 Chung-Shien Wu
Chung-Shien Wu Chung-I. Chen2
Chung-I. Chen2 Shu-Miaw Chaw
Shu-Miaw Chaw- 1Biodiversity Research Center, Academia Sinica, Taipei, Taiwan
- 2Department of Forestry, National Pingtung University of Science and Technology, Pingtung, Taiwan
Convolvulaceae, the morning glories or bindweeds, is a large family containing species of economic value, including crops, traditional medicines, ornamentals, and vegetables. However, not only are the phylogenetic relationships within this group still debated at the intertribal and intergeneric levels, but also plastid genome (plastome) complexity within Convolvulaceae is not well surveyed. We gathered 78 plastomes representing 17 genera across nine of the 12 Convolvulaceae tribes. Our plastid phylogenomic trees confirm the monophyly of Convolvulaceae, place the genus Jacquemontia within the subfamily Dicranostyloideae, and suggest that the tribe Merremieae is paraphyletic. In contrast, positions of the two genera Cuscuta and Erycibe are uncertain as the bootstrap support of the branches leading to them is moderate to weak. We show that nucleotide substitution rates are extremely variable among Convolvulaceae taxa and likely responsible for the topological uncertainty. Numerous plastomic rearrangements are detected in Convolvulaceae, including inversions, duplications, contraction and expansion of inverted repeats (IRs), and losses of genes and introns. Moreover, integrated foreign DNA of mitochondrial origin was found in the Jacquemontia plastome, adding a rare example of gene transfer from mitochondria to plastids in angiosperms. In the IR of Dichondra, we discovered an extra copy of rpl16 containing a direct repeat of ca. 200 bp long. This repeat was experimentally demonstrated to trigger effective homologous recombination, resulting in the coexistence of intron-containing and -lacking rpl16 duplicates. Therefore, we propose a hypothetical model to interpret intron loss accompanied by invasion of direct repeats at appropriate positions. Our model complements the intron loss model driven by retroprocessing when genes have lost introns but contain abundant RNA editing sites adjacent to former splicing sites.
Introduction
Convolvulaceae, commonly known as the morning glories or bindweeds, is a large family sister to Solanaceae (Stefanović et al., 2002) in the order Solanales (AGP IV, 2016). Morning glories comprise approximately 2,000 species in 60 genera (Simões et al., 2022), with a diverse range of morphological characteristics from herbs, shrubs, vines, to parasites Cuscuta (dodders). They mostly inhabit tropical areas and include many economically valuable crop (e.g., sweet potato), traditional medicine (e.g., dodders), ornamental (e.g., morning glory), and vegetable (e.g., water spinach) species. Using a few plastid and nuclear markers, early molecular phylogenetic studies classified 12 tribes in Convolvulaceae (Stefanović et al., 2002; Stefanovi´c et al., 2003), but controversies remain at the intertribal and intergeneric levels even when organellar (Lin et al., 2022) and nuclear (Simões et al., 2022) genomic data were used for comparative phylogenetic analyses.
Plastids are cellular organelles and have their own genomes, called plastomes. In general, seed plant plastomes are structurally conserved and contain two inverted repeats (IRs) separating the sequences into a large single copy (LSC) region and a small single copy (SSC) one. Integration of foreign DNA into plastomes was initially thought impossible due to the absence of active DNA import systems in plastids (Smith, 2011). However, plastid mitochondrial (mt) DNA-derived sequences (PTMTs) were later discovered in a handful of species (e.g., Iorizzo et al., 2012a; Iorizzo et al., 2012b; Straub et al., 2013; Ma et al., 2015; Rabah et al., 2017; Raman et al., 2019; Raman et al., 2021), suggesting that acquisition of foreign DNA that shapes plastome complexity is possible, although the mechanism is unclear. Plastomes also undergo rearrangements, including inversions and changes of gene content through deletions and duplications (see review in Wicke et al., 2011). Gene content changes due to IR contraction and expansion frequently occurred during plant evolution (Wang et al., 2008; Zhu et al., 2016). A comparison of plastid transcriptomes has shed light on the effects of inversions on gene expression in conifer plastids (Wu et al., 2021). However, to date there is little conclusive evidence as to the consequences of plastid gene content changes.
Plastids contain several group II introns. Unlike their eubacterial counterparts/ancestors, modern plastid group II introns have lost their mobility and rely on host-encoded factors for splicing (Lambowitz and Zimmerly, 2011). Group II introns are generally 400−800 bp long and possess six major domains (I−VI domains) that form a conserved tertiary structure and an active site for splicing (Kelchner, 2002; Lambowitz and Zimmerly, 2011). Precise deletions of introns have been proposed to be a consequence of retroprocessing operated in plastids (Downie et al., 1991), while localized retroprocessing appears to be a widespread mechanism underlying intron loss in plant mitochondria (Cuenca et al., 2016).
Recent studies of autotrophic plastomes in Convolvulaceae were mainly confined to the genus Ipomoea (Eserman et al., 2014; Park et al., 2018; Sun et al., 2019; Laux et al., 2022). However, Lin et al. (2022) compared plastomes across eight Convolvulaceous tribes and found several unusual features, such as atypical IRs, gene and intron losses, and insertions of foreign DNA of unknown origin. These studies suggest that Convolvulaceous plastomes are labile and their complexity might be underestimated. To further explore this variation, we gathered 78 plastomes representing 17 genera from nine of the 12 tribes in the morning-glory family, including Jacquemontia, considered to have the greatest sequence divergence among autotrophic Convolvulaceae (Stefanović et al., 2002). These data were used to construct phylogenetic trees, estimate nucleotide substitution rates, and characterize plastomic rearrangements. Furthermore, we report the first PTMT case in Convolvulaceae and propose a repeat-mediated model to interpret intron loss without retroprocessing.
Materials and methods
Taxon sampling, DNA and RNA extraction, and sequencing
We collected 19 Convolvulaceae taxa. Their voucher information is shown in Table S1. Total DNA was extracted from fresh leaves using the CTAB method described in Stewart and Via (1993). The extracted DNA was used for library construction using Celero™ DNA-Seq Library Preparation Kits. Total RNA was extracted from D. micrantha leaves using Plant Total RNA Purification Kits (GeneMark, Taiwan). After DNase I treatment, the NuQuant® Universal Plus mRNA-Seq Kit, designed for poly (A) selection, was used to prepare cDNA libraries. We used the Illumina HiSeq 4000 platform at Genomics BioSci & Tech (New Taipei City, Taiwan) to obtain approximately 10 Gb of paired-end (PE) reads per library.
Genome assembly and annotation
Adaptors and low-quality reads were trimmed using Trimmomatic v0.36 (Bolger et al., 2014) with default parameters. Plastomes were assembled using NOVOPlasty v4.3 (Dierckxsens et al., 2017) and GetOrganelle v1.7.6.1 (Jin et al., 2020) with our own databases containing available Ipomoea plastomes. We merged the results generated by the two assemblers and did base-scale corrections using Pilon v1.24 (Walker et al., 2014). The assembled scaffolds were annotated in Geneious Prime (https://www.geneious.com/ ), with the I. aquatica plastome (NC056300) as the reference. Protein-coding genes and tRNAs were further confirmed by aligning them to their orthologs and tRNAscan-SE v2.0 (Chan et al., 2021), respectively.
Sequence alignments and tree construction
Sequences of 78 plastid protein-coding genes were retrieved from our assemblies and other publicly available plastomes (Table S2). Sequence alignments were conducted using MUSCLE (Edgar, 2004) implemented in MEGA 7 (Kumar et al., 2016). Concatenations of the alignments were performed in Geneious Prime, yielding a supermatrix of 77,232 bp for downstream analyses. We used PartitionFinder v2.1.1 (Lanfear et al., 2017) to search the best scheme of data partitions under the Bayesian information criterion (BIC). This scheme was subsequently incorporated in building ML and BI trees using IQ-tree v2.2.0 (Minh et al., 2020) and MrBayes v3.2.7 (Ronquist et al., 2012), respectively. Supporting values for nodes of the ML tree were estimated from 5,000 ultrafast bootstraps. Two independent runs were conducted for 2 × 107 generations and one tree per 1,000 generations was sampled for BI tree construction. The initial 25% of the sampled trees were discarded as burin-in and Tracer v1.7.1 (Rambaut et al., 2018) was used to check if the effective sample size (ESS) exceeded 200 in all parameters.
