 Mélanie Massonnet
Mélanie Massonnet Summaira Riaz
Summaira Riaz Dániel Pap
Dániel Pap Rosa Figueroa-Balderas
Rosa Figueroa-Balderas Dario Cantu
Dario Cantu- Department of Viticulture and Enology, University of California Davis, Davis, CA, United States
Multiple grape powdery mildew (PM) genetic resistance (R) loci have been found in wild grape species. Little is known about the defense responses associated with each R locus. In this study, we compare the defense mechanisms associated with PM resistance in interspecific crosses segregating for a single R locus from Muscadinia rotundifolia (Run1, Run1.2b, Run2.1, Run2.2), Vitis cinerea (Ren2), V. romanetii (Ren4D and Ren4U), and the interspecific hybrid Villard blanc (Ren3). By combining optical microscopy, visual scoring, and biomass estimation, we show that the eight R loci confer resistance by limiting infection at different stages. We assessed the defense mechanisms triggered in response to PM at 1 and 5 days post-inoculation (dpi) via RNA sequencing. To account for the genetic differences between species, we developed for each accession a diploid synthetic reference transcriptome by incorporating into the PN40024 reference homozygous and heterozygous sequence variants and de novo assembled transcripts. Most of the R loci exhibited a higher number of differentially expressed genes (DEGs) associated with PM resistance at 1 dpi compared to 5 dpi, suggesting that PM resistance is mostly associated with an early transcriptional reprogramming. Comparison of the PM resistance-associated DEGs showed a limited overlap between pairs of R loci, and nearly half of the DEGs were specific to a single R locus. The largest overlap of PM resistance-associated DEGs was found between Ren3+, Ren4D+, and Ren4U+ genotypes at 1 dpi, and between Ren4U+ and Run1+ accessions at 5 dpi. The Ren3+, Ren4D+, and Ren4U+ were also found to have the highest number of R locus-specific DEGs in response to PM. Both shared and R locus-specific DEGs included genes from different defense-related categories, indicating that the presence of E. necator triggered distinct transcriptional responses in the eight R loci.
1 Introduction
Most cultivated grapevines (Vitis vinifera ssp. vinifera) are highly susceptible to powdery mildew (PM), a disease caused by the ascomycete Erysiphe necator Schwein (syn. Uncinula necator). Erysiphe necator is an obligate biotrophic pathogen that can infect any green tissue of the host. PM infections lead to reduced yield, and impaired fruit composition and wine quality (Calonnec et al., 2004; Stummer et al., 2005). Genetic resistance to PM has been found in several wild grapes, such as the North American grapes Muscadinia rotundifolia and Vitis cinerea, the Chinese Vitis species V. piasezkii, V. romanetii, V. quinquangularis and V. pseudoreticulata, and some Central Asian accessions of V. vinifera ssp. sylvestris (Dry et al., 2019). So far, thirteen PM resistance (R) loci have been genetically mapped (Dry et al., 2019; Karn et al., 2021) and named Resistance to Uncinula necator (Run) or Resistance to Erysiphe necator (Ren). Different allelic forms have been found for some of Run and Ren loci (Dry et al., 2019; Massonnet et al., 2022).
For most PM-resistant grape accessions, resistance to E. necator is associated with a programmed cell death (PCD)-mediated response (Qiu et al., 2015; Dry et al., 2019). Because PCD occurs in epidermal cells post-penetration of E. necator, the recognition of E. necator’s effectors by intracellular nucleotide-binding leucine-rich repeat (NLR) proteins is likely the trigger of PCD (Qiu et al., 2015; Dry et al., 2019). Only one NLR gene associated with PM resistance, MrRUN1, has been characterized in M. rotundifolia G52 (Feechan et al., 2013) and candidate NLR genes associated with Run1.2b and Run2.2 have been proposed for M. rotundifolia Trayshed (Massonnet et al., 2022).
In plants, NLR activation leads to multiple cellular responses, such as the generation of reactive oxygen species (ROS), calcium oscillations, kinase activation, and an extensive transcriptional reprogramming resulting in the activation of defense-related mechanisms, including the biosynthesis of pathogenesis-related (PR) proteins and antimicrobial compounds, and cell wall modifications (Dangl et al., 2013; Lolle et al., 2020). NLR-triggered immunity generally involves PCD at the site of infection, which inhibits the development of the pathogen (Dangl et al., 2013; Lolle et al., 2020). In grapes, few studies have investigated the transcriptomic responses to E. necator infection in PM-resistant accessions (Weng et al., 2014; Amrine et al., 2015; Jiao et al., 2021). For example, comparative transcriptomics revealed that E. necator infection leads to diverse whole-genome transcriptional responses among Central Asian V. vinifera accessions carrying different allelic forms of the Ren1 locus (Amrine et al., 2015). Whether the resistance conferred by different R loci depends on the activation of the same or different defense responses is still an open question. Understanding the functional differences and overlap between R loci will help select the most functionally diverse R loci to develop new V. vinifera cultivars that combine durable resistance to PM and high-quality fruit production (Michelmore et al., 2013). However, comparing genome-wide transcriptional changes in response to PM between R loci from different Vitis species is challenging. Comparative transcriptomics using RNA sequencing (RNA-seq) data relies on a reference transcriptome to evaluate transcript abundance. Recent studies showed that grape genomes are highly heterozygous and substantially differ between Vitis species (Zhou et al., 2019; Liang et al., 2019; Cochetel et al., 2021; Minio et al., 2022). A single haploid reference transcriptome from a PM-susceptible V. vinifera biases the alignment of the RNA-seq reads, underestimating the expression of alleles that are less similar to the reference, and confounding the subsequent testing of differential gene expression.
In this study, we aimed to identify the defense mechanisms associated with eight R loci (Ren2, Ren3, Ren4D, Ren4U, Run1, Run1.2b, Run2.1, and Run2.2) and to evaluate the functional differences and overlap between them. First, we monitored PM disease development on leaves of eight PM-resistant breeding lines, each representing a genetic R locus, as well as four PM-susceptible sib lines and two PM-susceptible V. vinifera parents. Defense mechanisms associated with each R locus were then assessed by profiling the leaf transcriptome with or without E. necator inoculations. To enable the comparison of the transcriptional modulation in response to PM, we constructed comparable reference transcriptomes incorporating both sequence variant information into the reference transcriptome of PN40024 and de novo assembled transcripts. We also refined the functional annotation of PN40024 predicted proteome to focus our analysis on genes involved in defense mechanisms. Defense-related genes differentially expressed in response to the pathogen were first compared between PM-resistant and PM-susceptible accessions to identify the genes with a transcriptional modulation associated with PM resistance. The latter ones were then compared between PM-resistant genotypes to evaluate the functional overlap between the different R loci.
2 Materials and methods
2.1 Plant material and evaluation of PM development
Fourteen grape accessions were used in this study: eight carrying one R locus, four sib lines without any R locus, and two susceptible V. vinifera parents. Information about the pedigree of each accession is provided in Supplementary Table 1.
PM susceptibility was evaluated with a detached leaf assay as described in Pap et al. (2016). Detached leaves were stained with Coomassie Brilliant Blue R-250 at 5 days post inoculation (dpi) as in Riaz et al. (2013). Visual disease susceptibility scores at 14 dpi were compared using a Kruskal-Wallis test followed by a post hoc Dunn’s test (P ≤ 0.05).
For transcriptional profiling, we followed the inoculation protocol described in Amrine et al. (2015). For each accession three plants were inoculated with E. necator C-strain (Jones et al., 2014) and three plants were mock-inoculated. Two leaves from each plant were collected 1 and 5 dpi and immediately frozen in liquid nitrogen. Leaves from an individual plant were pooled together and constitute a biological replicate. Three biological replications were obtained for each treatment.
2.2 RNA extraction, library preparation, and sequencing
RNA extraction and library preparation were performed as in Amrine et al. (2015). cDNA libraries were sequenced using Illumina HiSeq2500 and HiSeq4000 sequencers (DNA Technologies Core, University of California, Davis, CA, USA) as 50-bp single-end reads (Supplementary Table 2). Sequencing reads of the accessions e6-23 (Run1.2b+) and 08391-29 (Run2.2+) were retrieved from the NCBI BioProject PRJNA780568.
2.3 Reconstruction of accession-specific reference transcriptomes
For each accession, we constructed a reference transcriptome composed of a diploid synthetic transcriptome and de novo assembled transcripts. The diploid synthetic transcriptome was reconstructed using sequence variant information. First, adapter sequences were removed and RNA-seq reads were filtered based on their quality using Trimmomatic v.0.36 (Bolger et al., 2014) and these settings: LEADING:3 TRAILING:3 SLIDINGWINDOW:10:20 MINLEN:20. Quality-trimmed reads of the 12 samples from each accession were concatenated into a single file and mapped onto a combined reference genome composed of V. vinifera PN40024 V1 (Jaillon et al., 2007) and E. necator C-strain genome (Jones et al., 2014) following the STAR 2-pass mapping protocol (v.2.5.3a; Dobin et al., 2013; Engström et al., 2013). PCR and optical duplicates were removed with Picard tools (v.2.0.1 http://broadinstitute.github.io/picard/), and reads were split into exon segments using SplitNCigarReads from GATK v.3.5-0-g36282e4 (McKenna et al., 2010). GATK HaplotypeCaller was used to call sequence variants with the following parameters: -ploidy 2 -stand_call_conf 20.0 -stand_emit_conf 20.0 -dontUseSoftClippedBases. Variants were filtered using GATK VariantFiltration with these settings: -window 35 -cluster 3 -filterName FS -filter “FS > 30.0” -filterName QD -filter “QD < 2.0”. Variants passing all filters were selected using GATK SelectVariants with “–excludeFiltered” parameter. On average, 265,900 ± 39,570 variants were detected per genotype (Supplementary Table 3). Variants in grape protein-coding regions were extracted using bedtools intersect v.2.19.1 (Quinlan, 2014). Two separate genomes were reconstructed for each genotype using the vcf-consensus tool from vcftools (Danecek et al., 2011). The first genome was reconstructed by incorporating both homozygous and heterozygous alternative 1 (ALT1) variants relative to the PN40024 genome, while the second genome was reconstructed using both homozygous and heterozygous alternative 2 (ALT2) variants. A new annotation file was created for each genome from its corresponding variant information. CDS were extracted using gffread from Cufflinks v.2.2.1 (Trapnell et al., 2010).
