 Ruxue Huo1,2Yanshu Zhao2Tianxu Liu2Meng Xu2
Ruxue Huo1,2Yanshu Zhao2Tianxu Liu2Meng Xu2 Xiaohua Wang2Ping Xu2Shengjie Dai2Xiaoyu Cui2
Xiaohua Wang2Ping Xu2Shengjie Dai2Xiaoyu Cui2 Yonghua Han1
Yonghua Han1 Zhenning Liu2*
Zhenning Liu2* Zongyun Li1*
Zongyun Li1*- 1Jiangsu Key Laboratory of Phylogeny and Comparative Genomics, School of Life Sciences, Institute of Integrative Plant Biology, Jiangsu Normal University, Xuzhou, China
- 2College of Agriculture and Forestry Science, Linyi University, Linyi, China
Two-component system (TCS), which comprises histidine kinases (HKs), histidine phosphotransfer proteins (HPs), and response regulators (RRs), plays essential roles in regulating plant growth, development, and response to various environmental stimuli. TCS genes have been comprehensively identified in various plants, while studies on the genome-wide identification and analysis of TCS in sweet potato were still not reported. Therefore, in this study, a total of 90 TCS members consisting of 20 HK(L)s, 11 HPs, and 59 RRs were identified in the genome of Ipomoea batatas. Furthermore, their gene structures, conserved domains, and phylogenetic relationships were analyzed in detail. Additionally, the gene expression profiles in various organs were analyzed, and response patterns to adverse environmental stresses were investigated. The results showed that these 90 TCS genes were mapped on 15 chromosomes with a notably uneven distribution, and the expansion of TCS genes in sweet potato was attributed to both segmental and tandem duplications. The majority of the TCS genes showed distinct organ-specific expression profiles, especially in three types of roots (stem roots, fibrous roots, tuberous roots). Moreover, most of the TCS genes were either induced or suppressed upon treatment with abiotic stresses (drought, salinity, cold, heat) and exogenous phytohormone abscisic acid (ABA). In addition, the yeast-two hybrid system was used to reveal the HK-HP-RR protein-protein interactions. IbHP1, IbHP2, IbHP4, and IbHP5 could interact with three HKs (IbHK1a, IbHK1b, and IbHK5), and also interact with majority of the type-B RRs (IbRR20–IbRR28), while no interaction affinity was detected for IbHP3. Our systematic analyses could provide insights into the characterization of the TCS genes, and further the development of functional studies in sweet potato.
1. Introduction
The two-component system (TCS) mediated protein phosphorylation is a major signal transduction pathway in regulating the response to adverse environmental stimuli in bacteria and plants. This system was originally identified in Escherichia coli (Kamberov et al., 1994), which included a membrane-associated histidine protein kinase (HK) and a cytoplasmic response regulator (RR) with a receiver (REC) domain. When bacteria are exposed to stress conditions, the HK proteins primarily sense stress signals and autophosphorylation occurs in the histidine residue (H) of the HK domain. Thereafter, the phosphate is subsequently transferred to an aspartate residue (D) in the REC domain of the RR protein. RRs are transcription factors that can convert the external stimuli into internal signals by activating or suppressing downstream targets (Mizuno, 1997). In eukaryotes, such as yeast and plants, TCS have evolved a more complex multi-step TCS signaling system with additional His phosphotransfer (HP) proteins, which are responsible for the transfer of phosphates between HKs and RRs. The multi-step phosphorelay could provide more approaches for studying the signal transduction pathway, thereby further increasing the complexity and accuracy of its regulation (Huo et al., 2020).
To date, TCS genes have been identified at a genome-wide scale in several plant species, including Arabidopsis (Hwang et al., 2002), rice (Ito and Kurata, 2006; Du et al., 2007), maize (Chu et al., 2011), soybean (Mochida et al., 2010), Chinese cabbage (Liu et al., 2014), tomato (He et al., 2016a), cucumber (He et al., 2016b), and chickpea (Ahmad et al., 2020), among others. Classifications and characterizations of the TCS genes have been well established. The model plant Arabidopsis represents the most studied and widely understood TCS elements in plants. Genome-wide analysis revealed 55 TCS-related genes in Arabidopsis, including 16 AHKs, 6 AHPs, and 33 ARRs. The Arabidopsis HKs are comprised of three subfamilies: AHK, ethylene receptor, and phytochrome. Furthermore, the AHK subfamily contains three cytokinin receptors (AHK2, AHK3, and AHK4), AHK1, AHK5, and CKI1. AHPs (AHP1–AHP5) are characterized by a highly conserved XHQXKGSSXS motif with a His phosphorylation site. However, because AHP6 lacks the conserved His residue, it is known as a pseudo-HP protein. Based on conserved domains and phylogenetic analysis, ARRs could be further classified into four subgroups, type-A, type-B, type-C, and pseudo-RRs (Hwang et al., 2002; Huo et al., 2020).
The TCS signaling pathway plays essential roles in regulating plant growth, development, and response to various environmental stimuli. The cytokinin signaling pathway is a typical representative of TCS signaling system. Studies have shown that three cytokinin receptors act as positive regulators in cytokinin signal transduction. The ahk2 ahk3 ahk4 triple mutant exhibits reduced sensitivity to exogenous cytokinin, retarded root and leaf growth, and induced reproductive developmental defects (Higuchi et al., 2004; Nishimura et al., 2004; Riefler et al., 2006; Tran et al., 2007; Kinoshita-Tsujimura and Kakimoto, 2014). CKI1 is essential for central cell specification in embryo sacs as well as for vascular patterning (Hejatko et al., 2003; Hejatko et al., 2009; Deng et al., 2010; Yuan et al., 2016; Zhu et al., 2022). AHK5 acts as a negative regulator to control root elongation in an ETR1-dependent ethylene and ABA signaling pathway (Iwama et al., 2007). Additionally, there has been an increasing amount of evidence demonstrating that the TCS signaling pathway is involved in various stress responses. Transcripts of AHK2, AHK3, and AHK4 were all rapidly induced by dehydration, and the expression of AHK2 was upregulated with NaCl and ABA treatments. The expression of AHK3 was also induced under high salinity and cold stresses. Additional functional studies have revealed that these three cytokinin receptors act as negative regulators in the osmotic stress responses of Arabidopsis (Tran et al., 2007). AHK1 is a classical osmotic stress protein, which acts as positive regulators in drought and salt stress responses (Tran et al., 2007; Wohlbach et al., 2008; Lü et al., 2012). In addition, AHK5 positively regulates salt sensitivity and contributes to biotic pathogen resistance (Pham et al., 2012). Similarly, TCS genes in rice, soybean, and tomato have also been shown to participate in drought, high salinity, and extreme temperature stresses (Firon et al., 2012; Wen et al., 2015).
Sweet potato (Ipomoea batatas L.) is an economically important food crop that is widely cultivated. It is an important source of calories, proteins, vitamins, and minerals for humanity (Yan et al., 2022). However, studies on the TCS genes in sweet potato are still limited, and their respective functional roles have not yet been reported. In the present study, the putative TCS genes were identified in the sweet potato genome, and their gene structures, conserved domains, and phylogenetic relationships were analyzed in detail. Additionally, the gene expression profiles in various organs were analyzed, and the response patterns to adverse environmental stresses were investigated. Moreover, the gene duplications and evolutionary patterns of the TCS genes in I. batatas and its two wild relatives were determined, and the yeast-two hybrid assay was conducted to reveal the HK-HP-RR protein-protein interactions. Our systematic analyses could provide insight into the characterization of the TCS genes, and further the development of functional studies in sweet potato.
2. Materials and methods
2.1. Identification of the TCS genes in the sweet potato genome
To identify the TCS genes in the genome of sweet potato (I. batatas), database including 64295 whole protein sequences were downloaded primarily from the sweet potato genome website (https://sweetpotao.com/) (Jun et al., 2017). Then the local BLAST+ program in the R language was built based on the protein annotation database. Thereafter, Arabidopsis TCS protein sequences were used as seed sequences to conduct the BLAST P search with an expected e-value of 1×10-3 as the threshold. The protein sequences of the identified sweet potato TCS members were also used as queries to repeat the BLAST P search to ensure that no additional related genes were missing from the database. Finally, after removing the redundant sequences, all the retrieved TCS protein sequences were further analyzed using the Pfam database (http://pfam.janelia.org/) (Punta et al., 2012), the SMART database (http://smart.embl-heidelberg.de/) (Letunic et al., 2012), and the Conserved Domain Database of the NCBI (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (Marchler-Bauer et al., 2011). This was done to eliminate the sequences that did not contain the known conserved domains or motifs of the TCS elements, namely the HK, HATPase, REC, CHASE domain for cytokinin binding (CHASE), ethylene-binding domain (C2H4), and HPt domains. The TCS genes in the genome of two diploid wild relatives of cultivated sweet potato, Ipomoea trifida and Ipomoea trilobal, were identified using a BLAST P search against the databases of the Sweetpotato Genomics Resource (http://sweetpotato.uga.edu/) (Wu and Lau, 2018) with default expected values. The TCS genes in the genome of Ipomoea nil were identified using a BLAST P search against the NCBI databases. Protein sequences of the TCS members from Arabidopsis, rice, maize, tomato, and soybean were then obtained based on the published data.
2.2. Gene structure, motif recognition, multiple-sequence alignment, and phylogenetic analyses
The gene structure schematic with exon-intron organizations of the TCS genes was illustrated using the Gene Structure Display Server (http://gsds.gao-lab.org/) (Hu et al., 2015). Furthermore, the conserved motifs of the TCS protein sequences were identified using the online MEME program (https://meme-suite.org/meme/) (Bailey et al., 2015). The deduced amino acid sequences of the conserved HK, HATPase, REC, Myb, and CCT domains were aligned using the Clustal X program. For the phylogenetic relationships of HK, HP, and RR members, the identified HK, HP, and RR protein sequences were separately aligned with a gap opening penalty of 10 and a gap extension penalty of 0.2 using Clustal W in the MEGA7.0 software (http://www.megasoftware.net/) (Kumar et al., 2016). The phylogenetic trees were further constructed using the neighbor-joining (NJ) method, in which Poisson correction, pairwise deletion, and bootstrapping (1,000 replicates; random seeds) were considered as parameters (Liu et al., 2014).
2.3. Protein properties and putative promoter region predictions of TCS genes
The molecular weight (MW) and theoretical isoelectric point (pI) of the TCS proteins were calculated using the online tool ExPASy (http://web.expasy.org/compute_pi/) (Wilkins et al., 1999). The subcellular location was predicted with the WoLF PSORT (https://wolfpsort.hgc.jp/) (Horton et al., 2007). The upstream sequences (1,500 bp) of the transcriptional start site of each TCS gene were chosen as the putative promoter regions, and the PlantCARE website (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/search_CARE.html) (Lescot et al., 2002) was used to identify the abiotic stress-related and phytohormone-related cis-regulatory elements.
2.4. Chromosomal localization, gene duplications, and evolutionary analysis of the TCS genes
The TCS genes were mapped to the corresponding chromosomes based on the sweet potato genome database using the MapInspect software. Tandem duplications were defined if a pair of genes had >40% amino acid sequence similarity and was separated by fewer than five intervening genes (Nuruzzaman et al., 2010). Segmental duplications were analyzed using the MCScanX software with default parameters. The synteny relationship of the TCS genes between I. batatas and its wild related species was generated using the Circos software. The occurrence of duplication events and homologous gene divergence, as well as selective pressure on duplicated genes were estimated by calculating synonymous (Ks) and non-synonymous substitutions (Ka) per site between the duplicated gene pairs using the Codeml procedure of the PAML program (Yang, 1997). The divergence time was calculated at a neutral substitution rate of 1.5 × 10-8 substitutions per site per year for the chalcone synthase gene (Chs) (Koch et al., 2000).
2.5. Plant growth and treatments
The I. batatas cultivar Taizhong 6 was used for expression analysis. The plants were grown on an experimental farm at Linyi University. The stem roots, fibrous roots, tuberous roots, stems, leaves, and flowers were separately sampled to analyze the tissue- or organ-specific expression of the TCS genes. All samples were taken from at least 10 plants.
