
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 17 January 2023
Sec. Plant Bioinformatics
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1074697
 Yu Wang1,2†
Yu Wang1,2† Jing Xu3†
Jing Xu3† Bin Hu1,2,4†
Bin Hu1,2,4† Chunxing Dong1,2
Chunxing Dong1,2 Jin Sun1,2Zixian Li1,2
Jin Sun1,2Zixian Li1,2 Kangzhuo Ye1,2Fang Deng1,2
Kangzhuo Ye1,2Fang Deng1,2 Lulu Wang1,2,5,6
Lulu Wang1,2,5,6 Mohammad Aslam1,2,5,6Wenliang Lv3*
Mohammad Aslam1,2,5,6Wenliang Lv3* Yuan Qin1,2,4,7*
Yuan Qin1,2,4,7* Yan Cheng1,2,4,7*
Yan Cheng1,2,4,7*In the Convolvulaceae family, around 1650 species belonging to 60 genera are widely distributed globally, mainly in the tropical and subtropical regions of America and Asia. Although a series of chloroplast genomes in Convolvulaceae were reported and investigated, the evolutionary and genetic relationships among the chloroplast genomes of the Convolvulaceae family have not been extensively elucidated till now. In this study, we first reported the complete chloroplast genome sequence of Ipomoea pes-caprae, a widely distributed coastal plant with medical values. The chloroplast genome of I. pes-caprae is 161667 bp in length, and the GC content is 37.56%. The chloroplastic DNA molecule of I. pes-caprae is a circular structure composed of LSC (large-single-copy), SSC (small-single-copy), and IR (inverted repeat) regions, with the size of the three regions being 88210 bp, 12117 bp, and 30670 bp, respectively. The chloroplast genome of I. pes-caprae contains 141 genes, and 35 SSRs are identified in the chloroplast genome. Our research results provide important genomic information for the molecular phylogeny of I. pes-caprae. The Phylogenetic analysis of 28 Convolvulaceae chloroplast genomes showed that the relationship of I. pes-caprae with I. involucrata or I. obscura was much closer than that with other Convolvulaccae species. Further comparative analyses between the Ipomoea species and Cuscuta species revealed the mechanism underlying the formation of parasitic characteristics of Cuscuta species from the perspective of the chloroplast genome.
Ipomoea. pes-caprae, which belongs to the Convolvulaceae, grows on coastal beaches and dunes throughout the tropical and subtropical areas of the world (Devall, 1992). This species has a high-speed growth rate and long-trailing stems, being one of the earliest species to colonize newly deposited dunes, contributing to the initial stabilization of sand (Devall and Thien, 1989). I. pes-caprae is widely distributed in southeast coastal areas of China, is found on tropical and subtropical beaches, and belongs to associated mangrove plants. It is common in Zhejiang, Fujian, Guangdong, Guangxi, and Taiwan. I. pes-caprae is evergreen all year round, with a particular leaf shape, strong growth potential, long flowering, and fruiting period. It has flowers almost all year round with bright colors. Its capsule is spherical, its pericarp is leathery, and its leaves, flowers, and fruits are of high ornamental value. The root system of I. pes-caprae is deeply grown into the soil to be used for sand fixation or covering plants on the beach. Recently, the medicinal value of I. pes-caprae has attracted the attention of researchers. It has been reported that the chemical components from I. pes-caprae have a wide range of biological activities for their antioxidant, analgesic and anti-inflammatory, antispasmodic, antinociceptive, antihistaminic, immunostimulant, insulinogenic, hypoglycemic antimicrobial, antifungal, and antibacterial characteristics (Bragadeeswaran et al., 2010). Moreover, previous studies have reported that I. pes-caprae could be used to inhibit platelet aggregation, diarrhea, vomiting, and piles (Manigauha et al., 2015).
Chloroplast is a vital organelle for green plants on earth to convert light energy into chemical energy (Jagendorf and Uribe, 1966; Neuhaus and Emes, 2000). The photosynthesis processes in the chloroplast are strictly regulated by a complex group of genes (Price et al., 2012). In plants, three organelles contain their genomes, nucleus, mitochondria, and chloroplast. The chloroplast genomes are highly conserved in genome structure, gene order, gene content, and gene number (Mira et al., 2018). Therefore, the chloroplast genomes were widely used as a valuable information resource for investigating the evolutionary history and taxonomic confirmation of land plants (Timme et al., 2007; Dong et al., 2014; Curci et al., 2015; Ellegren et al., 2015; Ju and Gao, 2016; Mira et al., 2018). The chloroplast genome is a circular double-stranded DNA molecule with a size of 120-180 KB, which is circular and consists of a large single-copy (LSC) region and a small single-copy (SSC) region separated by a pair of inverted repeats (Ozeki et al., 1989; Jansen et al., 2005; Petit et al., 2005; Funk et al., 2007; Jansen and Ruhlman, 2012). The chloroplast genome of land plants contains protein-coding genes and non-protein-coding genes. The protein-coding genes are mainly involved in photosynthesis and protein translation, and only a few are related to the transcription in the chloroplast. The non-protein-coding genes are the tRNA genes, whose transcripts are the transporters of amino acids in the peptide elongation, and the rRNA genes, composed of the ribosome (Jansen et al., 2005). The chloroplast genome can replicate by itself inside the chloroplast organelle. However, information communication and substance exchange with cytosol are critical for this biological event, and the genetic orders from the nuclei supervise all the metabolism in the chloroplast (Bulychev and Komarova, 2015).
In this study, the complete chloroplast genome sequence of I. pes-caprae was assembled, annotated, and comparatively analyzed. The results show that the length of the chloroplast genome is 161,667 bp with a GC content of 37.56%. The chloroplast genome of I. pes-caprae has a canonical structure, which is circular and composed of LSC, SSC, and IR regions, containing 136 annotated genes. The chloroplast genomes of 26 Convolvulaceae species, including 14 Cuscuta species and 12 Ipomoea species, were used for phylogenetic analysis and comparative analyses in codon preference and gene number, and gene content. Phylogenetic analysis showed that the relationship of I. pes-caprae with I. involucrata or I. obscura was much closer than that with other Convolvulaccae species. The phylogenetic and gene content analyses of Convolvulaccae species also provided new insight into the evolution of parasitic characteristics of Cuscuta species.
The I. pes-caprae L. plants used for this study were naturally growing on the beach located in Changle, Fuzhou (Latitude 25°, 54’, 33˝ N, Longitude 119°, 40’, 42˝ E), Fujian, China. The leaf materials were used for DNA extraction using the modified CTAB method (Li et al., 2013).
The genome DNA samples were subject to SMART laboratory construction and then sequenced on the PacBio Sequel II sequencing platform. The CCS (Circular Consensus Sequence) reads corresponding to the chloroplast genome were extracted by mapping all the reads to the chloroplast genomes of all the Convolvulaceae species with Bowtie 2 (Langmead and Salzberg, 2012). Subsequently, the resulting CCS reads were considered to be derived from the chloroplast genome and were used for chloroplast genome assembly using Canu (V2.2) software (Nurk et al., 2020). The complete chloroplast genome sequences were annotated using the program PGA (Qu et al., 2019). Both tRNAs and rRNAs were identified by BLASTN and BLASTP by searching against the references composed of all the available chloroplast genomes of Convolvulaceae species. The annotation results were checked manually, and the codon positions were adjusted by comparing them to a previously homologous gene from various chloroplast genomes. The circular chloroplast genome map was drawn by the program Chloroplot (Zheng et al., 2020). The assembly and annotation of the chloroplast genome were submitted to NCBI (Accession No: MZ557416).
Repeat elements in chloroplast genomes of I. pes-caprae were investigated using two different programs. The program MicroSAtellite identification tool (Katoh et al., 2002), (Beier et al., 2017) was used to identify the SSR repeat, setting the parameters with thresholds of 10, 5, 4, 3, 3, and 3 repeat units for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. The program REPuter was used to detect the repeat sequences within the chloroplast genome (Kurtz et al., 2001). Four types of repeats, including forward repeats, reverse repeats, complement repeats, and palindromic repeats, were investigated in this analysis.
The chloroplast genome sequences of 26 Convolvulaccae species, including 14 Cuscuta species and 12 Ipomoea species, were used for phylogenetic analysis using three model species as outgroups (Arabidopsis thaliana, Amborella trichopoda, and Oryza sativa). The taxonomical details of the investigated species and the accession numbers of their chloroplast genome are listed in Supplementary Table 1. To accurately identify the phylogenetic position of I. pes-caprae, two phylogenetic trees were constructed based on the complete chloroplast genome sequences and the protein sequences, respectively. The alignment was conducted by MAFFT (Katoh et al., 2002), and the phylogenetic trees were constructed by MEGAX using Maximum Likelihood methods (Kumar et al., 2018).
To investigate the sequence divergence among Ipomoea species chloroplast genomes, The chloroplast genome of I. pre-caprae generated in this study, together with 11 released Ipomoea chloroplast genomes retrieved from NCBI, were used for comparative analysis. The sequences were aligned using the mVISTA program with Shuffle-LAGAN mode (https://pgrc.ipk-gatersleben.de/misa/) (Frazer et al., 2004).
To identify the SNPs and hypervariable regions within the chloroplast genome of I. pes-caprae in comparison with other Ipomoea species, the chloroplast genomes of other Ipomoea were aligned to the chloroplast genome of I. pes-caprae using MAFFT (Katoh and Standley, 2013). The nucleotide diversity (Pi) along the chloroplast genome was calculated using DnaSP version 5 software (Librado, 2009) with sliding window analysis. The window length was set to 800 base pairs, and the step size was set to 50 base pairs.