Read mapping
DNA and RNA mapping analyses were performed in Geneious Prime, with the option of “Map to Reference” and mappers = “Geneious” for DNA-seq and “Geneious RNA” for RNA-seq reads. Gaps were not allowed during mapping.
Calculation of absolute synonymous (RS) and nonsynonymous (RN) substitution rates
To facilitate calculations, the supermatrix mentioned above was trimmed to contain only 68 accessions of unique species. This trimmed matrix was then used to estimate synonymous (dS) and nonsynonymous (dN) trees using Codeml of the Paml 4.9j package (Yang, 2007) under a branch model and codon frequency = F3 × 4. Molecular dating was conducted using the Paml MCMCTree module. The constrained ages were based on the settings described in Eserman et al. (2014) with a few modifications: the crown group for Solanaceae (23.0–33.9), for Convolvulaceae (47.8–56.0), for Ipomoeeae s.l. + Merremia (41.2−47.8 MYA). Markov chain Monte Carlo was evaluated for 2 × 106 generations under a HKY85 + G5 model. We set the sampling frequency = 10 and burnin = 25%. Terminal branches leading to species were used to calculate species RS and RN from dividing dS and dN branch lengths by the duration of species evolution.
PCR assays
We verified the existence of mtDNA insertions using PCR and two specific primer sets (Set 1: F1, 5’-ACTTCT GGTTCCGGCGAACG-3’and R1, 5’-GCTAGAATTAGCATTATGAGCTGCTCG-3’; and Set 2: F2, 5’-CAATTCATTCGTAGTAGGATATTGAAACCTC-3’and R2, 5’-AAGCGCATAATTGGTTGAAGATCAC-3’). Targeted fragments were amplified in a 20 μl PCR reaction: 9.6 μl water, 3.2 μl dNTP mixture (2.5 mM each), 1 μl 10 μM of each primer, 0.2 μl TaKaRa LA Taq®, 2 μl 10X LA PCR buffer II, 2 μl 25 mM MgCl2, and 1 μl of genomic DNA (~25 ng). Amplification conditions were as follows: initial denaturation at 94°C for 3 min., followed by 30 cycles of 98°C for 15 sec., 58°C for 20 sec., and 68°C for 5 min. The PCR was finished with a final step at 72°C for 10.0 min. We also designed PCR primers to verify the coexistence of rpl16-LSC (rpl16-LSC-F: 5’-GAGAGTTTCTTCTCATCCAGCTCCTC-3’ and rpl16-LSC-R: 5’- CGGAACCTGTGAATGCAAAAGATC-3’), rpl16-IR (rpl16-IR-F: 5’- GATTAGGGTAAACCAGACCCATTCATAGT-3’ and rpl16-IR-R: 5’-ATTCTTCCTCTATGTTGTTTACGGAATCTG-3’), and rpl16-IR* (rpl16-IR*-F: 5’-CTTTTGATATAATTATCATTGCTATGCTTAGTCC-3’ and rpl16-IR*-R: 5’-AATTGAGTTCGTATAGGCATTTTGGATG-3’) copies. PCR conditions were similar to those used in examining mtDNA insertions, but the number of PCR cycles was gradually increased from 10 to 30. Electrophoresis was carried out for an hour at 50 V on 0.8% agarose TAE gels and PCR amplicons were visualized in a Quantum CX5 gel image system.
Results
Plastid phylogenomics of Convolvulaceae
The 19 newly sequenced plastomes are circular molecules. They exhibit typical quadripartite structure of a large single-copy (LSC) region, a small single-copy (SSC) region, and a pair of inverted repeats (IRs). Their sizes range from 152,365 to 165,459 bp and GC content from 37.3 to 38.8%. Using Solanaceae as the outgroup, our ML and BI trees show nearly identical topologies (Figure 1; Figure S1). Both trees suggest that the tribes Cardiochlamyeae and Erycibeae constitute the earliest diverged clade, followed by Cuscuteae, a solo parasitic tribe in Convolvulaceae. The tribes Cresseae s.l., Dichondreae s.l., and Jacquemontieae form a monophyletic clade sister to the clade comprising the tribes Convolvuleae, Ipomoeeae s.l., and Merremieae. Notably, our trees strongly support (BS = 100% and PP = 1) Merremieae as a paraphyletic tribe because the position of Merremia is closer to Ipomoeeae s.l. than to other Merremieae genera (Figure 1).
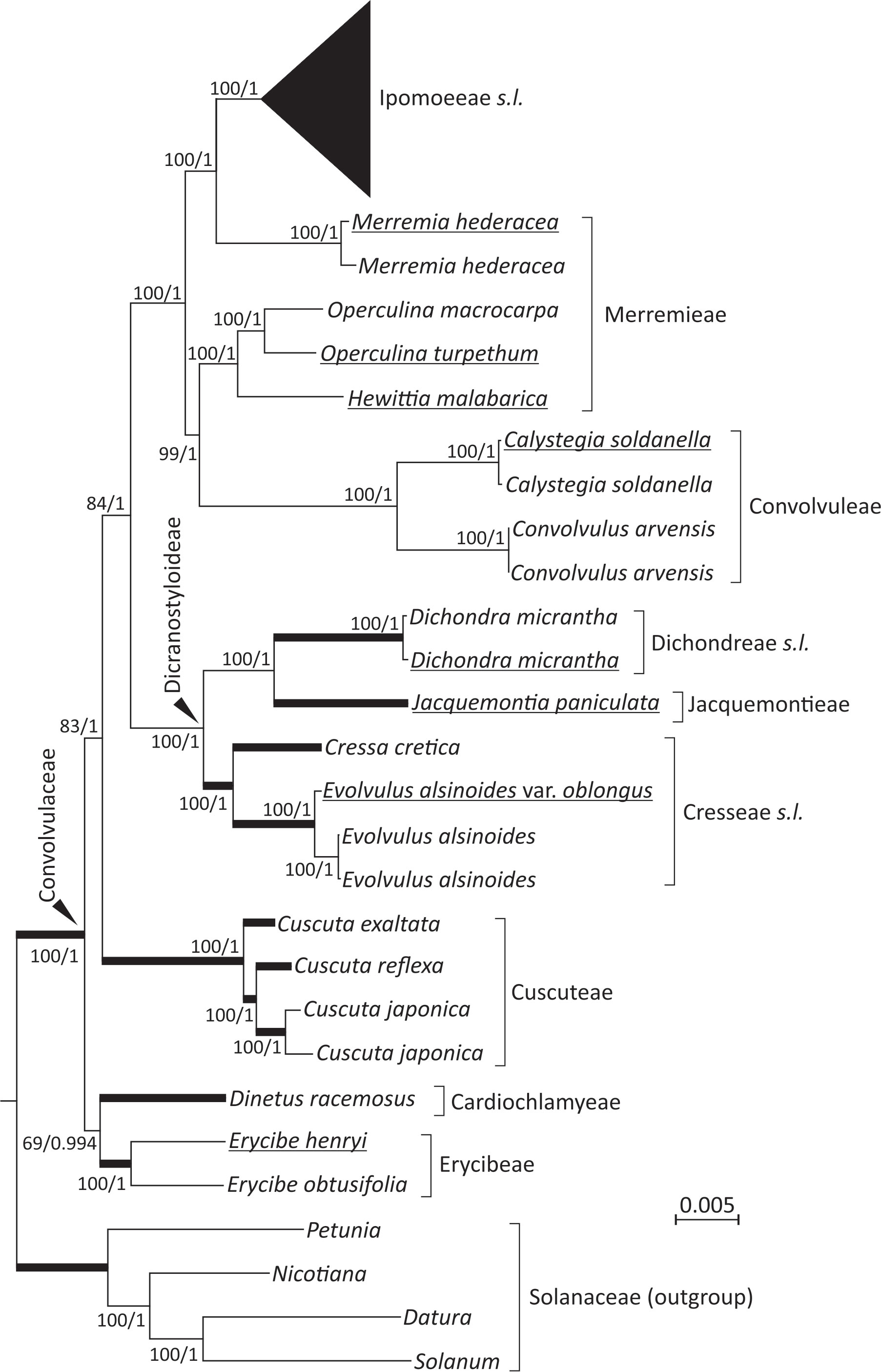
Figure 1 Plastid phylogenomics of representative Convolvulaceous genera and tribes. The topology is based on a ML tree inferred from a concatenation of 78 plastid protein-coding genes. Values along branches reflect bootstrap support (%) for ML trees and posterior possibilities for Bayesian inference (BI) trees. Thick branches are shortened to 1/10 of their real lengths. Underlined taxa were sequenced in this study.