For each genotype, quality-trimmed reads were mapped onto their respective diploid synthetic grape transcriptome and E. necator C-strain CDS (Jones et al., 2014) using Bowtie2 v.2.3.4.1 (Langmead and Salzberg, 2012) and the parameters –end-to-end –sensitive –un. For each grape accession, de novo assembly was performed using unmapped reads from the 12 RNA-seq libraries and TRINITY v.2.4.0 (Grabherr et al., 2011). A total of 509,960 sequences were reconstructed, corresponding to 36,426 ± 2,585 sequences per accession (Supplementary Table 4). To reduce sequence redundancy, reconstructed transcripts from all 14 genotypes were clustered using CD-HIT-EST v.4.6.8 (Li and Godzik, 2006) and an identity threshold of 90%. The longest representative sequence of each of the 98,340 transcript clusters was used as input for TransDecoder v.3.0.1 (https://github.com/TransDecoder/TransDecoder). For the 2,174 transcripts with an open reading frame protein containing a start and a stop codon, the longest CDS was selected. CDS redundancy was reduced by clustering using CD-HIT-EST v.4.6.8 (Li and Godzik, 2006), with coverage and identity thresholds of 100%. In total, 2,070 CDS were retained (Supplementary Data 1). Taxonomic analysis of the predicted proteins was performed using Megan v.6.12.5 (Huson et al., 2016) with default parameters after aligning predicted peptides against the RefSeq protein database (ftp://ftp.ncbi.nlm.nih.gov/refseq, retrieved 17 January 2017). The 103 peptides assigned to proteobacteria, opisthokonts, and viruses were considered microbial contaminants; the remaining 1,967 peptides were assigned to grape (Supplementary Table 5).
To evaluate the effect of the reference transcriptome on the mapping rate (i.e. the percentage of RNA-seq reads aligning onto the reference transcriptome), quality-trimmed RNA-seq reads were aligned onto two reference transcriptomes: (i) the combined protein-coding sequences of V. vinifera PN40024 V1 (Jaillon et al., 2007) and E. necator C-strain (Jones et al., 2014), (ii) the combined accession-specific diploid synthetic grape transcriptome, the de novo assembled CDS, and E. necator C-strain CDS (Jones et al., 2014), using Bowtie2 v.2.3.4.1 (Langmead and Salzberg, 2012) and the parameters –end-to-end –sensitive –un. For each accession, the difference of mapping rates between the two reference transcriptomes were tested using Kruskal-Wallis test followed by post hoc Dunn’s test (P ≤ 0.05).
2.4 Gene expression analysis
Transcript abundance was assessed using Salmon v.1.5.1 (Patro et al., 2017) and these parameters: –gcBias –seqBias –validateMappings. For each accession, a transcriptome index file was built using the accession’s diploid synthetic transcriptome combined with the de novo assembled transcripts and the E. necator C-strain transcriptome, PN40024 V1 and E. necator C-strain genomes as decoys, and a k-mer size of 13. Read counts were computed using the R package tximport v.1.20.0 (Soneson et al., 2015). Read counts of the two haplotypes of each gene locus of the diploid synthetic transcriptome were combined. Read-count normalization of the grape transcripts and statistical testing of differential expression were performed using DESeq2 v.1.16.1 (Love et al., 2014). The sample 14305-001_H_5dpi_1 was removed from the RNA-seq analysis because of its low mapping rate (Supplementary Table 2).
2.5 Functional annotation of the defense-related genes
To determine the defense mechanisms triggered by the presence of E. necator, we refined the functional annotation of the grape predicted proteins involved in the following processes: pathogen recognition by receptor-like kinases (RLKs) and intracellular receptors (NLRs), ROS production and scavenging, nitric oxide (NO) production, calcium oscillations, MAPK cascade, salicylic acid (SA), jasmonic acid (JA), ethylene (ET), and abscisic acid (ABA) signaling, pathogenesis-related (PR) protein and phytoalexin biosynthesis, and cell wall reinforcement. Functional annotation of the grape predicted proteins was assigned based on sequence homology with Arabidopsis thaliana predicted proteins involved in the aforementioned functional categories and protein domain composition. Grape proteins were aligned onto A. thaliana proteins (Araport11_genes.201606.pep.fasta; https://www.arabidopsis.org/download/index.jsp) using BLASTP v.2.6.0. Alignments with an identity greater than 30% and a reciprocal reference:query coverage between 75% and 125% were kept. For each grape protein, the alignment with the highest product of identity, query coverage, and reference coverage was selected to determine a homologous protein in A. thaliana. We verified that each grape protein and its assigned A. thaliana had a similar domain composition. Grape and Arabidopsis thaliana predicted proteins were scanned with hmmsearch from HMMER v.3.3.1 (http://hmmer.org/) and the Pfam-A Hidden Markov Models (HMM) database (El-Gebali et al., 2019; downloaded on 29 January 2021). Protein domains with an independent E-value less than 1.0 and an alignment covering at least 50% of the HMM were selected. Grape predicted proteins having a similar domain composition than their A. thaliana homologues were retained. The functional annotation of defense-related genes used in this study can be found in Supplementary Table 6.
3 Results
3.1 Powdery mildew resistance loci exhibit different intensity and timing of response to E. necator
PM disease severity was evaluated on detached leaves from fourteen grape accessions, including twelve interspecific accessions, and two PM-susceptible V. vinifera, Malaga Rosada and F2-35 (Table 1, Supplementary Table 1). The twelve interspecific hybrids included four pairs of siblings derived from backcrosses with one of the following resistant accessions: V. romanetii C166-043 (Ren4D+), V. romanetii C166-026 (Ren4U+), M. rotundifolia G52 (Run1+), M. rotundifolia Trayshed (Run2.2+) (Ramming et al., 2011; Riaz et al., 2011; Feechan et al., 2013). The four remaining interspecific accessions were 09390-023 (Ren2+), 07712-06 (Ren3+), e6-23 (Run1.2b+), and 09705-45 (Run2.1+); these inherited PM resistance from V. cinerea B9, the interspecific hybrid Villard blanc, M. rotundifolia Trayshed, and M. rotundifolia Magnolia, respectively (Dalbó et al., 2001; Riaz et al., 2011; Zyprian et al., 2016).
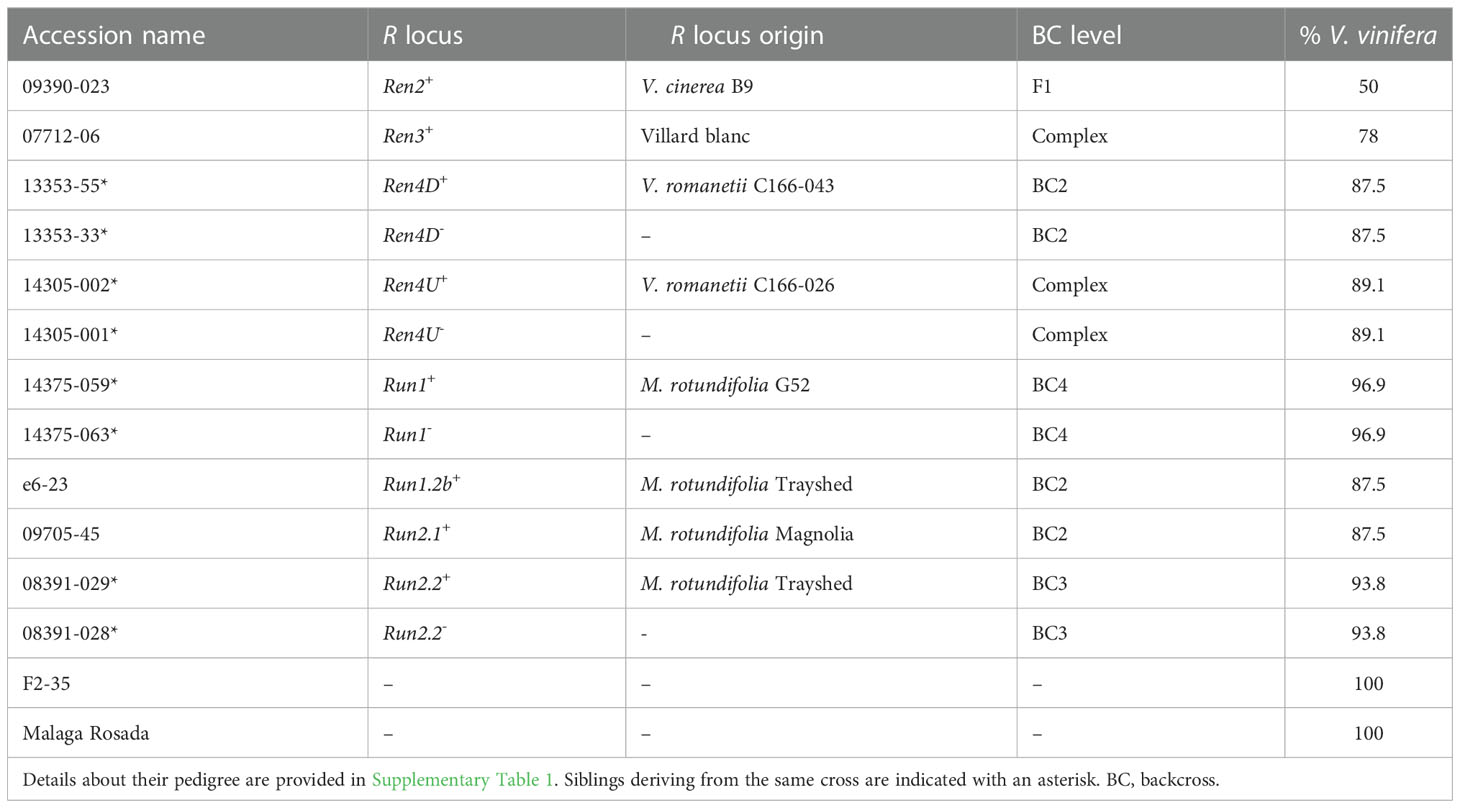
Table 1 Description of the fourteen grape genotypes used in this study.
At 5 dpi, extensive hyphal growth and conidiophores were observed on leaves from the two PM-susceptible V. vinifera cultivars, F2-35 and Malaga Rosada, and all the sib lines devoid of PM resistance loci (Figure 1A). Little or no hyphal growth was visible on the leaves of the accessions carrying Ren4D, Ren4U, Run1, and Run1.2b loci. Some secondary and tertiary hyphae were found on Ren2+, Ren3+, Run2.1+, and Run2.2+ genotypes (Figure 1A). PM infection was also assessed at an advanced disease development stage (14 dpi) using visual scoring (Supplementary Figure 1A). Infection rate at 5 and 14 dpi was similar for all genotypes except Ren2+ and Ren3+, which both had an extensive mycelium growth on their leaves at 14 dpi (Kruskal-Wallis test followed by post-hoc Dunn’s test; P ≤ 0.05).
