
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 02 November 2022
Sec. Plant Biotechnology
Volume 13 - 2022 | https://doi.org/10.3389/fpls.2022.1061196
This article is part of the Research TopicMulti-omics Strategies to Analyze Complex Agronomic Traits in PlantsView all 11 articles
 Chuanji Zhao1†‡
Chuanji Zhao1†‡ Li Yang1,2†‡
Li Yang1,2†‡ Minqiang Tang3‡
Minqiang Tang3‡ Lijiang Liu1
Lijiang Liu1 Junyan Huang1
Junyan Huang1 Chaobo Tong1
Chaobo Tong1 Yang Xiang4Shengyi Liu1Xiaohui Cheng1*
Yang Xiang4Shengyi Liu1Xiaohui Cheng1* Meili Xie1*‡
Meili Xie1*‡Rapeseed (Brassica napus) is an allotetraploid crop that is the main source of edible oils and feed proteins in the world. The ideal plant architecture breeding is a major objective of rapeseed breeding and determining the appropriate plant height is a key element of the ideal plant architecture. Therefore, this study aims to improve the understanding of the genetic controls underlying plant height. The plant heights of 230 rapeseed accessions collected worldwide were investigated in field experiments over two consecutive years in Wuhan, China. Whole-genome resequencing of these accessions yielded a total of 1,707,194 informative single nucleotide polymorphisms (SNPs) that were used for genome-wide association analysis (GWAS). GWAS and haplotype analysis showed that BnaA01g09530D, which encodes BRASSINOSTEROID-INSENSITIVE 2 and belongs to the GLYCOGEN SYNTHASE KINASE 3 (GSK3) family, was significantly associated with plant height in B. napus. Moreover, a total of 31 BnGSK3s with complete domains were identified from B. napus genome and clustered into four groups according to phylogenetic analysis, gene structure, and motif distribution. The expression patterns showed that BnGSK3s exhibited significant differences in 13 developmental tissues in B. napus, suggesting that BnGSK3s may be involved in tissue-specific development. Sixteen BnGSK3 genes were highly expressed the in shoot apical meristem, which may be related to plant height or architecture development. These results are important for providing new haplotypes of plant height in B. napus and for extending valuable genetic information for rapeseed genetic improvement of plant architecture.
Rapeseed (Brassica napus L., 2n = 38, AACC) is the main source of edible oils and feed proteins worldwide. However, the rapeseed industry is currently confronted with multiple bottlenecks, i.e. low yield, low planting density, low mechanization degree, large amount of fertilization, and high labor costs, which seriously impacts the sustainable development of the rapeseed industry. Shaping the ideal plant architecture of rapeseed is helpful to break through these bottlenecks, but the lack of a clear genetic basis and constituent elements has hindered the development of this research. Plant height is one of the most important determinants of ideal plant architecture. Since lodging is a common phenomenon and yield loss caused by lodging is severe (16.2%) in rapeseed production (Islam and Evans, 1994). Therefore, moderate dwarfing of crop plant height increased the harvest index.
Plant height is an agronomic trait with complex genetic basis. It is easily affected by environment and usually regulated by both major and minor genes. In recent years, with the rise of the green revolution in wheat, breeders have identified a large number of quantitative trait loci (QTLs) controlling wheat plant height on 21 chromosomes using different populations and markers (Chu et al., 2008; Buerstmayr et al., 2011; Guo et al., 2018). The green revolution in rice began with the application of a semi-dwarf gene sd1 (Monna et al., 2002; Sasaki et al., 2002; Spielmeyer et al., 2002). The discovery and utilization of dwarf mutants and corresponding genes have greatly promoted the development of new rice varieties. The cloned dwarf genes in rice are mainly involved in the biosynthesis and signal pathways of plant hormones (e.g., gibberellin, brassinolide, and strigolactone). Some of these genes contain special domains, including sd1 (Ye et al., 2015), D1 (Ferrero-Serrano et al., 2018; Sun et al., 2018), GID1 and GID2 (Hirano et al., 2010), OsDWARF4 (Fang et al., 2016), and OsTB1 (Fang et al., 2020). Currently, the only known gene responsible for ideal plant architecture gene in rice is IPA1, which encodes the squamosa-like promoter-binding protein OsSPL14. Mutations in OsSPL14 reduced tillering, increased grain number per ear and 1000-grain weight, thickened stem, and enhanced lodging resistance, thereby increasing the yield (Jiao et al., 2010; Miura et al., 2010).
In B. napus, the identification of QTLs highly related with plant height is an important task in genetic maps and genome-wide association analysis (GWAS). Fourteen QTLs for plant height were identified in different linkage groups using a recombinant inbred line (Cai et al., 2014). A major plant height QTL on chromosome A10, was identified by whole-genome resequencing (WGS) based genetic mapping (Dong et al., 2021). Using the Illumina Brassica 60 K Bead Chip Array and a diversity of 520 accessions, a total of 68 plant height-related loci were obtained by GWAS under six environments. Most of the genes in these loci were involved in gibberellin synthesis and signal pathway (Sun et al., 2016). In recent years, progress has been made in the exploitation of dwarf genetic resources and genes in B. napus. Most dwarf mutants belong to gibberellin, auxin, and brassinolide-insensitive mutants. In two B. napus dwarf mutants of approximately 70 cm height, their candidate genes were mapped on chromosomes A06 and C07, both of which encode DELLA proteins, a negative regulator of the gibberellin signal transduction pathway, and have missense mutations in the VHYNP domain (Liu et al., 2010; Zhao et al., 2017). Mutations at different sites of BnaC05g29300D, encoding an auxin signaling transport repressor, resulted in rapeseed plant heights of only 25 cm (Zhao et al., 2019; Zheng et al., 2019). Mutation of BnaA3.IAA7, which encodes an auxin-inducible protein, disrupted the conserved degradation motif GWPPV and reduced the affinity between BnaA3.IAA7 and the transport inhibitor in an auxin dose-dependent manner, thus inhibiting BnaA3.IAA7 degradation and auxin signaling in B. napus dwarf mutant sca (Li et al., 2019a). The dwarf locus BnDWARF2 was mapped to a 34.62 kb interval, in which BnaC04g41660D encoding a GLYCOGEN SYNTHASE KINASE 3 (GSK3-like) in the brassinosteroid signaling, was the causal gene controlling plant height in oilseed rape (Yang et al., 2021). In addition, other genes unrelated to plant hormones may also be involved in the regulation of plant height in B. napus; for example, the Octicosapeptide/Phox/Bem1p family protein encoding gene BnaC09g20450D contains a single nucleotide polymorphism (SNP) that co-segregates with the dwarf phenotype in df59 mutant (Wang et al., 2020a).
Although many plant height QTLs and dwarf genes have been identified, they have not been fully utilized in breeding, and cultivars with dwarf or semi-dwarf phenotypes are still the major objective in rapeseed breeding. This study aims to better understand the genetic control of plant height and to unearth more valuable information from the genome of polyploid rapeseed based on GWAS for plant height in 230 core rapeseed accessions around the world. We identified BnaA01g09530D, a BnGSK3 gene involved in the cross-talk between auxin and brassinosteroid signaling pathways, was significantly associated with plant height. We also analyzed the expression pattern in various tissues, overall distribution in the rapeseed genome, and phylogenetic analysis of the BnGSK3s family.
Extensive phenotypic variations of plant height were observed in 230 inbred accessions over two consecutive years (Table 1). The plant height ranged from 149.23–230.59 cm in 2017–2018 and from 115.12–189.28 cm in 2018–2019, suggesting that the environment factors had a great impact on plant height (Supplementary Figure 1A and Table 1). The plant heights in 2017–2018, 2018–2019, and the BLUP of the 230 rapeseed accessions displayed normal distributions (Supplementary Figure 1A). The coefficient of variation in 2018–2019 was 9.10%, which was higher than that in 2017–2018 (7.38%) and BLUP (7.69%) (Table 1). Nevertheless, no significant difference was observed between the phenotype of 2017-2018 and 2018-2019, as shown by the correlation analysis (R2 > 0.70) (Supplementary Figure 1B). These analyses revealed that the phenotype of 230 rapeseed accessions were reliable and feasible for association analysis.
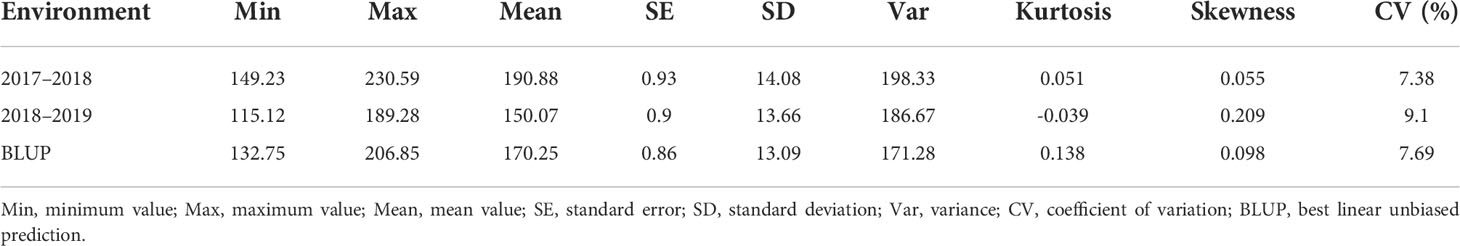
Table 1 Phenotypic variations of plant height in rapeseed natural population.
A total of 230 rapeseed accessions, consisting of 25 spring-, 33 winter-, and 172 semi-winter ecotypes, were employed for WGS (Supplementary Table 1). Approximately 1,097.37 Gb data were generated, with an average size of 4.77 Gb and an average depth of 6.46 × depth per accession (Supplementary Table 1). The average coverage of B. napus reference genome was 82.19% (Supplementary Table 1). A total of 1,707,194 informative SNPs were acquired with an average of 94,844 SNPs on each chromosome (Table 2). The density of SNPs on different chromosomes ranged from 1.01 to 4.86 SNP/kb, with chromosome C09 having the lowest density and A10 the highest (Table 2). These results suggested the reliability of SNP information and could be used for further analyses.