Using the PacBio Sequel II sequencing platform, 23252902 whole-genome long reads of I. pes-caprae were produced for genome assembly (unpublished project), and the mean read length is around 14 kb. Converted raw reads to CCS reads of the whole genome, the reads number from 23252902 to 1467275 (Supplementary Table 2). The chloroplast genome size of 161667 bp of I. pes-caprae was derived from the Canu program assembly. The I. pes-caprae complete chloroplast genome had a typical circular structure and with typical quadripartite organization consisting of the four conserved constitute regions, a pair of 30670 bp inverted repeats (IRs 30670 bp), an 88210 bp long single-copy regions (LSC), and a 12117 bp short single copy region (SSC) (Figure 1; Supplementary Table 3).
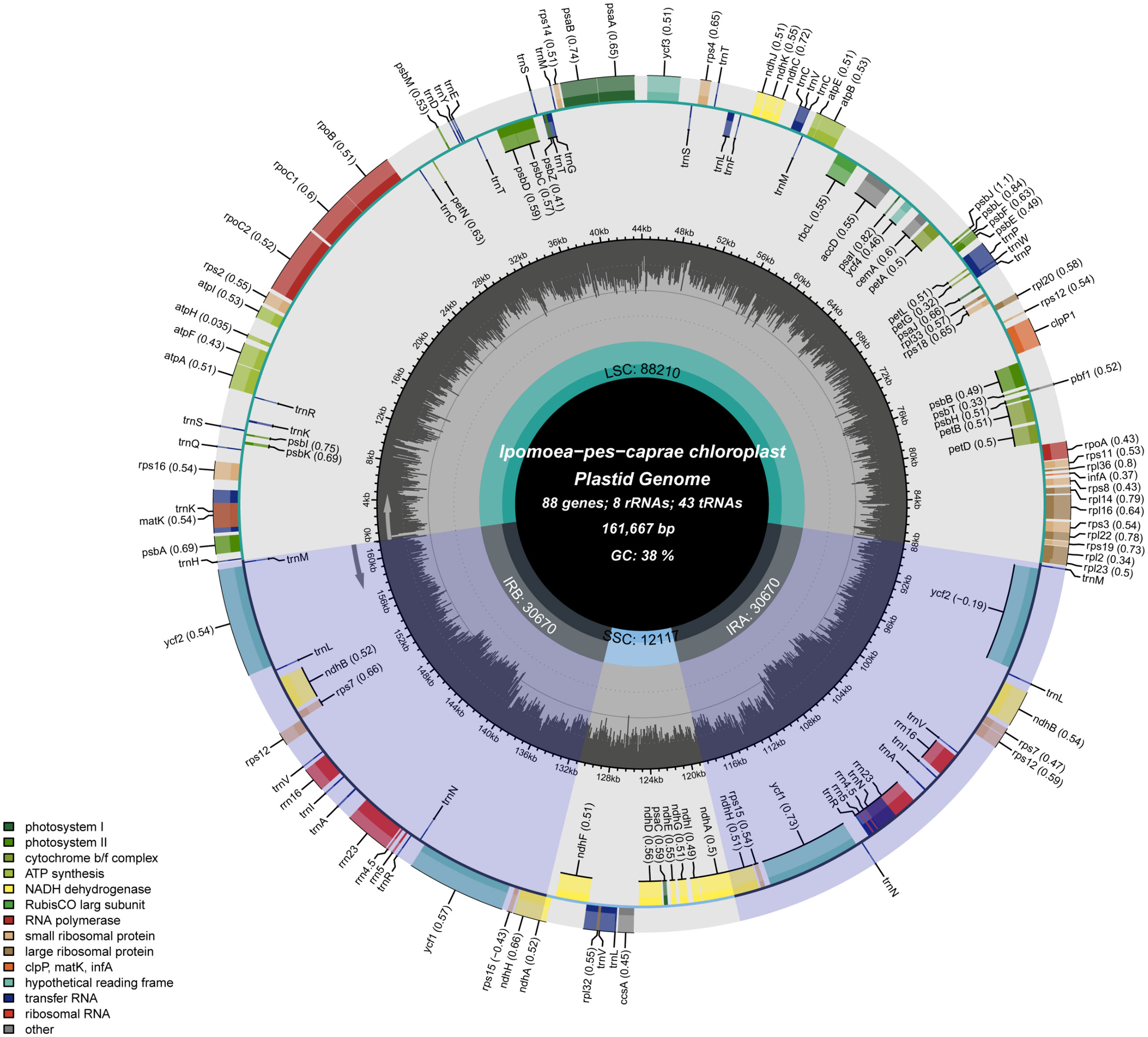
Figure 1 Circular gene map of I. pes-caprae chloroplast genome. Genes shown inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise. Genes belonging to different functional groups are color-coded. The darker gray color in the inner circle corresponds to the GC content, and the lighter gray color corresponds to the AT content. LSC, SSC, and IR are large single-copy regions, small single-copy regions, and inverted repeat regions, respectively.
A number of 136 functional genes were identified in the chloroplast genome of I. pes-caprae, including 86 protein-coding genes, 42 tRNA genes, and 8 rRNA genes (16S, 23S, 5S, and 4.5S) (Figure 1; Table 1). According to their functions, the 136 I. pes-caprae chloroplast genes were assigned into three categories: Most protein-coding genes are related to photosynthesis. There are 45 genes in this category, including genes encoding subunits of ATP synthase, subunits of photosystem II, subunits of cytochrome b/f complex, subunits of NADH-dehydrogenase, assembly/stability of photosystem I, subunits of photosystem I, subunits of photosystem II, cytochrome c synthesis (ccsA), photosystem biogenesis factor 1 (pbf1), and subunit of rubisco (rbcL). The second category consists of 86 genes associated with chloroplast transcription and translation, including protein-coding genes encoding the large subunits of the ribosome, the small subunits of ribosomal proteins, DNA-dependent RNA polymerase, and translational initiation factor, and two types of non-coding genes, i.e., rRNA genes, tRNA genes. And the rest of the four genes were classified as other genes because of their unique or unknown function, including matK with function in RNA processing and 3 conserved open reading frames (ycf1 and ycf2×2) encoding proteins of unknown function (Table 1; Supplementary File 3).
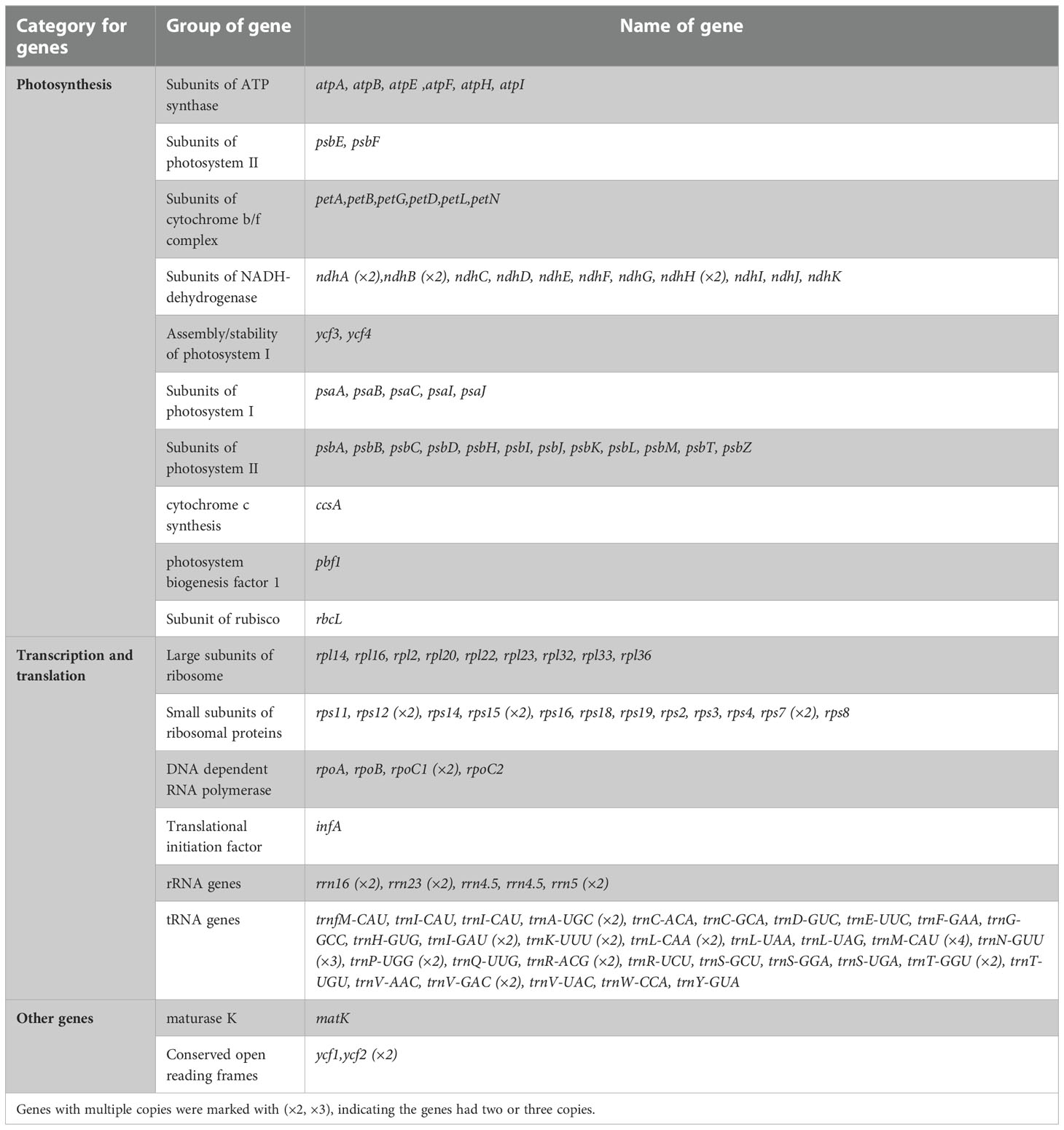
Table 1 The annotated genes in the chloroplast genome of I. pes-caprae.