The seedlings of the sweet potato cultivar Taizhong 6 were used for subsequent treatment. Seedlings of a uniform size were grown in Hoagland solution at 28/22°C under a photoperiod of 16 h light/8 h dark in a temperature-controlled greenhouse. About a week later, when the seedlings had 4–5 functional leaves and adventitious roots measuring 8 to 10 cm, they were subjected to stress treatment (Hou et al., 2021). For salt, drought, and ABA treatments, the adventitious roots of the seedlings were submerged in a solution containing 100 mM NaCl, 20% PEG 6000, and 100 µM ABA, respectively. For cold and heat treatments, the seedlings were placed under 10/4°C day/night and 35/35°C day/night conditions, respectively. All roots were separately collected at 0, 1, 6, 12, 24, and 48 h after treatment. All treatments were repeated three times, and each treatment contained at least 20 seedlings. After treatment, all samples were immediately frozen in liquid nitrogen and stored in a refrigerator at -80°C.
2.6. RNA extraction and qRT-PCR analysis
RNA extraction, reverse-transcription, and qRT-PCR reactions were performed as previously described (Xiao et al., 2012) using the primers listed in Supplementary Table S1. The primers were designed using the Primer (version 5.0) software. The specificity of the reactions was verified via melting curve analysis, and the products were further confirmed using agarose gel electrophoresis. qRT-PCR was performed using a Roche LightCycler® 480 II system under the following conditions: 95°C for 15 s; followed by 40 cycles of 95°C for 15 s, 55°C for 15 s, and 72°C for 15 s. There were two biological replicates and three technical replicates for each sample, as well as a negative control using the SYBR Premix ExTaq kit (TOYOBO, Osaka, Japan). The IbActin gene was used as the reference gene to study different organs or tissues and developmental stages. Notably, the IbARF gene has been used in abiotic stress and plant hormone treatment studies (Sung-Chul et al., 2012). The comparative ΔΔCT method was used to calculate the relative expression levels of different genes, and a heat map was generated in TBtools using the relative expression data of each gene.
2.7. Yeast two-hybrid assays
The matchmaker GAL4 Two-Hybrid System (Clontech) was used to determine the self-activation and protein-protein interactions. The full coding sequences of IbHK1a, IbHK1b, IbHK5, IbHP1–IbHP5, and IbRR20–IbRR28 were amplified using KOD DNA plomerase (TOYOBO) based on the gene sequences with primers listed in Supplementary Table S2. pGADT7 (AD) and pGBKT7 (BD) vectors were double digested with EcoRI and BamHI to prepare the linearized carriers, and then the purified IbHP1–IbHP5 fragments were inserted into a pGBKT7 vector with the In-Fusion Cloning Kit (TRANSGEN). The remaining IbHKs and IbRRs were inserted into the pGADT7 vertor. After the detection of bait vector self-activation, bait plasmids and prey plasmids were cotransformed into the yeast competent cell AH109 via the lithium acetate-mediated transformation method, according to the manufacturer’s instructions. The transformants were primarily cultured in SD/-Leu/-Trp and SD/-Ade/-Leu/-His/-Trp media, and the colonies were cultured in SD/-Ade/-Leu/-His/-Trp medium with X-α-gal. pGBKT7-53 and pGADT7-T were used as positive controls, while pGBKT7-Lam and pGADT7-T were used as negative controls.
3. Results
3.1. Identification of the TCS genes in I. batatas and its wild relatives
BLAST P searches were performed against the local BLAST+ program with the Arabidopsis TCS proteins as query sequences. After removing the redundant sequences and conserved domain elimination, 90 TCS members consisting of 20 HK(L)s, 11 HPs, and 59 RRs were identified in the I. batatas genome (Supplementary Table S3). Similarly, TCS genes in the genome of two wild relatives of cultivated sweet potato, I. trifida and I. trilobal, were identified. Seventy-one TCS members consisting of 21 HK(L)s, 6 HPs, and 44 RRs were obtained in the I. trifida genome (Supplementary Table S4); also, 68 TCS members consisting of 21 HK(L)s, 6 HPs, and 41 RRs were obtained in the I. trilobal genome (Supplementary Table S5). Additionally, 75 TCS members consisting of 21 HK(L)s, 10 HPs, and 44 RRs were retrieved in the genome of I. nil (Supplementary Table S6), which is in the same genus as cultivated sweet potato.
To show a clear and systematic understanding of the TCS gene numbers in plant genomes, we summarized the TCS genes that have already been identified in several representative plants (Table 1). The number of TCS genes in the I. batatas genome was only fewer than that in the Glycine max genome, which was approximately 1.5 times than that in the Arabidopsis thaliana genome. Comparatively, the I. batatas genome possessed more pseudo-HPs, type-A RRs, type-C RRs, and pseudo-RRs than A. thaliana, but the same amount of HK(L)s and type-B RRs, indicating their preferential gene duplications. Compared with its wild relatives, the I. batatas genome possessed more HPs and RRs, but this was not the case for HK(L)s. Interestingly, the large numbers of pseudo-HPs even exceed that of any plant species already reported. Therefore, their evolution mechanisms and possible functions require further investigations.
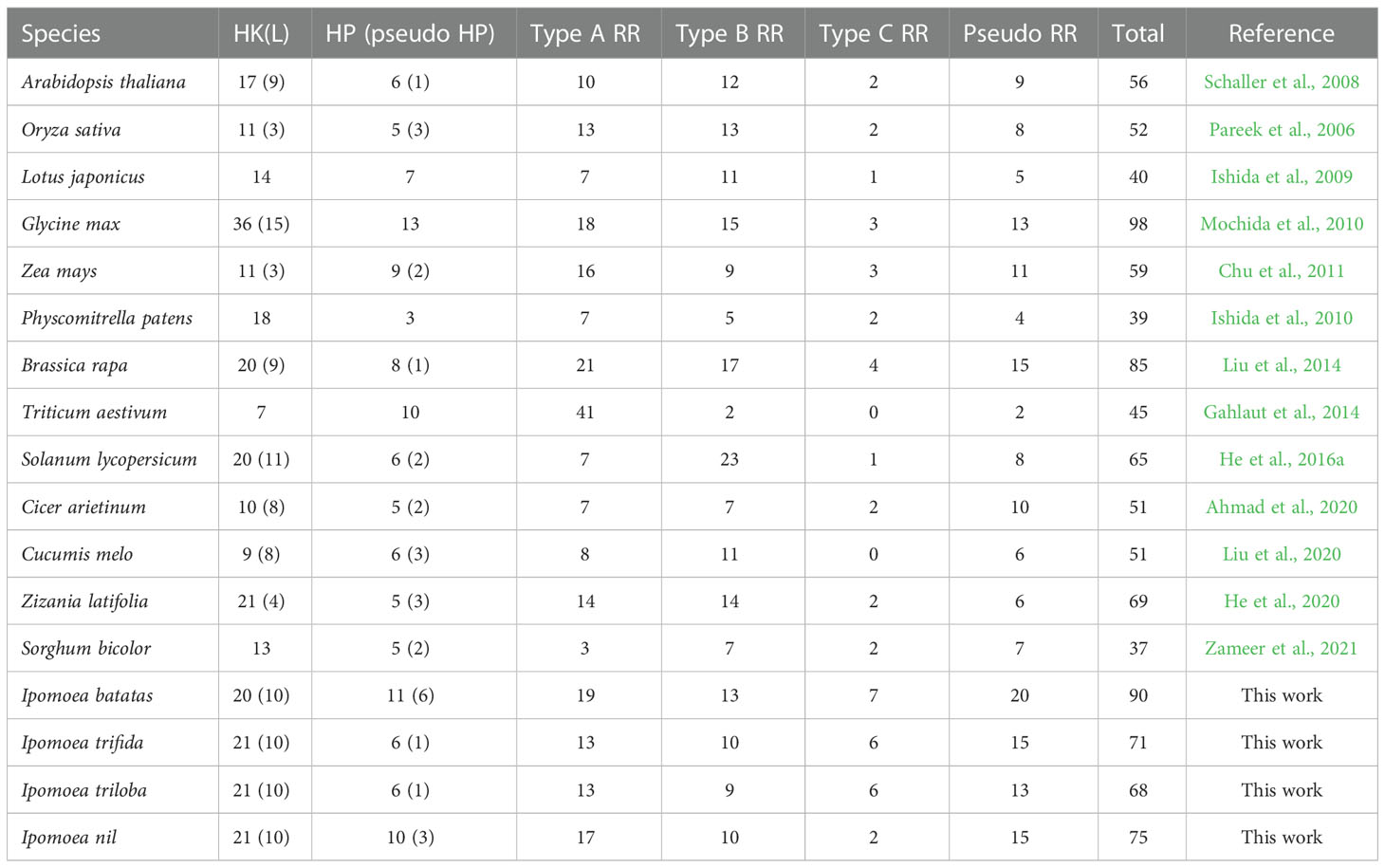
Table 1 Summary of the TCS gene numbers identified in plants. HK(L), HP, and RR represent His-kinase or like, histidine phosphotransfer, and response regulator protein, respectively.
3.2. HK proteins in I. batatas
Twenty HK(L) proteins identified in I. batatas were named according to the homologous genes in Arabidopsis. The conserved domains, ORF length, protein properties, and subcellular localization were predicted using bioinformatics. To further analyze their structural characteristics and conserved regions, we mapped the gene structures containing exons and introns, and also examined their conserved regions and motifs. Furthermore, their putative protein sequences were aligned and a phylogenetic tree was constructed (Supplementary Figure S1, S2).
The 20 HK(L) proteins identified in I. batatas could be categorized into 10 HKs and 10 HK-likes (HKLs) based on the presence of the conserved His-kinase transmitter (HK) domain. Among these, the 10 HKs were further classified into five subgroups: cytokinin receptor-like HKs, CKI1-like HKs, AHK1-like HKs, AHK5-like HKs, and ethylene receptor-like HKs (Supplementary Table S3). All these HKs possessed a conserved HK domain that contains five conserved signature motifs, namely, H, N, G1, F, and G2 (Supplementary Figure S1). The four cytokinin receptor-like HKs (IbHK2a, IbHK2b, IbHK3, and IbHK4) comprised HK, HATPase, REC, TM, and CHASE domains. Because the CHASE domain is specific for cytokinin receptor proteins, and necessary for cytokinin binding, this indicates that IbHK2a, IbHK2b, IbHK3, and IbHK4 might function as cytokinin receptors. Additionally, the presence of a TM domain in the cytokinin receptor proteins indicates the plasma membrane location of cytokinin-binding sites (Caesar et al., 2011; Wulfetange et al., 2011). The subcellular localization prediction showed that the four proteins mainly localize in the endoplasmic reticulum (ER) membrane and plasma membrane. The two AHK1-like HKs (IbHK1a, IbHK1b) contained HK, HATPase, REC, and TM domains, and were predicted to be localized in the plasma membrane. Both CKI1-like HK (IbCKI1) and AHK5-like HK (IbHK5) contained only one member with HK, HATPase, and REC; however, the IbHK5 lacked TM domain, and was deemed to possibly localize in the cytoplasm. Two ethylene receptor-like HKs (IbETR1 and IbERS1) contained the TM, C2H4, GAF, and HK domains. IbETR1 possessed the REC domain, while IbERS1 lacked the REC domain, which is consistent with those in other plants. C2H4 is an ethylene-binding domain, suggesting their potential function as ethylene receptors. Additionally, another 10 HKLs were divided into two subgroups: ethylene receptor-like HKLs and phytochrome-like HKLs, in which the H site of the HK domain is replaced by other amino acids. The five ethylene receptor-like HKLs (IbHKL1-5) were composed of TM, C2H4, GAF, HKL, and REC domains. Another five phytochrome-like HKLs (IbHKL6-10) contained HKL, HATPase, GAF, PHY, and PAS domains. However, all the phytochrome-like HKLs lacked the REC and TM domains, and were predicted to be localized in the cytoplasm and nucleus. The PHY domain is a chromophore-binding domain. The presence of chromophore-binding domains demonstrated that they were candidate proteins involved in phytochromes.
3.3. HP proteins in I. batatas
Eleven HP proteins were identified in sweet potato with a conserved His-containing phosphotransfer domain (HPt) corresponding to motif 1 (Supplementary Figure S3). The ORF lengths of the HP genes ranged from 339 bp to 961 bp, which encoded polypeptides of 113 aa to 287 aa with predicted molecular weights ranging from 12.8 kD to 32.8 kD. Furthermore, the theoretical pI ranged from 4.50 to 9.17. All the HP proteins were predicted to be localized in the cytoplasm and nucleus (Supplementary Table S3). Based on the phylogenetic analysis and protein sequence alignment results, these 11 HP proteins could be divided into two groups: HP and pseudo-HP. Five HP proteins (IbHP1-5) had a conserved HPt domain, whereas the other six HP proteins (IbHP6-11) had a pseudo-HPt that lacked the His phosphorylation site (Supplementary Figure S4).