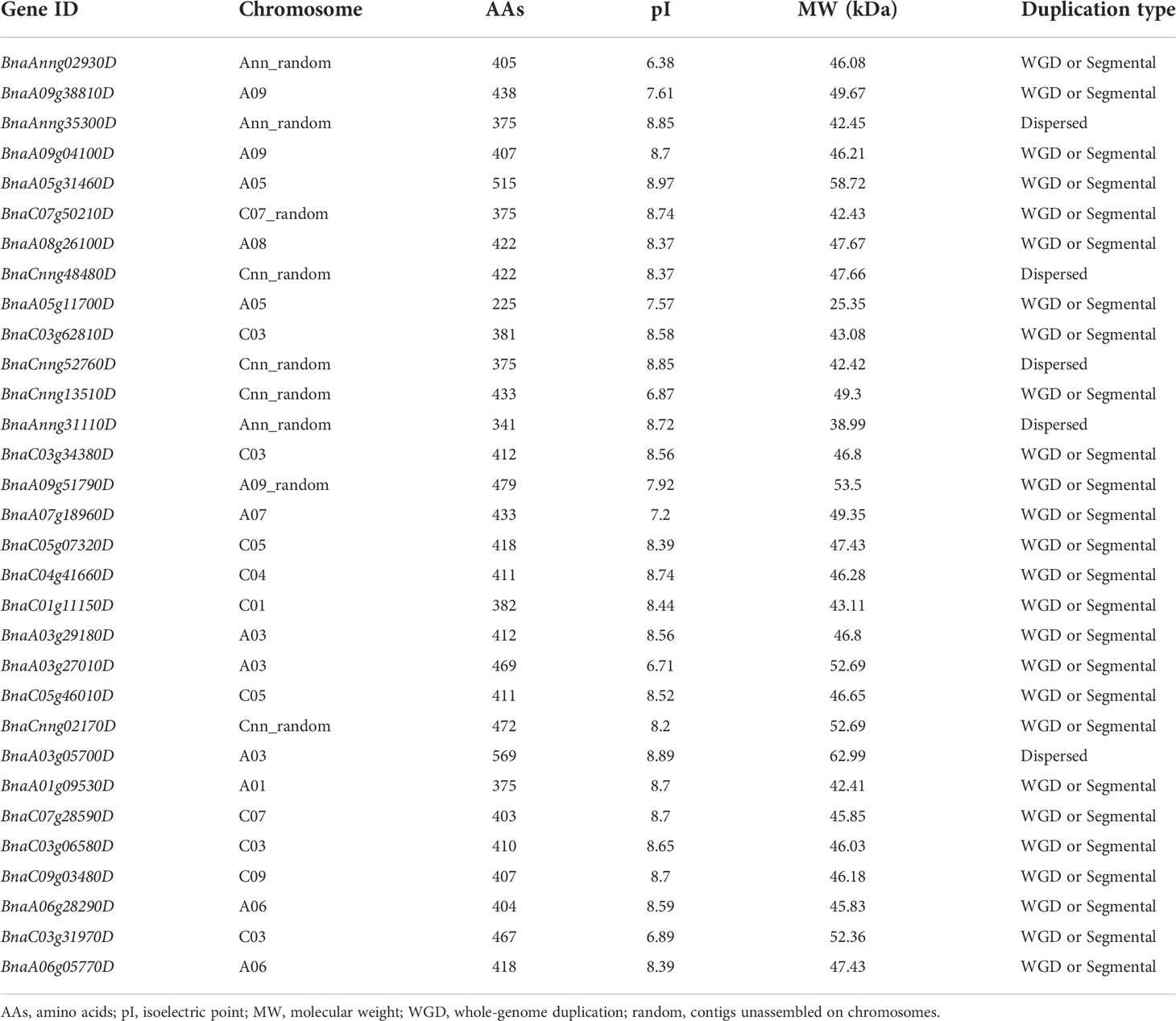
Table 2 Statistics of SNP number and density on each chromosome.
According to the Q+K model, the associated population could be divided into nine subgroups (Supplementary Figure 2A, B). More than 90% of the relative kinship coefficients among these accessions were found to be lower than 0.1, suggesting that most accessions in this population lacked or had weak genetic relatedness (Supplementary Figure 2C). The average linkage disequilibrium (LD) decay of the A and C sub-genomes were 4.1 and 120.3 kb, respectively. It was 33.4 kb for the whole genome (A + C) when r2 decayed to its half (Figure 1D).
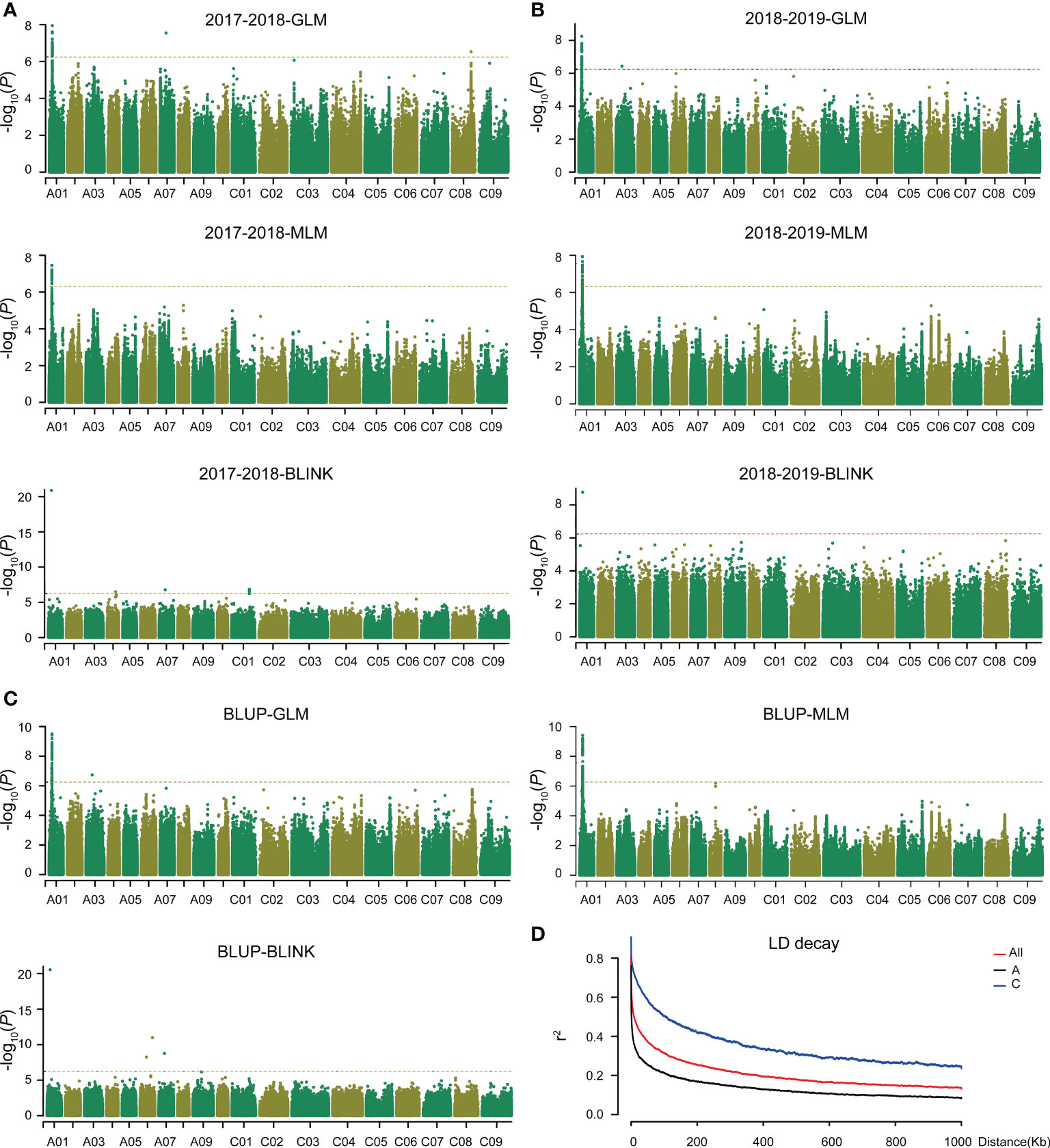
Figure 1 GWAS of plant height in Brassica napus and LD decay analysis. The threshold value is -log10(1/SNPs number). (A) GWAS of plant height in 2017–2018 based on GLM, MLM, and BILNK models. (B) GWAS of plant height in 2018–2019 based on GLM, MLM, and BILNK models. (C) GWAS of plant height for BLUP based on GLM, MLM, and BLINK models. (D) Linkage disequilibrium (LD) decay of A and C sub-genomes and whole genome.
To dissect the genetic control of plant height in B. napus, we performed GWAS in two consecutive years. A significant locus on chromosome A01 was simultaneously identified using GLM, MLM, and BLINK models (Figures 1A–C, and Supplementary Table 3). Quantile-quantile plots showed obvious deviations between the observed and expected values, indicating the selected models were correct and suitable for GWAS (Supplementary Figure 3). Within the significance interval, 806 SNPs were repeatedly identified in different environments and models (GLM and MLM) (Supplementary Table 3). According to the MLM model in BLUP and LD decay of A sub-genome (Figure 1), three genes (BnaA01g09530D, BnaA01g09540D, and BnaA01g09550D) near the significant SNPs were strongly associated (Figure 2A). Based on the annotation of the B. napus reference genome, BnaA01g09530D, encoding BRASSINOSTEROID-INSENSITIVE 2 (BIN2) and involving in the brassinosteroid signaling pathway, may be a candidate gene controlling plant height in B. napus. In addition, the position of co-identified SNP by BLINK model in different environments was 4,772,232 (Supplementary Table 3), which was far away from the co-identified significant SNPs in GLM and MLM, due to the different algorithm principle of BLINK. In the application of BLINK, a bin, containing all the linked SNPs in a region, is taken as a unit, rather than a single SNP as a unit like GLM and MLM (Huang et al., 2019), suggesting that BnaA01g09530D was also identified in the BLINK models. A total of ten SNPs variations were observed in the sequence of BnaA01.BIN2. Haplotype analysis of these ten SNPs revealed favorable allelic variation (Hap_II), conferring a significant reduction in plant height (Figure 2B).