The sequence repeats widely exist in eukaryotic genomes, and the simple sequence repeat (SSR) is the most abundant and typical repeat type. SSRs in chloroplast genomes exhibit high copy numbers, which play an essential role in genome rearrangement and recombination and are important molecular markers in plant phylogenetic and evolutionary studies (Kuang et al., 2011; Yang et al., 2011; Jiao et al., 2012; Xue et al., 2012). From the perspective of evolution, the differences in repetitive sequences among species resulted from natural selection (Huang et al., 2021). 35 SSRs were identified in the chloroplast genome of I. pes-caprae (Supplementary Table 4): 32 are A/T single nucleotide repeats, and one is the TA dinucleotide repeat. Notably, there was no di, tri-, tetra-, penta-, or hexanucleotide repeat detected in the chloroplast genome of I. pes-caprae. The longer sequence repeats within the chloroplast genome were identified using Reputer 2.0 software. As a result, 50 repeats consisting of 26 forward and 24 palindromic repeats were obtained, while no complement or reverse repeats were detected (Supplementary Table 5). The largest repeat unit with a size of 30670 bp is the inverted repeats of chloroplast, which is essential for the chloroplast structure organization. And the lengths of the rest of the repeat units ranged from 119 and 242 bp. The repeat information of the chloroplast genome of I. pes-caprae is valuable for developing genetic markers for phylogenetic and population studies (Nie et al., 2012).
In evolution, species are affected by natural selection pressure and genetic drift, resulting in differences in the use frequency of most genetic codons. Thus, the different genomes might have specific codon preferences (Hershberg and Petrov, 2008). In the complete chloroplast genome of I. pes-caprae, There are 53889 codons within the protein-coding genes. The I. pes-caprae chloroplast genome encoded all 20 amino acids, and 61 types of amino-acid codons were observed (Supplementary Table 6). The UU-started codons are found to be more frequent than the others (Supplementary Table 7). Among the 20 amino acids, Leucine was the most abundant (number of codons encoding Leucine = 5708, 10.59%), Serine was the second abundant (number of codons encoding Serine = 4961, 9.21%), while the rarest one is Tryptophan (713 codons, approximately 2.12%). Thirty-three codons were observed to be used more frequently than the expected usage at equilibrium (RSCU (Relative Synonymous Codon Usage) > 1), and 27 codons showed the codon usage bias (RSCU < 1). However, the frequency of use for the AUG (Methionine, start codon), UGG (Tryptophan), ACC (Threonine), and GGU (Glycine) showed no bias (RSCU = 1). In aspects of amino acids, most of them have codon preferences, except for Methionine and Tryptophan (Figure 2). And most of the 20 amino acids with at least two codons, and the acids Arginine, Leucine, and Serine have six codons. To investigate whether the codon usage preferences are conserved among the species in the Ipomoea genus, the protein-coding genes of the other 11 Ipomoea species, including I. batatas (53822 amino acids), I. hederifolia (53791), I. involucrata (53506), I. minutiflora (53729), I. murucoides (53327), I. nil (53965), I. obscura (53749), I. purpurea (54015), I. tricolor (53589), I. trifida (53711), and I. triloba (53916) were investigated. As shown in Supplementary File 1, the codon usage preferences of all the 11 investigated species showed a similar tendency as that of I. pes-caprae (Supplementary File 1), indicating the conservation of codon bias in the genus of Ipomoea, which might be because that the ancestors of those species underwent the shared evolutionary history.
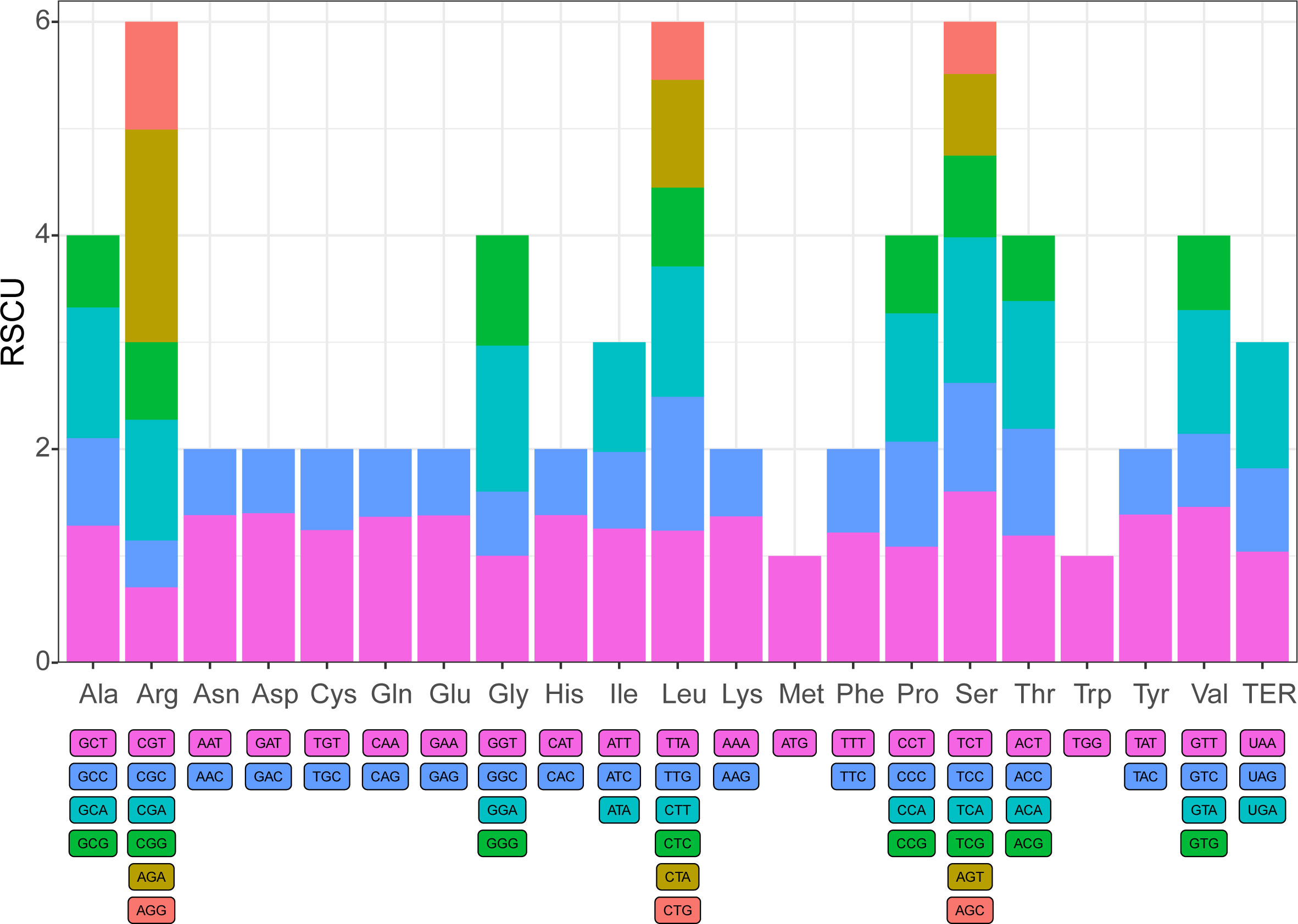
Figure 2 The Condon preference of the chloroplast genome of I. pes-caprae. Codon content of 20 amine acid and stop codons in all protein-coding genes of the chloroplast genome of I. pes-caprae.
The ENc plots are usually used to indicate the factors that affect the codon bias. To understand the relative importance of natural mutation and evolutionary selection in producing codon usage patterns, ENc (Effective Number of Codons) values of four Ipomoea species (I. pes-caprae, I. involucrata, I. murucoide, and I. tricolor) were estimated and plotted against the GC3s values (Figure 3). From the ENc plots, it is clear that the protein-coding genes of the four species showed similar codon bias patterns. Most of the genes were distributed on both sides of the standard curve, and more than half of the genes were below the curve, suggesting that the selection pressures predominantly influence codon bias in the chloroplast genome of the Ipomoea species. Photosynthesis-related genes are distributed most discretely, suggesting that some other factors might influence the codon bias, or maybe these genes are more conserved than self-replication and other group genes.