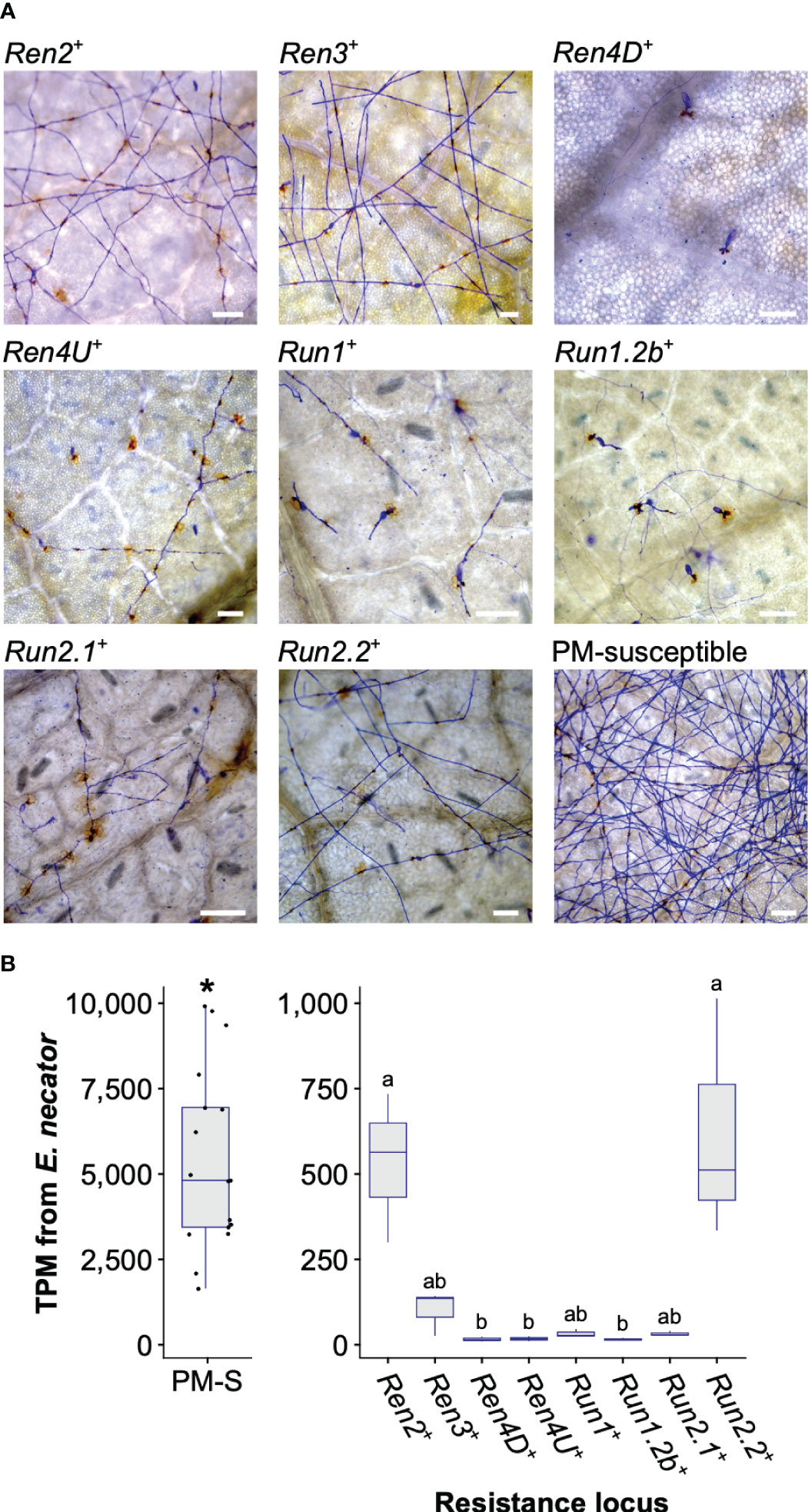
Figure 1 Powdery mildew disease development at 5 days post-inoculation. (A) Micrographs of detached leaves inoculated with E. necator. Scale = 100 μm. (B) Total Transcripts per Million (TPM) derived from E. necator transcriptome in PM-susceptible (PM-S) and PM-resistant accessions. Significant difference between grape accessions with and without a PM resistance locus is indicated by an asterisk (Kruskal-Wallis test, P = 4.0 x 10-8). Significant differences between PM resistance loci are indicated by different letters (Kruskal-Wallis test followed by post hoc Dunn’s test; P ≤ 0.05).
PM biomass on leaves was estimated by measuring E. necator transcript abundance at 5 dpi. As expected, significantly lower E. necator transcript counts were found in genotypes carrying a PM R locus (Figure 1B). Significant differences in E. necator transcripts were also observed between Ren2+, Run2.2+, and the other PM-resistant accessions (Kruskal-Wallis test followed by post hoc Dunn’s test; P ≤ 0.05). E. necator transcript abundance at 5 dpi and PM infection scores at 14 dpi correlated well (R2 = 0.71; Supplementary Figure 1B), which suggests that pathogen transcript abundance is a reliable measure of PM susceptibility.
These results show that all R loci confer resistance to PM in V. vinifera. However, PM resistance level varies between R loci, suggesting differences in the perception of the pathogen and/or in the efficiency of the defense responses.
3.2 Construction of comparable reference transcriptomes
To determine if the differences in PM development between R loci were associated with differences in the defense mechanisms induced, we profiled the leaf transcriptomes of the fourteen genotypes at 1 and 5 dpi using RNA-seq. Mock-inoculated samples were collected as controls. Because the genotypes have diverse and distant genetic backgrounds, we built comparable reference transcriptomes for each genotype by incorporating sequence variant information into the predicted transcriptome of PN40024 and by adding de novo assembled transcripts that are not found among the annotated CDS of PN40024.
RNA-seq reads were first used to identify sequence polymorphisms between all sequenced transcriptomes and the PN40024 reference (Jaillon et al., 2007). On average, 109,645 ± 17,463 single-nucleotide polymorphisms (SNPs) and 1,208 ± 119 short insertion-deletions (INDELs) were detected in the protein-coding regions of each grape accession (Figure 2; Supplementary Table 3). Although the number of homozygous SNPs was quite stable across genotypes (35,490 ± 4,667), the number of heterozygous SNPs varied extensively, ranging from 54,154 in V. vinifera cv. Malaga Rosada to 116,032 in the Ren2+ accession (Malaga Rosada x V. cinerea B9; Figure 2). Comparison of the SNPs detected in the fourteen leaf transcriptomes compared to V. vinifera PN40024 protein-coding sequences distinguished the Ren2+ accession from the other grape accessions, likely because it is the only F1 individual in this study (Table 1; Figure 2). Siblings and respective V. vinifera parents clustered together confirming the validity of the variant calling approach (Figure 2). Analysis of genetic relatedness also confirmed sibling and parent-offspring relationships (Supplementary Figure 2). Homozygous and heterozygous variants of each grape genotype were incorporated into the CDS of PN40024 to produce a diploid reference transcriptome for each genotype (Figure 3A).
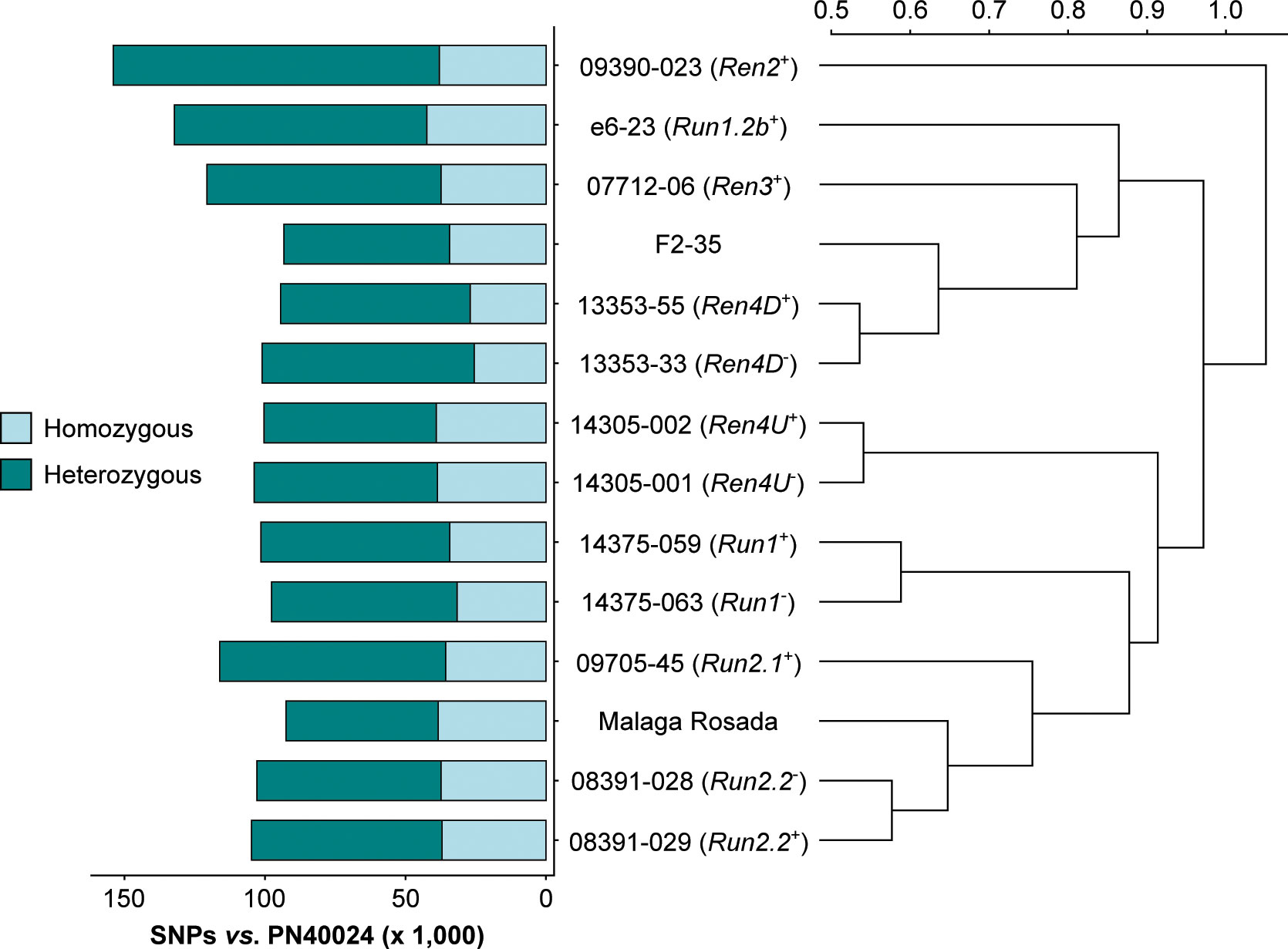
Figure 2 SNPs detected in the leaf transcriptome of the 14 grape accessions compared to the protein-coding sequences of Vitis vinifera PN40024. The Pearson correlation coefficients of the pairwise SNP comparison were converted into distance coefficients to define the height of the dendrogram branches.