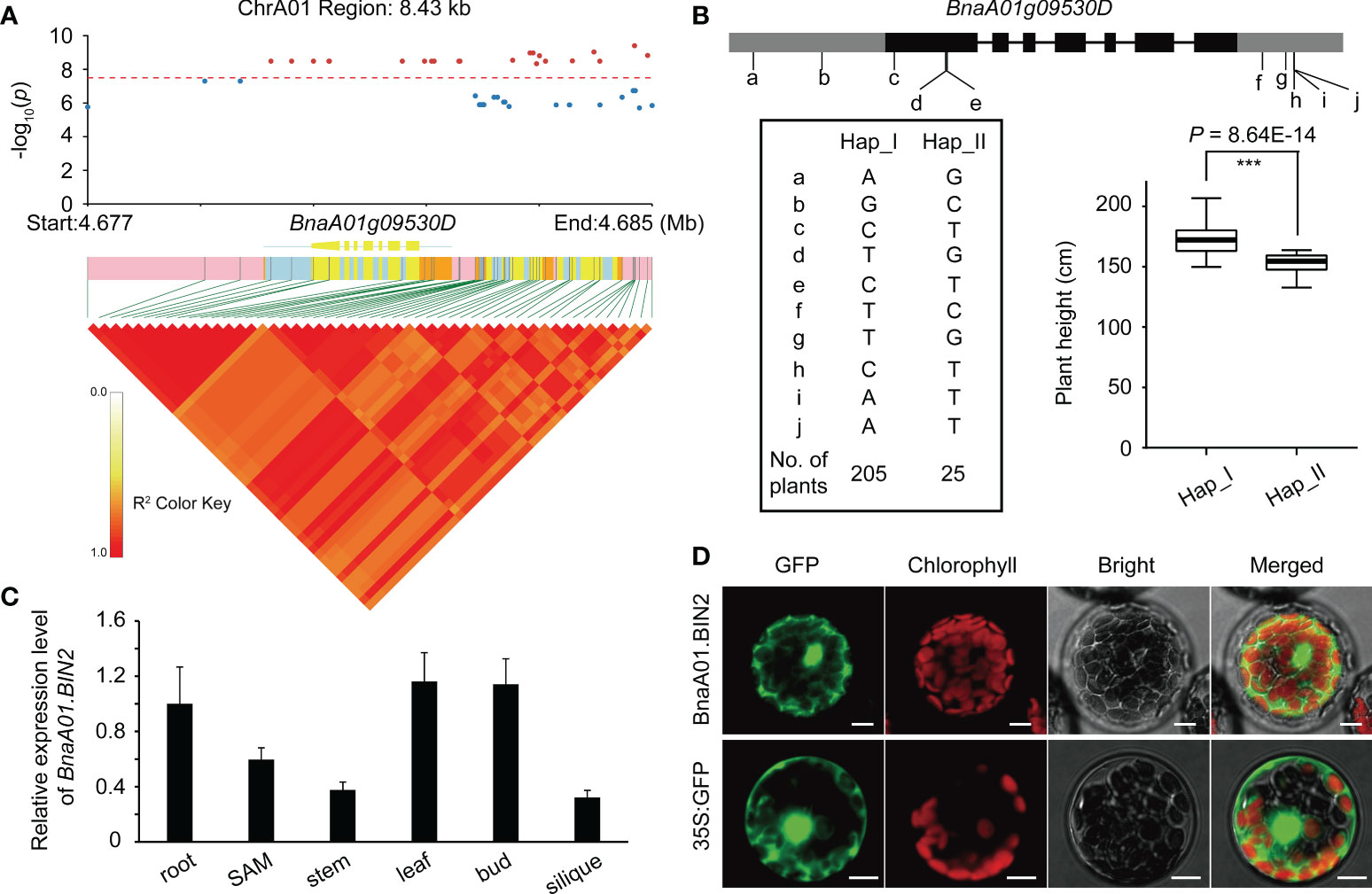
Figure 2 Integrated characteristic of BnA01.BIN2. (A) LD block analysis of significant SNPs in BLUP based on MLM models. The threshold value is -log10(0.05/SNPs number). (B) Gene structure of BnaA01.BIN2 and haplotype analysis. (C) Subcellular localization of BnaA01.BIN2 in Arabidopsis protoplasts. Green fluorescence, GFP; red fluorescence, chloroplast autofluorescence; Merged, merged images of all channels. Bar = 10 μm. (D) Expression pattern of BnaA01.BIN2.
The expression pattern of BnaA01.BIN2 showed that it was highly expressed in leaves, buds, and roots, followed by SAM, suggesting that it plays an important role in plant development. Subcellular localization, as indicated by green fluorescent protein (GFP), showed that BnaA01.BIN2 was localized in nucleus and cytoplasm (Figure 2D).
Candidate gene BnaA01.BIN2 belongs to the glycogen synthase kinase 3 (GSK3) gene family. Using protein sequences of AtGSK3s as the query of BLAST, a total of 38 BnGSK3s were identified in “Darmor-bzh” rapeseed genome, and 31 BnGSK3s with Pkinase domain were finally extracted (Table 3). Of these BnGSK3s, 16% (5) resulted from dispersed duplications and 84% (26) originated from whole-genome duplication (WGD) or segmental duplication (Table 3). Most AtGSK3s have several syntenic genes in B. napus, among which BnBIN2 contains six homologous genes and is the largest member of BnGSK3s (Supplementary Table 2). However, there were no homologous genes for AtBIL2 and AtSK42 in B. napus (Supplementary Table 2). We identified 15 BrGSK3s and 16 BoGSK3s according to the Brassica Database (BRAD) (http://brassicadb.cn/) and no homologous genes of AtBIL2 and AtSK42 were identified in the reference genomes of B. rapa (Brara_Chiifu_V3.5) and B. oleracea (Braol_JZS_V2.0) (Supplementary Table 2). These results suggested that the GSK3s family is highly conserved in Brassicaceae, whereas the loss of BIL2s and SK42s may occur prior to Brassicaceae speciation.
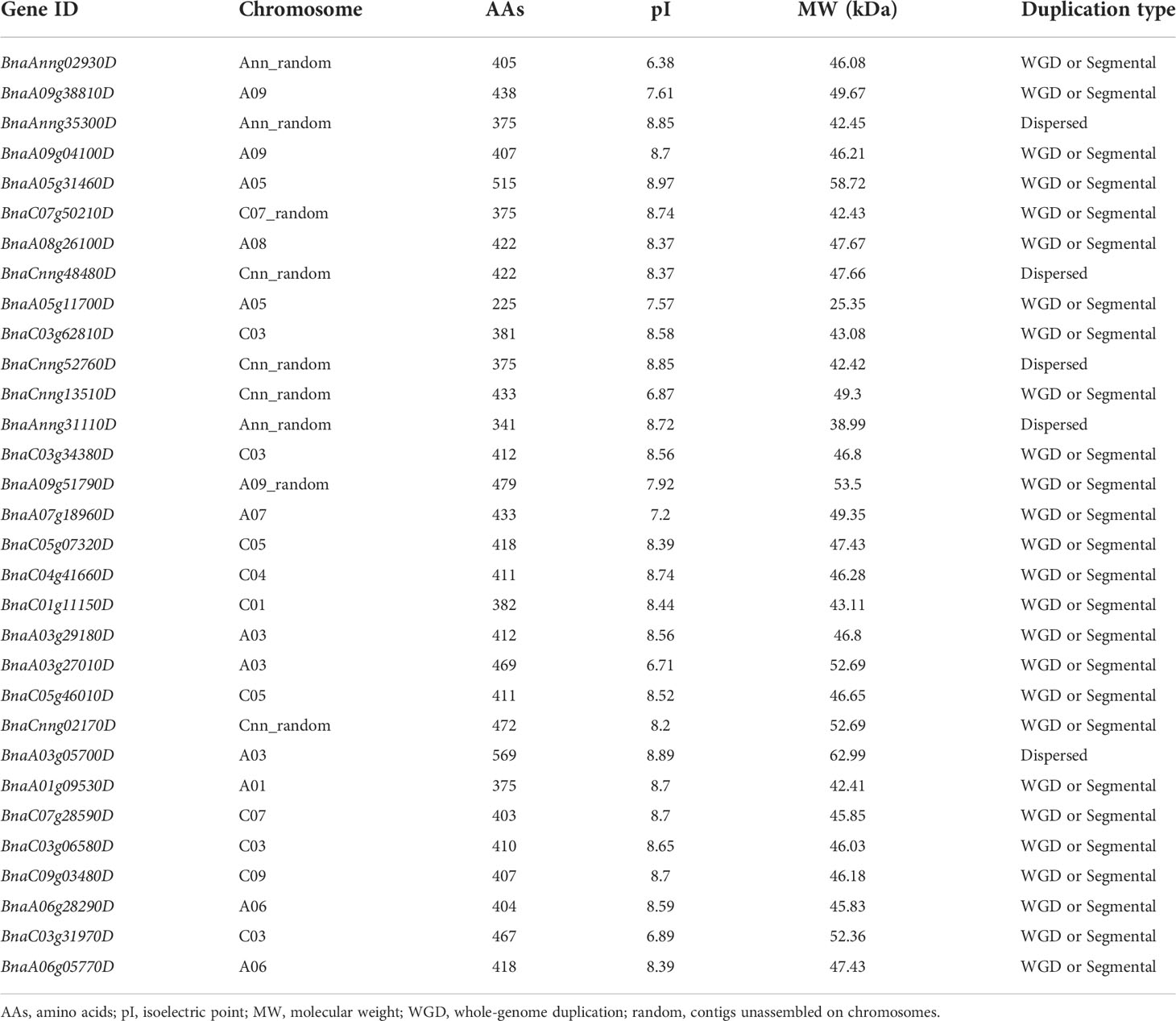
Table 3 The information of BnGSK3s family in rapeseed.
The 31 BnGSK3s were unevenly distributed in 13 chromosomes and four random chromosomes, 16 and 15 BnGSK3s were located on the A and C sub-genomes, respectively (Figure 3 and Table 3).