Within Ipomoeeae s.l., two separated clades are observed: Argyreiinae and Astripomoeinae, with the former containing the four genera Argyreia, Ipomoea, Stictocardia, and Turbina and the latter only Ipomoea (Figure S1). In the ML tree, the three I. aquatica accessions, including two cultivars, are monophyletic and sister to I. reptans with weak support (63%). Such relationships, however, are not present in the BI tree under the 50% majority rule (Figure S1). To date, I. reptans has been considered a synonym for I. aquatica. Here, we treat I. reptans as a separate species because of its semi-terrestrial habitat, purple stems, and corollaceous color differing from I. aquatica (Figure S1).
Extreme variability in plastid nucleotide substitution rates
To explicitly assess the variation in substitution rates across Convolvulaceae, synonymous (dS) and nonsynonymous (dN) trees were inferred from the concatenation of the 78 plastid genes. This concatenated gene set was also used to estimate divergence times among lineages. Our molecular dating suggests that Convolvulaceae split from Solanaceae at ca. 88.7 MYA and divergence of the Convolvulaceous tribes occurred in between 23.5 and 52.8 MYA (Figure S2). Figure 2 depicts absolute synonymous (RS) and nonsynonymous (RN) substitution rates that allow for rate comparisons on the same time scale. Notably, the RS and RN are strongly correlated across Convolvulaceae (R2 = 0.7395), suggesting their dependent evolution. We noticed that Ipomoeeae s.l., Merremieae, Convolvuleae, and Erycibe evolved at an equal rate but significantly slower than Dichondra, Jacquemontia, Cresseae s.l., Cuscuta, and Dinetus in RS (Mann Whitney test, P < 0.05). In particular, the mean RS in Cresseae s.l. is approximately seven times faster than in Ipomoeeae s.l. Significantly elevated RN is only detected in Cresseae s.l. and Cuscuta (Mann Whitney test, P < 0.05). Taken together, these results show that substitution rates at dS sites are highly variable across Convolvulaceae.
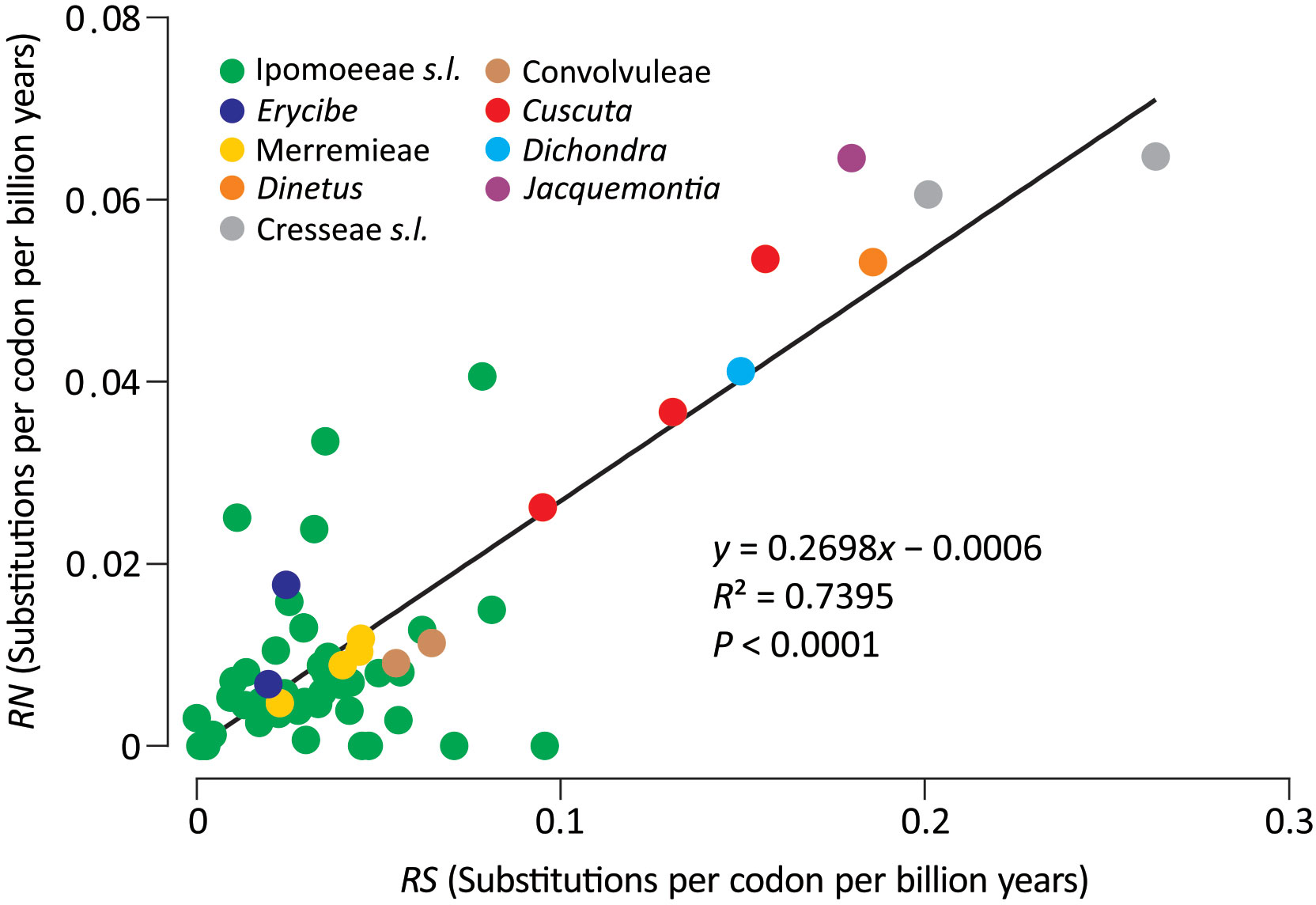
Figure 2 Comparisons of absolute synonymous (RS) versus nonsynonymous (RN) substitution rates across Convolvulaceae. The solid line is the regression between RN and RS.
Plastomic rearrangements and IR contraction/expansion
To sketch plastomic rearrangement profile across Convolvulaceae evolution, we labeled rearrangements in SC regions (hereafter called SC rearrangements) on the tree branches (Figure 3; left panel) and compared IR gene content among sampled taxa (Figure 3; right panel). SC Rearrangements in Cuscuta taxa are not included in the current study because they have been described previously (Braukmann et al., 2013; Banerjee and Stefanović, 2020). We discovered 13 SC rearrangements across the tree. All Convolvulaceous genera, including Cuscuta, lack the rpl2 intron and infA gene, and the latter is also absent in Solanaceae (Amiryousefi et al., 2018). This implicates two ancient loss events that occurred before (i.e., infA) and after (rpl2 intron) the split of Convolvulaceae from Solanaceae (Figure 3). In contrast, the remaining SC rearrangements are all genus specific. For instance, an inversion of the atpI−atpB region, duplication of psbT, and loss of rpl23 in Dinetus; integration of mtDNA between psaA and ycf3 in Jacquemontia; an inversion of the trnL−atpB region, loss of rpl23, and losses of rps16 and ycf3 introns in Dichondra; losses of rps16 and rpoC1 introns in Evolvulus; and loss of rpl23 in Operculina. Overall, our analyses suggest that rpl23 has been independently lost at least three times during Convolvulaceae evolution (Figure 3).
