
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 13 January 2023
Sec. Plant Development and EvoDevo
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1078248
This article is part of the Research Topic System Biology to Regulatory Grids: New Tools and Clues Aimed at Improving Plant Evolutionary-Developmental (Evo-Devo) Biology View all 6 articles
 Min Jiang1,2†
Min Jiang1,2† Jinjing Jian1
Jinjing Jian1 Chengchuan Zhou3
Chengchuan Zhou3 Linfeng Li1
Linfeng Li1 Yuguo Wang1
Yuguo Wang1 Wenju Zhang1
Wenju Zhang1 Zhiping Song1
Zhiping Song1 Ji Yang1,2*
Ji Yang1,2*The origin of seeds is one of the key innovations in land plant evolution. Ovules are the developmental precursors of seeds. The integument is the envelope structure surrounding the nucellus within the ovule and developing into the seed coat when ovules mature upon fertilization. The question of whether the integument arise de novo or evolve from elaboration of pre-existing structures has caused much debate. By exploring the origin and evolution of the key regulatory genes controlling integument development and their functions during both individual and historical developmental processes, we showed the widespread presence of the homologs of ANT, CUC, BEL1, SPL, C3HDZ, INO, ATS, and ETT in seedless plant genomes. All of these genes have undergone duplication-divergence events in their evolutionary history, with most of the descendant paralogous suffering motif gain and/or loss in the coding regions. Expression and functional characterization have shown that these genes are key components of the genetic program that patterns leaf-like lateral organs. Serial homology can thus be postulated between integuments and other lateral organs in terms of the shared master regulatory genes. Given that the genetic program patterning leaf-like lateral organs formed in seedless plants, and was reused during seed origin, the integument is unlikely to arise de novo but evolved from the stem segment-specific modification of pre-existing serially homologous structures. The master ‘switches’ trigging the modification to specify the integument identity remain unclear. We propose a successive transformation model of integument origin.
Seed formation is one of the key innovations in land plant evolution (Linkies et al., 2010; Baroux and Grossniklaus, 2019; Meade et al., 2021). The protective and nourishing structures of the seed enable a maternal care of the young sporophyte (embryo) and ensure seed dispersal over large areas for long time spans to colonize different environments, fueling seed plant radiation (Baroux and Grossniklaus, 2019). The origin of the seed is not a single innovation, but a unique ovule-centered reproductive syndrome involving a set of innovations that define the seed habit (Gerrienne and Meyer-Berthaud, 2007; Meyer-Berthaud et al., 2018; Hetherington, 2021; Meyer-Berthaud, 2022). The ovule is the developmental precursor of the seed. The molecular regulation mechanisms of ovule development have been systematically studied in model plants, such as Arabidopsis thaliana (Gasser et al., 1998; Gasser and Skinner, 2019; Barro-Trastoy et al., 2020). However, the comprehensive analysis of the evolutionary origin of the specialized structures of ovule that contain and protect the developing embryos is still limited, including the origin and evolution of the maternally derived integument — the one- or two-layer envelope surrounding the nucellus and developing into the seed coat when ovules mature after fertilization (D'Apice et al., 2021).
The integument is an important structure within the ovule, functioning to protect the internal tissues and define a route via the micropyle for the transfer of sperm from the pollen tube to the ovum (Linkies et al., 2010; Lora et al., 2019). The question of how the integument originate has caused much debate. Diverse theories have been put forward to explain the origin of integument. The telome theory is the most acceptable, which portrays the evolution of the integument through fusion of sterile branches or telomes around a terminal megasporangium (Herr, 1995). Distinct from the transformational hypotheses that integuments have originated through modification of pre-existing ancestral structures, such as the dichotomously branching axes (Zimmermann, 1952; Andrews, 1963; Smith, 1964) or sterilized sporangia of a synangium (Kenrick and Crane, 1997), an alternative hypothesis has been proposed based on developmental and genetic evidence, which suggests that ovules have characteristics of meristems (Gross-Hardt et al., 2002), and that integuments are lateral organs initiated by the nucellar meristems and are of de novo origin (Mathews and Kramer, 2012; Hetherington, 2021).
The paleontological perspective of the nature and origin of the integument is apparently inconsistent with the hypothesis deduced from the developmental genetic analysis of extant plants. The accumulated knowledge on genes and pathways involved in integument development, as well as the advent of massive genomic data from a wide range of species, opens the possibility to use master regulatory genes as markers to identify historically vs serially homologous structures by detecting the presence of well-characterized integument developmental genes throughout the evolutionary timeline and exploring whether the structures with different morphologies have been orchestrated by the same developmental system (Jaramillo and Kramer, 2007; Tomoyasu et al., 2017). Given the prominent role of transcription factors in driving morphological innovations, exploring the evolution of key transcription factor genes constructing the integument gene network may contribute to bridging the disconnects between fossils and genes (Zumajo-Cardona and Ambrose, 2020; Tomescu and Rothwell, 2022). Functional studies in the model species A. thaliana have identified a dozen transcription factors genes directing integument formation at different stages of ovule development, including ovule primordium initiation (ANT and CUC), ovule patterning (BEL1, SPL/NZZ and STK) and ovule morphogenesis (C3HDZ, INO, KAN, and ETT) (Figure 1). Of them, AINTEGUMENTA (ANT), a member of the APETALA2 (AP2) transcription factor gene family, participates in the positive regulation of the ovule and integument primordia initiation (Elliott et al., 1996). Single ant mutation can lead to the complete loss of integuments, as well as the reduction in ovule number with no concomitant reduction in pistil length (Elliott et al., 1996; Klucher et al., 1996). The two CUP SHAPED COTYLEDON genes (CUC2 and CUC3) that encode the transcription factors of the NAC family coordinate the pattern formation of ovules, and are mainly involved in the establishment of ovule primordia boundaries. The cuc2 and cuc3 double mutant harbors defects in ovule separation, producing fused ovule primordia with shared integuments that ultimately form fused seeds sharing seed coat (Galbiati et al., 2013; Goncalves et al., 2015). Mutations in CUC1 and CUC2 also lead to abnormal ovule spacing, which affects the number of normal ovule (Goncalves et al., 2015). The BELL1 (BEL1) gene encodes a homeodomain protein involved in the initiation of integument development. The bel1 mutant shows significant growth in the chalazal region where an amorphous structure develops instead of integuments (Robinsonbeers et al., 1992; Brambilla et al., 2007). SPOROCYTELESS/NOZZLE (SPL/NZZ) belongs to a transcription repressor family that is specific in embryophyte, promoting the formation of megasporocyte and integuments during ovule development (Wei et al., 2015). It has been shown that SPL/NZZ interacts with BEL1 to control chalaza and integument development. In the bel1 and spl/nzz double mutant, the ovules developed as finger-like structures without integuments (Balasubramanian and Schneitz, 2002; Bencivenga et al., 2012). It is noteworthy that SEEDSTICK (STK) is a negative regulator of funiculus development, and its mutants show drastically enlargement of the funiculi (Pinyopich et al., 2003). The adaxial functions of the class III homeodomain leucine zipper (C3HDZ) genes and the abaxial functions of the YABBY (YAB) and KANADI (KAN) family genes contribute to the integument shape and outgrowth. However, there exist differences in how these genes participate in this interaction (Gasser and Skinner, 2019). The C3HDZ genes PHABULOSA (PHB), PHAVOLUTA (PHV) and CORONA (CNA) are expressed in the adaxial layer of the inner integument, playing roles in defining adaxial regions of the planar integuments and causing reduced growth in both integuments when multiple family members are mutated (Kelley et al., 2009; Yamada et al., 2016). The other C3HDZ gene REVOLUTA (REV) is expressed across both integuments, promoting adaxial activity in the outer as well as inner integument (Kelley et al., 2009). INNER NO OUTER (INO) is the only YAB gene expressed in ovules and is essential for the formation of outer integuments (Villanueva et al., 1999). INO is expressed in the abaxial domains of the outer integument and its mutation leads to a complete absence of the outer integument (Villanueva et al., 1999). Recently it was shown that STIMPY (STIP; also known as WUSCHEL-RELATED HOMEOBOX 9) can regulate INO, functioning as a regulator of outer integument formation (Petrella et al., 2022). The KAN family genes KAN1 and KAN2 are also expressed in the abaxial region of the outer integument (McAbee et al., 2006). The kan1 and kan2 double mutant grows an amorphous structure in place of the outer integument (Eshed et al., 2001; McAbee et al., 2006). Another KAN gene, ABERRANT TESTA SHAPE (ATS) (also called KAN4), functions in regulating the inner integument development and the separation of two integuments. The ats mutants have a single integument, which is formed by fusing the inner and outer integuments (McAbee et al., 2006). ETTIN (ETT) (also called AUXIN RESPONSE FACTOR 3, ARF3) encodes a transcription factor physically interacting with ATS to define the boundary between integument primordia. Mutation of ETT generates the same integument fusion phenotype as seen in the ats mutant (Kelley et al., 2012).
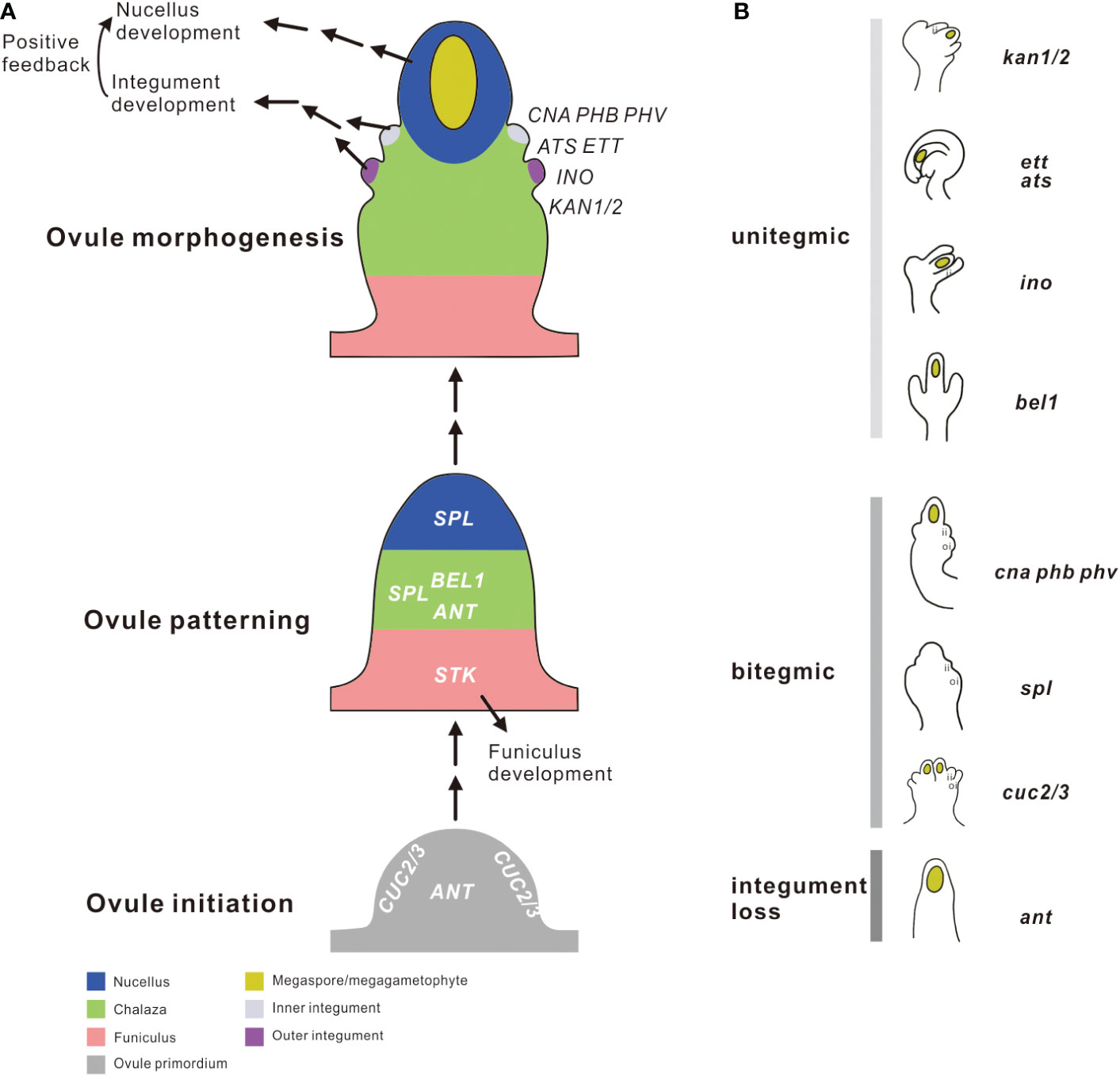
Figure 1 Schematic diagram demonstrating the genes regulating integument growth at different stages of ovule development. (A) The process of ovule development is divided into three stages in a bottom-up order: ovule initiation, ovule patterning, and ovule morphogenesis. Different tissue zones of the ovule are illustrated with different colors. Gene names are shown in the sites and developmental stages they are believed to act. (B) The integument phenotypes of different mutants. Refer to text for detailed explanation for each mutant. ii, inner integument; oi, outer integument; i, integument; unitegmic, possessing only a single integument; bitegmic, possessing two distinct integuments; integument loss, complete loss of two distinct integuments.