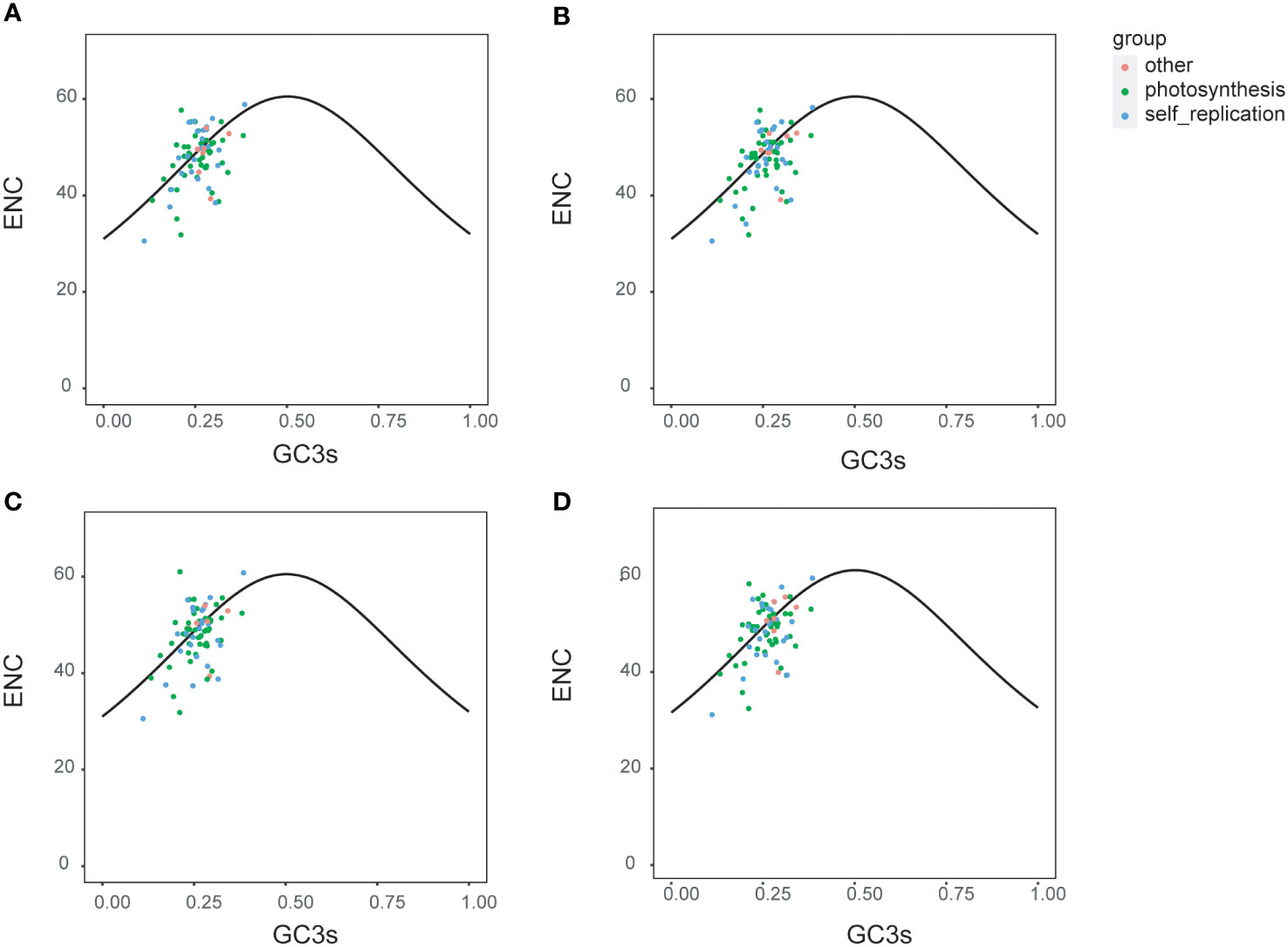
Figure 3 Enc plotted against GC3s of three Ipomoea species. The solid lines represented the expected curve of positions of genes when the codon usage was only determined by the GC3s composition. Enc and GC3s plots for four Ipomoea species, including I pes-caprae (A), I involucrate (B), I murucoides (C), and I tricolor (D).
Expansion and contraction of IR regions are common events that frequently happen in the evolutionary history of land plants (Raubeson et al., 2007; Kode et al., 2010; Yao, 2015). LSC/IR and IR/SSC junctions are sometimes regarded as an index of chloroplast genome evolution. The map illustrating the LSC/IRb (JLB), IRb/SSC (JSB), SSC/IRa (JSA), and IRa/LSC (JLA) boundaries/junctions of the 12 Ipomoea chloroplast genomes was constructed according to heir chloroplast genome sequences and annotation information (Figure 4). It is shown that the chloroplast genome organizations are highly conserved across the 12 Ipomoea species with only minor variations. The chloroplast genomes of 12 species have similar sizes ranging from 160072 bp (I. murucoides) to 161897 bp (I. nil). However, the sizes of LSC, SSC, and IR are varied significantly. For example, The lengths of IRs ranged from 8265 bp (I. tricolor) to 31061 bp (I. triloba), and the size of IR of I. pes-caprae was 30670 bp. The LSC/IR and SSC/IR boundaries in all 12 Ipomoea species were distributed with different genes, and only a few chloroplast genomes with consensus genes. The RS lines (the boundary lines between IRb/IRb and SSC) were mainly located between ycf1 and trnN.

Figure 4 Comparison of the borders of the IR, SSC, and LSC regions among 12 chloroplast genomes of Ipomoea species. JLB, JSB, JSA, and JLA represent the junctions of LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC, respectively.
On the contrary, the RL lines (the boundary lines between IRa/IRb and LSC) were more variable than RS lines. Compared to species of other genera, the LSC regions of I. involucrata, I. murucoides, I. obscura, and I. tricolor have the expansion of ycf2 to the IR LSC region, decreasing IR length. On the other hand, the SSC Regions of I. pes-caprae, I. nil, I. purpurea, and I. triloba were concentrated compared to those of other species, with the loss of ycf1 in the SSC regions. As previous studies suggested, the expansion and contraction detected in the IR regions might be a primary mechanism in the length variation of three regions (LSC, SSC, and IR) of the chloroplast genomes in Ipomoea species (Sun et al., 2019).
In order to understand the evolutionary relationships of I. pes-caprae with the Ipomoea genus, the chloroplast genomes of 28 Convolvulaccae species and three outgroup species (A. thaliana, A. trichopoda, and O. sativa) were used for phylogenetic analysis. The complete chloroplast genomes were downloaded from NCBI (National Center for Biotechnology Information database) and used for constructing the phylogenetic trees. We first constructed the phylogenetic trees based on the complete chloroplast genome sequences (Figure S1). The Convolvulaccae species were classified into the Ipomoea clade and the Cuscuta clade. The phylogenetic tree generated based on completed chloroplast sequences showed consistent relationships with taxonomical classification, suggesting confidence in the phylogenetic analysis based on the chloroplast genome. However, one of the outgroups species, A. trichopoda, the base angiosperm, was not in the basal position in the phylogenetic tree. One reason might be because of the sequence variation of the non-coding sequences. To this end, the sequences of orthologous single-copy genes were used to construct a maximum likelihood tree. As shown in Figure 5, the Basal Angiosperms, Monnocotes, and Diocts outgroups species showed a clear taxonomical relationship, and the 12 ipomoea species and 14 Cuscuta species were clustered into two clades. Our results showed that the chloroplast genomes could be used for constructing the phylogenetic tree reflecting the evolutionary relationships of land plants, and the orthologous single-copy genes sequence-based phylogenetic relationships based would be much more confident than the complete chloroplast genome sequences-based phylogenetic relationships.
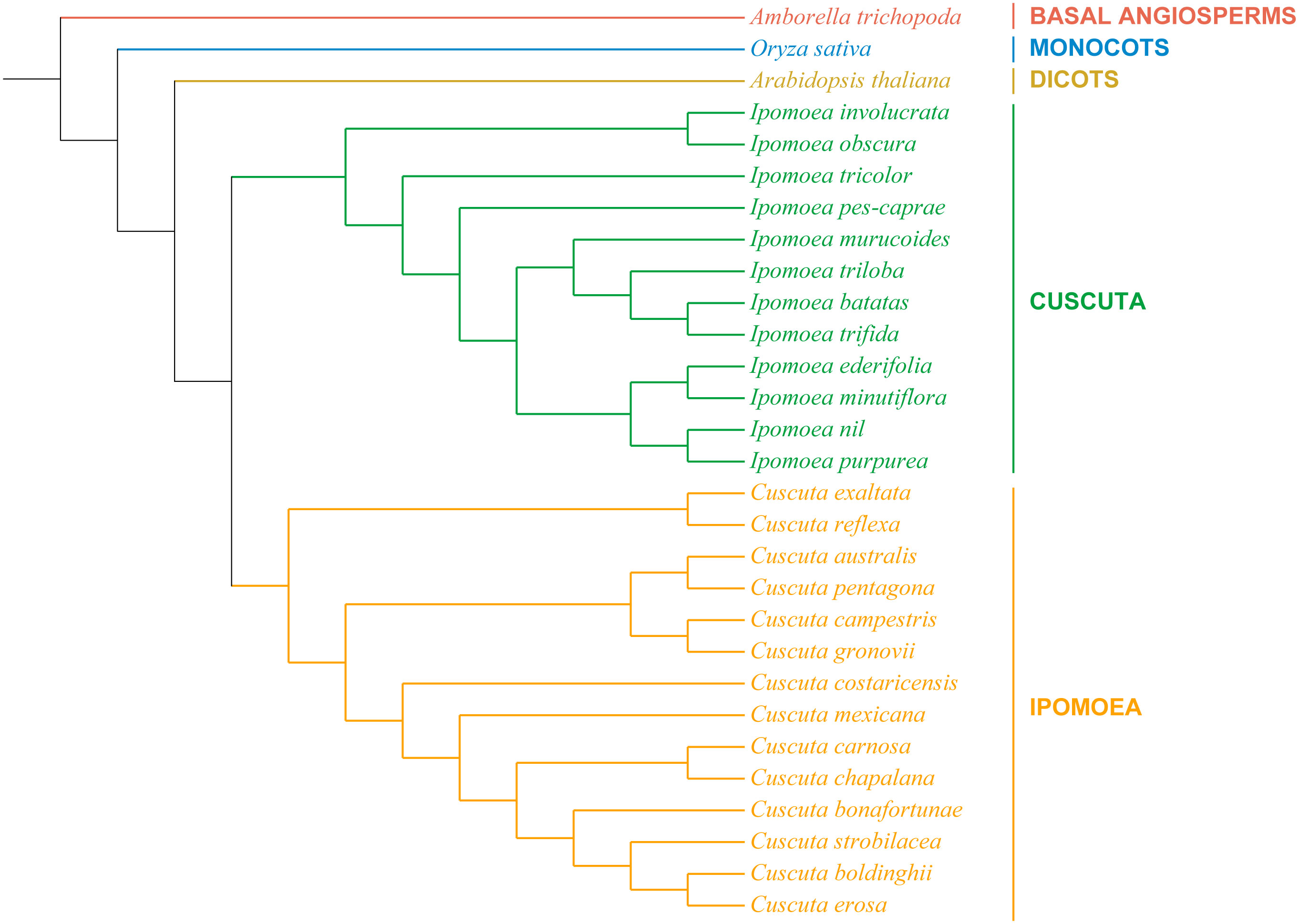
Figure 5 The phylogenetic relationships of the 26 Convolvulaceae species. Orthologous Single-copy genes phylogenetic tree of 26 Convolvulaceae family species and three outgroups constructed using maximum likelihood (ML) algorithm.
