 and RedS2, were used to amplify HygR, KanR, and DsRed gene, respectively. The three responding PCR products were mixed with pH35BTR1MCS added with XhoI, SalI, and T4 DNA ligase in 1×NEB rCutsmart buffer (with 1 mM ATP), respectively. The one-step digestion–ligation reaction was performed as follows: 10 cycles at 37°C for 3 min, at 16°C for 3 min, and at 12°C for 2 min, followed by at 37°C for 10 min. The generated three new vectors were named p35HygM, p35KanM, and p35RedM, respectively. The pHBTR1G and pHBTR2G were digested with HindIII and EcoRI, generated a fragment of 3846-bp and 3851-bp, respectively, and introduced into between HindIII and EcoRI of p35HygM, p35KanM, and p35RedM, and produced the corresponding promoter analysis vectors, pHBTR1/2PGUS, pKBTR1/2PGUS, and pRBTR1/2PGUS produced (Figure 2).
To construct pRBTR1PGUS-AtGCS, a 1178-bp upstream promoter region of the translation start site of the gamma-glutamylcysteine synthetase gene (GenBank accession no. AF068299.1) was amplified via PCR using primers AtGa4F and AtGa4R from A. thaliana Columbia (Col-0) genomic DNA as a template. The PCR products were cloned into pRBTR1PGUS using the protocol described in Figure 4 in this study.
Construction and verification of subcellular localization vectors
To construct subcellular localization vectors, pK35BTR1/2 was used as a backbone. The eGFP was amplified via PCR with a forward primer GFPB1 carrying a linker sequence, GGTGGATCCGGAGGTTCT, that codes a peptide linkage (GGSGGS) and a reverse primer GFPB2, using pNC-TRV2-GFP plasmid as a template (Yan et al., 2020). The eGFP gene was used to replace the expression cassette PnptⅡ::mScarlet-I in the pK35BTR1/2, generating the intermediate vector pKG according to the protocol in Figure 4. To introduce the expression cassette PnptⅡ::mScarlet-I into the pKG and produce the corresponding subcellular localization vector pK35BTR1/2GFP, the expression cassette PnptⅡ::mScarlet-I was amplified with primers mSXb1F/mSBam1 (to produce pK35BTR1GFP) and mSXb2F/mSBam2 (to produce pK35BTR2GFP), using pMRE-Tn5-155 plasmid as a template (Schlechter et al., 2018). The PCR products were digested with BamHI and XbaI, and directly ligated into pKG that was previously digested with BamHI and XbaI, thus producing pK35BTR1/2GFP (Figure 3).
The VvCEB1opt gene (Nicolas et al., 2013; Lim et al., 2018) was used to verify pK35BTR2GFP. VvCEB1opt without the stop codon was cloned into pK35BTR2GFP between the two BsaI cleavage sites to generate the pK35VvCEB-GFP fusion vector. VvCEB1opt was amplified via PCR with primers SuVvCF and SuVvCR from synthetic VvCEB1opt cDNA (kept in our lab) as a template. The digestion–ligation reaction was incubated for 10 cycles (37°C, 3 min; 16°C, 5 min; 12°C, 2 min) and then stayed at 37°C for 2 min (VvCEB1opt carrying no BsaI site).
Construction of binary expression vectors p-SBTR1/2 carrying no expression-related modules
Two PCR products were amplified with pR35BTR1 as a template by primers sets, mSHin3 and mSEco1, mSHi32 and mSEco2 (Table S1), and then were digested with HindIII and EcoRI for cloning into the same restriction sites of vectors, pK35Cas9, pH35Cas9, and pR35BTR1, respectively. The three newly generated vectors were named pSKBTR1/2, pSHBTR1/2 and pSRBTR1/2 (Figure S4).
Multiple fragments cloned into the pR35BTR2 to construct multiple expression vectors
To verify whether the pBTR1/2 vectors are suitable for constructing multiple expression vectors carrying different FDIs in a single reaction in a single transformation, four genes, Glyma.02g025400, Glyma.05g201700, Glyma.06g165700, and Glyma.17g095000, were cloned into pR35BTR2 according to the protocol described in Figure 4. The four genes were amplified using primers Gm2c4F/Gm2c4R, Gm5c7F/Gm5c7R, Gm6c7F/Gm6c7R, and Gm17c5F/Gm17c5R by PCR, respectively. The four PCR products were mixed (molar ratio=1:1:1:1) together with the pR35BTR2 vector (FDI: vector molar ratio=1:1). The digestion–ligation reaction was performed according to the protocol in Figure 4 with 10 cycles (37°C, 3 min; 16°C, 5 min; 12°C, 2 min) and then a holding period at 4°C for 1 h. All primers used in this study are listed in Table S1. All constructs were confirmed by Sanger sequencing. Entire sequences of all the pBTR vectors constructed in this paper are given in Supplementary Material Table S2 and the plasmids will be available via Addgene (https://bccm.belspo.be/deposit/public/plasmids).