The transcription factor genes mentioned above together establish a regulatory blueprint for integument primordia initiation and outgrowth during ovule development (Schneitz et al., 1995) (Figure 1). Although their functions have been extensively evaluated in different angiosperms with divergent ovule numbers and morphology (Yamada et al., 2001; Brown et al., 2010; Lora et al., 2011; Lora et al., 2015), questions remain unclear about how these genes evolved, how the timing of their appearance relates to the origination of integuments, and whether the wiring of these genes have changed over evolutionary time? In this study, we reconstructed the evolutionary history of these genes based on accumulated whole genome sequences. By examining the taxonomic distribution, structural diversification following duplication, and the spatiotemporal expression of these genes reported in diverse seed and seedless plants, we aim to extend our understanding of the function of the core regulatory genes described in model plants to ancestral lineages prior to the angiosperm diversification, to approach the fundamental nature of integuments and evaluate the competing explanations for the origin of the integument.
ANT belongs to the AP2/ERF gene family that are divided into two classes based on the number of the AP2 domain (repeated units 1 and 2, abbreviated as R1 and R2): the AP2-like class containing two AP2 domains and the ERF-like class containing one AP2 domain (Kim et al., 2006). AP2-like genes are further divided into two groups: the ANT group has a 10-aa insertion in the R1 domain and the euAP2 group without insertion. The ANT group consists of two subgroups: euANT and basalANT. There exist 10 ANT group members in the A. thaliana genome, which were used as queries to perform BLASTP searches against 49 plant genomes and transcriptomes (Supplementary Table S1) with an E-value threshold of 1e-5. The hit sequences with similarity to the query sequence were annotated with InterProScan (https://www.msi.umn.edu/sw/interproscan) (Quevillon et al., 2005), and sequences containing two conserved AP2 domains (R1 and R2) were retained and used for the next analysis. All the sequences were aligned using MAFFT v7.471 (Katoh and Standley, 2013) and trimmed by trimAL v1.4.1 (Capella-Gutierrez et al., 2009) with the threshold of -gt = 0.9 and -cons = 30. The phylogenetic tree was reconstructed using IQ-TREE (Nguyen et al., 2015) based on the JTT+G+F model selected by ProtTest3 (Darriba et al., 2011) with 2000 bootstraps.
The CUC1, CUC2 and CUC3 genes encode the NAC domain proteins, belonging to the subgroup II-3 of the NAC superfamily (Vroemen et al., 2003; Pereira-Santana et al., 2015; Maugarny-Cales et al., 2016). There are 11 members in the A. thaliana genome (Jensen et al., 2010), which were used as probes to perform BLASTP searches against 49 plant genomes and transcriptomes with an E-value threshold of 1e-50 (Supplementary Table S1). Sequences annotated to contain “NAC_dom” with InterProScan being retained and used for the next analysis. The NAC domain contain five conserved motifs: A, B, C, D and E (Puranik et al., 2012). All sequences were aligned using MAFFT v7.471 and trimmed using trimAL v1.4.1 with the thresholds of -gt = 0.9 and -cons = 25. The Maximum Likelihood (ML) phylogenetic tree was reconstructed using IQ-TREE with the JTT+I+G model selected by ProtTest3 with 2000 bootstraps.
BEL1 is a member of the BEL1-like homeodomain (BLH) family of the TALE (Three Amino acid Loop Extension) homeodomain superfamily (Mukherjee et al., 2009). There exist 13 BLH members in the A. thaliana genome, which were used as queries to perform BLASTP searches against 49 plant genomes and transcriptomes (Supplementary Table S1) with an E-value threshold of 1e-5. Sequences containing both ‘‘POX_dom’’ and “Homeobox_dom” were retained and used for the next analysis. The POX domain is composed of SKY and BEL-B domains (Mukherjee et al., 2009). All sequences were aligned using MAFFT v7.471 and trimmed by trimAL v1.4.1 with the threshold of -gt = 0.5. The ML phylogenetic tree was reconstructed using IQ-TREE based on the JTT+G+F model selected by ProtTest3 with 2000 bootstraps.
SPL/NZZ belongs to the SPEAR (SPL-like, EAR-containing proteins) family (Chen et al., 2014). There are five SPEAR members in A. thaliana genome, which were used as queries to perform BLASTP searches with an E-value threshold of 0.5 against 49 plant genomes and transcriptomes (Supplementary Table S1). The sequences were further annotated with InterProScan with the sequences containing the ‘‘NOZZLE’’ domain being retained and used for the next analysis. All sequences were aligned using MAFFT v7.471 and trimmed using trimAL v1.4.1 with the thresholds of -gt = 0.9 and -cons = 20. The ML phylogenetic tree was reconstructed by IQ-TREE based on the best substitution model JTT+G+F screened by ProtTest3, and with 2000 bootstrap iterations.
PHB, PHV, REV and CNA belong to C3HDZ gene family (Prigge et al., 2005). The A. thaliana genome contains five C3HDZ genes, which were used as queries for BLASTP searches with E-value of less than 1e-10 against 49 plant genomes and transcriptomes (Supplementary Table S1). After annotation with InterProScan, sequences containing “Homeobox_dom”, “START_dom” and “MEKHLA” domains were retained and used for the next analysis. All sequences were aligned by MAFFT v7.471, and then trimmed them by the trimAL v1.4.1 software under the control of -gt = 0.9 and -cons = 30. The trimmed sequences were used to reconstruct the ML phylogenetic tree using IQ-TREE based on the JTT+I+G+F model selected by ProtTest3 with 2000 bootstrapping events.
INO belongs to the YAB gene family (Finet et al., 2016). The A. thaliana genome contains six YAB members, which were used as queries to perform BLASTP searches against 49 plant genomes and transcriptomes (Supplementary Table S1) with an E-value threshold of 1e-10. The YAB homologs harbor the “YABBY” domain in addition to the C2-C2 zinc finger domain. The reported YAB sequence of Huperzia selago (Accession number: ASU87387) was collected from its transcriptome sequence (Evkaikina et al., 2017). The sequences were aligned by MAFFT v7.471 and trimmed by trimAL v1.4.1 with -gt = 0.9 and -cons = 20. The ML phylogenetic tree was reconstructed by IQ-TREE with the JTT+G+F model screened by ProtTest3 with 2000 bootstrapping events.
ATS belongs to the KAN gene family (McAbee et al., 2006). There exist four members in the A. thaliana genome, which were used as queries to perform BLASTP searches against 49 plant genomes and transcriptomes (Supplementary Table S1) with an E-value of 1e-23. The blast hits were annotated with InterProScan, and sequences containing the GARP (GOLDEN2, ARR-B Class, Par1 proteins) domain were retained and used for the next analysis. These sequences were aligned by MAFFT v7.471 and trimmed using trimAL v1.3 with the threshold of -gt = 0.55. The ML phylogenetic tree was reconstructed using IQ-TREE based on the best substitution model JTT+G+F tested by ProtTest3 with 2000 iterations of bootstraps.
ETT belongs to the ARF gene family (Finet et al., 2013). There exist 23 members in the A. thaliana genome, which were used as queries to perform BLASTP searches against 49 plant genomes and transcriptomes (Supplementary Table S1) with an E-value threshold of 1e-40. Sequences containing “B3”, “ARF” and “AUX/IAA” domains were designated as candidate ARF-class homologs. The AUX/IAA proteins contain two conserved C-terminal domains, referred to as III and IV (Finet et al., 2013). The sequences were aligned by MAFFT v7.471 and then trimmed using trimAL v1.4.1 with the thresholds of -gt = 0.9 and -cons = 20. The ML phylogenetic tree was reconstructed using IQ-TREE based on the JTT+G+F model selected by ProtTest3 with 2000 iterations.
The lineage-specific domains/motifs contained in the identified proteins were predicted using the online MEME program (Bailey et al., 2015) (https://meme-suite.org/meme/tools/meme) with the parameter settings as follows: the occurrence rate of a single motif was no greater than one per sequence; the motif width was between 6 and 50 amino acids; the maximum number of identified motifs was 25. Other parameters were set to default values. The identified domains/motifs and their positions within the amino acid sequence were then mapped to known conserved domains, those without homology to the conserved domains were identified as lineage-specific or unique.
The CodeML program implemented in the PAML v4.8 package was used to test shifts in substitution rates between specified foreground and background branches (Yang, 1997). Likelihood Ratio Tests (LRTs) were used to compare the one ratio model that assumes a constant ω (dN/dS = non-synonymous/synonymous substitutions) along all tree branches (ω0) against the two-ratio model that assumes a different ratio for the designated foreground branch (ωf) relative to the remaining background branches (ωb). The ANT clade in the AP2/ERF gene family, the clade C in the CUC family, the clade C in the BLH family, the SPL clade in the SPEAR family, the clades C and D in the C3HDZ family, the INO clade in the YAB family, the KAN and ATS clades in the KAN family, and the ETT clade in the ARF gene family were designated as foreground branches, respectively. A chi-squared distribution was assumed for 2Δℓ with the difference between np2 and np1 as the degree of freedom (difference between the parameter number of the one ratio and the two-ratio models) (Jeffares et al., 2015).
The gene expression data of Physcomitrella patens (E-MTAB-3069) (http://www.ebi.ac.uk/arrayexpress) (Ortiz-Ramirez et al., 2016), Adiantum capillus (PRJNA593361) (https://www.ncbi.nlm.nih.gov/sra) (Fang et al., 2021), Ginkgo biloba (T0001) (https://ginkgo.zju.edu.cn/project/T0001) (Bai et al., 2022), and rice (PRJNA591969) (https://www.ncbi.nlm.nih.gov/sra) (Zhao et al., 2020) were collected to investigate the spectrum of expression variation of key integument regulatory genes in various organs across species. The reference genome of A. capillus was downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/bioproject) under the BioProject accession number PRJNA593372 (Fang et al., 2021). Clean reads were mapped to their respective reference genomes using HISAT2 (Kim et al., 2019). Gene expression levels were quantified by TPM (transcripts per million) and calculated using the prepDE.py script implemented in the StringTie package (Pertea et al., 2015), and then presented with heatmaps plotted by the R package pheatmap (version 1.0.12).