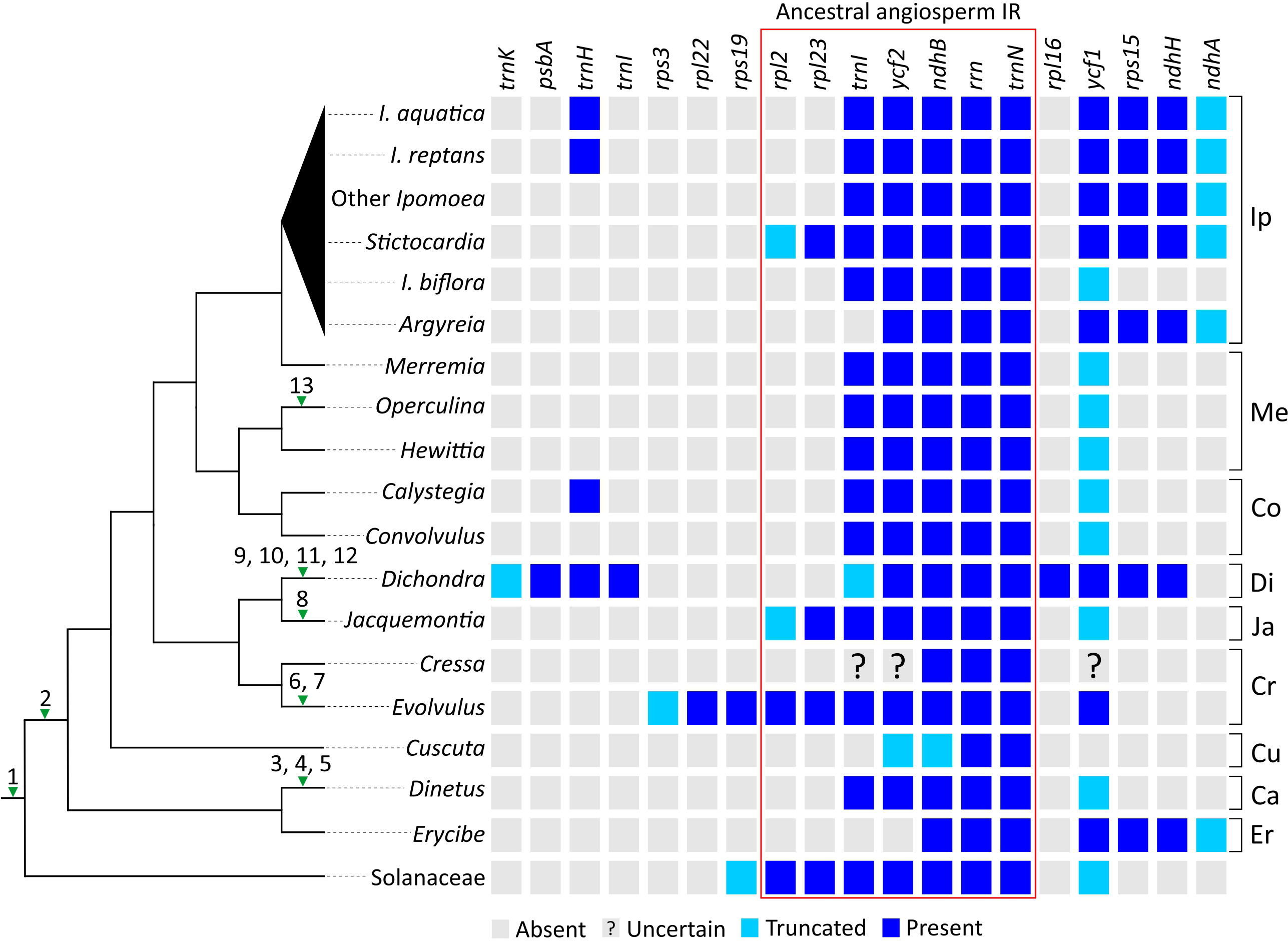
Figure 3 Evolution of plastomic rearrangements across Convolvulaceae. The right panel depicts presences/absences of genes in IRs. A “?” mark indicates that the gene state is uncertain due to poor sequence quality. Numerals on branches of the left panel tree denote specific rearrangements outside IRs. 1, loss of infA; 2, loss of the rpl2 intron; 3, inversion of the atpI−atpB region; 4, duplication of psbT; 5, loss of rpl23; 6, loss of the rps16 intron; 7, loss of the rpoC1 intron; 8, integration of PTMTs between psaA and ycf3; 9, inversion of the trnL−atpB region; 10, loss of rpl23; 11, loss of the rps16 intron; 12, loss of all introns from ycf3; 13, loss of rpl23. Ip, Ipomoeeae s.l.; Me, Merremieae; Co, Convolvuleae; Di, Dichondreae s.l.; Ja, Jacquemontieae; Cr, Cresseae s.l.; Cu, Cuscuteae; Ca, Cardiochlamyeae; Er, Erycibeae.
Using Solanaceae and ancestral angiosperm IRs (Zhu et al., 2016) as the references, we observed that IRs of most Convolvulaceous genera lack rpl2 and rpl23 (Figure 3). As a result, the most parsimonious scenario is that the common ancestor of Convolvulaceae experienced a contraction so that rpl2 and rpl23 were excluded from its IRs, followed by multiple rounds of lineage-specific expansion and contraction that shaped the IR diversity. For example, (1) Erycibe and Ipomoeeae s.l. have undergone a parallel expansion to include ycf1, rps15, ndhH, and partial ndhA in their IRs. This expansion is here characterized as a synapomorphic trait of Ipomoeeae s.l., despite a subsequent contraction removing rps15, ndhH, and partial ndhA from I. biflora’s IRs; (2) Dichondra has duplicated psbA, trnH, trnI, rpl16, ycf1, rps15, ndhH, and partial trnK in its IRs, manifesting a complex IR expansion history including genes originally located in the LSC and SSC regions; (3) IR expansions have caused duplications of rpl23, rpl2, rps19, rpl22, and partial rps3 in Evolvulus as well as rpl23 and partial rpl2 in Jacquemontia and Sticotocardia, whereas contractions have removed trnI and ycf2 from the Erycibe IRs as well as trnI from the Argyreia IRs; (4) Small IR expansions have led to parallel duplications of trnH in Calystegia, I. aquatica, and I. reptans.
Integration of mtDNA in the Jacquemontia plastome
We detected an unusual elongated intergenic spacer between psaA and ycf3 in the Jacquemontia plastome. This spacer is 9.1 kb long and approximately nine times longer than those (<1 kb) of other Convolvulaceous taxa. Furthermore, GC content of this intergenic spacer is elevated with cytosines and guanines accounting for 43.1% of all bases, in contrast to an average of 38.6% elsewhere (Figure 4A). Using this intergenic spacer sequence of Jacquemontia as the query, blast searches (NCBI nr database; access date: Jun/2022) yielded a mixture of fragments best matching mitogenomes of several taxa, including a basal angiosperm (Amborella) and other Convolvulaceous genera (Figure 4B). Such blast results, however, were not observed in other Convolvulaceous taxa whose syntenic sequences of this intergenic spacer always best matched plastomic sequences. Thus, this intergenic spacer between the psaA and ycf3 genes of Jacquemontia could have been mis-assembled with contaminants from endogenous mtDNA, or may be a true integrated PTMT.