Cuscuta spp plants are annual parasitic herbs, and most of them are leafless, lack chlorophyll, and have a wide range of hosts (Albert et al., 2008). By comparing the protein-coding genes between Cuscuta and Ipomoea species, it’s evident that many genes were lost in Cuscuta species (Supplementary Table 8). In Cuscuta species, only C. exaltata maintains a few ndh (Subunits of NADH-dehydrogenase) genes. The other 13 Cuscuta species missed all of the ndh genes related to the photosynthesis pathway (Figure 6; Supplementary File 2). The result of the comparative analysis showed that C. boldinghii, C. erosa, and C. strobilacea have the least number of genes, which are 31, 33, and 33, respectively, and C. exaltata has the most number of genes, which is 67. Except for C. exaltata, other Cuscuta species don’t have complete photosynthesis-related genes, such as the genes coding subunits of cytochrome b/f complex, subunits of photosystem I, and subunits of photosystem II. Therefore, it is concluded that the loss of genes involved in photosynthesis in Cuscuta chloroplast genomes happened gradually, and Cuscuta species lost the photosynthesis ability to various extents, which is consistent with their nutritional performance.
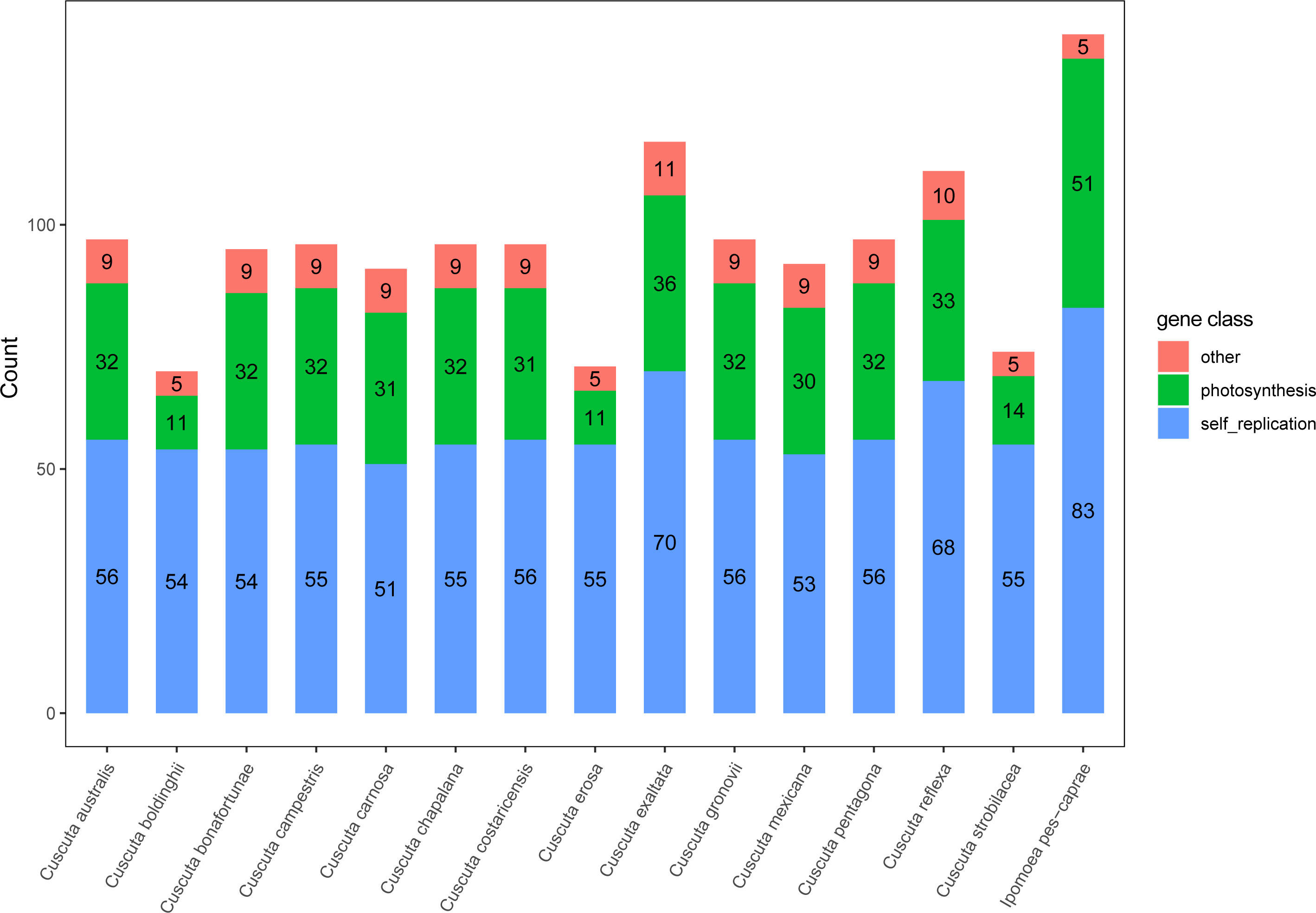
Figure 6 The number of three types of genes in the chloroplast genome of Cuscuta species. Red represents genes with other functions, green represents photosynthesis-related genes, and blue indicates genes involved in the self-replication of the chloroplast genome.
We also analyzed their chloroplast genomes differences to explore further the genetic relationship among the I. involucrata, I. murucoides, and I. tricolor. The mVISTA program was used for global alignment to exhibit the variation of the chloroplast genomes in different regions using the genome sequence and annotation of I. pes-caprae chloroplast as the reference (Figure 7). The gene organization was highly conserved across the four chloroplast genomes, with few variation regions, consistent with previous studies (Nguyen et al., 2021). The results also exhibited that the divergences in LSC and SSC regions were higher than in IR regions. Besides, the sequences in the coding regions tended to be more conserved, whereas most of the variations detected were found in conserved non-coding sequences (NCS). The sequences of exons had almost 100% similarity throughout the four taxa. Among the coding genes, the highly disparate sequences are the regions harbouring rpl2, ycf2, ndhK, ndhD, and ycf1 genes.