A total of 361 ANT homologs were identified from 49 plant genomes and transcriptomes (Supplementary Table S2). Seven sequences were excluded from the phylogenetic analyses for they could not be aligned properly. The phylogenetic tree showed that 354 ANT homologs divided into two main groups: the basalANT group which was further split into clade A (ERF) and clade B (WRI1 and WRI3/4), and the euANT group consisting of clade C (AIL2, AIL3/4, AIL5, and AIL6/7) and clade D (including ANT and AIL1) (Supplementary Figure S1-1). The homologous sequences of both the basalANT and euANT groups can be traced back to bryophyte genomes. However, the ANT orthologs were only present in angiosperm plants, which originated via gene duplications and subsequent diversification of an ancestral gymnosperm gene. Comparing gene structure between ANT orthologs and other genes revealed two additional motifs (motif 14 and 19) in the ANT orthologs (Supplementary Figures S1-1, S2-1). Substitution rate tests revealed the significant rate difference between ANT and other lineages (p < 0.05) (Supplementary Table S3).
A total of 269 CUC homologs were identified and used to reconstruct the phylogenetic tree (Supplementary Table S2). The tree showed that the CUC homologs fell into three main clades: clade A, clade B, and clade C (Supplementary Figure S1-2). The CUC homologous sequences can be traced back to the origin of terrestrial plants. However, the CUC1/2/3 orthologs were only present in angiosperm plants, which originated via angiosperm-specific gene duplication events. Comparing gene structures between different CUC homologs revealed one additional motif (motif 18) in CUC1/2 orthologs (Supplementary Figures S1-2, S2-2). Substitution rate tests did not show rate differences between different lineages (Supplementary Table S3).
A total of 363 BLH homologs were identified from 49 plant genomes and transcriptomes (Supplementary Table S2) and used to reconstruct the phylogenetic tree (Supplementary Figure S1-3). The tree showed that the BLH homologs could be divided into three main clades: clade A, clade B, and clade C (Supplementary Figure S1-3). The homologous sequences of BLH were found in bryophytes, but the BEL1 orthologs were only present in angiosperm plants. Compared to its ancestral sequence, BEL1 orthologs underwent both motifs gain and loss events (Supplementary Figures S1-3, S2-3). Substitution rate tests revealed a significant difference between BEL1 and other lineages (p < 0.01) (Supplementary Table S3).
A total of 128 SPL/NZZ homologs were identified from 49 representative plant genomes and transcriptomes (Supplementary Table S2). Eight sequences were excluded from phylogenetic analyses for they could not be aligned properly. The phylogenetic tree displayed that 120 SPL/NZZ homologs split into four major clades: clade A (gymnoSPEAR), clade B (SPEAR2/4), clade C (SPEAR1/3), and clade D (SPL) (Supplementary Figure S1-4). The homologous sequences of SPL/NZZ have been present in mosses, with the SPL/NZZ orthologs being only present in angiosperm plants. Compared to its paralogs, SPL/NZZ lineages lost several motifs following duplication (Supplementary Figures S1-4, S2-4). Substitution rate tests revealed a significant difference between SPL/NZZ and other lineages (p < 0.05) (Supplementary Table S3).
A total of 215 C3HDZ homologs were identified from 49 plant genomes and transcriptomes examined (Supplementary Table S2) and used to reconstruct the phylogenetic tree (Supplementary Figure S1-5). The tree clear indicated that the C3HDZ homologs fell into four main clades: clade A (HDZ1), clade B (HDZ2), clade C (including CNA, and HB8), and clade D [(PHX including PHB and PHV), and REV] (Supplementary Figure S1-5). The orthologs of PHB, PHV and REV were angiosperm-specific, but their homologs were already present in charophytes. The homologs HDZ1 and HDZ2 were restrictedly present in non-flowering vascular plants and have lost in angiosperms. The genes of the C3HDZ family were highly conserved without structural variation between different members (Supplementary Figures S1-5, S2-5). Substitution rate tests revealed a significant difference between C3HDZ and other lineages (p < 0.01) (Supplementary Table S3).
A total of 251 YAB homologs were identified from 49 representative plant genomes and transcriptomes examined (Supplementary Table S2 and Supplementary Figure S1-6) and used to reconstruct the phylogenetic tree (Figure 2A). The YAB homologs formed three major clades in the tree: clade A (including FIL and INO lineages), clade B (YAB homologs from gymnosperms) and clade C (including YAB2, YAB5, and CRC lineages) (Figure 2A). The homologous sequences of YAB were present in chlorophytes, mosses and lycophytes, but were absent in monilophytes. The INO orthologs were only present in angiosperm plants and had two additional motifs (motif 23 and 25) compared to its paralogs (Figure 2B and Supplementary Figure S2-6). Substitution rate tests revealed a significant rate difference between INO and other lineages (p < 0.01) (Supplementary Table S3).
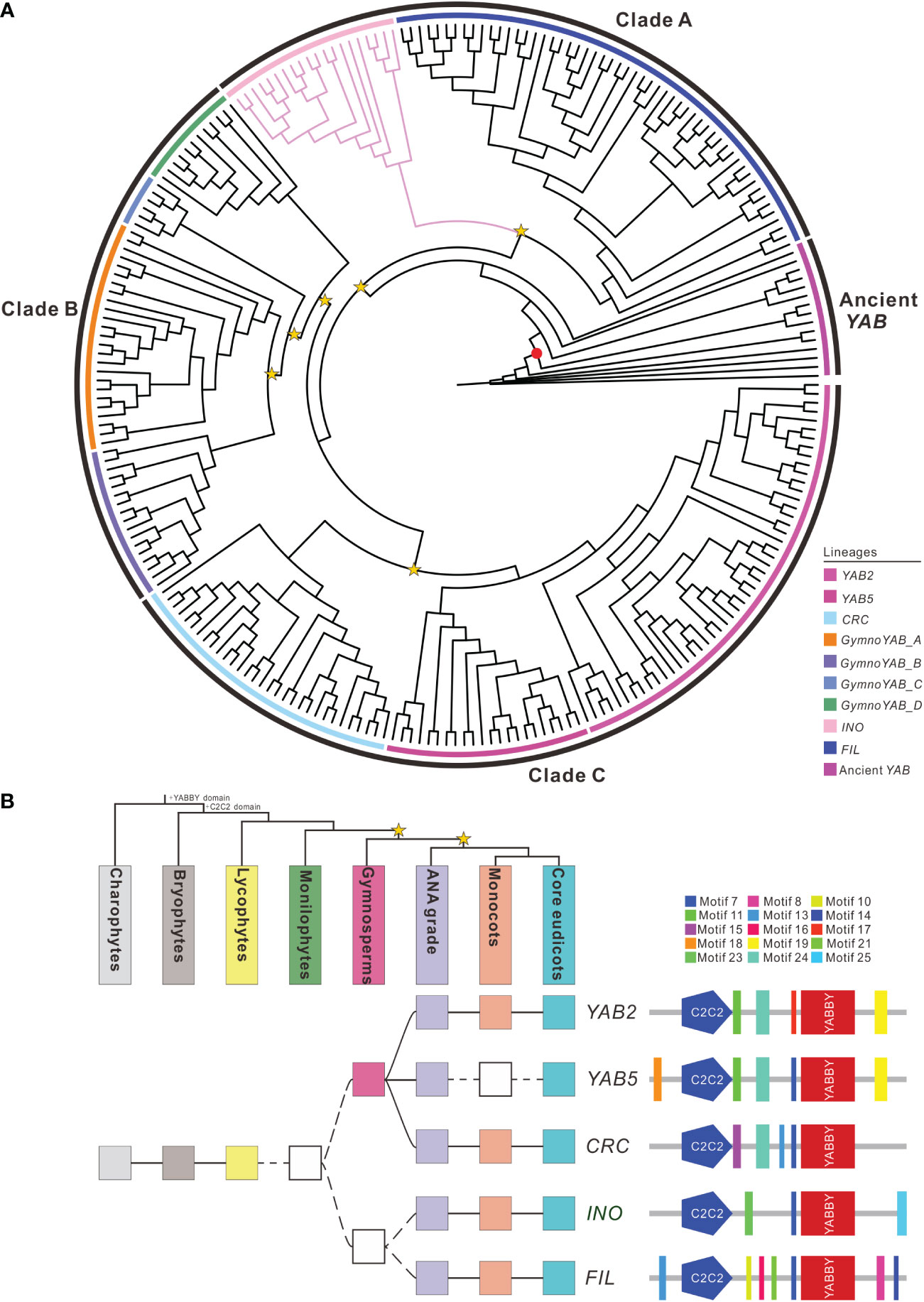
Figure 2 Phylogeny and domain architecture of the YABBY homologs. (A) Phylogenetic tree of 251 YABBY homologs identified from diverse green plants. The red dot indicates the branch support values of BP > 85, while the yellow star indicates the large-scale duplication events. Branches marked with pink indicate the INO orthologs. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. The support values for each node are shown in Supplementary Figure S1-6. (B) Duplication history and domain architecture of the YABBY homologs. Filled squares indicate the presence of the corresponding members, open squares indicate lack of data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: C2-C2 zinc finger (C2C2) and the YABBY domain. The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-6.
A total of 199 KAN homologs were identified from 49 plant genomes and transcriptomes (Supplementary Table S2) and used subsequently to reconstruct the phylogenetic tree (Supplementary Figure S1-7). The KAN homologs clustered into three main clades: clade A (including gymnoKAN_A and gymnoKAN_B lineages), clade B (ATS lineage), and clade C (including KAN1 and KAN2/3 lineages) (Supplementary Figure S1-7). The presence of the KAN homologous sequences was limited to land plants, suggesting an origin of this gene family along with the invasion of the land by plants. However, the ATS orthologs were only present in angiosperms and lost several motifs following duplication compared to its paralogs (Supplementary Figures S1-7, S2-7). Substitution rate tests displayed a significant rate difference between ATS and other lineages (p < 0.01) (Supplementary Table S3).
A total of 685 ARF homologs were identified (Supplementary Table S2) and used subsequently to reconstruct the phylogenetic tree. In the tree, the ARF homologs were grouped into three major clades: clade A (including ARF1, ARF2, ETT/ARF4, and ARF9), clade B (including ARF5/7 and ARF6/8), and clade C (including ARF10/16 and ARF17) (Supplementary Figure S1-8). The ARF homologous sequences could be traced back to charophytes, suggesting an early origin of this gene family. The ETT orthologs were only present in angiosperm plants, and lost the Aux/IAA dimerization domain and several motifs following duplication (Supplementary Figures S1-8, S2-8). Substitution rate tests revealed a significant rate difference between ETT and other lineages (p < 0.01) (Supplementary Table S3), possibly due to repeated domain/motif losses.
Tracking the expression pattern of key genes associated with integument development in various organs and plants showed that the homologs of key integument regulatory genes were not only expressed in the ovules of G. biloba and rice but also in the leaf-like organs of A. capillus, even in P. patens tissues (Figure 3 and Supplementary Figure S3). For instance, the CUC, BEL1, C3HDZ, KAN, and ETT orthologs in A. capillus were highly expressed in the gametophyte with embryo (EG), the vegetative growth leaf (VGL), the leaf bearing green sporangium (GSL), the leaf bearing juvenile sporangium (JSL), the leaf bearing mature sporangium MSL), and the leaf bearing dehiscent sporangium (DSL) (Figure 3 and Supplementary Figure S3). Moreover, ANT, CUC, C3HDZ, KAN, and ETT orthologs of P. patens have high expression in the protonema and gametophyte tissues (Supplementary Figure S3). These results indicated that the developmental regulatory program composed by the co-expression of these genes was not unique to the integument, but was common to different leaf-lateral organs, and had been formed prior to seed-formation.