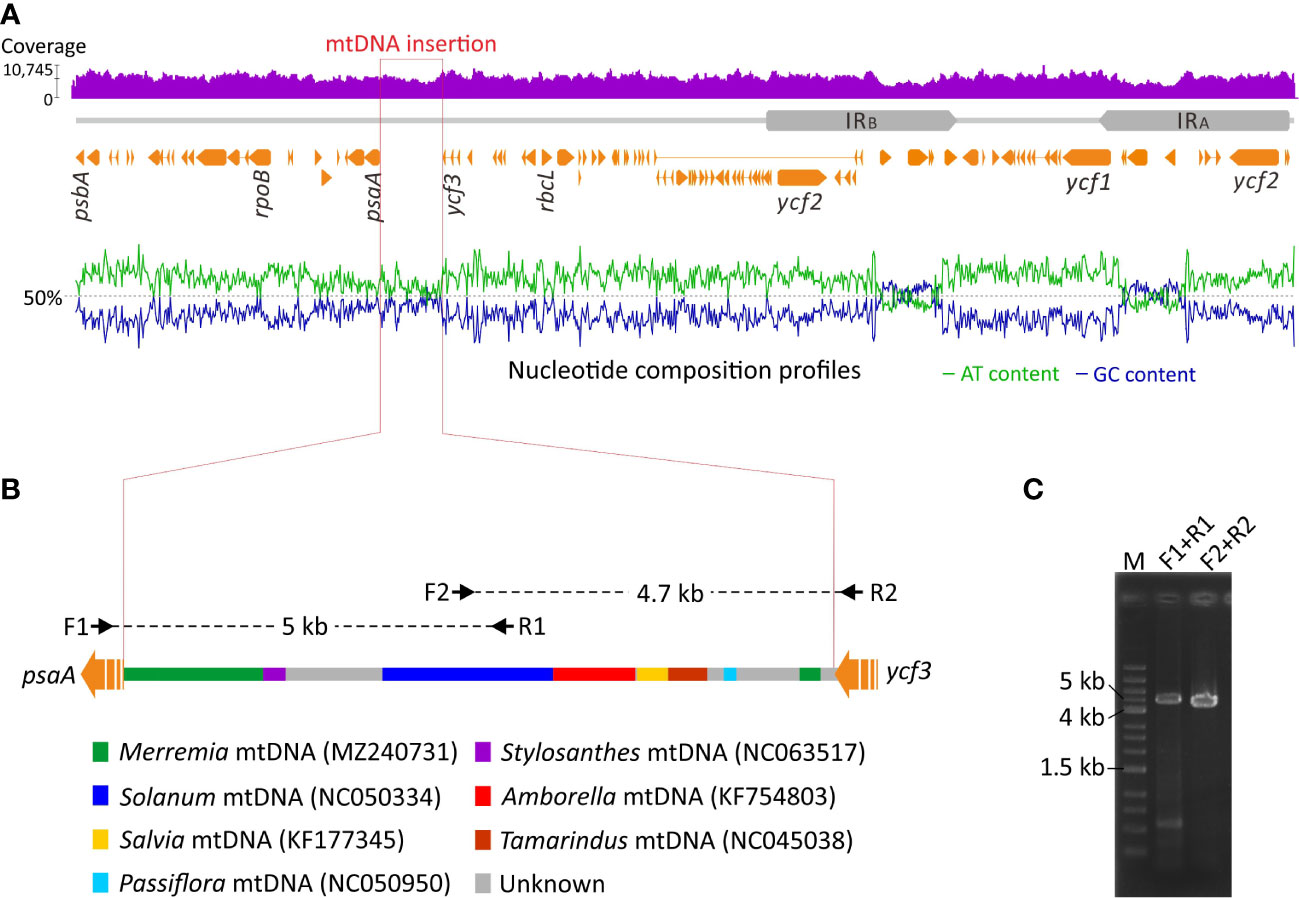
Figure 4 Schematic diagrams of PTMT integration in Jacquemontia. (A) Analyses of the DNA-seq read coverage and nucleotide composition across the whole plastome. (B) Blast results of the integrated PTMT showing a mixture of fragments best matching mtDNAs of some taxa with color codes given below. Black arrows are PCR primers designed in this study. (C) PCR verification of the integrated PTMT.
To verify the accuracy of our assembly, DNA-seq reads were mapped to the Jacquemontia plastome. Given the remarkably different copy numbers of plastomes compared to mitogenomes in plant cells (Johnston, 2019), we would expect drastic decreases of the read coverage in mis-assembled regions. However, our mapping analysis yielded a similar degree of the read coverage across the entire plastome (Figure 4A), thus rejecting the assumption that our assembly had endogenous mtDNA contamination. Both NOVOPlasty (Dierckxsens et al., 2017) and GetOrganelle (Jin et al., 2020) assemblers, though based on different assembly strategies, obtained identical results that again confirmed the presence of a PTMT in Jacquemontia. We also designed specific primers to amplify the region across the intergenic spacer between psaA and ycf3. Our PCR successfully yielded fragments of the expected intergenic size (9.1 Kb; Figure 4C). Collectively, our data demonstrate an unprecedented case of PTMTs in Convolvulaceae.
Short, domain-lacking, but repeat-containing introns in duplicated rpl16 genes
As mentioned above, Dichondra has duplicated rpl16 in its IRs, resulting in three rpl16 copies in our assembly. One of these copies is located at the typical locus between rpl14 and rps3 in the LSC (hereafter designated as rpl16-LSC), while the others are located between trnN and ycf1 in the IRs (hereafter designated as rpl16-IR). The two types of the rpl16 copies differ in intron composition but share nearly identical coding sequences (% identical sites between different copies = 99.1). The intron of rpl16-LSC is 966 bp long with I−VI domains forming a typical group II intron structure (Figure 5A). In contrast, the rpl16-IR intron is relatively short (429 bp long) and completely lacks domains II−VI and EBS1 and EBS2 motifs in domain I. It is noteworthy that we detected a pair of direct repeats ─ one copy (205 bp long) in the region encompassing the exon 1 and its upstream region and the other (201 bp) at the 3’-end of the intron (Figure 5A). Their pairwise sequence identity is 97.6%.
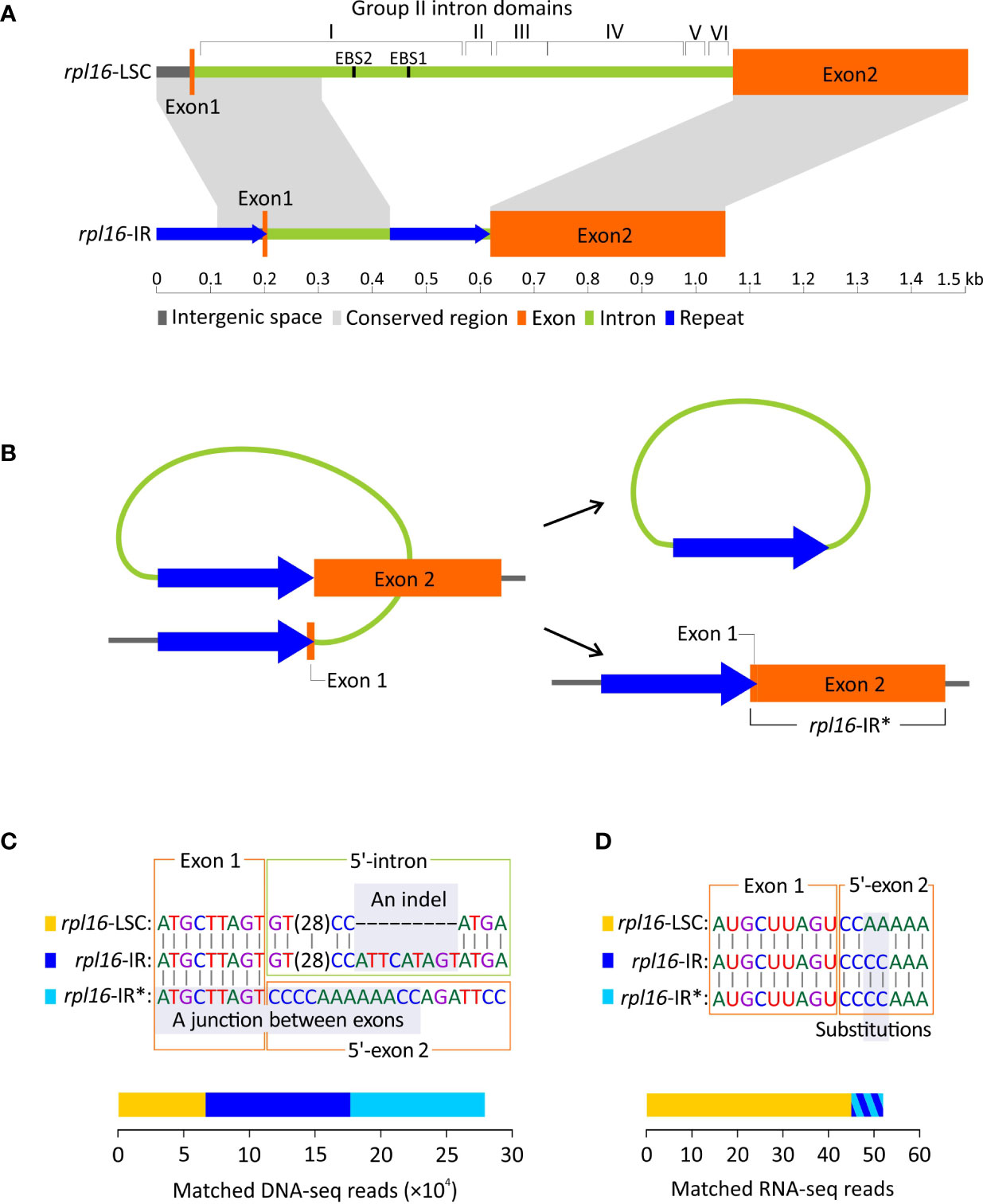
Figure 5 Duplication of rpl16 in Dichondra. (A) Comparisons of rpl16 copies residing in the LSC (rpl16-LSC) and IR (rpl16-IR) regions. The rpl16-LSC harbors a typical group II intron containing I−VI domains, while the rpl16-IR is short and degraded with only partial sequences of the domain I (B) A hypothetical scenario for loss of the rpl16-IR intron via homologous recombination between direct repeats. The intron-lacking rpl16-IR is therefore designated as rpl16-IR*. (C) DNA-seq mapping analyses showing the coexistence of three rpl16 copies (color-coded) in Dichondra. Regions enabling copy discrimination are highlighted with grey and indicated with the terms “An indel” and “A junction between exons”. Stacked bars are the number of reads matching specific copies. (D) RNA-seq reads mapped to putative transcripts of the three rpl16 copies after splicing. The bars for rpl16-IR and rpl16-IR* are added together because they are not distinguishable by highlighted substitutions.