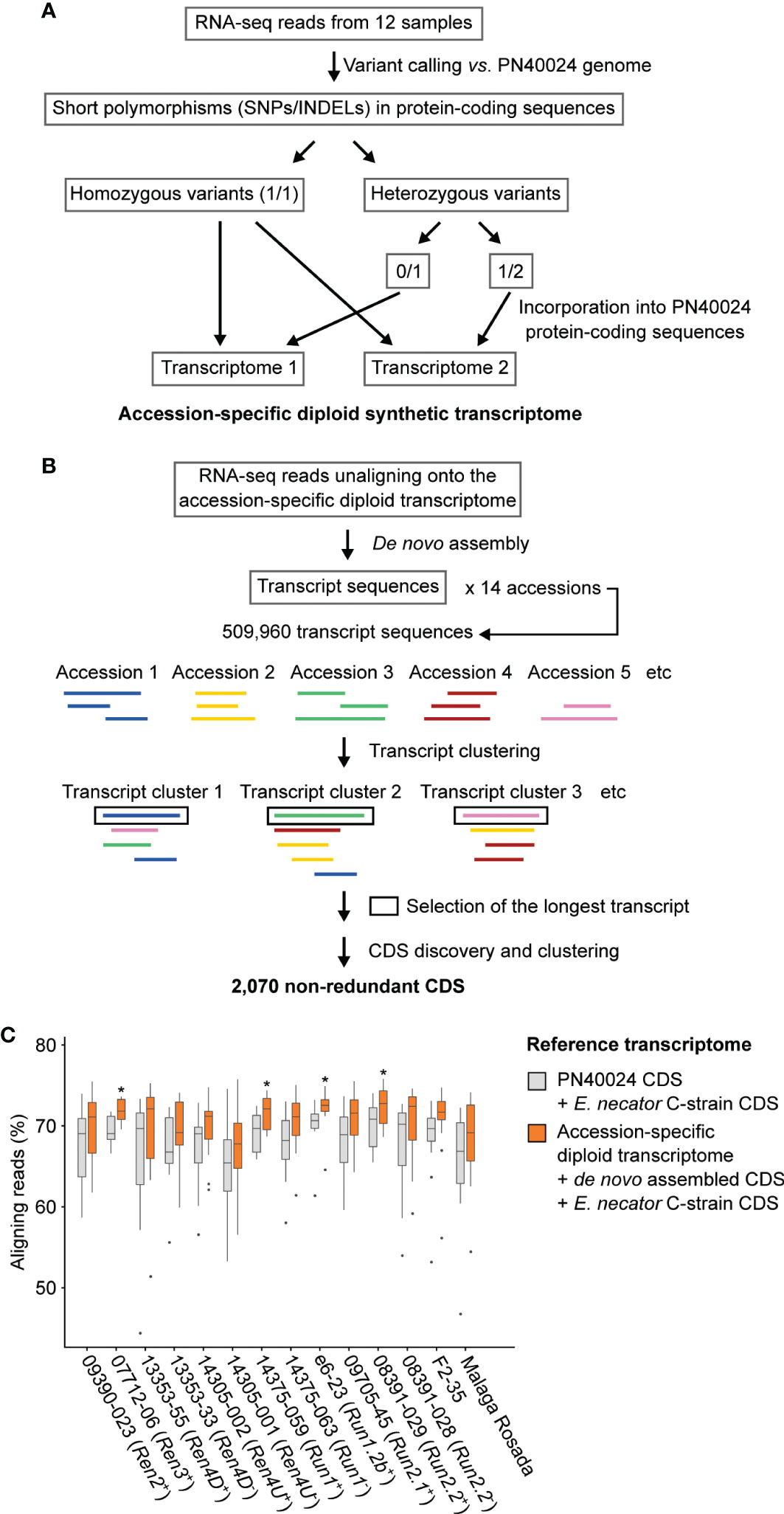
Figure 3 Adapting the grape transcriptome to each accession enhances the alignment of RNA-seq reads. Schematic representations of the bioinformatics approaches used to reconstruct the diploid synthetic transcriptome of each grapevine accession (A) and to cluster the de novo assembled transcripts (B). (C) Effect of the reference transcriptome on the percentage of aligning reads. For each genotype, difference of mapping rate with the combined transcriptomes of PN40024 and E. necator was tested using a Kruskal-Wallis test. * indicates P value ≤ 0.05.
The RNA-seq reads that did not align onto their respective diploid transcriptome reference were de novo assembled in order to construct transcripts that are not present in the PN40024 transcriptome (Figure 3B). All de novo assembled transcripts from the fourteen accessions were clustered to obtain a non-redundant dataset. After sequence clustering, CDS discovery, and removal of non-plant sequences, 1,967 non-redundant plant CDS were identified (See methods; Supplementary Table 5). Based on transcript clustering, each grape accession possessed on average 1,201 ± 31 of the grape de novo assembled CDS. As expected, siblings grouped together based on shared CDS (Supplementary Figure 3). Incorporating the sequence polymorphisms and de novo assembled CDS into the reference transcriptome increased the percentage of aligning RNA-seq reads by 2.5 ± 0.9% per sample (Figure 3C) and likely also increased the mapping specificity.
3.3 Annotation of the grape defense-related genes
To determine the defense mechanisms involved in the response to E. necator, we refined the functional annotation of the grape predicted proteins based on protein domain composition and homology with Arabidopsis thaliana predicted proteins. We identified 2,694 grape genes involved in the following processes: pathogen recognition by receptor-like kinases (RLKs) and intracellular receptors (NLRs), ROS production and scavenging, nitric oxide (NO) production, calcium oscillations, MAPK cascade, salicylic acid (SA), jasmonic acid (JA), ethylene (ET), and abscisic acid (ABA) signaling pathways, pathogenesis-related (PR) protein and phytoalexin biosynthesis, and cell wall reinforcement (Table 2; Supplementary Table 6). These included 22 de novo assembled CDS, including nine NLRs, a glutaredoxin, three pathogenesis-related (PR) protein 14-like proteins, and three ethylene-responsive transcription factors (ERFs) (Supplementary Table 6).
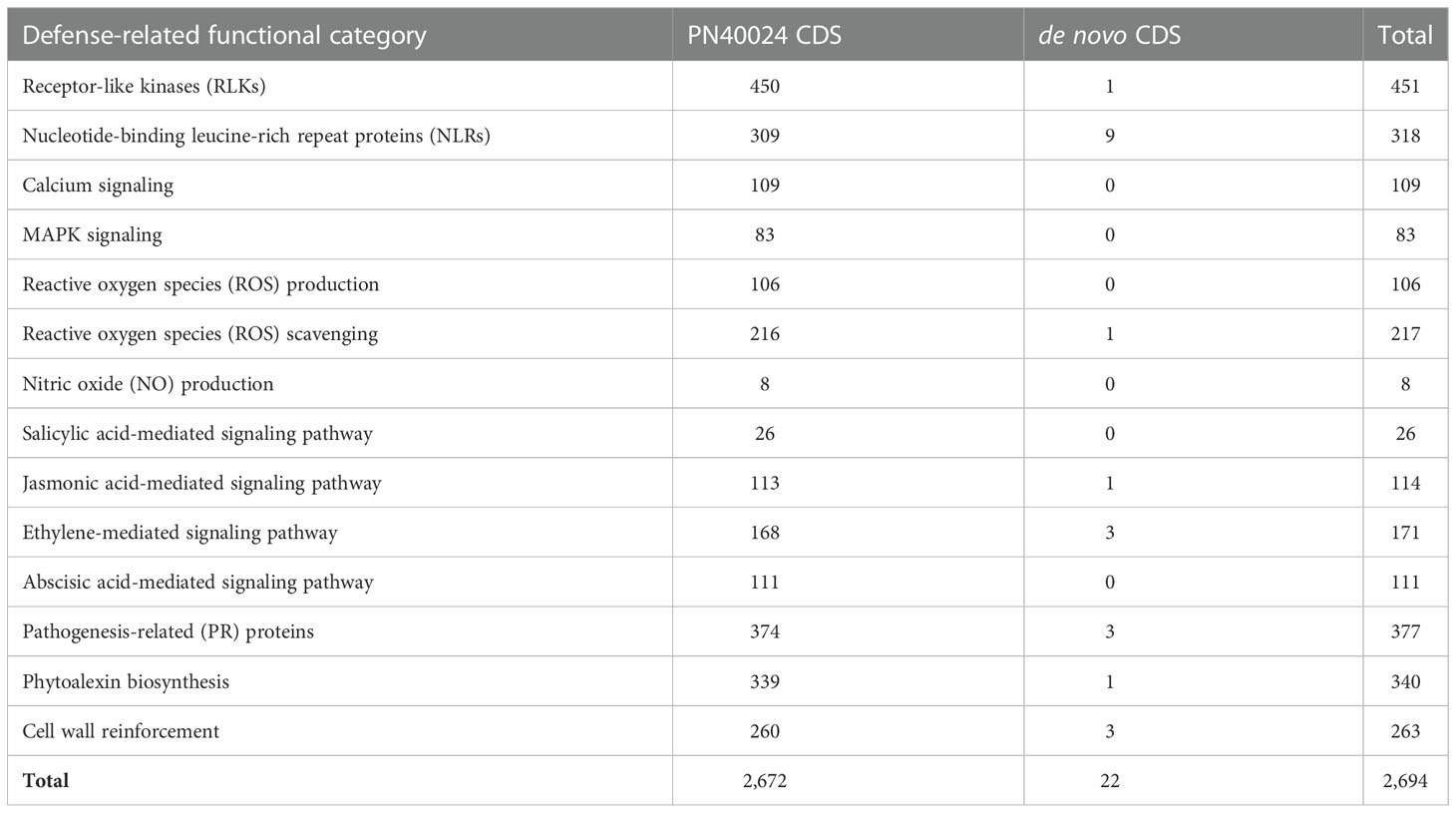
Table 2 Number of defense-related genes identified among the predicted proteins of PN40024 CDS and the de novo assembled CDS.
3.4 Assessment of the transcriptional modulations associated with powdery mildew resistance
PM- and mock-inoculated leaf transcriptomes were compared at 1 and 5 dpi to identify the defense-related genes that were differentially expressed in response to E. necator (Figure 4; Supplementary Table 7). In the PM-resistant vines Ren3+, Ren4D+, Ren4U+, and Run1.2b+, up- and down-regulated genes were more numerous at 1 dpi relative to 5 dpi, while only few differentially expressed genes (DEGs) were detected in Run2.1+ leaves. The opposite pattern was observed in the Run2.2+ accession as well as five of the six PM-susceptible plants. These results suggest that disease resistance is mostly associated with an early transcriptional reprogramming.