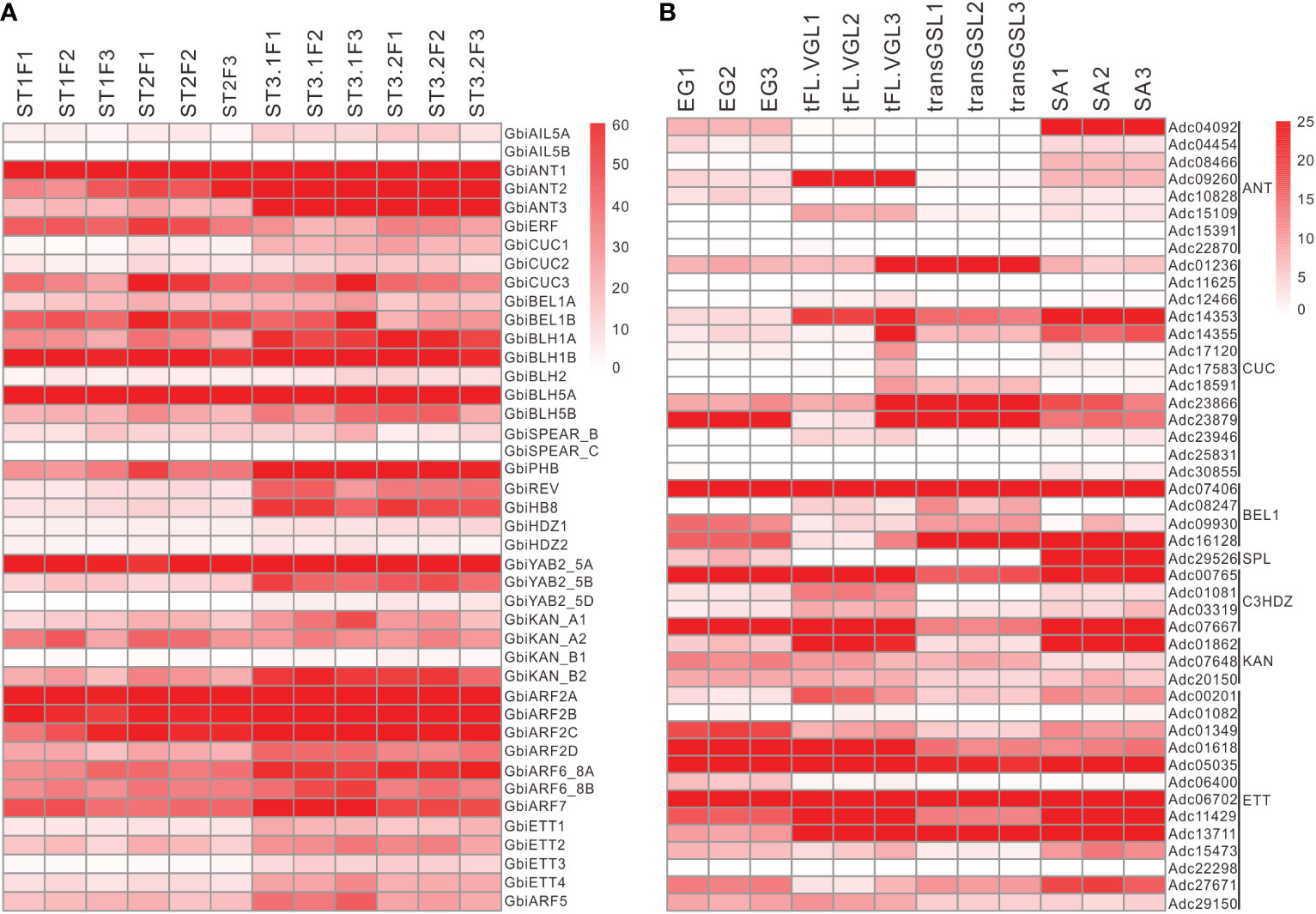
Figure 3 Expression of key genes related to integument development in Ginkgo biloba (A) and Adiantum capillus (B). The four developmental stages of G. biloba included the vegetative (ST1), the initial differentiation (ST2), the early exuberant differentiation (ST3-1) and the late exuberant differentiation (ST3-2) stages. The four tissues of A. capillus included the embryo gametophyte (EG), the transFL vegetative growth leaf (tFL-VGL), the transGSL green sporangium leaf (transGSL) and the stem apical (SA). Each stage or tissue contained three independent biological replicates.
Reconstruction of gene phylogenies revealed that all the genes of interest have experienced duplication-divergence events in their evolutionary history. The ancestral genes of ANT, BEL1, C3HDZ and ETT have been duplicated in bryophyte and/or seedless vascular plant genomes, prior to the origin of seed plants (Supplementary Figure S1 and Table 1). In contrast, the ancestor genes of CUC, SPL, INO and ATS were duplicated along with the origin of gymnosperms, followed by another round of duplication in angiosperm genomes (Supplementary Figure S1). INO is usually recognized to be angiosperm-specific, which originated from the duplication and diversification of the YAB ancestor in flowering plant genomes without orthologs in non-flowering plants. Strictly speaking, however, the other genes including ANT, CUC, BEL1, SPL, ATS and ETT, were also angiosperm-specific because they were all derived from the angiosperm-specific duplication events. Subsequent diversification in gene structure and expression patterns have been detected between these genes and their paralogs in angiosperm genomes, as well as their gymnosperm homologs (co-orthologs) that predate those duplications (Table 1). For instance, AIL1 has orthologs in gymnosperm genomes. It duplicated in the ancestor of angiosperms followed by diverging in gene structure and functions between paralogs, and finally gave rise to ANT (Supplementary Figure S1-1). Unlike other genes, the paralogs of the C3HDZ genes, PHB, REV and CNA, were relatively conserved in gene structure, maintaining the same motif pattern after duplication (Supplementary Figure S1-5 and Table 1). Subfunctionalization of these paralogs most likely resulted from variations of the regulatory motif in the noncoding region. The results of this study thus not only demonstrate that different core regulatory genes involved in integument development which are normally considered typical or even unique in seed plants have actually homologs in seedless plants, but also indicate that gene duplication followed by subfunctionalization of duplicates play a decisive role in the origin and evolution of the integument development genes.
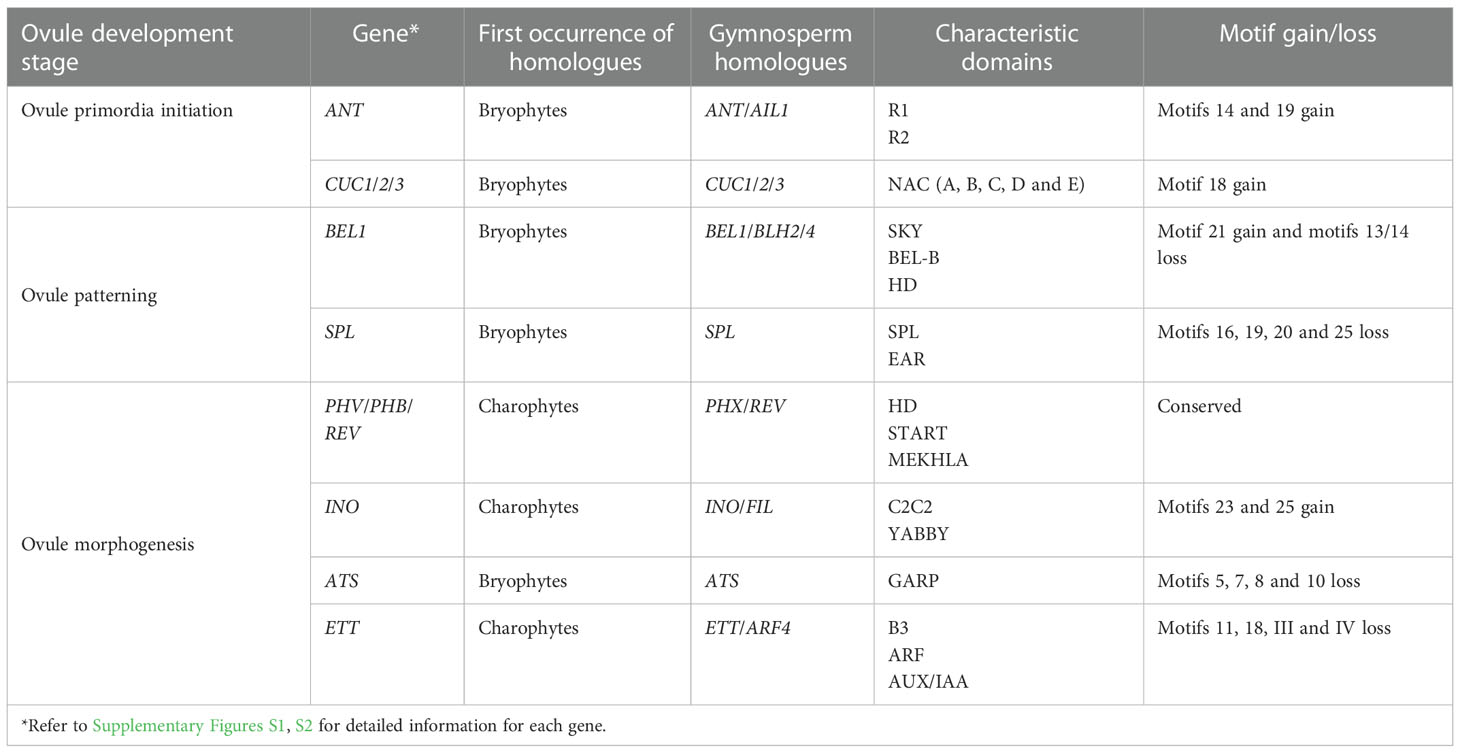
Table 1 Summary table showing the characteristics of the genes associated with integument development.
ANT, CUC, BEL1, SPL, C3HDZ, INO, ATS and ETT function as master regulators of integument development in angiosperms. Their homologs, however, play varied roles in seedless plants. The ANT/AIL homologs have been found to be expressed in the emerging gametophore apical cells, which was in accordance with our study (Supplementary Figure S3-3), in P. patens to determine stem cell identity (Aoyama et al., 2012). They were also expressed in the emerging fertile fronds in Ceratopteris richardii (Bui et al., 2017) and A. capillus (Figure 3 and Supplementary Figure S3-2). Horst et al. reported that the BEL1 homolog functioned as a master regulator for the gametophyte-to-sporophyte transition in P. patens; loss of function of PpBELL1 generated bigger egg cells unable to form embryos (Horst et al., 2016). The SPL/NZZ homolog has been shown to act as a transcription repressor to regulate auxin homeostasis across embryophytes and participate in controlling sporogenesis (Chen et al., 2014; Wei et al., 2015) and lateral organ morphogenesis (Li et al., 2008). Duplication and neofunctionalization of the C3HDZ genes have been revealed to occur in the ancestor of euphyllophytes, with a functional shift from regulating sporangium development to initiate of lateral primordia and leaf development (Vasco et al., 2016). The identification of an INO/YAB homolog in H. selago (Lycopodiales) suggests that the ancestral INO/YAB gene has evolved in the common ancestor of the vascular plants to control leaf formation (Evkaikina et al., 2017). The ATS/KAN homologs seem to be involved not only in establishing leaf polarity in Selaginella moellendorffii but also in the initiation of sporangium development (Zumajo-Cardona et al., 2019). As one of the core components of the nuclear auxin pathway, the ETT/ARF homologs have participated in regulating many aspects of plant growth and development since the early stages of green plant evolution by regulating transcription of auxin responsive genes (Kato et al., 2018; Martin-Arevalillo et al., 2019). The most notable is that these distinct transcription factors have assembled and acted cooperatively to form a genetic framework controlling the initiation and patterning of leaves during the origin and early evolution of euphyllophytes (Fang et al., 2021). The framework seems to have maintained throughout the development of leaf-like lateral organs in ferns and seed plants.