The above-mentioned short, domain-lacking, but repeat-containing intron prompted us to ask two questions: (1) Is the intron in rpl16-IR still actively splicing? Specifically, we want to know if formation of an open reading frame is achieved after intron splicing. (2) Does homologous recombination between repeats occur and result in an intron-lacking rpl16-IR copy in Dichondra (Figure 5B)? The intron-lacking copy (hereafter designated as rpl16-IR*) is separated from other two genomic rpl16 copies by the junction of two exons (Figure 5C). Furthermore, the rpl16-LSC and rpl16-IR copies are distinguishable from each other by a 9 bp long indel at the 5’-end of their introns (Figure 5C). Our mapping analyses reveal 6,665, 10,228, and 11,032 DNA-seq reads that match rpl16-LSC, rpl16-IR, and rpl16-IR*, respectively (Figure 5C; Figure S3). These results confirm that (1) at least three different rpl16 copies coexist in the Dichondra plastomes, (2) the genomic copy numbers of rpl16-IR and rpl16-IR* are approximately equal but two-fold higher than rpl16-LSC, consistent with the fact that the former two are in the IR, while the latter is single in the LSC, and (3) the equal numbers of matched reads between rpl16-IR and rpl16-IR* copies suggest effective repeat-mediated recombination. The coexistence and copy number divergence were further verified by semi-quantitative PCR that shows specific duplicate amplicons appearing earlier than the single-copy locus (Figure S3).
To examine splicing capability, RNA-seq reads were mapped to putative mature transcripts of the three rpl16 copies (Figure 5D). We then detected 45 RNA reads that not only spanned exon junctions but also contained substitutions specific to rpl16-LSC. Eight RNA reads were found to match both rpl16-IR and rpl16-IR* because the mature transcripts of these two copies are identical, leading to difficulty in identification of which copies contributed to the matched reads. Therefore, it remains uncertain whether the rpl16-IR’s intron is spliced. If it is, the splicing efficiency should be much lower than that in rpl16-LSC.
Discussion
To date, the genus Ipomoea is significantly overrepresented among sequenced morning glory plastomes. This bias holds back advances in understanding the evolution and utility of plastomes across the family. Our study compared plastomes representing nine of the 12 tribes, and provides new insights into phylogeny, plastome structural variation, PTMTs, and mechanisms underlying intron loss in the family.
Monophyletic status of the family, as well as phylogenetic placements of several of its tribes and genera, are still subject to debate. We adopted the classification system modified by Stefanovi´c et al. (2003), who recognized twelve tribes within Convolvulaceae rather than nine in Austin’s system (1998). Notably, Austin (1998) raised the parasitic genus Cuscuta to the status of a monogeneric family (Cuscutaceae), but Stefanovi´c et al. (2003) considered it a monogeneric tribe (Cuscuteae). The present phylogenetic analyses reveal that Cuscuta is nested within Convolvulaceae, in line with the viewpoint that Convolvulaceae, including Cuscuta, is monophyletic (Stefanović et al., 2002; Stefanović and Olmstead, 2004; Lin et al., 2022; Simões et al., 2022). This monophyly is further reinforced by a common absence of the rpl2 intron unique to Convolvulaceae among the subclass Asterideae (Stefanović et al., 2002; Lin et al., 2022; this study). However, our trees placed Cuscuta as a sister to other Convolvulaceous taxa except for the two genera Dinetus and Erycibe (Figure 1). This placement has never been reported before, even in the most recent plastid phylogenomic analysis by Lin et al. (2022). As our sampled taxa are denser and broader than those (25 taxa across eight tribes) in Lin et al. (2022), we conclude that the phylogenetic position of Cuscuta is unsettled and that expanded taxonomic sampling will be required to resolve Cuscuta’s evolutionary status.
Our analyses yielded strong support for a close relationship between Ipomoeeae s.l. and Merremia (a genus in the tribe Merremieae), confirming non-monophyly of the tribe Merremieae and calling for its revision. Furthermore, both of our ML (BS = 100%) and BI (PP = 1) trees resolve the clade comprising four genera: Dichondra, Jacquemontia, Cressa, and Evolvulus. Taxa in this clade, except Jacquemontia, possess deeply divided styles, the so-called “bifid style” clade or the subfamily Dicranostyloideae (Stefanović et al., 2002). Recently, the placement of Jacquemontia within Dicranostyloideae was recovered with full support in coalescent trees based on multiple nuclear loci (Simões et al., 2022). As a result, the placement of Jacquemontia within Dicranostyloideae is confirmed by a variety of methods and molecular data sets. However, the position of the genus Erycibe appears to be discordant among plastid, mitochondrial, and nuclear trees. For example, a sisterhood between Dinetus and Erycibe is weakly supported (BS=69%; PP = 0.994) in our trees (Figure 1), but such relationship was not observed in the trees inferred from mitochondrial CDSs and nuclear 45S (Lin et al., 2022). Simões et al. (2022) noted shifts of the Erycibe position with and without incorporation of Cuscuta in their analytical dataset, implicating the influence of taxonomic sampling. Collectively, our results affirm some but not all relationships in Convolvulaceae. The unresolved (or weakly supported) relationships are likely due to highly variable rates in nucleotide substitutions among taxa as demonstrated in Figure 2.
We identified numerous plastomic rearrangements in the LSC and IR regions (Figure 3). However, more than half (7/13 = 0.54) of the LSC rearrangements occurred in the subfamily Dicranostyloideae, indicating an asymmetric distribution of plastomic rearrangements across Convolvulaceae. Taxa in Dicranostyloideae also exhibit variable IRs and accelerated nucleotide substitution rates (Figure 2), implying an association between plastomic rearrangements and nucleotide substitution rates. This association likely results from aberrant mutation rates and/or improper DNA repair systems proposed for some Geraniaceae lineages whose plastomic rearrangements and nucleotide substitution rates are co-elevated (Guisinger et al., 2011; Blazier et al., 2016). IRs might act to stabilize plastomes (Palmer and Thompson, 1982). This hypothetical function should be ineffective in Convolvulaceae. Blazier et al. (2016) suggested that plastome stabilization by IRs is evident when repeat content is low. Indeed, we have detected a pair of direct repeats and shown its capability of efficiently triggering recombination (Figure 5). We propose that multiple independent rounds of IR contraction and expansion have shaped the evolution of gene content in the IRs of Convolvulaceae. Illegitimate recombination between IR and single copy regions might have led to IR contraction and expansion (Goulding et al., 1996). Although not involved in gene losses, IR size fluctuation has resulted in copy-number variation of some genes in some Convolvulaceae plastomes (Figure 3). The low expression level we observed in rpl16-IR and rpl16-IR* implies down-regulation of these duplicates (Figure 5). Whether expression regulation also acts on other duplicate IR resident loci to prevent (or mediate) dosage effects remains to be elucidated.
In this study we report the first PTMT case in Convolvulaceae (Figure 4). This PTMT replaces the plastid intergenic sequences between psaA and ycf3 in Jacquemontia. Replacements of plastid sequences with PTMTs were previously documented in Daucus (Iorizzo et al., 2012a; Iorizzo et al., 2012b), Pariana (Ma et al., 2015), and Convallaria (Raman et al., 2021). In Jacquemontia, the PTMT is inserted upstream of the psaA operon consisting of psaA, psaB, and rps14 (Meng et al., 1988). This insertion has interrupted the transcription of the psaA operon driven by the original plastid promoters (Figure 5). Therefore, the expression machinery of the psaA operon must be reshaped after the PTMT insertion to ensure regular transcription of psaA and psaB, for their products are essential for the assembly of the PSI complex. In Daucus, replacements of plastid promoters with PTMTs were hypothesized to alter the expression pattern of the corresponding genes (Iorizzo et al., 2012a). A recent study suggested that mis-regulation of the plastid psbB operon results in hybrid incompatibility that may ultimately lead to speciation in Oenothera (Zupok et al., 2021). The PTMT we identified in Jacquemontia provides an opportunity to assess the association between PTMT insertions and speciation.