3.4. RR proteins in I. batatas
Fifty-nine RR proteins, including 19 type-A RRs, 13 type-B RRs, 7 type-C RRs, and 20 pseudo-RRs were identified in I. batatas (Table 1). The type-A RRs (IbRR1-19) had relatively short ORF lengths with only one conserved REC domain corresponding to motifs 1, 2, and 3 as identified using MEME (Supplementary Figure S5). The type-B RRs are usually transcription factors characterized by one REC domain in the N terminal end and one Myb domain in the C terminal end. Eleven type-B RRs (IbRR20-30) possessed both the REC domain and Myb domain (motif 4), whereas IbRR31 and IbRR32 had only the REC domain. Seven type-C RRs (IbRR33-39) also possessed only one REC domain with shorter ORF lengths, which is similar to the structures of type-A RRs. In addition, 20 pseudo-RRs (PRRs) could be further divided into two subgroups: Clock PRR and type-B PRR. Compared with authentic RRs, PRRs had a pseudo-REC domain, in which the conserved D site was substituted with other amino acid residues (Supplementary Figure S6). Clock PRRs are featured with a CCT domain, which plays an important role in regulating circadian rhythm and controlling flowering time (Hayama and Coupland, 2003). Nine clock PRRs (IbPRR1-9), except for IbPRR1 and IbPRR4, contained the conserved CCT domain (motif 5) and pseudo-REC domain. Eleven pseudo-RRs (IbPRR10-20) were assigned to type-B PRRs. IbPRR10, 11, 12, 13, 16, and 17 possessed both one pseudo-REC domain and one Myb domain, which were predicted to have transcription factor functions. However, IbPRR14, 15, 18, 19, and 20 had only the pseudo-REC domain.
3.5. Phylogenetic analysis of plant TCS proteins
To further reveal the phylogenetic relationships among TCS genes, 179 HK(L)s, 72 HPs, and 373 RRs from Arabidopsis (Hwang et al., 2002), rice (Pareek et al., 2006), maize (Chu et al., 2011), soybean (Mochida et al., 2010), tomato (He et al., 2016a), sweet potato, and sweet potato’s wild relatives were used to perform multiple alignments and construct phylogenetic trees. The 179 HK(L) proteins from the nine land plant species were perfectly divided into six distinct subfamilies (Figure 1), namely, cytokinin receptors, CKI1, AHK1, AHK5, ethylene receptors, and phytochromes. This is consistent with previously reported classification results (Hwang et al., 2002; Schaller et al., 2008; Huo et al., 2020). Each subfamily contained representative HK(L) proteins from the nine species, indicating that the subfamilies were evolutionarily conserved and already formed before the divergence of monocotyledon and dicotyledon. I. batatas have the nearest relationships with I. trifida, I. trilobal, and I.nil, their HK(L) proteins were clustered in a single clade among each distinct subfamily. Overall, the HK(L) proteins in the Ipomoea genus were more closely related to those in dicotyledons than those in monocotyledons.
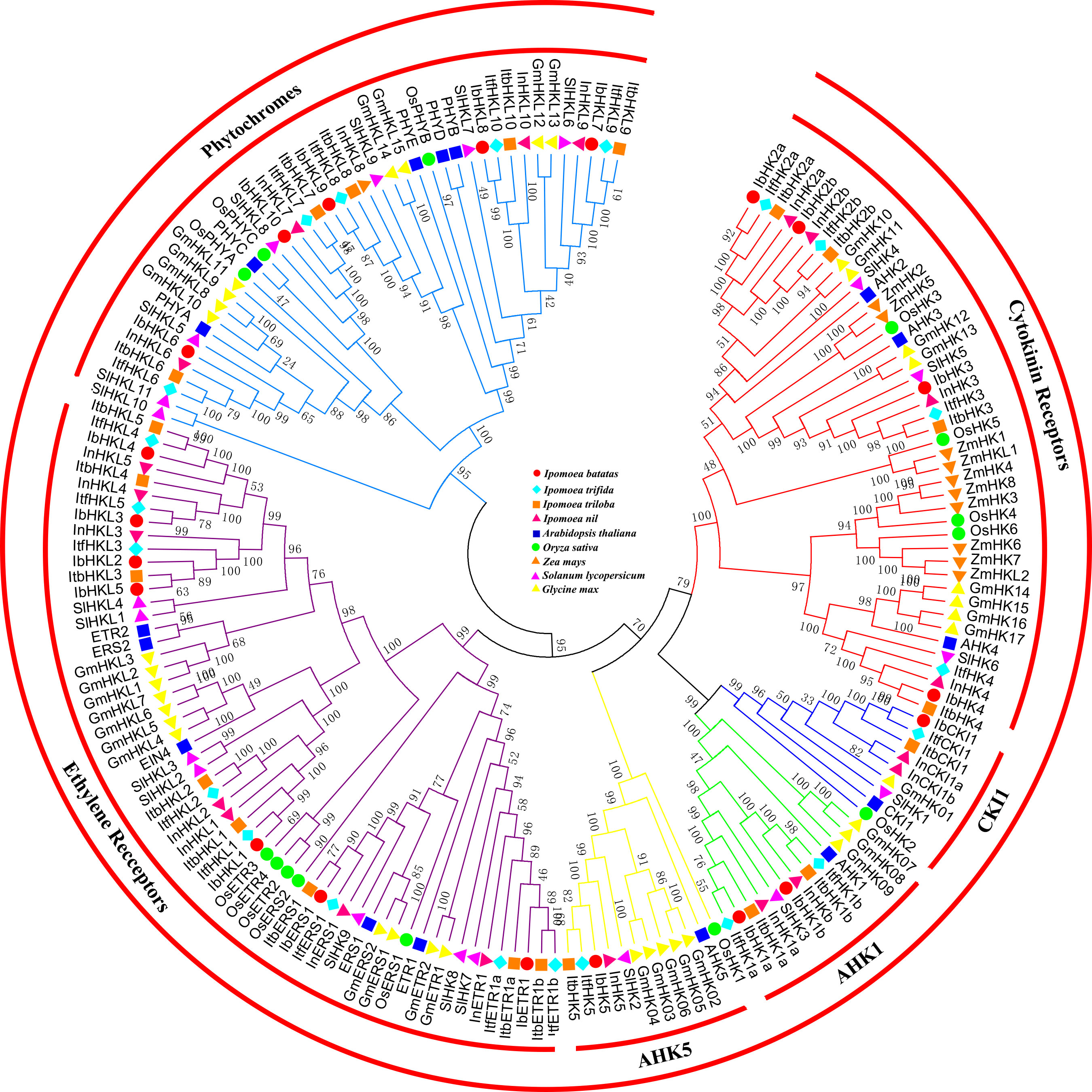
Figure 1 Phylogenetic relationships of HK(L)s in Arabidopsis, rice, maize, tomato, soybean and sweet potato. The phylogenetic tree was constructed using the neighbor-joining method with MEGA 7.0 software. HK(L)s in various species were marked with different labels and diverse subgroups of HK(L)s were highlighted by different colors.
All the authentic and pseudo-HP proteins from these nine species mentioned above were mainly divided into two subfamilies, which were labeled with red and blue lines in Figure 2. The authentic HP proteins of IbHP1-3 that were homologous to Arabidopsis (AHP1, AHP2, AHP3, and AHP5) occupied an independent clade, which may also act as positive regulators in cytokinin signaling (Hutchison et al., 2006). However, IbHP4 and IbHP5 were clustered into the same group with AHP4, which showed relatively distinct genetic relationships from the other authentic HP proteins. The pseudo-HP proteins showed more scattered distributions in the phylogenetic tree; specifically, the pseudo-HP proteins in the monocotyledon and dicotyledon plants occupied two different clades. Notably, IbHP7-11, together with ItfHP6, InHP7, and InHP8, occupied a single distinct clade, considering these extra members lacked the homologous counterparts in Arabidopsis, their evolutionary mechanisms and functional roles require further investigation.
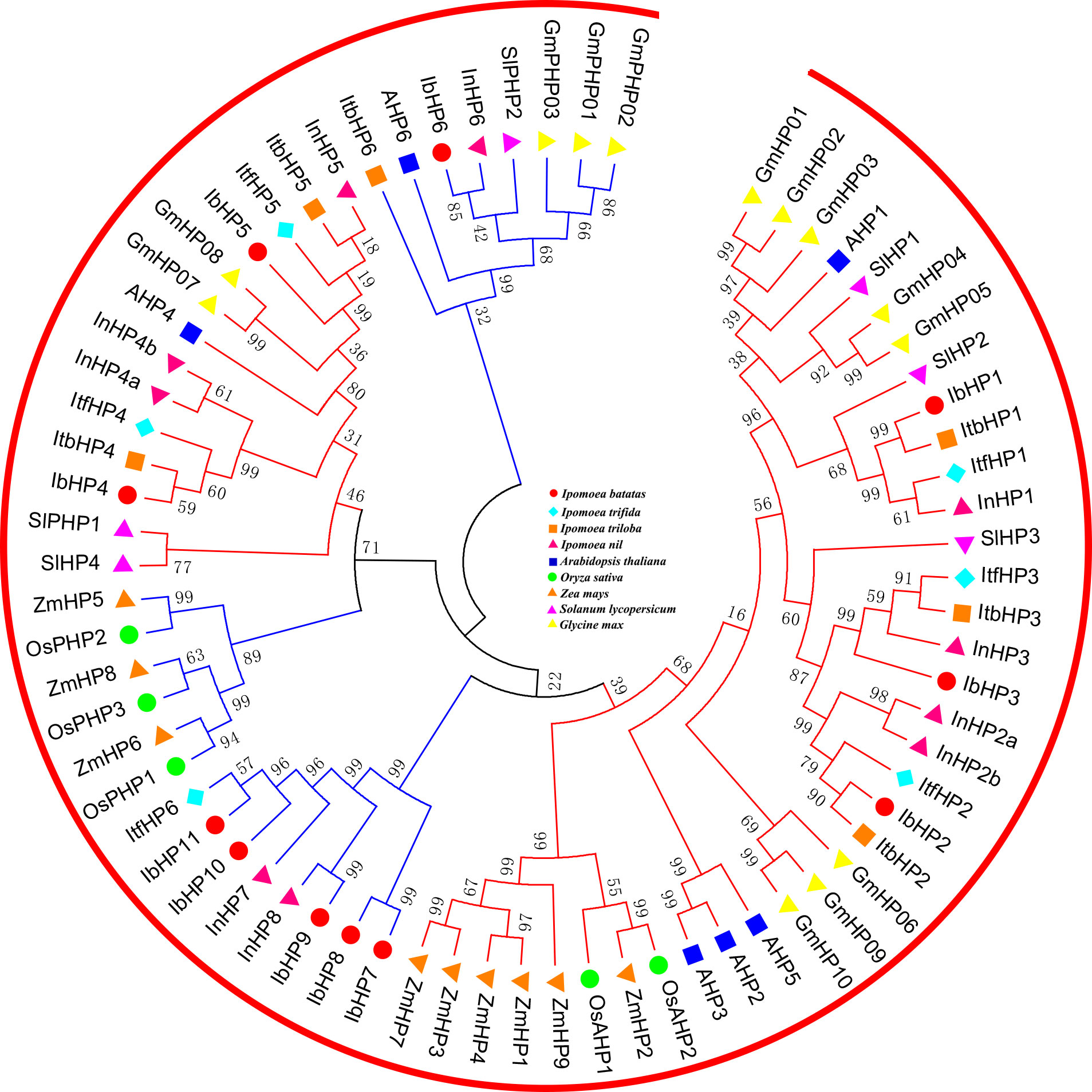
Figure 2 Phylogenetic relationships of HPs in Arabidopsis, rice, maize, tomato, soybean and sweet potato. For other details, see Figure 1.