Hairy root transformation mediated by A. rhizogenes and Arabidopsis transformation mediated by A. tumefaciens
The vectors pK35BTR1-AtMyb75 and pK35BTR2-GUSPlus were introduced into A. rhizogenes strain K599 by electroporation. Composite soybean and tomato plants were generated by one-step A. rhizogenes-mediated transformation as published previously (Fan et al., 2020a) with minor modification. To inoculate the K599 harboring the plasmid of interest, the step of watering the K599 with the transformed plasmid was removed. The pRBTR2PGUS-AtGCS construct described above was introduced into wild-type A. thaliana plants using the floral dip method (Clough and Bent, 1998).
GUS staining and subcellular localization assays
Histochemical staining of GUS activity assay followed the protocol (Jefferson et al., 1987; Fan et al., 2017; Fan et al., 2020c). Plant tissues materials were stained in the GUS staining solution (100 mM sodium phosphate at pH 7.0, 0.1% Triton X-100, 1 mg/mL X-Gluc, 1 mM potassium ferricyanide, and 1 mM potassium ferrocyanide) in the dark at 37°C for 5-10 h. The roots stained were rinsed in 70% ethanol for 10-20 min. Transient expression with subcellular localization vector was analyzed in the transgenic N. benthamiana following the protocol (Sparkes et al., 2006). pK35VvCEB-GFP was transformed into A. tumefaciens strain GV3101 by electroporation. The GFP fluorescence of tobacco leaves was observed and photographed using a confocal laser scanning microscope (Zeiss LSM 700, Germany) with a Fluar ×10/0.50 M27 objective lens and SP640 filter after 48 h cultivation (Carl Zeiss).
Reverse transcription-polymerase chain reaction analysis
The total RNA was isolated, first-strand cDNA was synthesized, and RT-PCR amplification was performed as previously described (Lü et al., 2010; Fan et al., 2020a). To analyze the expression of GUSPlus and AtMYB75 in the transgenic hairy roots of tomato and soybean transformed with pK35BTR1-AtMyb75 and pK35BTR2-GUSPlus, RT-PCR reaction was carried out in a reaction mixture to amplify the endogenous gene and AtMYB75 (tomato roots transformed with pK35BTR1-AtMyb75) or GUSPlus (soybean roots transformed with pK35BTR2-GUSPlus), respectively. The tomato SlEF (Solyc06g005060) (Liao et al., 2015) and soybean endogenous gene GmActin (Fan et al., 2020a) were used as a endogenous gene control, respectively. To analysis the transcript of GUSPlus in the 7-day-old seedlings of A. thaliania transformed with pRBTR1PGUS-AtGCS, RT-PCR reaction was performed in a reaction mixture to amplify the A. thaliania endogenous gene ACTIN (At3g18780) and exogenous GUSPlus. The gene-specific primer pairs SlEFF and SlEFR for SlEF (Liao et al., 2015), GmActinF and GmActinR for GmActin (Fan et al., 2020a), AtActinF and AtActinR for A. thaliania endogenous gene ACTIN, RTGusF and RTGusR for GUSPlus (Fan et al., 2020a), At751 and At752 for AtMyb75 were used. RT-PCR experiments were performed with three replicates. All primers sequences used in this paper are listed in Supplemental Table S1.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
YF and SHL designed the experiments and wrote the paper. XW, CT, HW, SL, HX, WP, QL, HH, and QYL performed the research work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (No. 31271751); Partial financial support was received from Industrial upgrading project of Shandong Agricultural Science and Technology (2019YQ035); and Innovation Project for an undergraduate to QYL (no. CYCY2022412).