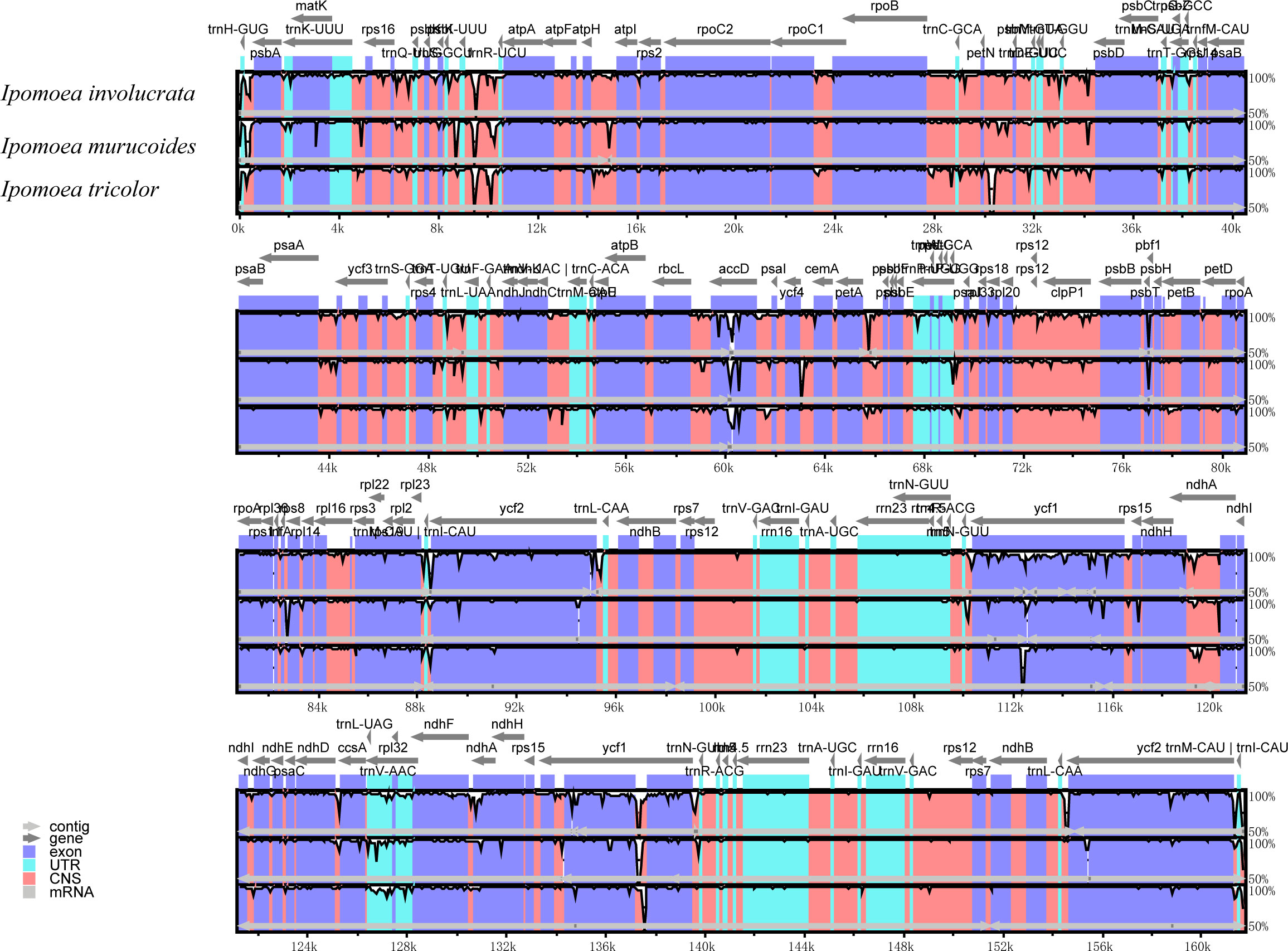
Figure 7 The sequence diversity of the chloroplast genomes of three Ipomoea species. The sequence identity plot shows the diversity along the chloroplast genome of I. involucrate, I. murucoides, and I. tricolor with I. pes-caprae as a reference using mVISTA. Gray arrows and thick black lines above the alignment indicate genes with their orientation and the position of the IRs, respectively. The colors indicate the exon, UTR, and conserved non-coding sequences (CNS) and mRNA regions. A cutoff of 70% identity was used for the plots, and the Y-scale represents the identity percentage ranging from 50 to 100%.
To visualize the overall sequence divergence of the 12 Ipomoea species, the pi values of the chloroplast genome sequence were calculated with a slide window length of 600 bp and a step size of 100 bp. The sliding window pi plots showed that the average pi value of the LSC (Pi = 0.008101) and SSC (Pi = 0.054394) regions was much higher than that in the IR (Pi = 0.001942) regions, which showed that LSC and SSC regions contained the most of the variation (Figure 8). The plots also showed that the SSC region is hypervariable. Further investigation revealed that most of the SSC region of I. pes-caprae was much shorter than other Ipomoea species, indicating an invasion of IRs to this region. These regions with higher pi values are more variables that might experience rapid nucleotide substitution and could be used to develop molecular markers for identification and phylogenetic analysis (Lyu et al., 2020).
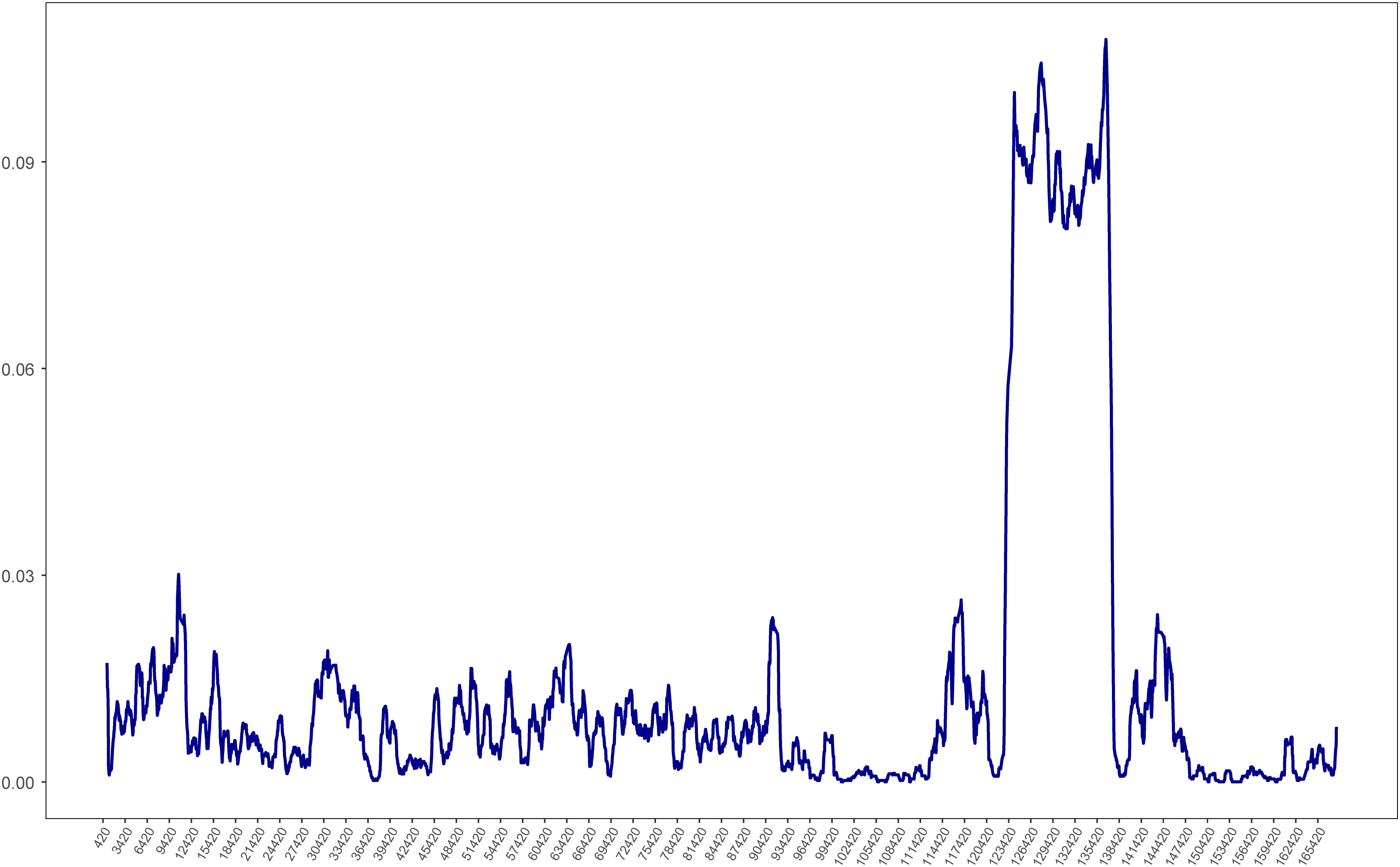
Figure 8 Chloroplast genome comparative analysis between I. pes-caprae and other 11 Ipomoea species. Sliding window plots of nucleotide diversity (π) across the complete cp genomes of I pes-caprae and other 11 Ipomoea species (window length: 600 bp, step size: 100 bp). Y-axes: nucleotide diversity (π) of each window; X-axes: the position of the midpoint of a window.
The chloroplasts are the semi-autonomous organelles in green plants, algae, and cyanobacteria. The main function of chloroplast is to carry out photosynthesis converting the light energy to chemical energy, which is critical for the autotrophic characteristics of those species (Howe et al., 2003; Daniell et al., 2021). Convolvulaceae is a family of about 60 genera and more than 1,650 species of mostly herbaceous vines, trees, shrubs, herbs, and the sweet potato and a few other food tubers (van Ooststroom and Hoogland, 1953; Stefanović et al., 2003). In this family, Ipomoea is the largest genus, with over 600 large diversity species with common names such as morning glory, water convolvulus or kangkung, sweet potato, bindweed, and moonflower (Gunn, 1972). The Ipomoea species includes food species. For example, I. batatas and I. aquatica are important food sources for humans and animals (Meira et al., 2012; Mohanraj and Sivasankar, 2014). Some other Ipomoea species, for example, I. Carnea, I. quamoclit, I. jalapa, and I. simulans, are renowned for their properties in folk medicine and herbalism (Sharma and Bachheti, 2013; Paul and Sinha, 2016); Cuscuta is another typical genus in the Convolvulaceae family, well-known for their parasitism characteristics, It is composed of over 201 species of yellow, orange, or red (rarely green) parasitic plants, comely found throughout the temperate and tropical regions of the world, with the greatest species diversity in subtropical and tropical regions (Machado and Zetsche, 1990). Since the Cuscuta plants could not conduct photosynthesis and had to uptake nutrition from the host plants, they became an ideal model system for studying the communication between plants recently (Hettenhausen et al., 2017; Shahid et al., 2018; Vogel et al., 2018; Sun et al., 2018; Zhuang et al., 2018; Li et al., 2020). In this study, the chloroplast genome of I. pes-caprae was assembled and annotated, and comparative analyses of the chloroplast genome of Convolvulaceae were conducted.