All the RR proteins from these nine species were grouped into four subfamilies: type-A RR, type-B RR, type-C RR, and PRR (Figure 3). Type-A RR proteins occupied a single clade, with the majority of the members in a lager subclade and several members from monocotyledon plants in the other clade. Type-B RR proteins could be further subdivided into three subgroups. The type B-I RR subgroup was most predominant, and contained the RRs from all the nine species. The type B-II RR subgroup contained only five Arabidopsis members (ARR13, ARR19, ARR20, ARR21, and ARR23), and no homologs of other plants were clustered in this clade. Previously, we had analyzed the phylogenetic tree containing the species of the Chinese cabbage. While corresponding homologs were identified in Chinese cabbage, no members from rice and soybean were clustered in this subgroup, which was consistent with these results (Liu et al., 2014). We proposed that the type B-II RR subgroup members occurred only after the diversification of Cruciferae and other families. Instead, the type B-III RR subgroup contained several members from the monocotyledon plants, rice and maize, indicating that this group most likely occurred after the diversification of monocotyledon and dicotyledon. Type-C RRs shared a similar structure with type-A RRs, but their genetic distance was not closely related. Previous studies have suggested that type-C RRs might be the oldest RRs, and type-A RRs might have evolved from type-C RRs due to mutations in their promoters (Pils and Heyl, 2009). Pseudo-RR members were mainly divided into two groups, i.e., clock PRRs and type-B PRRs. These Pseudo-RR members had relatively closer phylogenetic relationships with type-B RR members. Similarly, the PRR subfamily contained members from all the nine species. It is important to note that IbPRR18, IbPRR19, and IbPRR20 were not grouped into any subfamily or clade, most likely due to the lower amino acid sequence similarity.
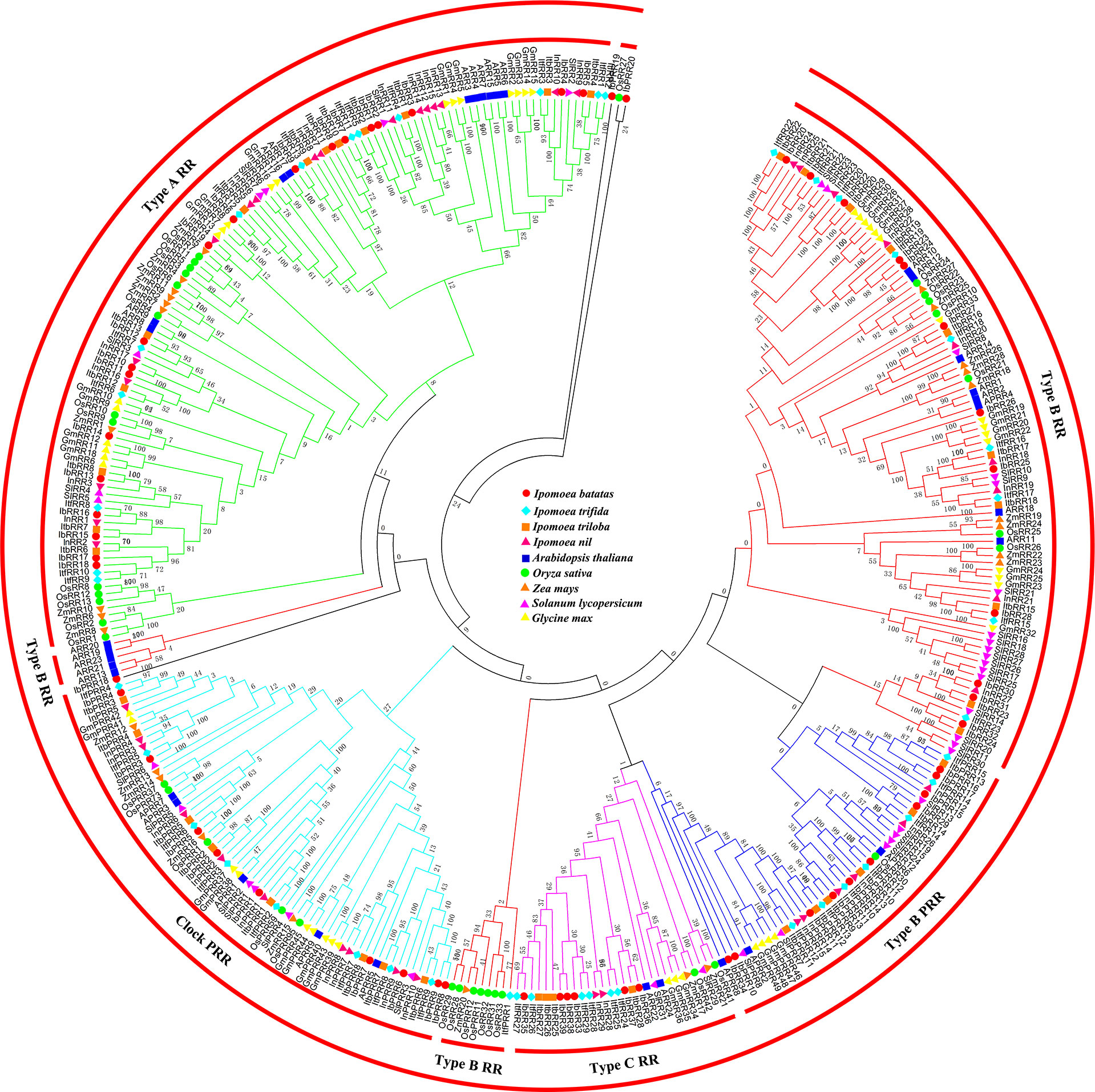
Figure 3 Phylogenetic relationships of RRs in Arabidopsis, rice, maize, tomato, soybean and sweet potato. For other details, see Figure 1.
3.6. Genomic distribution and gene duplication in sweet potato and its wild relatives
The chromosomal location of each TCS gene was determined based on the genomic information of I. batatas, and 90 TCS genes were mapped on the 15 chromosomes with a notably uneven distribution (Figure 4, Supplementary Figure S7). Among which Chr02, Chr04, Chr05, Chr02, Chr06, Chr08, Chr12, and Chr14 possessed relatively more TCS genes. Gene duplication events play critical roles in gene family evolution, especially the segmental and tandem duplications are the two dominated approaches for gene family members expansion and novel gene functions generation in plants (Cannon et al., 2004; Kong et al., 2007). The gene duplication events were investigated in the sweet potato TCS gene family, and the results revealed 20 segmental duplication gene pairs involved 32 genes, including HK(L)s, HPs, and RRs (Table 2, Supplementary Table S7). Specifically, five segmental duplication gene pairs that involved nine genes were present in the HK(L) gene subfamily, with a proportion of 45%. Moreover, the percentage of gene numbers involved in segmental duplications for the HP, type-A RR, type-B RR, and pseudo-RR subfamilies were approximately 30%. However, no segmental duplicated genes were found in the type-C RR subfamily. In addition, nine tandem duplication gene pairs that involved 21 genes were also identified (Supplementary Table S8). The tandem duplicated genes were only present in the RR subfamily. Most notably, the percentage of gene numbers involved in tandem duplications for the type-A RR and type-C RR subfamilies were 47.4% and 42.9%, respectively. These results confirmed that the expansion of TCS genes in I. batatas was mainly attributed to both segmental and tandem duplications. In contrast, the expansion of HK(L)s and HPs mainly depended on segmental duplications, whereas the expansion of type-C RRs mainly depended on tandem duplications.
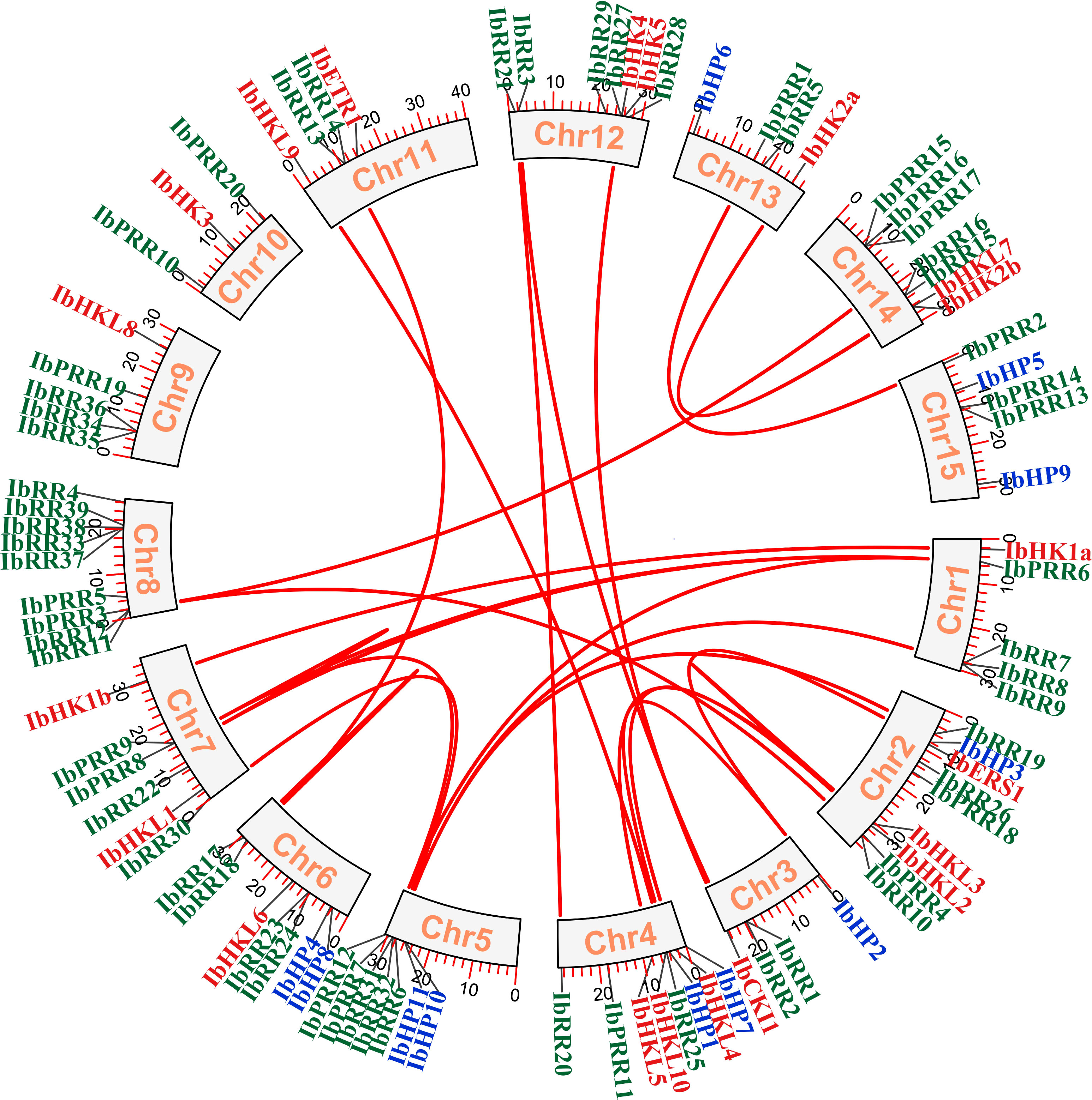
Figure 4 Synteny relationships of duplicated TCS genes in Ipomoea batatas. Lines between the chromosomes indicate duplicated gene pairs. Bars located in the chromosomes represent the chromosome length (cM). HK(L)s, HP and RR gene family members were highlighted with different color fonts.

Table 2 Comparisons of segmental and tandem duplicated TCS genes in Ipomoea batatas.
The synonymous rate (Ks), non-synonymous rate (Ka), and Ka/Ks of these duplicates were calculated, and divergence time was determined using the Ks values (Supplementary Table S7). The Ks values ranged from 0.01278–3.422052, corresponding to the divergence time from 0.423–114.07 MYA (million years ago). The long time span suggests that TCS gene expansion and evolution occurred over millions of years, and not only at specific time points. Furthermore, the Ka/Ks values of the segmental duplications were less than 1, indicating that they all underwent purification selection.
To further reveal the TCS gene evolution in sweet potato and its two wild relatives, I. trifida and I. trilobal, the synteny relationships between their TCS genes were analyzed (Figure 5). In total, 73 segmental duplicated gene pairs were identified between I. batatas and I. trifida, and 83 segmental duplicated gene pairs were identified between I. batatas and I. trilobal (Supplementary Table S9, S10). This finding indicates their close relationships and conserved evolution. Likewise, the Ks and Ka modes for these segmental duplicated paralogs were also determined. Results showed the Ka/Ks values of the segmental duplications between I. batatas and its two wild relatives, I. trifida and I. trilobal, were all less than 1, indicating that these paralogs also underwent purification selection.