A variety of mechanisms can underlie PTMT insertions. The presence of short repeats and transcriptase-like sequences leads to the conclusion that the PTMT in Daucus was a consequence of transposition events governed by non-LTR retrotransposons (Iorizzo et al., 2012a; Iorizzo et al., 2012b). In Asclepias, migrations of mtDNA to plastids were achieved by homologous recombination between mitochondrial plastid-derived DNA and plastid sequences (Straub et al., 2013). Previously, deletions adjacent to insertion sites were considered to be signatures of homologous recombination facilitating PTMT integration in Anacardium (Rabah et al., 2017). Nevertheless, neither transposable elements nor deletions were detected in the PTMT or its flanking regions in Jacquemontia. Surprisingly, our blast searches revealed that the Jacquemontia PTMT contains several regions that best match mitogenomes of diverse species across five taxonomic orders (Amborellales, Fabales, Lamiales, Malpighiales, and Solanales; Figure 4). It is implausible to assume multiple rounds of horizontal gene transfer (HGT) from different origins into the same plastid intergenic region. Several studies have shown that PTMTs preserve the ancestral state of their mitochondrial donors (e.g., Iorizzo et al., 2012a; Iorizzo et al., 2012b; Straub et al., 2013; Ma et al., 2015). Unfortunately, we did not obtain any matches when the PTMT of Jacquemontia was used to blast against the mtDNA scaffolds we generated from conspecific PE reads. It is known that land plant mitogenomes can acquire abundant foreign DNA from intercellular gene transfer (IGT), HGT, or both (Knoop, 2004; Alverson et al., 2010; Rice et al., 2013; Park et al., 2014). Accordingly, we propose a straightforward scenario that the Jacquemontia PTMT has retained the ancestral state of its mitochondrial donor that once harbored an array of foreign DNA of different origins but later experienced reduction by purging the sequences homologous to this PTMT.
We found a short group II intron in the rpl16-IR copy (Figure 5). This intron is degraded and likely not functional since it lacks most of the domains, including the domain V for binding catalytic Mg2+ during intron splicing (Lambowitz and Zimmerly, 2011). This degraded intron is flanked by direct repeats capable of triggering homologous recombination to precisely remove it. This repeat-mediated recombination happens with a rate of 0.52 [11,032/(10,228 + 11,032)] conversions per copy and its associated product is transcribable, albeit at a low level. These findings lead us to speculate that these direct repeats might have been selected to complement the degraded intron since the intron is nonfunctional.
Retroprocessing is a well-known mechanism driving intron loss in plant organelles through conversion between retro-transcribed cDNA molecules and their corresponding DNA fragments. This mechanism is especially prominent when transcripts of target genes lack RNA editing in regions adjacent to former splicing sites (Ran et al., 2010; Sloan et al., 2010; Grewe et al., 2011; Cuenca et al., 2016). In addition, HGT and sequential gene conversion have created a chimeric cox2 gene without introns (Hepburn et al., 2012). Here, we provide a different model for intron loss after invasion of direct repeats at appropriate positions in intron-containing genes. This model better interprets intron loss from non-chimeric genes that have abundant RNA editing sites in regions adjacent to the former intron splicing sites.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI, LC729542-LC729560 and the SRA bioproject: PRJNA876072.
Author contributions
S-MC and C-SW conceived and designed the study. C-SW performed experiments and data analyses. C-SW and C-IC collected the plant materials. C-SW and S-MC wrote the manuscript. All the authors checked and approved the final version. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by research grants from National Science and Technology Council, Taiwan (108-2621-B-001-006-) and Biodiversity Research Center, Academia Sinica (AS), Taiwan, and partially from AS-23-23 of AS president office to S-MC.
Acknowledgments
We thank Taipei Botanical Garden for providing some plant materials. We also thank Wayne Y. Lin and two reviewers for their critical reading and constructive comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1061174/full#supplementary-material
Supplementary Figure 1 | Plastid phylogenomics of the tribe Ipomoeeae s.l. The tree details the simplified area depicted in . A 50% majority rule was used to condense the tree topology. Values along branches are bootstrap supports (%) and posterior possibilities for ML and Bayesian inference (BI) trees, respectively. Conflicting topologies are highlighted with grey.
Supplementary Figure 2 | Dating of taxon divergence time during Convolvulaceae evolution. Black circles indicate nodes with constrained ages. Blue bars denote a range of the 95% highest posterior density (HPD).
Supplementary Figure 3 | Read-mapping and Semi-quantitative PCR demonstrating coexistence of the three different rpl16 copies and discrepancies in their copy numbers in Dichondra. Primers, rpl16-LSC-F and rpl16-LSC-R, were designed to amplify the region containing intronic II−IV domains that are lacking in the rpl16-IR copy.
References
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., Palmer, J. D. (2010). Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 27, 1436−1448. doi: 10.1093/molbev/msq029
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). The chloroplast genome sequence of bittersweet (Solanum dulcamara): Plastid genome structure evolution in solanaceae. PloS One 13, e0196069. doi: 10.1371/journal.pone.0196069
Angiosperm Phylogeny Group (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc 181, 1–20. doi: 10.1111/boj.12385
Austin, D. F. (1998). “Parallel and convergent evolution in the convolvulaceae,” in Biodiversity and taxonomy of tropical flowering plants. Eds. Mathews, P., Sivadasan, M. (Calicut: Mentor Books), 201–234.
Banerjee, A., Stefanović, S. (2020). Reconstructing plastome evolution across the phylogenetic backbone of the parasitic plant genus Cuscuta (Convolvulaceae). Biol. J. Linn. Soc Lond. 194, 423–438. doi: 10.1093/botlinnean/boaa056
Blazier, J. C., Jansen, R. K., Mower, J. P., Govindu, M., Zhang, J., Weng, M. L., et al. (2016). Variable presence of the inverted repeat and plastome stability in Erodium. Ann. Bot. 117, 1209–1220. doi: 10.1093/aob/mcw065
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Braukmann, T., Kuzmina, M., Stefanović, S. (2013). Plastid genome evolution across the genus Cuscuta (Convolvulaceae): two clades within subgenus Grammica exhibit extensive gene loss. J. Exp. Bot. 64, 977−989. doi: 10.1093/jxb/ers391
Chan, P. P., Lin, B. Y., Mak, A. J., Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077−9096. doi: 10.1093/nar/gkab688
Cuenca, A., Ross, T. G., Graham, S. W., Barrett, C. F., Davis, J. I., Seberg, O., et al. (2016). Localized retroprocessing as a model of intron loss in the plant mitochondrial genome. Genome Biol. Evol. 8, 2176−2189. doi: 10.1093/gbe/evw148
Dierckxsens, N., Mardulyn, P., Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi: 10.1093/nar/gkw955
Downie, S. R., Olmstead, R. G., Zurawski, G., Soltis, D. E., Soltis, P. S., Watson, J. C., et al. (1991). Six independent losses of the chloroplast DNA rpl2 intron in dicotyledons: molecular and phylogenetic implications. Evolution. 45, 1245−1259. doi: 10.1111/j.1558-5646.1991.tb04390.x
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792−1797. doi: 10.1093/nar/gkh340
Eserman, L. A., Tiley, G. P., Jarret, R. L., Leebens-Mack, J. H., Miller, R. E. (2014). Phylogenetics and diversification of morning glories (tribe ipomoeeae, convolvulaceae) based on whole plastome sequences. Am. J. Bot. 101, 92–103. doi: 10.3732/ajb.1300207
Goulding, S. E., Olmstead, R. G., Morden, C. W., Wolfe, K. H. (1996). Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 252, 195−206. doi: 10.1007/BF02173220
Grewe, F., Herres, S., Viehöver, P., Polsakiewicz, M., Weisshaar, B., Knoop, V. (2011). A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucleic Acids Res. 39, 2890−2902. doi: 10.1093/nar/gkq1227