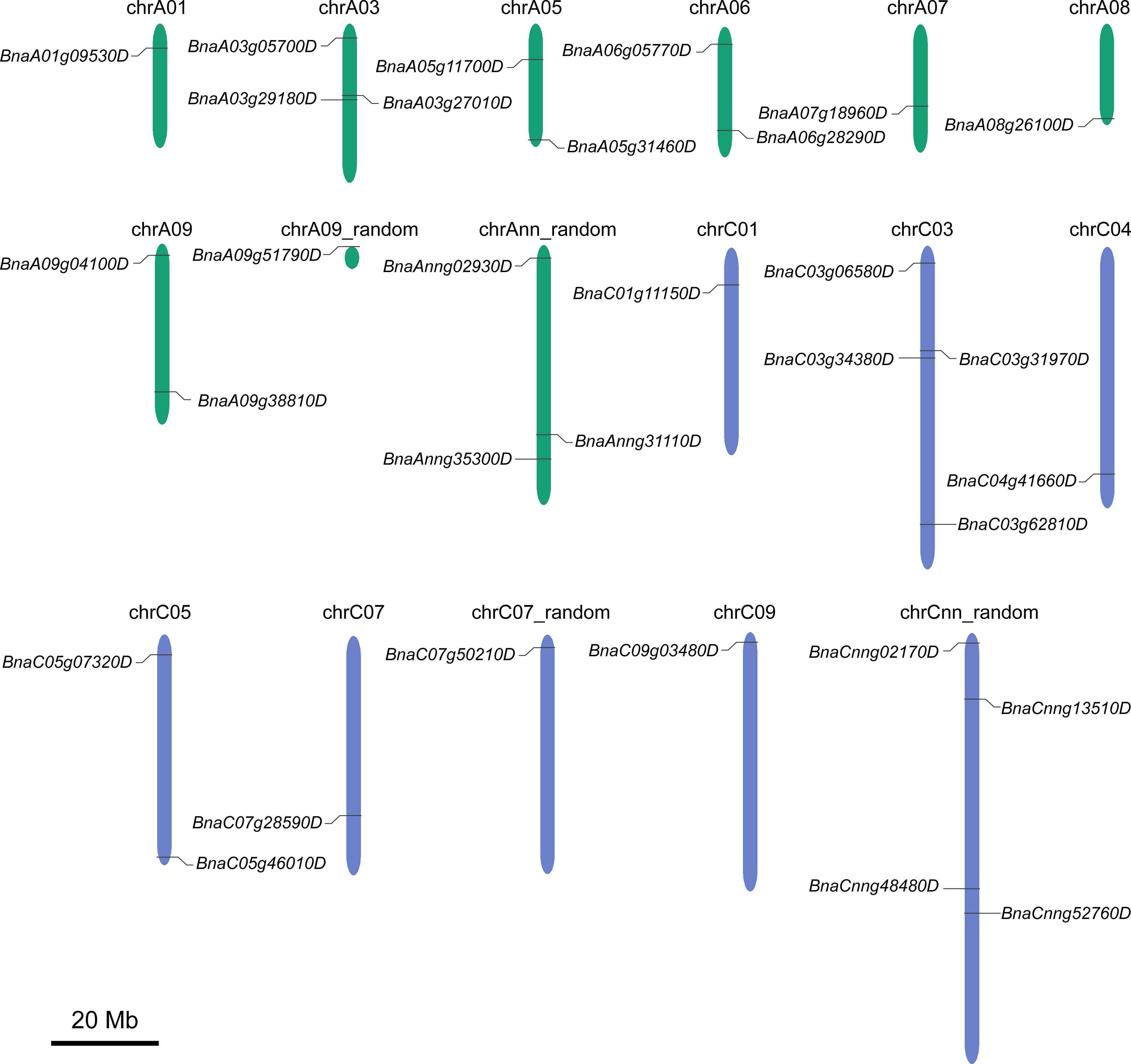
Figure 3 Chromosomal distribution of BnGSK3s in the genome of Brassica Napus. Green and blue chromosomes represent A and C sub-genome of B. napus, respectively.
To explore the phylogenetic relationship of GSK3s family, we constructed a phylogenetic tree using GSK3s protein sequences from Arabidopsis and B. napus. The 10 AtGSK3s and 31 BnGSK3s were divided into four groups: Group I (SK11, SK12, and SK13), Group II (BIN2, BIL1, and BIL2), Group III (SK31 and SK32), and Group IV (SK41 and SK42) (Figure 4A and Supplementary Table 2). Group I had 22 GSK3s, including 11 BnGSK3s, 5 BrGSK3s, and 6 BoGSK3s, accounting for the largest group. Group IV was the smallest, with only eight GSK3s (Figure 4A and Supplementary Table 2). This suggests that Group I of GSK3s was more expanded compared to that of Group IV. Within each group, BnGSK3s belonging to the A and C sub-genomes in B. napus, along with the AtGSK3s in Arabidopsis, clustered into a small clade (Figure 4A), suggesting that the phylogenetic relationship of GSK3 was consistent with the evolution of rapeseed. The syntenic analysis between AtGSK3s and BnGSK3s showed that most of AtGSK3s have over two syntenic genes in B. napus (Figure 4B), which is consistent with phylogenetic relationship of GSK3s.
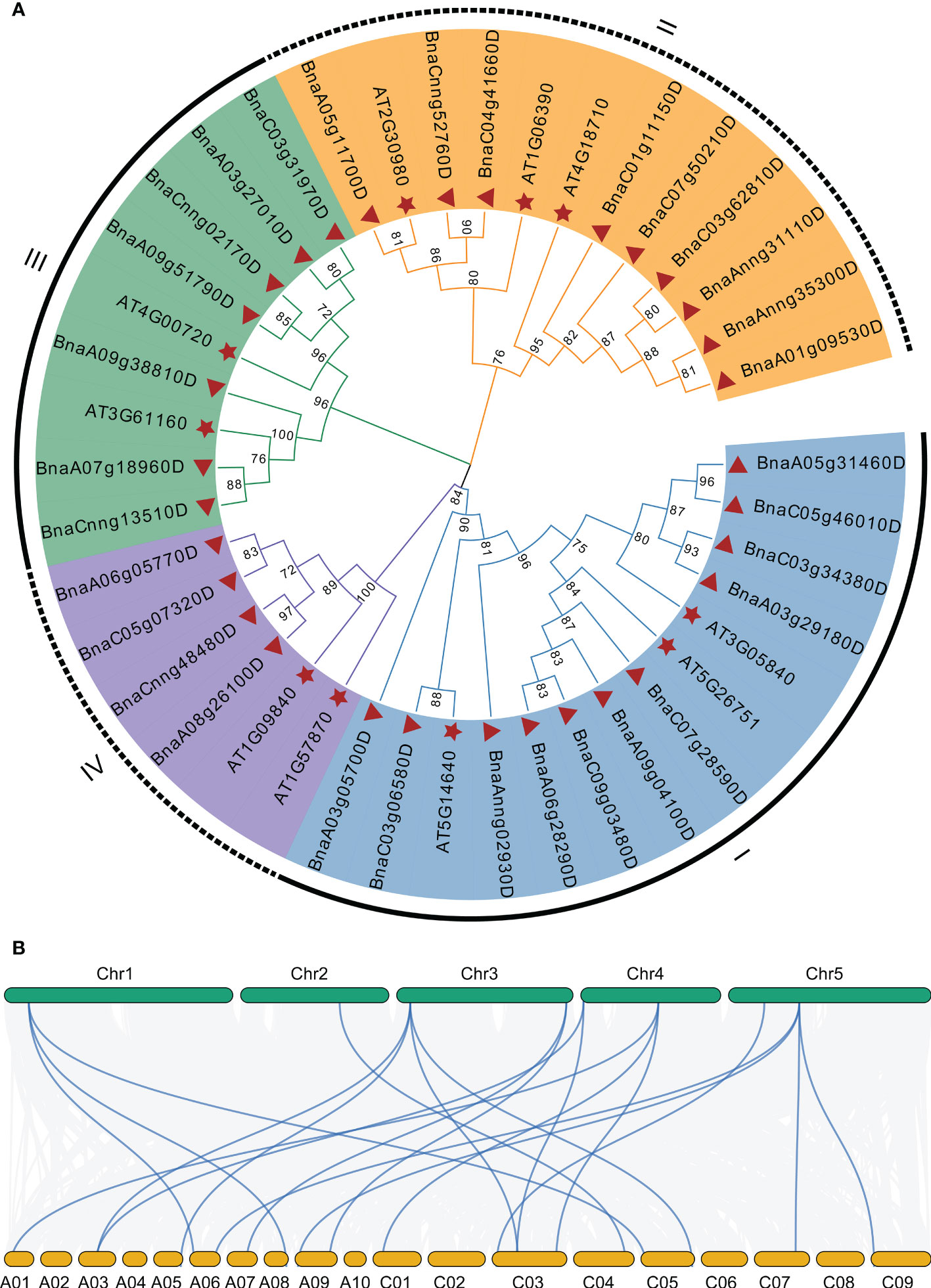
Figure 4 Phylogenetic and syntenic analysis of AtGSK3s and BnGSK3s. (A) Phylogenetic analysis. (B) syntenic analysis. The green and yellow blocks represent Arabidopsis and B. napus chromosome, respectively.
To explore the conservation of BnGSK3s, gene structure and protein motifs were analyzed (Figure 5). In general, gene structures of the 31 BnGSK3s differed obviously between different groups. Among them, the syntenic genes showed relatively similar gene structures (Figures 5A, C). The gene structures of approximately 74% of BnGSK3s (23) exhibited 5’ and 3’ untranslated regions (UTR) (Figure 5C). Six BnGSK3s (BnA05g31460D, BnA03g05700D, BnA09g38810D, BnA09g51790D, BnCnng52760D, and BnAnng35300D) only had 5-’ or 3-UTR (Figure 5C). In addition, all BnGSK3s contained exons and introns (Figure 5C). As for motif analysis, except BnaA05g11700D possessed seven conserved motifs, the remaining BnGSK3s had ten conserved motifs (Figure 5B). These results suggested that the core sequences of the BnGSK3s were conserved.