Did the integument arise de novo, or evolve from elaboration of pre-existing structures? Providing a reasonable answer to this question depends largely on the clarification of the fundamental nature of integuments because there is little consensus between these two hypotheses in regards to the genetic basis of origin and the evolutionary route.
Gene expression analyses have clearly shown the expression of the ANT/AIL homologs in developing integuments in G. biloba (Wang et al., 2016; Zumajo-Cardona et al., 2021; D'Apice et al., 2022), the conifer Pinus thunbergii (Shigyo and Ito, 2004; Yamada et al., 2008) and different species of Gnetum (Yamada et al., 2008; Zumajo-Cardona and Ambrose, 2021). Larsson et al. and Schlögl et al. showed the roles of the CUC homologs in regulating the differentiation of the shoot apical meristem (SAM) and embryogenesis in Picea abies (Larsson et al., 2012) and Araucaria angustifolia (Schlogl et al., 2012), respectively, as consistent with the roles in Arabidopsis (Chakraborty and Roy, 2019). A strong expression of the BEL1 homolog has been detected in the integument in G. biloba (Zumajo-Cardona et al., 2021; D'Apice et al., 2022). The homologs of C3HDZ and ATS/KAN were expressed throughout ovule development in G. biloba (Zumajo-Cardona et al., 2021; D'Apice et al., 2022) and the Gnetum species (Zumajo-Cardona and Ambrose, 2021). The INO/YAB homologs were also found to be expressed in the ovule integument in Ephedra distachya (Finet et al., 2016). Functional characterization of these genes in model plants has revealed that they are mostly the key components of the genetic program that patterns the laminar structure of leaf-like lateral organs (Mathews and Kramer, 2012). These genes participate in the establishment of abaxial-adaxial polarity during the development of integuments and other leaf-like lateral organs. Not only at the molecular level, integuments and leaves also share a number of defining features at the morphological level, including similar modes of organ initiation, determinant growth, and bilateral symmetry. Given the shared features of patterning and morphogenesis, serial homology has been postulated between leaves and integuments (Kelley and Gasser, 2009). Serial homology denotes the similarity of repetitive structures within the same organism (within-individual homology) (Minelli and Fusco, 2013), which is distinct from the historical (phylogenetic) homology that means the similarity of structures in two or more taxa descended from a common ancestor (between-species homology) (Mabee et al., 2020). The idea of serial homology is often rejected by traditional comparative biologists because for them homology is about the comparison of different species (Brigandt, 2003). However, serial homology is widely accepted and utilized in evolutionary developmental biology, which intends to account for the origin of similar structures both within and between organisms and for structural identity in ontogeny and phylogeny (Brigandt, 2003; DiFrisco et al., 2022; Fusco, 2022).
Two structures in the same organism are serial homologs if they share a large proportion of their genetic architecture and developmental pathways. The sharing of core regulatory genes supports the inference of integuments as leaf serial homologues. The results of this study demonstrate that the genetic regulatory network composed of the shared genes had been assembled at the early stage of euphyllophyte evolution, patterning a leaf by establishing polarities prior to the origin of seed plants. The underlying mechanism is reused in integument patterning while seed formation. The integument is thus unlikely to arise de novo from the perspective of developmental regulation, but evolved from the modification of the pre-existing genetic regulatory apparatus. It could be argued that the reuse or sharing of the leaf patterning gene network in integument development does not necessarily indicate a duplication of serial homologs. The genetic overlap may also be due to co-option and deep homology. Both terms are commonly used to describe the repeated use of the same genes or gene networks even in phylogenetically distant lineages, without specification of the implications for the corresponding phenotype (DiFrisco et al., 2022). Deep homology is often seen as an evolutionary residue of co-option events, with the shared genes or conserved gene networks being convergently recruited into different developmental processes to build morphologically and phylogenetically disparate features (Shubin et al., 1997; DiFrisco et al., 2022). The resulting morphological structures are usually not thought to be homologous (Tschopp and Tabin, 2017). The sharing of the leaf patterning gene network in integument translates to acquisition of the laminar organ identity at different stem segments within the same individual organism, strongly suggesting the origin of the integument via reusing and subsequent individuation of serially homologous structures but not due to co-option or ordinary gene sharing and pleiotropy which is often referred to as homocracy, i.e. shared patterns of regulatory gene expression among organs (Nielsen and Martinez, 2003). The recognition of ovules as meristematic axes provides further support for the stem segment-specific modification hypothesis of integument origin. Gross-Hardt et al. showed that the meristematic gene WUSCHEL (WUS) was expressed in the nucellus to regulate integument initiation in the chalaza by generating a downstream signal (Gross-Hardt et al., 2002). Just like the formation of leaf primordia in apical meristems, integuments arise from the nucellar meristem as novel lateral organs (Mathews and Kramer, 2012). This view is consistent with the axial theory proposed by several botanists in the nineteenth century that the nucellus is of the nature of a bud bearing the integuments as lateral foliar appendages (Worsdell, 1904).
The master ‘switches’ trigging the modification of the genetic program to specify the integument identity and development during the emergence of gymnosperms remain unclear. Identification of the upstream “selector” genes that are tightly linked to the identity of integument and contribute to distinct character development relative to other lateral organs is crucial for evaluating the serial homology hypothesis (DiFrisco et al., 2022). Duplication and subfunctionalization of known development regulatory genes might also facilitate the evolution of the regulatory network in tissue-developmental stage specificity. For instance, AIL is a pleiotropic gene which is involved not only in initiation and development of integument but also in initiation and growth of all plant organs except roots through control of cell proliferation (Horstman et al., 2014). This pleiotropic effect is the result of its control of cell proliferation during organogenesis, which seems to be a conserved function in the entire core eudicotyledons. The specialization of the descendant paralogous is not entirely dependent on changes to cis-regulatory DNA because there is growing evidence that changes to the coding regions of transcription factors play a much larger role in the evolution of developmental gene regulatory networks than originally imagined (Jarvela and Hinman, 2015). After duplication, relaxed constraints allow paralogous genes to diverge through mutations or exon shuffling to gain or loss motifs/domains. Just as cis-regulatory changes avoid pleiotropy by modulating the binding site composition, the changes in coding region can also lead functional specialization of paralogs by affecting alternative splicing, post-translational modification and protein-protein interaction. Nearly all the genes surveyed in this study exhibited structural variations in the coding regions, except C3HDZ (Table 1 and Supplementary Figure S1-5). Of them, ANT acquired two additional motifs following duplication (Supplementary Figure S1-1), while CUC, SPL, ATS and ETT lost motifs compared to their paralogs (Supplementary Figure S1). BEL1 and INO have undergone both motifs gain and loss events (Figure 2B and Supplementary Figure S1). Meister et al. (2005) and Gallagher and Gasser (2008) have shown that the C-terminal regions of INO which contain the novel motifs acquired following duplication were essential for its function in controlling the initiation and asymmetric growth of the outer integument by interacting with SUP (Meister et al., 2005; Gallagher and Gasser, 2008).
The extant integuments are morphologically quite distinct from that of fossil plants, making it difficult to decide whether they are historically homologous structures orchestrated by the same developmental system. There exist two alternative perspectives. The morphological distinctions possibly reflect the difference in their derivation with the lobed structure surrounding ovules in ancestral seed plants derived from transformation of sterile branches or sterilized sporangia, as predicted by the telome theory. The other perspective is that the envelopes encircling nucellus in the ovules of both extinct and extant taxa are historically (phylogenetically) homologous, possessing the common nature as lateral organs but undergoing differing degrees of evolutionary modification. It is impossible to examine how development of the ancestral structure might have been regulated. However, the latest research on the most primitive Genomosperma ovules showed that the organization of the lobed integument was highly plastic, with the lobe number and the degree of fusion varying considerably among individual fossils, exhibiting strong ontogenic (developmental) and polymorphic signals (Meade et al., 2021). Considering their findings above, the authors speculated that the lobes of the Genomosperma integument developed similarly to a whorl of extant floral organs and have been regulated by similar mechanisms to extant seed plant reproductive organs (Meade et al., 2021). The results of evolutionary and functional analysis of multiple integument regulatory genes seem to support their speculations. The ancestral integument structure and extant integuments, different though they seem, might share a gene network that formed via the stem segment-specific modification of the pre-existing genetic program for other lateral organs. The integument very likely evolved as a consequence of successive transformations from the modification of terminal branches (telomes) to leaf-like lateral organs and then into integuments (Figure 4).
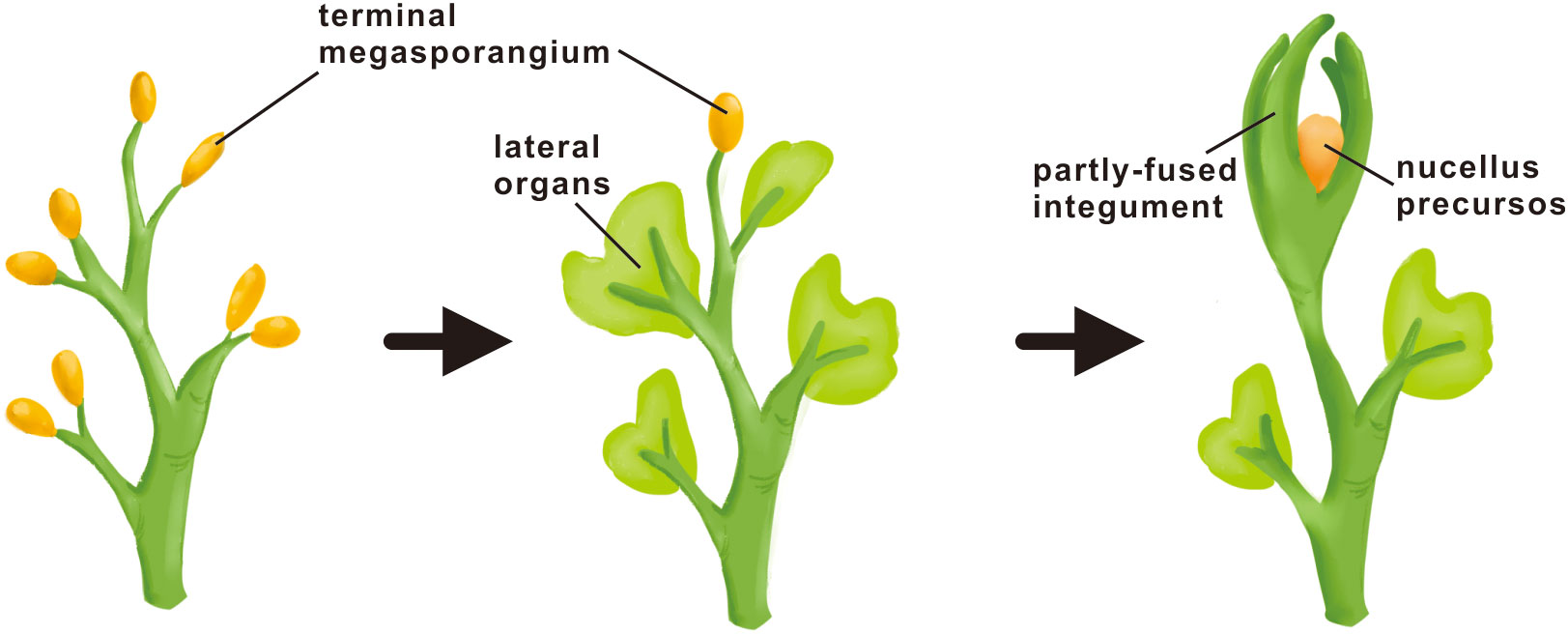
Figure 4 Hypothetical mode of integument origin. The origin of the integument is neither a direct transformation process nor a de novo process, but a successive transformation process. In early plant evolution, the sterilized telomes (branches) were first transformed into leaf-like lateral organs in seedless plants, and then the leaf-like lateral organs were further transformed into the integument at the time of seed occurs.