Guisinger, M. M., Kuehl, J. V., Boore, J. L., Jansen, R. K. (2011). Extreme reconfiguration of plastid genomes in the angiosperm family geraniaceae: rearrangements, repeats, and codon usage. Mol. Biol. Evol. 28, 583–600. doi: 10.1093/molbev/msq229
Hepburn, N. J., Schmidt, D. W., Mower, J. P. (2012). Loss of two introns from the Magnolia tripetala mitochondrial cox2 gene implicates horizontal gene transfer and gene conversion as a novel mechanism of intron loss. Mol. Biol. Evol. 29, 3111−3120. doi: 10.1093/molbev/mss130
Iorizzo, M., Grzebelus, D., Senalik, D., Szklarczyk, M., Spooner, D., Simon, P. (2012a). Against the traffic: The first evidence for mitochondrial DNA transfer into the plastid genome. Mob. Genet. Elements 2, 261–266. doi: 10.4161/mge.23088
Iorizzo, M., Senalik, D., Szklarczyk, M., Grzebelus, D., Spooner, D., Simon, P. (2012b). De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12, 61. doi: 10.1186/1471-2229-12-61
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Johnston, I. G. (2019). Tension and resolution: dynamic, evolving populations of organelle genomes within plant cells. Mol. Plant 12, 764−783. doi: 10.1016/j.molp.2018.11.002
Kelchner, S. A. (2002). Group II introns as phylogenetic tools: structure, function, and evolutionary constraints. Am. J. Bot. 89, 1651−1669. doi: 10.3732/ajb.89.10.1651
Knoop, V. (2004). The mitochondrial DNA of land plants: peculiarities in phylogenetic perspective. Curr. Genet. 46, 123–139. doi: 10.1007/s00294-004-0522-8
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870−1874. doi: 10.1093/molbev/msw054
Lambowitz, A. M., Zimmerly, S. (2011). Group II introns: mobile ribozymes that invade DNA. cold spring harb. Perspect. Biol. 3, a003616. doi: 10.1101/cshperspect.a003616
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772−773. doi: 10.1093/molbev/msw260
Laux, M., Oliveira, R. R. M., Vasconcelos, S., Pires, E. S., Lima, T. G. L., Pastore, M., et al. (2022). New plastomes of eight ipomoea species and four putative hybrids from Eastern Amazon. PloS One 17, e0265449. doi: 10.1371/journal.pone.0265449
Lin, Y., Li, P., Zhang, Y., Akhter, D., Pan, R., Fu, Z., et al. (2022). Unprecedented organelle genomic variations in morning glories reveal independent evolutionary scenarios of parasitic plants and the diversification of plant mitochondrial complexes. BMC Biol. 20, 49. doi: 10.1186/s12915-022-01250-1
Ma, P. F., Zhang, Y. X., Guo, Z. H., Li, D. Z. (2015). Evidence for horizontal transfer of mitochondrial DNA to the plastid genome in a bamboo genus. Sci. Rep. 5, 11608. doi: 10.1038/srep11608
Meng, B. Y., Tanaka, M., Wakasugi, T., Ohme, M., Shinozaki, K., Sugiura, M. (1988). Cotranscription of the genes encoding two P700 chlorophyll a apoproteins with the gene for ribosomal protein CS14: determination of the transcriptional initiation site by in vitro capping. Curr. Genet. 14, 395−400. doi: 10.1007/BF00419998
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530−1534. doi: 10.1093/molbev/msaa015
Palmer, J. D., Thompson, W. F.. (1982). Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell. 29, 537‒550. doi: 10.1016/0092-8674(82)90170-2
Park, S., Ruhlman, T. A., Sabir, J. S., Mutwakil, M. H., Baeshen, M. N., Sabir, M. J., et al. (2014). Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics 15, 405. doi: 10.1186/1471-2164-15-405
Park, I., Yang, S., Kim, W. J., Noh, P., Lee, H. O., Moon, B. C. (2018). The complete chloroplast genomes of six ipomoea species and indel marker development for the discrimination of authentic pharbitidis semen (seeds of I. nil or I. purpurea). Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00965
Rabah, S. O., Lee, C., Hajrah, N. H., Makki, R. M., Alharby, H. F., Alhebshi, A. M. (2017). Plastome sequencing of ten nonmodel crop species uncovers a large insertion of mitochondrial DNA in cashew. Plant Genome. 10. doi: 10.3835/plantgenome2017.03.0020
Raman, G., Lee, E. M., Park, S. (2021). Intracellular DNA transfer events restricted to the genus Convallaria within the asparagaceae family: Possible mechanisms and potential as genetic markers for biographical studies. Genomics 113, 2906−2918. doi: 10.1016/j.ygeno.2021.06.033
Raman, G., Park, S., Lee, E. M., Park, S. (2019). Evidence of mitochondrial DNA in the chloroplast genome of Convallaria keiskei and its subsequent evolution in the asparagales. Sci. Rep. 9, 5028. doi: 10.1038/s41598-019-41377-w
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., Suchard, M. A.. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901‒904. doi: 10.1093/sysbio/syy032
Ran, J. H., Gao, H., Wang, X. Q. (2010). Fast evolution of the retroprocessed mitochondrial rps3 gene in conifer II and further evidence for the phylogeny of gymnosperms. Mol. Phylogenet. Evol. 54, 136−149. doi: 10.1016/j.ympev.2009.09.011
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science. 342, 1468−1473. doi: 10.1126/science.1246275
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539−542. doi: 10.1093/sysbio/sys029
Simões, A. R. G., Eserman, L. A., Zuntini, A. R., Chatrou, L. W., Utteridge, T. M. A., Maurin, O., et al. (2022). A bird's eye view of the systematics of convolvulaceae: novel insights from nuclear genomic data. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.889988
Sloan, D. B., MacQueen, A. H., Alverson, A. J., Palmer, J. D., Taylor, D. R. (2010). Extensive loss of RNA editing sites in rapidly evolving Silene mitochondrial genomes: selection vs. retroprocessing as the driving force. Genetics 185, 1369−80. doi: 10.1534/genetics.110.118000
Smith, D. R. (2011). Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 3, 743–748. doi: 10.1093/gbe/evr068
Stefanović, S., Austin, D. F., Olmstead, R. G. (2003). Classification of convolvulaceae: a phylogenetic approach. Syst. Bot. 28, 791–806. doi: 10.1043/02-45.1
Stefanović, S., Krueger, L., Olmstead, R. G. (2002). Monophyly of the convolvulaceae and circumscription of their major lineages based on DNA sequences of multiple chloroplast loci. Am. J. Bot. 89, 1510–1522. doi: 10.3732/ajb.89.9.1510
Stefanović, S., Olmstead, R. G. (2004). Testing the phylogenetic position of a parasitic plant (Cuscuta, convolvulaceae, asteridae): Bayesian inference and the parametric bootstrap on data drawn from three genomes. Syst. Biol. 53, 384−399. doi: 10.1080/10635150490445896
Stewart, C. N., Jr., Via, L. E. (1993). A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. Biotechniques 14, 748–750.
Straub, S. C., Cronn, R. C., Edwards, C., Fishbein, M., Liston, A. (2013). Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (Apocynaceae). Genome Biol. Evol. 5, 1872–1885. doi: 10.1093/gbe/evt140
Sun, J., Dong, X., Cao, Q., Xu, T., Zhu, M., Sun, J., et al. (2019). A systematic comparison of eight new plastome sequences from ipomoea l. Peer J. 7, e6563. doi: 10.7717/peerj.6563
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8, 36. doi: 10.1186/1471-2148-8-36
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273−297. doi: 10.1007/s11103-011-9762-4
Wu, C. S., Sudianto, E., Chaw, S. M. (2021). Tight association of genome rearrangements with gene expression in conifer plastomes. BMC Plant Biol. 21, 33. doi: 10.1186/s12870-020-02809-2
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zhu, A., Guo, W., Gupta, S., Fan, W., Mower, J. P. (2016). Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol. 209, 1747−1756. doi: 10.1111/nph.13743
Keywords: convolvulaceae, plastome, phylogenomics, gene transfer, intron loss
Citation: Wu C-S, Chen C-I and Chaw S-M (2022) Plastid phylogenomics and plastome evolution in the morning glory family (Convolvulaceae). Front. Plant Sci. 13:1061174. doi: 10.3389/fpls.2022.1061174
Received: 04 October 2022; Accepted: 23 November 2022;
Published: 20 December 2022.
Edited by:
Wenpan Dong, Beijing Forestry University, ChinaReviewed by:
Zhiqiang Wu, Chinese Academy of Agricultural Sciences, ChinaXiao-Jian Qu, Shandong Normal University, China
Copyright © 2022 Wu, Chen and Chaw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-Miaw Chaw, c21jaGF3QHNpbmljYS5lZHUudHc=