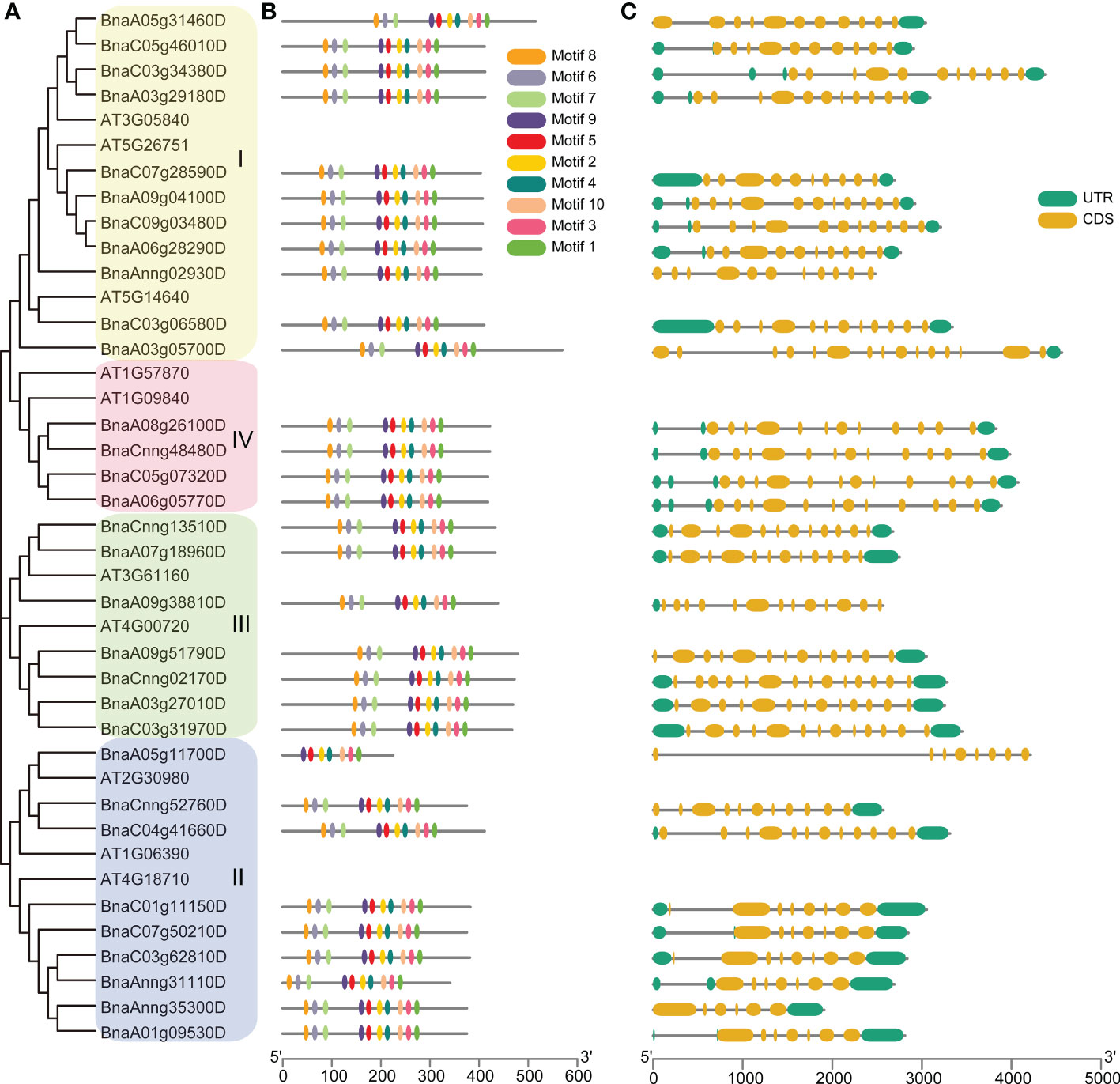
Figure 5 Gene structure and conserved motif analyses of AtGSK3s and BnGSK3s. (A) Phylogenetic tree of AtGSK3s and BnGSK3s. (B) Conserved motifs of AtGSK3s and BnGSK3s. (C) Gene structures of AtGSK3s and BnGSK3s.
Based on published transcriptome data (Li et al., 2019b; Dong et al., 2021), the expression patterns of the 31 BnGSK3s in 13 tissues of ZS11 were analyzed, which showed that the BnGSK3s were expressed in different tissues (Figure 6A). However, a set of homologous genes, including BnSK31, BnBIL1, and BnBIN2, showed similar expression patterns, suggesting a potential redundancy of function (Figure 6A). Different expression patterns were observed within the same group, suggesting functional divergence in BnGSK3s. For example, BnBIL1 was highly expressed in SAM, whereas BnBIN2 was highly expressed in roots (Figure 6A). In addition, BnSK13s were prone to express in pistils and buds, suggesting that these genes may be involved in flower development (Figure 6A). Thirteen BnGSK3s were highly expressed in SAM, indicating that BnGSK3s have a certain effect on the development of plant architecture. In addition, we selected eight BnGSK3s from different groups to perform qRT-PCR in six tissues, which suggested that the expression pattern was consistent with the RNA-seq data (Figure 6B).
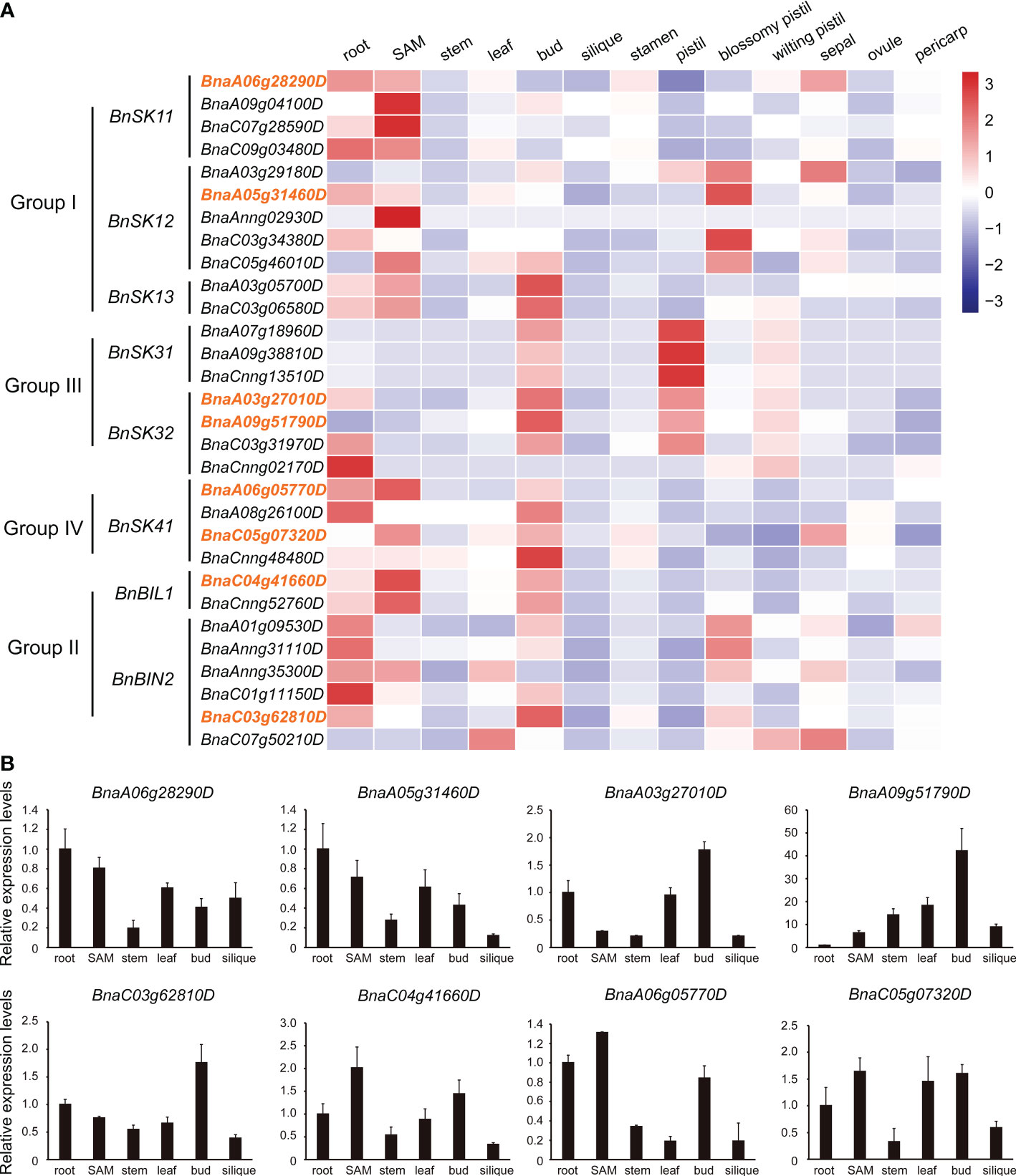
Figure 6 Expression pattern of BnGSK3s. (A) Expression pattern of the 31 BnGSK3s in 13 tissues of B. napus cv. ZS11 based on RNA-seq. Bar represents the normalized transformed counts of FPKM. The expression of eight genes marked in orange were verified by qRT-PCR.(B) Expression patterns of eight BnGSK3s in six tissues of B. napus cv. ZS11 based on qRT-PCR.
Since the “Green Revolution” in the 1960s, researchers have carried out extensive research to come up with ideal plant architecture models for many crops (Teichmann and Muhr, 2015; Liu et al., 2020; Pearce, 2021). Several novel genes controlling aboveground plant architecture have been identified and their regulatory mechanisms have been expounded, laying the foundation for breeding new high-yielding varieties of rice (Monna et al., 2002; Sasaki et al., 2002; Wang and Li, 2008; Wang et al., 2020b), wheat (Chai et al., 2022; Xiong et al., 2022), maize (Phillips et al., 2011; Li et al., 2020a), and soybean (Guo et al., 2020; Chen et al., 2021b). Many researchers have proposed models of ideal rapeseed plant architecture (Liu et al., 2022; Zheng et al., 2022). However, these are only concepts and cannot solve actual problems in production. There are several difficulties in studying the ideal plant architecture of rapeseed: 1) the lack of materials with good plant architecture materials; 2) the uncertainty of proper index used for the research of rapeseed plant architecture; 3) severe environmental impact on plant architecture-related traits; 4) the lack of clear genetic basis. For many crops, such as rice and wheat, plant height has been used as a breakthrough point to study plant architecture (Peng et al., 1999; Hedden, 2003). Therefore, plant height is essential in shaping the ideal plant architecture of crops. Although research progress has been made in the study of plant height traits of rapeseed (Liu et al., 2010; Wang et al., 2016a; Wang et al., 2016b), the genetic basis of rapeseed plant height remains unclear.
Currently, a single genetic resource cannot effectively improve the present plant architecture of rapeseed. GWAS is often used as an effective method to unravel the genetic architecture of complex agronomic traits in crops. Combined with association analysis and linkage analysis, 61 SNPs significantly associated with low zinc tolerance and 15 QTLs were identified in maize. Expression and haplotype analyses were used to mine the favorable allele conferring low zinc tolerance (Xu et al., 2022). Similar study could be found in Guo et al. (2021), in which 63 loci related to stem strength and yield were identified and favorable alleles for both high stem strength and high yield were discovered using 524 rice germplasm resources and 193 recombinant inbred lines (Guo et al., 2021). Based on GWAS and a transcriptome-wide association study, 15 stable QTLs and 1,854 candidate genes were detected in B. napus, which were significantly associated with seed glucosinolate content. Haplotype analysis showed that seed low glucosinolate was mainly resulted by the co-action of multiple favorable alleles (Tan et al., 2022). In this study, GWAS was performed on plant height of 230 B. napus accessions using three models (GLM, MLM, and BLINK). An unreported gene, BnaA01.BIN2, was simultaneously identified by all three models (Figures 1, 2), which increased the confidence of the results. However, no other loci or reported genes were co-identified, probably due to population structure constraints. These results provide insights for subsequent adjustment of population structure to more effectively detect available loci, genes, or favorable alleles.