The origin of evolutionary novelty and the mechanics of innovation are central topics in evolutionary developmental biology. However, the definition of novelty and its relationship to homology are not well defined. If a new morphological trait has been shown to originate through the use and modification of pre-existing gene regulatory networks, it becomes unclear whether the trait is truly novel or merely modified serial homolog. Given the sharing of a homologous core set of genes, the integument was postulated to be a serially homologous structure in this study, but not arise de novo. From the structural and functional perspective, however, the integument drastically diverged from other lateral foliar appendages, facilitating survive and spread of seed plants. The definition of novelty and homology and the criteria of their application seem to remain controversial across the biological hierarchy. Comparing the character states, development processes and the underlying gene networks of different lateral organs within the context of serial vs historical homology may contribute to a more comprehensive understanding of how organs be transformed into one another during ontogeny and phylogeny, which has implication for our understanding of the key developmental events and regulators leading to the origin and evolution of the integument.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
JY conceived and designed the work. MJ collected and analyzed the data. JJ, CZ, LL, YW, WZ and ZS participated in data analysis. MJ drafted the manuscript. JY revised the manuscript. All authors read and approved the final manuscript.
This study was supported by the grant from Science and Technology Commission of Shanghai Municipality (14DZ2260400).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1078248/full#supplementary-material
Supplementary Table 1 | 49 representative genomes or transcriptomes selected in this study for identifying homologous genes. Species, family and the corresponding database are included.
Supplementary Table 2 | List of sequences included in the phylogenetic analyses for the ANT, CUC, BEL1, SPL, C3HDZ, YABBY, KANADI, and ARF gene families, respectively. Species name, gene name, and the accession numbers are included.
Supplementary Table 3 | The likelihood ratio tests are performed for the eight gene families investigated in the study. The ANT clade in the AP2/ERF gene family, the clade C in the CUC family, the clade C in the BLH family, the SPL clade in the SPEAR family, the clades C and D in the C3HDZ family, the INO clade in the YAB family, the KAN and ATS clades in the KAN family, and the ETT clade in the ARF gene family were designated as foreground branches, respectively.
Supplementary Figure 1 | Phylogeny and domain architecture of different gene families. Support values are shown for nodes. The scale bar indicates the number of changes per site. Different gene lineages are marked with different colors. (S1-1) Phylogeny and domain architecture of the ANT homologs. (A) Phylogenetic tree of 354 ANT homologs identified from diverse land plants. The red dot indicates the branch support value of BP > 85, while the yellow stars indicate the large-scale duplication events. Branches marked with pink indicate the ANT orthologs. The outer black circles represent the range of different clades, and the inner colored circles indicate sublineages within each clade. (B) Duplication history and domain architecture of the ANT homologs. Filled squares indicate the presence of the corresponding members, open squares indicate absence data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: euANT1, euANT2, euANT3, euANT4, the AP2 domain (R1 and R2) and the linker regions (L). The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-1. (C) Phylogenetic tree of ANT homologs with the support values at each node. (S1-2) Phylogeny and domain architecture of the CUC homologs. (A) Phylogenetic tree of 266 CUC homologs identified from diverse land plants. The red dot indicates the branch support value of BP > 85, while the yellow star indicates the large-scale duplication events. Branches marked with purple indicate the CUC orthologs. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. (B) Duplication history and domain architecture of the CUC homologs. Filled squares indicate the presence of the corresponding members, open squares indicate lack of data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: five subdomains of the NAC domain A, B, C, D, and E, the V motif (V), the L motif (L) and the W motif (W). The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-2. (C) Phylogenetic tree of CUC homologs with the support values at each node. (S1-3) Phylogeny and domain architecture of the BLH homologs. (A) Phylogenetic tree of 363 BLH homologs identified from diverse land plants. The red dot indicates the branch support values of BP > 85, while the yellow star indicates the large-scale duplication events. Branches marked with pink indicate the BEL1 orthologs. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. (B) Duplication history and domain architecture of the BLH homologs. Filled squares indicate the presence of the corresponding members, open squares indicate absence data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: BEL-A (SKY), BEL-B, BEL-C, ZIBEL and the homeodomain (HD). The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-3. (C) Phylogenetic tree of BLH homologs with the support values at each node. (S1-4) Phylogeny and domain architecture of the SPEAR homologs. (A) Phylogenetic tree of 120 SPEAR homologs identified from diverse land plants. The red dot indicates the branch support values of BP > 85, while the yellow star indicates the large-scale duplication events. Branches marked with pink indicate the SPL orthologs. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. (B) Duplication history and domain architecture of the SPEAR homologs. Filled squares indicate the presence of the corresponding members, open squares indicate absence data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: the nuclear localization signal domain (NLS), the SPL-motif and the EAR motif (LxLxL). The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-4. (C) Phylogenetic tree of SPEAR homologs with the support values at each node. (S1-5) Phylogeny and domain architecture of the C3HDZ homologs. (A) Phylogenetic tree of 215 C3HDZ homologs identified from diverse green plants. The red dot indicates the branch support values of BP > 85, while the yellow star indicates the large-scale duplication events. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. All members of Arabidopsis (angiosperms) are associated with integument development. (B) Duplication history and domain architecture of the C3HDZ homologs. Filled squares indicate the presence of the corresponding members, open squares indicate lack of data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: homeodomain (HD), leucine-zipper (LZ), CESVV, START, miR165/166 complementary site, HD-SAD (HD-START associated domain) and the MEKHLA domain. (C) Phylogenetic tree of C3HDZ homologs with the support values at each node. (S1-6) Phylogeny and domain architecture of the YABBY homologs. (A) Phylogenetic tree of 251 YABBY homologs identified from diverse green plants. The red dot indicates the branch support values of BP > 85, while the yellow star indicates the large-scale duplication events. Branches marked with pink indicate the INO orthologs. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. (B) Duplication history and domain architecture of the YABBY homologs. Filled squares indicate the presence of the corresponding members, open squares indicate lack of data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: C2-C2 zinc finger (C2C2) and the YABBY domain. The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-6. (C) Phylogenetic tree of YABBY homologs with the support values at each node. (S1-7) Phylogeny and domain architecture of the KANADI homologs. (A) Phylogenetic tree of 199 KANADI homologs identified from diverse land plants. The red dot indicates the branch support values of BP > 85, while the yellow star indicates the large-scale duplication events. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. All members from Arabidopsis (angiosperms) are associated with integument development. (B) Duplication history and domain architecture of the KANADI homologs. Filled squares indicate the presence of the corresponding members, open squares indicate absence data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: the GARP (GOLDEN2, ARR-B Class, Par1 proteins) domain and the L-rich motif. The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-7. (C) Phylogenetic tree of KANADI homologs with the support values at each node. (S1-8) Phylogeny and domain architecture of the ARF homologs. (A) Phylogenetic tree of 658 ARF homologs identified from diverse green plants. The red dot indicates the branch support values of BP > 85, while the yellow star indicates the large-scale duplication events. Branches marked with blue indicate the ETT orthologs. The outer black circles represent the range of different clades and the inner colored circles indicate sublineages within each clade. (B) Duplication history and domain architecture of the ARF homologs. Filled squares indicate the presence of the corresponding members, open squares indicate lack of data. The color of squares is corresponding to the top organismal tree. The yellow stars in the tree indicate whole-genome duplication events. The diagram on the right demonstrates the domain/motif composition of different duplicates. The known conserved domains/motifs included: the dimerization domain (DD), the B3 DNA binding domain (B3), the ARF and Aux/IAA domain (Motif III and IV are consensus sequences shared by Aux/IAA proteins). The unnamed domains/motifs are linage-specific and are marked with the colors corresponding to the sequence logos in Supplementary Figure S2-8. (C) Phylogenetic tree of ARF homologs with the support values at each node.
Supplementary Figure 2 | Sequence logos of the conserved (A) and unique (B) domains/motifs of different gene families. The height of the letter indicates its relative frequency at the given position (x -axis) in the domain/motif.
Supplementary Figure 3 | Expression of key genes related to integument development in rice (S3-1), Adiantum capillus (S3-2) and Physcomitrella patens (S3-3). The 33 various developmental stages or tissues of A. capillus (S3-2) included the embryo gametophyte (EG), gametophyte (GA), the transUL vegetative growth leaf (tUL-VGL), the transLCGS green sporangium leaf (tCGS-GSL), the transLCMS mature sporangium leaf (tCMS-MSL), the transLCJS juvenile sporangium leaf (tCJS-JSL), the transLCDS dehiscent sporangium leaf (tCDS-DSL), the transCL vegetative growth leaf (tCL-VGL), the transFL vegetative growth leaf (tFL-VGL), the AnotUL vegetative growth leaf (AUL-VGL), the AnotFL vegetative growth leaf (AFL-VGL), the AnotCL vegetative growth leaf (ACJS-JSL), the AnotLCGS vegetative growth leaf (ACGS-DSL), the AnotLCTS sporangium leaf (ACTS-SL), the AnotLCMS mature sporangium leaf (ACMS-MSL), the AnotLCDS dehiscent sporangium leaf (ACDS-DSL), the transMS mature sporangium stage (tMS-MSS), the transGS green sporangium stage (tGS-GSS), the transJS juvenile sporangium stage (tJS-JSS), the AnotJS juvenile sporangium stage (AJS-JSS), the AnotGS green sporangium stage (ATS-SS), the AnotMS mature sporangium stage (AMS-MSS), the transMSL mature sporangium leaf (transMSL), the transGSL green sporangium leaf (transGSL), transDSL dehiscent sporangium leaf (transDSL), transJSL juvenile sporangium stage (transJSL), the AnotJSL juvenile sporangium stage (AnotJSL), the AnotGSL green sporangium leaf (AnotGSL), AnotTSL sporangium leaf (AnotTSL), the AnotMSL mature sporangium leaf (AnotMSL), the AnotDSL dehiscent sporangium leaf (AnotDSL), the transYS young sporophyte (transYS) and the stem apical (SA).