Acknowledgments
We thank Prof. Yaoguang Liu and Letian Chen (South China Agricultural University) for providing the vectors pYLsgRNA-AtU3d and pYLCRISPR/Cas9Pubi-B/H/N, Prof. Qijun Chen (China Agricultural University) for pHSE401/pKSE401, and Dr. Pu Yan (Chinese Academy of Tropical Agricultural Sciences) for pNC-TRV2-GFP.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1104905/full#supplementary-material
Supplementary Figure 1 | Verification of positive recombinants in E. coli transformed with pR35BTR2-GmbHLH293 and analysis of cloning efficiency. Electrophoretogram of enzyme digestion of pR35BTR2-GmbHLH293 recombinant plasmids with HindIII. M, DL5000 DNA marker (bought from Tsingke Co., Qingdao, China); Lane 1, pR35BTR2 plasmid used as a control; Lanes 2–9, eight randomly selected pR35BTR2-GmbHLH293 plasmids isolated from white E. coli colonies (A). An example of Sanger sequencing analysis to verify the presence of GmbHLH293 fragment in the pR35BTR2 vector (partial sequences shown) (B). Analysis of cloning efficiency in E. coli transformed with pK35BTR1-AtMyb75 (C) and pK35BTR2-GUSPlus (D).
Supplementary Figure 2 | RT-PCR assays on transgenic hairy roots/plants expressing AtMyb75/PAP1 or GUS. Expression of AtMyb75/PAP1 in transgenic hairy roots. Lanes 1-2, No Purple/red colored anthocyanin accumulated on non-transgenic root inoculated K599 with pK35BTR1-AtMyb75 plasmid; Lane 3, pK35BTR1-AtMyb75 plasmid; Lanes 4-8, Purple/red colored anthocyanin accumulated on independent transgenic root inoculated K599 with pK35BTR1-AtMyb75 plasmid; Lane 9, A. rhizogenesis K599; Lane 10, ddH2O (A); RT-PCR analysis on transgenic hairy roots expressing GUSPlus. Lanes 1-2, GUS-negative non-transgenic root inoculated K599 with pK35BTR2-GUSPlus plasmid; Lane 3, pK35BTR2-GUSPlus plasmid; Lanes 4-8, GUS-positive independent transgenic root inoculated K599 with pK35BTR2-GUSPlus plasmid; Lane 9, A. rhizogenesis K599; Lane 10, ddH2O (B); RT-PCR analysis on GUSPlus transgene expression in the A. thaliana plants transformed with pRBTR1PGUS-AtGCS. Lane 1-2, ddH2O; Lanes 3-7, Independent GUS-positive transgenic A. thaliana line; Lane 8, GUS-negative transgenic A. thaliana line; Lane 9, pRBTR1PGUS-AtGCS plasmid (C).
Supplementary Figure 3 | Validation of multiple fragments cloned into the pR35BTR2 expression vector. Electrophoretogram of enzyme digestion of plasmid DNA extracted from 10 white colonies digested with HindIII. M, DL5000 DNA marker; Lanes 1, 8, pR35BTR2-Glyma.06g165700; Lanes 2, 5, 7, pR35BTR2-Glyma.05g201700; Lanes 3, 6, 10, pR35BTR2-Glyma.17g095000; Lanes 4, 9, pR35BTR2-Glyma.02g025400 (A). Examples of Sanger sequencing analyses of candidate FDIs cloned into the pR35BTR2 expression vector (B).
Supplementary Figure 4 | Diagram of p-SBTR1/2 vectors.
Abbreviations
- FDI, foreign DNA fragment of interest; RFP/GFP, red/ green fluorescent protein; GUS, β-glucuronidase; KanR, kanamycin resistance gene; HygR, hygromycin resistance gene; BarR, basta resistance gene; pBTR1/2, pBTR1 and pBTR2.