GSK3 is a group of highly conserved cytoplasmic serine/threonine protein kinases that are widely present in animal and plant cells. These proteins perform their functions mainly by phosphorylating key substrate proteins of different signaling pathways. GSK3 is regulated by a variety of post-translational modification mechanisms. BnaA01.BIN2, identified by GWAS in this study (Figures 1, 2), encodes BRASSINOSTEROID-INSENSITIVE 2 (BIN2) and, belongs to the BnGSK3 family. AtBIN2 plays a role in the crosstalk between auxin and brassinosteroid signaling pathways (https://www.arabidopsis.org/index.jsp). In B. napus, BnaC04.BIL1, which has been isolated from the dwarf mutant Bndwarf2 (Yang et al., 2021), encodes BIN2-LIKE 1, and is also a member of GSK3s.
As a core member of GSK3s, BIN2 is a constitutively active kinase in plants, whose activity is affected by various regulatory mechanisms, including nucleocytoplasmic distribution, protein-protein interaction strength, phosphorylation and dephosphorylation, acetylation, and ubiquitination (Mao and Li, 2020). The direct function of BIN2 is to participate in the signal transduction pathway of brassinolide, which plays an important role in plant development (Anne et al., 2015). BIN2 directly controls the transcriptional regulatory complex composed of WEREWOLF (WER), transcription factor GLABRA3 (GL3), and WD40 repeat protein TRANSPARENT TESTA GLABRA1 (TTG1), It can phosphorylates GL3 and TTG1 in the WER-GL3-TTG1 complex to inhibit their transcriptional activity, thereby regulating root hair development (Cheng et al., 2014). BIN2 participates in photomorphogenesis by interacting with HY5, an important transcription factor for photomorphogenesis (Li et al., 2020b). In addition, BIN2 is involved in osmotic stress and adverse effects, and can promote lateral root development by phosphorylating auxin-responsive factor ARF7 (Cho et al., 2014). BIN2 is also involved in abscisic acid signal transduction to regulate the osmotic stress response (Wang et al., 2018) and enhances plant drought tolerance by phosphorylating RSPONSIVE OT DESICCATION 26, NAC family transcription factor (Jiang et al., 2019).
GSK3 is involved in the regulation of plant growth and development. However, only one gene has been reported to be related with plant height in rapeseed (Yang et al., 2021). In this study, to determine the relationship between BnGSK3s and plant height in allotetraploid rapeseed, we investigated the BnGSK3s family, which consists of 16 homologs in A sub-genome and 15 in C sub-genome (Figure 3 and Table 3). Based on the transcriptome data of 13 tissues in ZS11, the expression pattern of BnGSK3s were found to show obvious expression preference difference in rapeseed (Figure 6), in which 16 genes were highly expressed in SAM and three were highly expressed in the pistil (Figure 6A). Moreover, we identified favorable allelic variations in BnaA01.BIN2 among 230 B. napus accessions, whereas we failed to detect any SNP variation in the corresponding syntenic gene BnaC01.BIN2 (BnaC01g11150D). This could be caused by the limited numbers of accessions used for GWAS in this study. As such, more rapeseed genetic resources should be collected to dissect more favorable allelic variations in BnGSK3s for plant height and plant architecture.
In this study, GWAS was performed on plant heights of a bio-panel of 230 rapeseed accessions in two consecutive years based on three models. The results showed that BnaA01.BIN2 belonging to BnGSK3s family, was significantly associated with plant height in B. napus. A total of 31 BnGSK3s were identified and clustered into four groups. Expression pattern analysis suggests that BnGSK3s may be involved in tissue-specific development. Sixteen BnGSK3 genes were highly expressed in SAM, which may be related to plant height development. These findings are important for the genetic improvement of plant height and architecture in rapeseed.
A total of 230 rapeseed cultivars or inbred lines (Supplementary Table 1) were collected worldwide, representing the genetic diversity of B. napus for GWAS of plant height. Field trials were conducted by a randomized design with three replications. For each accession, 45 individuals were grown in a 2.0 × 1.0 m2 plot with three rows in each environment (2017–2018, 2018–2019, winter-spring growing season) in the Yangluo experimental field of Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences, Wuhan, China. The R script lme4 (CRAN-Package lme4 (r-project.org)) and lsmeans were used to calculate the best linear unbiased prediction (BLUP) of each inbred line in the natural population (Zhao et al., 2022).
At the mature stage, 10 plants with good growth and development were randomly selected from each plot for phenotype investigation. The length from the cotyledon node to the apical position of the whole plant was measured and recorded as plant height. The statistics of the phenotype variation and frequency distribution were calculated using SPSS 22 (IBM SPSS, Armonk, NY, United States) (Zhao et al., 2022). The Pearson’s product-moment correlation analysis of plant height between 2017-2018 and 2018-2019 was carried out by R Package.
Total genomic DNA from fresh young leaf tissue of each inbred line (230 accessions) was extracted using a Hi-DNAsecure Plant Kit (TIANGEN, Beijing). DNA libraries were constructed with high-quality genomic DNA and whole-genome resequencing (WGS) was performed using the Illumina NovaSeq 6000 system. Clean data (clean reads) were obtained by filtering the raw data. All clean reads were mapped to the B. napus reference genome (Darmor-bzh V5, https://www.genoscope.cns.fr/brassicanapus/data/) using the Burrows-Wheeler Aligner software (Li and Durbin, 2009; Chalhoub et al., 2014). SAMTools (parameter: -q 30; http://samtools.sourceforge.net/) and Sentieon Genomics (parameter: –algo Dedup –rmdup) software were used to filter alignment duplications (Li et al., 2009; Freed et al., 2017). GATK (version 4.1.4.0) and vcftools (version 4.2) were used for SNP identification and filtration (parameters: MQ < 50.0 || QD < 2.0, -min-alleles 2 -max-alleles 2 -maf 0.05 -max-missing 0.9, and -cluster-size 3 -cluster-window size 10) (McKenna et al., 2010; Danecek et al., 2011). At last, a total of 1,707,194 informative SNPs were acquired, and the original SNPs were obtained from published data of our lab (Tang, 2019; Ding et al., 2020).
To analyze the natural population structure and linkage disequilibrium (LD) decay, ADMIXTURE (Version 1.3.0) (Alexander et al., 2009), Q+K model, and PopLDdecay (Zhang et al., 2018) were performed according to detailed descriptions from previous studies (Zhao et al., 2022). Three software and models were used, including the general linear model (GLM) in trait analysis by association, evolution, and linkage (TASSEL, Version 5.0) (http://www.maizegenetics.net/tassel); mixed linear model (MLM) in Efficient Mixed-Model Association eXpedited (EMMAX); and Bayesian information and Linkage-disequilibrium Iteratively Nested Keyway (BLINK) (Huang et al., 2019). TASSEL was used to calculate the kinship of 230 B. napus accessions (Yu et al., 2006). The LD block was displayed using LDBlockShow software (Dong et al., 2020).
Complete coding sequence of BnaA01.GSK3 was amplified from the B. napus cv. Zhongshuang11 (ZS11). The purified DNA fragment was fused with green fluorescent protein (GFP) in the backbone vector pBWA(V)HS-gfp, resulting in the plasmid 35S:BnaA01.BIN2-GFP via the ClonExpressMultiS One Step Cloning Kit C113-01 (Vazyme). The 35S:GFP plasmid was used as the mock control. These plasmids were transiently transformed into Arabidopsis protoplast cells using the Agrobacterium-mediated method. The subcellular localization of BnaA01.BIN2 was determined by observing GFP using a Nikon C2-ER confocal microscope (Nikon, Japan) (Zhao et al., 2021). The primers used for amplification of BnaA01.BIN2 are listed in Supplementary Table 4.
The amino acid sequences of AtGSK3s family were obtained from the database “The Arabidopsis Information Resource (TAIR; https://www.arabidopsis.org/),” which were used to build a Hidden Markov Model, and HMMER3.0 was used to search the annotation and genome information of B. napus “Darmor-bzh” in the Brassicaceae Database (BRAD) (Mistry et al., 2013; Chalhoub et al., 2014; Chen et al., 2021a). The isoelectric point (pI) and molecular weight (MW) of BnGSK3s proteins were predicted using ProtParam online software (https://web.expasy.org/protparam/).
The National Center for Biotechnology Information (NCBI) Conserved Domain Database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and the SMART database (http://smart.embl.de/) were performed to verify the candidate BnGSK3s (Lu et al., 2020; Letunic et al., 2021). Chromosomal locations of the candidate BnGSK3s were visualized via MapGene2Chromosome V2 (MG2C, http://mg2c.iask.in/mg2c_v2.0/).
The sequences of BnaGSK3s were downloaded from BRAD and the gene structures were displayed by Tbtools. The conserved motifs were analyzed by Multiple Expectation Maximization for Motif Elicitation (MEME, http://meme-suite.org) (Bailey et al., 2015; Chen et al., 2020).
The alignment of the amino acid sequences of AtGSK3s and BnGSK3s was performed by ClustalW (Larkin et al., 2007). Phylogenetic tree was constructed and visualized using the neighbor-joining (NJ) method in MEGA11 software with 1,000 bootstrap replications (Tamura et al., 2021), and visualized by Evolview7 software (He et al., 2016). The syntenic analysis of GSK3s between AtGSK3s and BnGSK3s were obtained from the BRAD database.