The chloroplast genome phylogenetic trees were usually used to describe the taxonomical and evolutionary relationships among the plant species. The chloroplast genome phylogeny revealed that 2 Cuscuta species (C. exaltata and C. reflexa) were closely related to the Ipomoea species (Figure S1), which conflicted with their taxonomic positions. While constructing phylogenetic analysis, we found that the length of their chloroplast genome severely influenced the positions of specific species on the phylogenetic tree. The chloroplasts with a similar genome size tend to have a closer phylogenetic relationship. Since the chloroplast genomes of land plants are conserved in gene order and organization, while the order of four regions might be different for using different assembly and annotation strategies, we reconstructed a phylogenetic tree with a specific order (LSC-IRb-SSC-IRa). The new phylogenetic tree was slightly different from the previous one. However, two Cuscuta species, C. exaltata, and C. reflexa, were still clustered close to Ipomoea species. The coding sequences are much more conserved than the non-coding regions during evolution. Therefore, the conservative single-copy genes of the chloroplast genome were extracted and used to construct the phylogenetic tree. As shown in Figure 5, C. exaltata and C. reflexa were clustered together with other Cuscuta species, and the phylogenetic relationships of the investigated species highly correspond to their taxonomic relationships. These analyses show that phylogenetic trees constructed based on the conservative single-copy gene sequences are more credible than the complete genome.
The diversity along the chloroplast genome was investigated through pi plotting. The results showed that SSC regions of the Ipomoea chloroplast were significantly diverse compared to the other areas. Since there are two orientations of the SSC in plant chloroplast genomes (Cheng et al., 2020), which will interfere with calculating the pi value, we extensively checked the SSC orientation of the 12 Ipomoea chloroplast genomes. The alignments map showed that the SSC orientation of I. pes-caprae chloroplast was opposite from these of other Ipomoea species (Supplementary Figure S2). Therefore, we reversed the SSC region of I. pes-caprae and recalculated the pi value of the 12 Ipomoea chloroplast genomes. The pi plots still showed a significant peak in the SSC regions. This analysis indicates that high diversity in the SSC region is indeed excited in the Ipomoea species. The mVISTA analysis showed the sequence variation among three Ipomoea species, I. involucrata, I. murucoides, and I. tricolor (Figure 7). There were many small blocks with low identity in the SSC regions, especially in genes ycf1 and rpl32. We can conclude that the SSC regions are highly diverse, attributed to the diversity of the genes in these regions, such as ycf1 and rpl32.
The comparative analysis of the chloroplast genomes of Ipomoea and Cuscuta species showed that Cuscuta species belong to parasitic plants, and their chloroplast genomes were shorter than their close relative Ipomoea species. Therefore the missing genes of Cuscuta species could provide clues to deciphering the evolutionary history of the parasite plants. In the Cuscuta species, the CDS number is ranged from 31 to 67, with an average of 54.85. However, the CDS number of Ipomoea species is varied from 85 to 87, with an average of 85.4. There were 16 CDSs (pbf1, ndhB, ndhH, ndhE, infA, ndhJ, ndhG, ndhC, ndhA, ndhF, ycf15, rpl23, rps16, ndhK, ndhI, and ndhD) existing in Ipomoea species but not existing in Cuscuta species. These CDSs coding genes are mainly involved in photosynthesis. That might explain the loss of the photosynthesis function of Cuscuta species during evolution (Funk et al., 2007).
The data presented in the study are deposited in the NCBI repository, accession number MZ557416.
YC, YQ, and WL conceived and designed the research. YW, JX, BH, CD, JS, ZL, KY, and FD performed the experiments. LW and MA helped with a critical discussion on the work. YC and YW wrote the paper. MA, WL, and YQ revised the paper All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (32170380 to YC, and 31970333 to YQ) and the Science and Technology Innovation Project of Pingtan Institute of Science and Technology (PT2021001 to YC, and PT2021007 to YQ).
We thank the editors and revivers for revising this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YH declared a shared affiliation with the authors YW, BH, CD, JS, ZL, YK, FD, LW, MA, WL, YQ, YC to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1074697/full#supplementary-material
Albert, M., Belastegui-Macadam, X. M., Bleischwitz, M., Kaldenhoff, R. (2008). Cuscuta spp: “Parasitic Plants in the Spotlight of Plant Physiology, Economy and Ecology.” In Lüttge, U., Beyschlag, W., Murata, J. (Eds.), Progress in Botany (pp. 267–277). Springer. doi: 10.1007/978-3-540-72954-9_11
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33 (16), 2583–2585. doi: 10.1093/bioinformatics/btx198
Bragadeeswaran, S., Prabhu, K., Rani, S. S., Priyadharsini, S., Vembu, N. (2010). Biomedical application of beach morning glory ipomoea pes-caprae. Int. J. Trop. Med. 5 (4), 81–85. doi: 10.3923/ijtmed.2010.81.85
Bulychev, A. A., Komarova, A. V. (2015). Photoinduction of cyclosis-mediated interactions between distant chloroplasts. Biochim. Biophys. Acta (BBA)-Bioenerg. 1847 (4-5), 379–389. doi: 10.1016/j.bbabio.2015.01.004
Cheng, Y., Zhang, L., Qi, J., Zhang, L. (2020). Complete chloroplast genome sequence of hibiscus cannabinus and comparative analysis of the malvaceae family. Front. Genet. 11, 227. doi: 10.3389/fgene.2020.00227
Curci, P. L., Domenico, D. P., Donatella, D., Vendramin, G. G., Gabriella, S. (2015). Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other asteraceae. PloS One 10 (3), e0120589-. doi: 10.1371/journal.pone.0120589
Daniell, H., Jin, S., Zhu, X. G., Gitzendanner, M. A., Soltis, D. E., Soltis, P. S. (2021). Green giant–a tiny chloroplast genome with mighty power to produce high-value proteins: history and phylogeny. Plant biotechnology journal 19, 3, 430–447. doi: 10.1111/pbi.13556
Devall, M. S. (1992). The biological flora of coastal dunes and wetlands. 2. ipomoea pes-caprae (L.) Roth. J. Coast. Res. 8 (2), 442–456. https://www.jstor.org/stable/4297988
Devall, M. S., Thien, L. B. (1989). Factors influencing the reproductive success of ipomoea pes-caprae (convolvulaceae) around the gulf of mexico. Am. J. Bot. 76 (12), 1821–1831. doi: 10.1002/j.1537-2197.1989.tb15171.x
Dong, W., Liu, H., Xu, C., Zuo, Y., Chen, Z., Zhou, S. (2014). A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 15(1), 1–8. doi: 10.1186/s12863-014-0138-z
Nadachowska-Brzyska, K., Li, C., Smeds, L., Zhang, G., Ellegren, H. (2015). Temporal Dynamics of Avian Populations during Pleistocene Revealed by Whole-Genome Sequences. Current Biology. 25(10), 1375–1380. doi: 10.1016/j.cub.2015.03.047
Frazer, K. A., Lior, P., Alexander, P., Rubin, E. M., Inna, D. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32 (Web Server issue), W273. doi: 10.1093/nar/gkh458
Funk, H. T., Berg, S., Krupinska, K., Maier, U. G., Krause, K. (2007). Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, cuscuta reflexa and cuscuta gronovii. BMC Plant Biol. 7 (1), 1–12. doi: 10.1186/1471-2229-7-45
Gunn, C. R. J. B. (1972). Moonflowers, ipomoea section calonyction, in temperate north America. Brittonia 24 (2), 150–168. doi: 10.2307/2805866
Hershberg, R., Petrov, D. A. (2008). Selection on codon bias. Annu. Rev. Genet. 42, 287–299. doi: 10.1146/annurev.genet.42.110807.091442
Hettenhausen, C., Li, J., Zhuang, H., Sun, H., Xu, Y., Qi, J., et al. (2017). Stem parasitic plant cuscuta australis (dodder) transfers herbivory-induced signals among plants. Proceedings of the National Academy of Sciences 114 (32), E6703–E6709. doi: 10.1073/pnas.1704536114
Howe, C. J., Barbrook, A. C., Koumandou, V. L., Nisbet, R. E. R., Symington, H. A., Wightman, T. F. (2003). Evolution of the chloroplast genome. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 358 (1429), 99–107. doi: 10.1098/rstb.2002.1176
Huang, X., Tan, W., Li, F., Liao, R., Guo, Z., Shi, T., et al. (2021). The chloroplast genome of prunus zhengheensis: Genome comparative and phylogenetic relationships analysis. Gene 793, 145751. doi: 10.1016/j.gene.2021.145751
Jagendorf, A. T., Uribe, E. (1966). ATP formation caused by acid-base transition of spinach chloroplasts. Proc. Natl. Acad. Sci. U.S.A. 55 (1), 170–177. doi: 10.1073/pnas.55.1.170