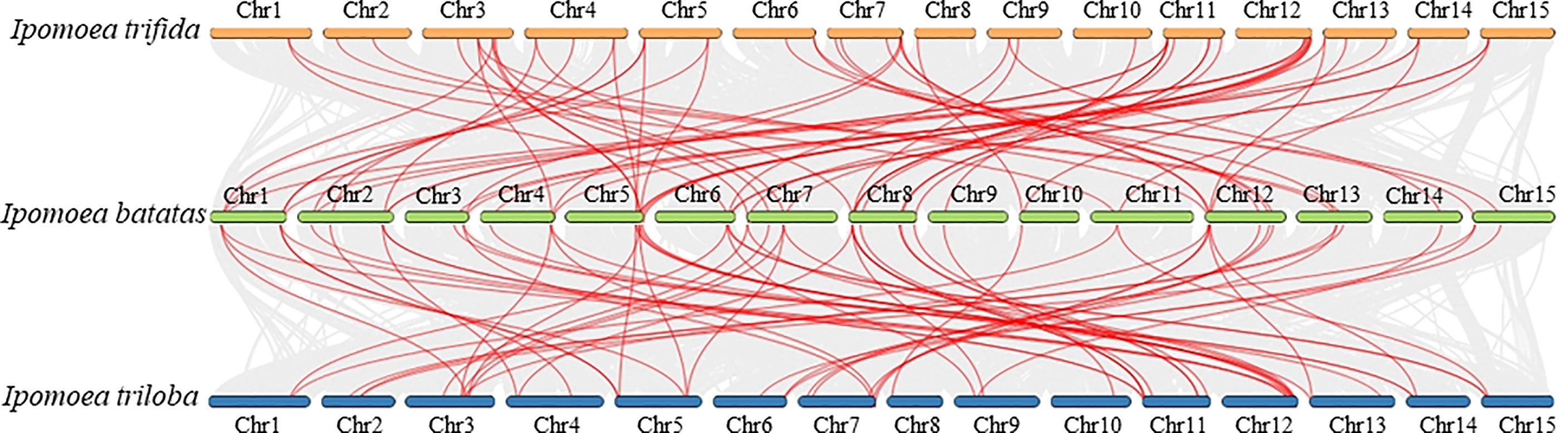
Figure 5 Synteny relationships of duplicated TCS genes in Ipomoea batatas, Ipomoea trifida and Ipomoea trilobal. Lines between the chromosomes indicate duplicated gene pairs.
3.7. Analysis of cis-elements in the putative promoter regions of TCS genes in I. batatas
Cis-regulatory elements, which are located upstream of genes and act as binding sites for TFs, have essential functions in determining the tissue-specific or stress-responsive expression patterns of genes (Le et al., 2012). To further understand the transcriptional regulation and potential functions of these TCS genes, 1,500 bp regions upstream of the transcriptional start site were selected to identify cis-regulatory elements. A number of abiotic stress-related (e.g., drought, high salinity, low temperature, and wound) and hormone-related (e.g., ABA, auxin, ethylene, GA, MeJA, and SA) cis-elements were found in the putative promoters of TCS genes in I. batatas. These cis-elements are counted and classified in Supplementary Table S11. The occurrence of these cis-elements suggests that these TCS genes might have functions in abiotic stress adaptations and various hormone signaling processes.
3.8. Expression profiles of TCS genes in various organs
To obtain first insight into the functions of TCS genes during the vegetative and reproductive developmental stages of the sweet potato, qRT-PCR was performed to analyze the expression patterns in various organs or tissues such as the stem roots, fibrous roots, tuberous roots, stems, leaves, and flowers (Supplementary Figure S8). This study only focused on the HKs, HPs, and type-B RRs. IbRR23 and IbRR24 were analyzed together as one gene because they shared high similarity and transcripts were not distinguished by qRT-PCR. The expression profiles of 27 TCS genes were clustered in a heat map, but the transcripts of IbHK2b and IbRR29 were not detected and thus not included here. As shown in Figure 6, the majority of the TCS genes showed relatively high expression levels in three types of roots. In particular, the IbHK1a, IbHK5, IbRR22, IbHK1b, IbRR25, IbRR23&24, and IbHP1 were grouped together and were mainly expressed in both the stem roots and fibrous roots. Consistently, Arabidopsis HK1 and HK5 were also mainly expressed in the roots, where functional studies revealed their essential roles in various abiotic stress responses (Tran et al., 2007; Wohlbach et al., 2008; Pham et al., 2012; Kumar et al., 2013). The corresponding Arabidopsis homologs in sweet potato (IbHK1a, IbHK1b, and IbHK5) were also most likely involved in stress adaptations. Moreover, IbHP5, IbERS1, IbRR21, IbHKL1–IbHKL4 showed relatively higher expression levels in tuberous roots, indicating their potential functions in tuberous root expansion and yield formation. IbHKL5, IbHP2, IbHP3, IbHP5, and IbRR22 were expressed abundantly in stems, while two cytokinin receptors (IbHK3 and IbHK4) exhibited the highest expressions in leaves. IbCKI1, IbERS1, IbRR21, and IbHKL2–IbHKL4 had relatively higher expressions in flowers, suggesting their potential roles in reproductive development. Notably, IbCKI1 was exclusively expressed in flowers, and IbCKI1 was homologous with Arabidopsis CKI1. Previous studies showed that AtCKI1 was expressed almost exclusively in the ovules, and is involved in regulating female gametophyte development (Deng et al., 2010; Yuan et al., 2016). Similar expression profiles may suggest similar functions, thus IbCKI1 was also proposed to regulate female gametophyte development in sweet potato.
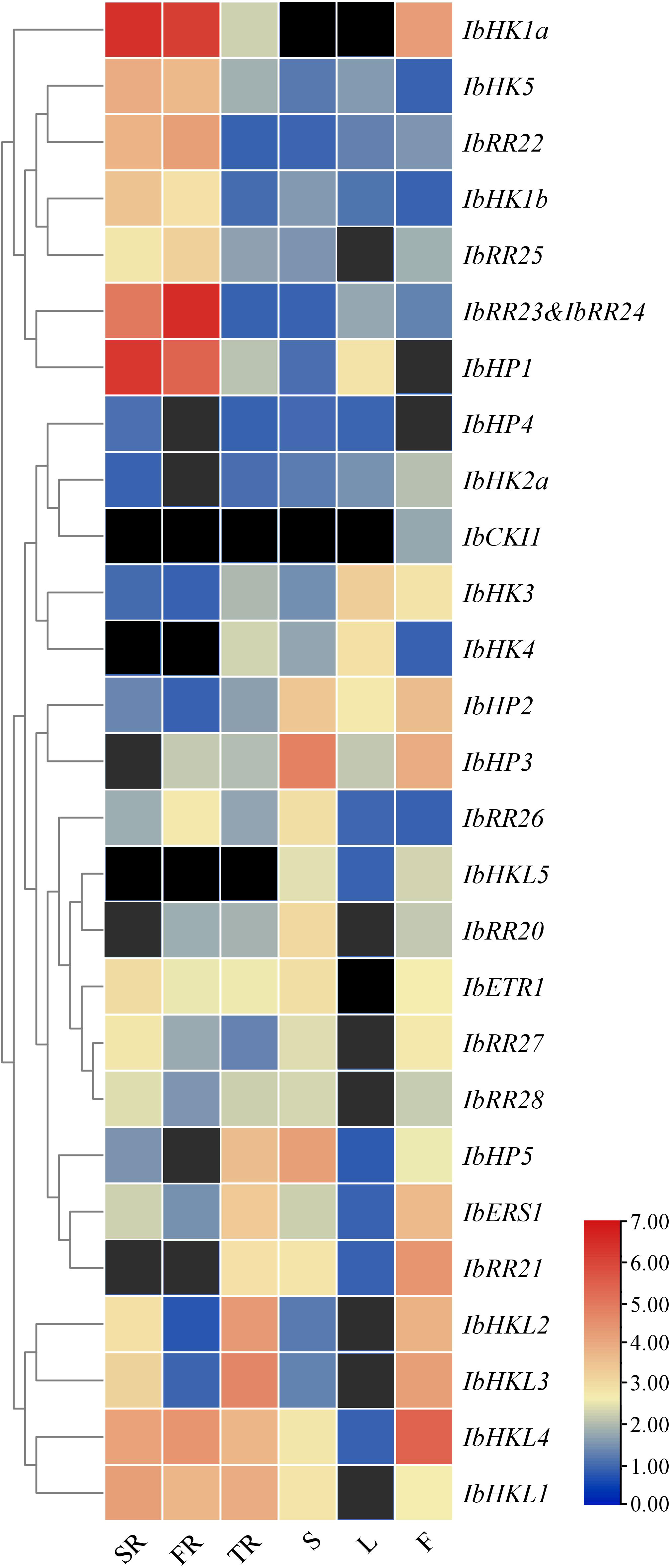
Figure 6 Heat map representation for the organ-specific TCS gene expression profiles in Ipomoea batatas. SR, stem roots, FR, fibrous roots, TR, tuberous roots, S, stems, L, leaves and F, flowers. The expression levels of genes are presented using fold-change values transformed to Log2 format. The Log2 (fold-change values) and the color scale are shown at the bottom of heat map. Genes that were not expressed were shown with black boxes.
3.9. Expression profiles of TCS genes in response to various abiotic stresses
Mounting evidence has shown that TCS is involved in the adaptation of plants to various environmental stresses. Because roots were generally considered as the primary organs involved in these stresses, the above mentioned 27 TCS genes examined were also used to study the response patterns to various abiotic stresses such as drought, high salinity, heat, cold, and ABA in the roots. In total, 25 root-expressed genes showing an abiotic stress response were clustered in a heat map with the exception of two unexpressed genes, IbCKI1 and IbHP1. As shown in Figure 7A, seven genes (IbHKL5, IbHP5, IbHK1a, IbHKL2, IbHKL3, IbRR21, and IbHK2a) were clustered in a clade, and their expressions were clearly upregulated upon PEG treatment. Meanwhile, another clade contained three genes (IbHK1b, IbHP4, and IbHP3) whose expressions were continuously suppressed and remained at low levels at 48 h. For the high salinity treatment, the transcripts of IbHP3 were consistently suppressed and even decreased to ~10% of the control level. The expressions of IbHK1a and IbHK1b were primarily downregulated at 1 h, then continuously upregulated and peaked at 24 h, before decreasing rapidly at 48 h. In contrast, IbHKL5, IbHP5, IbRR22, IbHK3, and IbRR28 were primarily induced with upregulated expressions, then suppressed at 12 h or 24 h, and finally decreased to almost basal levels at 48 h (Figure 7B). For the high temperature treatment, most of the TCS genes under examination responded to heat treatment and exhibited upregulated expressions (Figure 7C), especially IbHK1a, IbHKL4, IbHK3, IbHP4, IbHK5, IbRR28, IbHK1b, and IbHP3. This suggests their positive functions under high temperature conditions. However, the IbHP5 transcript was primarily induced, subsequently repressed after 6 h treatment, and almost undetectable at 48 h. Unlike those for the heat treatment, the response patterns to the cold treatment for the TCS genes seemed to be more diverse. As shown in Figure 7D, IbHKL5, IbHK2a, IbHK3, IbHK4, and IbHP2 responded positively to the heat treatment with notably upregulated expressions. Contrarily, some representative genes, such as IbRR21, IbHK1b, IbHKL2, IbHKL3, and IbRR22, their transcripts were clearly suppressed with downregulated expressions. Interestingly, the response patterns to the heat and cold treatments of the TCS genes were generally opposite. For example, the transcripts of IbHK1a, IbHK1b, IbHK5, IbERS1, IbHKL2, IbHKL3, IbHKL4, IbHP4, IbHP5, and IbRR28 were observably induced upon heat treatment but they were suppressed in cold treatment conditions. ABA is an important phytohormone involved in regulating various environmental stress responses. Studies have also suggested the presence of intensive interactions and crosstalk between cytokinins and ABA, as well as their signaling pathways (Ha et al., 2012). In light of this, we also examined the effects of exogenous ABA on the TCS gene expressions in sweet potato. As shown in Figure 7E, most of the examined TCS genes were generally upregulated upon ABA treatment, which was consistent with the results that ABA-responsive elements (ABRE) were widely identified in the putative promoters of TCS genes in sweet potato.