The RNA-seq data generated from 13 tissues of ZS11, including roots, SAM, stems, leaves, buds, siliques, stamens, pistils, blossomy petals, wilting petals, sepals, ovules, and pericarps, was previously published in our lab (Sequence Read Archive accession: PRJNA474576 in NCBI and CNP0001630 in China National GeneBank DataBase) were used for the expression pattern analysis of the BnGSK3s family (Li et al., 2019b; Dong et al., 2021). FastPure Plant Total RNA Isolation Kit RC401 (Vazyme) was used to extracted total RNA from three biological replicates of different tissues of ZS11, including the roots, SAM, stems, leaves, buds, and siliques using the. First-strand cDNA was generated using a HiScript III 1st Strand cDNA Synthesis Kit (+gDNA wiper) R312 (Vazyme). Quantitative real-time PCR (qRT-PCR) was performed according to a previously described protocol, and the BnActin gene was used as an internal control to quantify the relative expression levels of target genes (Zhao et al., 2021). Gene-specific primers for BnGSK3s used for qRT-PCR were obtained from the qPrimerDB qPCR Primer Database (Lu et al., 2018) and the corresponding sequences were listed in Supplementary Table 4. The heatmap is illustrated using OmicShare Tools (https://www.omicshare.com/tools/).
Publicly available datasets were analyzed in this study. This data can be found here: https://bnaomics.ocri-genomics.net/tools/jb-dev/?data=data%2FBna_darmor_v4.1.
CZ, MX, and XC designed this study and provided the funding. SL supervised the study. CZ and MX performed experiments and wrote the manuscript. LY, XC, MT, YX, and LL provided the plant materials and collected the data. LY assisted in data analysis. LY and MX revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (32101813, 32070217), Central Public-interest Scientific Institution Basal Research Fund (CAAS-OCRI-XKPY-202104), China Agriculture Research System of MOF and MARA (CARS-12), and the Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2013-OCRI). Precursor projects of Guizhou province for biological breeding supporting by science and technology in 2022 (Fine identification and evaluation of crop germplasm resources). LY was supported by China Scholarship Council (201903250085).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1061196/full#supplementary-material
Supplementary Figure 1 | Phenotype distribution and correlation of plant height in two consecutive years in B. napus. (A) Phenotype distribution of plant height in 2017-2018, 2018-2019, and BLUP. (B) Correlation of plant height between 2017-2018 and 2018-2019.
Supplementary Figure 2 | Population structure of 230 B. napus accessions. (A) Cross−validation error under different K values. (B) Model-based population structure under K = 9. The y axis represents clusters memberships and the x axis represents the 230 B. napus accessions. (C) Relative kinship of 230 rapeseed accessions.
Supplementary Figure 3 | Quantile-quantile plots of GWAS in two consecutive years under three models.
Alexander, D. H., Novembre, J., Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Anne, P., Azzopardi, M., Gissot, L., Beaubiat, S., Hématy, K., Palauqui, J. C. (2015). OCTOPUS negatively regulates BIN2 to control phloem differentiation in Arabidopsis thaliana. Curr. Biol. 25, 2584–2590. doi: 10.1016/j.cub.2015.08.033
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Buerstmayr, M., Lemmens, M., Steiner, B., Buerstmayr, H. (2011). Advanced backcross QTL mapping of resistance to fusarium head blight and plant morphological traits in a triticum macha x t. aestivum population. Theor. Appl. Genet. 123, 293–306. doi: 10.1007/s00122-011-1584-x
Cai, D., Xiao, Y., Yang, W., Ye, W., Wang, B., Younas, M., et al. (2014). Association mapping of six yield-related traits in rapeseed (Brassica napus l.). Theor. Appl. Genet. 127, 85–96. doi: 10.1007/s00122-013-2203-9
Chai, L., Xin, M., Dong, C., Chen, Z., Zhai, H., Zhuang, J., et al. (2022). A natural variation in ribonuclease h-like gene underlies Rht8 to confer “Green revolution” trait in wheat. Mol. Plant 15, 377–380. doi: 10.1016/j.molp.2022.01.013
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A. P., Tang, H., Wang, X., et al. (2014). Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, Y., Zhu, W., Chen, Y., Ito, S., Asami, T., Wang, X. (2014). Brassinosteroids control root epidermal cell fate via direct regulation of a MYB-bHLH-WD40 complex by GSK3-like kinases. Elife 3, e02525. doi: 10.7554/eLife.02525.022
Chen, H., Wang, T., He, X., Cai, X., Lin, R., Liang, J., et al. (2021a). BRAD V3.0: an upgraded brassicaceae database. Nucleic Acids Res. 50, D1432–D1441. doi: 10.1093/nar/gkab1057
Chen, L., Yang, H., Fang, Y., Guo, W., Chen, H., Zhang, X., et al. (2021b). Overexpression of GmMYB14 improves high-density yield and drought tolerance of soybean through regulating plant architecture mediated by the brassinosteroid pathway. Plant Biotechnol. J. 19, 702–716. doi: 10.1111/pbi.13496
Cho, H., Ryu, H., Rho, S., Hill, K., Smith, S., Audenaert, D., et al. (2014). A secreted peptide acts on BIN2-mediated phosphorylation of ARFs to potentiate auxin response during lateral root development. Nat. Cell Biol. 16, 66–76. doi: 10.1038/ncb2893
Chu, C. G., Xu, S. S., Friesen, T. L., Faris, J. D. (2008). Whole genome mapping in a wheat doubled haploid population using SSRs and TRAPs and the identification of QTL for agronomic traits. Mol. Breed. 22, 251–266. doi: 10.1007/s11032-008-9171-9
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., Depristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Ding, L. N., Li, M., Guo, X. J., Tang, M. Q., Cao, J., Wang, Z., et al. (2020). Arabidopsis GDSL1 overexpression enhances rapeseed sclerotinia sclerotiorum resistance and the functional identification of its homolog in Brassica napus. Plant Biotechnol. J. 18, 1255–1270. doi: 10.1111/pbi.13289
Dong, Z., Alam, M. K., Xie, M., Yang, L., Liu, J., Helal, M. M. U., et al. (2021). Mapping of a major QTL controlling plant height using a high-density genetic map and QTL-seq methods based on whole-genome resequencing in Brassica napus. G3-GENES Genom. Genet. 11, jkab118. doi: 10.1093/g3journal/jkab118
Dong, S.-S., He, W.-M., Ji, J.-J., Zhang, C., Guo, Y., Yang, T.-L. (2020). LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 22, bbaa227. doi: 10.1093/bib/bbaa227
Fang, Z., Ji, Y., Hu, J., Guo, R., Sun, S., Wang, X. (2020). Strigolactones and brassinosteroids antagonistically regulate the stability of the D53-OsBZR1 complex to determine FC1 expression in rice tillering. Mol. Plant 13, 586–597. doi: 10.1016/j.molp.2019.12.005
Fang, N., Xu, R., Huang, L., Zhang, B., Duan, P., Li, N., et al. (2016). SMALL GRAIN 11 controls grain size, grain number and grain yield in rice. Rice 9, 64. doi: 10.1186/s12284-016-0136-z
Ferrero-Serrano, Á., Su, Z., Assmann, S. M. (2018). Illuminating the role of the gα heterotrimeric G protein subunit, RGA1, in regulating photoprotection and photoavoidance in rice. Plant Cell Environ. 41, 451–468. doi: 10.1111/pce.13113
Freed, D., Aldana, R., Weber, J. A., Edwards, J. S. (2017). The sentieon genomics tools - a fast and accurate solution to variant calling from next-generation sequence data. BioRxiv 115717. doi: 10.1101/115717
Guo, W., Chen, L., Herrera-Estrella, L., Cao, D., Tran, L.-S. P. (2020). Altering plant architecture to improve performance and resistance. Trends Plant Sci. 25, 1154–1170. doi: 10.1016/j.tplants.2020.05.009
Guo, Z., Liu, X., Zhang, B., Yuan, X., Xing, Y., Liu, H., et al. (2021). Genetic analyses of lodging resistance and yield provide insights into post-Green-Revolution breeding in rice. Plant Biotechnol. J. 19, 814–829. doi: 10.1111/pbi.13509
Guo, J., Shi, W., Zhang, Z., Cheng, J., Sun, D., Yu, J., et al. (2018). Association of yield-related traits in founder genotypes and derivatives of common wheat (Triticum aestivum l.). BMC Plant Biol. 18, 38. doi: 10.1186/s12870-018-1234-4
Hedden, P. (2003). The genes of the green revolution. Trends Genet. 19, 5–9. doi: 10.1016/S0168-9525(02)00009-4
He, Z., Zhang, H., Gao, S., Lercher, M. J., Chen, W. H., Hu, S. (2016). Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 44, W236–W241. doi: 10.1093/nar/gkw370
Hirano, K., Asano, K., Tsuji, H., Kawamura, M., Mori, H., Kitano, H., et al. (2010). Characterization of the molecular mechanism underlying gibberellin perception complex formation in rice. Plant Cell 22, 2680–2696. doi: 10.1105/tpc.110.075549
Huang, M., Liu, X., Zhou, Y., Summers, R. M., Zhang, Z. (2019). BLINK: a package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 8, giy154. doi: 10.1093/gigascience/giy154
Islam, N., Evans, E. J. (1994). Influence of lodging and nitrogen rate on the yield and yield attributes of oilseed rape (Brassica napus l.). Theor. Appl. Genet. 88, 530–534. doi: 10.1007/BF01240914
Jiang, H., Tang, B., Xie, Z., Nolan, T., Ye, H., Song, G. Y., et al. (2019). GSK3-like kinase BIN2 phosphorylates RD26 to potentiate drought signaling in Arabidopsis. Plant J. 100, 923–937. doi: 10.1111/tpj.14484
Jiao, Y., Wang, Y., Xue, D., Wang, J., Yan, M., Liu, G., et al. (2010). Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 42, 541–544. doi: 10.1038/ng.591
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Letunic, I., Khedkar, S., Bork, P. (2021). SMART: recent updates, new developments and status in 2020. Nucleic. Acids Res. 49, D458–D460. doi: 10.1093/nar/gkaa937