References
Aslanidis, C., de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18 (20), 6069–6074. doi: 10.1093/nar/18.20.6069
Barton, K. A., Binns, A. N., Matzke, A. J., Chilton, M. D. (1983). Regeneration of intact tobacco plants containing full length copies of genetically engineered T-DNA, and transmission of T-DNA to R1 progeny. Cell 32 (4), 1033–1043. doi: 10.1016/0092-8674(83)90288-x
Belknap, W., Rockhold, D., McCue, K. (2008). pBINPLUS/ARS: an improved plant transformation vector based on pBINPLUS. Biotechniques 44 (6), 753–756. doi: 10.2144/000112731
Benoit, R. M., Wilhelm, R. N., Scherer-Becker, D., Ostermeier, C. (2006). An improved method for fast, robust, and seamless integration of DNA fragments into multiple plasmids. Protein Expr. Purif. 45, 66–71. doi: 10.1016/j.pep.2005.09.022
Bevan, M. (1984). Binary Agrobacterium vectors for plant transformation. Nucleic Acids Res. 12 (22), 8711–8721. doi: 10.1093/nar/12.22.8711
Bindels, D. S., Haarbosch, L., van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14 (1), 53–56. doi: 10.1038/nmeth.4074
Clough, S. J., Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16 (6), 735–743. doi: 10.1046/j.1365-313x.1998.00343.x
Curtis, M. D., Grossniklaus, U. (2003). Gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol. 133 (2), 462–469. doi: 10.1104/pp.103.027979
Emami, S., Yee, M., Dinneny, J. R. (2013). A robust family of golden gate Agrobacterium vectors for plant synthetic biology. Front. Plant Sci. 4. doi: 10.3389/fpls.2013.00339
Engler, C., Kandzia, R., Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PloS One 3 (11), e3647. doi: 10.1371/journal.pone.0003647
Fan, Y. L., Liu, J. G., Lyu, S. H., Wang, Q., Yang, S. M., Zhu, H. Y. (2017). The soybean Rfg1 gene restricts nodulation by Sinorhizobium fredii USDA193. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01548
Fan, Y. L., Wang, X. Y., Li, H. Y., Liu, S. R., Jin, L. S., Lyu, Y. Y., et al. (2020b). Anthocyanin, a novel and user-friendly reporter for convenient, non-destructive, low cost, directly visual selection of transgenic hairy roots in the study of rhizobia-legume symbiosis. Plant Methods 16, 94. doi: 10.1186/s13007-020-00638-w
Fan, Y. L., Xu, F. L., Zhou, H. Z., Liu, X. X., Yang, X. Y., Weng, K. X., et al. (2020c). A fast, simple, high efficient and one-step generation of composite cucumber plants with transgenic roots by Agrobacterium rhizogenes-mediated transformation. Plant Cell Tiss. Org. (PCTOC) 141, 207–216. doi: 10.1007/s11240-020-01781-x
Fan, Y. L., Zhang, X. H., Zhong, L. J., Wang, X. Y., Lyu, S. H. (2020a). One-step generation of composite soybean plants with transgenic roots by Agrobacterium rhizogenes-mediated transformation. BMC Plant Biol. 20 (1), 208. doi: 10.1186/s12870-020-02421-4
Fu, J., Bian, X., Hu, S., Wang, H. L., Huang, F., Seibert, P. M., et al. (2012). Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 30, 440. doi: 10.1038/nbt.2183
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., 3rd., Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6 (5), 343–345. doi: 10.1038/nmeth.1318
Hajdukiewicz, P., Svab, Z., Maliga, P. (1994). The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol. Biol. 25 (6), 989–994. doi: 10.1007/BF00014672
Hao, H. J., Jiang, Y. Q., Zheng, Y. L., Ma, R., Yu, D. W. (2005). Improved stability and yield of fv targeted superantigen by introducing both linker and disulfide bond into the targeting moiety. Biochimie 87 (8), 661–667. doi: 10.1016/j.biochi.2005.04.005
Hartley, J. L., Temple, G. F., Brasch, M. A. (2000). DNA Cloning using in vitro site-specific recombination. Genome Res. 10 (11), 1788–1795. doi: 10.1101/gr.143000
Hellens, R. P., Edwards, E. A., Leyland, N. R., Bean, S., Mullineaux, P. M. (2000). pGreen: a versatile and flexible binary Ti vector for Agrobacterium-mediated plant transformation. Plant Mol. Biol. 42 (6), 819–832. doi: 10.1023/a:1006496308160
Jefferson, R., Kavanagh, T., Bevan, M. (1987). GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907. doi: 10.1002/j.1460-2075.1987.tb02730.x