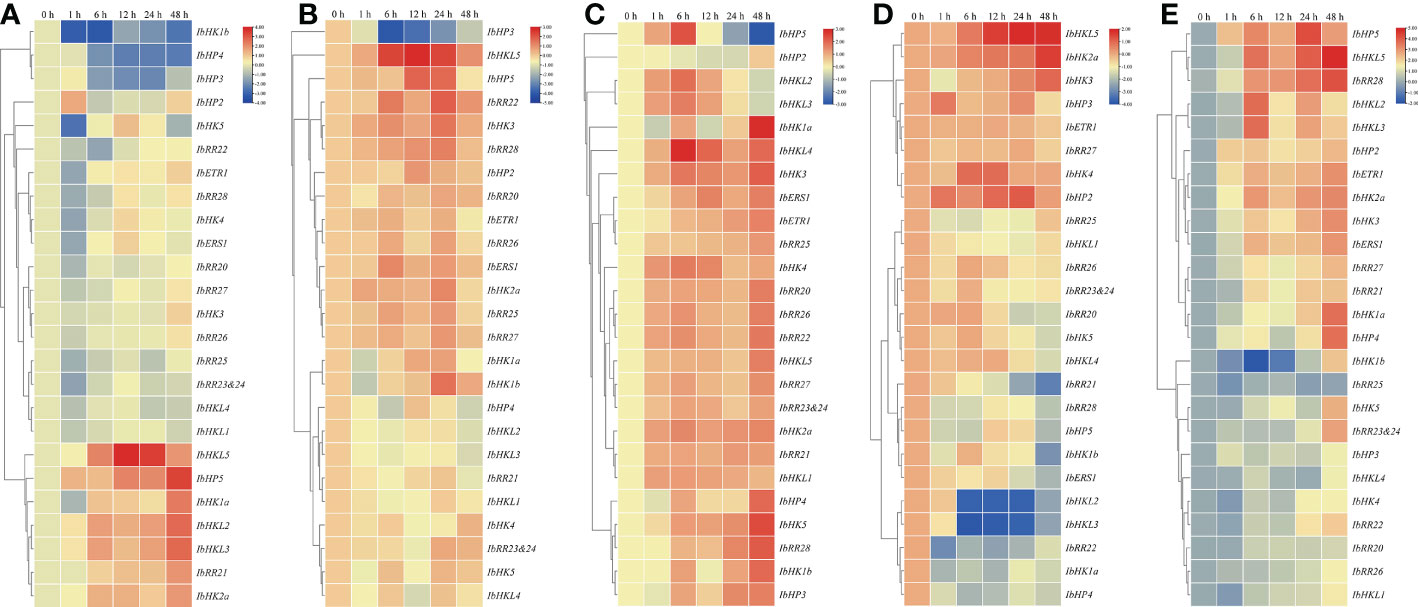
Figure 7 Heat map representation for the response patterns to drought (A), salt stress (B), heat (C), cold stress (D), and exogenous ABA (E) treatment TCS genes in Ipomoea batatas. The expression levels of genes are presented using fold-change values transformed to Log2 format compared to control.
3.10. The protein-protein interaction network of TCS elements in I. batatas
Since the discovery of the two-component multi-step phosphorylation system in plants, the protein-protein interactions of the TCS elements were investigated to reveal the phosphorylation signal transduction network in Arabidopsis, rice, and polar (Dortay et al., 2008; Sharan et al., 2017; Héricourt et al., 2019). To gain first insight into the interaction network of the TCS proteins in sweet potato, three osmosensing HKs (IbHK1a, IbHK1b, IbHK5) involved in the abiotic stress response were chosen to identify the downstream HPs-RRs counterparts using yeast two hybrid assays. The bait vector self-activation was primarily detected but no self-activation was detected. HK-HP interactions were first studied, and the results showed that IbHK1a, IbHK1b, and IbHK5 could interact with IbHP1, IbHP2, IbHP4, and IbHP5; no interactions were detected for IbHP3 (Figure 8). The interaction assays between five HPs (IbHP1–IbHP5) and nine type-B RRs (IbRR20–IbRR28) were also conducted. As shown in Figure 9, IbHP1 could interact with IbRR20–IbRR25 and also IbRR27–IbRR28, even though only relatively weak interaction activities were detected between IbHP1 and IbRR22. However, no interactions were detected between IbHP1 and IbRR26. Moreover, no interaction was detected between IbHP5 and IbRR20, IbRR22, and IbRR26, but IbHP5 could interact well with the other six type-B RRs. Additionally, IbHP2 and IbHP4 could interact with all nine type-B RRs while showing strong interaction activities. Remarkably, interactions were never detected between IbHP3 and type-B RRs with repeated trials. Considering that no interaction activity was detected between both upstream HKs and downstream type-B RRs, we assumed that IbHP3 was most likely not involved in the TCS signal transduction system in sweet potato. Based on these results, a protein-protein interaction map was created to present a set of IbHK-IbHP-IbRR phosphorylation signal transduction pathways in sweet potato (Figure 10).
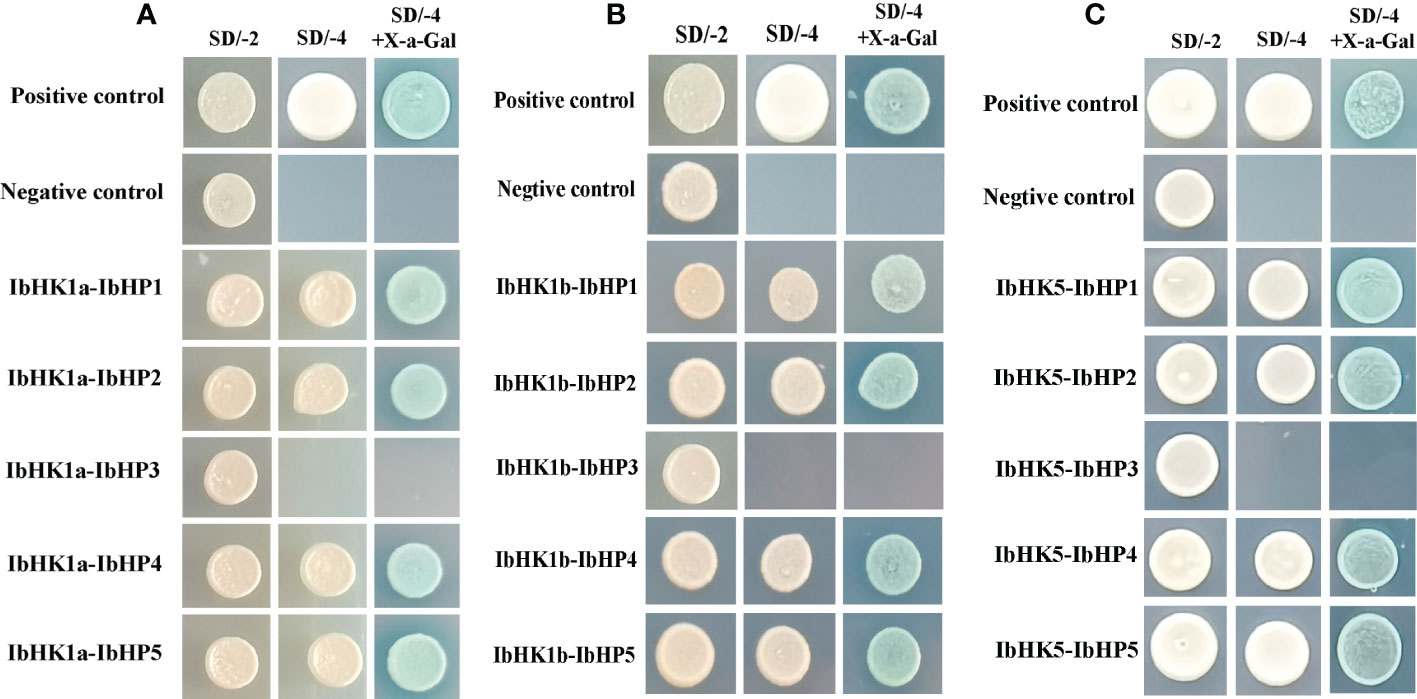
Figure 8 Protein-protein interaction studies among IbHKs and IbHP1~IbHP5 with yeast-two hybrid assays. (A) Interactions between IbHK1a and IbHP1~IbHP5, (B) Interactions between IbHK1b and IbHP1~IbHP5, (C) Interactions between IbHK5 and IbHP1~IbHP5.

Figure 9 Protein-protein interaction studies among IbHP1~IbHP5 and IbRR20~IbRR28 with yeast-two hybrid assays. (A) Interactions between IbHP1 and IbRR20~IbRR28, (B) Interactions between IbHP2 and IbRR20~IbRR28, (C) Interactions between IbHP3 and IbRR20~IbRR28, (D) Interactions between IbHP4 and IbRR20~IbRR28, (E) Interactions between IbHP5 and IbRR20~IbRR28.
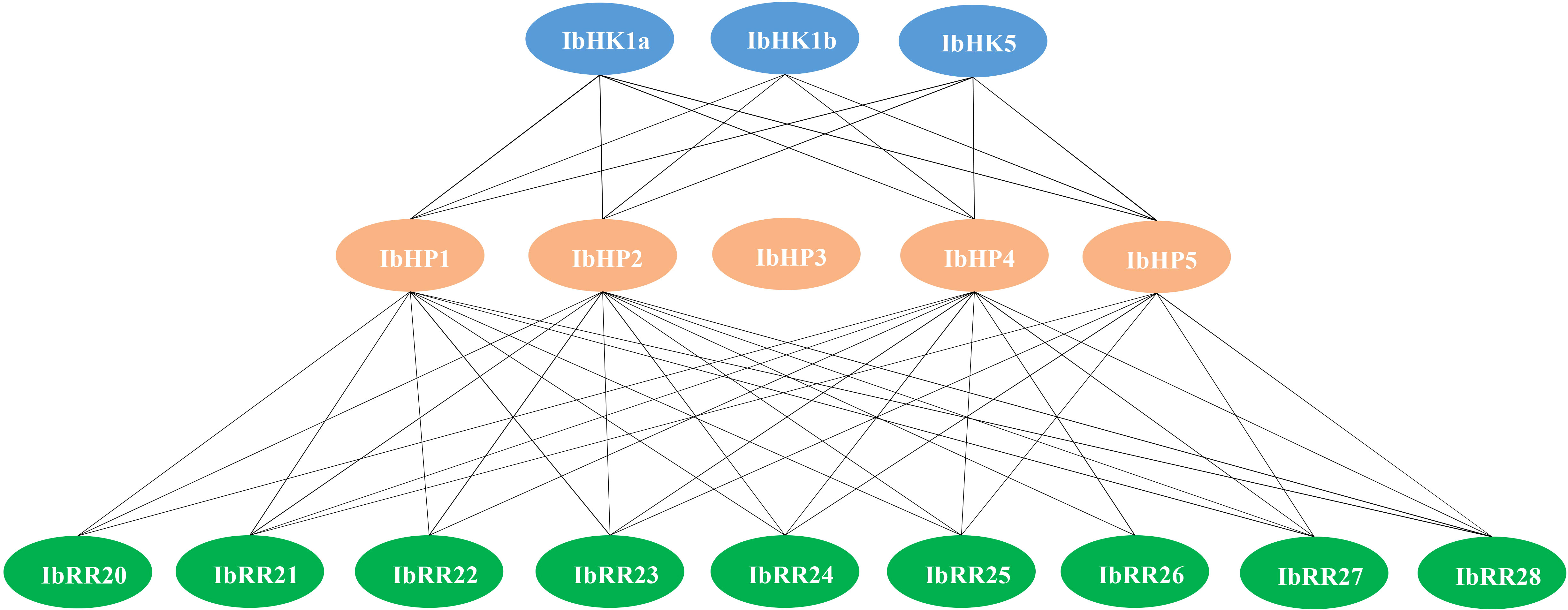
Figure 10 Schematic presentation of interacted TCS proteins in Ipomoea batatas. Lines indicate the direct protein-protein Interactions detected with yeast-two hybrid assays in this study.
4. Discussions
In this study, 90 TCS members consisting of 20 HK(L)s, 11 HPs, and 59 RRs were identified in the sweet potato (I. batatas) genome. Furthermore, 71 and 68 TCS members were found in its two wild relatives, I. trifida and I. trilobal. In addition, 75 TCS members existed in the genome of I. nil. According to previous studies, the calculated genome size of I. batatas, I. trifida, I. trilobal, and I. nil is 851.4 Mb, 476.4 Mb, 495.9 Mb, and 734.8 Mb, respectively (Hirakawa et al., 2015; Hoshino et al., 2016; Yang and Moeinzadeh, 2017; Wu and Lau, 2018; Li et al., 2019; Yan et al., 2022). The I. batatas genome possessed relatively more genes than the other three Ipomoea genus relatives, which was consistent with their genome size. Comparatively, the I. batatas genome possessed more HPs, RRs, and particularly large numbers of pseudo-HPs, but not more HK(L)s.