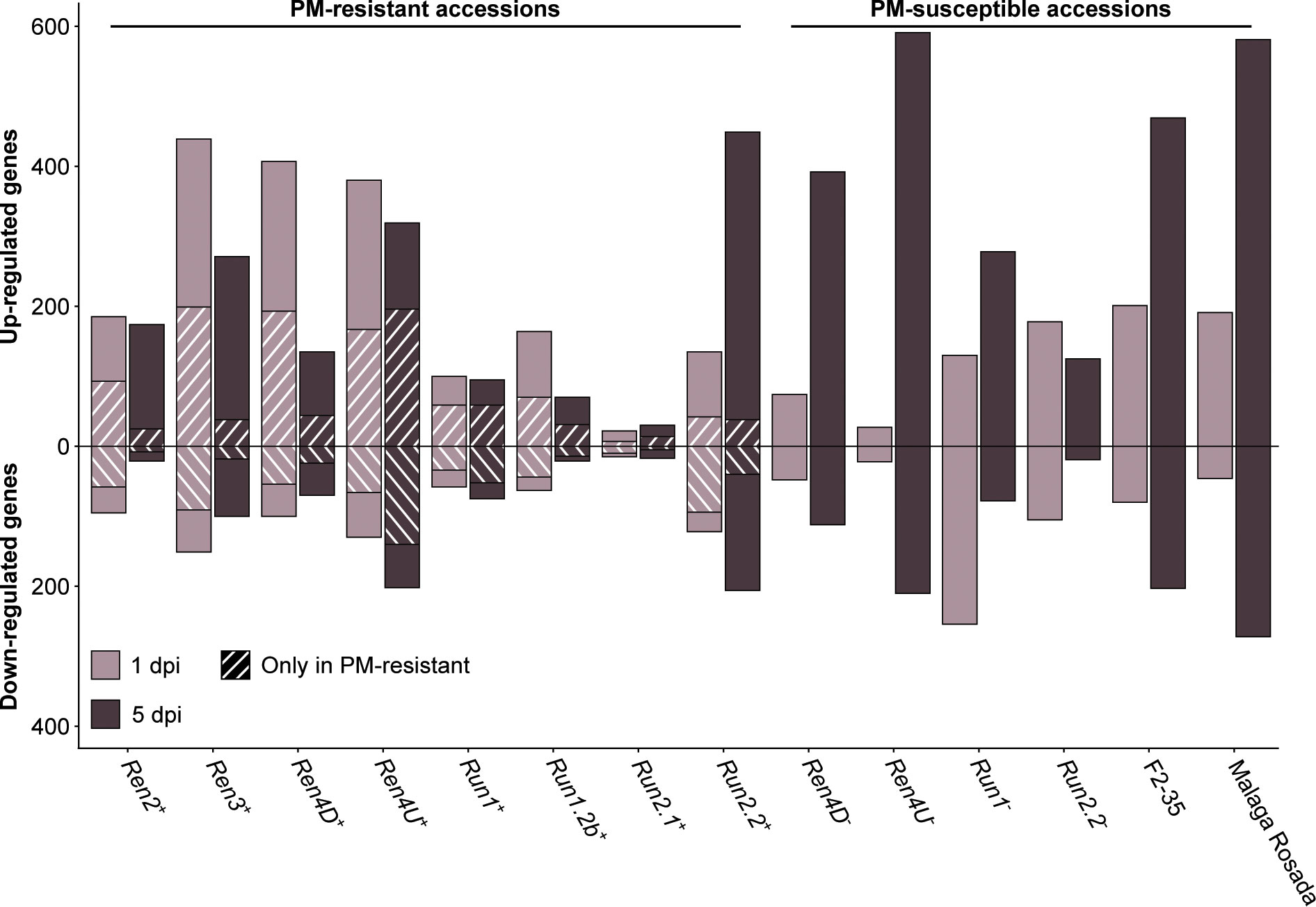
Figure 4 Defense-related genes differentially expressed in response to E. necator at 1 and 5 dpi. The genes uniquely differentially expressed by E. necator in the grape accessions possessing a R locus were identified by comparing the up- and down-regulated genes in PM-resistant leaves with the ones detected in PM-susceptible vines at each time point.
To determine the transcriptional modulations associated with PM resistance, defense-related DEGs in PM-resistant accessions were compared with all the DEGs found in the six PM-susceptible individuals at each time point. On average, 39.7% ± 16.6% of the up-regulated genes and 52.7% ± 19.0% of the down-regulated genes detected in PM-resistant accessions were not found in PM-susceptible plants (Figure 4). PM resistance-associated DEGs were more numerous at 1 dpi compared to 5 dpi in all PM-resistant genotypes except the Ren4U+ vine. This supports the hypothesis that an early transcriptional reprogramming caused by the prompt perception of E. necator by intracellular NLR is crucial for PM resistance. However, the PM-resistant accessions exhibited different amounts of defense-related DEGs in response to E. necator at 1 dpi. The highest number of up-regulated genes associated with disease resistance at 1 dpi was detected in Ren3+, Ren4D+, and Ren4U+ vines (199, 193, and 167 genes, respectively), while a lower number of up-regulated genes was found in Ren2+, Run1+, Run1.2b+, Run2.2+ leaves (93, 59, 70, and 42 genes, respectively). The Run2.1+ accession was the PM-resistant with the fewest up-regulated genes associated with PM resistance, with only 7 and 14 genes at 1 and 5 dpi, respectively. Similar patterns were observed for the down-regulated genes detected only in PM-resistant vines.
3.5 Overlap in transcriptional modulation associated with powdery mildew resistance between R loci
PM resistance-associated DEGs were compared across PM-resistant accessions to evaluate the overlap in defense responses between the different PM resistance loci (Figure 5, 6; Supplementary Figures 4-7). On average, the R loci shared 52.9 ± 10.4% and 38.0 ± 13.6% of their defense-related DEGs associated with disease resistance with at least another R locus at 1 and 5 dpi, respectively. However, the overlap of PM resistance-associated DEGs between two R loci was limited (8.4 ± 10.0%). The largest pairwise overlaps were found among the up-regulated genes at 1 dpi between the Ren3+, Ren4D+, and Ren4U+ genotypes (Figure 5A, Supplementary Figure 4). The Ren3+ and Ren4D+ accessions shared 64 up-regulated genes, while 49 and 44 defense-related genes were up-regulated in both Ren3+ and Ren4U+ genotypes, and both Ren4D+ and Ren4U+ vines, respectively. The overlap of up-regulated genes between Ren3+ and Ren4D+ accessions encompassed the largest number of extra- and intracellular receptor genes (15 RLK and 10 NLR genes), MAPK-signaling genes (7), and phytoalexin biosynthesis-related genes (11) including six stilbene synthase genes (Figure 5A). Eight genes involved in ROS production were found up-regulated in several R loci: a copper amine oxidase, three respiratory burst oxidase homolog (RBOH), and four class III cell wall peroxidase (Prx) genes. Regarding ROS scavenging, two catalase genes were up-regulated in both Ren3+, Ren4D+, and Ren4U+ leaves, as well as seven glutathione S-transferase genes in Ren3+ and Ren4U+ accessions. Some hormone-mediated signaling genes were also found up-regulated in several R loci at 1 dpi, including one gene involved in SA signaling, 12 genes in JA signaling, 13 genes in ET signaling, and 11 genes in ABA signaling. The shared JA-signaling genes encompassed 8 genes involved in JA biosynthesis, including an allene oxide synthetase (AOS) gene (VIT_18s0001g11630) that was more highly expressed in Ren3+, Ren4D+, Ren4U+, Run1.2b+ leaves in response to E. necator. The gene VviJAZ4 (VIT_09s0002g00890) encoding a jasmonate-ZIM-domain protein was also up-regulated by the same PM resistance loci. Ectopic expression of VqJAZ4 from V. quinquangularis was showed to enhance resistance to PM in A. thaliana, suggesting a role of the jasmonate-ZIM-domain protein in grape PM resistance (Zhang et al., 2019). Furthermore, VviMYC2 (VIT_02s0012g01320) was up-regulated in Ren2+, Ren3+, Ren4D+, and Ren4U+ accessions at 1 dpi but not in any genotype possessing a R locus from M. rotundifolia. The same pattern was observed for VviJAR1 (VIT_15s0046g01280) which was up-regulated only in Ren3+, Ren4D+, and Ren4U+ accessions. In A. thaliana, JAR1 encodes a jasmonate-amido synthetase that catalyzes the formation of the biologically active jasmonyl-isoleucine (JA-Ile) conjugate, while MYC2 is a basic helix-loop-helix (bHLH) transcription factor that is involved in JA signaling but also in ABA- and SA-mediated signaling (Abe et al., 2003; Staswick and Tiryaki, 2004; Dombrecht et al., 2007; Gautam et al., 2021). Regarding cell wall reinforcement, a dirigent protein-encoding gene (VIT_06s0004g01020) was up-regulated in five PM-resistant accessions at 1 dpi: Ren3+, Ren4D+, Ren4U+, Run1+ and Run2.2+ genotypes at 1 dpi. Dirigent proteins participate in the biosynthesis of lignans and lignin which both play a role in plant defense. Lignans inhibit microbe-derived degradative enzymes while lignin accumulation in the plant cell wall forms a physical barrier against pathogens (Paniagua et al., 2017). In addition, the callose synthase genes VviCalS1 and VviCalS3 (Yu et al., 2016) were up-regulated in only Ren4D+ and Ren4U+ accessions at 1 dpi and 5 dpi, respectively (Figures 5A, 6A, Supplementary Figures 4 and 6). These results support previous observations which suggested that Ren4-mediated resistance relies on the encasement of the pathogen haustorium in callose in addition to PCD of the infected cells (Qiu et al., 2015).
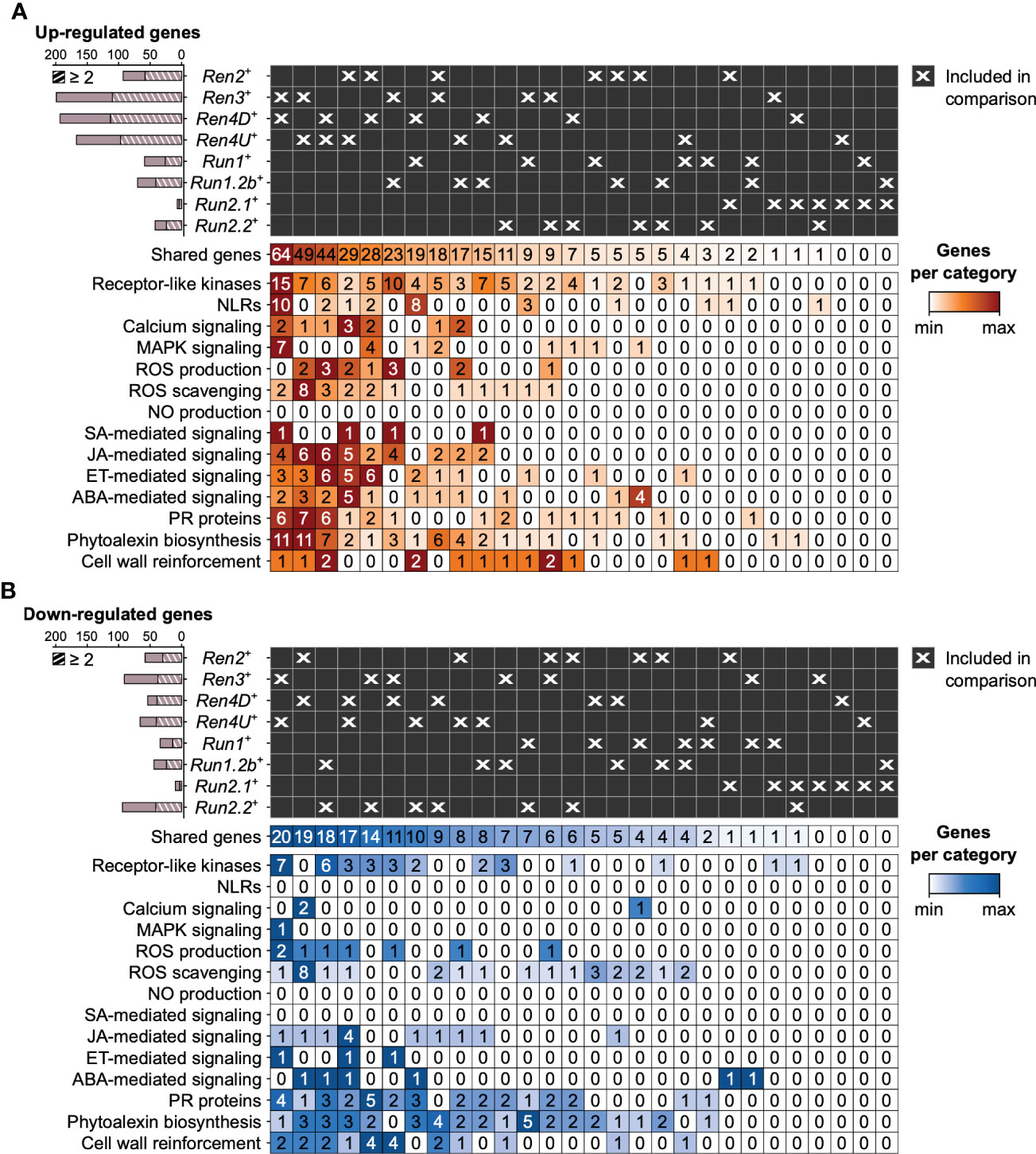
Figure 5 Pairwise comparison of the defense-related genes found up- (A) and down-regulated (B) in response to E. necator at 1 dpi in only the eight R loci. Striped bar plots depict the number of defense-related genes differentially expressed in at least two R loci.