Aoyama, T., Hiwatashi, Y., Shigyo, M., Kofuji, R., Kubo, M., Ito, M., et al. (2012). AP2-type transcription factors determine stem cell identity in the moss Physcomitrella patens. Development 139, 3120–3129. doi: 10.1242/dev.076091
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Bai, P. P., Lin, H. Y., Sun, Y., Wu, J. J., Gu, K. J., Zhao, Y. P. (2022). Temporal dynamic transcriptome landscape reveals regulatory network during the early differentiation of female strobilus buds in Ginkgo biloba. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.863330
Balasubramanian, S., Schneitz, K. (2002). NOZZLE links proximal-distal and adaxial-abaxial pattern formation during ovule development in Arabidopsis thaliana. Development 129, 4291–4300. doi: 10.1242/dev.129.18.4291
Baroux, C., Grossniklaus, U. (2019). Seeds-an evolutionary innovation underlying reproductive success in flowering plants. Curr. Top. Dev. Biol. 131, 605–642. doi: 10.1016/bs.ctdb.2018.11.017
Barro-Trastoy, D., Gomez, M. D., Tornero, P., Perez-Amador, M. A. (2020). On the way to ovules: the hormonal regulation of ovule development. Crit. Rev. Plant Sci. 39, 431–456. doi: 10.1080/07352689.2020.1820203
Bencivenga, S., Simonini, S., Benkova, E., Colombo, L. (2012). The transcription factors BEL1 and SPL are required for cytokinin and auxin signaling during ovule development in Arabidopsis. Plant Cell 24, 2886–2897. doi: 10.1105/tpc.112.100164
Brambilla, V., Battaglia, R., Colombo, M., Masiero, S., Bencivenga, S., Kater, M. M., et al. (2007). Genetic and molecular interactions between BELL1 and MADS box factors support ovule development in Arabidopsis. Plant Cell 19, 2544–2556. doi: 10.1105/tpc.107.051797
Brigandt, I. (2003). Homology in comparative, molecular, and evolutionary developmental biology: The radiation of a concept. J. Exp. Zool B Mol. Dev. Evol. 299, 9–17. doi: 10.1002/jez.b.36
Brown, R. H., Nickrent, D. L., Gasser, C. S. (2010). Expression of ovule and integument-associated genes in reduced ovules of santalales. Evol. Dev. 12, 231–240. doi: 10.1111/j.1525-142X.2010.00407.x
Bui, L. T., Pandzic, D., Youngstrom, C. E., Wallace, S., Irish, E. E., Szovenyi, P., et al. (2017). A fern AINTEGUMENTA gene mirrors BABY BOOM in promoting apogamy in Ceratopteris richardii. Plant J. Cell Mol. Biol. 90, 122–132. doi: 10.1111/tpj.13479
Capella-Gutierrez, S., Silla-Martinez, J. M., Gabaldon, T. (2009). trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chakraborty, R., Roy, S. (2019). Evaluation of the diversity and phylogenetic implications of NAC transcription factor members of four reference species from the different embryophytic plant groups. Physiol. Mol. Biol. Pla 25, 347–359. doi: 10.1007/s12298-018-0581-9
Chen, G. H., Sun, J. Y., Liu, M., Liu, J., Yang, W. C. (2014). SPOROCYTELESS is a novel embryophyte-specific transcription repressor that interacts with TPL and TCP proteins in Arabidopsis. J. Genet. Genomics 41, 617–625. doi: 10.1016/j.jgg.2014.08.009
D'Apice, G., Moschin, S., Araniti, F., Nigris, S., Di Marzo, M., Muto, A., et al. (2021). The role of pollination in controlling Ginkgo biloba ovule development. New Phytol. 232, 2353–2368. doi: 10.1111/nph.17753
D'Apice, G., Moschin, S., Nigris, S., Ciarle, R., Muto, A., Bruno, L., et al. (2022). Identification of key regulatory genes involved in the sporophyte and gametophyte development in Ginkgo biloba ovules revealed by in situ expression analyses. Am. J. Bot. 109, 1–12. doi: 10.1002/ajb2.1862
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
DiFrisco, J., Wagner, G. P., Love, A. C. (2022). Reframing research on evolutionary novelty and co-option: Character identity mechanisms versus deep homology. Semin. Cell Dev. Biol 126. doi: 10.1016/j.semcdb.2022.03.030
Elliott, R. C., Betzner, A. S., Huttner, E., Oakes, M. P., Tucker, W. Q., Gerentes, D., et al. (1996). AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell 8, 155–168. doi: 10.1105/tpc.8.2.155
Eshed, Y., Baum, S. F., Perea, J. V., Bowman, J. L. (2001). Establishment of polarity in lateral organs of plants. Curr. Biol. CB 11, 1251–1260. doi: 10.1016/s0960-9822(01)00392-x
Evkaikina, A. I., Berke, L., Romanova, M. A., Proux-Wera, E., Ivanova, A. N., Rydin, C., et al. (2017). The Huperzia selago shoot tip transcriptome sheds new light on the evolution of leaves. Genome Biol. Evol. 9, 2444–2460. doi: 10.1093/gbe/evx169
Fang, Y., Qin, X., Liao, Q., Du, R., Luo, X., Zhou, Q., et al. (2021). The genome of homosporous maidenhair fern sheds light on the euphyllophyte evolution and defences. Nat. Plants 8, 1024–1037. doi: 10.1038/s41477-022-01222-x
Finet, C., Berne-Dedieu, A., Scutt, C. P., Marletaz, F. (2013). Evolution of the ARF gene family in land plants: old domains, new tricks. Mol. Biol. Evol. 30, 45–56. doi: 10.1093/molbev/mss220
Finet, C., Floyd, S. K., Conway, S. J., Zhong, B. J., Scutt, C. P., Bowmanb, J. L. (2016). Evolution of the YABBY gene family in seed plants. Evol. Dev. 18, 116–126. doi: 10.1111/ede.12173
Galbiati, F., Sinha Roy, D., Simonini, S., Cucinotta, M., Ceccato, L., Cuesta, C., et al. (2013). An integrative model of the control of ovule primordia formation. Plant J. Cell Mol. Biol. 76, 446–455. doi: 10.1111/tpj.12309
Gallagher, T. L., Gasser, C. S. (2008). Independence and interaction of regions of the INNER NO OUTER protein in growth control during ovule development. Plant Physiol. 147, 306–315. doi: 10.1104/pp.107.114603
Gasser, C. S., Broadhvest, J., Hauser, B. A. (1998). Genetic analysis of ovule development. Annu. Rev. Plant Phys. 49, 1–24. doi: 10.1146/annurev.arplant.49.1.1
Gasser, C. S., Skinner, D. J. (2019). Development and evolution of the unique ovules of flowering plants. Curr. Top. Dev. Biol. 131, 373–396. doi: 10.1016/bs.ctdb.2018.10.007
Gerrienne, P., Meyer-Berthaud, B. (2007). The proto-ovule runcaria heinzelinii stockmans 1968 emend. gerrienne et al. 2004 (mid-givetian, belgium): Concept and epitypification. Rev. Palaeobot Palyno 145, 321–323. doi: 10.1016/j.revpalbo.2006.12.003
Goncalves, B., Hasson, A., Belcram, K., Cortizo, M., Morin, H., Nikovics, K., et al. (2015). A conserved role for CUP-SHAPED COTYLEDON genes during ovule development. Plant J. Cell Mol. Biol. 83, 732–742. doi: 10.1111/tpj.12923
Gross-Hardt, R., Lenhard, M., Laux, T. (2002). WUSCHEL signaling functions in interregional communication during Arabidopsis ovule development. Genes Dev. 16, 1129–1138. doi: 10.1101/gad.225202
Hetherington, A. J. (2021). New views on old seeds: a new description of Genomosperma sheds light on early seed evolution. New Phytol. 229, 1189–1191. doi: 10.1111/nph.16875
Horst, N. A., Katz, A., Pereman, I., Decker, E. L., Ohad, N., Reski, R. (2016). A single homeobox gene triggers phase transition, embryogenesis and asexual reproduction. Nat. Plants 2, 15209. doi: 10.1038/nplants.2015.209
Horstman, A., Willemsen, V., Boutilier, K., Heidstra, R. (2014). AINTEGUMENTA-LIKE proteins: hubs in a plethora of networks. Trends Plant Sci. 19, 146–157. doi: 10.1016/j.tplants.2013.10.010
Jaramillo, M. A., Kramer, E. M. (2007). The role of developmental genetics in understanding homology and morphological evolution in plants. Int. J. Plant Sci. 168, 61–72. doi: 10.1086/509078
Jarvela, A. M. C., Hinman, V. F. (2015). Evolution of transcription factor function as a mechanism for changing metazoan developmental gene regulatory networks. Evodevo 6, 3. doi: 10.1186/2041-9139-6-3
Jeffares, D. C., Tomiczek, B., Sojo, V., dos Reis, M. (2015). A beginners guide to estimating the non-synonymous to synonymous rate ratio of all protein-coding genes in a genome. Methods Mol. Biol. 1201, 65–90. doi: 10.1007/978-1-4939-1438-8_4
Jensen, M. K., Kjaersgaard, T., Nielsen, M. M., Galberg, P., Petersen, K., O'Shea, C., et al. (2010). The Arabidopsis thaliana NAC transcription factor family: Structure-function relationships and determinants of ANAC019 stress signalling. Biochem. J. 426, 183–196. doi: 10.1042/Bj20091234
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kato, H., Nishihama, R., Weijers, D., Kohchi, T. (2018). Evolution of nuclear auxin signaling: lessons from genetic studies with basal land plants. J. Exp. Bot. 69, 291–301. doi: 10.1093/jxb/erx267
Kelley, D. R., Arreola, A., Gallagher, T. L., Gasser, C. S. (2012). ETTIN (ARF3) physically interacts with KANADI proteins to form a functional complex essential for integument development and polarity determination in Arabidopsis. Development 139, 1105–1109. doi: 10.1242/dev.067918
Kelley, D. R., Gasser, C. S. (2009). Ovule development: Genetic trends and evolutionary considerations. Sex Plant Reprod. 22, 229–234. doi: 10.1007/s00497-009-0107-2
Kelley, D. R., Skinner, D. J., Gasser, C. S. (2009). Roles of polarity determinants in ovule development. Plant J. Cell Mol. Biol. 57, 1054–1064. doi: 10.1111/j.1365-313X.2008.03752.x
Kenrick, P., Crane, P. R. (1997). The origin and early diversification of land plants: A cladistic study (Washington DC: Smithsonian Institution Press).