To further reveal the mechanisms of TCS gene family members expansion in I. batatas genome, gene duplication events were investigated. Twenty segmental duplicated gene pairs involved 32 genes and nine tandem duplicated gene pairs involved 21 genes were identified, suggesting that the expansion of TCS genes in I. batatas was attributed to both segmental and tandem duplications. He et al. (2016a) analyzed the gene duplications among TCS genes in tomato, and also found that segmental duplication and tandem duplication events both contributed to the expansion of these TCS gene members (He et al., 2016a). However, 10 segmental duplicated gene pairs were identified in Arabidopsis, accounting for 35.71% of the total TCS genes. Furthermore, 66 out of the 98 TCS genes in soybean were involved in segmental duplication events, whereas the tandem duplication of TCS genes was not found in Arabidopsis or soybean (Mochida et al., 2010; Liu et al., 2014). Similarly, in Chinese cabbage, 61 of the 85 TCS genes were found to be segmental duplications, whereas only one tandem duplicated gene pair was found (Hirakawa et al., 2015). These results suggest that segmental duplication was the main mechanism of TCS gene expansion in Arabidopsis, Chinese cabbage, and soybean. Notably, only three pairs of tandem duplications and one pair of segmental duplications were identified in the Cucurbitaceae crops, cucumber and watermelon (He et al., 2016b), which indicates that mechanisms of TCS gene expansions in various plants may be quite distinctive.
I. trifida is assumed to be the diploid progenitor of I. batatas, after the B2 subgenome specialization at an estimated 1.3 MYA, I. batatas alone underwent two whole-genome duplication events at approximately 0.8 MYA and 0.5 MYA (Yang and Moeinzadeh, 2017). We analyzed the synteny relationships of TCS genes between I. batatas and I. trifida, and found 73 segmental duplicated gene pairs. This demonstrates that the evolution of TCS genes between these two species was fairly conserved; more importantly, the gene expansions mainly occurred before their specification.
The TCS signaling pathway plays essential roles in regulating plant growth, development, and response to various environmental stimuli. The expression profiles of TCS genes in stem roots, fibrous roots, tuberous roots, stems, leaves, and flowers of the sweet potato were analyzed, and their response patterns under drought, high salinity, heat, cold, and ABA in roots were also conducted. The majority of TCS genes showed relatively high expression levels in roots, which was consistent with TCS gene expression profiles in other plants, such as Arabidopsis (Huo et al., 2020), tomato (He et al., 2016a), soybean (Mochida et al., 2010), and Chinese cabbage (Liu et al., 2014). This is most likely due to the fact that cytokinins are mainly synthesized in the roots. Roots are believed to be the most important primary organs in the response to drought and high salinity stresses. IbHK1a, IbHK5, IbRR22, IbHK1b, IbRR25, IbRR23&24, and IbHP1 were grouped together and were mainly expressed in the stem roots and fibrous roots. In fact, Arabidopsis HK1 and HK5 were also mainly expressed in roots, and functional studies revealed their essential roles in various abiotic stress responses (Tran et al., 2007; Wohlbach et al., 2008; Pham et al., 2012; Kumar et al., 2013). Coincidentally, transcripts of homologous genes in sweet potato could also be induced or suppressed with PEG or NaCl treatment, suggesting their potential functional roles in environmental stress adaptations. Our expression analysis results could provide valuable information to further elucidate the functional roles of sweet potato TCS genes under abiotic stress conditions.
Information on the protein-protein interactions of TCS elements was indispensable for the correct or precise phosphorylation signal transduction, protein interaction map was already depicted in Arabidopsis, rice, and polar (Dortay et al., 2008; Sharan et al., 2017; Héricourt et al., 2019). To gain first insight into the protein-protein interaction network of the TCS signal transduction pathway in sweet potato, three osmosensing proteins (IbHK1a, IbHK1b, and IbHK5) were chosen to identify the downstream HPs-RRs counterparts using yeast two hybrid assays. The protein-protein interaction network was schematically presented. The results showed that IbHK1a, IbHK1b, and IbHK5 could interact with IbHP1, IbHP2, IbHP4, and IbHP5, but no interactions were detected for IbHP3. Moreover, interactions were never detected between IbHP3 and nine type-B RRs. Still it remains to be resolved whether IbHP3 could interact with other HK proteins or type-A RRs. Similarly, AHP4 could not interact with any of the AHKs, and is a non-functional protein in Arabidopsis (Dortay et al., 2008). Thus, we proposed that it is likely that IbHP3 was not involved in the TCS signal transduction in sweet potato. In this study, only three osmosensing proteins were chosen for the HK-HP interaction assays in sweet potato, more efforts are needed to include other HKs and type-A RRs in the protein interaction network.
5. Conclusion
This study found 90 putative members of the TCS family in the genome of I. batatas, including 20 HK(L)s, 11 HPs, and 59 RRs. Additionally, 71, 68, and 75 TCS genes were found in I. trifida, I. trilobal, and I. nil, respectively. A comparative analysis of diverse plant species revealed that the evolution of TCS members was highly conserved. Twenty segmental duplication gene pairs and nine tandem duplication gene pairs were identified, suggesting that both segmental and tandem duplications contributed to the expansion of TCS genes in I. batatas, and this expansion occurred before the specification of I. batatas and its diploid progenitor I. trifida. The majority of TCS genes were specifically or preferentially expressed in certain organs, especially in the three types of roots (stem roots, fibrous roots, tuberous roots). The promoter regions of TCS genes exhibited numerous abiotic stress-related and hormone-related cis-elements, indicating their potential roles in abiotic stress adaptations and various hormone signaling processes. Moreover, their response patterns to PEG, NaCl, cold, heat, and exogenous ABA were verified. Protein-protein interactions were conducted among three osmosensing proteins (IbHK1a, IbHK1b, IbHK5), five HPs (IbHP1–IbHP5), and nine type-B RRs (IbRR20–IbRR28); thereafter, the interaction network was preliminary established. These results could provide insight into the characterization of the TCS genes in sweet potato, and further the development of functional studies on these TCS genes under abiotic stress conditions.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
RH performed the experiments, analyzed the data, and drafted the manuscript. YZ and TL participated in qRT-PCR experiments and data analysis. MX, XW, and PX contributed to bioinformatic analysis, XC and SD participated in abiotic stress treatments, ZNL, ZYL, and YH conceived the study and its design and assisted with revisions to the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX19_2203), the China Agriculture Research System of sweetpotato (CARS-10-GW04), The Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the National Science Foundation of China (32070344, 32201868), and the Innovation Team of Youth Technology Project of High School in Shandong Province (2021KJ055).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1091620/full#supplementary-material
References
Ahmad, B., Azeem, F., Ali, M. A., Nawaz, M. A., Nadeem, H., Abbas, A., et al. (2020). Genome-wide identification and expression analysis of two component system genes in cicer arietinum. Genomics 112, 1371–1383. doi: 10.1016/j.ygeno.2019.08.006
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). "The MEME suite". Nucleic Acids Res. 43 (W1), W39–W49. doi: 10.1093/nar/gkv416
Caesar, K., Thamm, A. M. K., Witthoft, J., Elgass, K., Huppenberger, P., Grefen, C., et al. (2011). Evidence for the localization of the arabidopsis cytokinin receptors AHK3 and AHK4 in the endoplasmic reticulum. J. Exp. Bot. 62, 5571–5580. doi: 10.1093/jxb/err238
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., May, G. (2004). The roles of segmental and tandem gene duplication in the evolution of large gene families in arabidopsis thaliana. BMC. Plant Biol. 4, 10. doi: 10.1186/1471-2229-4-10
Chu, Z. X., Ma, Q., Lin, Y. X., Tang, X. L., Zhou, Y. Q., Zhu, S. W., et al. (2011). Genome-wide identification, classification, and analysis of two-component signal system genes in maize. Genet. Mol. Res. 10, 3316–3330. doi: 10.4238/2011.December.8.3
Deng, Y., Dong, H., Mu, J., Ren, B., Zheng, B., Ji, Z., et al. (2010). Arabidopsis histidine kinase CKI1 acts upstream of histidine phosphotransfer proteins to regulate female gametophyte development and vegetative growth. Plant Cell. 22, 1232–1248. doi: 10.1105/tpc.108.065128
Dortay, H., Gruhn, N., Pfeifer, A., Schwerdtner, M., Schmülling, T., Heyl, A. (2008). Toward an interaction map of the two-component signaling pathway of arabidopsis thaliana. J. Proteome. Res. 7, 3649–3660. doi: 10.1021/pr0703831
Du, L., Jiao, F., Chu, J., Jin, G., Chen, M., Wu, P. (2007). The two-component signal system in rice (Oryza sativa l.): a genome-wide study of cytokinin signal perception and transduction. Genomics 89, 697–707. doi: 10.1016/j.ygeno.2007.02.001
Firon, N., Pressman, E., Meir, S., Khoury, R., Altahan, L. (2012). Ethylene is involved in maintaining tomato (Solanum lycopersicum) pollen quality under heat-stress conditions. AoB. Plants. 2012, pls024. doi: 10.1093/aobpla/pls024
Gahlaut, V., Mathur, S., Dhariwal, R., Khurana, J. P. (2014). A multi-step phosphorelay two-component system impacts on tolerance against dehydration Stress in common wheat. Funct. Integr. Genomics. 14, 707–716. doi: 10.1007/s10142-014-0398-8
Ha, S., Vankova, R., Yamaguchi-Shinozaki, K., Shinozaki, K., Tran, L. S. (2012). Cytokinins: metabolism and function in plant adaptation to environmental stresses. Trends. Plant Sci. 17, 172–179. doi: 10.1016/j.tplants.2011.12.005
Hayama, R., Coupland, G. (2003). Shedding light on the circadian clock and the photoperiodic control of flowering. Curr. Opin. Plant Biol. 6, 13–19. doi: 10.1016/s1369-5266(02)00011-0
Hejatko, J., Pernisova, M., Eneva, T., Palme, K., Brzobohaty, B. (2003). The putative sensor histidine kinase CKI1 is involved in female gametophyte development in arabidopsis. Mol. Genet. Genomics 269, 443–453. doi: 10.1007/s00438-003-0858-7
Hejatko, J., Ryu, H., Kim, G. T., Dobesova, R., Choi, S., Choi, S. M., et al. (2009). The histidine kinases CYTOKININ-INDEPENDENT1 and ARABIDOPSIS HISTIDINE KINASE2 and 3 regulate vascular tissue development in arabidopsis shoots. Plant Cell. 21, 2008–2021. doi: 10.1105/tpc.109.066696
He, Y., Liu, X., Ye, L., Pan, C., Chen, L., Zou, T., et al. (2016a). Genome-wide identification and expression analysis of two-component system genes in tomato. Int. J. Mol. Sci. 17 (8), 1204. doi: 10.3390/ijms17081204
He, Y., Liu, X., Zou, T., Pan, C., Qin, L., Chen, L., et al. (2016b). Genome-wide identification of two-component system genes in cucurbitaceae crops and expression profiling analyses in cucumber. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00899
Héricourt, F., Larcher, M., Chefdor, F., Koudounas, K., Carqueijeiro, I., Lemos Cruz, P., et al. (2019). New insight into HPts as hubs in poplar cytokinin and osmosensing multistep phosphorelays: cytokinin pathway uses specific HPts. Plants (Basel). 8, 591. doi: 10.3390/plants8120591
He, L. L., Zhang, F., Wu, X. Z., Hu, Y. M., Dong, L. L., Dewitte, W., et al. (2020). Genome-wide characterization and expression of two-component system genes in cytokinin-regulated gall formation in zizania latifolia. Plants (Basel) 9 (11), 1409. doi: 10.3390/plants9111409