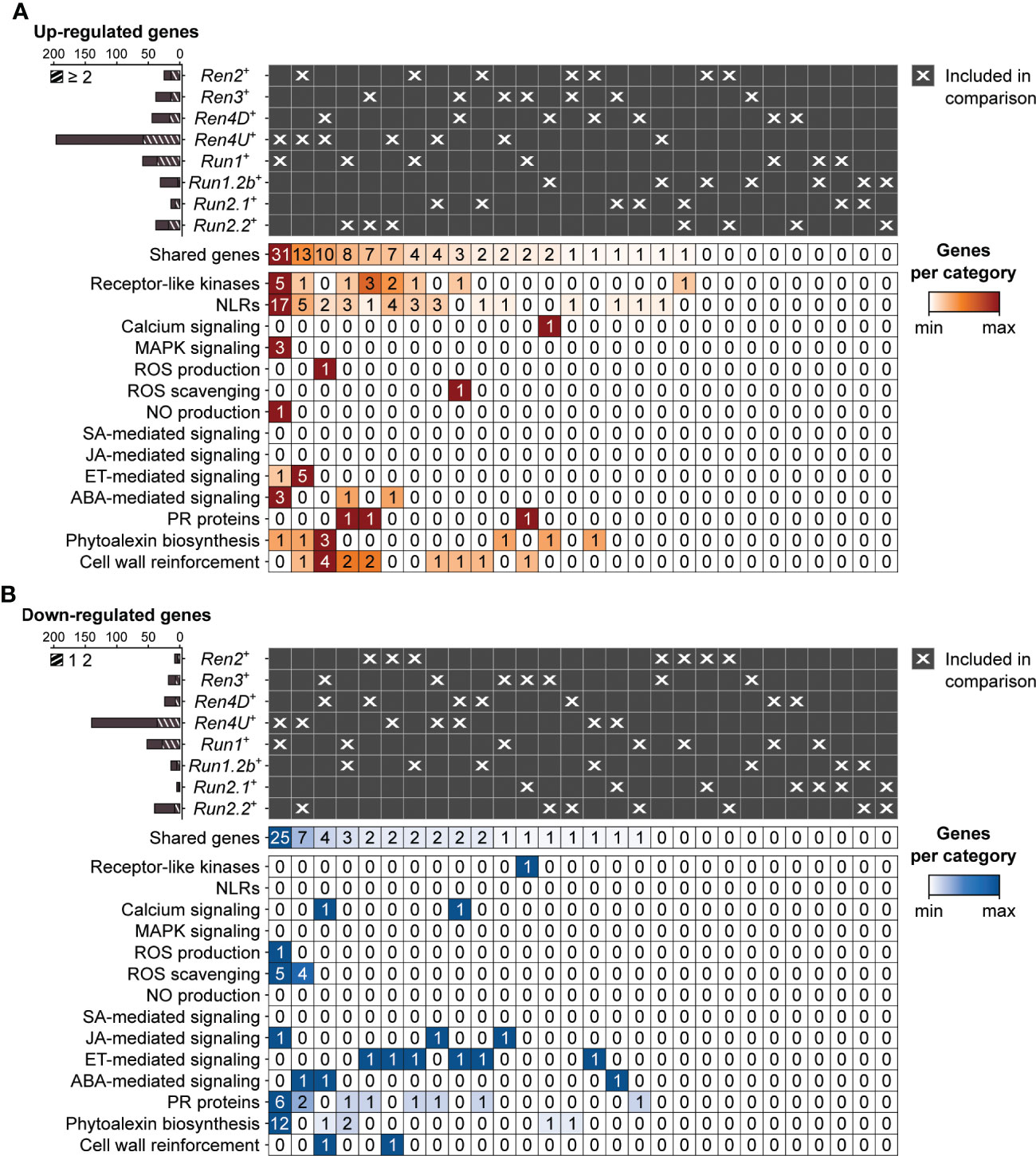
Figure 6 Pairwise comparison of the defense-associated genes found up- (A) and down-regulated (B) in response to E. necator at 5 dpi in only the eight accessions carrying a R locus. Striped bar plots represent the defense-related genes differentially expressed in at least two R loci.
Regarding the down-regulated genes associated with PM resistance at 1 dpi, Ren3+ and Ren4U+ accessions shared the highest number of genes with a lower gene expression in response to E. necator (20), followed by the Ren2+ and Ren4D+ vines (19), the Run1.2b+ and Run2.2+ plants (18), and the Ren4D+ and Ren4U+ genotypes (17; Figure 5B). The overlap in down-regulated genes between the Ren3+ and Ren4U+ accessions comprised 7 RLK genes, 2 Prx genes, and 2 cellulose synthase-like genes. Both Ren2+ and Ren4D+ leaves exhibited a lower gene expression of 8 ROS scavenging-related genes in response to E. necator at 1 dpi. Four fatty acid desaturase genes involved in JA biosynthesis were down-regulated in the Ren4D+ and Ren4U+ genotypes, and an ERF gene was down-regulated in Ren3+, Ren4D+ and Ren4U+ plants.
At 5 dpi, the highest overlap of DEGs associated with PM resistance was found between Ren4U+ and Run1+ genotypes, with 31 and 25 common up- and down-regulated genes, respectively (Figure 6). The shared up-regulated genes between Ren4U+ and Run1+ accessions encompassed 5 RLKs and 17 NLRs, 3 MAPKK genes, 3 genes involved in ABA signal transduction, and the gene encoding the exoribonuclease 4 (XRN4/EIN5; VIT_14s0030g01580) involved in ethylene response (Figure 6A). Five additional ethylene-signaling genes were found up-regulated in Ren2+ and Ren4U+ accessions at 5 dpi: a 1-aminocyclopropane-1-carboxylate synthase (ACS) gene (VIT_02s0025g00360) and four ERF genes. In contrast, the 25 down-regulated genes shared by Ren4U+ and Run1+ genotypes included six genes associated with ROS production (1) and scavenging (5), and 10 genes involved in the biosynthesis of anthocyanins and condensed tannins (Figure 6B).
3.6 Resistance locus-specific transcriptional modulations in response to E. necator
Nearly half of the PM resistance-associated DEGs were found in only one PM resistance locus (47.1 ± 10.4% and 62.0 ± 13.6% at 1 and 5 dpi, respectively). Although the Ren3+, Ren4D+, and Ren4U+ genotypes exhibited the greatest overlap of PM resistance-associated up-regulated genes at 1 dpi, the three PM resistance loci also had the highest number of genes (89, 80, and 70 genes, respectively) that were up-regulated in a R locus-specific manner (Figure 7). Regarding the down-regulated genes at 1 dpi, Ren3+ and Run1.2b+ plants had the most numerous R locus-specific ones (53 genes each). At 5 dpi, the largest number of R locus-specific DEGs was identified in Ren4U+ leaves, with 138 up-regulated and 104 down-regulated genes.
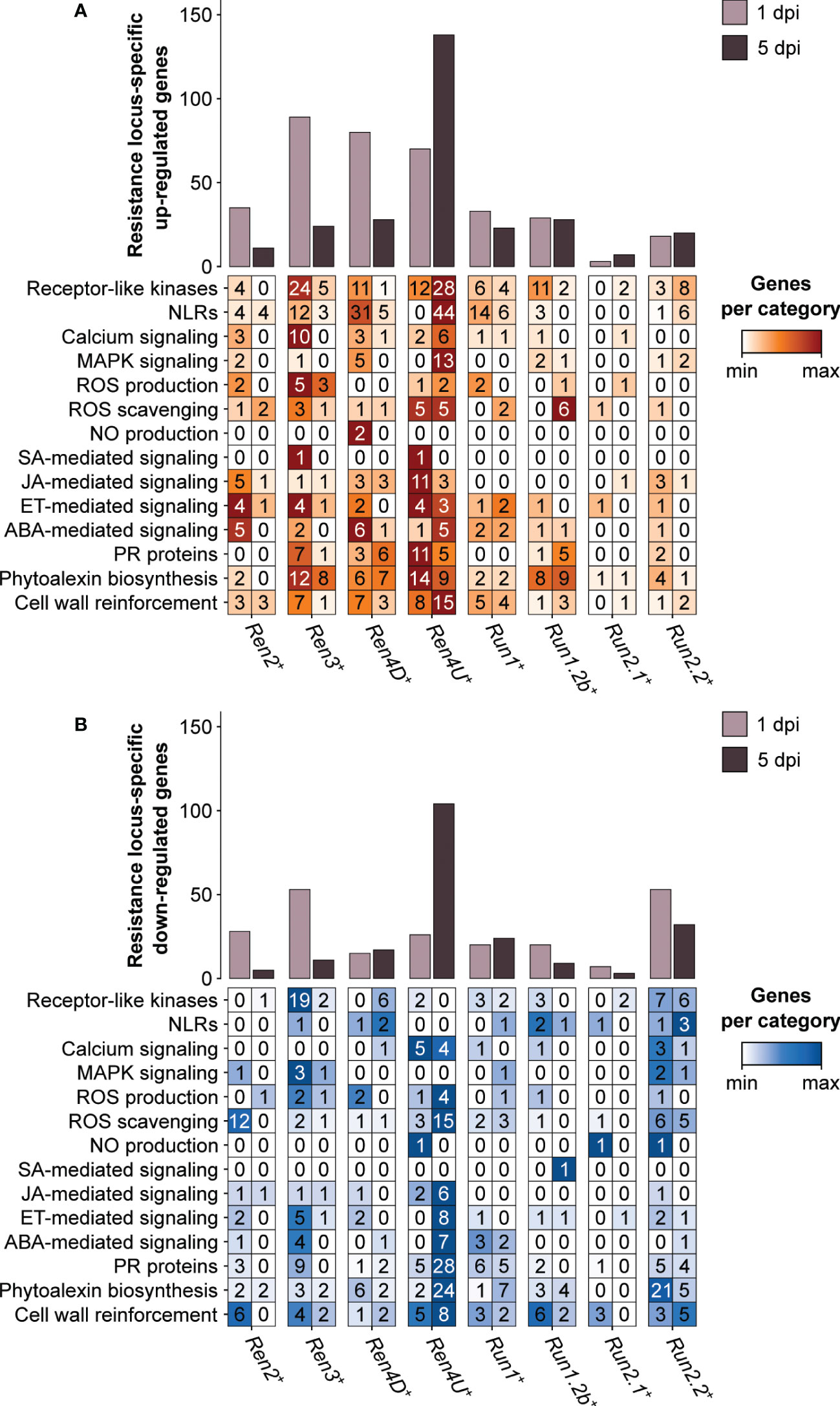
Figure 7 Defense-related categories among the genes identified as up-regulated (A) and down-regulated (B) by only one single PM resistance locus in response to E. necator.