Kamal, N., Tsardakas, R. N., Bentzer, J., Gundlach, H., Haberer, G., Juhász, A., et al. (2022). The mosaic oat genome gives insights into a uniquely healthy cereal crop. Nature 606 (7912), 113–119. doi: 10.1038/s41586-022-04732-y
Karimi, M., Depicker, A., Hilson, P. (2007). Recombinational cloning with plant gateway vectors. Plant Physiol. 145 (4), 1144–1154. doi: 10.1104/pp.107.106989
Lampropoulos, A., Sutikovic, Z., Wenzl, C., Maegele, I., Lohmann, J. U., Forner, J. (2013). GreenGate- a novel, versatile, and efficient cloning system for plant transgenesis. PloS One 8 (12), e83043. doi: 10.1371/journal.pone.0083043
Liao, D., Chen, X., Chen, A., Wang, H., Liu, J., Liu, J., et al. (2015). The characterization of six auxin-induced tomato GH3 genes uncovers a member, SlGH3.4, strongly responsive to arbuscular mycorrhizal symbiosis. Plant Cell Physiol. 56 (4), 674–687. doi: 10.1093/pcp/pcu212
Li, M. Z., Elledge, S. J. (2007). Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4 (3), 251–256. doi: 10.1038/nmeth1010
Lim, S. D., Yim, W. C., Liu, D., Hu, R. B., Yang, X. H., Cushman, J. C. (2018). A Vitis vinifera basic helix–loop–helix transcription factor enhances plant cell size, vegetative biomass and reproductive yield. Plant Biotechnol. J. 16, 1595–1615. doi: 10.1111/pbi.12898
Liu, S., Wang, X. Y., Li, Q. Q., Peng, W. T., Zhang, Z. M., Chu, P. F., et al. (2022). AtGCS promoter-driven CRISPR/Cas9 highly efficiently generates homozygous/biallelic mutations in the transformed roots by Agrobacterium rhizogenes-mediated transformation. Front. Plant Sci. 18. doi: 10.3389/fpls.2022.952428
Lü, S. H., Fan, Y. L., Liu, L. K., Liu, S. J., Zhang, W. H., Meng, Z. (2010). Ectopic expression of TrPI, a Taihangia rupestris (Rosaceae) PI ortholog, causes modifications of vegetative architecture in Arabidopsis. J. Plant Physiol. 167 (18), 1613–1621. doi: 10.1016/j.jplph.2010.06.028
Lu, J., Luo, M., Wang, L., Li, K., Yu, Y., Yang, W., et al. (2021). The Physalis floridana genome provides insights into the biochemical and morphological evolution of physalis fruits. Hortic. Res. 8, 244. doi: 10.1038/s41438-021-00705-w
Marks, R. A., Hotaling, S., Frandsen, P. B., VanBuren, R. (2021). Representation and participation across 20 years of plant genome sequencing. Nat. Plants 7, 1571–1578. doi: 10.1038/s41477-021-01031-8
Ma, X., Zhang, Q., Zhu, Q., Liu, W., Chen, Y., Qiu, R., et al. (2015). A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 8 (8), 1274–1284. doi: 10.1016/j.molp.2015.04.007
Mccullock, T. W., Maclean, D. M., Kammermeier, P. J. (2020). Comparing the performance of mScarlet-I, mRuby3, and mCherry as FRET acceptors for mNeonGreen. PloS One 15 (2), e0219886. doi: 10.1371/journal.pone.0219886
Mosberg, J. A., Lajoie, M. J., Church, G. M. (2010). Lambda red recombineering in Escherichia coli occurs through a fully single-stranded intermediate. Genetics 186, 791–799. doi: 10.1534/genetics.110.120782
Motohashi, K. (2015). A simple and efficient seamless DNA cloning method using SLiCE from Escherichia coli laboratory strains and its application to SLiP site-directed mutagenesis. BMC Biotechnol. 15, 47. doi: 10.1186/s12896-015-0162-8
Nakagawa, T., Kurose, T., Hino, T., Tanaka, K., Kawamukai, M., Niwa, Y., et al. (2007). Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J. Biosci. Bioeng. 104 (1), 34–41. doi: 10.1263/jbb.104.34
Nicolas, P., Lecourieux, D., Gomès, E., Delrot, S., Lecourieux, F. (2013). The grape berry-specific basic helix–loop–helix transcription factor VvCEB1 affects cell size. J. Exp. Bot. 64 (4), 991–1003. doi: 10.1093/jxb/ers374
Peng, Y., Yan, H., Guo, L., Deng, C., Wang, C., Wang, Y., et al. (2022). Reference genome assemblies reveal the origin and evolution of allohexaploid oat. Nat. Genet. 54 (8), 1248–1258. doi: 10.1038/s41588-022-01127-7
Schlechter, R. O., Jun, H., Bernach, M., Oso, S., Boyd, E., Muñoz-Lintz, D. A., et al. (2018). Chromatic bacteria - a broad host-range plasmid and chromosomal insertion toolbox for fluorescent protein expression in bacteria. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.03052