Higuchi, M., Pischke, M. S., Mahonen, A. P., Miyawaki, K., Hashimoto, Y., Seki, M., et al. (2004). In planta functions of the arabidopsis cytokinin receptor family. Proc. Natl. Acad. Sci. 101, 8821–8826. doi: 10.1073/pnas.0402887101
Hirakawa, H., Okada, Y., Tabuchi, H., Shirasawa, K., Watanabe, A., Tsuruoka, H., et al. (2015). Survey of genome sequences in a wild sweet potato, ipomoea trifida (H. b. k.) g. don. DNA Res. 22, 171–179. doi: 10.1093/dnares/dsv002
Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Hoshino, A., Jayakumar, V., Nitasaka, E., Toyoda, A., Noguchi, H., Itoh, T., et al. (2016). Genome sequence and analysis of the Japanese morning glory ipomoea nil. Nat. Commun. 7, 13295. doi: 10.1038/ncomms13295
Hou, F., Du, T., Qin, Z., Xu, T., Li, A., Dong, S., et al. (2021). Genome-wide in silico identification and expression analysis of beta-galactosidase family members in sweetpotato [Ipomoea batatas (L.) lam]. BMC Genomics 22, 140. doi: 10.1186/s12864-021-07436-1
Hu, B., Jin, J. P., Guo, A. Y., Zhang, H., Luo, J. C., Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31 (8), 1296–1297. doi: 10.3390/ijms21144898
Huo, R. X., Liu, Z. N., Yu, X. L., Li, Z. Y. (2020). The interaction network and signaling specificity of two-component system in Arabidopsis. Int J Mol Sci. 21, 4898. doi: 10.3390/ijms21144898
Hutchison, C. E., Li, J., Argueso, C., Gonzalez, M., Lee, E., Lewis, M. W., et al. (2006). The arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell. 18, 3073–3087. doi: 10.1105/tpc.106.045674
Hwang, I., Chen, H. C., Sheen, J. (2002). Two-component signal transduction pathways in arabidopsis. Plant Physiol. 129, 500–515. doi: 10.1104/pp.005504
Ishida, K., Niwa, Y., Yamashino, T., Mizuno, T. (2009). A genome-wide compilation of the two-component systems in Lotus japonicus. DNA Res. 16, 237–247. doi: 10.1093/dnares/dsp012
Ishida, K., Yamashino, T., Nakanishi, H., Mizuno, T. (2010). Classification of the genes involved in the two-component system of the moss Physcomitrella patens. Biosci. Biotechnol. Biochem. 74, 2542–2545. doi: 10.1271/bbb.100623
Ito, Y., Kurata, N. (2006). Identification and characterization of cytokinin-signalling gene families in rice. Gene 382, 57–65. doi: 10.1016/j.gene.2006.06.020
Iwama, A., Yamashino, T., Tanaka, Y., Sakakibara, H., Kakimoto, T., Sato, S., et al. (2007). AHK5 histidine kinase regulates root elongation through an ETR1-dependent abscisic acid and ethylene signaling pathway in arabidopsis thaliana. Plant Cell Physiol. 48, 375–380. doi: 10.1093/pcp/pcl065
Jun, Y., Moeinzade, M.-H., Heiner, K., Johannes, H., Peng, X., Stefan, H., et al. (2017). Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants. 3 (9), 696–703. doi: 10.1038/s41477-017-0002-z
Kamberov, E. S., Atkinson, M. R., Chandran, P., Ninfa, A. J. (1994). Effect of mutations in escherichia coli glnL (ntrB), encoding nitrogen regulator II (NRII or NtrB), on the phosphatase activity involved in bacterial nitrogen regulation. J. Biol. Chem. 269 (45), 28294–28299. doi: 10.1016/S0021-9258(18)46927-X
Kinoshita-Tsujimura, K., Kakimoto, T. (2014). Cytokinin receptors in sporophytes are essential for male and female functions in arabidopsis thaliana. Plant Signal Behav. 6, 66–71. doi: 10.4161/psb.6.1.13999
Koch, M. A., Haubold, B., Mitchell-Olds, T. (2000). Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in arabidopsis, arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 17, 1483–1498. doi: 10.1093/oxfordjournals.molbev.a026248
Kong, H., Landherr, L. L., Frohlich, M. W., Leebens-Mack, J., Ma, H., dePamphilis, C. W. (2007). Patterns of gene duplication in the plant SKP1 gene family in angiosperms: evidence for multiple mechanisms of rapid gene birth. Plant J. 50, 873–885. doi: 10.1111/j.1365-313X.2007.03097.x
Kumar, M. N., Jane, W. N., Verslues, P. E. (2013). Role of the putative osmosensor arabidopsis histidine kinase1 in dehydration avoidance and low-water-potential response. Plant Physiol. 161, 942–953. doi: 10.1104/pp.112.209791
Kumar, S., Stecher., G., Tamura., K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Le, D. T., Nishiyama, R., Watanabe, Y., Vankova, R., Tanaka, M., Seki, M., et al. (2012). Identification and expression analysis of cytokinin metabolic genes in soybean under normal and drought conditions in relation to cytokinin levels. PloS One 7 (8), e42411. doi: 10.1371/journal.pone.0042411
Lescot, M., Dehais, P., Thijs, G., Marchal, K., Moreau, Y., Peer, Y. V., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., Doerks, T., Bork, P. (2012). SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305. doi: 10.1093/nar/gkr931
Liu, P. J., Wang, S. S., Wang, X. F., Yang, X. Y., Li, Q., Wang, C., et al. (2020). Genome-wide characterization of two-component system (TCS) genes in melon (Cucumis melo l.). Plant Physiol. Biochem. 151, 197–213. doi: 10.1016/j.plaphy.2020.03.017
Liu, Z., Zhang, M., Kong, L., Lv, Y., Zou, M., Lu, G., et al. (2014). Genome-wide identification, phylogeny, duplication, and expression analyses of two-component system genes in Chinese cabbage (Brassica rapa ssp. pekinensis). DNA Res. 21, 379–396. doi: 10.1093/dnares/dsu004
Li, M., Yang, S., Xu, W., Pu, Z., Feng, J., Wang, Z., et al. (2019). The wild sweetpotato (Ipomoea trifida) genome provides insights into storage root development. BMC Plant Biol. 19 (1), 119. doi: 10.1186/s12870-019-1708-z
Lü, D., Wang, W., Miao, C. (2012). ATHK1 acts downstream of hydrogen peroxide to mediate ABA signaling through regulation of calcium channel activity in arabidopsis guard cells. Chin. Sci. Bull. 58, 336–343. doi: 10.1007/s11434-012-5207-y
Marchler-Bauer, A., Lu, S. N., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229. doi: 10.1093/nar/gkq1189
Mizuno, T. (1997). Compilation of all genes encoding two-component phosphotransfer signal transducers in the genome of escherichia coli. DNA Res. 4, 161–168. doi: 10.1093/dnares/4.2.161
Mochida, K., Yoshida, T., Sakurai, T., Yamaguchi-Shinozaki, K., Shinozaki, K., Tran, L. S. (2010). Genome-wide analysis of two-component systems and prediction of stress-responsive two-component system members in soybean. DNA Res. 17, 303–324. doi: 10.1093/dnares/dsq021
Nishimura, C., Ohashi, Y., Sato, S., Kato, T., Tabata, S., Ueguchi, C. (2004). Histidine kinase homologs that act as cytokinin receptors possess overlapping functions in the regulation of shoot and root growth in arabidopsis. Plant Cell. 16, 1365–1377. doi: 10.1105/tpc.021477
Nuruzzaman, M., Manimekalai, R., Sharoni, A. M., Satoh, K., Kondoh, H., Ooka, H., et al. (2010). Genome-wide analysis of NAC transcription factor family in rice. Gene 465, 30–44. doi: 10.1016/j.gene.2010.06.008
Pareek, A., Singh, A., Kumar, M., Kushwaha, H. R., Lynn, A. M., Singla-Pareek, S. L. (2006). Whole-genome analysis of oryza sativa reveals similar architecture of two-component signaling machinery with arabidopsis. Plant Phy. 142, 380–397. doi: 10.1104/pp.106.086371
Pham, J., Liu, J., Bennett, M. H., Mansfield, J. W., Desikan, R. (2012). Arabidopsis histidine kinase 5 regulates salt sensitivity and resistance against bacterial and fungal infection. New Phytol. 194, 168–180. doi: 10.1111/j.1469-8137.2011.04033.x
Pils, B., Heyl, A. (2009). Unraveling the evolution of cytokinin signaling. Plant Phy. 151, 782–791. doi: 10.1104/pp.109.139188
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2012). The pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkaa913
Riefler, M., Novak, O., Strnad, M., Schmulling, T. (2006). Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell. 18, 40–54. doi: 10.1105/tpc.105.037796
Schaller, G. E., Kieber, J. J., Shiu, S. H. (2008). Two-component signaling elements and histidyl-aspartyl phosphorelays. Arabidopsis Book. 6, e0112. doi: 10.1199/tab.0112
Sharan, A., Soni, P., Nongpiur, R. C., Singla-Pareek, S. L., Pareek, A. (2017). Mapping the ‘Two-component system’network in rice. Sci. Rep. 7, 1–13. doi: 10.1038/s41598-017-08076-w
Sung-Chul, P., Yun-Hee, K., Yoon, J. C., Seyeon, P., Cheol, J. J., Haeng-Soon, L., et al. (2012). Stable internal reference genes for the normalization of real-time PCR in different sweetpotato cultivars subjected to abiotic stress conditions. PloS One 7, e51502. doi: 10.1371/journal.pone.0051502
Tran, L. S., Urao, T., Qin, F., Maruyama, K., Kakimoto, T., Shinozaki, K., et al. (2007). Functional analysis of AHK1/ATHK1 and cytokinin receptor histidine kinases in response to abscisic acid, drought, and salt stress in arabidopsis. Proc. Natl. Acad. Sci. 104, 20623–20628. doi: 10.1073/pnas.0706547105
Wen, F., Qin, T., Wang, Y., Dong, W., Zhang, A., Tan, M., et al. (2015). OsHK3 is a crucial regulator of abscisic acid signaling involved in antioxidant defense in rice. J. Integr. Plant Biol. 57, 213–228. doi: 10.1111/jipb.12222
Wilkins, M. R., Gasteiger, E., Bairoch, A., Sanchez, J. C., Williams, K. L., Appel, R. D., et al. (1999). Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552. doi: 10.1385/1-59259-584-7:531
Wohlbach, D. J., Quirino, B. F., Sussman, M. R. (2008). Analysis of the arabidopsis histidine kinase ATHK1 reveals a connection between vegetative osmotic stress sensing and seed maturation. Plant Cell. 20, 1101–1117. doi: 10.1105/tpc.107.055871
Wu, S., Lau, K. H. (2018). Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun. 9, 4580. doi: 10.1038/s41467-018-06983-8
Wulfetange, K., Lomin, S. N., Romanov, G. A., Stolz, A., Heyl, A., Schmulling, T. (2011). The cytokinin receptors of arabidopsis are located mainly to the endoplasmic reticulum. Plant Physio. 156, 1808–1818. doi: 10.1104/pp.111.180539
Xiao, D., Zhang, N. W., Zhao, J. J., Bonnema, G., Hou, X. L. (2012). Validation of reference genes for real-time quantitative PCR normalisation in non-heading Chinese cabbage. Funct. Plant Biol. 39, 342. doi: 10.1071/FP11246
Yang, Z. H. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556. doi: 10.1093/bioinformatics/13.5.555
Yang, J., Moeinzadeh, M. H. (2017). Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants. 3, 696–703. doi: 10.1038/s41477-017-0002-z
Yan, M., Nie, H., Wang, Y., Wang, X., Jarret, R., Zhao, J., et al. (2022). Exploring and exploiting genetics and genomics for sweetpotato improvement: Status and perspectives. Plant Commun. 3, 100332. doi: 10.1016/j.xplc.2022.100332
Yuan, L., Liu, Z., Song, X., Johnson, C., Yu, X., Sundaresan, V. (2016). The CKI1 histidine kinase specifies the female gametic precursor of the endosperm. Dev. Cell. 37, 34–46. doi: 10.1016/j.devcel.2016.03.009
Zameer, R., Sadaqat, M., Fatima, K., Fiaz, S., Rasul, S., Zafar, H., et al. (2021). Two-component system genes in sorghum bicolor: genome-wide identification and expression profiling in response to environmental stresses. Front. Genet. 12. doi: 10.3389/fgene.2021.794305
Keywords: two-component system, sweet potato, phylogeny, evolution, expression profiles
Citation: Huo R, Zhao Y, Liu T, Xu M, Wang X, Xu P, Dai S, Cui X, Han Y, Liu Z and Li Z (2023) Genome-wide identification and expression analysis of two-component system genes in sweet potato (Ipomoea batatas L.). Front. Plant Sci. 13:1091620. doi: 10.3389/fpls.2022.1091620
Received: 07 November 2022; Accepted: 28 December 2022;
Published: 12 January 2023.
Edited by:
Shaopei Gao, China Agricultural University, ChinaReviewed by:
Li Yuan, Northwest A&F University, ChinaFarrukh Azeem, Government College University, Faisalabad, Pakistan
Muhammad Ali, Zhejiang University, China
Copyright © 2023 Huo, Zhao, Liu, Xu, Wang, Xu, Dai, Cui, Han, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenning Liu, bGl1emhlbm5pbmdAbHl1LmVkdS5jbg==; Zongyun Li, em9uZ3l1bmxpQGpzbnUuZWR1LmNu