Differences in defense-related categories among the R locus-specific DEGs could be observed between the eight R loci and the two time points (Figure 7). The Ren3+ accession exhibited the greatest number of R locus-specific DEGs encoding extracellular receptors (RLKs) at 1 dpi, with 24 up-regulated and 19 down-regulated genes. In contrast, the presence of E. necator led to the up-regulation of 28 RLK and 44 NLR genes in only the Ren4U+ leaves at 5 dpi. In addition to the intra- and extracellular receptors, the R locus-specific DEGs encompassed several PR protein genes as well as genes involved in phytoalexin biosynthesis and cell wall rearrangement. Regarding the PR protein genes, the Ren4U+ accession showed the largest number of up-regulated genes at 1 dpi (11) but also the most numerous down-regulated genes at 5 dpi (28). The Ren3+ genotype had also some R locus-specific DEGs encoding PR proteins at 1 dpi (7 up-regulated and 9 down-regulated genes), while the Ren4D+ and Run1.2b+ vines had six and five specific up-regulated ones at 5 dpi, respectively. The PM resistance locus-specific DEGs encoding PR proteins included PR1-like proteins, beta-1,3-glucanases (PR2), basic chitinases (PR3), thaumatin-like proteins (PR5), subtilisin-like endoproteinases (PR7), chitinases type I (PR11) and III (PR8), Bet v 1 homologs (PR10), lipid-transfer proteins (PR14), and germin-like proteins (PR16). PM resistance loci also exhibited differences in R locus-specific DEGs involved in the biosynthesis of antimicrobial compounds. Like the PR proteins, the Ren4U+ accession had the highest number of genes that were up-regulated at 1 dpi (14) and down-regulated at 5 dpi (24) in a PM resistance locus-specific way. In addition to the Ren4U+ accession, several phytoalexin biosynthesis-related genes were found up-regulated only in the Ren3+, Ren4D+, and Run1.2b+ genotypes at both time points. In contrast, 21 genes involved in the biosynthesis of phytoalexins were found down-regulated specifically in the Run2.2+ plants at 1 dpi, including 18 genes from the phenylpropanoid biosynthesis pathway. The Ren3+-specific up-regulated genes at 1 dpi comprised three alkaloid-related genes (berberine bridge enzymes), three genes from the phenylpropanoid biosynthesis pathway: two cinnamate 4-hydroxylases (C4H) and one 4-coumarate–CoA ligase (4CL), and two stilbene synthases. Genes involved in the biosynthesis of terpenoids and triterpenoids were also found up-regulated in only one R locus at 1 dpi, such as two and three terpene synthase genes in Ren4D+ and Ren4U+ accessions, respectively, as well as two and three oxidosqualene cyclase genes in Run2.2+ and Ren4D+ vines, respectively. Concerning the R locus-specific DEGs involved in cell wall rearrangement, the Ren4U+ genotype had the largest number of both up- and down-regulated genes. The Ren4U+-specific up-regulated genes at 5 dpi included five cellulose synthase genes, six cellulose synthase-like genes and two callose synthase genes: VviCalS7 (VIT_12s0028g00400) and VviCalS10 (VIT_17s0000g10010). Another callose synthase gene, VviCalS11 (VIT_00s0265g00050), was also found up-regulated in Run1+ plants at 5 dpi. This result suggests that VviCalS11 could play a role in the accumulation of callose deposits described in Run1+ plants (Feechan et al., 2015).
4 Discussion
4.1 Powdery mildew resistance loci confer different levels of resistance to E. necator
Evaluation of PM development using microscopy, visual scoring, and total E. necator transcript abundance, revealed differences of intensity and timing of the response to E. necator at 5 and 14 dpi (Figure 1; Supplementary Figure 1). In particular, the Ren2, Ren3 and Run2.2 loci conferred a lower level of resistance to PM compared to the five other R loci. Similar disease phenotypes have been described for these R loci in previous studies (Qiu et al., 2015; Dry et al., 2019). The absence of primary hypha on the leaves of the Ren4D+, Ren4U+, Run1+, and Run1.2b+ accessions and the very low transcript abundance for E. necator at 5 dpi suggest that restraint of the pathogen growth occurs rapidly in these vines, likely after haustorium formation at approximately 1 dpi (Leinhos et al., 1997). In contrast, the presence of some secondary and tertiary hyphae on Ren2+, Ren3+, Run2.1+, and Run2.2+ genotypes’ leaves suggest that restriction of E. necator happens at later time, between 1 and 2 dpi (Leinhos et al., 1997). In addition, fungal structures covered most of the leaves of Ren2+ and Ren3+ vines at 14 dpi, suggesting that these two R loci are less efficient in restricting the pathogen growth. Assessing the development of E. necator at additional time points post-inoculation and monitoring PCD would help to narrow down the difference of timing in the response to PM between the R loci. Furthermore, repeating the disease evaluation with additional breeding lines for each R locus would allow to confirm the observed phenotypes; while repeating the inoculation with additional E. necator isolates would help determine if the observed phenotypes are strain-specific.
4.2 Adapting the reference transcriptome allows comparing the transcriptional modulations of defense-related genes between different Vitis backgrounds
This study encompassed fourteen accessions with different genetic backgrounds making the comparison of the gene expression from orthologous genes challenging. To cope with this challenge, we incorporated small sequence polymorphisms (SNPs and INDELs) compared to the grape PN40024 CDS into a diploid synthetic transcriptome for each grape accession and performed a de novo assembly of the unaligned RNA-seq reads. The sequence variant analysis revealed an elevated number of heterozygous SNPs in the leaf transcriptomes of interspecific and intergeneric hybrids compared to pure V. vinifera cultivars, reflecting the genetic diversity among Vitis species and between M. rotundifolia and Vitis genera. The construction of the fourteen accession-specific reference transcriptomes improved the alignment of the RNA-seq reads (Figure 3C). Complete genome and/or transcriptome references of each grape accession would provide a more comprehensive representation of the defense-related genes and potentially a more accurate sequence for read alignment.
To assess the functional overlap among defense mechanisms between PM resistance loci, we focused our study on the transcriptional modulation of 2,694 defense-related genes in response to PM. These genes were selected based on functional domain composition and sequence similarity with proteins of A. thaliana. Although the defense-related genes identified might not be exhaustive and not definitive, the refinement of the functional annotation of PN40024 that was performed in this study represents a valuable resource for the present and future transcriptomic studies of grape-microbe interactions.
4.3 Powdery mildew resistance loci trigger the transcriptional modulation of different defense-related genes in response to E. necator
By comparing the transcriptomes of PM- and mock-inoculated leaves of eight PM-resistant and six PM-susceptible grape accessions, we identified the defense-related DEGs associated with disease resistance (Figure 4). Although the comparison of the PM resistance-associated DEGs showed that the R loci shared DEGs to some extent, the overlap between two R loci was restricted (8.4 ± 10.0%). It is worth noting that no defense-related gene was found differentially expressed in response to E. necator in more than five PM resistance loci (Supplementary Figures 4-7). In addition, around half of the PM resistance-associated DEGs of each R locus was found specific (Figures 5, 6). Comparison of the R locus-specific DEGs showed that the eight R loci extensively differ in their transcriptional response to E. necator through the transcriptional modulation of specific defense-related genes (Figure 7). Interestingly, different allelic forms of the Ren4, Run1, and Run2 genetic loci exhibited different responses to E. necator and even shared a larger number of DEGs with another genetic locus rather than with their respective haplotype (Figures 5, 6).
To our knowledge, this study corresponds to the first attempt of comparing the transcriptional modulations in response to PM between multiple R genetic loci. Previous studies focused on a single R locus, a single PM-resistant accession (Fung et al., 2008; Weng et al., 2014; Jiao et al., 2021), or multiple haplotypes of a single genetic locus (Amrine et al., 2015). Comparisons with previous studies are difficult because different protocol of inoculation and different strains of E. necator were used. Repeating the transcriptome profiling of additional recombinant lines for each R locus, both PM-resistant and PM-susceptible, combined with whole-genome sequencing would help dissect the R loci and identify the genes essential for resistance. In addition, using breeding lines with a higher level of backcrossing with V. vinifera would help eliminate the transcriptional responses triggered by the genetic background of the wild Vitis species that are not involved in PM resistance, including responses activated by potential additional disease resistance loci. Including other time points may provide additional information to understand gene expression modulation earlier and later than what considered in this study. For instance, few DEGs were detected for the Run2.1+ accession, suggesting either that PM resistance is associated with the expression modulation of few genes or that the transcriptional reprogramming in response to E. necator occurs at a different time for the Run2.1 locus.
Finally, this dataset represents a valuable resource for future breeding perspectives as it could help select the most functionally diverse R loci to introgress into V. vinifera.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. RNA sequencing data are accessible through NCBI under the BioProject PRJNA897013.