Jansen, R. K., Raubeson, L. A., Boore, J. L., Depamphilis, C. W., Chumley, T. W., Haberle, R. C., et al. (2005). Methods for Obtaining and Analyzing Whole Chloroplast Genome Sequences. In Methods in Enzymology (Vol. 395, pp. 348–384). Academic Press. doi: 10.1016/S0076-6879(05)95020-9
Jansen, R. K., Ruhlman, T. A. (2012). Plastid genomes of seed plants. Genomics Chloroplasts Mitochondria. (pp. 103–126). Springer, Dordrecht. doi: 10.1007/978-94-007-2920-9_5
Jiao, Y., Jia, H-m., Li, X-w., Chai, M-l., Jia, H-j., Chen, Z., et al. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genomics 13 (1), 201. doi: 10.1186/1471-2164-13-201
Gao, J., Gao, L. (2016). The complete chloroplast genome sequence of the phyllostachys sulphurea (Poaceae: Bambusoideae). Mitochondrial DNA Part A 27 (2), 983–985. doi: 10.3109/19401736.2014.926516
Katoh, K., Misawa, K., Ki, K., Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30 (14), 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kode, V., Mudd, E. A., Iamtham, S., Day, A. (2010). The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44 (2), 237–244. doi: 10.1111/j.1365-313X.2005.02533.x
Kuang, D. Y., Wu, H., Wang, Y. L., Gao, L. M., Lu, L. (2011). Complete chloroplast genome sequence of magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54 (8), 663–673. doi: 10.1139/g11-026
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29 (22), 4633–4642. doi: 10.1093/nar/29.22.4633
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Librado, R. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinf. (Oxford England) 25 (11), 1451–1452. doi: 10.1093/bioinformatics/btp187
Li, J., Wang, S., Yu, J., Wang, L., Zhou, S. (2013). A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 48 (1), 72. doi: 10.3724/SP.J.1259.2013.00072
Li, S., Zhang, J., Liu, H., Liu, N., Shen, G., Zhuang, H., et al. (2020). Dodder-transmitted mobile signals prime host plants for enhanced salt tolerance. Journal of Experimental Botany 71 (3), 1171–1184. doi: 10.1093/jxb/erz481
Lyu, Y., Dong, X., Sun, H., Huang, L. (2020). Characterization of the complete chloroplast genome sequence of koelreuteria bipinnata. Mitochondrial DNA Part B 5 (3), 3324–3325. doi: 10.1080/23802359.2020.1806129
Machado, M. A., Zetsche, K. (1990). A structural, functional and molecular analysis of plastids of the holoparasites Cuscuta reflexa and Cuscuta europaea. Planta 181 (1), 91–96. doi: 10.1007/BF00202329
Manigauha, A., Kharya, M. D., Ganesh, N. (2015). In vivo antitumor potential of ipomoea pes-caprae on melanoma cancer. Pharmacogn. Mag. 11 (42), 426–433. doi: 10.4103/0973-1296.153099
Meira, M., Silva, E. P., da, David, J. M., David, J. P. (2012). Review of the genus Ipomoea: Traditional uses, chemistry and biological activities. Revista Brasileira de Farmacognosia 22, 682–713. doi: 10.1590/S0102-695X2012005000025
Mira, P., Hyun, P., Hyoungseok, L., Byeong-Ha, L., Jungeun, L. (2018). The complete plastome sequence of an Antarctic bryophyte sanionia uncinata (Hedw.) loeske. Int. J. Mol. Sci. 19 (3), 709. doi: 10.3390/ijms19030709
Mohanraj, R., Sivasankar, S. (2014). Sweet potato (Ipomoea batatas [L.] lam)-a valuable medicinal food: A review. Journal of Medicinal Food 17(7), 733–741. doi: 10.1089/jmf.2013.2818
Neuhaus, H., Emes, M. J. NONPHOTOSYNTHETIC METABOLISM IN PLASTIDS - annual review of plant physiology and plant molecular biology. Starch Fatty Acids Nitrogen Metabolite Transp. Gene Expression 51 (1), 111. doi: 10.1146/annurev.arplant.51.1.111
Nguyen, H. Q., Nguyen, T. N. L., Doan, T. N., Nguyen, T. T. N., Phạm, M. H., Le, T. L., et al. (2021). Complete chloroplast genome of novel adrinandra megaphylla hu species: molecular structure, comparative and phylogenetic analysis. Sci. Rep. 11 (1), 11731. doi: 10.1038/s41598-021-91071-z
Nie, X., Lv, S., Zhang, Y., Du, X., Wang, L., Biradar, S. S., et al. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PloS One 7 (5), e36869. doi: 10.1371/journal.pone.0036869
Nurk, S., Walenz, B. P., Rhie, A., Vollger, M. R., Logsdon, G. A., Grothe, R., et al. (2020). HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome research 30(9), 1291–1305. doi: 10.1101/2020.03.14.992248
Ozeki, H., Umesono, K., Inokuchi, H., Kohchi, T., Ohyama, K. (1989). The chloroplast genome of plants: a unique origin. Genome. 31(1), 169–174. doi: 10.1139/g89-029
Paul, D., Sinha, S. N. (2016). An update on biological activities of medicinal plant Ipomoea quamoclit L. Trop Plant Res. 3 (1), 186–190. https://www.tropicalplantresearch.com/archives/2016/vol3issue1/25.pdf
Petit, R., Duminil, J., Fineschi, S., Hampe, A., Vendramin, G. G. (2005). Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mo-lecular Ecology 14, 689–701. doi: 10.1111/j.1365-294X.2004.02410.x
Price, D. C., Chan, C. X., Yoon, H. S., Yang, E. C., Qiu, H., Weber, A. P., et al. (2012). Cyanophora paradoxa genome elucidates origin of photosynthesis in algae and plants. Science 335(6070), 843–847. doi: 10.1126/science.1213561
Qu, X.-J., Moore, M., Li, D.-Z., Yi, T. (2019). PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 15, 1–12. doi: 10.1186/s13007-019-0435-7
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms nuphar advena and ranunculus macranthus. BMC Genomics 8 (1), 174. doi: 10.1186/1471-2164-8-174
Shahid, S., Kim, G., Johnson, N. R., Wafula, E., Wang, F., Coruh, C., et al. (2018). MicroRNAs from the parasitic plant cuscuta campestris target host messenger RNAs. Nature 553(7686), 82–85. doi: 10.1038/nature25027
Sharma, A., Bachheti, R. (2013). A review on ipomoea carnea. International Journal of Pharma and Bio Sciences 4(4), 363–377. https://www.cabdirect.org/globalhealth/abstract/20143080424
Stefanović, S., Austin, D. F., Olmstead, R. G. (2003). Classification of convolvulaceae: a phylogenetic approach 28 (4), 791–806. doi: 10.1043/02-45.1
Sun, J., Dong, X., Cao, Q., Xu, T., Zhu, M., Sun, J., et al. (2019). A systematic comparison of eight new plastome sequences from ipomoea L. PeerJ 7, e6563. doi: 10.7717/peerj.6563
Sun, G., Xu, Y., Liu, H., Sun, T., Zhang, J., Hettenhausen, C., et al. (2018). Large-Scale gene losses underlie the genome evolution of parasitic plant cuscuta australis. Nature Communications 9 (1), 1–8. doi: 10.1038/s41467-018-04721-8
Timme, R. E., Kuehl, J. V., Boore, J. L., Jansen, R. K. (2007). A comparative analysis of the lactuca and helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am. J. Bot. 94 (3), 302–312. doi: 10.3732/ajb.94.3.302
van Ooststroom, S. J., Hoogland, R. J. F. M. (1953). Convolvulaceae. Flora Malesiana 4, 439–454. https://www.cabdirect.org/cabdirect/welcome/?target=%2fcabdirect%2fabstract%2f20057008230
Vogel, A., Schwacke, R., Denton, A. K., Usadel, B., Hollmann, J., Fischer, K., et al. (2018). Footprints of parasitism in the genome of the parasitic flowering plant cuscuta campestris. Nature Communications 9(1), 1–11. doi: 10.1038/s41467-018-04344-z
Xue, J., Wang, S., Zhou, S.-L. (2012). Polymorphic chloroplast microsatellite loci in nelumbo (Nelumbonaceae). Am. J. Bot. 99 (6), e240–e244. doi: 10.3732/ajb.1100547
Yang, A.-H., Zhang, J.-J., Yao, X.-H., Huang, H.-W. (2011). Chloroplast microsatellite markers in liriodendron tulipifera (Magnoliaceae) and cross-species amplification in l. chinense. Am. J. Bot. 98 (5), e123–e126. doi: 10.3732/ajb.1000532
Yao, X. (2015). The first complete chloroplast genome sequences in actinidiaceae: Genome structure and comparative analysis. PloS One 10 (10), e0129347. doi: 10.1371/journal.pone.0129347
Zheng, S., Poczai, P., Hyvönen, J., Tang, J., Amiryousefi, A. (2020). Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 11. doi: 10.3389/fgene.2020.576124
Keywords: chloroplast genome, Convolvulaceae, gene loss, parasitic plants, photosynthesis
Citation: Wang Y, Xu J, Hu B, Dong C, Sun J, Li Z, Ye K, Deng F, Wang L, Aslam M, Lv W, Qin Y and Cheng Y (2023) Assembly, annotation, and comparative analysis of Ipomoea chloroplast genomes provide insights into the parasitic characteristics of Cuscuta species. Front. Plant Sci. 13:1074697. doi: 10.3389/fpls.2022.1074697
Received: 19 October 2022; Accepted: 21 December 2022;
Published: 17 January 2023.
Edited by:
Qing-Yong Yang, Huazhong Agricultural University, ChinaReviewed by:
Dayong Zhang, Nanjing Agricultural University, ChinaCopyright © 2023 Wang, Xu, Hu, Dong, Sun, Li, Ye, Deng, Wang, Aslam, Lv, Qin and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Cheng, Y2hlbmd5YW4xMjIwQGhvdG1haWwuY29t; Yuan Qin, eXVhbnFpbkBmYWZ1LmVkdS5jbg==; Wenliang Lv, d2VubGlhbmdfbHZAaGJ0Y20uZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.