Li, Y., Dong, C., Hu, M., Bai, Z., Tong, C., Zuo, R., et al. (2019b). Identification of flower-specific promoters through comparative transcriptome analysis in Brassica napus. Int. J. Mol. Sci. 20, 5949. doi: 10.3390/ijms20235949
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, H., Li, J., Song, J., Zhao, B., Guo, C., Wang, B., et al. (2019a). An auxin signaling gene BnaA3.IAA7 contributes to improved plant architecture and yield heterosis in rapeseed. New Phytol. 222, 837–851. doi: 10.1111/nph.15632
Li, J., Terzaghi, W., Gong, Y., Li, C., Ling, J. J., Fan, Y., et al. (2020b). Modulation of BIN2 kinase activity by HY5 controls hypocotyl elongation in the light. Nat. Commun. 11, 1592. doi: 10.1038/s41467-020-15394-7
Liu, S., Raman, H., Xiang, Y., Zhao, C., Huang, J., Zhang, Y. (2022). De novo design of future rapeseed crops: Challenges and opportunities. Crop J. 10, 587–596. doi: 10.1016/j.cj.2022.05.003
Liu, C., Wang, J., Huang, T., Wang, F., Yuan, F., Cheng, X., et al. (2010). A missense mutation in the VHYNP motif of a DELLA protein causes a semi-dwarf mutant phenotype in Brassica napus. Theor. Appl. Genet. 121, 249–258. doi: 10.1007/s00122-010-1306-9
Liu, S., Zhang, M., Feng, F., Tian, Z. (2020). Toward a “Green revolution” for soybean. Mol. Plant 13, 688–697. doi: 10.1016/j.molp.2020.03.002
Li, H., Wang, L., Liu, M., Dong, Z., Li, Q., Fei, S., et al. (2020a). Maize plant architecture is regulated by the ethylene biosynthetic gene ZmACS7. Plant Physiol. 183, 1184–1199. doi: 10.1104/pp.19.01421
Lu, K., Li, T., He, J., Chang, W., Zhang, R., Liu, M., et al. (2018). qPrimerDB: a thermodynamics-based gene-specific qPCR primer database for 147 organisms. Nucleic Acids Res. 46, D1229–D1236. doi: 10.1093/nar/gkx725
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Mao, J., Li, J. (2020). Regulation of three key kinases of brassinosteroid signaling pathway. Int. J. Mol. Sci. 21, 4340. doi: 10.3390/ijms21124340
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121. doi: 10.1093/nar/gkt263
Miura, K., Ikeda, M., Matsubara, A., Song, X. J., Ito, M., Asano, K., et al. (2010). OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 42, 545–549. doi: 10.1038/ng.592
Monna, L., Kitazawa, N., Yoshino, R., Suzuki, J., Masuda, H., Maehara, Y., et al. (2002). Positional cloning of rice semidwarfing gene, sd-1: Rice “Green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 9, 11. doi: 10.1093/dnares/9.1.11
Pearce, S. (2021). Towards the replacement of wheat ‘Green revolution’ genes. J. Exp. Bot. 72, 157–160. doi: 10.1093/jxb/eraa494
Peng, J., Richards, D. E., Hartley, N. M., Murphy, G. P., Devos, K. M., Flintham, J. E., et al. (1999). ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 400, 256. doi: 10.1038/22307
Phillips, K. A., Skirpan, A. L., Liu, X., Christensen, A., Slewinski, T. L., Hudson, C., et al. (2011). Vanishing tassel2 encodes a grass-specific tryptophan aminotransferase required for vegetative and reproductive development in maize. Plant Cell 23, 550–566. doi: 10.1105/tpc.110.075267
Sasaki, A., Ashikari, M., Ueguchitanaka, M., Itoh, H., Nishimura, A., Swapan, D., et al. (2002). Green revolution: a mutant gibberellin-synthesis gene in rice. Nature 416, 701–702. doi: 10.1038/416701a
Spielmeyer, W., Ellis, M. H., Chandler, P. M. (2002). Semidwarf (sd-1), “green revolution” rice, contains a defective gibberellin 20-oxidase gene. P. Natl. Acad. Sci. U.S.A. 99, 9043–9048. doi: 10.1073/pnas.132266399
Sun, S., Wang, L., Mao, H., Shao, L., Li, X., Xiao, J., et al. (2018). A G-protein pathway determines grain size in rice. Nat. Commun. 9, 851. doi: 10.1038/s41467-018-03141-y
Sun, C., Wang, B., Yan, L., Hu, K., Liu, S., Zhou, Y., et al. (2016). Genome-wide association study provides insight into the genetic control of plant height in rapeseed (Brassica napus l.). Front. Plant Sci. 7, 1102. doi: 10.3389/fpls.2016.01102
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tang, M. Q. (2019). Population genome variations and subgenome asymmetry in brassica napus l (Wuhan, China: Huazhong Agricultural University), D50–D59.
Tan, Z., Xie, Z., Dai, L., Zhang, Y., Zhao, H., Tang, S., et al. (2022). Genome- and transcriptome-wide association studies reveal the genetic basis and the breeding history of seed glucosinolate content in Brassica napus. Plant Biotechnol. J. 20, 211–225. doi: 10.1111/pbi.13707
Teichmann, T., Muhr, M. (2015). Shaping plant architecture. Front. Plant Sci. 6, 233. doi: 10.3389/fpls.2015.00233
Wang, Y., Chen, W., Chu, P., Wan, S., Yang, M., Wang, M., et al. (2016a). Mapping a major QTL responsible for dwarf architecture in Brassica napus using a single-nucleotide polymorphism marker approach. BMC Plant Biol. 16, 178. doi: 10.1186/s12870-016-0865-6
Wang, Y., He, J., Yang, L., Wang, Y., Chen, W., Wan, S., et al. (2016b). Fine mapping of a major locus controlling plant height using a high-density single-nucleotide polymorphism map in brassica napus. Theor. Appl. Genet. 129, 1479–1491. doi: 10.1007/s00122-016-2718-y
Wang, Y., Li, J. (2008). Molecular basis of plant architecture. Annu. Rev. Plant Biol. 59, 253–279. doi: 10.1146/annurev.arplant.59.032607.092902
Wang, Y., Shang, L., Yu, H., Zeng, L., Hu, J., Ni, S., et al. (2020b). A strigolactone biosynthesis gene contributed to the green revolution in rice. Mol. Plant 13, 923–932. doi: 10.1016/j.molp.2020.03.009
Wang, H., Tang, J., Liu, J., Hu, J., Liu, J., Chen, Y., et al. (2018). Abscisic acid signaling inhibits brassinosteroid signaling through dampening the dephosphorylation of BIN2 by ABI1 and ABI2. Mol. Plant 11, 315–325. doi: 10.1016/j.molp.2017.12.013
Wang, X., Zheng, M., Liu, H., Zhang, L., Chen, F., Zhang, W., et al. (2020a). Fine-mapping and transcriptome analysis of a candidate gene controlling plant height in Brassica napus l. Biotechnol. Biofuels. 13, 42. doi: 10.1186/s13068-020-01687-y
Xiong, H., Zhou, C., Fu, M., Guo, H., Xie, Y., Zhao, L., et al. (2022). Cloning and functional characterization of Rht8, a “Green revolution” replacement gene in wheat. Mol. Plant 15, 373–376. doi: 10.1016/j.molp.2022.01.014
Xu, J., Ni, Z., Chen, F., Fu, X., Yu, F. (2022). Integrated linkage mapping and genome-wide association study to dissect the genetic basis of zinc deficiency tolerance in maize at seedling stage. Crop J. doi: 10.1016/j.cj.2022.05.004
Yang, M., He, J., Wan, S., Li, W., Chen, W., Wang, Y., et al. (2021). Fine mapping of the BnaC04.BIL1 gene controlling plant height in Brassica napus L. BMC Plant Biol. 21, 359. doi: 10.1186/s12870-021-03137-9
Ye, H., Feng, J., Zhang, L., Zhang, J., Mispan, M. S., Cao, Z., et al. (2015). Map-based cloning of seed Dormancy1-2 identified a gibberellin synthesis gene regulating the development of endosperm-imposed dormancy in rice. Plant Physiol. 169, 2152–2165. doi: 10.1104/pp.15.01202
Yu, J., Pressoir, G., Briggs, W. H., Vroh Bi, I., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Zhang, C., Dong, S.-S., Xu, J.-Y., He, W.-M., Yang, T.-L. (2018). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. doi: 10.1093/bioinformatics/bty875
Zhao, B., Li, H., Li, J., Wang, B., Dai, C., Wang, J., et al. (2017). Brassica napus DS-3, encoding a DELLA protein, negatively regulates stem elongation through gibberellin signaling pathway. Theor. Appl. Genet. 130, 727–741. doi: 10.1007/s00122-016-2846-4
Zhao, C., Safdar, L. B., Xie, M., Shi, M., Dong, Z., Yang, L., et al. (2021). Mutation of the PHYTOENE DESATURASE 3 gene causes yellowish-white petals in Brassica napus. Crop J. 9, 1124–1134. doi: 10.1016/j.cj.2020.10.012
Zhao, B., Wang, B., Li, Z., Guo, T., Zhao, J., Guan, Z., et al. (2019). Identification and characterization of a new dwarf locus DS-4 encoding an Aux/IAA7 protein in brassica napus. Theor. Appl. Genet. 132, 1435–1449. doi: 10.1007/s00122-019-03290-8
Zhao, C., Xie, M., Liang, L., Yang, L., Han, H., Qin, X., et al. (2022). Genome-wide association analysis combined with quantitative trait loci mapping and dynamic transcriptome unveil the genetic control of seed oil content in Brassica napus l. Front. Plant Sci. 13, 929197. doi: 10.3389/fpls.2022.929197
Zheng, M., Hu, M., Yang, H., Tang, M., Zhang, L., Liu, H., et al. (2019). Three BnaIAA7 homologs are involved in auxin/brassinosteroid-mediated plant morphogenesis in rapeseed (Brassica napus l.). Plant Cell Rep. 38, 883–897. doi: 10.1007/s00299-019-02410-4
Keywords: plant height, genome-wide association study (GWAS), rapeseed (B. napus L.), RNA sequencing (RNA-Seq), GSK3 gene family
Citation: Zhao C, Yang L, Tang M, Liu L, Huang J, Tong C, Xiang Y, Liu S, Cheng X and Xie M (2022) Genome-wide association study reveals a GLYCOGEN SYNTHASE KINASE 3 gene regulating plant height in Brassica napus. Front. Plant Sci. 13:1061196. doi: 10.3389/fpls.2022.1061196
Received: 04 October 2022; Accepted: 20 October 2022;
Published: 02 November 2022.
Edited by:
Guo-Fei Tan, Guizhou Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Shengwu Hu, Northwest A&F University, ChinaCopyright © 2022 Zhao, Yang, Tang, Liu, Huang, Tong, Xiang, Liu, Cheng and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaohui Cheng, Y2hlbmd4aWFvaHVpQGNhYXMuY24=; Meili Xie, eGllbWVpbGkwMTAxQDE2My5jb20=
†These authors have contributed equally to this work
‡ORCID ID: Chuanji Zhao, orcid.org/0000-0002-2100-2510
Li Yang, orcid.org/0000-0002-9360-4617
Minqiang Tang, orcid.org/0000-0002-8590-1814
Meili Xie, orcid.org/0000-0003-1679-4012
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.