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Kim, S., Soltis, P. S., Wall, K., Soltis, D. E. (2006). Phylogeny and domain evolution in the APETALA2-like gene family. Mol. Biol. Evol. 23, 107–120. doi: 10.1093/molbev/msj014
Klucher, K. M., Chow, H., Reiser, L., Fischer, R. L. (1996). The AINTEGUMENTA gene of Arabidopsis required for ovule and female gametophyte development is related to the floral homeotic gene APETALA2. Plant Cell 8, 137–153. doi: 10.1105/tpc.8.2.137
Larsson, E., Sundstrom, J. F., Sitbon, F., von Arnold, S. (2012). Expression of PaNAC01, a Picea abies CUP-SHAPED COTYLEDON orthologue, is regulated by polar auxin transport and associated with differentiation of the shoot apical meristem and formation of separated cotyledons. Ann. Bot. 110, 923–934. doi: 10.1093/aob/mcs151
Linkies, A., Graeber, K., Knight, C., Leubner-Metzger, G. (2010). The evolution of seeds. New Phytol. 186, 817–831. doi: 10.1111/j.1469-8137.2010.03249.x
Li, L. C., Qin, G. J., Tsuge, T., Hou, X. H., Ding, M. Y., Aoyama, T., et al. (2008). SPOROCYTELESS modulates YUCCA expression to regulate the development of lateral organs in Arabidopsis. New Phytol. 179, 751–764. doi: 10.1111/j.1469-8137.2008.02514.x
Lora, J., Hormaza, J. I., Herrero, M. (2015). Transition from two to one integument in Prunus species: expression pattern of INNER NO OUTER (INO), ABERRANT TESTA SHAPE (ATS) and ETTIN (ETT). New Phytol. 208, 584–595. doi: 10.1111/nph.13460
Lora, J., Hormaza, J. I., Herrero, M., Gasser, C. S. (2011). Seedless fruits and the disruption of a conserved genetic pathway in angiosperm ovule development. Proc. Natl. Acad. Sci. United States America 108, 5461–5465. doi: 10.1073/pnas.1014514108
Lora, J., Laux, T., Hormaza, J. I. (2019). The role of the integuments in pollen tube guidance in flowering plants. New Phytol. 221, 1074–1089. doi: 10.1111/nph.15420
Mabee, P. M., Balhoff, J. P., Dahdul, W. M., Lapp, H., Mungall, C. J., Vision, T. J. (2020). A logical model of homology for comparative biology. Syst. Biol. 69, 345–362. doi: 10.1093/sysbio/syz067
Martin-Arevalillo, R., Thevenon, E., Jegu, F., Vinos-Poyo, T., Vernoux, T., Parcy, F., et al. (2019). Evolution of the auxin response factors from charophyte ancestors. PloS Genet. 15, e1008400. doi: 10.1371/journal.pgen.1008400
Mathews, S., Kramer, E. M. (2012). The evolution of reproductive structures in seed plants: A re-examination based on insights from developmental genetics. New Phytol. 194, 910–923. doi: 10.1111/j.1469-8137.2012.04091.x
Maugarny-Cales, A., Goncalves, B., Jouannic, S., Melkonian, M., Wong, G. K. S., Laufs, P. (2016). Apparition of the NAC transcription factors predates the emergence of land plants. Mol. Plant 9, 1345–1348. doi: 10.1016/j.molp.2016.05.016
McAbee, J. M., Hill, T. A., Skinner, D. J., Izhaki, A., Hauser, B. A., Meister, R. J., et al. (2006). ABERRANT TESTA SHAPE encodes a KANADI family member, linking polarity determination to separation and growth of Arabidopsis ovule integuments. Plant J. Cell Mol. Biol. 46, 522–531. doi: 10.1111/j.1365-313X.2006.02717.x
Meade, L. E., Plackett, A. R. G., Hilton, J. (2021). Reconstructing development of the earliest seed integuments raises a new hypothesis for the evolution of ancestral seed-bearing structures. New Phytol. 229, 1782–1794. doi: 10.1111/nph.16792
Meister, R. J., Oldenhof, H., Bowman, J. L., Gasser, C. S. (2005). Multiple protein regions contribute to differential activities of YABBY proteins in reproductive development. Plant Physiol. 137, 651–662. doi: 10.1104/pp.104.055368
Meyer-Berthaud, B. (2022). First ovules integument: What roles? Natl. Sci. Rev. 9, nwab224. doi: 10.1093/nsr/nwab224
Meyer-Berthaud, B., Gerrienne, P., Prestianni, C. (2018). Letters to the twenty-first century botanist. second series: "what is a seed?"-3. how did we get there? palaeobotany sheds light on the emergence of seed. Bot. Lett. 165, 434–439. doi: 10.1080/23818107.2018.1505547
Minelli, A., Fusco, G. (2013). “Homology,” in Kampourakis, K. (ed.), The Philosophy of Biology: A Companion for Educators, Springer. pp. 1–289, Philosophy and theory of the life sciences. doi: 10.1007/978-94-007-6537-5_15
Mukherjee, K., Brocchieri, L., Burglin, T. R. (2009). A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol. Biol. Evol. 26, 2775–2794. doi: 10.1093/molbev/msp201
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nielsen, C., Martinez, P. (2003). Patterns of gene expression: Homology or homocracy? Dev. Genes Evol. 213, 149–154. doi: 10.1007/s00427-003-0301-4
Ortiz-Ramirez, C., Hernandez-Coronado, M., Thamm, A., Catarino, B., Wang, M., Dolan, L., et al. (2016). A transcriptome atlas of Physcomitrella patens provides insights into the evolution and development of land plants. Mol. Plant 9, 205–220. doi: 10.1016/j.molp.2015.12.002
Pereira-Santana, A., Alcaraz, L. D., Castano, E., Sanchez-Calderon, L., Sanchez-Teyer, F., Rodriguez-Zapata, L. (2015). Comparative genomics of NAC transcriptional factors in angiosperms: implications for the adaptation and diversification of flowering plants. PloS One 10, e0141866. doi: 10.1371/journal.pone.0141866
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Petrella, R., Gabrieli, F., Cavalleri, A., Schneitz, K., Colombo, L., Cucinotta, M. (2022). Pivotal role of STIP in ovule pattern formation and female germline development in Arabidopsis thaliana. Development 149, dev201184. doi: 10.1242/dev.201184
Pinyopich, A., Ditta, G. S., Savidge, B., Liljegren, S. J., Baumann, E., Wisman, E., et al. (2003). Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 424, 85–88. doi: 10.1038/nature01741
Prigge, M. J., Otsuga, D., Alonso, J. M., Ecker, J. R., Drews, G. N., Clark, S. E. (2005). Class III homeodomain-leucine zipper gene family members have overlapping, antagonistic, and distinct roles in Arabidopsis development. Plant Cell 17, 61–76. doi: 10.1105/tpc.104.026161
Puranik, S., Sahu, P. P., Srivastava, P. S., Prasad, M. (2012). NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 17, 369–381. doi: 10.1016/j.tplants.2012.02.004
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Robinsonbeers, K., Pruitt, R. E., Gasser, C. S. (1992). Ovule development in wild-type Arabidopsis and two female-sterile mutants. Plant Cell 4, 1237–1249. doi: 10.1105/tpc.4.10.1237
Schlogl, P. S., dos Santos, A. L. W., Vieira, L. D., Floh, E. I. S., Guerra, M. P. (2012). Cloning and expression of embryogenesis-regulating genes in Araucaria angustifolia (Bert.) o. kuntze (Brazilian pine). Genet. Mol. Biol. 35, 172–181. doi: 10.1590/S1415-47572012005000005
Schneitz, K., Hulskamp, M., Pruitt, R. E. (1995). Wild-type ovule development in Arabidopsis thaliana: a light-microscope study of cleared whole-mount tissue. Plant J. Cell Mol. Biol. 7, 731–749. doi: 10.1046/j.1365-313X.1995.07050731.x
Shigyo, M., Ito, M. (2004). Analysis of gymnosperm two-AP2-domain-containing genes. Dev. Genes Evol. 214, 105–114. doi: 10.1007/s00427-004-0385-5
Shubin, N., Tabin, C., Carroll, S. (1997). Fossils, genes and the evolution of animal limbs. Nature 388, 639–648. doi: 10.1038/41710
Smith, D. L. (1964). The evolution of the ovule. Biol. Rev. 39, 137–159. doi: 10.1111/j.1469-185X.1964.tb00952.x
Tomescu, A. M. F., Rothwell, G. W. (2022). Fossils and plant evolution: Structural fingerprints and modularity in the evo-devo paradigm. Evodevo 13, 8. doi: 10.1186/s13227-022-00192-7
Tomoyasu, Y., Ohde, T., Clark-Hachtel, C. (2017). What serial homologs can tell us about the origin of insect wings. F1000Research 6, 268. doi: 10.12688/f1000research.10285.1
Tschopp, P., Tabin, C. J. (2017). Deep homology in the age of next-generation sequencing. Philos. Trans. R Soc. Lond B Biol. Sci. 372, 20150475. doi: 10.1098/rstb.2015.0475
Vasco, A., Smalls, T. L., Graham, S. W., Cooper, E. D., Wong, G. K., Stevenson, D. W., et al. (2016). Challenging the paradigms of leaf evolution: Class III HD-zips in ferns and lycophytes. New Phytol. 212, 745–758. doi: 10.1111/nph.14075
Villanueva, J. M., Broadhvest, J., Hauser, B. A., Meister, R. J., Schneitz, K., Gasser, C. S. (1999). INNER NO OUTER regulates abaxial- adaxial patterning in Arabidopsis ovules. Genes Dev. 13, 3160–3169. doi: 10.1101/gad.13.23.3160
Vroemen, C. W., Mordhorst, A. P., Albrecht, C., Kwaaitaal, M. A. C. J., de Vries, S. C. (2003). The CUP-SHAPED COTYLEDON3 gene is required for boundary and shoot meristem formation in Arabidopsis. Plant Cell 15, 1563–1577. doi: 10.1105/tpc.012203
Wang, L., Lu, Z. G., Li, W. X., Xu, J., Luo, K. G., Lu, W. C., et al. (2016). Global comparative analysis of expressed genes in ovules and leaves of Ginkgo biloba l. Tree Genet. Genomes 12, 29. doi: 10.1007/s11295-016-0989-8
Wei, B. Y., Zhang, J. Z., Pang, C. X., Yu, H., Guo, D. S., Jiang, H., et al. (2015). The molecular mechanism of SPOROCYTELESS/NOZZLE in controlling Arabidopsis ovule development. Cell Res. 25, 121–134. doi: 10.1038/cr.2014.145
Worsdell, W. C. (1904). The structure and morphology of the 'ovule': An historical sketch. Ann. Bot. 18, 57–86. doi: 10.1093/oxfordjournals.aob.a088955
Yamada, T., Hirayama, Y., Imaichi, R., Kato, M. (2008). AINTEGUMENTA homolog expression in Gnetum (gymnosperms) and implications for the evolution of ovulate axes in seed plants. Evol. Dev. 10, 280–287. doi: 10.1111/j.1525-142X.2008.00237.x
Yamada, T., Imaichi, R., Kato, M. (2001). Developmental morphology of ovules and seeds of nymphaeales. Am. J. Bot. 88, 963–974. doi: 10.2307/2657077
Yamada, T., Sasaki, Y., Hashimoto, K., Nakajima, K., Gasser, C. S. (2016). CORONA, PHABULOSA and PHAVOLUTA collaborate with BELL1 to confine WUSCHEL expression to the nucellus in Arabidopsis ovules. Development 143, 422–426. doi: 10.1242/dev.129833
Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. CABIOS 13, 555–556. doi: 10.1093/bioinformatics/13.5.555
Zhao, H., Guo, M., Yan, M., Cheng, H., Liu, Y., She, Z., et al. (2020). Comparative expression profiling reveals genes involved in megasporogenesis. Plant Physiol. 182, 2006–2024. doi: 10.1104/pp.19.01254
Zimmermann, W. (1952). Main results of the "Telome theory". J. Palaeosciences 1, 456–470. doi: 10.54991/jop.1952.423
Zumajo-Cardona, C., Ambrose, B. A. (2020). Phylogenetic analyses of key developmental genes provide insight into the complex evolution of seeds. Mol. Phylogenet Evol. 147, 106778. doi: 10.1016/J.Ympev.2020.106778
Zumajo-Cardona, C., Ambrose, B. A. (2021). Deciphering the evolution of the ovule genetic network through expression analyses in gnetum gnemon. Ann. Bot. 128, 217–230. doi: 10.1093/aob/mcab059
Zumajo-Cardona, C., Little, D. P., Stevenson, D., Ambrose, B. A. (2021). Expression analyses in Ginkgo biloba provide new insights into the evolution and development of the seed. Sci. Rep Uk 11, 21995. doi: 10.1038/S41598-021-01483-0
Keywords: seed formation, integument origin, successive transformation, regulatory gene, molecular evolution, evo-devo, serial homology
Citation: Jiang M, Jian J, Zhou C, Li L, Wang Y, Zhang W, Song Z and Yang J (2023) Does integument arise de novo or from pre-existing structures? ── Insights from the key regulatory genes controlling integument development. Front. Plant Sci. 13:1078248. doi: 10.3389/fpls.2022.1078248
Received: 24 October 2022; Accepted: 29 December 2022;
Published: 13 January 2023.
Edited by:
Stefan de Folter, Center for Research and Advanced Studies (CINVESTAV), MexicoReviewed by:
Günter Theißen, Friedrich Schiller University Jena, GermanyCopyright © 2023 Jiang, Jian, Zhou, Li, Wang, Zhang, Song and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji Yang, aml5YW5nQGZ1ZGFuLmVkdS5jbg==
†ORCID: Min Jiang, orcid.org/0000-0002-0757-9217
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.