Author contributions
DC, MW, SR, and MM designed the project. SR and DP performed the sample inoculation, collection, microscopy, and visual scoring. RF-B extracted RNA and prepared sequencing libraries. MM performed the data analyses. MM and DC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially funded by the American Vineyard Foundation grant #2017–1657 and the US Department of Agriculture (USDA)-National Institute of Food and Agriculture (NIFA) Specialty Crop Research Initiative award #2017-51181-26829, and partially supported by funds to DC from Louis P. Martini Endowment in Viticulture.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1096862/full#supplementary-material
References
Abe, H., Urao, T., Ito, T., Seki, M., Shinozaki, K., Yamaguchi-Shinozaki, K. (2003). Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15 (1), 63–78. doi: 10.1105/tpc.006130
Amrine, K. C., Blanco-Ulate, B., Riaz, S., Pap, D., Jones, L., Figueroa-Balderas, R., et al. (2015). Comparative transcriptomics of central Asian Vitis vinifera accessions reveals distinct defense strategies against powdery mildew. Hortic. Res. 2, 15037. doi: 10.1038/hortres.2015.37
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Calonnec, A., Cartolaro, P., Poupot, C., Dubourdieu, D., Darriet, P. (2004). Effects of Uncinula necator on the yield and quality of grapes (Vitis vinifera) and wine. Plant Pathol. 53, 434–445. doi: 10.1111/j.0032-0862.2004.01016.x
Cochetel, N., Minio, A., Massonnet, M., Vondras, A. M., Figueroa-Balderas, R., Cantu, D. (2021). Diploid chromosome-scale assembly of the Muscadinia rotundifolia genome supports chromosome fusion and disease resistance gene expansion during Vitis and Muscadinia divergence. G3 (Bethesda) 11 (4), jkab033. doi: 10.1093/g3journal/jkab033
Dalboí, M. A., Ye, G. N., Weeden, N. F., Wilcox, W. F., Reisch, B. I. (2001). Marker-assisted selection for powdery mildew resistance in grapes. J. Amer Soc. Hort Sci. 126 (1), 83–89. doi: 10.21273/JASHS.126.1.83
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi: 10.1093/bioinformatics/btr330
Dangl, J. L., Horvath, D. M., Staskawicz, B. J. (2013). Pivoting the plant immune system from dissection to deployment. Science 341 (6147), 746–751. doi: 10.1126/science.1236011
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. doi: 10.1093/bioinformatics/bts635
Dombrecht, B., Xue, G. P., Sprague, S. J., Kirkegaard, J. A., Ross, J. J., Reid, J. B., et al. (2007). MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell. 19 (7), 2225–2245. doi: 10.1105/tpc.106.048017
Dry, I., Riaz, S., Fuchs, M., Sosnowski, M., Thomas, M. (2019). “Scion breeding for resistance to biotic stresses,” in The grape genome. Eds. Cantu, D., Walker, M. A. (Switzerland: Springer Cham), Switzerland 319–347. doi: 10.1007/978-3-030-18601-2
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The pfam protein family’s database in 2019. Nucleic Acids Res. 47 (D1), D427–D432. doi: 10.1093/nar/gky995
Engström, P. G., Steijger, T., Sipos, B., Grant, G. R., Kahles, A., Rätsch, G., et al. (2013). Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 10 (12), 1185–1191. doi: 10.1038/nmeth.2722
Feechan, A., Anderson, C., Torregrosa, L., Jermakow, A., Mestre, P., Wiedemann-Merdinoglu, S., et al. (2013). Genetic dissection of a TIR-NB-LRR locus from the wild north American grapevine species Muscadinia rotundifolia identifies paralogous genes conferring resistance to major fungal and oomycete pathogens in cultivated grapevine. Plant J. 76 (4), 661–674. doi: 10.1111/tpj.12327
Feechan, A., Kocsis, M., Riaz, S., Zhang, W., Gadoury, D. M., Walker, M. A., et al. (2015). Strategies for RUN1 deployment using RUN2 and REN2 to manage grapevine powdery mildew informed by studies of race specificity. Phytopathology 105 (8), 1104–1113. doi: 10.1094/PHYTO-09-14-0244-R
Fung, R. W., Gonzalo, M., Fekete, C., Kovacs, L. G., He, Y., Marsh, E., et al. (2008). Powdery mildew induces defense-oriented reprogramming of the transcriptome in a susceptible but not in a resistant grapevine. Plant Physiol. 146 (1), 236–249. doi: 10.1104/pp.107.108712
Gautam, J. K., Giri, M. K., Singh, D., Chattopadhyay, S., Nandi, A. K. (2021). MYC2 influences salicylic acid biosynthesis and defense against bacterial pathogens in Arabidopsis thaliana. Physiol. Plant 173 (4), 2248–2261. doi: 10.1111/ppl.13575
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi: 10.1038/nbt.1883
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition - interactive exploration and analysis of Large-scale microbiome sequencing data. PloS Comput. Biol. 12 (6), e1004957. doi: 10.1371/journal.pcbi.1004957
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449 (7161), 463–467. doi: 10.1038/nature06148
Jiao, C., Sun, X., Yan, X., Xu, X., Yan, Q., Gao, M., et al. (2021). Grape transcriptome response to powdery mildew infection: comparative transcriptome profiling of Chinese wild grapes provides insights into powdery mildew resistance. Phytopathology 111 (11), 2041–2051. doi: 10.1094/PHYTO-01-21-0006-R
Jones, L., Riaz, S., Morales-Cruz, A., Amrine, K. C., McGuire, B., Gubler, W. D., et al. (2014). Adaptive genomic structural variation in the grape powdery mildew pathogen, Erysiphe necator. BMC Genomics 15 (1), 1081. doi: 10.1186/1471-2164-15-1081
Karn, A., Zou, C., Brooks, S., Fresnedo-Ramírez, J., Gabler, F., Sun, Q., et al. (2021). Discovery of the REN11 locus from Vitis aestivalis for stable resistance to grapevine powdery mildew in a family segregating for several unstable and tissue-specific quantitative resistance loci. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.733899
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Leinhos, G. M. E., Gold, R. E., Düggelin, M., Guggenheim, R. (1997). Development and morphology of Uncinula necator following treatment with the fungicides kresoxim-methyl and penconazole. Mycol Res. 101 (9), 1033–1046. doi: 10.1017/S0953756297003651
Liang, Z., Duan, S., Sheng, J., Zhu, S., Ni, X., Shao, J., et al. (2019). Whole-genome resequencing of 472 Vitis accessions for grapevine diversity and demographic history analyses. Nat. Commun. 10 (1), 1190. doi: 10.1038/s41467-019-09135-8
Li, W., Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22 (13), 1658–1659. doi: 10.1093/bioinformatics/btl158
Lolle, S., Stevens, D., Coaker, G. (2020). Plant NLR-triggered immunity: from receptor activation to downstream signaling. Curr. Opin. Immunol. 62, 99–105. doi: 10.1016/j.coi.2019.12.007
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Massonnet, M., Vondras, A. M., Cochetel, N., Riaz, S., Pap, D., Minio, A., et al. (2022). Haplotype-resolved powdery mildew resistance loci reveal the impact of heterozygous structural variation on NLR genes in Muscadinia rotundifolia. G3 (Bethesda) 12 (8), jkac148. doi: 10.1093/g3journal/jkac148
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi: 10.1101/gr.107524.110
Michelmore, R. W., Christopoulou, M., Caldwell, K. S. (2013). Impacts of resistance gene genetics, function, and evolution on a durable future. Annu. Rev. Phytopathol. 51, 291–319. doi: 10.1146/annurev-phyto-082712-102334
Minio, A., Cochetel, N., Massonnet, M., Figueroa-Balderas, R., Cantu, D. (2022). HiFi chromosome-scale diploid assemblies of the grape rootstocks 110R, kober 5BB, and 101-14 mgt. Sci. Data 9 (1), 660. doi: 10.1038/s41597-022-01753-0
Paniagua, C., Bilkova, A., Jackson, P., Dabravolski, S., Riber, W., Didi, V., et al. (2017). Dirigent proteins in plants: modulating cell wall metabolism during abiotic and biotic stress exposure. J. Exp. Bot. 68 (13), 3287–3301. doi: 10.1093/jxb/erx141
Pap, D., Riaz, S., Dry, I. B., Jermakow, A., Tenscher, A. C., Cantu, D., et al. (2016). Identification of two novel powdery mildew resistance loci, Ren6 and Ren7, from the wild Chinese grape species Vitis piasezkii. BMC Plant Biol. 16 (1), 170. doi: 10.1186/s12870-016-0855-8
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14 (4), 417–419. doi: 10.1038/nmeth.4197
Qiu, W., Feechan, A., Dry, I. (2015). Current understanding of grapevine defense mechanisms against the biotrophic fungus (Erysiphe necator), the causal agent of powdery mildew disease. Hortic. Res. 2, 15020. doi: 10.1038/hortres.2015.20
Quinlan, A. R. (2014). BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinf. 47, 11.12.1–11.12.34. doi: 10.1002/0471250953.bi1112s47
Ramming, D. W., Gabler, F., Smilanick, J., Cadle-Davidson, M., Barba, P., Mahanil, S., et al. (2011). A single dominant locus, ren4, confers rapid non-race-specific resistance to grapevine powdery mildew. Phytopathology 101 (4), 502–508. doi: 10.1094/PHYTO-09-10-0237
Riaz, S., Lejkina, I., Gubler, W., Walker, M. A. (2013). Report of a new grape powdery mildew morphotype with branched conidiophores. Plant Pathol. Quar. 3, 19–27. doi: 10.5943/ppq/3/1/4
Riaz, S., Tenscher, A. C., Ramming, D. W., Walker, M. A. (2011). Using a limited mapping strategy to identify major QTLs for resistance to grapevine powdery mildew (Erysiphe necator) and their use in marker-assisted breeding. Theor. Appl. Genet. 122, 1059–1073. doi: 10.1007/s00122-010-1511-6
Soneson, C., Love, M. I., Robinson, M. D. (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. doi: 10.12688/f1000research.7563.2
Staswick, P. E., Tiryaki, I. (2004). The oxylipin signal jasmonic acid is activated by an enzyme that conjugates it to isoleucine in Arabidopsis. Plant Cell. 16 (8), 2117–2127. doi: 10.1105/tpc.104.023549
Stummer, B. E., Francis, I. L., Zanker, T., Lattey, K. A., Scott, E. S. (2005). Effects of powdery mildew on the sensory properties and composition of Chardonnay juice and wine when grape sugar ripeness is standardised. Aust. J. Grape Wine Res. 11, 66–76. doi: 10.1111/j.1755-0238.2005.tb00280.x
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28 (5), 511–515. doi: 10.1038/nbt.1621
Weng, K., Li, Z. Q., Liu, R. Q., Wang, L., Wang, Y. J., Xu, Y. (2014). Transcriptome of Erysiphe necator-infected Vitis pseudoreticulata leaves provides insight into grapevine resistance to powdery mildew. Hortic. Res. 1, 14049. doi: 10.1038/hortres.2014.49
Yu, Y., Jiao, L., Fu, S., Yin, L., Zhang, Y., Lu, J. (2016). Callose synthase family genes involved in the grapevine defense response to downy mildew disease. Phytopathology. 106 (1), 56–64. doi: 10.1094/PHYTO-07-15-0166-R
Zhang, G., Yan, X., Zhang, S., Zhu, Y., Zhang, X., Qiao, H., et al. (2019). The jasmonate-ZIM domain gene VqJAZ4 from the Chinese wild grape Vitis quinquangularis improves resistance to powdery mildew in arabidopsis thaliana. Plant Physiol. Biochem. 143, 329–339. doi: 10.1016/j.plaphy.2019.09.018
Zhou, Y., Minio, A., Massonnet, M., Solares, E., Lv, Y., Beridze, T., et al. (2019). The population genetics of structural variants in grapevine domestication. Nat. Plants. 5 (9), 965–979. doi: 10.1038/s41477-019-0507-8
Keywords: grape, genetic disease resistance, powdery mildew, comparative transcriptomics, disease evaluation
Citation: Massonnet M, Riaz S, Pap D, Figueroa-Balderas R, Walker MA and Cantu D (2022) The grape powdery mildew resistance loci Ren2, Ren3, Ren4D, Ren4U, Run1, Run1.2b, Run2.1, and Run2.2 activate different transcriptional responses to Erysiphe necator. Front. Plant Sci. 13:1096862. doi: 10.3389/fpls.2022.1096862
Received: 12 November 2022; Accepted: 02 December 2022;
Published: 19 December 2022.
Edited by:
David Gramaje, Institute of Vine and Wine Sciences, Spanish National Research Council (CSIC), SpainCopyright © 2022 Massonnet, Riaz, Pap, Figueroa-Balderas, Walker and Cantu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dario Cantu, ZGFjYW50dUB1Y2RhdmlzLmVkdQ==