Sleight, S. C., Bartley, B. A., Lieviant, J. A., Sauro, H. M. (2010). In-fusion BioBrick assembly and re-engineering. Nucleic. Acids Res. 38 (8), 2624–2636. doi: 10.1093/nar/gkq179
Sparkes, I. A., Runions, J., Kearns, A., Hawes, C. (2006). Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1 (4), 2019–2025. doi: 10.1038/nprot.2006.286
Sun, Y., Shang, L., Zhu, Q. H., Fan, L., Guo, L. (2022). Twenty years of plant genome sequencing: achievements and challenges. Trends Plant Sci. 27 (4), 391–401. doi: 10.1016/j.tplants.2021.10.006
Thole, V., Worland, B., Snape, J. W., Vain, P. (2007). The pCLEAN dual binary vector system for Agrobacterium-mediated plant transformation. Plant Physiol. 145 (4), 1211–1219. doi: 10.1104/pp.107.108563
van der Krol, A. R., Chua, N. H. (1991). The basic domain of plant b-ZIP proteins facilitates import of a reporter protein into plant nuclei. Plant Cell 3, 667–675. doi: 10.1105/tpc.3.7.667
van Engelen, F. A., Molthoff, J. W., Conner, A. J., Nap, J. P., Pereira, A., Stiekema, W. J. (1995). pBINPLUS: an improved plant transformation vector based on pBIN19. Transgenic Res. 4 (4), 288–290. doi: 10.1007/BF01969123
Walhout, A. J., Temple, G. F., Brasch, M. A., Hartley, J. L., Lorson, M. A., van den Heuvel, S., et al. (2000). GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 328, 575–592. doi: 10.1016/s0076-6879(00)28419-x
Watson, J. F., García-Nafría, J. (2019). In vivo DNA assembly using common laboratory bacteria: A re-emerging tool to simplify molecular cloning. J. Biol. Chem. 294 (42), 15271–15281. doi: 10.1074/jbc.REV119.009109
Xing, H. L., Dong, L., Wang, Z. P., Zhang, H. Y., Han, C. Y., Liu, B., et al. (2014). A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 14, 327. doi: 10.1186/s12870-014-0327-y
Yan, P., Zeng, Y., Shen, W., Tuo, D., Li, X., Zhou, P. (2020). Nimble cloning: A simple, versatile, and efficient system for standardized molecular cloning. Front. Bioeng. Biotech. 7. doi: 10.3389/fbioe.2019.00460
Zhang, Y., Werling, U., Edelmann, W. (2012). SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic. Acids Res. 40, e55. doi: 10.1093/nar/gkr1288
Zheng, S. J., Henken, B., Sofiari, E., Jacobsen, E., Krens, F. A., Kik, C. (2001). Molecular characterization of transgenic shallots (Allium cepa l.) by adaptor ligation PCR (AL-PCR) and sequencing of genomic DNA flanking T-DNA borders. Transgenic Res. 10 (3), 237–245. doi: 10.1023/a:1016633410041
Zhu, B., Cai, G., Hall, E. O., Freeman, G. J. (2007). In-fusion assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. Biotechniques 43 (3), 354–359. doi: 10.2144/000112536
Keywords: plant binary expression vector, mScarlet-I gene, visible selection marker, golden gate cloning, one-step digestion-ligation reaction
Citation: Wang X, Teng C, Wei H, Liu S, Xuan H, Peng W, Li Q, Hao H, Lyu Q, Lyu S and Fan Y (2023) Development of a set of novel binary expression vectors for plant gene function analysis and genetic transformation. Front. Plant Sci. 13:1104905. doi: 10.3389/fpls.2022.1104905
Received: 22 November 2022; Accepted: 21 December 2022;
Published: 12 January 2023.
Edited by:
Kaijun Zhao, Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Xingguo Ye, Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, ChinaZhe Yan, Chinese Academy of Agricultural Sciences (CAAS), China
Zeng-Fu Xu, Guangxi University, China
Copyright © 2023 Wang, Teng, Wei, Liu, Xuan, Peng, Li, Hao, Lyu, Lyu and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shanhua Lyu, bHZzaGFuaHVhQGxjdS5lZHUuY24=; bHZzaGFuaHVhQGZveG1haWwuY29t; Yinglun Fan, ZmFueWluZ2x1bkBsY3UuZWR1LmNu
ORCID: Shanhua Lyu, orcid.org/0000-0002-1895-3614
Yinglun Fan, orcid.org/0000-0002-3760-2502
†Present address: Huitian Wei, Faculty of Veterinary and Agricultural Sciences, University of Melbourne, Parkville VIC, Australia
‡These authors have contributed equally to this work
§These authors share senior authorship